Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism
- Division of Pharmacology, Physiology and Neuroscience, School of Life Sciences, Queen’s Medical Centre, University of Nottingham, United Kingdom
- Centre of Membrane Protein and Receptors, Universities of Birmingham and Nottingham, United Kingdom
- Computational Chemistry and Molecular Biophysics Unit, National Institute on Drug Abuse - Intramural Research Program, National Institutes of Health, United States
- Drug Discovery Biology, Department of Pharmacology and Medicinal Chemistry, Monash Institute of Pharmaceutical Sciences, Monash University, Australia
- Department of Psychiatry, Vagelos College of Physicians and Surgeons, Columbia University, United States
- Division of Molecular Therapeutics, New York State Psychiatric Institute, United States
- Vagelos College of Physicians and Surgeons, Columbia University, United States
Abstract
By analyzing and simulating inactive conformations of the highly homologous dopamine D2 and D3 receptors (D2R and D3R), we find that eticlopride binds D2R in a pose very similar to that in the D3R/eticlopride structure but incompatible with the D2R/risperidone structure. In addition, risperidone occupies a sub-pocket near the Na+ binding site, whereas eticlopride does not. Based on these findings and our experimental results, we propose that the divergent receptor conformations stabilized by Na+-sensitive eticlopride and Na+-insensitive risperidone correspond to different degrees of inverse agonism. Moreover, our simulations reveal that the extracellular loops are highly dynamic, with spontaneous transitions of extracellular loop 2 from the helical conformation in the D2R/risperidone structure to an extended conformation similar to that in the D3R/eticlopride structure. Our results reveal previously unappreciated diversity and dynamics in the inactive conformations of D2R. These findings are critical for rational drug discovery, as limiting a virtual screen to a single conformation will miss relevant ligands.
eLife digest
Almost a third of prescribed drugs work by acting on a group of proteins known as GPCRs (short for G-protein coupled receptors), which help to transmit messages across the cell’s outer barrier. The neurotransmitter dopamine, for instance, can act in the brain and body by attaching to dopamine receptors, a sub-family of GPCRs. The binding process changes the three-dimensional structure (or conformation) of the receptor from an inactive to active state, triggering a series of molecular events in the cell.
However, GPCRs do not have a single ‘on’ or ‘off’ state; they can adopt different active shapes depending on the activating molecule they bind to, and this influences the type of molecular cascade that will take place in the cell. Some evidence also shows that classes of GPCRs can have different inactive structures; whether this is also the case for the dopamine D2 and D3 receptors remained unclear. Mapping out inactive conformations of receptors is important for drug discovery, as compounds called antagonists can bind to inactive receptors and interfere with their activation.
Lane et al. proposed that different types of antagonists could prefer specific types of inactive conformations of the dopamine D2 and D3 receptors. Based on the structures of these two receptors, the conformations of D2 bound with the drugs risperidone and eticlopride (two dopamine antagonists) were simulated and compared. The results show that the inactive conformations of D2 were very different when it was bound to eticlopride as opposed to risperidone. In addition D2 and D3 showed a very similar conformation when attached to eticlopride. The two drugs also bound to the inactive receptors in overlapping but different locations. These computational findings, together with experimental validations, suggest that D2 and D3 exist in several inactive states that only allow the binding of specific drugs; these states could also reflect different degrees of inactivation. Overall, the work by Lane et al. contributes to a more refined understanding of the complex conformations of GPCRs, which could be helpful to screen and develop better drugs.
Introduction
G-protein-coupled receptors (GPCRs) are important therapeutic targets for numerous human diseases. Our understanding of GPCR functional mechanisms has evolved from a simple demarcation of single active and inactive states to the appreciation and detection of multiple active states responsible for partial or biased agonism (Latorraca et al., 2017; Venkatakrishnan et al., 2013; Weis and Kobilka, 2018). High-resolution crystal structures of these proteins are vital for structure-based (rational) drug discovery (RDD) efforts designed to tailor selectivity and efficacy (Congreve et al., 2014; Michino et al., 2015a). While considerable efforts have been directed at the development of biased agonists that couple preferentially to a particular effector pathway (Free et al., 2014; Manglik et al., 2016; McCorvy et al., 2018), less attention has been dedicated to the possibility that different antagonist scaffolds with differing efficacy of inverse agonism might lead to different receptor conformations and hence different ‘inactive’ states. Such a possibility could have a major impact on RDD for antagonists, since a GPCR crystal structure stabilized by a particular antagonist might represent an invalid docking target for an antagonist that prefers a different inactive conformation. Although substantial differences in antagonist binding mode and position of the binding pockets have been revealed among different aminergic receptors, no conformational differences has been detected for the inactive state in any individual aminergic receptor (Michino et al., 2015a). In particular, although a number of antagonists derived from different scaffolds have been co-crystallized with the β2 adrenergic receptor, conformational differences among these crystal structures are minimal (Michino et al., 2015a).
Curiously, the inactive state structures of the highly homologous dopamine D2 and D3 receptors (D2R and D3R) revealed substantial differences on the extracellular side of the transmembrane domain, especially in TM6 (Figure 1), when bound with antagonists derived from different scaffolds (Chien et al., 2010; Wang et al., 2018). Specifically, the D3R structure is in complex with eticlopride, a substituted benzamide (PDB: 3PBL) (Chien et al., 2010), while the D2R structure is bound with risperidone, a benzisoxazole derivative (PDB: 6CM4) (Wang et al., 2018). The binding poses of the two ligands differ substantially. Risperidone is oriented relatively perpendicular to the membrane plane with its benzisoxazole ring penetrating into a hydrophobic pocket beneath the orthosteric binding site (OBS) of D2R; in contrast, eticlopride is oriented relatively parallel to the membrane plane and contacts the extracellular portion of TM5 in D3R, a sub-pocket that risperidone does not occupy in D2R (Sibley and Shi, 2018; Wang et al., 2018). Nemonapride, another substituted benzamide, binds in the OBS of the slightly divergent D4R (PDB: 5WIV) (Wang et al., 2017) in a manner very similar to that of eticlopride in the D3R (Sibley and Shi, 2018).
Figure 1 with 1 supplement see all
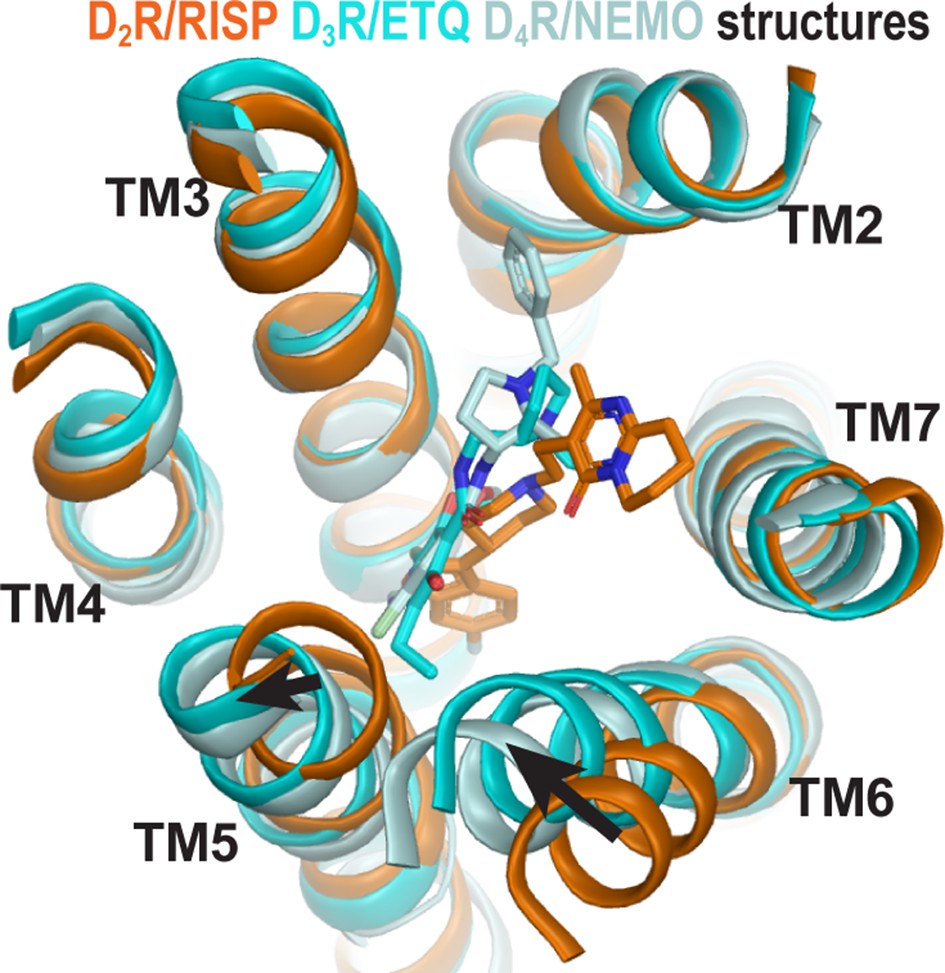
The structures of homologous D2R, D3R, and D4R show different conformations in the extracellular vestibules.
Superpositioning of D2R, D3R, and D4R structures shows that the binding of eticlopride (ETQ, cyan) in D3R and nemonapride (NEMO, pale cyan) in D4R result in outward and inward rearrangements of the extracellular portions of TM5 and TM6, respectively, compared to the binding of risperidone (RISP, orange) in D2R.
Importantly, the co-crystalized ligands (risperidone, eticlopride, and nemonapride) display little subtype selectivity across D2R, D3R, and D4R (Chien et al., 2010; Hirose and Kikuchi, 2005; Silvestre and Prous, 2005; Wang et al., 2017) (also see PDSP database; Roth et al., 2000). Given the high homology among these D2-like receptors, especially between D2R and D3R, the drastic conformational differences between the inactive state structures of these receptors may be better explained by different binding poses of antagonists bearing different scaffolds rather than inherent differences in the receptors. Thus, we hypothesized that different antagonist scaffolds may favor distinct inactive conformations of D2R. To test this hypothesis, we carried out extensive molecular dynamics (MD) simulations of D2R in complex with non-selective antagonists derived from different scaffolds to characterize the plasticity of the OBS and the extracellular loop dynamics in the inactive conformational state.
Results
The Ile3.40 sub-pocket is occupied by risperidone and spiperone but not eticlopride in D2R
Compared to eticlopride bound in the D3R structure, risperidone in the D2R structure penetrates deeper into the binding site, with its benzisoxazole moiety occupying a sub-pocket that eticlopride does not reach. By examining the D2R/risperidone structure, we found that the benzisoxazole moiety is enclosed by eight residues in D2R, which are identical among all D2-like receptors (i.e. D2R, D3R, and D4R): Cys1183.36 (superscripts denote Ballesteros-Weinstein numbering Ballesteros and Weinstein, 1995), Thr1193.37, Ile1223.40, Ser1975.46, Phe1985.47, Phe3826.44, Trp3866.48, and Phe3906.52. Notably, three of these residues (Ile1223.40, Phe1985.47, and Phe3826.44) on the intracellular side of the OBS that we previously defined (Michino et al., 2015a), accommodate the F-substitution at the tip of the benzisoxazole ring in a small cavity (termed herein as the Ile3.40 sub-pocket) (Figure 2a). Both Ile1223.40 and Phe3826.44 of this Ile3.40 sub-pocket are part of the conserved Pro5.50-Ile3.40-Phe6.44 motif that undergoes rearrangement upon receptor activation (Rasmussen et al., 2011), and we have found that the I1223.40A mutation renders D2R non-functional (Klein Herenbrink et al., 2019; Wang et al., 2018). Interestingly, this Ile3.40 sub-pocket is collapsed in both the D3R and D4R structures (Sibley and Shi, 2018; Figure 2b,c). We noted that this collapse is associated with rotation of the sidechain of Cys3.36: In the D2R/risperidone structure, the sidechain of Cys3.36 faces the OBS, whereas in the D3R/eticlopride and D4R/nemonapride structures, it rotates downwards to partially fill the Ile3.40 sub-pocket (Figure 2a–c).
