Har-P, a short P-element variant, weaponizes P-transposase to severely impair Drosophila development
- Boston University School of Medicine, Boston University, United States
- Brandeis University, United States
- Genome Science Institute, Boston University School of Medicine, United States
Abstract
Without transposon-silencing Piwi-interacting RNAs (piRNAs), transposition causes an ovarian atrophy syndrome in Drosophila called gonadal dysgenesis (GD). Harwich (Har) strains with P-elements cause severe GD in F1 daughters when Har fathers mate with mothers lacking P-element-piRNAs (i.e. ISO1 strain). To address the mystery of why Har induces severe GD, we bred hybrid Drosophila with Har genomic fragments into the ISO1 background to create HISR-D or HISR-N lines that still cause Dysgenesis or are Non-dysgenic, respectively. In these lines, we discovered a highly truncated P-element variant we named ‘Har-P’ as the most frequent de novo insertion. Although HISR-D lines still contain full-length P-elements, HISR-N lines lost functional P-transposase but retained Har-P’s that when crossed back to P-transposase restores GD induction. Finally, we uncovered P-element-piRNA-directed repression on Har-P’s transmitted paternally to suppress somatic transposition. The Drosophila short Har-P’s and full-length P-elements relationship parallels the MITEs/DNA-transposase in plants and SINEs/LINEs in mammals.
eLife digest
DNA provides the instructions needed for life, a role that relies on it being a very stable and organized molecule. However, some sections of DNA are able to move from one place in the genome to another. When these “mobile genetic elements” move they may disrupt other genes and cause disease. For example, a mobile section of DNA known as the P-element causes a condition called gonadal dysgenesis in female fruit flies, leading to infertility.
Only certain strains of fruit flies carry P-elements and the severity of gonadal dysgenesis in their daughters varies. For example, when male fruit flies of a strain known as Harwich (or Har for short) is crossed with female fruit flies that do not contain P-elements, all of their daughters develop severe gonadal dysgenesis and are infertile. However, if the cross is done the other way around, and female Har flies mate with males that do not contain P-elements, the daughters are fertile because the Har mothers provide their daughters with protective molecules that silence the P-elements. But it was a mystery as to why the P-elements from the Har fathers always caused such severe gonadal dysgenesis in all the daughters.
Here, Srivastav et al. bred fruit flies to create offspring that had different pieces of Har DNA in a genetic background that was normally free from P-elements; they then analyzed the ‘hybrid’ offspring to identify which pieces of the Har genome caused gonadal dysgenesis in the daughter flies. These experiments showed that Har flies possess a very short variant of the P-element (named “Har-P”) that is more mobile than other variants. However, the Har-P variants still depended on an enzyme known as P-transposase encoded by the full-length P-elements to move around the genome. Further experiments showed that other strains of fruit flies that cause severe gonadal dysgenesis also had very short P-element variants that were almost identical to Har-P.
These findings may explain why Har and some other strains of fruit flies drive severe gonadal dysgenesis. In the future, it may be possible to transfer P-transposase and Har-P into mosquitoes, ticks and other biting insects to make them infertile and help reduce the spread of certain diseases in humans.
Introduction
The sterility syndrome of ‘P-M’ hybrid dysgenesis in Drosophila melanogaster (Engels and Preston, 1979; Kidwell et al., 1977) is due to uncontrolled P-element transposition that damages ovarian development and induces female sterility (Bingham et al., 1982 and reviewed in Kelleher, 2016). This gonadal dysgenesis (GD) phenotype occurs in hybrid F1 daughters whose paternal genome comes from a father possessing active P-elements (a ‘P’ strain) and a maternal genome unable to express P-element piRNAs (an ‘M’ strain) (Brennecke et al., 2008; Khurana et al., 2011). The fascinating nature of this genetic syndrome is complete fertility in daughters from the reciprocal cross because the mother possessing active P-elements contribute P-element-derived Piwi-interacting RNAs (piRNAs) to silence these transposons in daughters (Brennecke et al., 2008; Khurana et al., 2011). Thus, despite identical genetic makeup in daughters between the reciprocal crosses, the epigenetic maternal transmission of transposon repression by piRNAs starkly defines female fertility (illustrated in Figure 1A).
Figure 1

No correlation between paternally-induced gonadal dysgenesis (GD) rate and P-element copy number.
(A) Illustration of the P-element-induced GD phenomenon, where two different types of crosses with one parent lacking P-elements while the other parents containing P-elements can result in genetically identical daughters having very different gonadal phenotypes. (B) GD rates from paternal genome strains mated with ISO1 females; at least 100 F1 daughters per cross were assayed. (C) Genomic quantitative PCR assessment of P-element load of strains, normalized to Har at 100%. (D) Scatterplot comparing TIDAL counts of P-element insertions to the GD rate reflects the lack of correlation.
-
Figure 1—source data 1
Spreadsheets with the tabulation of gonadal dysgenesis assays and raw values from the qPCR experiments.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig1-data1-v1.xlsx
Between fertility and complete sterility lies a spectrum of GD induction variation amongst different strain crosses that may be attributed to differential P-element copy numbers in different strain genomes (Anxolabéhère et al., 1985; Bergman et al., 2017; Biémont et al., 1990; Bingham et al., 1982; Boussy et al., 1988; Kidwell et al., 1981; Ronsseray et al., 1989; Srivastav and Kelleher, 2017; Yoshitake et al., 2018), and capacity to generate piRNAs (Wakisaka et al., 2017). In addition, there are many non-autonomous P-element variants that can be mobilized by P-transposases, including very short elements from the pi[2] strain (Bingham et al., 1982; O'Hare and Rubin, 1983) that actually assemble in vitro with the P-transposase tetramer complex >100X more efficiently than the full-length P-element (Tang et al., 2007). However, many earlier studies perceived truncated variants such as the ‘KP2’ variants as inhibitors of transposition by acting to titrate P-transposase since P-element piRNAs were unknown at the time (Black et al., 1987; Gloor et al., 1993; Jackson et al., 1988; Robertson and Engels, 1989; Simmons et al., 2002a). Most studies of GD were typically calibrated with a strong paternal inducer ‘P’-strain like Harwich (Har) or pi[2] when mated with ‘M’ strain females lacking P-elements (Bingham et al., 1982; Brennecke et al., 2008; Kidwell et al., 1977; Rubin et al., 1982). Despite over 40 years of study, what defines a strong paternal inducer of GD has remained a mystery.
Although P-element copy numbers in Har are significant (120–140 copies; Khurana et al., 2011), strains with even more copies like OreR-MOD do not induce GD whereas other strong inducer strains that have >75% fewer P-element copies than Har can also trigger complete GD (Figure 1B and C). Thus, there is a lack of correlation between P-element copy number and GD induction (Figure 1D) that we and others have previously observed (Bergman et al., 2017; Ronsseray et al., 1989; Srivastav and Kelleher, 2017). Since P-element copy numbers do not explain GD severity, we hypothesized that a special P-element variant or insertion locus might underlie the strong GD phenotype in certain strong ‘P’ strains like Har. To discover this P-element variant, we undertook a reductionist approach to find specific P-element variant(s) required for GD induction that revealed unexpectedly a short variant from the Har strain that may act together with the full-length P-transposase to drive strong GD.
Results
GD retention and loss in hybrid Drosophila lines with reduced P-element copy numbers
To genetically isolate the causative transposon strongly inducing GD and facilitate discovery by whole genome sequencing (WGS), we generated hybrid lines where only a minor fraction of the Har genome is within the background of the ISO1 reference genome sequence. We first conducted several fertility-permissive backcrosses between female Har and male ISO1, selecting hybrid progeny that propagated a red-eye phenotype which we attributed to the ‘red’ eyes due to Har alleles replacing the cn, bw, sp, alleles on Chromosome 2R (Chr2R) of ISO1 (Figure 2A-abridged scheme, Figure 2—figure supplement 1-detailed scheme). We then performed an initial GD validation screen with many vials of individual hybrid males crossed to ISO1 females and selecting for lines that caused 100% GD from this cross. Lines were propagated with additional self-crosses and further in-bred with single-sibling pairs. We then subjected multiple independent Har-ISO1-Selfed-Red (HISR) lines to a second GD assay. Finally, we conducted qPCR to identify the lines with the greatest reduction of P-element copy numbers (Figure 2B) and settled on four lines each that either retained severe paternally-induced GD (HISR-D) or had lost this capacity (HISR-N) (Figure 2C).
