Ecdysone steroid hormone remote controls intestinal stem cell fate decisions via the PPARγ-homolog Eip75B in Drosophila
- Institute of Genetics, Heinrich-Heine-University, Germany
Abstract
Developmental studies revealed fundamental principles on how organ size and function is achieved, but less is known about organ adaptation to new physiological demands. In fruit flies, juvenile hormone (JH) induces intestinal stem cell (ISC) driven absorptive epithelial expansion balancing energy uptake with increased energy demands of pregnancy. Here, we show 20-Hydroxy-Ecdysone (20HE)-signaling controlling organ homeostasis with physiological and pathological implications. Upon mating, 20HE titer in ovaries and hemolymph are increased and act on nearby midgut progenitors inducing Ecdysone-induced-protein-75B (Eip75B). Strikingly, the PPARγ-homologue Eip75B drives ISC daughter cells towards absorptive enterocyte lineage ensuring epithelial growth. To our knowledge, this is the first time a systemic hormone is shown to direct local stem cell fate decisions. Given the protective, but mechanistically unclear role of steroid hormones in female colorectal cancer patients, our findings suggest a tumor-suppressive role for steroidal signaling by promoting postmitotic fate when local signaling is deteriorated.
Introduction
Reproduction is an energetically costly process triggering multiple physiological adaptations of organs such as liver, pancreas and gastrointestinal tract upon pregnancy in various species (Hammond, 1997; Roa and Tena-Sempere, 2014). As a part of the hormonal response to mating and increased metabolic energy consumption, the female Drosophila melanogaster midgut is remodeled in size and physiology by stimulating intestinal stem cell (ISC) driven epithelial expansion to achieve an even energy balance (Cognigni et al., 2011; Klepsatel et al., 2013; Reiff et al., 2015).
Since the discovery of adult intestinal stem cells (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006), local signaling pathways such as Notch (N), Jak/Stat, EGFR, Wnt/wingless, Insulin-receptor, Hippo/Warts and Dpp-signaling were shown to contribute to intestinal homeostasis under physiological and challenged conditions like bacterial infections (Miguel-Aliaga et al., 2018)(and references therein). The midgut epithelium is maintained by ISC giving rise to only two types of differentiated cells: enteroendocrine cells (EE) and absorptive enterocytes (EC) (Figure 1A). Pluripotent ISC are able to self-renew or divide asymmetrically into either committed EC precursor cells called enteroblasts (EB) or enteroendocrine precursor (EEP) cells. EEP, upon timely activation of scute in ISC, divide once more prior to terminal differentiation yielding a pair of EE (Chen et al., 2018; Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007). Nine out of ten ISC mitosis give rise to EB specified by N-activation in EB daughters (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007). Post-mitotic EB retain a certain degree of plasticity by: (1) delaying their terminal differentiation through mesenchymal-to-epithelial transition (MET) (Antonello et al., 2015a), (2) changing their fate to EE upon loss of the transcription factor klu (klumpfuss) and (3) undergoing apoptosis as an additional homeostatic mechanism (Korzelius et al., 2019; Reiff et al., 2019).
Figure 1 with 1 supplement see all

The Ecdysone receptor in intestinal progenitors controls tissue homeostasis.
(A) Scheme of the adult Drosophila melanogaster gastrointestinal tract with cartoon depicting the midgut epithelial monolayer composed of intestinal stem cells (ISC), enteroblasts (EB), enterocytes (EC) and enteroendocrine cells (EE) colored according to the lineage tracing system ReDDM with esg-Gal4 (Antonello et al., 2015a). (B) Schematic of esgReDDM tracing including full genotype. ReDDM differentially marks cells having active or inactive Gal4 expression. Combined with esg-Gal4, active in ISC and EB, esgReDDM double marks ISC and EB driving the expression of UAS-CD8::GFP (membrane CD8::GFP, green), UAS-H2B::RFP (nuclear H2B::RFP, red) and further UAS-driven transgenes (UAS abbreviated as >hereafter in Figure panels). Newly differentiated EC and EE with inactive esg-Gal4 are RFP+-only owing to protein stability of H2B::RFP. Flies are grown at permissive 18°C in which transgene expression is repressed by ubiquitous tubulin-driven Gal80ts. By shifting adult females to the restrictive temperature of 29°C, Gal80ts is destabilized, in turn enabling ReDDM-tracing marking progeny (EE and EC with H2B::RFP nuclear stain) and in parallel manipulation by allowing transactivation of UAS-sequences through esg-driven Gal4-expression (Antonello et al., 2015a). Posterior midguts (PMG) after seven days of esgReDDM tracing of control (crossed with w1118) adult MF (D) show mating dependent addition of new EC compared to control VF (C) (Reiff et al., 2015). (E) Knockdown of EcR using UAS-driven RNAi abolishes mating induced new EC generation in MF. (F+G) Overexpression of >EcR.B2 in VF (F) and >EcRFlyORFFlyORF840 (G) does not induce proliferation or differentiation of progenitors (ISC+EB). (H–I) Quantification of progenitor numbers (H) and traced progeny encompassing EC and EE (I) in R5 PMG (n = 24,17,17,17, 8). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001). (J) Cartoon depicting experimental manipulations on EcR signaling pathway investigated with esgReDDM. Scale bars = 100 µm.
-
Figure 1—source data 1
Data from Figure 1.
Panel G,H experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig1-data1-v3.xlsx
Apart from aforementioned local signaling pathways, systemic hormones are released into the hemolymph and act on distant organs (Figueroa-Clarevega and Bilder, 2015; Kwon et al., 2015; Reiff et al., 2015). Upon mating, JH released by the neuroendocrine corpora allata is able to control ISC proliferation through heterodimers of Met (methoprene-tolerant) and gce (germ cells expressed) nuclear hormone receptors (Reiff et al., 2015). JH coordinates Drosophila larval development in concert with the steroid hormone 20-hydroxy-ecdysone (20HE) and both hormones stimulate egg production in adult females (Bownes et al., 1984; Gilbert et al., 2002; Kozlova and Thummel, 2000; Truman and Riddiford, 2002). In mated adult female flies, we confirmed an increase of 20HE titers in ovary and detected a similar increase of hemolymph 20HE titers compared to virgin females (Ameku and Niwa, 2016; Gilbert and Warren, 2005; Harshman et al., 1999). The anatomical proximity of ovaries and the posterior midgut (PMG) prompted us to investigate a role for the Ecdysone-receptor (EcR) signaling cascade in organ plasticity during reproduction. Downstream of EcR activation, we detected upregulation of Ecdysone-induced protein 75B (Eip75B) protein isoforms ensuring absorptive EC production upon mating. Using the established Notch tumor paradigm, we found that 20HE through Eip75B/PPARγ remote controls EB differentiation and suppresses N-loss of function driven hyperplasia. The mechanism identified in this study not only plays a role in the physiology of mating, but also contributes to our understanding of the protective effects of steroid hormone signaling in the pathophysiology of human colorectal cancer.
Results
EcR controls intestinal stem cell proliferation and progenitor differentiation
The female fly intestine undergoes various physiological post-mating adaptations including a size increase of the absorptive epithelium (Cognigni et al., 2011; Klepsatel et al., 2013; Reiff et al., 2015). Intrigued by post-mating increases of 20HE titers (Ameku and Niwa, 2016; Harshman et al., 1999), we explored a role for EcR-signaling in mating adaptations of the adult Drosophila melanogaster intestine.
EcR encodes for three different splice variants: EcR.A, EcR.B1 and EcR.B2 (Cherbas et al., 2003; Talbot et al., 1993), which we detected by PCR in intestinal tissue with highest expression for EcR.B2 (Figure 1—figure supplement 1A). Using EcR antibodies detecting all splice variants, we found EcR in ISC (positive for the Notch ligand Delta+), EB (Notch responsive element, NRE-GFP+) and EC (Discs-large-1, Dlg-1+, Figure 1—figure supplement 1B–D’’’ and Figure 1A for an overview). To investigate a role for the EcR in intestinal tissue homeostasis, we first manipulated EcR function in ISC and EB using the ‘ReDDM’ (Repressible Dual Differential Marker, Figure 1B) tracing method to observe its overall impact on tissue renewal (Antonello et al., 2015a). Briefly, ReDDM differentially marks cells having active or inactive Gal4 expression with fluorophores of different stability over a defined period of time. Combined with the enhancer trap esg-Gal4, active in progenitors (ISC and EB), esgReDDM double marks ISC and EB driving the expression of UAS-CD8::GFP (>CD8::GFP) with short half-life and >H2B::RFP with long half-life. Upon epithelial replenishment, CD8::GFP signal is lost and new terminally differentiated EC (Dlg-1+) and EE (Prospero, Pros+) stemming from ISC divisions retain a RFP+-nuclear stain due to fluorophore stability (Figure 1A,B; Antonello et al., 2015a). Crosses are grown at 18°C in which transgene expression is repressed by ubiquitous tubulin-driven temperature sensitive Gal80ts. By shifting adult females to 29°C, Gal80ts is destabilized, in turn enabling spatiotemporal control of esgReDDM-tracing and additional UAS-driven transgenes in progenitors (Figure 1B).
After seven days of esgReDDM-tracing, mated females (MF, Figure 1D) showed increases of progenitor numbers (Figure 1H) and newly generated progeny (EE+EC, Figure 1I) in the R5 region of the PMG over virgin females (VF, Figure 1C) confirming previous observations (Reiff et al., 2015). Reducing EcR-levels with two different >EcR RNAi stocks in MF resulted in a reduction of ISC/EB numbers (Figure 1E,H, Figure 1—figure supplement 1L) and newly generated progeny (Figure 1E,I, Figure 1—figure supplement 1L) to levels comparable to VF controls (Figure 1C,H,I). We confirmed knockdown efficiency of >EcR RNAi in esgReDDM by measuring fluorescence intensity of EcR in progenitor cells in situ and found EcR-protein levels significantly decreased in both RNAi lines (Figure 1—figure supplement 1E–H).
Independent of EcR-abundance, we found that expressing dominant-negative EcR.B2 isoforms or EcR-heterozygosity using EcRM554fs, a well described loss-of-function (LOF) allele, phenocopy >EcR RNAi (Figure 1—figure supplement 1I–K). Generally, EcR LOF leads to a similar phenotype as JH-receptor knockdown in MF (Reiff et al., 2015). To investigate whether EcR-levels affect progenitor behavior, we overexpressed wildtype EcR.B2 and pan-EcR using esgReDDM in VF. We found neither induction of progenitor numbers nor an increase in new EC (Figure 1F–I), suggesting that EcR-dependent proliferation and differentiation of progenitors might be limited by 20HE availability (Figure 1J).