Figure 2 with 1 supplement see all
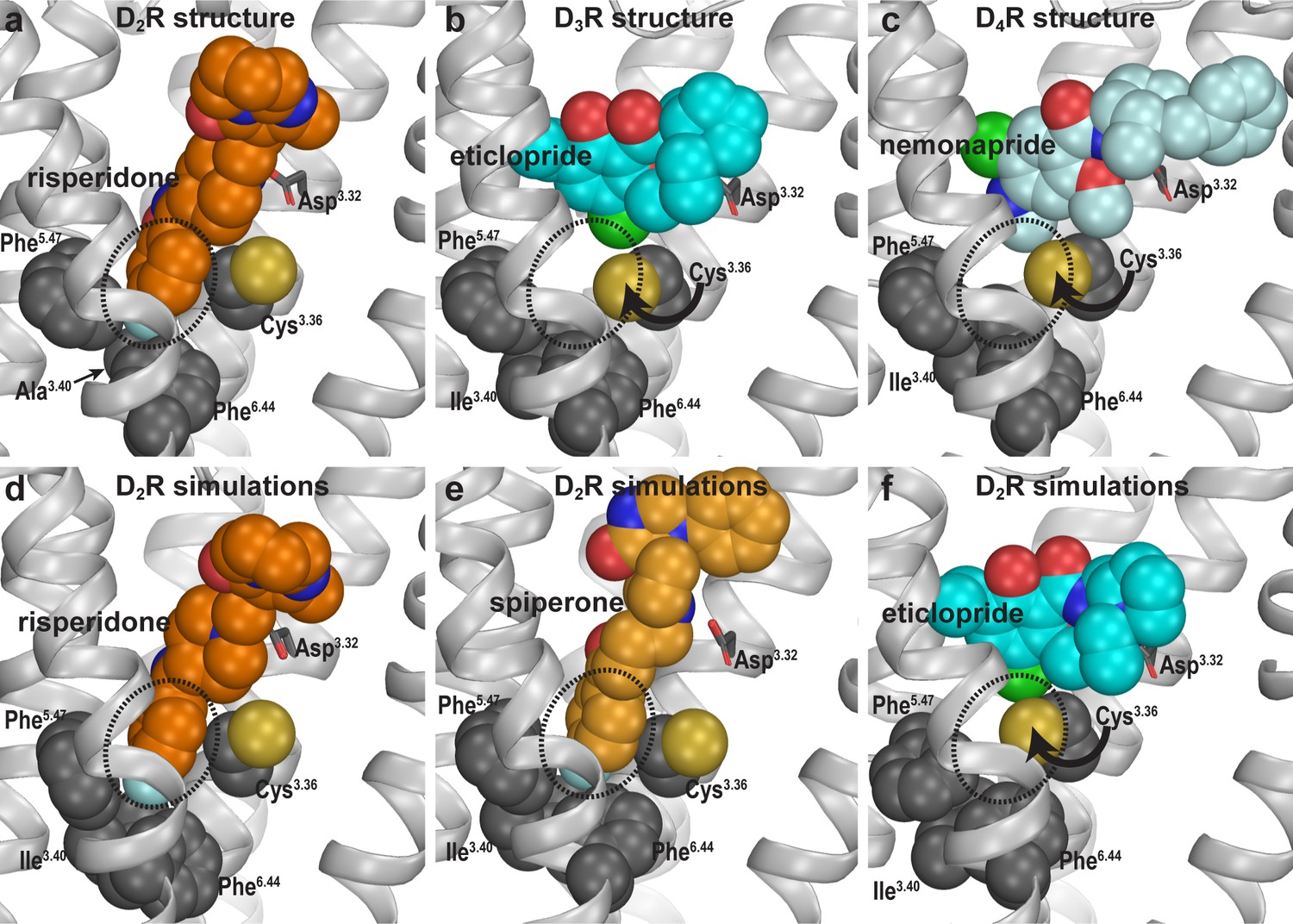
Divergent occupations of the Ile3.40 sub-pocket by non-selective ligands from different scaffolds.
In the D2R structure (a), the F-substitution on the benzisoxazole ring of risperidone occupies the Ile3.40 sub-pocket (dotted circle) enclosed by conserved Ile3.40 (mutated to Ala in the crystal structure to thermostabilize the receptor), Phe5.47, and Phe6.44. The same viewing angle shows that in the D3R (b) and D4R (c) structures, Cys3.36 rotates to fill in the Ile3.40 sub-pocket, and the substituted benzamides eticlopride and nemonapride cannot occupy the aligned sub-pockets. In our D2R/risperidone simulations (d), risperidone maintains its pose revealed by the crystal structure. In the D2R/spiperone simulations (e), the Ile3.40 sub-pocket is similarly occupied as in D2R/risperidone. In the D2R/eticlopride simulations (f), the Ile3.40 sub-pocket is collapsed as in the D3R (b) and D4R (c) structures (this trend is independent of the force field being used in the simulations).
To test our hypothesis that these observed differences in the crystal structures are due to the binding of antagonists bearing different scaffolds but not intrinsic divergence of D2-like receptors, we compared the binding modes of three non-selective antagonists in D2R. We reverted three thermostabilizing mutations introduced for crystallography (I1223.40A, L3756.37A, and L3796.41A) back to their WT residues, established WT D2R models in complex with risperidone, spiperone, or eticlopride, and carried out extensive MD simulations (see Materials and methods, Figure 1—figure supplement 1 and Table 1).
Table 1
Summary of molecular dynamics simulations.
| Receptor | Ligand | Bound na+ | Number of OPLS3e trajectories | Number of CHARMM36 trajectories | Accumulated simulation time (ns) |
|---|---|---|---|---|---|
| D2R | Risperidone | + | 12 | 28410 | |
| - | 11 | 42240 | |||
| Spiperone | + | 22 | 42000 | ||
| - | 17 | 29550 | |||
| Eticlopride | + | 5 | 12 | 51540 | |
| - | 7 | 11280 | |||
| (-)-Sulpiride | + | 3 | 4500 | ||
| - | 3 | 3600 | |||
| Aripiprazole | + | 40 | 66660 | ||
| D3R | Eticlopride | + | 3 | 13200 | |
| - | 4 | 6240 | |||
| R22 | + | 7 | 33600 | ||
| S22 | - | 7 | 59400 | ||
| Total | 120 | 33 | 392220 | ||
In our prolonged MD simulations of the WT D2R/risperidone complex (>65 µs, Table 1), we observed that risperidone stably maintains the binding pose captured in the crystal structure, even without the thermostabilizing mutations (Figure 2d). Thus, the I1223.40A mutation has minimal impact on the binding pose of risperidone. Interestingly, in the simulations of the WT D2R model in complex with spiperone, a butyrophenone derivative, the F-substitution on the butyrophenone ring similarly occupies the Ile3.40 sub-pocket as risperidone (Figure 2e). Note that the F-substitutions in risperidone and spiperone are located at similar distances to the protonated N atoms that interact with Asp3.32 (measured by the number of carbon atoms between them, Figure 1—figure supplement 1) and these two ligands appear to be optimized to occupy the Ile3.40 sub-pocket.
In contrast, in our simulations of the D2R/eticlopride complex, the eticlopride pose revealed in the D3R structure (PDB: 3PBL) is stable throughout the simulations and does not protrude into the Ile3.40 sub-pocket (Figure 2f). Consistent with the difference in the crystal structures noted above (Figure 2a,b), when risperidone and spiperone occupy the Ile3.40 sub-pocket, the sidechain of Cys1183.36 rotates away with its χ1 rotamer in gauche-, while in the presence of the bound eticlopride, this rotamer is stable in trans (Figure 2—figure supplement 1).
To validate these computational findings regarding the occupation of the Ile3.40 sub-pocket, we mutated Ile1223.40 of WT D2R to both Trp and Ala and characterized how these mutations affect the binding affinities for spiperone, risperidone, and eticlopride (Table 2). We hypothesized that the bulkier sidechain of Trp at position 3.40 would hamper the binding of spiperone and risperidone since they occupy the Ile3.40 sub-pocket but have no effect on eticlopride binding, while the smaller Ala should not affect the binding of spiperone or risperidone. Consistent with this hypothesis, the I122W mutation decreased the binding affinities of risperidone (13-fold) and spiperone (6-fold) compared to WT but had no effect on that of eticlopride. In contrast, the I122A mutation did not affect the affinities of spiperone or risperidone, which is consistent with our simulation results that show the I122A mutation has minimal impact on risperidone binding. In contrast, I122A caused a threefold increase in the affinity of eticlopride, suggesting that the I122A mutation may promote an inactive conformation of D2R that favors eticlopride binding. Together these results support our proposal that different antagonist scaffolds may favor distinct inactive conformations of D2R.
Table 2
The effect of mutations on the binding affinities of selected D2R ligands.
The affinities of [3H]spiperone were determined in saturation experiments at WT or mutant SNAP-tagged D2SRs stably expressed in FlpIn CHO cells. Binding affinity values for risperidone and eticlopride were obtained in competition binding experiments. Means of n independent experiments performed in triplicate are shown with 95% confidence intervals.
| [3H]spiperone saturation binding | [3H]spiperone competition binding | |||||
|---|---|---|---|---|---|---|
| SNAP-D2SR | pKd (Kd, nM) (95% CI) | N | Risperidone pKi (Ki, nM) (95% CI) | N | Eticlopride pKi (Ki, nM) (95% CI) | N |
| WT | 9.74 (0.18) (9.36–10.14) | 3 | 8.55 (2.8) (8.07–9.04) | 8 | 9.84 (0.14) (9.10–10.58) | 3 |
| WT -Na+ | 9.70 (0.20) (9.09–10.32) | 3 | 8.96 (1.1) (8.84–9.08) | 6 | - | |
| I1223.40A | 9.74 (0.18) (9.09–10.38) | 3 | 8.14 (7.9) (7.97–8.32) | 8 | 10.33 (0.04) (10.22–10.44) | 3 |
| I1223.40W | 8.95 (1.15) (8.59–9.30) | 3 | 7.43 (37) (7.11–7.75) | 5 | 9.61 (0.25) (9.33–9.89) | 4 |
Occupation of the Ile3.40 sub-pocket confers insensitivity to Na+ in antagonist binding
Ligand binding in D2-like receptors can be modulated by Na+ bound in a conserved allosteric binding pocket coordinated by Asp2.50 and Ser3.39 (Michino et al., 2015b; Neve, 1991; Wang et al., 2017). Note that the aforementioned Cys3.36 and Ile3.40 are adjacent to the Na+ coordinating Ser3.39; thus, we further hypothesized that the occupation of the Ile3.40 sub-pocket by spiperone or risperidone makes them insensitive to Na+. To test this hypothesis, we simulated D2R/risperidone, D2R/spiperone, D2R/eticlopride, and D2R/(-)-sulpiride complexes in the presence versus absence of bound Na+ (Table 1). Interestingly, the occupancy of the Ile3.40 sub-pocket by either spiperone or risperidone was unaffected by the presence or absence of bound Na+ (Figure 2—figure supplement 1). In contrast, while the poses of eticlopride and (-)-sulpiride are highly stable in the presence of bound Na+, they oscillated between different poses in the absence of Na+. These oscillations are associated with the sidechain of Cys3.36 swinging back and forth between the two rotamers, suggesting an important role of Na+ binding in stabilizing the poses of eticlopride and (-)-sulpiride and the configuration of the Ile3.40 sub-pocket (Figure 2—figure supplement 1). Interestingly, the previous MD simulations described by Wang et al. indicated that nemonapride’s binding pose in D4R is more stable in the presence of bound Na+ as well (Wang et al., 2017).
Consistent with these computational results, we have previously shown that spiperone binding is insensitive to the presence of Na+, while the affinities of eticlopride and sulpiride are increased in the presence of Na+ (Michino et al., 2015b). In this study, we performed binding experiments in the absence or presence of Na+ and found the affinity of risperidone to be unaffected, in accordance with this hypothesis (Table 2).
Together these findings support our hypothesis that the ability of a ligand to bind the Ile3.40 sub-pocket relates with its sensitivity to Na+ in binding, due to allosteric connections between the sub-pocket and the Na+ binding site.