Figure 2 with 2 supplements see all
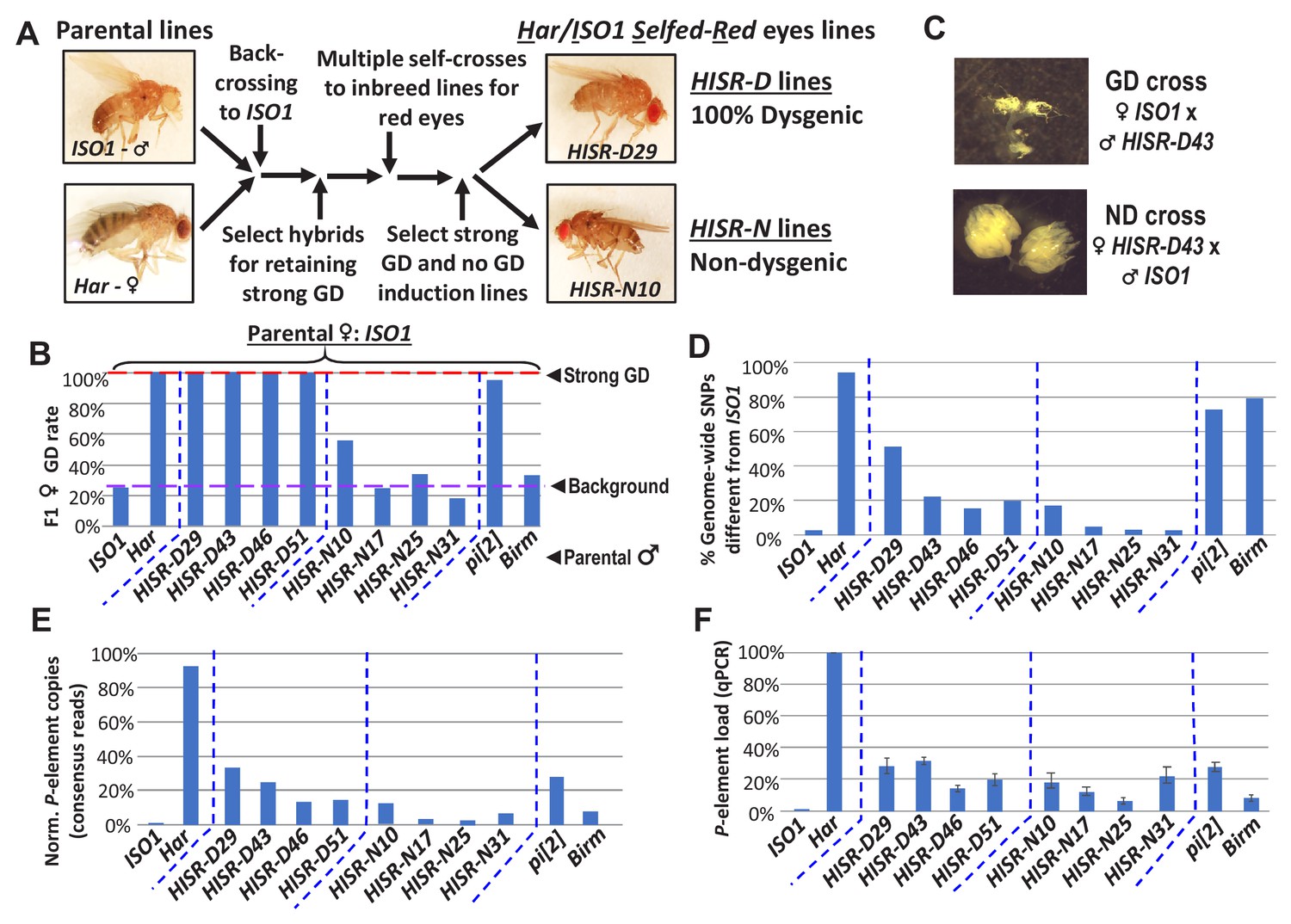
HISR lines retaining or losing strong gonadal dysgenesis (GD) induction.
(A) Abridged scheme for generating hybrid lines retaining a small fraction of the Har genome in the ISO1 background. Full scheme is Figure 2—figure supplement 1. (B) GD rates from paternal genome lines and strains mated with ISO1 females. (C) Ovarian atrophy phenotype only observed from paternal induction of GD. (D) Genome-wide single nucleotide polymorphism profile differences distinct from ISO1 genome. (E) Normalized counts of P-element copies by consensus read mapping of genomic libraries. (F) qPCR assessment of total genomic P-element load.
-
Figure 2—source data 1
Spreadsheets with the tabulation of gonadal dysgenesis assays, quantitation of genomewide SNP profiles, calculations of P-element genomic loads, and raw values from the qPCR experiments.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig2-data1-v1.xlsx
Genomic PCR genotyping of deletion loci of Har compared to ISO1 in HISR lines indicated that these lines carried mostly ISO1 genomes (Figure 2—figure supplement 2). Therefore, we performed WGS of the parental Har and ISO1 strains, the 8 HISR lines, and the pi[2] and Birmingham (Birm) strains, two classic strains with similar numbers of P-elements but diametric capacity to induce GD (Engels et al., 1987; Simmons et al., 1987). Single-nucleotide polymorphism (SNP) profiles of HISR lines confirmed that only a small percentage of the Har genome was retained in mostly an ISO1 background (Figure 2D). Quantification of P-element copies from WGS with the TIDAL program (Transposon Insertion and Depletion AnaLyzer, Figure 2E) (Rahman et al., 2015) was also consistent with qPCR measurements (Figure 2F).
HISR-D lines produce similar levels of P-element piRNAs as the parental Har strain
To determine how substantial reduction in P-element copy numbers in HISR lines affected P-element-directed piRNA production, we generated and sequenced highly-consistent ovarian small RNA libraries (Figure 3A) and confirmed the expected presence and absence of P-element piRNAs in Har and ISO1 ovaries, respectively (Figure 3B). Surprisingly, there were similar-to-increased levels of P-element piRNAs between HISR-D and Har strains, whereas amongst the HISR-N lines, only HISR-N10 retained P-element piRNAs (Figure 3C). Our own mapping analysis indicated a common 3' end antisense bias of P-element piRNAs that we also confirmed with an independent piRNA analysis pipeline (Han et al., 2015). These mapping patterns are consistent with piRNAs silencing transposons and suppressing hybrid GD (Brennecke et al., 2008; Erwin et al., 2015; Khurana et al., 2011) as well as correlating with all the HISR-D’s and HISR-N10’s immunity to strong GD induction when these females are mated to Har males (Figure 3D).
Figure 3 with 2 supplements see all
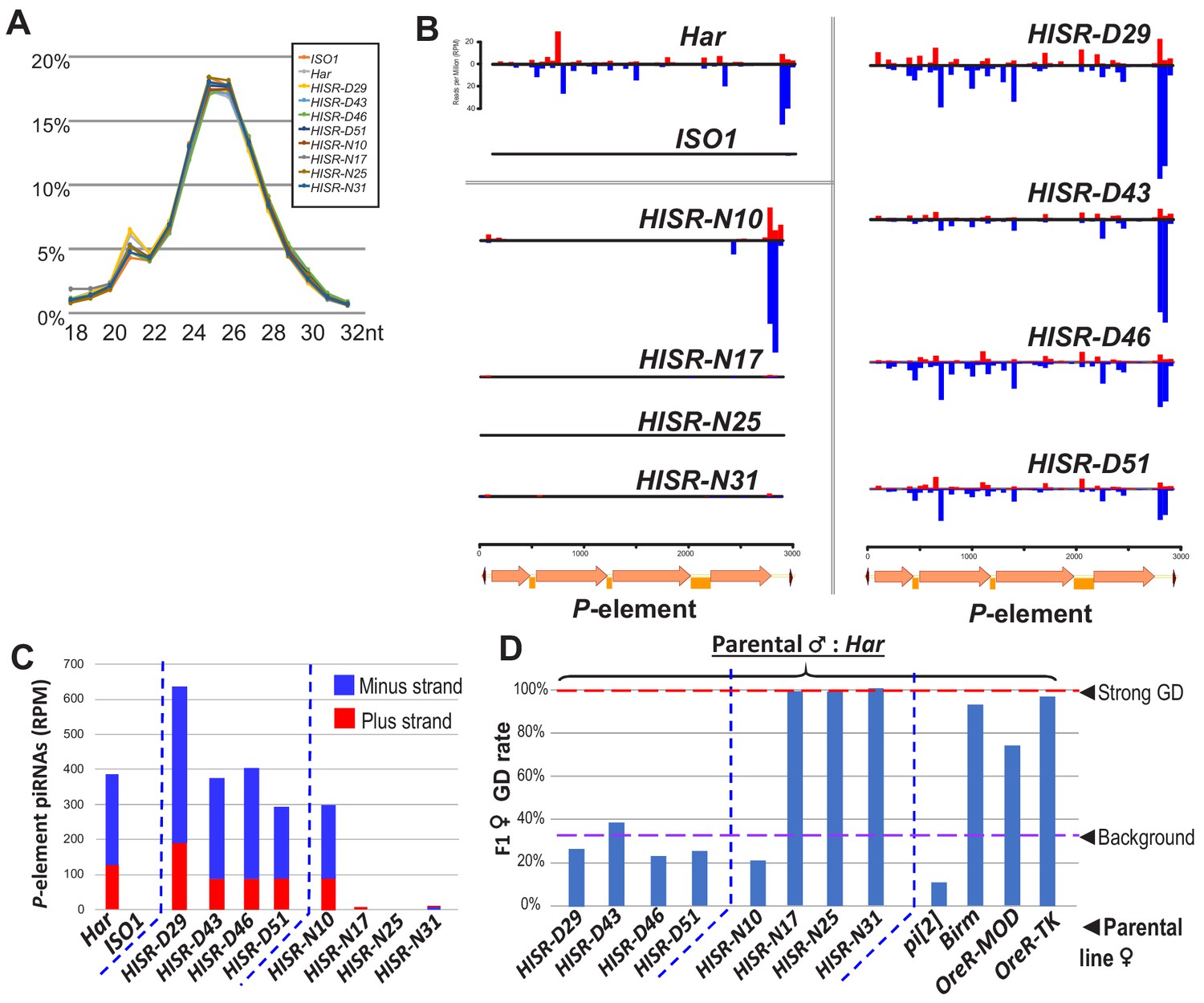
P-element directed piRNA production in HISR strains ovaries.