Ecdysone titers are increased upon mating and actively transported into progenitors to adapt intestinal physiology
In close anatomical proximity to the PMG, the ovaries are an established ecdysteroidogenic tissue. Determining 20HE titers 48 hr after mating using enzyme immunoassays, we confirmed previous reports of mating dependent increases of ovarian 20HE titers (Figure 2A; Ameku and Niwa, 2016; Harshman et al., 1999) and observed a similar increase in the hemolymph (Figure 2A). As a study investigating the ecdysoneless mutants suggested that the ovary is the only source for 20HE in adult females (Garen et al., 1977), we sought to diminish 20HE titers in adult females. Therefore, we genetically ablated the ovaries using the dominant sterile ovoD1 allele in which egg production is blocked prior to vitellogenesis (Busson et al., 1983; Oliver et al., 1987; Reiff et al., 2015; Figure 2D). 20HE titers in the hemolymph of ovoD1 females are reduced around 40–50% compared to wild-type females (Figure 2B,A). Interestingly, 20HE titers in hemolymph and remnants of the ovaries are still significantly increased upon mating of ovoD1 females (Figure 2C). Consequently, 20HE titers in sterile esgReDDM/ovoD1 MF increase the number of progenitors (Figure 2E) and progeny (Figure 2F). This suggests that remaining 20HE levels are sufficient to elicit mating related midgut adaptations.
Figure 2 with 1 supplement see all
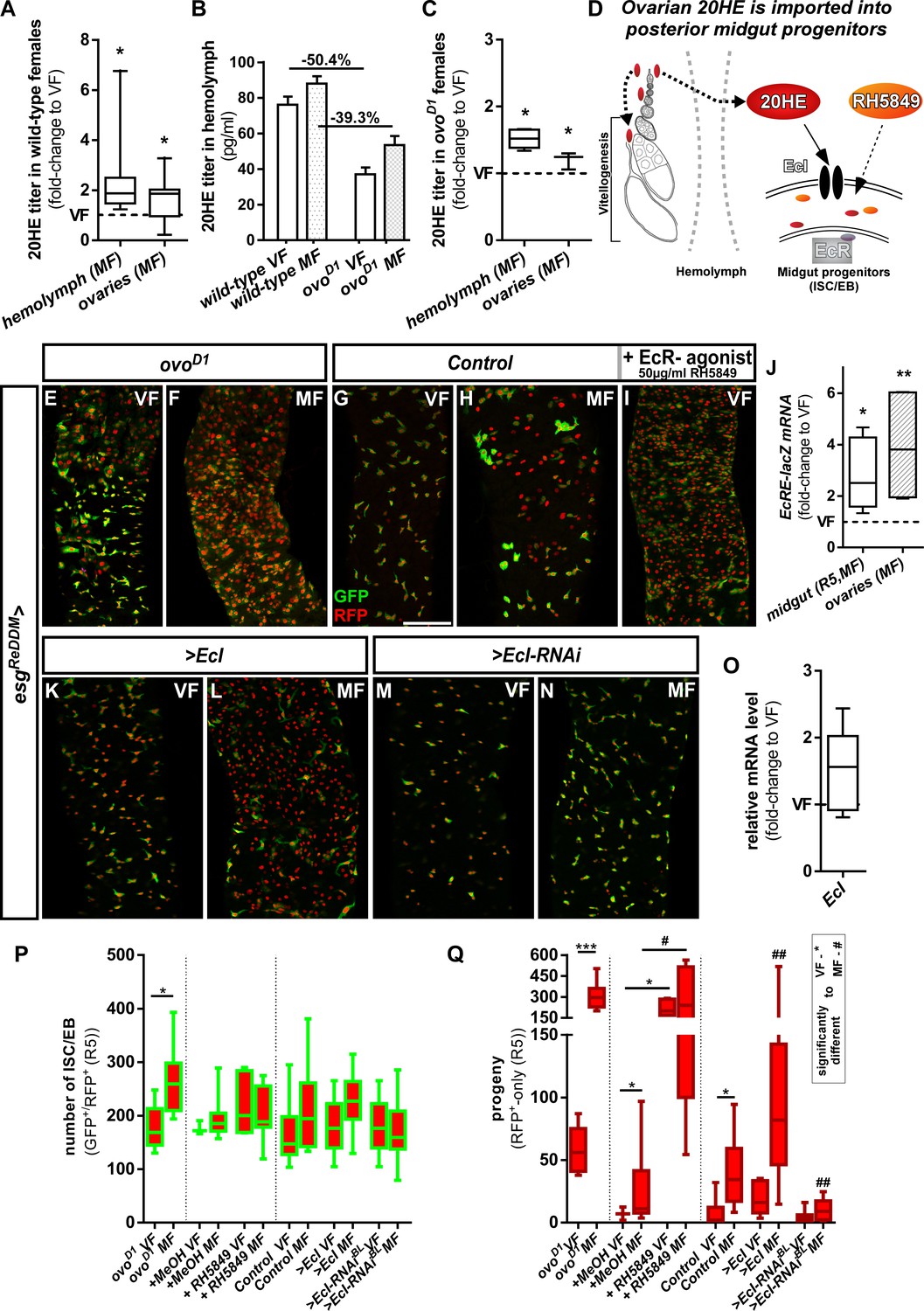
Intracellular 20-Hydroxy-ecdysone levels control ecdysone response through the ecdysone importer.
(A–C) Determination of 20HE titers in ovaries and hemolymph of wild-type (A,B) and ovoD1 (B,C) adult VF and MF 48 hr after mating. (A,C) show fold-change increases over VF titer (dotted line at y = 1). (D) Cartoon depicting ovarian 20HE release to ISC/EB in the adjacent PMG. Please note that in wild-type females, 20HE is incorporated into developing eggs during vitellogenesis, whereas in ovoD1 vitellogenesis is absent and might lead to higher (proportional) release into the hemolymph. 20HE from the hemolymph is absorbed by ISC/EB in the PMG, where the cartoon illustrates specific genetical and pharmacological manipulations on the EcR-signaling pathway. (E–I) Representative images of adult PMG after seven days of esgReDDM tracing of ovoD1 VF (E), ovoD1 MF (F) and control VF (G) and MF (H). (I) Control VF PMG after oral administration of RH5849 (50 µg/ml). (J) Quantitative RT-PCR on EcRE (Ecdysone responsive elements, [Schwedes et al., 2011]) driving lacZ expression on intestinal cDNA from VF and MF control flies. Values are normalized to VF levels (dotted line at y = 1) and statistically analysed using student´s t-test (*p<0.05, **p<0.01; ***p<0,001;). (K–N) Up- and downregulation of EcI in VF (K,M) and MF (L,N) using UAS driven transgenes after seven days of tracing with esgReDDM. (O) Quantitative RT-PCR of EcI on intestinal cDNA from VF and MF control flies. Values are normalized to VF levels (dotted line at y = 1) and statistically analysed using student´s t-test (*p<0.05, **p<0.01; ***p<0,001;). (P,Q) Quantification of progenitor numbers (P) and traced progeny encompassing EC and EE (Q) in R5 PMG (n = 7,9/3,9,5,13/16,13,8,10,10,19). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001). Scale bars = 100 µm.
-
Figure 2—source data 1
Data from Figure 2.
Panel A,B,C,J,O,P,Q experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig2-data1-v3.xlsx
20HE is a polar steroid, which disperses through hemolymph and binds to EcR to activate target gene transcription (Figure 2D; Bownes et al., 1984; Gilbert et al., 2002). Using an established reporter for the activation of 20HE signaling, we found mating induced increases of EcR activity in PMG and ovaries using qPCR (Figure 2J). As orally administered 20HE is metabolized and cleared of rapidly, we used the potent non-steroidal EcR agonist RH5849. RH5849 is used as pest control due to its stability, specificity and a 30–60 times higher efficacy compared to 20HE (Robinson et al., 1987; Wing et al., 1988). Testing concentrations from 1 to 100 µg/ml in timed egg-layings, we observed expected larval molting defects for concentrations from 50 µg/ml upwards (Figure 2—figure supplement 1A). Feeding control esgReDDM VF 50 µg/ml with RH5849, we traced intestinal progenitors with pharmacologically activated EcR-signaling for seven days. RH5849 strongly induces ISC mitosis, reflected by a tenfold increase in newly generated progeny over mating induction (Figure 2G–I,Q). Interestingly, RH5849 mediated EcR-activation leads to no accumulation of progenitors (Figure 2P) as observed in oncogenic manipulations or disruptions of intestinal homeostasis, suggesting a role for EcR in both, proliferation and differentiation (Antonello et al., 2015a; Chen et al., 2016; Patel et al., 2015; Reiff et al., 2019). Given the role of 20HE induced programmed cell death (PCD) in larval metamorphosis and the recently discovered role of EB PCD in adult midgut homeostasis (Jiang et al., 1997; Jiang et al., 2000; Reiff et al., 2019), we addressed PCD by activated caspase-3 staining, but found no increase of PCD by RH5849 (data not shown).
Cellular 20HE uptake was recently shown to depend on Ecdysone Importer (EcI) belonging to the evolutionary conserved SLCO superfamily of solute carrier transporters. EcI LOF causes phenotypes indistinguishable from 20HE and EcR deficiencies in vivo (Okamoto et al., 2018). To investigate a role of EcI, we confirmed membrane localization of >EcI tagged with HA by immunostaining in progenitors using esgReDDM (Figure 2—figure supplement 1B). After seven days, forced expression of >EcI using esgReDDM in VF lead to no increase in progenitor numbers and new EC, underlining that VF 20HE levels are low (Figure 2K,P,Q). Further supporting this hypothesis,>EcI in MF lead to an increase of newly generated EC exceeding typical mating induction of MF controls by 4.8 fold (Figure 2L,P,Q). Blocking 20HE uptake by EcI-RNAi in MF abolished ecdysone induced tissue expansion to VF control levels (Figure 2N,P,Q). In addition, we tested a function of the EcI gene in mating induction, but found no change in EcI expression upon mating (Figure 2O). Both, pharmacological and genetic experiments, suggest that 20HE titer and import control EcR-activity upon mating (Figure 2D).