Functional consequences of distinct antagonist-bound inactive conformations
To further investigate the functional impact of these conformational differences surrounding the OBS, we used a bioluminescence resonance energy transfer (BRET) assay, which measures conformational changes of the Go protein heterotrimer following activation by D2R (Michino et al., 2017), to evaluate the inverse agonism activities of several representative D2R ligands. These ligands can be categorized into two groups according to their sensitivities to Na+ in binding at D2R, which have been characterized either in our current study or in previous studies (Michino et al., 2015b; Neve, 1991; Newton et al., 2016). While risperidone, spiperone, and (+)-butaclamol have been found to be insensitive to Na+ in binding, (-)-sulpiride, eticlopride, and raclopride show enhanced binding affinities in the presence of Na+. Using quinpirole as a reference full agonist, we found that the Na+ insensitive ligands display significantly greater inverse agonism (< −30% that of the maximal response of quinpirole) relative to the Na+-sensitive ligands (> −15% that of the maximal response of quinpirole, Figure 3). These observations are consistent with findings from earlier [35S]GTPγS binding experiments of Roberts and Strange in which (+)-butaclamol, risperidone, and spiperone were found to inhibit significantly more [35S]GTPγS binding than raclopride and (-)-sulpiride (Roberts and Strange, 2005). Of note, these [35S]GTPγS-binding experiments were performed in the absence of Na+.
Figure 3
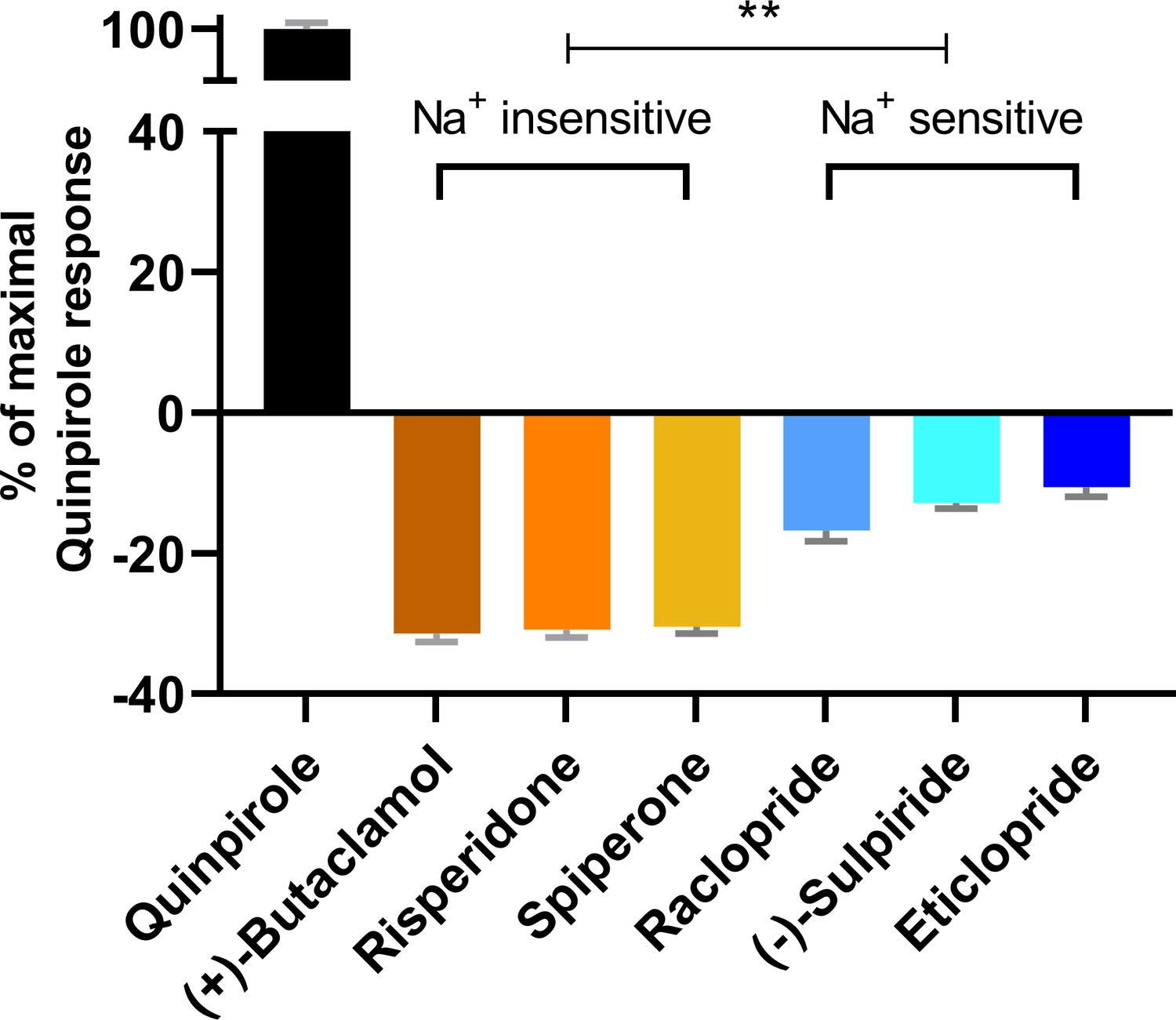
The extent of inverse agonism is negatively related with the Na+ sensitivity of ligand binding.
In a D2R-Go BRET assay, the maximal responses of the indicated ligands are normalized to that of the reference full agonist quinpirole. The ligands that are insensitive to Na+ in D2R binding display significantly higher inverse agonism (in each case, **p<0.0001 using ordinary one-way ANOVA followed by Tukey’s multiple comparisons test) than the Na+-sensitive ligands; however, within the Na+-sensitive group, raclopride is significantly different from eticlopride (p=0.005).
Based on these functional data together with the different binding modes revealed by our computational simulations, we propose that ligands that occupy the Ile3.40 sub-pocket exhibit a greater level of inverse agonism as compared to those that do not. Therefore, across the tested inverse agonists there is a negative relation between ligand sensitivity to Na+ and the extent of inverse agonism at D2R. The differential occupation of the Ile3.40 sub-pocket is the structural basis for the Na+ sensitivity, which contributes significantly to the extent of inverse agonism of the tested ligands.
Plasticity of the ligand-binding site propagates to affect the overall receptor conformation
By occupying the Ile3.40 sub-pocket, the benzisoxazole moiety of risperidone pushes the conserved Phe6.52 away from the binding site in the D2R/risperidone structure compared to its position in the D3R/eticlopride structure. This interaction is responsible for positioning the aromatic cluster of TM6 and TM7 (Trp6.48, Phe6.51, Phe6.52, His6.55, and Tyr7.35) in D2R differently from its configurations in the D3R and D4R structures, resulting in an overall outward positioning of the extracellular portion of TM6 in D2R (Figure 4—figure supplement 1). On the extracellular side of the OBS, the space near Ser5.42 and Ser5.43 that accommodates the bulky substitutions of the benzamide rings of the bound eticlopride and nemonapride in the D3R and D4R structures is not occupied by risperidone in D2R, which is likely associated with the inward movement of the extracellular portion of TM5 in D2R relative to those in the D3R and D4R structures (Figure 1).
To evaluate whether these conformational rearrangements are due to the minor divergence in these regions of the receptors or to the ligand-binding site plasticity that accommodates ligands bearing different scaffolds, we compared the resulting conformations of D2R bound with risperidone or eticlopride. We observed the same trend of rearrangements of the transmembrane segments surrounding the OBS in the resulting receptor conformations from our D2R/risperidone and D2R/eticlopride simulations (Figure 4a), that is, an inward movement of TM6 and outward movement of TM5 in the presence of the bound eticlopride (Figure 4b,c). Without such movements in D2R/eticlopride, Ser1935.42 and Ser1945.43 would clash with the bound eticlopride (Figure 4a). These findings further support our inference that differences between the D2R and D3R inactive structures are largely due to the different scaffolds of the bound non-selective ligands.
Figure 4 with 1 supplement see all

The different conformations in the extracellular vestibules of D2R and D3R are likely due to binding of non-selective ligands from different scaffolds.
(a) Compared to the comparison of the crystal structures shown in Figure 1, superpositioning of representative frames of the D2R/ETQ and D2R/RISP simulations shows a similarly trend of the outward and inward movements of TM5 and TM6, respectively, in the presence of the bound ETQ, even when the simulations were started from the D2R conformation stabilized by RISP. Note Ser1935.42 and Ser1945.43 would clash with the bound eticlopride if there was no conformational adjustment. (b, c) PIA-GPCR analysis (see Materials and methods) comparing the D2R/ETQ and D2R/RISP conformations. The analysis of the pairwise-distance differences among the subsegments (b) indicates that TM6e moves inward (smaller distance to TM2e, dark red pixel), while TM5e moves outward (larger distances to TM7e, dark blue pixel) in the D2R/ETQ simulations. The analysis of pairwise-distance differences among the Cα atoms of the ligand-binding residues (c) indicates significant changes near residues Phe1895.38, Ser1935.42, Asn3676.58, and Ile3686.59 (darker colored pixels).
The extracellular loop 2 (EL2) of D2R/risperidone can spontaneously unwind
In addition to differences in the transmembrane segments surrounding the OBS, there are also substantial differences in the configuration of EL2 in the D2R and D3R structures. EL2 between TM4 and TM5 is connected to TM3 via a disulfide bond formed between CysEL2.50 (see Materials and methods and Figure 5—figure supplement 1 for the indices of EL1 and EL2 residues) and Cys3.25. The conformation of EL2, the sequence of which is not conserved among aminergic GPCRs, is expected to be dynamic. Indeed, in the D2R/risperidone structure, the sidechains of residues 176EL2.40, 178EL2.46, 179EL2.47, and 180EL2.48, which are distal to the OBS were not solved, likely due to their dynamic nature. Curiously, the portion of EL2 C-terminal to Cys182EL2.50 (residues 182EL2.50-186EL2.54), which forms the upper portion of the OBS that is in contact with ligand, is in a helical conformation in the D2R/risperidone structure.
Strikingly, in our MD simulations of D2R complexes, we found that this helical region showed a tendency to unwind (Video 1). The unwinding of EL2 involves a drastic rearrangement of the sidechain of Ile183EL2.51, which dissociates from a hydrophobic pocket formed by the sidechains of Val1113.29, Leu1704.60, Leu174EL2.38, and Phe1895.38. Specifically, the unwinding process is initiated by the loss of a hydrogen-bond (H-bond) interaction between the sidechain of Asp1083.26 and the backbone amine group of Ile183EL2.51 formed in the D2R/risperidone structure (Figure 5—figure supplement 2b, step (i). When this interaction is broken, the orientation of residues 182EL2.50-186EL2.54 deviates markedly from that of the crystal structure, losing its helical conformation (see below). Subsequently, the sidechain of Ile183EL2.51 rotates outwards and passes a small steric barrier of Gly173EL2.37 (Figure 5—figure supplement 2b, step (ii), and in some trajectories makes a favorable hydrophobic interaction with the sidechain of Ala177EL2.45. In a few long trajectories, Ile183EL2.51 rotates further toward the extracellular vestibule where it can make favorable interactions with hydrophobic or aromatic residues from the N terminus, or the bound risperidone (Video 1). Consequently, residues 182EL2.50-186EL2.54 are in a fully extended loop conformation while Ile184EL2.52 tilts under EL2 (Figure 5—figure supplement 2b, step (iii).
Video 1
A movie of a 4.2 µs D2R/risperidone trajectory collected using the OPLS3e force field shows spontaneous unwinding of EL2.
The conformation of EL2 gradually transitions to an extended configuration similar to that in the D3R structure. See Figure 5—figure supplement 2 for the pathway of unwinding. Note that the extended conformation of EL2 stabilizes Trp100EL1.50. The Cα atom of Gly173EL2.37, the sidechains of Trp100EL1.50, Ile183EL2.51, and Ile184EL2.52 and the bound risperidone are shown as spheres. Asp1083.26 and the disulfide bond between Cys1073.25 and Cys182EL2.50 are shown as sticks. The carbon atoms of Gly173EL2.37 and Ile184EL2.52 are colored in cyan, those of Ile183EL2.51 are in green, those of Trp100EL1.50, Cys1073.25, Asp1083.26, Asn175EL2.39, and Cys182 EL2.50 are in dark gray; those of the bound ligand risperidone are in orange.