(A) Nearly identical size distributions of small RNAs from parental and HISR ovaries. (B) P-element piRNAs coverage plots and (C) quantitation of the P-element piRNAs mapping to plus and minus strands, in reads per million (RPM). (D) Assays for repression of P-element induced GD for HISR strains (N ≥ 100 females) are a good proxy for production of piRNAs silencing P-elements.
-
Figure 3—source data 1
Spreadsheets with the calculations for P-element piRNA coverage and tabulation of gonadal dysgenesis assays.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig3-data1-v1.xlsx
Additional piRNAs broadly cover the full length of P-element in Har and HISR-D lines (Figure 3B-top and right), but the notable depletion of internal P-element piRNAs in HISR-N10 (Figure 3B-middle left) prompted us to conjecture which of its 23 TIDAL-mapped P-elements might be stimulating this novel piRNA pattern. We only found one euchromatic P-element insertion in HISR-N10 that specifically coincided with an increase of local piRNAs (Figure 3—figure supplement 1A). This P-element inserted into the 5' UTR of DIP1, adjacent to the enhancer and promoter region of Flamenco, the major piRNA cluster located in a pericentromeric region of the X-chromosome (Brennecke et al., 2007). However, when we selected just the HISR-N10 X-chromosome balanced with the FM7a balancer chromosome (Figure 3—figure supplement 1B), this X-chromosome locus did not generate enough P-element piRNAs to provide full GD immunity. It is possible for additional P-elements to have inserted into major piRNA cluster loci like 42AB, Flamenco and TAS-regions as part of the endogenizing process (Khurana et al., 2011; Moon et al., 2018), but the intractable repetitiveness of piRNA cluster regions prevents bioinformatic programs from pinpointing P-element insertions in these regions. Interestingly, all the P-element piRNAs detected in HISR-N10, -D29, and parental Har appeared to be expressed in the germline due to the detection of clear ping-pong piRNA biogenesis signatures (Figure 3—figure supplement 2). Finally, the P-element piRNA patterns in HISR-N10 can be explained by the abundant P-element variant that will be discussed below.
Dispersed P-element landscapes indicate de novo transposition in HISR lines
The selection for ‘red’ eyes of Har alleles in HISR lines should have replaced the cn, bw, sp, alleles on Chromosome 2R (Chr2R) of ISO1, therefore we had hoped that WGS of HISR line genomes might to point to a specific set of P-elements responsible for inducing strong GD. Unexpectedly, the P-element insertions were not confined to Chr2R, but rather were dispersed across the entire genomes of all HISR lines (Figure 4A), seemingly defying the genomic PCR genotyping and WGS-SNP profiling that indicated sufficient backcrossing to favor mostly the ISO1 genetic background (Figure 4—figure supplement 1).
Figure 4 with 1 supplement see all

P-elements are mobilized de novo during generation of HISR lines.
(A) TIDAL program counts of novel P-element insertions, left Y-axis and colored bars. Right Y-axis, black dots and dashed line are the total distinct transposon insertions in the unique-mapping portion of genome. (B) Lineage analysis of the Har P-elements retained in the HISR lines, colored by the major chromosomal segments. (C) Comparison of shared P-elements between Har and HISR lines, with total number of P-elements called by TIDAL in the top row and first column. Color shade reflects degree of shared P-elements between the two strains being compared.
-
Figure 4—source data 1
Spreadsheets with the calculations of P-element locus comparisons amongst the HISR strains and the records of the de novo P-element insertions amongst the HISR strains.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig4-data1-v1.xlsx
To explore this conundrum, we examined how many of the original P-elements in the Har genome were conserved in the HISR lines’ genomes (Figure 4B). As expected for HISR-D29 whose P-element copy numbers was closest to Har, this line conserved the highest share of parental Har P-elements compared to other HISR lines. However, there were also 35 novel P-element insertions (~45%) in HISR-D29 absent from Har. Surprisingly, the vast majority of the P-element insertions across all HISR lines were also de novo P-element insertions (Figure 4C), with each line clearing out nearly all parental Har P-element insertions and developing unique landscapes of P-element insertions. These data suggest that during the course of stabilizing the HISR lines, there were bursts of new P-element transpositions resulting in novel transposon landscapes that are completely distinct from the parental Har genome.
Although this dispersion of de novo P-elements in HISR lines’ genomes stymied our goal to pinpoint a particular Har locus strongly inducing GD, we next cloned and sequenced genomic PCR amplicons of all P-elements from the various P-element-containing strains. By using a single oligonucleotide that primes from both the 5' and 3' Terminal Inverted Repeats (TIRs), we amplified full-length P-elements as well as several additional truncation variants (Figure 5A) that have been missed in other genomic PCR assays using internal primers (Wakisaka et al., 2017). The most abundant variant accounting for more P-element copies in OreR-MOD and OreR-TK strains compared to Har were the ‘KP’ variant shown to encode a dominant negative protein that inhibits full-length P-transposase activity (Jackson et al., 1988; Simmons et al., 1990) (Figure 5A–5C), thus explaining the innocuous accumulation of these P-element variants in these OreR strains. Full-length P-elements were also sequenced from Har, pi[2] and Lerik-P strains, but there were no sequence differences in these clones from the original full-length P-element sequence in GenBank that might suggest a superlative quality to the full-length P-element in these strong GD-inducing strains.
Figure 5 with 2 supplements see all
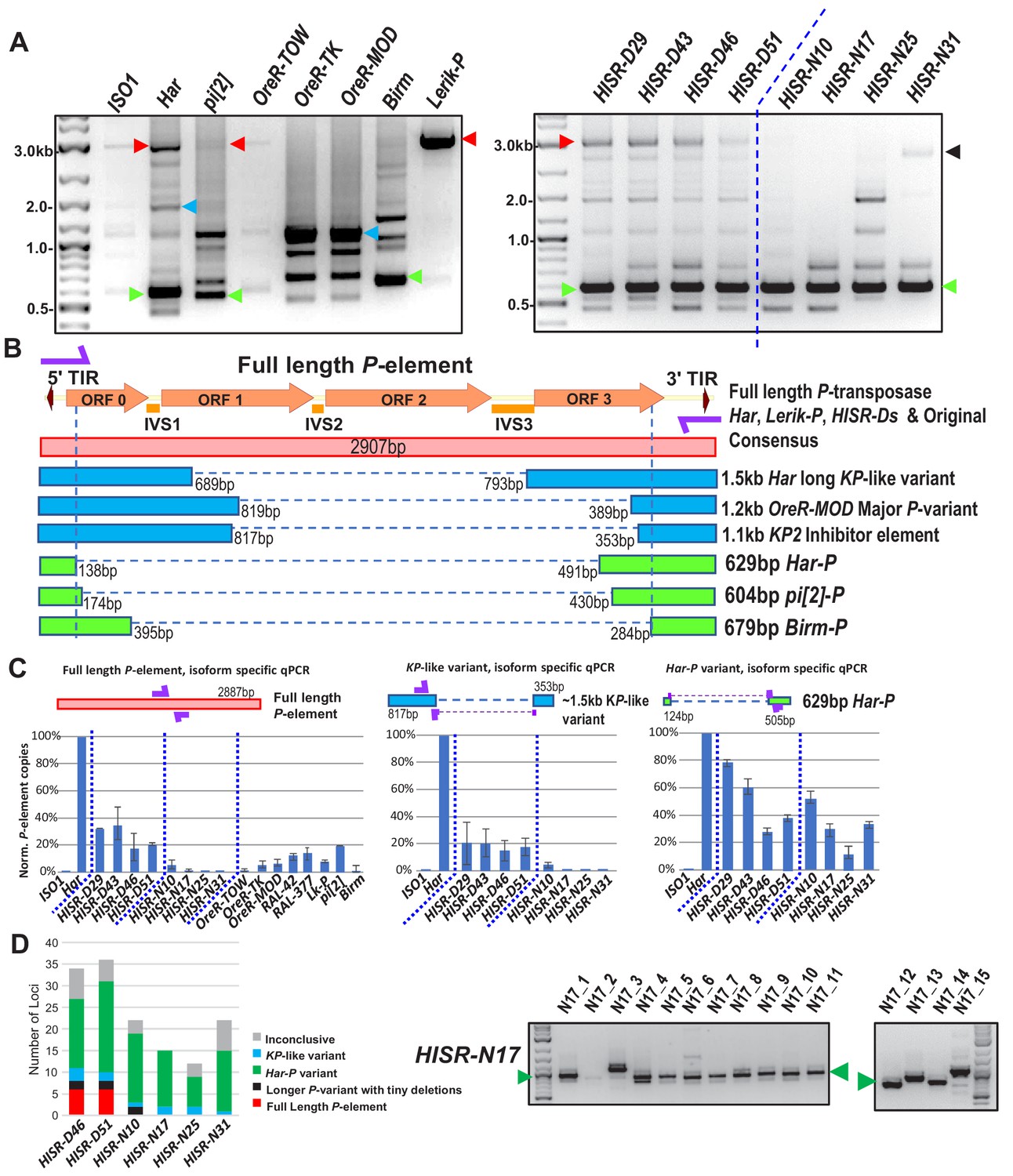
Har-P is a short and highly mobile P-element variant in strains used in P-element GD assays.