Upon mating, absorptive EC undergo metabolic adaptations upregulating genes known for lipid uptake (Reiff et al., 2015). We found EC immunoreactive for EcR (Figure 1—figure supplement 1D) and investigated a function for EcR in mating related upregulation of lipid uptake by measuring the activity of sterol regulatory element-binding protein (Srebp) (Reiff et al., 2015). Therefore, we used a GFP-reporter (Srebp >CD8::GFP) that is subjected to the same proteolytic processing as Srebp (Athippozhy et al., 2011; Reiff et al., 2015). Confirming previous observations, Srebp-activity increases 2.2 fold upon mating (mean fluorescence intensity, Figure 2—figure supplement 1C,D; Reiff et al., 2015). Using the Srebp-reporter to drive >EcR RNAi in MF, we found reduced Srebp-activity comparable VF controls (Figure 2—figure supplement 1E–F,H), whereas feeding VF with RH5849 induced Srebp-activity 2.4 fold (Figure 2—figure supplement 1G,H). Next, we directly addressed lipid uptake with OilRedO-staining on PMG using the EC driver Mexts. In accordance with Srebp-activity (Figure 2—figure supplement 1C,D,G), feeding flies with RH5849 (Figure 2—figure supplement 1K) and forced expression of EcI (Figure 2—figure supplement 1L) significantly induced lipid uptake (Figure 2—figure supplement 1O) over controls (Figure 2—figure supplement 1I,J). Mexts > EcI RNAi and >EcR RNAi did not result in a reduction of lipid uptake below control levels (Figure 2—figure supplement 1M,N,I), confirming previous observations of direct fatty acid incorporation into newly produced eggs (Reiff et al., 2015).
These data support the idea that mating related synergistic effects of JH- and 20HE-signaling drive adaptation of intestinal homeostasis and physiology. The JH-signal is transduced by the transcription factor Krüppel-homolog 1 (Kr-h1) in adult MF intestines, which prompted us to explore a function for the ‘classical’ Ecdysone target genes (Jindra et al., 2013; Reiff et al., 2015).
Ecdysone-induced protein 75b protein isoforms Eip75B-A and Eip75B-C control enteroblast differentiation
During Drosophila development, 20HE pulses lead to direct binding of EcR to regulatory regions of early ecdysone response genes Ecdysone-induced protein 74A (Eip74EF) and Ecdysone-induced protein 75B (Eip75B, Figure 3A; Bernardo et al., 2014; Karim and Thummel, 1991; Segraves and Hogness, 1990). First, we performed conventional PCR to analyze JH- and Ecdysone target gene expression. Signal for Kr-h1-A (Kr-h1 in the following), but not Kr-h1-B, and for all isoforms of Eip74EF and Eip75B was found (Figure 3B). Using quantitative real time PCR (qPCR) analysis, we confirmed an increase of JH-pathway activity upon mating using Kr-h1 as control (Figure 3C; Reiff et al., 2015). To our surprise, we found Eip74EF expression unchanged, in contrast to its prominent role in the control of germline stem cell (GSC) proliferation in oogenesis (Ables and Drummond-Barbosa, 2010). Instead, we found induction of Eip75B-A and -C isoforms (Figure 3C). Eip75B encodes for three protein isoforms (Eip75B-A, -B and –C) that differ in their N-terminal domain structure (Segraves and Hogness, 1990) and Eip75B-B lacks one of the two zinc-finger DNA binding domains rendering it incapable of direct DNA binding (White et al., 1997).
Figure 3 with 2 supplements see all
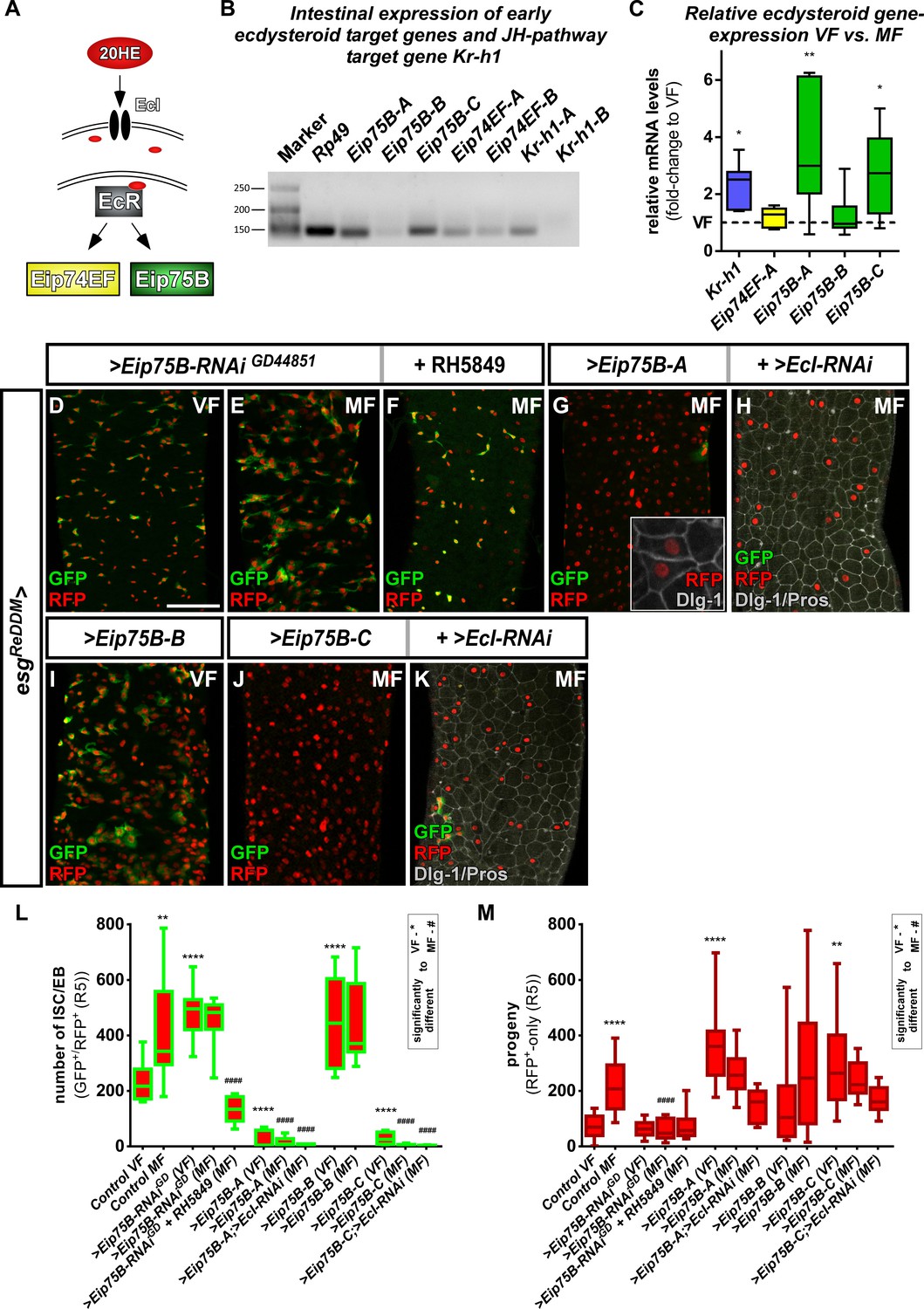
Ecdysone induced protein 75B is upregulated upon mating and controls progenitor differentiation.
(A) Cartoon depicting EcR signaling cascade activating early ecdysteroid target genes. (B) Expression analysis of Ecdysone- and JH-signaling target genes including protein isoforms on cDNA transcribed from mRNA isolations from whole midgut dissections of MF. (C) Quantitative RT-PCR on early ecdysteroid genes on intestinal cDNA from VF and MF control flies. Values are normalized to VF levels (horizontal line = 1) and statistically analyzed using student´s t-test (n = 6; *p<0.05, **p<0.01;). (D–E) RNAi-mediated downregulation of Eip75B in VF (D), MF (E) and MF fed with RH5849 (F) after seven days of tracing with esgReDDM. (G–K) Representative images of adult PMG with forced expression of Eip75B isoforms Eip75B-A (G), Eip75B-A and EcI-RNAi (H), Eip75B-B (I), Eip75B-C (J), Eip75B-C and EcI-RNAi (K) after seven days of tracing with esgReDDM. Inset in (F) depicts epithelial integration of newly generated Dlg-1+/RFP+-EC. (I–J) Quantification of progenitor numbers (I) and traced progeny encompassing EC and EE (J) in R5 PMG (n = 12,13,10,12,11,11,14,8,10,10,10,5,11). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001, identical p-values are marked by # when compared to MF). Scale bars = 100 µm.
-
Figure 3—source data 1
Data from Figure 3.
Panel C,L,M experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig3-data1-v3.xlsx
Intrigued by mating increases of Eip75B-A /- C levels, we manipulated Eip75B function using esgReDDM. Reducing Eip75B levels with RNAi, we found a significant increase in ISC and EB numbers (Figure 3D,E,L), but not in EC generation compared to VF controls (Figure 3M). In MF, Eip75B knockdown reduced newly generated EC compared to MF controls, pointing to a role of Eip75B in EB differentiation downstream of EcR-activation (Figure 3E,M). Interestingly, feeding flies with RH5849 lacking Eip75B in their intestinal progenitors (Figure 3F) did not result in newly generated progeny (Figure 3M compare to Figure 2Q), suggesting a central role for Eip75B in the RH5849 response. Additionally, we investigated homozygous Eip75B-A mutants using MARCM clonal analysis with a known LOF allele (Rabinovich et al., 2016). MARCM clones of Eip75B-A (Eip75BA81) are significantly larger compared to controls (Figure 3—figure supplement 1A–D; Lee and Luo, 1999). Eip75BA81 deficient clones contain few differentiated EC (GFP+/Dlg-1+, Figure 3—figure supplement 1C) suggesting disturbed EB to EC differentiation as observed for esgReDDM >Eip75 B-RNAi (Figure 3E). Together, these LOF experiments hint to a role for mating induced Eip75B-A /- C isoforms in EC differentiation of EB downstream of EcR.