In the D3R structure, the aligned residue for Asp1083.26 of D2R is conserved as Asp1043.26; its sidechain forms an interaction not with Ile182EL2.51 but rather with the sidechain of Asn173EL2.39, which is also conserved in D2R as Asn175EL2.39. In the D4R, the aligned two residues (Asp1093.26 and Asn175EL2.39) are conserved as well, their sidechains are only 4.3 Å away in the D4R structure, a distance slightly larger than the 3.2 Å in the D3R structure. Even though these residues are conserved in D2R, the interaction in D3R (and potentially in D4R), between Asp3.26-AsnEL2.39, is not present in the D2R structure in which the aligned Asn175EL2.39 faces lipid (Figure 5—figure supplement 2a). However, in a few of our long D2R simulations, Asn175EL2.39 gradually moves inwards and approaches Asp1083.26 (Figure 5—figure supplement 2b, step (iv). At this point, the EL2 conformation of D2R is highly similar to that of D3R (Figure 5—figure supplement 2c), suggesting that EL2 is dynamic and can exist in both conformations.
We evaluated the tendency of the EL2 helix to unwind in each of the simulated D2R complexes by measuring the stability of the backbone H-bond between Ile183EL2.51 and Asn186EL2.54, a key stabilizing force of the helix (Figure 5a). When we plotted the Ile183EL2.51-Asn186EL2.54 distance against the Asp1083.26-Ile183EL2.51 distance for each D2R complex (Figure 5b), we found that the loss of the Asp1083.26-Ile183EL2.51 interaction increases the probability of breaking the Ile183EL2.51-Asn186EL2.54 H-bond, that is the unwinding of EL2. Interestingly, in all our simulated D2R complexes, EL2 has a clear tendency to unwind, regardless of the scaffold of the bound ligand (Figure 5c,d, Videos 1–3). Note that in the D3R/eticlopride simulations, the aligned residues Ser182EL2.51 and Asn185EL2.54 do not form such a H-bond, and EL2 is always in an extended conformation (Figure 5b–d). This tendency of EL2 to transition toward the extended conformation is also present in our simulations of D2R in complex with a partial agonist, aripiprazole, whereas EL2 in the D3R complexes with partial agonists (R22 and S22) remains in the extended conformation (Table 1 and Figure 5—figure supplement 3). Interestingly, Asp1043.26 and Ser182EL2.51 can move into interacting range in the D3R/eticlopride simulations, and the Ser182EL2.51-Asn185EL2.54 interaction can sporadically form in the D3R/R22 simulations – both raise the possibility that the extended conformation of D3R EL2 may transition to a helical conformation.
Figure 5 with 6 supplements see all
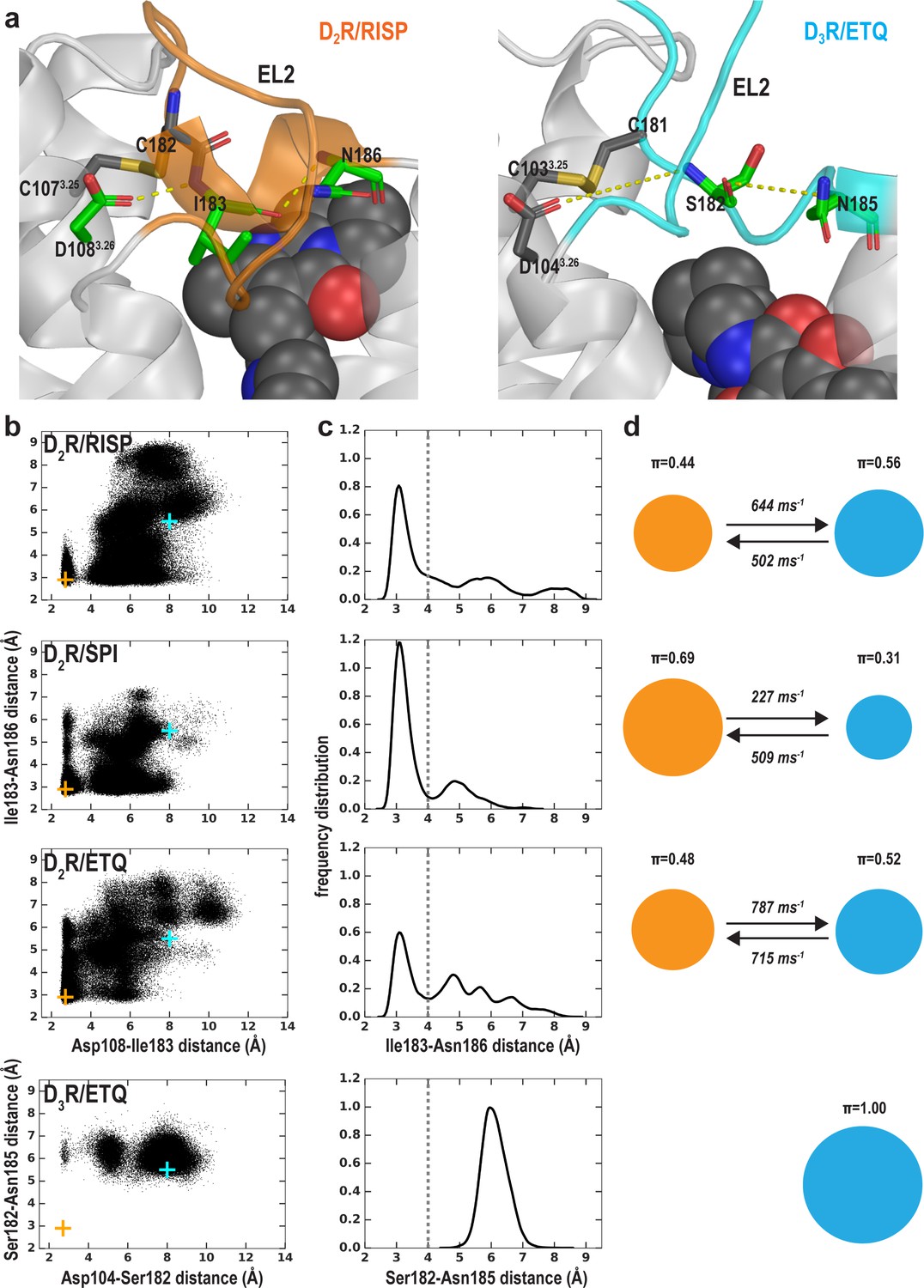
The helical conformation of EL2 in the D2R/risperidone structure has a tendency to unwind in our simulations, regardless of the bound ligand.
(a) The Ile183EL2.51-Asn186EL2.54 backbone H-bond and the Ile183EL2.51-Asp1083.26 interaction in D2R and their aligned interactions in D3R. (b) The scatter plots of the two distances in the indicated D2R and D3R complexes. The orange and cyan crosses indicated the distances in the D2R/risperidone and D3R/eticlopride structures, respectively. (c) The distributions of the EL2.51-EL2.54 distances in the indicated simulations. These distances were used to evaluate the tendency to unwind using Markov state model (MSM) analysis in d). (d) The MSM analysis of the transition between the helical and extended conformational states of EL2. The area of each disk representing a state is proportional to the equilibrium probability (π) in each simulated condition. The values from the maximum likelihood Bayesian Markov model for π and transition rates from 500 Bayesian Markov model samples are shown. Thus, EL2 in all the D2R complexes show significant tendencies to unwind, while that in D3R/eticlopride remains extended.
Video 2
A movie of a 4.2 µs D2R/eticlopride trajectory shows the dynamics of Trp100EL1.50 when the C-terminal portion of EL2 is in a helical conformation.
Note that Trp100EL1.50 can be stabilized by interacting with the disulfide bond. The presentation and color scheme are similar to those in Video 1, except that the bound carbon atoms of the ligand eticlopride are colored in cyan.
Video 3
A movie of a 3.6 µs D2R/eticlopride trajectory collected using the CHARMM36 force field shows another example of unwinding of EL2.
Thus, considering the similar unwinding pathway as that in Video 1 (Figure 5—figure supplement 2), the unwinding does not depend on the force field used in the simulations or the identity of the antagonist bound in the OBS. Note the sidechain of Asn175EL2.39 rotates inward and approaches Asp1083.26 in this trajectory. The presentation and color scheme are the same as those in Video 2.
Interestingly, in one of our long MD trajectories of the D2R/risperidone complex, EL2 evolved into a conformation that has a helical N-terminal portion and an extended C-terminal portion (Video 4 and Figure 5—figure supplement 4). This conformation is not observed in either of the D2R/risperidone and D3R/eticlopride structures but is similar to that of the 5-HT2AR/risperidone structure, further demonstrating the dynamics of this loop region (Figure 5—figure supplement 4).
Video 4
A movie of a 4.5 µs D2R/risperidone trajectory shows the N-terminal portion of EL2 can transition into a helical conformation when the C-terminal portion is extended.
This is a novel EL2 conformation that has not been revealed by the D2R, D3R or D4R structures but similar to those in the 5-HT2AR/risperidone (Figure 5—figure supplement 4f), β1AR and β2AR structures. The presentation and color scheme are the same as those in Video 1.
In marked contrast to the obvious trend toward unwinding of EL2 in all our simulated D2R complexes, in our recent simulations of MhsT, a transporter protein with a region found by crystallography to alternate between helical and unwound conformations (Malinauskaite et al., 2014), we failed to observe any spontaneous unwinding over a similar simulation timescale (with the longest simulations being ~5–6 µs) when the region was started from the helical conformation (Abramyan et al., 2018; Stolzenberg et al., 2017). This shows how difficult it can be to capture known dynamics in simulations and suggests that the C-terminal helical conformation of EL2 in D2R represents a higher energy state than the extended conformation, which allows for observation of the transitions in a simulation timescale not usually adequate to sample folding/unfolding events (Piana et al., 2011).
Both the EL2 conformation and ligand scaffold affect the EL1 conformation
We have previously shown that the divergence in both the length and number of charged residues in EL1 among D2R, D3R, and D4R is responsible for the selectivity of more extended ligands (Michino et al., 2013; Newman et al., 2012). Another striking difference in the D2R, D3R, and D4R structures is the position of the conserved TrpEL1.50 in EL1. Trp100EL1.50 is in a much more inward position in the D2R structure, making a direct contact with the bound risperidone (Figure 6a), Trp101EL1.50 in D4R interacts with the bound nemonapride that has an extended structure, whereas Trp96EL1.50 in D3R is not in contact with eticlopride (Figure 6b). Thus, we asked whether these distinct positions of TrpEL1.50 are due to the divergence in EL1 among these receptors (Michino et al., 2013) or due to the multiple inactive conformations that differentially accommodate the binding of non-selective ligands of divergent scaffolds.