(A) P-element variant amplicons generated with TIR primers were cloned and sequenced as marked by colored arrows for the sequenced diagrams in (B). (B) Diagram of P-element variants cloned and sequenced from genomic PCR amplicons shown above. (C) Genomic qPCR quantifications of three P-element variants in Harwich and Harwich-derived HISR lines. Relative quantifications (in percentage) were calculated from ΔΔCt with rp49 as reference gene. (D) Proportions of the P-element variants verified by locus-specific PCR from TIDAL predictions of all HISR-N and HISR-D strains with <40 P-element insertions. The gel for HISR-N17 is on the right, while remaining gels are in Figure 5—figure supplement 1.
-
Figure 5—source data 1
Spreadsheets with the raw values from the qPCR experiments and calculations of the percentage of the insertions corresponding to the three P-element variant types.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig5-data1-v1.xlsx
Interestingly, we sequenced short ~630 bp P-element variants that were all very similar in configuration in Har, pi[2], and Birm strains, which only retains ~130 bp of the 5' end and ~500 bp of the 3' end of the P-element (Figure 5A and B). By retaining functional TIRs, these short elements can still be detected by TIDAL in WGS, can mobilize during crosses with the pi[2] strain (Bingham et al., 1982; Mullins et al., 1989; O'Hare and Rubin, 1983); and were previously shown to be able to assemble in vitro with the P-transposase tetramer complex >100X more efficiently than the full-length P-element (Tang et al., 2007). In addition, these short P-element variants seemed unlikely to translate into a protein due to multiple premature stop codons introduced by the massive internal deletion.
In all HISR-D lines that retain strong GD induction, we detected this short P-element variant and the full-length P-element encoding P-transposase, whereas the HISR-N lines retained the short variant but appeared to have lost the full-length P-element (Figure 5A, right panel). With the smaller number of TIDAL-predicted P-element insertions in HISR-N lines, we confirmed by locus-specific PCR the absence of full-length P-elements and that the majority of P-element insertions (~55–95%) were these de novo short P-element insertions (Figure 5D and Figure 5—figure supplement 1). We name this short variant ‘Har-Ps’ (Harwich P’s) in homage to Harpies, highly mobile hybrid bird-human creatures from the Greek mythological stories of the Argonauts.
To further support the conclusion that Har-P-like elements are the ammunition to drive severe GD, we examined additional fly lines from the DGRP collection (Mackay et al., 2012) which are of completely independent origin from Har, pi[2], and Birm strains. Indeed, the strains RAL-42 and RAL-377 that cause severe GD despite having a fraction of the load of P-elements as Har also possessed Har-P-like short variants (Figure 5—figure supplement 2). Meanwhile, two other P-element-containing strains RAL-508 and RAL-855 did not induce GD because only the longer KP-like elements were present (Figure 5—figure supplement 2).
Restoring GD when Har-P is crossed with P-transposase expressed in the germline
We hypothesized that Har-Ps combined with P-transposase from full-length P-elements could be the drivers of strong GD induction from pi[2], Har, and HISR-D strains. To test this hypothesis, we used negative-control yw-background females that lack P-transposase and transgenic H{CP}3 females that only express P-transposase in the germline (Simmons et al., 2002b) in crosses with males that either lack Har-P copies (ISO1, Lerik-P, OreR-MOD) or contain many Har-P copies (Har, HISR-N’s, Birm)(Figure 6A). GD induction was only restored in the F1 daughters of this cross in strains with many Har-Ps (Figure 6B). To avoid silencing of P-transposase by maternal P-element piRNAs in these strains, these crosses specifically used males that should only contribute paternal chromatin without contributing piRNAs (Figure 6A). Notably, the KP-length and full-length P-elements in OreR-MOD and Lerik-P, respectively, did not restore GD (Figure 6B, right most bars of left graph). These results suggest P-transposase act upon Har-P loci rather than longer P-element variants to induce GD and support the observation for Har-P loci making up the majority of the de novo P-element insertions in HISR-N lines (Figure 6C). Our data now genetically explain a previously described biochemical result showing that P-transposase assembles much more efficiently in vitro on short P-elements compared to the full-length P-element (Tang et al., 2007).
Figure 6 with 1 supplement see all
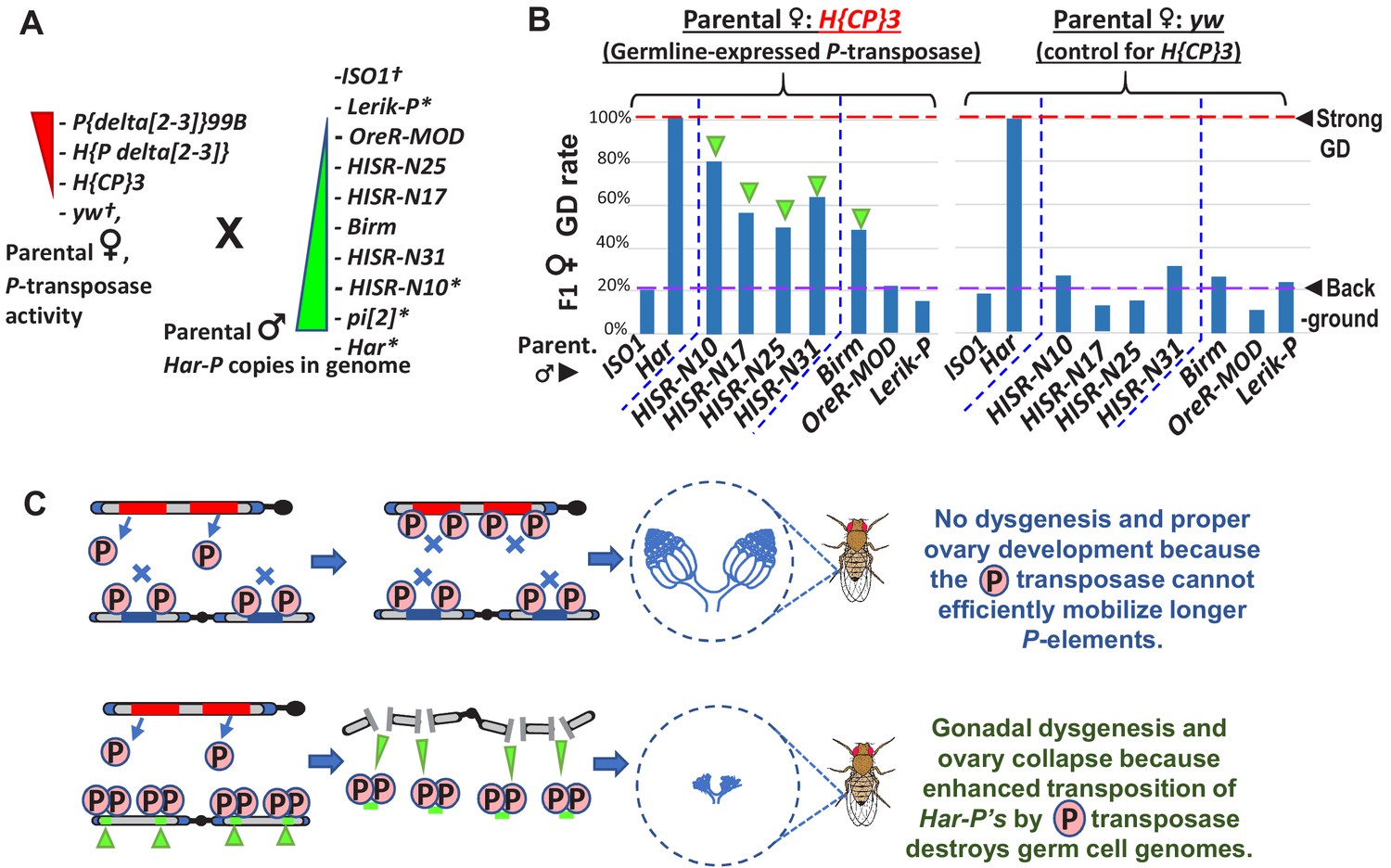
In absence of piRNA silencing, Har-P crossed with P-transposase restores severe GD.