Forced expression of Eip75B variants with esgReDDM resulted in three prominent phenotypes: (1) Eip75B-A and Eip75B-C strongly drive differentiation into EC (Dlg-1+, Figure 3G,J,M) depleting the entire progenitor pool (Figure 3G,J,M). (2) The Eip75B-B isoform induces ISC proliferation in VF (Figure 3I, Figure 3—figure supplement 1F) and raised progenitor numbers suggesting slowed down but not inhibited terminal differentiation (Figure 3I,L,M). (3) All Eip75B manipulations strongly reduce the number of newly differentiated EE (Figure 3—figure supplement 1E). This suggests that EE differentiation upon Eip75B-RNAi and forced expression of the Eip75B-B isoform is blocked (Figure 3I,L, Figure 3—figure supplement 1E). Eip75B-A and Eip75B-C are sufficient to instantaneously drive progenitors into EC differentiation (Figure 3G,H,J,K) even in the absence of 20HE import (Figure 3H,K). The immediate strong differentiation stimulus prevents further ISC division and as a consequence also reduces new EE (Figure 3L,M, Figure 3—figure supplement 1E). Disrupting intestinal homeostasis alters midgut length (Hudry et al., 2016) and in line with this, Eip75B manipulations result in a shorter midgut as EC make up around 90% of the whole midgut epithelium (Figure 3—figure supplement 1G). Eip75B affects intestinal homeostasis by either slowing down terminal EB to EC differentiation (Eip75B-B and Eip75B-RNAi) or depleting the ISC and EB pool (Eip75B-A /– C, Figure 3—figure supplement 1G) preventing sufficient EC production along the gut. When EC production is blocked using >N RNAi, the midgut shrinks around one third over a period of seven days (Chen et al., 2018; Guo and Ohlstein, 2015; Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007; Patel et al., 2015). Thus, all Eip75B manipulations ultimately lead to an insufficient number of new absorptive EC resulting in a shorter midgut.
Eip75B is a long term predicted PPARγ-homologue (peroxisome proliferator-activated receptor gamma) in Drosophila. A close functional homology got recently confirmed in pharmacogenetic approaches demonstrating that Eip75B-mutant flies are irresponsive to PPARγ activating drugs (Joardar et al., 2015; King-Jones and Thummel, 2005). This homology is of particular interest, as human PPARγ plays a role in i) pregnancy related adaptations of lipid metabolism (Waite et al., 2000) and ii) as target in colorectal cancer (CRC) (Sarraf et al., 1999). Thus, we tested pharmacological activation of Eip75B/PPARγ with the established agonist Pioglitazone, a drug used in Diabetes mellitus treatment (Gillies and Dunn, 2000; Jafari et al., 2007). Flies fed with food containing Pioglitazone show a strong increase in the number of progenitor cells (Figure 4B,E) and newly generated EC (Figure 4F) compared to controls (Figure 4A). Strikingly, knockdown of Eip75B led to irresponsiveness to Pioglitazone (Figure 4C,D,F) suggesting that Pioglitazone activates Eip75B in intestinal progenitors.
Figure 4
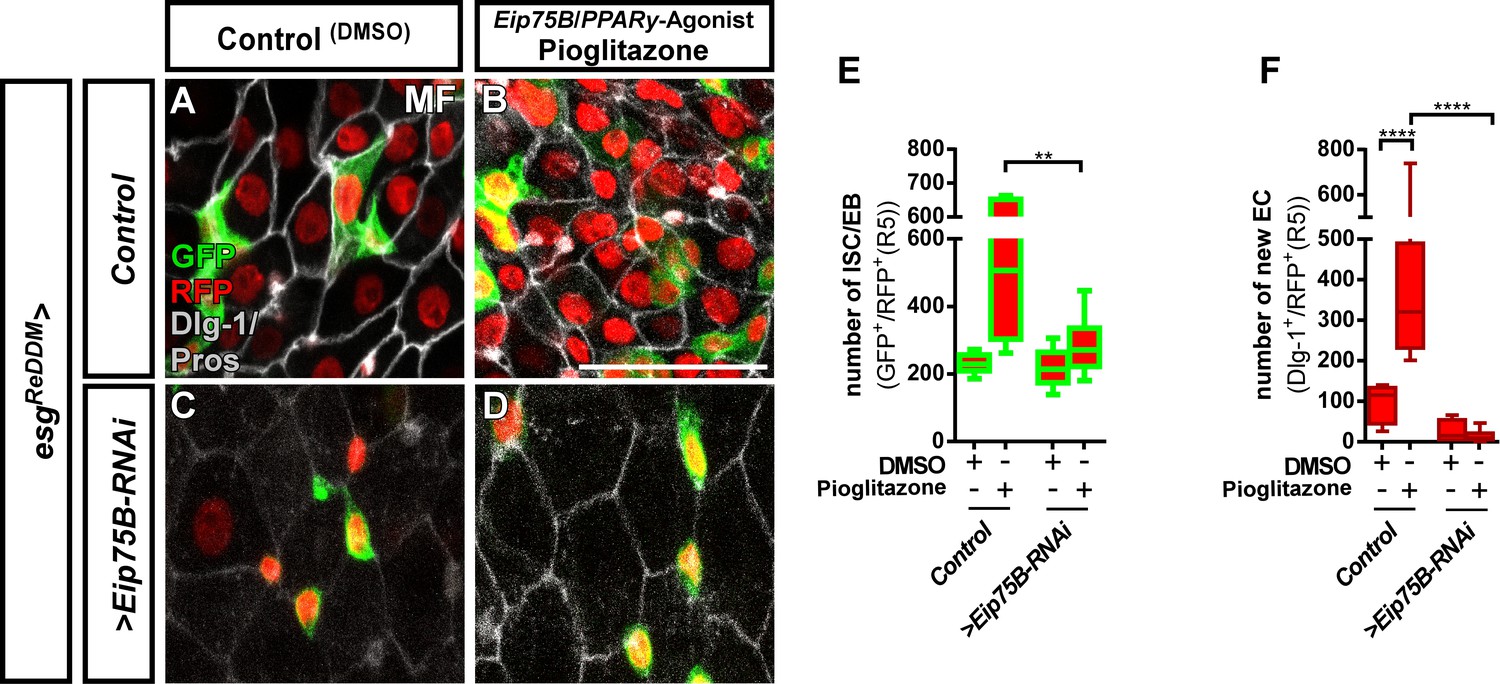
The Eip75B/PPARγ agonist Pioglitazone acts through Eip75B to stimulate progenitor differentiation.
(A–D) Representative images of adult PMG after seven days of esgReDDM tracing of control MF (A,B) and >Eip75 B-RNAi MF (C,D) fed with DMSO as control (A,C, 2.5 µl/ml food) and Pioglitazone (B,D; 0,002 mg in DMSO/ml food). (E+F) Quantification of progenitor numbers (E) and newly generated EC (F) in R5 PMG (n = 7,6,10,11). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001,. Scale bars = 100 µm.
-
Figure 4—source data 1
Data from Figure 4.
Panel E,F experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig4-data1-v3.xlsx
Taken together, these findings strongly suggested a role for Eip75B in EB differentiation. EB are specified by N-signaling and their lineage is maintained by the transcription factor klumpfuss (klu). klu+-EB retain some plasticity to change fate to EE upon reduction of klu-levels (Korzelius et al., 2019; Reiff et al., 2019). We used kluReDDM to explore the function of EcR and Eip75B in EB lineage identity (Figure 3—figure supplement 2A). No changes in EB fate decisions towards EE differentiation were observed upon knockdown of EcR and Eip75B (Figure 3—figure supplement 2A,B–D). Eip75B-A and Eip75B-C expression with kluReDDM phenocopied EB to EC differentiation effects observed in esgReDDM (Figure 3—figure supplement 2G,I,J,K) without non-autonomously inducing proliferation in wild-type ISC of VF and MF (Figure 3—figure supplement 2J,K). Expressing Eip75B-B in kluReDDM lead to delayed EB differentiation similar to esgReDDM and non-autonomous induction of ISC mitosis (Figure 3I, Figure 3—figure supplement 2J,L; Patel et al., 2015; Reiff et al., 2019). Having identified Eip75B-A /- C as mating induced differentiation effector of 20HE signaling, we hypothesized and investigated a synergism of 20HE and JH hormonal signaling pathways controlling epithelial expansion upon mating.
The interplay between JH and 20HE in intestinal progenitors
Our data suggest that mating induced JH and 20HE signaling affect ISC proliferation and EB differentiation through their effectors Kr-h1 and Eip75B-A /- C (Figure 5K; Reiff et al., 2015). In VF lacking mating induction of both hormones (Figure 2A–C; Ameku and Niwa, 2016; Harshman et al., 1999; Reiff et al., 2015), forced >Kr h1 expression doubles progenitor numbers reproducing previous results (Figure 5B,I; Reiff et al., 2015). Initially, we aimed to perform a full genetic epistasis analysis for Eip75B and Kr-h1, but this analysis was hampered as we failed to recombine Kr-h1-RNAi with Eip75B isoform expressing stocks.