Figure 6 with 2 supplements see all
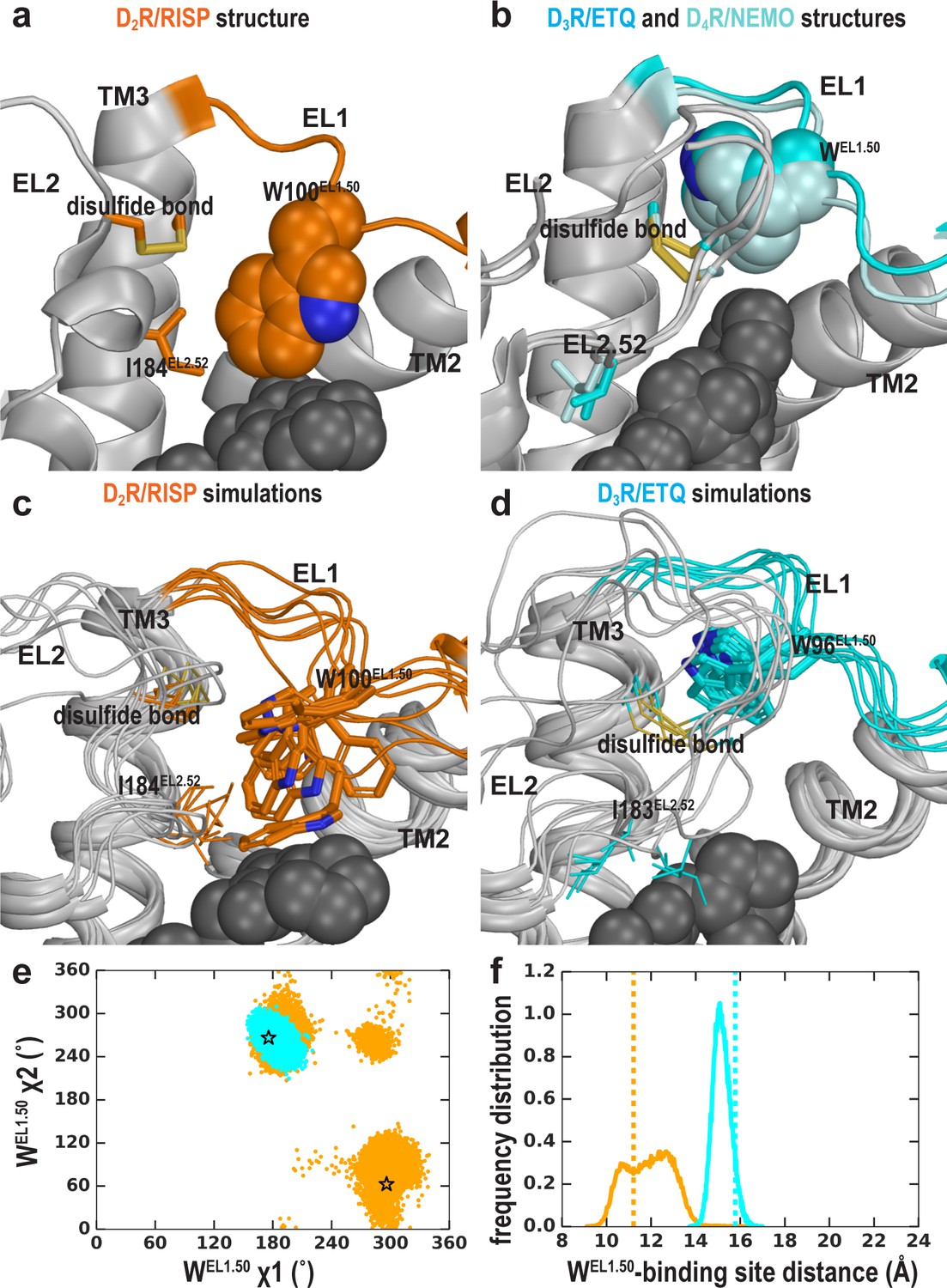
The EL2 conformation affects the EL1 conformation.
Divergent EL1-EL2 interfaces among the D2R (a), D3R, and D4R (b) structures. In the D2R structure, the Trp100EL1.50 in EL1 forms a weak interaction with Ile184EL2.52; while the aligned Trp96EL1.50 of D3R and Trp101EL1.50 in D4R are stabilized by their interactions with the disulfide bond – their passages toward the position of Trp100EL1.50 in D2R are blocked by the extended EL2. In our simulations, Trp100EL1.50 in D2R shows significant flexibility and can adopt multiple positions and orientations in D2R/risperidone (c), while Trp96EL1.50 in D3R is highly stable in D3R/eticlopride (d). (e) The χ1 and χ2 dihedral angles of Trp100EL1.50 in the subset of the D2R/risperidone simulations in which EL2 is still in a helical conformation (orange), are more widely distributed than those of Trp96EL1.50 in the D3R/eticlopride simulations in which EL2 remains in extended conformations (cyan). These dihedral angle values in the D2R and D3R structures are indicated with the orange and cyan stars, respectively. (f), For the same two sets of simulations in e, the distance between the center of mass (COM) of the sidechain heavy atoms of Trp100 in D2R and the COM of the Cα atoms of the ligand-binding site residues (excluding Trp100, see Materials and methods for the list of the residues) has wider distributions than the corresponding distance between Trp96EL1.50 in D3R and its ligand binding site. These distances in the D2R and D3R structures are indicated with the orange and cyan dotted lines, respectively.
When residues 182EL2.50-186EL2.54 of EL2 are in a helical conformation, in the D2R/risperidone simulations, we found that there is more room in the extracellular vestibule and the position of Trp100EL1.50 is flexible and can adopt several positions and orientations (Figure 6c,e,f). In the D2R/eticlopride simulations, Trp100EL1.50, which cannot interact with eticlopride, shows more flexibility than that observed in the presence of risperidone and can move to a similar position like that of Trp96EL1.50 in the D3R structure (Figure 6—figure supplement 1 and Video 2). Interestingly, in this position, the conformation of TrpEL1.50 can be stabilized by the disulfide bond of EL2 (Ioerger et al., 1999) (as shown in Video 2) or by interaction with the N terminus, which was truncated in the receptor construct used in the determination of the crystal structure. In the D2R/spiperone simulations, the phenyl substitution on the triazaspiro[4.5]decane moiety protrudes toward the interface between TM2 and TM3, and contacts Trp100EL1.50, which is flexible as well and can adopt a position that is even further away from the OBS than that of Trp96EL1.50 in the D3R structure (Figure 6—figure supplement 1).
In contrast, when EL2 is in an extended conformation like that in D3R, it restricts the flexibility of Trp100EL1.50 (Video 3). This trend is consistent with the D3R/eticlopride simulations in which we do not observe any significant rearrangement of Trp96EL1.50 (Figure 6d,e,f).
Thus, we infer that the distinct conformation of Trp100EL1.50 in the D2R structure is a combined effect of the helical EL2 conformation and the favored interaction that Trp100EL1.50 can form with the bound risperidone in the crystal structure, the latter of which however, has a limited influence on the binding affinity of risperidone (Wang et al., 2018), consistent with the unstable interaction between risperidone and Trp100EL1.50 in our simulations (Figure 6, Video 2). Indeed, in the fully extended EL2 conformation in which Ile183EL2.51 rotates to face the extracellular vestibule, Ile183EL2.51 makes a direct contact with the bound risperidone, whereas Trp100EL1.50 loses its interaction with the ligand entirely (Video 1). Nevertheless, risperidone retains all other contacts in the OBS. In the recently reported 5-HT2AR/risperidone structure (PDB: 6A93) Kimura et al. (2019), risperidone has a very similar pose in the OBS as that in the D2R structure, occupying the Ile3.40 sub-pocket as well. However, on the extracellular side of the OBS, EL2 in the 5-HT2AR/risperidone complex is in an extended conformation and the EL2 residue Leu228EL2.51 contacting risperidone aligns to Ile183EL2.51 of D2R, whereas the conserved Trp141EL1.50 does not interact with risperidone in the 5-HT2AR. It is tempting to speculate that the EL2 and EL1 dynamics we observe in the D2R/risperidone simulations represents a more comprehensive picture, as the divergent interactions shown in the extracellular loops of the 5-HT2AR/risperidone and D2R/risperidone structures may not result from differences in the protein sequences of this dynamic region between these two receptors but rather two different static snapshots due to differences in the crystallographic conditions (Note risperidone has similarly high affinities for both D2R and 5HT2AR; Kimura et al., 2019; Wang et al., 2018).
Thus, the plasticity of the OBS and the dynamics of the extracellular loops appear to be two relatively separated modules in ligand recognition. To the extent of our simulations, we did not detect strong ligand-dependent bias in the EL2 dynamics as we did for the OBS. However, when EL2 is helical, the EL1 dynamics are sensitive to the bound ligand (compare Figure 6 and Figure 6—figure supplement 1); when EL2 is extended, it restricts EL1 dynamics (Figure 6).
The Ile184EL2.52-Trp100EL1.50 interaction is not critical for risperidone binding
To further investigate the dynamics and coordination of EL2 and EL1 loops, we mutated Leu942.64, Trp100EL1.50, and Ile184EL2.52, and evaluated the effects of the L94A, W100A, and I184A, mutations on the binding affinities of eticlopride, risperidone, and spiperone. As shown in Figure 6—figure supplement 2, Leu942.64 and Trp100EL1.50 are closely associated in both the D2R and D3R structures, while Ile184EL2.52 interacts with Trp100EL1.50 only in the D2R structure. In our time-resolved energy transfer (Tr-FRET) binding experiments, using a fluorescently labeled spiperone derivate (spiperone-d2) as a tracer ligand, we found that both L94A and W100A significantly reduced the affinities of all tested antagonists, whereas I184A only reduced the affinity of eticlopride while it improved that of risperidone (Table 3). Thus, the effects of the L94A and W100A mutations have similar trends, which appear independent of the effect of I184A. Indeed, for Trp100 to switch between the positions in the D2R and D3R structures, it must pass the steric hinderance of the sidechain of Leu94; thus, some effects of the L94A mutation may reflect its perturbation of the positioning of Trp100, and vice versa.
Table 3
The effect of mutations on the binding affinities of selected D2R ligands as determined in Tr-FRET-binding experiments.
The affinities of the fluorescently labeled spiperone derivative (Spiperone-d2) or unlabeled antagonists were determined in saturation experiments at WT or mutant SNAP-tagged D2SRs stably expressed in FlpIn CHO cells. Binding affinity values for risperidone and eticlopride were obtained in competition binding experiments. Means of n independent experiments are shown with 95% confidence intervals (CIs).
| Spiperone-d2 saturation binding | Spiperone-d2 competition binding | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| SNAP-D2SR | pKd (Kd, nM) (95% CI) | N | Mut/WT | Eticlopride pKi (Ki, nM) (95% CI) | N | Mut/WT | Risperidone pKi (Ki, nM) (95% CI) | N | Mut/WT | Spiperone pKi (Ki, nM) (95% CI) | N | Mut/WT |
| WT | 8.54 (2.88) (8.32–8.77) | 9 | 1.0 | 10.06 (0.09) (9.90–10.21) | 8 | 1.0 | 8.47 (3.34) (8.15–8.80) | 7 | 1.0 | 9.96 (0.11) (9.76–10.18) | 8 | 1.0 |
| L94A | 7.71 (19.5) (7.41–8.00)* | 5 | 6.8 | 9.08 (0.83) (8.91–9.23)* | 4 | 9.2 | 8.02 (9.54) (7.86–8.17)* | 5 | 2.9 | 8.36 (4.37) (8.21–8.50)* | 5 | 39.7 |
| W100A | 7.39 (40.7) (7.21–7.56)* | 9 | 14.1 | 8.06 (8.71) (7.78–8.32)* | 4 | 96.8 | 7.60 (25.1) (7.41–7.79)* | 7 | 7.5 | 8.39 (4.07) (8.19–8.59)* | 7 | 37.0 |
| I184A | 8.79 (1.62) (8.58–9.00) | 5 | 0.6 | 9.34 (0.45) (8.94–9.75)* | 4 | 5 | 9.33 (0.47) (9.18–9.48)* | 5 | 0.1 | 9.78 (0.17) (9.51–10.05) | 5 | 1.6 |
-
*=significantly different from WT value, p<0.05, one-way ANOVA with Dunnett’s post-hoc test.
These findings support our conclusions that the close interaction between Ile184EL2.52 and Trp100EL1.50 revealed by the D2R/risperidone crystal structure is not necessary for the stabilization of the risperidone pose. Indeed, in our simulations, EL2 has significant intrinsic dynamics and transitions from the helical to unwound conformation independent of the bound ligands (see above). When it is in an extended conformation, Ile184 is dissociated from Trp100.
The clustering of the binding site conformations
Virtual screening has been widely used as an initial step in drug discovery for novel ligand scaffolds. To this end, we found that D2R can significantly change its binding site shape to accommodate antagonists bearing different scaffolds, while EL2 is intrinsically dynamic. Thus, it is necessary to comprehensively consider the binding site conformations in virtual screening campaigns against D2R, because limiting the screening to only a single conformation will miss relevant ligands. Indeed, the strategy of ensemble docking, in which each ligand is docked to a set of receptor conformers, has been adapted in recent virtual screening efforts (Amaro et al., 2018).