(A) Strains for testing Har-P genetic interaction with P-transposase activity. *-strains with piRNA silencing; †-strains lacking any P-elements. (B) The H{CP}3 strain’s moderately-expressed maternal dose of P-transposase crossed with paternal HISR-N strains and Birm strain restores GD in F1 daughters, but strains with longer and full-length P-elements like OreR-MOD and Lerik-P lack the GD phenotype. (C) Model for P-transposase mobilizing Har-Ps to cause catastrophic transposition.
-
Figure 6—source data 1
Spreadsheet with the tabulation of gonadal dysgenesis assays.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig6-data1-v1.xlsx
We noticed that GD severity in crossing HISR-N with the H{CP}3 transgenic line was not completely penetrant like GD assays with the parental Har (Figure 6B versus Figure 1B) because Har contributes both multiple copies of full-length P-elements and Har-P loci versus the single copy of the natural P-element transgene in H{CP}3 (Simmons et al., 2002b). In addition, natural P-element translation is inhibited by strong somatic splicing inhibition of the native P-element’s third intron (IVS3) containing a premature stop codon and only inefficient splicing in the Drosophila germline that is further suppressed by piRNAs (Siebel et al., 1994; Teixeira et al., 2017). We also confirmed that IVS3 intron splicing was the main alteration that increased P-element expression in ovaries from a dysgenic cross between Har and ISO1, whereas Open Reading Frame (ORF) parts of the P-element transcript are only modestly increased (Figure 6—figure supplement 1A). We believe this sufficient expression of P-transposase promotes the preferred mobilization of Har-P short variants in dysgenic cross ovaries, but the cut-and-paste transposition mechanism of P-transposase should theoretically conserve the total copy number of P-elements. By using digital droplet PCR to precisely quantity total P-element copy numbers, we confirmed that total P-element copy numbers were stable across ovaries of daughters from two sets of dysgenic and non-dysgenic crosses (Figure 6—figure supplement 1B).
Somatic expression of P-transposase with Har-P’s causes pupal lethality
To test whether a stronger expressing P-transposase transgene could induce the complete GD in crosses with HISR-N lines, we turned to the delta[2-3] P-transposase transgenes that lack the IVS3 intron to enable strong somatic and germline P-transposase activity (Robertson et al., 1988). When we crossed two different delta[2-3] female strains to males of HISR-N17, -N25, and -N31 which lack P-element piRNA expression but have Har-Ps, we were unable to assay GD because of extensive pupal lethality (Figure 7A). We also confirmed extensive pupal lethality in crosses between delta[2-3] and the Birm strain (Figure 7A) as previously described (Engels et al., 1987; Simmons et al., 1987). Since we also detected very short P variants in Birm that are similar to Har-P (Figure 5A and B) we conclude that somatically expressed P-transposase acting only on the Har-Ps in Birm, HISR-N17, -N25, and -N31 is sufficient to disrupt pupal development.
Figure 7 with 2 supplements see all

Somatic expression of P-transposase triggers pupal lethality with Har-P loci that are not silenced by P-element piRNAs.
(A) Green triangles denote crosses showing pupal lethality from stronger somatic expression of P-transposase acting on Har-Ps in HISR-N and Birm strains lacking P-element piRNAs. Red hexagons denote crosses with strains expressing P-element piRNAs that suppress pupal lethality through a paternally transmitted epigenetic imprint. (B) The paternal P-element piRNA imprinting on Har-Ps in Har and HISR-N10 cannot suppress GD in F1 daughters, as marked by green triangles. The longer P variants in OreR-MOD do not result in GD with the delta[2-3] P-transposase. (C) Revised P Dysgenesis paradigm proposing a paternally-transmitted piRNA-directed epigenetic mark that resists P-transposase activity in the soma, but this mark is erased during oogenesis.
-
Figure 7—source data 1
Spreadsheets with the tabulation of pupal lethality and gonadal dysgenesis assays.
- https://cdn.elifesciences.org/articles/49948/elife-49948-fig7-data1-v1.xlsx
Unexpectedly, the pupal lethality was suppressed when delta[2-3] females were crossed with Har-P-containing males that also expressed P-element piRNAs, such as Har, pi[2], the four HISR-D lines, and HISR-N10 (Figure 7A). These hybrid F1 progeny developed into adults, but the adult females of Har and HISR-N10 hybrids with delta[2-3] still exhibited severe GD (Figure 7B). In addition, we also observed severe pupal lethality when delta[2-3] females were crossed to RAL-42 but not RAL-377, although strong GD was still observed with RAL-377 (Figure 7—figure supplement 1). These RAL strains of independent origin from Har, pi[2], and HISR lines provide convincing support for the conclusion that P-element piRNAs impart a paternally-transmitted imprint on Har-P loci that resists mobilization with somatically-expressed P-transposase and enables development to adulthood. However, this imprint is either erased in ovaries or insufficient to prevent ovarian GD. Finally, the notable P-element piRNA pattern of HISR-N10 perfectly matches the Har-P structure since many internal piRNAs are absent (Figure 3B), but overall P-element piRNAs in HISR-N10 are equivalent to Har and HISR-D lines (Figure 3C), and therefore are sufficient to repress Har-P’s epigenetically from being mobilized in the soma by the delta[2-3] P-transposase.
Discussion
After a Drosophila strain has silenced an invading transposon through the Piwi/piRNA pathway, the neutered transposon will naturally decay into various truncations that are presumed to be neutral or even beneficial to host fitness (Kelleher, 2016), such as natural KP2 truncation variants that inhibit P-element transposition (Jackson et al., 1988; Simmons et al., 1990). However, we discovered one such truncation we call Har-P via our unbiased genetic and molecular approach that can actually be detrimental to the host. Our findings resonate with the previous finding that P-transposase assembles in vitro much more efficiently on very short natural P-element variants (Tang et al., 2007), therefore we propose a new model for catastrophic P-element transposition in strong GD inducer strains like pi[2] and Har (Figure 6C).
When a P-element truncates to a ~630 bp Har-P variant, this non-autonomous variant dominates as the main mobilizing P-element during a dysgenic cross to induce strong GD. Thus, previous studies examining GD variability across other Drosophila strains and isolates may now be explained by whether these genomes contain both full length and very short P-elements (Bergman et al., 2017; Kozeretska et al., 2018; Ronsseray et al., 1989; Srivastav and Kelleher, 2017; Wakisaka et al., 2017; Yoshitake et al., 2018). Moreover, this particular deletion size of a ~630 bp P-element variant that arose in at least four completely independent lines has persisted without detriment to these animals either because of piRNA silencing (i.e. Har, pi[2], RAL-377) or because the P-transposase was separated (i.e. HISR-N and Birm). The short configuration must be special because additional sequence lengths such as P-element-based transgenes that were mobilized by P-transposase into transgenic strains are not strong triggers of GD like the Har-P elements (Figure 7—figure supplement 1C).
Although our future goal will be to determine which specific epigenetic marks are deposited at full length P-elements and Har-P’s by piRNAs, we believe a chromatin mark resisting P-transposase activity is more likely than somatic piRNAs or siRNAs (Chung et al., 2008; Ghildiyal et al., 2008; Kawamura et al., 2008) silencing the delta[2-3] P-transposase in our pupal lethality crosses because we confirmed robust P-transposase mRNA expression regardless of the expression of P-element piRNAs (Figure 7—figure supplement 2A). A second future goal will be to generate transgenic flies with single or multiple synthetic Har-P copies to determine the precise dosage of Har-P’s that would trigger GD or pupal lethality. However, in addition to copy number, genomic location may also influence host tolerance of Har-P’s, because we observed a significant rescue of viable pupae in crosses between delta[2-3] P-transposase and a derivative strain of HISR-N17 with Har-P’s only on Chromosome 3 with six P-elements, while no pupae survived with delta[2-3] P-transposase and Har-P’s on Chromosome 2 with nine P-elements (Figure 7—figure supplement 2B).