Figure 5
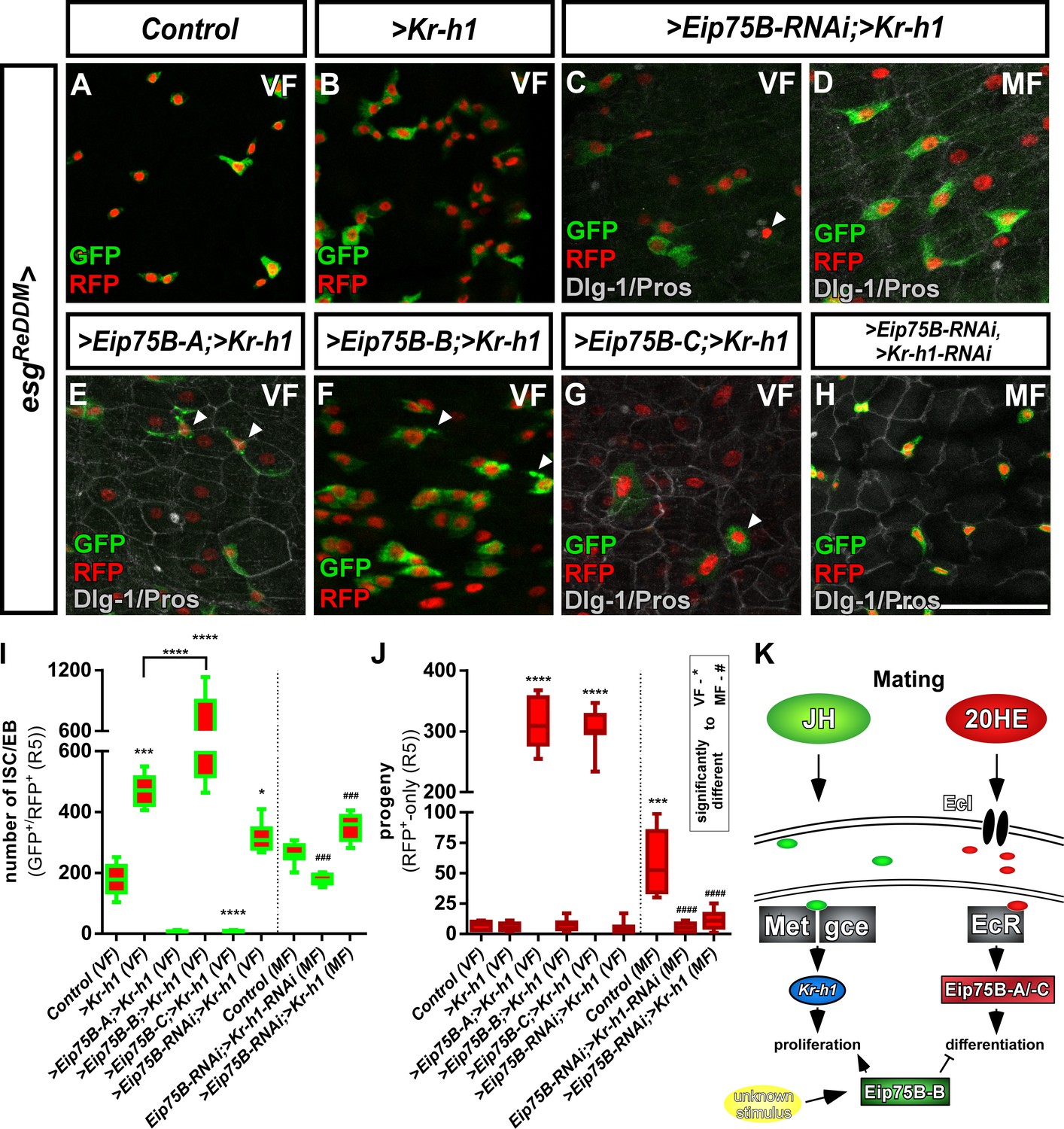
Crosstalk between JH- and Ecdysone-signaling pathways controlling intestinal progenitor proliferation and differentiation.
(A–B) Images of adult PMG of control VF (A) and forced expression of > Kr h1 (B) traced for seven days with esgReDDM. (C–H) Images of adult PMG with forced expression of > Kr h1 with > Eip75 B-RNAi in VF and MF (C,D) and expression of Eip75B isoforms > Eip75 B-A (E),>Eip75 B-B (F),>Eip75 B-C (G) and double > Eip75B-RNAi/>Kr-h1-RNAi (H) after seven days of tracing with esgReDDM. Please note that genotypes of (C–G) were accompanied with semi-lethality even at permissive 18°C, suggesting for example background transgene expression or position effects most probably caused by the total number of six transgenes including esgReDDM. PMG of > Kr h1 combinations also showed some progenitor lethality indicated by membrane-blebbing and irregularities (arrowheads, (E–G) as described in Reiff et al., 2019. (J) Quantification of progenitor numbers (I) and traced progeny encompassing EC and EE (J) in R5 PMG (n = 12,5,9,8,7,13,8,8,13). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001, identical p-values are marked by # when compared to MF). Scale bars = 50 µm (K) Cartoon depicting transcriptional effectors of JH- and Ecdysone signaling pathways. The JH receptor is formed by a heterodimer of Methoprene tolerant (Met) and germ cells expressed (gce). Ligand bound receptor activates the transcription of krüppel homolog 1 (Kr-h1) mediating mating effects in the adult intestine (Reiff et al., 2015) Forced expression of Eip75B-B affects proliferation and differentiation through an yet unknown stimulus.
-
Figure 5—source data 1
Data from Figure 5.
Panel I,J experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig5-data1-v3.xlsx
However, we were able to analyze midguts of MF with forced Kr-h1-expression and simultaneous Eip75B-RNAi (Figure 5C,D), which increases progenitor numbers (Figure 5I) with only few newly generated EC (Figure 5J). This finding supports the requirement of Eip75B in EB differentiation (Figure 3E) and Kr-h1 in ISC proliferation (Figure 5A). Simultaneous double knockdown of >Kr-h1-RNAi/Eip75B-RNAi (Figure 5H) in MF did not show mating induction. Progenitor numbers and differentiated progeny are reduced (Figure 5I,J) phenocopying single >Kr h1-RNAi (Reiff et al., 2015). Surprisingly, we observed a strong increase of progenitor numbers forcing Kr-h1 and Eip75B-B expression (Figure 5F,I,J). These results indicate an additive role of forced Eip75B-B and Kr-h1-expression on ISC proliferation, whereas the latter transduces the mating related proliferation response (Figure 3C). The stimulus inducing Eip75B-B expression yet needs to be identified (Figure 5K).
Most interestingly, Eip75B-A and –C, despite of raised Kr-h1-levels, drive progenitors into EC differentiation largely phenocopying sole Eip75B-A and -C expression (compare Figures 3G,J and 5E,G,I,J). Our data on Eip75B-A and -C corroborate a potent role for Ecdysone-induced EB differentiation. ISC fate decisions in the adult midgut rely on N-signaling (Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007). During Drosophila follicle cell development N opposes EcR-function (Sun et al., 2008), which prompted us to investigate Eip75B-A /- C as effectors of EcR-signaling in the context of N-dependent EB differentiation.
Ecdysone signaling through Eip75B-A and Eip75B-C controls enteroblast differentiation in a notch deficiency tumor model
In the Drosophila and mammalian intestinal stem cell niche, N-signaling specifies EE and EC fate (Jensen et al., 2000; Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007; VanDussen et al., 2012). In Drosophila, mitosis of N mutant ISC generates only ISC-like progenitor cells and EE, which results in an intestinal epithelium accumulating a lack of newly produced EC and compensatory proliferation (Figure 6A,B; Chen et al., 2018; Guo and Ohlstein, 2015; Micchelli and Perrimon, 2006; Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007; Patel et al., 2015).
Figure 6 with 1 supplement see all
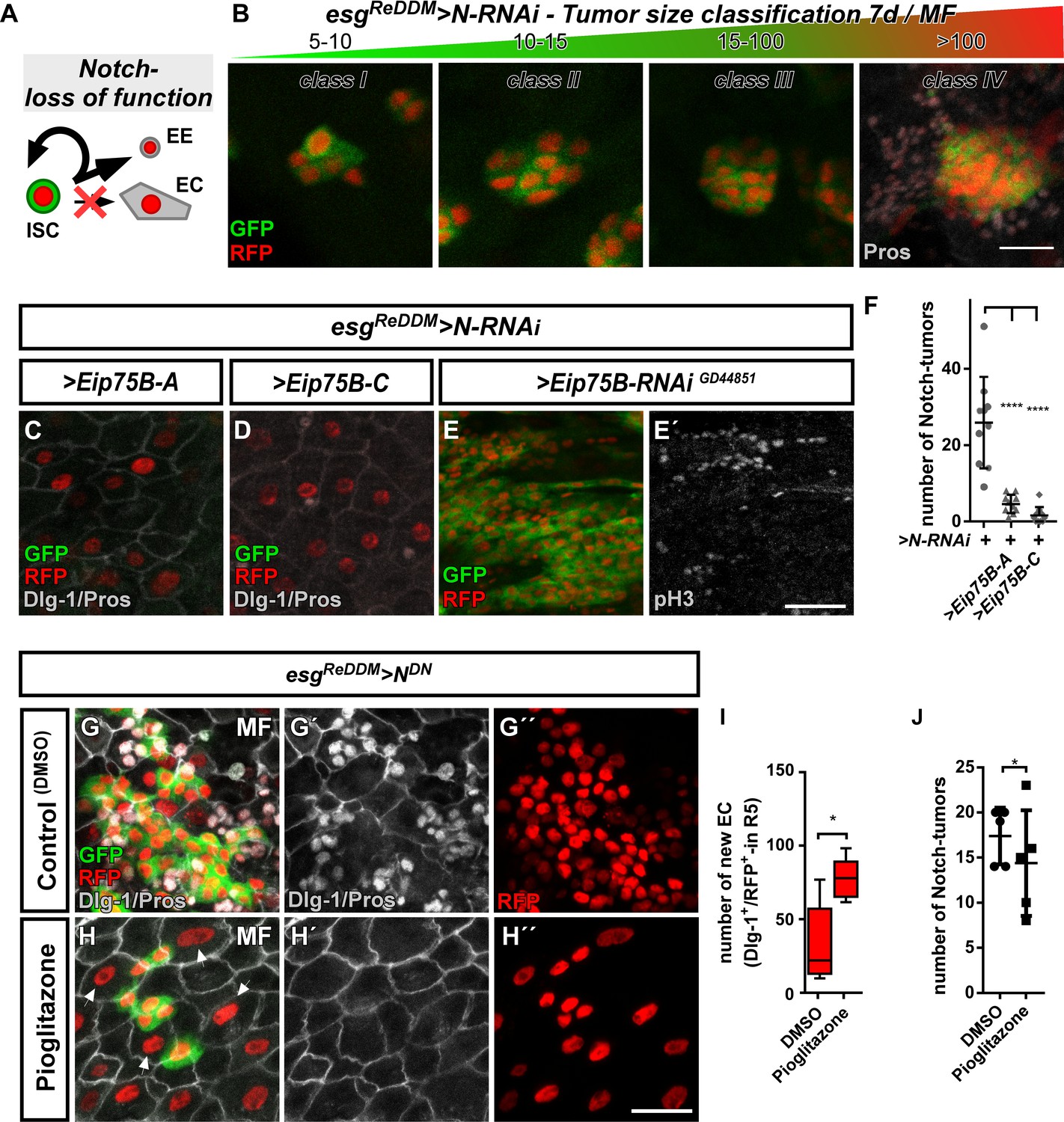
Ecdysone induced protein 75B promotes EB differentiation in a Notch tumor paradigm.