To characterize the OBS conformational ensemble sampled by D2R in complex with ligands bearing different scaffolds in the context of EL2 dynamics, we clustered the OBS conformations in our representative D2R/eticlopride and D2R/risperidone MD trajectories in which EL2 transitioned from helical to unwound conformations (see Materials and methods). As expected, the OBS conformations in these two complexes are significantly different and can be easily separated into distinct clusters. For the clustering results shown in Table 4, the average pairwise RMSDs of the OBS residues (apRMSDs, see Materials and methods) between the D2R/eticlopride and D2R/risperidone clusters are >1.1 Å, which are similar to that between the D2R and D3R structures (1.2 Å), while the apRMSDs within each cluster is smaller than those between any two clusters (Figure 7). Interestingly, at this level of clustering, when the two clusters for each complex are ~0.8–0.9 Å apRMSD away from each other, the extended and helical conformations of EL2 are always mixed in a cluster (Table 4). This observation suggests that the helical versus extended EL2 conformations are not closely associated with the OBS conformations.
Figure 7
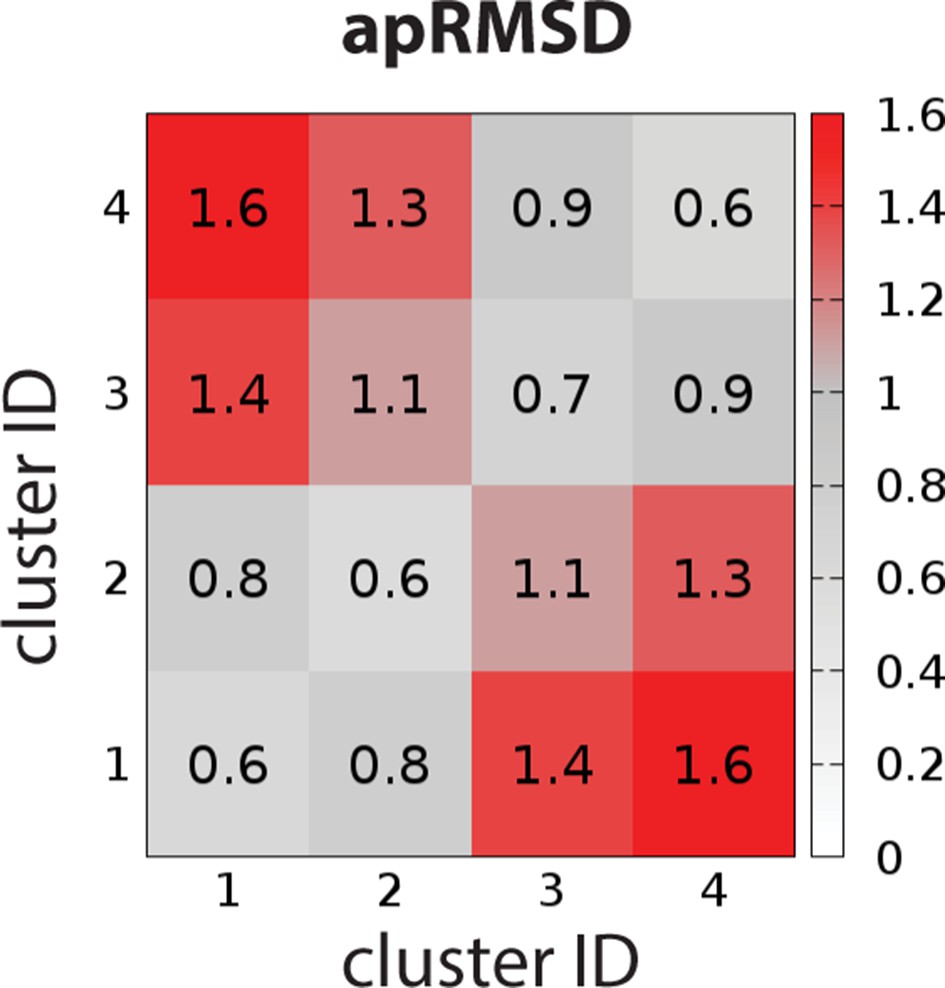
The average pairwise RMSDs of the clusters of the OBS conformations.
The clustering level was chosen to be 4, so that the average pairwise RMSDs (apRMSDs) between the D2R/eticlopride clusters (1 and 2, see Table 4 for the composition of each cluster) and D2R/risperidone clusters (3 and 4) are similar to that between D2R and D3R structures (1.2 Å), while all the apRMSDs within a cluster are smaller than those between any given two clusters.
Table 4
Clustering results of the OBS conformations sampled in the D2R/eticlopride and D2R/risperidone simulations.
The compositions in each cluster are shown as percentages of the frames randomly extracted for each complex (see Materials and methods), when sorted by either receptor/ligand complex or EL2 conformation.
| Cluster ID | Percentage (%) | |||||||
|---|---|---|---|---|---|---|---|---|
| Complex | EL2 conformation | |||||||
| D2R/eticlopride | D2R/risperidone | Extended | Helical | |||||
| Mean | Sd | Mean | Sd | Mean | Sd | Mean | Sd | |
| 1 | 38.4 | 0.7 | 0.0 | 0.0 | 4.9 | 0.4 | 33.5 | 0.5 |
| 2 | 61.6 | 0.7 | 0.0 | 0.0 | 45.1 | 0.4 | 16.5 | 0.6 |
| 3 | 0.0 | 0.0 | 43.7 | 1.0 | 2.5 | 0.4 | 41.3 | 0.8 |
| 4 | 0.0 | 0.0 | 56.3 | 1.0 | 47.5 | 0.4 | 8.7 | 0.8 |
Thus, while the centroid frames from each cluster can form an ensemble for future virtual screening for the primary scaffold occupying the OBS, in order to discover novel extended ligands that protrude out of the OBS to interact with EL2 and EL1 residues (Michino et al., 2015a), additional frames that cover both helical and extended EL2 conformations from each cluster will have to be used to screen for the optimal extensions of the primary scaffold.
Discussion
Our results highlight unappreciated conformational complexity of the inactive state of GPCRs and suggest that the risperidone bound D2R structure represents only one of a number of possible inactive conformations of D2R. Critically, this conformation is incompatible with the binding of other high-affinity D2R ligands such as eticlopride. While distinct conformational states responsible for functional selectivity have garnered great attention, the potential existence of divergent inactive conformations is of critical importance as well. By combining in silico and in vitro findings, we propose that occupation of the Ile3.40 sub-pocket by antagonists confers a distinct D2R conformation that is associated with both a greater degree of inverse agonism and Na+ insensitivity in binding, such that Na+ sensitivity is negatively related with the extent of inverse agonism for the tested ligands. However, other structural elements may also contribute to the extent of inverse agonism (Zhang et al., 2014). Regardless, the distinct inactive conformations stabilized by antagonists with different scaffolds may reflect different degrees of inactivation.
In addition to advancing our mechanistic understanding of receptor function, our findings have implications for high-throughput virtual screening campaigns, as important hits would be missed by focusing on a single inactive state captured in a crystal structure that is stabilized by an antagonist bearing a specific scaffold. Moreover, rational lead optimization requires rigorous physical description of molecular recognition (Beuming and Shi, 2017), which depends on adequate understanding of the conformational boundary and flexibility of the targeted state. We have shown previously that both dopamine receptor subtype selectivity and modulation of agonist efficacy can be achieved through the design of ligands that extend from the OBS into an extracellular secondary binding pocket (SBP) (Michino et al., 2015a; Newman et al., 2012). We now show that one might consider the occupation of the Ile3.40 sub-pocket in the process of decorating an D2R antagonist scaffold to attain a desired level of inverse agonism. Our findings also reveal allosteric communication between the IIe3.40 sub-pocket and the Na+-binding site. Thus, Na+ sensitivity in antagonist binding may provide useful mechanistic insights as part of such efforts.
The mutation of Trp100EL1.50 in D2R to alanine, leucine or phenylalanine cause substantial increases in both the association and dissociation rate of risperidone (Wang et al., 2018). Curiously, both the dissociation and association rates of D2R antagonists used as antipsychotics have been proposed to determine their propensity to cause extrapyramidal side-effects and hyperprolactinaemia (Seeman, 2014; Sykes et al., 2017). Our results indicate that both the EL2 conformation and antagonist scaffolds may influence the dynamics of Trp100EL1.50, which in turn controls ligand access and egress to and from the OBS. Thus, understanding the relationship between the distinct inactive D2R conformations stabilized by different antagonist scaffolds and these kinetic parameters will likely be important to facilitate the design of D2R antagonists with an optimal kinetic profile that minimizes the risk of side effects.
Previously, using the substituted-cysteine accessibility method (SCAM) in D2R (Javitch et al., 2000; Shi and Javitch, 2004), we found that G173EL2.37C, N175 EL2.39C, and I184EL2.52C were accessible to charged MTS reagents and that this accessibility could be blocked by the bound Na+-sensitive antagonist sulpiride, consistent with their water accessibility and involvement in ligand binding and not with a static orientation facing lipid, whereas A177EL2.45C and I183EL2.51C were accessible but not protected by sulpiride. Curiously, in the D2R/risperidone structure, Ile184EL2.52 is only marginally in contact with the ligand, Ile183EL2.51 blocks the accessibility of Gly173EL2.37 to the OBS and is itself buried in a hydrophobic pocket, whereas Asn175EL2.39 faces lipid, where it would be much less reactive. In the D3R/eticlopride structure, Ile183EL2.52 is in close contact with the bound ligand, Ser182EL2.51 faces the extracellular vestibule, whereas the sidechain of Asn173EL2.39 is oriented toward the OBS (Figure 5—figure supplement 5). Thus, our analysis shows that the accessibility pattern of EL2 revealed by previous SCAM studies in D2R are more consistent with the extended EL2 conformation revealed by the D3R/eticlopride structure but not with the D2R/risperidone structure. Indeed, we observed spontaneous transitions of EL2 from a helical to extended conformation in our D2R simulations, which suggests that EL2 of D2R exists in an ensemble of structured and unwound conformations, with substantial occupation of the configuration found in the D3R structure. Such dynamics of EL2 suggest that the drastically different conformations between the D2R and D3R structures near EL2 are not related to the divergence of the receptors. Thus, the D2R EL2 appears to have quite dramatic dynamics that are not captured by the crystal structure.
Taken together, our findings reveal that both the plasticity of the transmembrane domain in accommodating different scaffolds and the dynamics of EL2 and EL1 are important considerations in RDD targeting the inactive conformation of D2R.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Cricetulus griseus) | FlpIn CHO | Invitrogen | Cat# R75807 | |
| Transfected construct (human) | SNAP-D2SR | Cisbio | Cat# pSNAPD2 | |
| Transfected construct (human) | D2R GαoA-RLuc8 Gβ1 Gγ2-Venus | Michino et al., 2017 | N/A | |
| Commercial assay or kit | Spiperone-d2 SNAP-Lumi4-Tb 5x SNAP/CLIP labeling medium | Cisbio | Cat# L0002RED Cat# SSNPTBX Cat# LABMED | |
| Chemical compound, drug | Na bisulfite Glucose (+)-Butaclamol Risperidone Haloperidol | Sigma Aldrich | Cat# 243973 Cat# D9434 Cat# D033 Cat# R3030 Cat# H1512 | |
| Chemical compound, drug | Spiperone | Cayman chemicals | Cat# 19769 | |
| Chemical compound, drug | Eticlopride HCl Raclopride (-)-Sulpiride Quinpirole | Tocris Bioscience | Cat# 1847 Cat# 1810 Cat# 0895 Cat# 1061 | |
| Chemical compound, drug | [3H]spiperone | Perkin Elmer | Cat# NET1187250UC | |
| Chemical compound, drug | Polyethylenimine | Polysciences | Cat# 23966 | |
| Chemical compound, drug | Coelenterazine-h | NanoLight Technology | Cat# 301–5 | |
| Software, algorithm | Prism | GraphPad | v7.0 and v8.2.1 |
Residue indices in EL1 and EL2
Request a detailed protocolBased on a systematic analysis of aminergic receptors, we found a Trp in the middle of EL1 and the disulfide-bonded Cys in the middle of EL2 are the most conserved residues in each segment, and defined their residue indices as EL1.50 and EL2.50, respectively (Michino et al., 2015a), In this study, for the convenience of comparisons among D2R, D3R, and D4R, and 5-HT2AR, based on the alignments of EL1 And EL2 shown in Figure 5—figure supplement 1, we index the EL1 and EL2 residues of each receptor in the same way as the Ballesteros-Weinstein numbering, for example the residues before and after the EL2.50 are EL2.49 and EL2.51, respectively. Note the indices for the shorter sequences are not necessarily be consecutive, given the gaps in the alignment.