The Drosophila P-element system of hybrid GD mainly affects female sterility and requires maternally contributed P-element piRNAs to propagate transgenerational P-element silencing in daughters via trimethylation of histone H3 lysine 9 (H3K9me3) (Josse et al., 2007; Le Thomas et al., 2014). Although previous studies of dysgenic crosses focused on complete GD in females (Bingham et al., 1982; Brennecke et al., 2008; Khurana et al., 2011; Rubin et al., 1982), sons respond differently because they are fertile despite presumed somatic P-element excision (Wei et al., 1991). Since previous studies of P-M hybrid dysgenesis above never considered a paternal imprint on P-elements that our findings now suggest is being propagated (Figure 7C), future studies with HISR-N strains will enable us to dissect a paternally-transmitted small RNA-directed silencing effect in Drosophila that harkens to also a similar paternally-transmitted RNAi effects observed earlier in nematodes (Grishok et al., 2000).
Mouse piRNAs bound by MIWI2 direct the re-establishment of DNA methylation marks on transposons like L1 and IAP (Aravin et al., 2008), which may propagate in sperm, but Drosophila DNA methylation is not prominent (Krauss and Reuter, 2011). Similar to other metazoans, Drosophila sperm also undergoes histone exchange with protamines (ProtB), with little contribution of paternal cytoplasm (Rathke et al., 2007). However, recent data do support the retention of some H3K9me3 in sperm (Yamaguchi et al., 2018), which might underlie the paternal imprint of piRNA-silencing of P-elements that will be investigated further in future studies.
This interplay between the truncated Har-Ps and full-length P-element DNA transposon resembles other examples in nature, such as the extreme proliferation of MITEs (miniature inverted repeat transposable element) in rice scavenging other transposases to mobilize (Yang et al., 2009), and short mammalian SINEs retrotransposons taking advantage of the transposition machinery of longer LINEs, since SINEs persist in much greater numbers than their longer LINE counterparts (Hancks and Kazazian, 2012). However, while the full impact of MITEs and SINEs on organism development is still obscure, our study indicates that Har-Ps combined with the P-transposase to trigger transposition events so efficiently to be detrimental to ovarian and pupal development. Notwithstanding, the high efficiency of Har-P mobilization by P-transposase may also be engineered into a new generation of transposon-based mutagenesis approaches.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source reference | Identifier | Additional information |
|---|---|---|---|---|
| Strain, strain background (Drosophila melanogaster) | ISO1 | Susan Celnicker Lab | PMID: 25589440 | iso-1 : y[1]; Gr22b[1] Gr22d[1] cn[1] CG33964[R4.2] bw[1] sp[1]; LysC[1] MstProx[1] GstD5[1] Rh6[1] |
| Strain, strain background (Drosophila melanogaster) | Harwich | William Theurkauf Lab | PMID: 22196730 | |
| Strain, strain background (Drosophila melanogaster) | HISR-D29 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-D43 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-D46 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-D51 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-N10 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-N17 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-N25 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | HISR-N31 | Created in Lau Lab | This paper | |
| Strain, strain background (Drosophila melanogaster) | H{KP} | BDSC | Stock No. 64175 | y[1] w[67c23]; H{w[+mC]=KP}H |
| Strain, strain background (Drosophila melanogaster) | pi[2] | BDSC | Stock No.2384 | |
| Strain, strain background (Drosophila melanogaster) | OreR-MOD | BDSC | Stock No.25211 | |
| Strain, strain background (Drosophila melanogaster) | OreR-TK | BDSC | Stock No.2376 | |
| Strain, strain background (Drosophila melanogaster) | OreR-TOW | Terry Orr-Weaver's Lab | PMID: 21177974 | |
| Strain, strain background (Drosophila melanogaster) | RAL-42 | BDSC | Stock No.28127 | |
| Strain, strain background (Drosophila melanogaster) | RAL-377 | BDSC | Stock No.28186 | |
| Strain, strain background (Drosophila melanogaster) | RAL-508 | BDSC | Stock No.28205 | |
| Strain, strain background (Drosophila melanogaster) | RAL-855 | BDSC | Stock No.28251 | |
| Strain, strain background (Drosophila melanogaster) | H{CP}3 | BDSC | Stock No.64160 | y[1] w[67c23]; H{w[+mC]=hsp/CP}3 PMID: 12019234 |
| Strain, strain background (Drosophila melanogaster) | yw | John Abrams's Lab | PMID: 26701264 | |
| Strain, strain background (Drosophila melanogaster) | P{delta[2-3]}99B | BDSC | Stock No.3629 | w[*]; wg[Sp-1]/CyO; ry[506] Sb[1] P{ry[+t7.2]=Delta2-3}99B/TM6B, Tb[+] PMID: 3000622 |
| Strain, strain background (Drosophila melanogaster) | H{P delta[2-3]} | BDSC | Stock No.64161 | y[1] w[67c23]; H{w[+mC]=w[+].Delta2- 3.M}6 |
| Strain, strain background (Drosophila melanogaster) | P{TubGal80}Chr2 | BDSC | Stock No.7108 | |
| Strain, strain background (Drosophila melanogaster) | P{TubGal80}Chr3 | BDSC | Stock No.7017 | |
| Strain, strain background (Drosophila melanogaster) | P{Elav-Gal4}Chr2 | BDSC | Stock No.8765 | |
| Strain, strain background (Drosophila melanogaster) | Sp/CyO;TM6b/Sb | Michael Rosbash's Lab | ||
| Strain, strain background (Drosophila melanogaster) | Lerik-P | Stephane Ronsseray's Lab | PMID: 1660427 | Lk-P(1A)-SL2 |
| Strain, strain background (Drosophila melanogaster) | Birm | BDSC | Stock No. 2359 | Birm Chr2; PMID: 2835286 |
| software, algorithm | QuantaSoft Analysis Pro | Bio-Rad | ||
| software, algorithm | Applied Biosystems 7500/7500 Fast Real-Time PCR System v2.0 | Applied Biosystems | ||
| software, algorithm | BWA MEM | Li and Durbin, 2010 | PMID: 20080505 | |
| software, algorithm | TIDAL-Fly | Rahman et al., 2015 | PMID: 26578579 | |
| software, algorithm | GATK | McKenna et al., 2010 | PMID: 20644199 | |
| commerical assay or kit | TOPO PCR Cloning Kit | ThermoFischerSci | Cat No. 450031 | |
| commerical assay or kit | Luna Universal qPCR master mix | New England Biolabs Inc | Cat No. M3003 | |
| commerical assay or kit | NEB Ultra II FS DNA library prep | New England Biolabs Inc | Cat No. E7805 | |
| commerical assay or kit | First Strand cDNA Synthesis Kit using ProtoScript II | New England Biolabs Inc | Cat No. M0368 | |
| commerical assay or kit | NEBNext Small RNA Library Prep Set for Illumina | New England Biolabs Inc | Cat No. E7330 | |
| commerical assay or kit | QX200 ddPCR EvaGreen Supermix | Bio-Rad | Cat No. 1864034 | |
| Chemical compound, drug | Tri-reagent | Molecular Research Center Inc,OH | ||
| Chemical compound, drug | Q Sepharose Fast Flow, 300 mL | GE HealthCare | Cat No. 17051001 |
Fly strains
Request a detailed protocolAll strains were maintained on standard cornmeal medium at 22°C. Because the ISO1(BDSC#2057) stock had accumulated >180 new transposon insertions relative to the original stock sequenced in the Berkeley Drosophila genome project (Adams et al., 2000; Rahman et al., 2015), we obtained the ISO1 strain from Susan Celniker’s lab (ISO1-SC). The Har strain was obtained from (Har-WET) was obtained from the William Theurkauf’s lab (Khurana et al., 2011). Three Oregon-R strains were obtained from Terry Orr-Weaver’s lab, OreR-TOW, OreR-TK (Kaufman, BDSC#2376) and OreR-MOD (BDSC#25211). The Lerik-P strain was obtained from Stephane Ronserray’s lab (Josse et al., 2007; Marin et al., 2000). All the following strains were also directly obtained from the BDSC – RAL-42 (#28127), RAL-377 (#28186), RAL-508 (#28205), RAL-855 (#28251), pi[2] (#2384), y[1] w[67c23]; H{w[+mC]=hsp/CP}3 (#64160), Birmingham; Sb[1]/TM6 (#2539), w[*]; wg[Sp-1]/CyO; ry[506] Sb[1] P{ry[+t7.2]=Delta2-3}99B/TM6B, Tb[+] (#3629), y[1] w[67c23]; H{w[+mC]=w[+].Delta2-3.M}6 (#64161), H{w[+mC]=KP} (#64175), P{TubGal80}Chr2 (#7108), P{TubGal80}Chr3 (#7017), and P{Elav-Gal4}Chr2 (#8765). Sp/CyO;TM6b/Sb was obtained from Michael Rosbash’s lab.