(A) Cartoon depicting cell fate changes upon N-LOF combined with esgReDDM coloring (Ohlstein and Spradling, 2006; Ohlstein and Spradling, 2007). (B) > N RNAi driven by esgReDDM leads to different tumor sizes after seven days of tracing that were classified in four classes according to size. Note, ISC-like/EE clusters up to four cells are not quantified as they occasionally occur in controls too. Progenitors are double labelled (GFP+/RFP+), where newly generated EE are identified by immunostaining for Prospero (Pros) and H2B::RFP trace from esgReDDM. (C–D) Co- expression of Eip75B isoforms > Eip75 B-A (C) and > Eip75 B-C (D) after seven days of tracing with esgReDDM > N RNAi in MF. Note additional Dlg-1+-immunoreactivity in (grey,C+D) demonstrating epithelial integration of newly generated cells as EC (Dlg-1+/RFP+). (E+E’) RNAi-mediated downregulation of Eip75B in MF shows confluent N-tumors (E) accompanied by increased mitosis (E’). (F) Quantification of ISC progeny encompassing ISC-like and EE total N-tumor number in R5 PMG (n = 10,10,10). (G–J) Feeding of the Eip75B/PPARy agonist Pioglitazone to esgReDDM > NDN flies leads to the generation of newly generated EC ((H,I), Dlg-1+/RFP+) after seven days of tracing compared to DMSO controls (G,I). (I–J) Quantification of ISC progeny encompassing ISC-like and EE total N-tumor number in R5 PMG (n = 5,5). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; *** p$$BOX_TXT_END$$. <0,001; ****p<0.0001). Scale bars = 25 µm.
-
Figure 6—source data 1
Data from Figure 6.
Panel F,I,J experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig6-data1-v3.xlsx
Using N-LOF tumors as an experimental paradigm lacking EC production, we sought to investigate the differentiation inducing properties of Eip75B-A /- C. Reduction of N by RNAi or a dominant-negative N-receptor using esgReDDM lead to tumors of different sizes, which we quantified and classified according to their number (Figure 6B). We predominantly found tumors of 5–10 cells (Figure 6B, Figure 6—figure supplement 1A), but also few tumors containing more than 100 ISC/EE-like cells with indistinguishable single or multiple ISC origin (class IV, Figure 6B, Figure 6—figure supplement 1A).
Co-expression of Eip75B-A /- C with N-LOF strongly reduced total tumor number (Figure 6C,D,F) and smaller class I, II and III tumors (Figure 6—figure supplement 1A), whereas Eip75B-RNAi results in confluent class IV tumors all along the PMG (Figure 6E). Although tumor number is strongly reduced (Figure 6F), occasional class IV tumors (<1/PMG) are able to escape suppressive Eip75B-A /- C showing no reduction in tumor size (Figure 6—figure supplement 1A). Additional tumorigenic mechanisms such as Upd- or EGF-ligand upregulation may counteract the tumor suppressive function of 20HE signaling in class IV tumors (Patel et al., 2015).
Since Eip75B-A and Eip75B-C re-enable EC differentiation despite the lack of N (Figure 6C,D; Dlg-1+/RFP+), we investigated whether the EcR-pathway and pharmacological Eip75B-activation are able to trigger EC fate in a N-LOF context. Activation of Eip75B using Pioglitazone led to more newly generated EC (Figure 6H,I) compared to controls (Figure 6G). In addition, we observed a slight, but significant reduction in the number of N-tumors upon feeding Pioglitazone (Figure 6J).
Given the protective role of steroid hormone signaling in colorectal cancer (CRC), we sought to investigate N tumor numbers upon EcR-signaling activation (Chen et al., 1998; Hendifar et al., 2009; Lin et al., 2012). We activated EcR-signaling with RH5849 (Figure 7B) or by raising intracellular levels of 20HE expressing > EcI in esgReDDM(Figure 7D). Either manipulation resulted in numerous confluent tumors reflecting the previously observed role of 20HE in proliferation (Figure 7F). In line with this,>EcI RNAi- and >EcR RNAi significantly reduced tumor burden (Figure 7C,E,F).
Figure 7 with 1 supplement see all
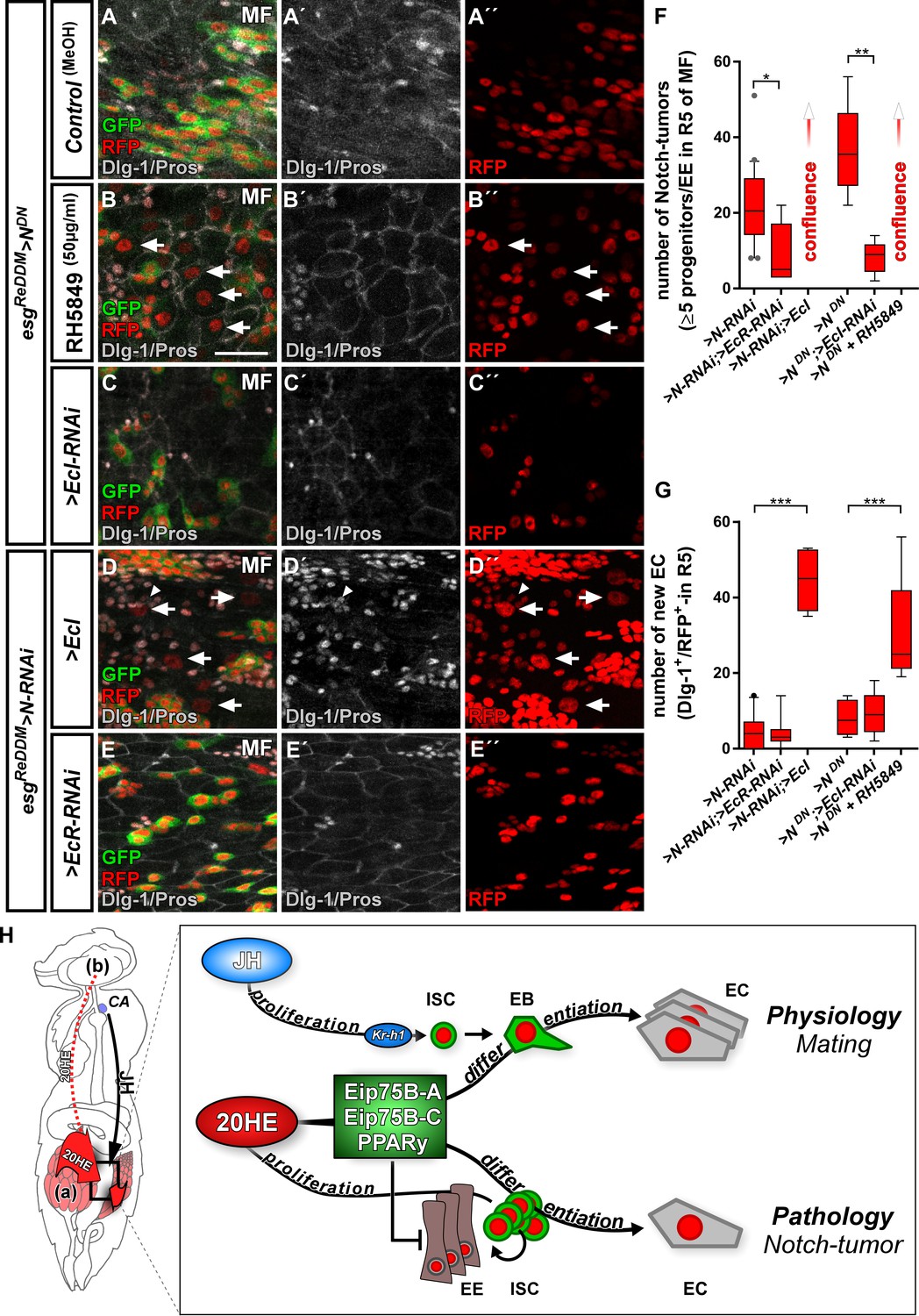
Ecdysone signaling promotes EC-fate in a Notch tumor paradigm.
(A–E’’) Pharmacological and genetic manipulation of Ecdysone signaling. Adult PMG of MF with N-LOF (>N RNAi or > NDN) treated with MeOH as control (A–A’’) or RH5849 (B–B’’) to activate Ecdysone signaling. Arrowheads highlight newly generated Dlg-1+/RFP+-EC (B+B’’) after seven days of esgReDDM > NDN. (C–C’’) RNAi-mediated downregulation of EcI in esgReDDM > NDN, forced expression of >EcI (D–D’’) and >EcR RNAi (E–E’’) in esgReDDM > N RNAi MF after seven days of tracing. Arrowheads highlight newly generated Dlg-1+-EC (D+D’’), scale bars = 25 µm. (F) Quantification of ISC progeny encompassing ISC-like, EC and EE classified after tumor size in R5 PMG (n = 13,5,7,4,6,9). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001). (G) Quantification of newly generated EC (Dlg-1+/RFP+) in R5 PMG (n = 13,5,7,4,6,9). Error bars are Standard Error of the Mean (SEM) and asterisks denote significances from one-way ANOVA with Bonferroni's Multiple Comparison Test (*p<0.05, **p<0.01; ***p<0,001; ****p<0.0001). (H) Model of 20HE and JH hormonal pathways influencing physiological and pathological turnover in the intestine. Upon mating, JH from the neuroendocrine CA (corpora allata, [Reiff et al., 2015]) as well as ovarian 20HE (a) synergize on progenitor cells in the posterior midgut of adult female flies. The source of 20HE could not be definitely determined in this current study and 20HE might as well stem from the brain (b). JH induces ISC proliferation through Kr-h1, whereas 20HE signaling transduced by Eip75B-A /- C/PPARγ, ensures that newly produced EB differentiate into EC. New EC lead to a net increase in absorptive epithelium and thus ensures physiological adaptation of intestinal size to the new metabolic energy requirements of pregnancy. In the adult intestine, early steps of tumor-pathology are recapitulated when EC fate is inhibited by the lack of Notch signaling in progenitors (Patel et al., 2015). Notch mutant ISC divisions only produces ISC-like progenitors and EE. We show in this study that 20HE signaling, through Eip75B-A /- C/PPARγ, is capable to alleviate Notch tumor growth by driving intestinal progenitors into post-mitotic absorptive EC fate.
-
Figure 7—source data 1
Data from Figure 7.