Molecular modeling and docking
Request a detailed protocolThe D2R models in this study are based on the corrected crystal structure of D2R bound to risperidone (PDB: 6CM4) (Wang et al., 2018). We omitted T4 Lysozyme fused into intracellular loop 3. Three thermostabilizing mutations (Ile1223.40A, L3756.37A, and L3796.41A) were reverted to their WT residues. The missing N terminus in the crystal structure was built de novo using Rosetta (Bradley et al., 2005), and then integrated with the rest of the D2R model using Modeller (John and Sali, 2003). Using Modeller, we also extended two helical turns at the TM5 C terminus and three residues at the TM6 N terminus of the structure and connected these two ends with a 9 Gly loop, similar to our experimentally validated treatment of D3R models (Michino et al., 2017). The position of the Na+ bound in the canonical Na+-binding site near the negatively charged Asp2.50 was acquired by superimposing the Na+-bound structure of adenosine A2A receptor (Liu et al., 2012) to our D2R models.
The binding poses of risperidone and eticlopride were taken according to their poses in the D2R (Wang et al., 2018) and D3R (Chien et al., 2010) structures, respectively. Docking of spiperone in our D2R model was performed using the induced-fit docking (IFD) protocol (Sherman et al., 2006) in the Schrodinger software (release 2017–2; Schrodinger, LLC: New York NY). Based on our hypothesis regarding the role of the Ile3.40 sub-pocket in the Na+ sensitivity (see text), from the resulting poses of IFD, we choose the spiperone pose with the F-substitution on the butyrophenone ring occupying the Ile3.40 sub-pocket. Note that in risperidone and spiperone the F-substitutions have similar distances to the protonated N atoms that interact with Asp3.32 (measured by the number of carbon atoms between them, Figure 1—figure supplement 1).
Molecular dynamics (MD) simulations
Request a detailed protocolMD simulations of the D2R and D3R complexes were performed in the explicit water and 1-palmitoyl-2-oleoylphosphatidylcholine (POPC) lipid bilayer environment using Desmond MD System (version 4.5; D. E. Shaw Research, New York, NY) with either the OPLS3e force field (Roos et al., 2019) or the CHARMM36 force field (Best et al., 2012; Klauda et al., 2010; MacKerell et al., 1998; MacKerell et al., 2004) and TIP3P water model. For CHARMM36 runs, the eticlopride parameters were obtained through the GAAMP server (Huang and Roux, 2013), with the initial force field based on CGenFF assigned by ParamChem (Vanommeslaeghe et al., 2010). The system charges were neutralized, and 150 mM NaCl was added. Each system was first minimized and then equilibrated with restraints on the ligand heavy atoms and protein backbone atoms, followed by production runs in an isothermal–isobaric (NPT) ensemble at 310 K and one atm with all atoms unrestrained, as described previously (Michino et al., 2017; Michino et al., 2015b). We used Langevin constant pressure and temperature dynamical system (Feller et al., 1995) to maintain the pressure and the temperature, on an anisotropic flexible periodic cell with a constant-ratio constraint applied on the lipid bilayer in the X-Y plane. For each condition, we collected multiple trajectories, the aggregated simulation length is ~392 μs (Table 1).
While the majority of our D2R simulations in this study used the OPLS3e force field, to compare with the D3R simulations using CHARMM36 that have been continued from the previously reported shorter trajectories (Michino et al., 2017; Michino et al., 2015b), we carried out the D2R/eticlopride simulations using both the OPLS3e and CHARMM36 force fields (see Table 1). We did not observe significant differences and pooled their results together for the analysis.
Conformational analysis
Request a detailed protocolDistances and dihedral angles of MD simulation results were calculated with MDTraj (version 1.8.2) (McGibbon et al., 2015) in combination with in-house Python scripts.
To characterize the structural changes in the receptor upon ligand binding, we quantified differences of structural elements between the D2R/eticlopride and D2R/risperidone conditions (using last 600 ns from a representative trajectory for each condition), by applying the previously described pairwise interaction analyzer for GPCR (PIA-GPCR) (Michino et al., 2017). The subsegments on the extracellular side of D2R were defined as following: TM1e (the extracellular subsegment (e) of TM1, residues 31–38), TM2e (residues 92–96), TM3e (residues 104–113), TM4e (residues 166–172), TM5e (residues 187–195), TM6e (residues 364–369), and TM7e (residues 376–382).
For the PIA-GPCR analysis in Figure 4 and the distance analysis in Figure 6, we used the set of ligand-binding residues previously identified by our systematic analysis of GPCR structures. Specifically, for D2R, they are residues 91, 94, 95, 100, 110, 111, 114, 115, 118, 119, 122, 167, 184, 189, 190, 193, 194, 197, 198, 353, 357, 360, 361, 364, 365, 367, 368, 376, 379, 380, 383, 384, 386, and 387; for D3R, they are residues 86, 89, 90, 96, 106, 107, 110, 111, 114, 115, 118, 165, 183, 188, 189, 192, 193, 196, 197, 338, 342, 345, 346, 349, 350, 352, 353, 362, 365, 366, 369, 370, 372, and 373.
For the clustering of the OBS conformations, we used representative D2R/eticlopride and D2R/risperidone MD trajectories in which EL2 transitioned from the helical to unwound conformations. For each complex, using the Ile183-Asn186 distance as a criterion to differentiate the EL2 conformation (Figure 5), 1000 MD frames with helical EL2 conformations and another 1000 frames with extended EL2 conformations were randomly selected. For these 4000 frames, the pairwise RMSD of the backbone heavy atoms of the OBS residues defined in Michino et al. (2015a), except for Ile184EL2.52, were calculated. The resulting 4000 × 4000 matrix was used to cluster these frames using the k-mean algorithm implemented in R. We chose nstart to be 20 to assure the convergence of cluster centroids and boundaries. We chose the clustering level to be 4, so that the average pairwise RMSDs (apRMSDs) between the D2R/eticlopride and D2R/risperidone clusters are similar to that between D2R and D3R structures (1.2 Å), while all the apRMSDs within a cluster are smaller than those between any given two clusters. The same frame selection and clustering procedure was repeated to 20 times. The averages of these 20 runs for the compositions of each cluster were reported in Table 4.
Markov State Model (MSM) analysis
Request a detailed protocolThe MSM analysis was performed using the pyEMMA program (version 2.5.5) (Scherer et al., 2015). To characterize the dynamics of EL2 of D2R, specifically the transitions between helical and extended conformations of its C-terminal portion, we focused on a key hydrogen bond formed in the helical conformation between the backbone carbonyl group of Ile183 and the backbone amine group of Asn186. Thus, for each of the simulated conditions, the distance of Ile183-Asn186 (Ser182-Asn185 in D3R) was used as an input feature for the MSM analysis. We discretized this feature into two clusters – distances below and above 4 Å (i.e. EL2 forming a helical conformation and unwinding). Implied relaxation timescale (ITS) (Swope et al., 2004) for the transition between these clusters was obtained as a function of various lag times. Convergences of ITS for the MSMs for all conditions was achieved at a lag time of 300 ns (Figure 5—figure supplement 6), which we further used to estimate Bayesian Markov models with 500 transition matrix samples (Trendelkamp-Schroer and Noé, 2013). The maximum likelihood transition matrix was used to calculate the transition and equilibrium probabilities (π) shown in Figure 5 and Figure 5—figure supplement 3.
Cell culture and cell line generation
Request a detailed protocolSite-directed mutagenesis was performed using the Quickchange method using pEF5/DEST/FRT plasmid encoding FLAG-SNAP-D2SR as the DNA template. The mutagenesis was confirmed, and the full coding region was checked using Sanger sequencing at the DNA Sequencing Laboratory (University of Nottingham). Stable cell lines were generated using the Flp-In recombination system (Invitrogen).
[3H]spiperone binding assay
Request a detailed protocolFlpIn CHO cells (Invitrogen) stably expressing WT or mutant SNAP-D2s cells were cultured before the preparation of cell membrane as described before (Klein Herenbrink et al., 2019). All stable cell lines were confirmed to be mycoplasma free. For saturating binding assays cell membranes (Mutant or WT SNAP-D2s-FlpIn CHO, 2.5 µg) were incubated with varying concentrations of [3H]spiperone and 10 µM haloperidol as a non-specific control, in binding buffer (20 mM HEPES, 100 mM NaCl, 6 mM MgCl2, 1 mM EGTA, and 1 mM EDTA, pH 7.4) to a final volume of 200 μL and were incubated at 37°C for 3 hr. For competition binding assays, cell membranes (SNAP-D2s-FlpIn CHO, 2.5 µg) were incubated with varying concentrations of test compound in binding buffer containing 0.2 nM of [3H]spiperone to a final volume of 200 µL and were incubated at 37°C for 3 hr. Binding was terminated by fast-flow filtration using a Uniplate 96-well harvester (PerkinElmer) followed by five washes with ice-cold 0.9% NaCl. Bound radioactivity was measured in a MicroBeta2 LumiJET MicroBeta counter (PerkinElmer). Data were collected from at least three separate experiments performed in triplicate and analysed using non-linear regression (Prism 7, Graphpad software). For radioligand saturation binding data, the following equation was globally fitted to nonspecific and total binding data:
(1)
where Y is radioligand binding, Bmax is the total receptor density, [A] is the free radioligand concentration, KA is the equilibrium dissociation constant of the radioligand, and NS is the fraction of nonspecific radioligand binding. The Bmax of the SNAP-tagged D2SRs we as follows; WT = 7.95 ± 1.63 pmol.mg−1, 6.39 ± 1.04 pmol.mg−1, 4.37 ± 0.92 pmol.mg−1, 2.61 ± 0.50 pmol.mg−1.
For competition binding assays, the concentration of ligand that inhibited half of the [3H]spiperone binding (IC50) was determined by fitting the data to the following equation:
(2)
where Y denotes the percentage specific binding, Top and Bottom denote the maximal and minimal asymptotes, respectively, IC50 denotes the X-value when the response is midway between Bottom and Top, and nH denotes the Hill slope factor. IC50 values obtained from the inhibition curves were converted to Ki values using the Cheng and Prusoff equation. No statistical methods were used to predetermine sample size.
Bioluminescence resonance energy transfer (BRET) assay
Request a detailed protocolThe Go-protein activation assay uses a set of BRET-based constructs previously described (Michino et al., 2017). Briefly, HEK293T cells were transiently co-transfected with pcDNA3.1 vectors encoding (i) D2R, (ii) GαoA fused to Renilla luciferase 8 (Rluc8; provided by Dr. S. Gambhir, Stanford University, Stanford, CA) at residue 91, (iii) untagged Gβ1, and (iv) Gγ2 fused to mVenus. Transfections were performed using polyethyleneimine (PEI) at a ratio of 2:1 (PEI:total DNA; weight:weight), and cell culture was maintained as described previously (Bonifazi et al., 2019). After ~48 hr of transfection, cells were washed with PBS and resuspended in PBS + 0.1% glucose + 200 µM Na Bisulfite buffer. Approximately 200,000 cells were then distributed in each well of the 96-well plates (White Lumitrac 200, Greiner bio-one). 5 μM Coelenterazine H, a luciferase substrate for BRET, was then added followed by addition of vehicle and test compounds using an automated stamp transfer protocol (Nimbus, Hamilton Robotics) from an aliquoted 96-well compound plate. Following ligands were used – quinpirole, eticlopride, raclopride, and (-)-sulpiride (Tocris Bioscience), (+)-butaclamol, dopamine, and risperidone (Sigma Aldrich), and Spiperone (Cayman chemicals). mVenus emission (530 nm) over RLuc 8 emission (485 nm) were then measured after 30 min of ligand incubation at 37°C using a PHERAstar FSX plate reader (BMG Labtech). BRET ratio was then determined by calculating the ratio of mVenus emission over RLuc eight emission.