Crosses, gonadal dysgenesis and pupal lethality assays
Request a detailed protocolAll crosses were set up with 3–5 virgin females and 2–4 young males per replicate on standard cornmeal medium at 25°C and parents were purged after 5 days of egg laying (Srivastav and Kelleher, 2017). For GD assays, F1 females aged to 4–5 days at 25°C were examined for GD using food dye and GD % shown is average of 3 replicate crosses with total minimum of 100 F1 females assayed (Simmons et al., 2007). Somatic pupal lethality was recorded by counting dead (uneclosed) and empty pupae (eclosed) 6 days after first eclosion was observed in respective control cross (P{delta[2-3]}99B x ISO1 or H{P delta[2-3]} x ISO1) (Engels et al., 1987). Pupal lethality percentage shown is average of two or more replicate crosses that obtained at least total of 50 F1 pupae each.
Crossing scheme to generate HISR lines
Request a detailed protocolThe detailed crossing scheme is illustrated in Figure 2—figure supplement 1. After a first cross between virgin Har females and ISO1 males, three more backcrosses of virgin Har/ISO1 hybrid progeny females mated to ISO1 males were performed and following the progeny with red eyes to select for the Har segment segregating with the cn, bw, sp, alleles on Chromosome 2R. We hoped that a particular set of P-elements that drive strong GD induction would co-segregate with red eye color. We then performed a ‘Validation Cross’ with the F4 hybrid males individually mated to ISO1 females. We screened >100 individual groups of F4 males for their GD induction, where the early-hatching 3 day old daughters were screened via the squash assay for 100% GD. Only the F5 vials showing 100% GD from F4 males crossed to ISO1 females were kept, and then were allowed to age and self-crossed and propagated in 11 more generations to attempt to create recombining-inbred-lines (RILs).
Selecting only flies with red eyes required purging any flies emerging with the ‘white’ eyes of ISO1 and discarding many vials that failed to generate progeny due to genotoxic collapse from inability to silence P-element transposition. At the F16 stage, Har/ISO1 Selfed Red (HISR) lines males were rescreened in a Validation Cross with ISO1 females, this time keeping lines that still caused 100% GD and designated as HISR-D (Dysgenic) lines. We also selected additional lines that had now lost GD and allowing for >50% of females to generate egg chambers, and these were designated HISR-N (Non-dysgenic) lines. We performed 2 rounds of single-sibling pair mating to further inbreed these lines in an attempt to stabilize the genotypes, and we maintained 4 lines of each HISR-D and HISR-N for true propagation of just the red or cinnabar eyes and speck phenotype.
Genomic DNA extraction, PCR, quantitative-PCR and Droplet Digital PCR
Request a detailed protocolGenomic DNA was prepared from 10 young female flies by homogenizing tissues with plastic pestle in 300 µL Lysis buffer (10 mM Tris pH-8.0, 100 mM NaCl, 10 mM EDTA, 0.5% SDS, and Proteinase K at 50 µg/ml) and incubated at 65°C overnight followed by treatment of RNase A at 100 µg/ml at 37°C for 30 mins. 200 µL of 0.5M NaCl was added followed by one volume of Phenol:CHCl3:IAA (at 25:24:1) and spun at 14,000 rpm for 10 min to isolate DNA in aqueous phase. Aqueous phase was extracted again with one volume of CHCl3:IAA (at 24:1) and supplemented with one volume of 5M LiCl and incubated at −20°C and then spun at 15,000 rpm for 15 mins to precipitate RNA. Supernatant was isolated and supplemented with 2 volumes of 100% ethanol and incubated in −20°C for 2 hr and then spun at 15,000 rpm for 20 mins. DNA pellet was washed with chilled 70% ethanol and dissolved in nuclease free water. DNA integrity checked (>10 kb) by running 1 µg on 1% agarose gel with EtBr.
Genomic PCR reactions to characterize P-element structural variation were set up in 30 µL reactions of 1X NEB GC buffer, 300 µM dNTPs, 0.5M Betaine, 2.5 mM MgCl2, 0.25 µM of IR primer (Rasmusson et al., 1993), 1 µL of Phusion polymerase and 50 ng of genomic DNA and cycled at 94°C for 1 min, 62°C for two mins, 72°C for 4 mins for 27 cycles and followed by 72°C for 15 min. Genomic PCR reactions to characterize P-element structural variation in HISR lines, predicted by TIDAL were also set up similarly using P-element insertion locus specific primers. Genomic PCR reactions for genotyping of HISR-N lines were set up similarly but cycled at 94°C for 30 s, 60°C for 15 s, 72°C for 30 s for 27 cycles and followed by 72°C for 5 min.
Genomic qPCR experiments were performed in three biological replicates with two 20 µL technical reactions replicates each, using Luna Mastermix (NEB), primers at 0.5 µM and 20 ng of genomic DNA per reaction in real time quantitative PCR. P-element load was calculated from 2^(-∆∆Ct) normalized to Har at 100% and ∆Ct from RP49. All primers used for are listed in Supplementary file 1.
For the Droplet Digital PCR (ddPCR), we utilized the Evagreen Mastermix (Biorad) and conducted on a QX500 ddPCR machine with manual setting of droplet signal thresholds. 10–15 pairs of ovaries and corresponding carcass from 4 to 5 day old F1 females were dissected from dysgenic and non-dysgenic crosses of Har and HISR-D51 with ISO1 strain at 18 °C. DNA was extracted from the ovaries and carcass and quantified using Qubit 2.0 Fluorometer. Digital PCR probe assays were conducted in 40 µL droplet reactions, generated from 25 µL digital PCR reaction and 70 µL droplet oil each. 25 µL digital PCR reactions were set up with BioRad ddPCR probe supermix, P-element7a (FAM) and rp49 (HEX) probes each at 250 nM and 200 pg of DNA. Reactions were cycled at 95 °C for 10 mins followed by 95 °C for 30 s and 58 °C for 1 min for 40 cycles, and 98 °C for 10 mins. Copies/µL values were extracted from QuantaSoft (BioRad) software and P-element copies per genome were calculated normalized to rp49.
P-element amplicon cloning and sequencing
Request a detailed protocolP-elements amplified from IR PCR were purified from 1% agarose gel using QIAquick Gel extraction kit and cloned into pCR4-TOPO vector using Zero Blunt TOPO PCR Cloning Kit at RT, followed by transformation of chemically competent DH10β cells, which were then grown on LB plates with 0.05 mg/ml Kanamycin overnight. 5–10 colonies were screened by PCR and two colonies positive for P-element cloned were chosen for plasmid mini-prep and sequenced using M13 forward and reverse primers for all variants in addition to internal primers to complete the sequencing of full-length P-elements.
Whole genome sequencing, SNP profile analysis, and TIDAL analysis
Request a detailed protocolGenomic DNA libraries were prepared using NEB Ultra II FS kit E7805. Briefly, 500 ng of genomic DNA (>10 kb) was fragmented at 37°C for 12 min, followed by adaptor ligation and loop excision according to kit manual protocol. Size selection was performed with two rounds of AmpureXP beads addition to select for insert size 150–250 bp as per kit manual. Library PCR amplification was also carried out as per manual instructions for six cycles and purified using one round of AmpureXP beads addition at 0.9X volume. Individual barcoded libraries were quantified on NanoDrop and each diluted to 2 nM and then pooled to produce equimolar concentration.
Whole genome sequencing was performed on an Illumina NextSeq 500 with paired-end reads of 75 bp x 75 bp in the Rosbash lab at Brandeis University. Reads were demultiplexed and trimmed by Trimmomatic to remove low quality bases, and then reads were analyzed by the TIDAL program (Rahman et al., 2015). TIDAL outputs were sorted for P-element insertions and the insertion coordinates were compared across the HISR lines using SQL queries in MS-Access. To calculate the Single Nucleotide Polymorphism (SNP) profiles, paired-end reads were mapped to the Dm6 ISO1 genome with ‘BWA MEM’(Li and Durbin, 2010) using default parameters. PCR duplicates are removed with Picard and SNPs are called with GATK HaplotypeCaller (Danecek et al., 2011; DePristo et al., 2011; McKenna et al., 2010). We then generated the nucleotide distribution for each SNP to ensure that there are at least 20 reads supporting each SNP. Then, we created a unified SNP list by using the union of SNPs from all libraries and carefully noted if each SNP is present in each library. The SNP counts were binned by 5 kb segments and converted into a graphical representation as differences between the reference genome and strain/line in Figure 4—figure supplement 1.