Panel F,G experimental data.
- https://cdn.elifesciences.org/articles/55795/elife-55795-fig7-data1-v3.xlsx
More importantly, ecdysone-pathway activation led to significantly higher numbers of newly generated EC (arrowheads in Figure 7B,D,G), suggesting an upregulation of Eip75B-A and Eip75B-C in Notch tumors upon EcR-activation through RH5849 and > EcI. To exclude remaining N activity, we generated MARCM clones mutant for N (N55e11) (Guo and Ohlstein, 2015). In agreement with our previous observations, activating 20HE signaling in N clones led to a tenfold increase in EC numbers over controls (Figure 7—figure supplement 1A–C) and forced expression of > E75 A/-C to instant EC formation (Figure 7—figure supplement 1D–E). Interestingly, in the presence of N, its activity is increased upon RH5849 feeding (Figure 7—figure supplement 1F), which suggests an interplay between Notch and EcR-signaling that might converge on Eip75B (this study) and klu (Korzelius et al., 2019; Reiff et al., 2019).
Taken together, our data establish 20HE as a systemic signal controlling reproductive adaptations in synergism with JH (Figure 7H). Focusing on EcR-signaling in ISC and EB, we found the mating induced Eip75B-A /- C isoforms acting as differentiation factors in EB. Eip75B-A /- C induces EC fate even in Notch-deficient midgut progenitors, an experimental paradigm used to recapitulate early steps of tumorigenesis. Notch dependent fate decisions in midgut precursor cells are conserved between flies and humans. Thus, our findings suggest a new mechanism how steroid hormone signaling might suppress tumor growth by promoting post-mitotic cell fate in intestinal neoplasms marked by a lack of fate specification.
Discussion
Systemic hormones and intestinal remodeling upon mating
Growing offspring involves significant metabolic adaptations to raised energy demands in mothers. Drosophila melanogaster uses a classical r-selected reproductive strategy with high number of offspring and minimal parental care (Pianka, 1970). Upon mating, egg production is increased tenfold supported by metabolic and behavioral adaptations such as food intake and transit, nutrient preference and a net increase of absorptive tissue in the PMG depending on JH release (Carvalho et al., 2006; Cognigni et al., 2011; Reiff et al., 2015; Ribeiro and Dickson, 2010).
Here, we describe a role for the steroid-hormone ecdysone controlling intestinal tissue remodeling upon mating through EcR-signaling. The unliganded heterodimer of EcR and ultraspiracle (USP) binds its DNA consensus sequences serving as a repressor. Upon ligand binding, the EcR/USP heterodimer turns into an activator inducing the expression of early response genes Eip74EF, Eip75B and broad (br) (Schwedes et al., 2011; Uyehara and McKay, 2019). Initially, we investigated knockdown of USP, br and Eip74EF in intestinal progenitors and observed effects on progenitor survival independent of mating status (Figure 3C, Zipper and Reiff, unpublished data).
Our data support the idea that 20HE and JH synergistically concert intestinal adaptations balancing nutrient uptake to the increased energy demands after mating (Figures 1–5). We confirmed that mating induces ovarian ecdysteroid biosynthesis stimulating egg production by ovarian GSC (Figure 2A; Ables and Drummond-Barbosa, 2010; Ameku and Niwa, 2016; Ameku et al., 2017; Harshman et al., 1999; König et al., 2011; Morris and Spradling, 2012; Uyehara and McKay, 2019). In accordance with an inter-organ signaling role for 20HE, we also detected increased 20HE titers in the hemolymph (Figure 2A). Surprisingly, we found this mating dependent increase of 20HE titers still present upon partial genetic ablation of the ovaries using ovoD1 (Figure 2B; Reiff et al., 2015). This can be explained by either (i) other source(s) for 20HE in adult females like the brain (Chen et al., 2014; Itoh et al., 2011) or (ii) 20HE release from remaining germarial cells in stage 1–4 eggs of ovoD1 VF and MF. Indeed, germarial cells have been shown to express the ecdysteroidogenic Halloween genes (Ameku and Niwa, 2016; Ameku et al., 2017). In addition, ovoD1 MF lack ovarian 20HE uptake during vitellogenesis (Figure 2D) of later egg stages, which potentially contributes to 20HE titer in the hemolymph of ovoD1 females and intestinal epithelium expansion (Figure 2D,E,J,K; Enya et al., 2014).
The epithelial expansion upon mating of ovoD1 females has been observed before (Reiff et al., 2015) and might be comparably high due to the different genetic background of ovoD1 flies compared to w1118 (Figure 2Q). While this work was in review, genetic ablation of ovarian ecdysteroidogenic enzymes was performed in female flies and resulted in a reduction of mitotic ISC. However, a direct assessment of 20HE titers was not performed and a mating dependent context was not analyzed (Ahmed et al., 2020). Taken together, current data cannot clearly dissect whether the ovary is the exclusive source of 20HE (Garen et al., 1977). In future experiments, 20HE titers have to be directly addressed to investigate whether other sources like the brain might be involved in 20HE release upon mating and other physiological stimuli (Chen et al., 2014; Itoh et al., 2011).
In the adjacent posterior midgut, we describe the impact of 20HE on Eip75B-A /- C expression (Figure 7H) and show that 20HE import triggers EcR-activation: i) in VF with low ovarian 20HE levels, overexpression of wild-type EcR and EcI are unable to elicit neither ISC proliferation nor differentiation effects (Figures 1–2). ii) pharmacological activation through RH5849 and Pioglitazone (Figures 2 and 4) as well as (iii) forced EcI expression induce proliferation and EB differentiation in MF mediated by Eip75B-A /- C (Figures 2, 3, 6 and 7). Together with the previous findings on JH upon mating (Reiff et al., 2015), our current data suggest a concerted role for JH on proliferation and 20HE on differentiation of midgut progenitors.
The interplay between Eip75B and Kr-h1 controls intestinal size adaptation
Downstream of the EcR, we found differential regulation of Eip75B isoforms upon mating. Eip75B activates egg production in ovarian GSC (Ables and Drummond-Barbosa, 2010; König et al., 2011; Morris and Spradling, 2012; Uyehara and McKay, 2019) and its knockdown reduces germarial size and number. In accordance with differentiation defects found in EB in our work, the number of cystoblasts, the immediate daughters of GSC, is reduced as well (Ables and Drummond-Barbosa, 2010; Morris and Spradling, 2012).
In our study, we dissected specific roles for Eip75B isoforms. Eip75B-B expression is at the lower detection limit (Figure 3B) and unchanged upon mating (Figure 3C). Thus, our finding that forced Eip75B-B expression raised ISC mitosis in VF (Figure 3—figure supplement 1F) might be due to ectopic expression. Eip75B-B mutants are viable and fertile and Eip75B-B is interacting with DNA only upon forming heterodimers with Hormone receptor 3 (Hr3) temporarily repressing gene expression (Bialecki et al., 2002; Sullivan and Thummel, 2003; White et al., 1997). Further studies of Eip75B-B, especially its expression pattern and transcriptional regulation, are necessary to elucidate in which physiological context, else than mating, Eip75B-B controls ISC proliferation.
Mating induction of Eip75B-A /- C, but not Eip75B-B, underlines their differential transcriptional regulation (Segraves and Hogness, 1990). This idea is supported by the finding that larval epidermis from Manduca sexta exposed to heightened JH-levels is sensitized to 20HE, which results in a 10-fold increase of Eip75B-A, but not of Eip75B-B expression (Dubrovskaya et al., 2004; Zhou et al., 1998). An in silico analysis of 4 kb regulatory regions upstream of Kr-h1 using JASPAR revealed several EcR/USP and Met consensus sequences (Khan et al., 2018), suggesting that both hormonal inputs may also converge on Kr-h1 driving ISC proliferation (Figure 5B; Reiff et al., 2015). We propose a synergistic interplay between JH- and 20HE-signaling upon mating, in which the JH pathway effector Kr-h1 increases proliferation in ISC and the EcR effectors Eip75B-A /- C drive EB into differentiation. The combined action of Kr-h1 and Eip75B-A /- C ensures mating induced organ size growth (Figures 5K and 7H). The experimental investigation of Eip75B and Kr-h1 regulatory regions incorporating endocrine signals from at least two different organs and leading to differential activity and function in ISC and EB will be a fascinating topic for future studies.
Eip75B/PPARγ and progenitor fate commitment in physiology and pathology
A key discovery of our study is that systemic 20HE directs local intestinal progenitor differentiation. Upon EcR activation, we found Eip75B-A /- C promoting EB to EC differentiation. When tissue demand arises, EB are thought to physically separate from ISC and become independent of N. Detached EB acquire motility and are able to postpone their terminal epithelial integration after initial specification (Antonello et al., 2015a; Antonello et al., 2015b; Martin et al., 2018; Siudeja et al., 2015). Epithelial integration of progenitors can be initiated experimentally by nubbin (nub) mediated downregulation of esg and the nub-RB isoform is sufficient to initiate EC formation downstream of Notch signaling (Korzelius et al., 2014; Tang et al., 2018).
Our data suggest, that the ecdysone pathway acting through Eip75B-A /- C provides a similar, remotely inducible signal for terminal EB differentiation. This idea is supported by several lines of evidence: i) EB-specific knockdown of Eip75B stalls their differentiation using kluReDDM (Figure 3—figure supplement 2E), suggesting Eip75B functions after initial N-input. ii) klu+-EB do not change fate upon Eip75B knockdown, suggesting a more mature EB differentiation status that already lost plasticity (Figure 3—figure supplement 2A–D; Korzelius et al., 2019; Reiff et al., 2019). iii) Forced Eip75B-A /- C expression in EB leads to their immediate differentiation independent of N-input (Figure 6C,D, Figure 7—figure supplement 1D,E). In addition, Eip75B was found to be highly expressed in EB in a recent study from the Perrimon lab (Hung et al., 2018), raising the question about its transcriptional regulation.