Data were collected from at least nine independent experiments and analyzed using Prism 7 (GraphPad Software). Drug-induced BRET, defined by BRET ratio difference in the presence and absence of compounds, was calculated. Concentration response curves (CRCs) were generated using a non-linear sigmoidal dose-response analyses using Prism 7 (GraphPad Software). CRCs are presented as mean drug-induced BRET ± SEM. Emax bar graphs are plotted as the percentage of maximal drug-induced BRET by quinpirole ± SEM.
Tr-FRET ligand binding
Materials: Spiperone-d2, SNAP-Lumi4-Tb and 5x SNAP/CLIP labeling medium were purchased from Cisbio Bioassays. Eticlopride hydrochloride was purchased from Tocris Bioscience. Saponin was purchased from Fluka/Sigma-Aldrich. Bromocriptine, haloperidol, risperidone, spiperone, pluronic-F127, Gpp(NH)p, DNA primers, Hanks Balanced Salt Solution H8264 (HBSS) and phosphate buffered saline (PBS) was purchased from Sigma-Aldrich.
Terbium cryptate labeling and membrane preparation
Request a detailed protocolTerbium cryptate labeling of the SNAP-tagged receptors and membrane preparation was performed with minor changes to previously described methods (Klein Herenbrink et al., 2016). Flp-In CHO-K1 cells stably expressing the mutant SNAP-D2SR constructs were grown in T175 flasks to approximately 90% confluency. Cell media was aspirated, and the cells were washed twice with 12 mL PBS. The cells were then incubated with terbium cryptate labeling reagent in 1xSNAP/CLIP labeling medium for 1 hr at in a humidified cell culture incubator with 5% CO2 at 37°C. The terbium cryptate labeling reagent was then removed and the cells were washed once with 12 mL PBS. The labeled cells were then harvested in 10 mL PBS by cell scraping. Harvested cells were then collected by centrifugation at 300 g for 5 min and removal of the supernatant. The cell pellets were then frozen at −80°C for later membrane preparation. For cell membrane preparation, each cell pellet was removed from the −80°C freezer and thawed on ice. The pellet was then resuspended in 10 mL of ice-cold Buffer 1 (10mM HEPES 10 mM EDTA pH7.4). The pellet was then homogenised (IKA works T 10 basic Ultra-Turrax homogeniser) with eight bursts of 3 s on setting 4. The homogenized cells were transferred to an ultra-fast centrifuge tube and an additional 10 mL of Buffer one was added. The tube was then centrifuged at 48,000 g for 30 min at 4°C. The supernatant was discarded, 20 mL of Buffer one was added and the pellet was resuspended. The resuspension was then centrifuged a second time at 48,000 g for 30 min at 4°C. The supernatant was then removed, and the cell membrane pellet was collected by resuspension in 2 mL ice-cold Buffer 2 (10mM HEPES 0.1 mM EDTA pH 7.4). The resuspended membranes were then put through a syringe with a BD precision glide 26-gauge needle to make the solution uniform. Membrane protein concentration was determined by bicinchonic acid (BCA) assay detecting the absorbance at 562 nm on a CLARIOstar plate reader (BMG Labtech) using bovine serum albumin (BSA) as the protein standard. The cell membrane solution was then aliquoted and frozen at −80°C.
TR-FRET binding assay
Request a detailed protocolAll ligands were diluted in Binding Buffer (Hanks Balanced Salt Solution (Sigma H8264), 20 mM HEPES, 0.02% Pluronic-F127, 1% dimethyl sulfoxide, pH 7.4 (with KOH)). For competition binding experiments; 10 μL of spiperone-d2 in Binding Buffer was added to each well of a 384-well white optiplate LBS coated (PerkinElmer) at varied concentrations depending on the SNAP-D2SR mutant. 10 μL of increasing concentrations of unlabeled ligands were then added into the 10 μL of fluorescent ligand and mixed. A final concentration of 100 μM haloperidol was used to determine non-specific binding. Cell membranes were diluted to 0.075 μg/μL in Binding Buffer supplemented with 50 μg/mL saponin and 100 μM Gpp(NH)p.
TR-FRET measurements were acquired on a PHERAstar FS plate reader (BMG Labtech) at 37°C. The optiplate containing the ligand cocktails in the wells was incubated in the instrument for 6 min. The cell membrane solution was primed into the on-board injection system and incubated for 5 min. 20 μL of cell membrane solution was injected at 400 μL/s into the ligand cocktail wells to initiate the binding reaction. After 30-min incubation, the HTRF optic filter module was used to perform an excitation at 337 nm and simultaneous dual emission detection at 620 nm (terbium cryptate donor) and 665 nm (fluorescent ligand acceptor). The focal height was set to 10.4 mm. All experiments were performed in singlet wells. The TR-FRET binding values were determined by dividing the by the fluorescent ligand acceptor channel values by the terbium cryptate donor channel values and multiplying by 10,000. These values were then subtracted by the non-specific binding values determined in each experiment to give the specific HTRF ratio x 10,000. The data was then analysed with GraphPad Prism 8.2.1 using Equations 1 and 2.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Exploring substrate binding in the extracellular vestibule of MhsT by atomistic simulations and markov modelsJournal of Chemical Information and Modeling 58:1244–1252.https://doi.org/10.1021/acs.jcim.8b00175
-
Ensemble docking in drug discoveryBiophysical Journal 114:2271–2278.https://doi.org/10.1016/j.bpj.2018.02.038
-
Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral anglesJournal of Chemical Theory and Computation 8:3257–3273.https://doi.org/10.1021/ct300400x
-
Editorial: computer aided Structure-based lead optimizationCurrent Topics in Medicinal Chemistry 17:2575–2576.https://doi.org/10.2174/156802661723170808161306
-
Novel and potent dopamine D2 Receptor Go-Protein Biased AgonistsACS Pharmacology & Translational Science 2:52–65.https://doi.org/10.1021/acsptsci.8b00060
-
Structure-based drug design for G protein-coupled receptorsProgress in Medicinal Chemistry 53:1–63.https://doi.org/10.1016/B978-0-444-63380-4.00001-9
-
Constant pressure molecular dynamics simulation: the langevin piston methodThe Journal of Chemical Physics 103:4613–4621.https://doi.org/10.1063/1.470648
-
Aripiprazole, a novel antipsychotic agent: dopamine D2 receptor partial agonistThe Journal of Medical Investigation 52:284–290.https://doi.org/10.2152/jmi.52.284
-
Automated force field parameterization for nonpolarizable and polarizable atomic models based on ab initio target dataJournal of Chemical Theory and Computation 9:3543–3556.https://doi.org/10.1021/ct4003477
-
Comparative protein structure modeling by iterative alignment, model building and model assessmentNucleic Acids Research 31:3982–3992.https://doi.org/10.1093/nar/gkg460
-
Structures of the 5-HT2A receptor in complex with the antipsychotics risperidone and zotepineNature Structural & Molecular Biology 26:121–128.https://doi.org/10.1038/s41594-018-0180-z
-
Update of the CHARMM all-atom additive force field for lipids: validation on six lipid typesThe Journal of Physical Chemistry B 114:7830–7843.https://doi.org/10.1021/jp101759q
-
The role of kinetic context in apparent biased agonism at GPCRsNature Communications 7:10842.https://doi.org/10.1038/ncomms10842
-
Molecular determinants of the intrinsic efficacy of the antipsychotic aripiprazoleACS Chemical Biology 14:1780–1792.https://doi.org/10.1021/acschembio.9b00342
-
GPCR dynamics: structures in motionChemical Reviews 117:139–155.https://doi.org/10.1021/acs.chemrev.6b00177
-
All-atom empirical potential for molecular modeling and dynamics studies of proteinsThe Journal of Physical Chemistry B 102:3586–3616.https://doi.org/10.1021/jp973084f
-
Improved treatment of the protein backbone in empirical force fieldsJournal of the American Chemical Society 126:698–699.https://doi.org/10.1021/ja036959e
-
A mechanism for intracellular release of na+ by neurotransmitter/sodium symportersNature Structural & Molecular Biology 21:1006–1012.https://doi.org/10.1038/nsmb.2894
-
Structure-inspired design of β-arrestin-biased ligands for aminergic GPCRsNature Chemical Biology 14:126–134.https://doi.org/10.1038/nchembio.2527
-
MDTraj: a modern open library for the analysis of molecular dynamics trajectoriesBiophysical Journal 109:1528–1532.https://doi.org/10.1016/j.bpj.2015.08.015
-
Structural basis for na+-sensitivity in dopamine D2 and D3 receptorsChemical Communications 51:8618–8621.https://doi.org/10.1039/c5cc02204e
-
Toward understanding the structural basis of partial agonism at the dopamine D3 ReceptorJournal of Medicinal Chemistry 60:580–593.https://doi.org/10.1021/acs.jmedchem.6b01148
-
Regulation of dopamine D2 receptors by sodium and pHMolecular Pharmacology 39:570–578.
-
Molecular determinants of selectivity and efficacy at the dopamine D3 receptorJournal of Medicinal Chemistry 55:6689–6699.https://doi.org/10.1021/jm300482h
-
Mechanisms of inverse agonist action at D2 dopamine receptorsBritish Journal of Pharmacology 145:34–42.https://doi.org/10.1038/sj.bjp.0706073
-
OPLS3e: extending force field coverage for Drug-Like small moleculesJournal of Chemical Theory and Computation 15:1863–1874.https://doi.org/10.1021/acs.jctc.8b01026
-
PyEMMA 2: a software package for estimation, validation, and analysis of markov modelsJournal of Chemical Theory and Computation 11:5525–5542.https://doi.org/10.1021/acs.jctc.5b00743
-
Clozapine, a fast-off-D2 antipsychoticACS Chemical Neuroscience 5:24–29.https://doi.org/10.1021/cn400189s
-
Novel procedure for modeling ligand/receptor induced fit effectsJournal of Medicinal Chemistry 49:534–553.https://doi.org/10.1021/jm050540c
-
Research on adverse drug events. I. Muscarinic M3 receptor binding affinity could predict the risk of antipsychotics to induce type 2 diabetesMethods and Findings in Experimental and Clinical Pharmacology 27:289–304.https://doi.org/10.1358/mf.2005.27.5.908643
-
Describing Protein Folding Kinetics by Molecular Dynamics Simulations. 1. Theory †The Journal of Physical Chemistry B 108:6571–6581.https://doi.org/10.1021/jp037421y
-
Efficient bayesian estimation of markov model transition matrices with given stationary distributionThe Journal of Chemical Physics 138:164113.https://doi.org/10.1063/1.4801325
-
CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fieldsJournal of Computational Chemistry 31:671–690.https://doi.org/10.1002/jcc.21367
-
The molecular basis of G Protein-Coupled receptor activationAnnual Review of Biochemistry 87:897–919.https://doi.org/10.1146/annurev-biochem-060614-033910
-
Constitutive activities and inverse agonism in dopamine receptorsAdvances in Pharmacology 70:175–214.https://doi.org/10.1016/B978-0-12-417197-8.00007-9
Article and author information
Author details
Funding
National Institutes of Health (Z1A DA000606-03)
- Lei Shi
National Institutes of Health (MH54137)
- Jonathan A Javitch
National Health and Medical Research Council (APP1049564)
- J Robert Lane
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Support for this research was provided by the National Institute on Drug Abuse–Intramural Research Program, Z1A DA000606-03 (LS), NIH grant MH54137 (JAJ) and National Health and Medical Research Council (NHMRC) Project Grant APP1049564 (JRL). We thank Jackie Glenn for technical support in generating membrane preparations.
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 2,335
- views
-
- 375
- downloads
-
- 34
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 34
- citations for umbrella DOI https://doi.org/10.7554/eLife.52189
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Distinct inactive conformations of the dopamine D2 and D3 receptors correspond to different extents of inverse agonism
eLife 9:e52189.
https://doi.org/10.7554/eLife.52189




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}