Ovary small RNA sequencing and analysis
Request a detailed protocolTo remove the 2S rRNA from Drosophila ovaries, we adapted a protocol from our previous Q-sepharose beads matrix technique (Lau et al., 2009). About 50 ovaries per parental Har and ISO1 strains and HISR lines were dissected from young adult females. Ovaries were then lysed in ice cold 500 ul Elution Buffer (20 mM Hepes pH 7.9 (with KOH), 10% glycerol, 400 mM KOAc, 0.2 mM EDTA, 1.5 mM MgCl2, 1.0 mM DTT, 1X Roche Complete EDTA-free Protease Inhibitor Cocktail) using one freeze-thaw cycle and pulverizing with a blue plastic pestle. A 1.5 ml aliquot of Q-Sepharose FF matrix suspension was washed 1X in water, then 3X in Elution buffer, then incubated for 10 min with the ovaries lysate with occasional agitation in cold room. Ribosomal RNA gets bound by the Q-sepharose, while small RNA RNPs remains in the elution buffer. Elution buffer was removed and then subjected to small RNA extraction with the Tri-reagent protocol. The precipitated small RNAs where then converted into Illumina libraries using the NEBNext Small RNA Library Construction kit. One modification we employed during the overnight linker ligation is to supplement the reactions to 12.5% PEG 8000 to reduce the potential sequence biases from T4 RNA ligase activity.
Small RNA libraries were sequenced as 75 bp single end reads on the NextSeq550. Adapters for the small RNA libraries were removed with CutAdapt and then mapped to the Drosophila transposon consensus sequences from RepBase and Flybase using Bowtie v1 with up to two mismatches and R plotting scripts as applied in our previous published studies on Drosophila piRNAs (Clark et al., 2017; Sytnikova et al., 2014).
RT-qPCR analysis of P-element expression in gonadal dysgenesis and pupal lethality
Request a detailed protocolFor this assay, 5–10 pairs of ovaries were dissected from 3 to 5 day old F1 females of dysgenic and non-dysgenic cross with Har and ISO1, as well as with Har and HISR-D46. RNA was extracted from such ovaries and integrity checked by running 1 µg RNA at 2% Agarose II gel (Fischer BioReagents). 3 µg was reverse transcribed using Protoscript RT enzyme (NEB) as per manufacturer’s protocol and negative RT control was carried out similarly without RT enzyme. 50 ng of cDNA was used for setting up rp49 PCR reactions (as described above) from RT and corresponding negative RT reactions to evaluate DNA contamination. qPCR reactions for P-element ORF2, ORF3, IVS3 were also carried out as genomic qPCR reactions with 20 ng cDNA input and ΔCt were calculated similarly using rp49 RNA levels.
In the RT-qPCR analysis of H{P delta[2-3]} gene expression in gonadal dysgenesis and pupal lethality, 5–10 pairs of ovaries and corresponding carcass were dissected from F1 females of pupal lethality crosses conducted at 18 °C. RNA extraction, reverse transcription, PCR and qPCR reactions were carried out similarly as above. ΔCt were calculated similarly using rp49 RNA levels. Fold change values were obtained from normalizing F1 carcass P-element RNA levels to H{P delta[2-3]} carcass and F1 ovary P-element RNA levels were normalized to H{P delta[2-3]} carcass in Figure 7—figure supplement 2.
Isolation of HISR-N17 autosomes for modulating Har-P genomic dosage
Request a detailed protocolHISR-N17 autosomes were isolated first by crossing virgin HISR-N17 females with Sp/CyO;TM6b/Sb stock males and using virgin F1 females with CyO and TM6 to cross again with Sp/CyO;TM6b/Sb. F2 males with either HISR-N17 Chr2 or HISR-N17 Chr3 were crossed to virgin H{P delta[2-3]}. All crosses were performed in triplicates at 25 °C. F3 pupal lethality was recorded on 16th day of the H{P delta[2-3]} crosses.
Data availability
The high-throughput sequencing data in our study #SRP178563 can be accessed here: http://www.ncbi.nlm.nih.gov/bioproject/?term=PRJNA514796.
-
NCBI SRAID SRP178563. Har-P is a short P-element variant that collaborates with P transposase to induce strong gonadal dysgenesis.
References
-
Molecular analysis of the P-M gonadal dysgenesis cline in eastern Australian Drosophila MelanogasterGenetics 119:889–902.
-
Drosophila PAF1 modulates PIWI/piRNA silencing capacityCurrent Biology 27:2718–2726.https://doi.org/10.1016/j.cub.2017.07.052
-
The variant call format and VCFtoolsBioinformatics 27:2156–2158.https://doi.org/10.1093/bioinformatics/btr330
-
Somatic effects of P element activity in Drosophila Melanogaster: pupal lethalityGenetics 117:745–757.
-
Hybrid dysgenesis in Drosophila Melanogaster: the biology of female and male sterilityGenetics 92:161–174.
-
Active human retrotransposons: variation and diseaseCurrent Opinion in Genetics & Development 22:191–203.https://doi.org/10.1016/j.gde.2012.02.006
-
Amplification of KP elements associated with the repression of hybrid dysgenesis in Drosophila MelanogasterGenetics 120:1003–1013.
-
Hybrid dysgenesis in DROSOPHILA MELANOGASTER: a syndrome of aberrant traits including mutation, sterility and male recombinationGenetics 86:813–833.
-
DNA methylation in Drosophila--a critical evaluationProgress in Molecular Biology and Translational Science 101:177–191.https://doi.org/10.1016/B978-0-12-387685-0.00003-2
-
P-Element repression in Drosophila Melanogaster by a naturally occurring defective telomeric P copyGenetics 155:1841–1854.
-
A robust Transposon-Endogenizing response from germline stem cellsDevelopmental Cell 47:660–671.https://doi.org/10.1016/j.devcel.2018.10.011
-
cis-acting DNA sequence requirements for P-element transpositionGenes & Development 3:729–738.https://doi.org/10.1101/gad.3.5.729
-
Unique transposon landscapes are pervasive across Drosophila Melanogaster genomesNucleic Acids Research 43:10655–10672.https://doi.org/10.1093/nar/gkv1193
-
Repression of hybrid dysgenesis in Drosophila Melanogaster by individual naturally occurring P elementsGenetics 133:605–622.
-
A stable genomic source of P element transposase in Drosophila MelanogasterGenetics 118:461–470.
-
Modified P elements that mimic the P cytotype in Drosophila MelanogasterGenetics 123:815–824.
-
The influence of nonautonomous P elements on hybrid dysgenesis in Drosophila MelanogasterGenetics 117:671–685.
-
Repression of P element-mediated hybrid dysgenesis in Drosophila MelanogasterGenetics 124:663–676.
-
Regulation of P-element transposase activity in Drosophila Melanogaster by hobo transgenes that contain KP elementsGenetics 161:205–215.
-
A hobo transgene that encodes the P-element transposase in Drosophila Melanogaster: autoregulation and cytotype control of transposase activityGenetics 161:195–204.
-
Analysis of P element transposase protein-DNA interactions during the early stages of transpositionJournal of Biological Chemistry 282:29002–29012.https://doi.org/10.1074/jbc.M704106200
-
Gonadal dysgenesis reveals sexual dimorphism in the embryonic germline of DrosophilaGenetics 129:203–210.
Article and author information
Author details
Funding
National Institutes of Health (R01-AG052465)
- Nelson C Lau
National Institutes of Health (R21-HD088792)
- Nelson C Lau
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank W Theurkauf, S Celniker, S Ronsseray, T Orr-Weaver, and the BDSC (NIH grant P40OD018537) for fly strains; M Rosbash and Brandeis University for deep sequencing and fly food; and D Schwarz, R McCrae, A Grishok and D Cifuentes for comments. SPS and NCL conceived and conducted the experiments, RR and QM provided bioinformatics analyses, JP and SB contributed final experiments, and NCL wrote the paper. This work was supported by NIH grants R01-AG052465 and R21-HD088792 to NCL. Sequencing data are deposited in the NCBI SRA as Study #SRP178563.
Copyright
© 2019, Srivastav et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,713
- views
-
- 163
- downloads
-
- 23
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 23
- citations for umbrella DOI https://doi.org/10.7554/eLife.49948
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Har-P, a short P-element variant, weaponizes P-transposase to severely impair Drosophila development
eLife 8:e49948.
https://doi.org/10.7554/eLife.49948




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}