A plethora of studies established Eip75B as an early response gene downstream of the Ecdysone pathway (Ables and Drummond-Barbosa, 2010; König et al., 2011; Morris and Spradling, 2012; Thummel, 1996; Uyehara and McKay, 2019). Another pathway stimulating Eip75B expression in EB might be the N pathway. N activation leads to immediate EC differentiation of progenitors and thus phenocopies Eip75B-A /- C expression (Hudry et al., 2016; Reiff et al., 2019). Indeed, 20HE- and Notch-signaling have been shown to converge on target genes in various tissues and functions (Mitchell et al., 2013; Sun et al., 2008; Xu et al., 2018) and both pathways acetylate H3K56 modifying multiple regulatory genomic regions including Eip75B (Skalska et al., 2015). We observed that 20HE-signaling leads to the differentiation of progenitors to EC in a N LOF context (Figures 5 and 6, Figure 7—figure supplement 1). This is probably the result from strong genetic and pharmacological stimuli (Figures 6 and 7, Figure 7—figure supplement 1) acting through Eip75B. 20HE signaling might facilitate the expression of differentiation factors like Eip75B as well as klu in the presence of N signaling under normal physiological conditions (Figure 7—figure supplement 1F). Future in-depth analysis of Eip75B regulatory regions and the generation of Eip75B reporter flies will help to understand the interplay of 20HE and N signaling acting on Eip75B.
Confirming a close relationship between fly Eip75B and human PPARγ, we found midgut progenitors responding to the known PPARγ agonist Pioglitazone. In patients, PPARγ plays a role in i) pregnancy related adaptations of lipid metabolism (Waite et al., 2000) and ii) as target in colorectal cancer (CRC) (Sarraf et al., 1999). Decreased PPARγ levels are associated with development of insulin resistance and failure of lipolysis in obese pregnant women (Rodriguez-Cuenca et al., 2012; Vivas et al., 2016). Mating upregulates PPARγ/Eip75B (Figure 3) and we also describe increased lipid uptake upon EcR-activation (Figure 2—figure supplement 1) in absorptive EC. Srebp, a gene with known function in fatty acid metabolism, is also activated (Horton et al., 2003; Reiff et al., 2015; Seegmiller et al., 2002). Studying this interplay between SREBP, PPARγ and steroid hormone signaling will shed light on to which extent adaptation of intestinal size and lipid metabolism contribute to pathophysiological conditions like diabetes.
In CRC, PPARγ loss-of-function mutations are observed in around 8% of patients (Sarraf et al., 1999). PPARγ expression correlates with good prognosis and PPARγ is epigenetically silenced during CRC progression (Ogino et al., 2009; Pancione et al., 2010). In agreement with our in vivo experiments on Eip75B-A /- C in EB differentiation (Figures 3–7), in vitro studies identified PPARγ promoting differentiation in various colon cancer cell lines (Cesario et al., 2006; Shimizu and Moriwaki, 2008; Yamazaki et al., 2007; Yoshizumi et al., 2004).
In a CRC mouse model, biallelic loss of PPARγ leads to a 4-fold increase tumor incidence and reduced survival in female mice over males (ApcMin/+/PPARγ-/-) (McAlpine et al., 2006), whereas males develop around three times more colon tumors with wild type PPARγ (Cooper et al., 2005). In line with this, ovariectomized ApcMin/+ mice develop significantly more tumors and have decreased PPARγ expression, highlighting a possible link between PPARγ and estrogen signaling. Meta-analyses of hormone replacement studies have shown that estrogen confers a lower risk to develop CRC and better survival rates for female CRC patients (Chen et al., 1998; Hendifar et al., 2009; Lin et al., 2012).
It is tempting to speculate that this mechanism of steroid hormones signaling through EcR/ER (estrogen receptor) to activate Eip75B/PPARγ is conserved from flies to humans. Indeed, human estradiol and 20HE activate the EcR with similar affinity and elicit Eip75B transcription by binding to EcRE (Ecdysone-Responsive Elements). EcRE can be converted to functional ERE (estrogen responsive elements) by a simple change of nucleotide spacing suggesting high conservation (Martinez et al., 1991; Schwedes et al., 2011).
Taken together, our findings reveal that mating induced steroid hormone release, which signals to adjacent intestinal stem cells, controls their proliferation and, more importantly, differentiation of committed precursor cells through Eip75B/PPARγ. Ecdysone control of cell fate ensures the production of absorptive enterocytes during mating related intestinal adaptations and induces enterocyte fate in a Drosophila intestinal tumor model marked by loss of enterocyte differentiation. Mechanistically, we propose that Eip75B/PPARγ exerts an anti-neoplastic role by promoting progenitor differentiation into postmitotic enterocyte fate, thereby reducing the pool of mitotically active cells. Our findings might be a first step towards understanding the protective, but so far mechanistically unclear tumor suppressive role of steroid hormones in female colorectal cancer patients. Downstream of steroidal signaling, Eip75B/PPARγ promotes postmitotic cell fate when local signaling is deteriorated and thus might reflect a promising target for future studies in colorectal cancer models.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (Drosophila melanogaster) | esgReDDM | Antonello et al., 2015a DOI: 10.15252/embj.201591517 | Figures 1–7; Figure 1—figure supplement 1; Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | esgReDDM > Eip75B-A | Rabinovich et al., 2016 DOI: 10.1016/j.cell.2015.11.047 | Figure 3; Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | esgReDDM > Eip75B-B | Rabinovich et al., 2016 DOI: 10.1016/j.cell.2015.11.047 | Figure 3; Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | esgReDDM > Eip75B-C | Rabinovich et al., 2016 DOI: 10.1016/j.cell.2015.11.047 | Figure 3; Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | Srebp > CD8::GFP | Reiff et al., 2015 DOI: 10.7554/eLife.06930 | Figure 2—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | Mex > ts | Phillips and Thomas, 2006 DOI: 10.1242/jcs.02839 | Figure 2—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | MARCM (FRT2A) | Lee and Luo, 1999 DOI: 10.1016/S0896-6273 (00)80701–1 | Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | Eip75A81-MARCM (FRT2A) | Rabinovich et al., 2016 DOI: 10.1016/j.cell.2015.11.047 | Figure 3—figure supplement 1 | |
| Genetic reagent (D. melanogaster) | kluReDDM | Reiff et al., 2019 DOI: 10.15252/embj.2018101346 | Figure 3—figure supplement 2 | |
| Genetic reagent (D. melanogaster) | N55e11-MARCM (FRT19A) | Guo and Ohlstein, 2015 DOI: 10.1126/science.aab0988 | Figure 7—figure supplement 1 | |
| Chemical compund, drug | RH5849 | DrEhrenstorfer | DRE-C16813000 | 340 µM final concentration |
| Chemical compund, drug | Pioglitazone | Sigma-Aldrich | Sigma-Aldrich 112529-15-4 | 14 µM final concentration |
Genetics and fly husbandry/fly strains
Request a detailed protocolOregonR and w1118 flies served as wild-type controls. The following transgenes and mutations were employed: esgReDDM (Antonello et al., 2015a), kluReDDM (Reiff et al., 2019), UAS-EcI-Flag-HA (Okamoto et al., 2018), UAS-EcI (Okamoto et al., 2018), UAS-EcI-RNAi (Okamoto et al., 2018), UAS-Eip75B-A-Flag (Rabinovich et al., 2016), UAS-Eip75B-B-Flag (Rabinovich et al., 2016), UAS-Eip75B-C-Flag (Rabinovich et al., 2016), UAS-NDN (J. Treisman), Dl::GFP (F. Schweisguth), N55e11 FRT19A (Guo and Ohlstein, 2015), Srebp-GAL4 (Kunte et al., 2006), Mex-Gal4 (Phillips and Thomas, 2006), Gbe+Su(H)dsRed (T. Klein). From Bloomington Drosophila Stock Center (BDSC): UAS-EcR-RNAi (BL58286), UAS-EcR.B2 (BL4934), UAS-EcR.B2W650A (BL9449), UAS-EcR.B2F645A (BL9450), EcRM554fs (BL4894), EcRE-lacZ (BL4516), Eip75BA81(BL23654), UAS-CD8::GFP (BL5137), ovoD1 (BL1309), NRE::GFP (BL30727), NRE::GFP (BL30728), Dlg-1::GFP (BL59417), From Kyoto Drosophila Stock Center: UAS-Kr-h1 (DGRC120052). From FlyORF, Switzerland: UAS-EcR-HA (F000480), UAS-Kr-h1 (F000495). From Vienna Drosophila Resource Center (VDRC) UAS-EcR-RNAi (GD37059), UAS-Eip75B-RNAi (GD44851), UAS-Eip75B-RNAi (KK108399), UAS-N-RNAi (GD14477).
MARCM clones
Request a detailed protocolMosaic analysis with repressible cell marker (MARCM; Lee and Luo, 1999) clones were induced in midguts by Flippase under control of a heat-shock promoter. Expression of the Flippase was activated for 45 min in a 37°C-water bath to induce positively marked clones. Guts were dissected 5 days after induction. Clones in experimental and control flies were induced in parallel.
Food composition and fly keeping
Request a detailed protocolFly food contained 1424 g corn meal, 900 g malt extract, 800 g sugar beet syrup, 336 g dried yeast, 190 g soy fluor, 100 g agarose, 90 ml propionic acid and 30 g NIPAGIN powder (antimycotic agent) in 20 l H2O. Food was cooked for about an hour to reduce bioburden, then filled into small plastic vials and cooled down to RT. Flies were kept at 25°C except for crosses with temperature-sensitive GAL80ts (GAL4 repressor) which were kept at 18°C (permissive temperature) until shifted to 29°C (restrictive temperature) to activate GAL4-mediated transgene expression. Crosses with esgReDDM and kluReDDM were carried out as described previously (Antonello et al., 2015a; Reiff et al., 2015; Reiff et al., 2019). Due to persisting problems with mucous formation on food surface in vials with VF, all experiments distinguishing between mated and virgin female flies were run on food with twice the amount of NIPAGIN. Mucous formation was avoided because of massive induction of tissue renewal by pathogenic stress.
Hormone analogue treatments
Request a detailed protocolA vial of fly food was reheated in the microwave until it became liquid, the hormone analogues were added, thoroughly mixed and filled into a new vial. For each ml of food 5 µl of RH5849 (340 µM final concentration; 20 µg/µl stock solution, diluted in MeOH; DRE-C16813000, DrEhrenstorfer) was added. As a control, an equivalent volume of carrier solution (MeOH) was added to the food. Hormone analogue treatments were performed for the period of the 7 days ReDDM (Antonello et al., 2015a) shift.
PPARγ agonist treatments
Request a detailed protocolA vial of fly food was reheated in the microwave until it became liquid, the PPARγ agonist was added, thoroughly mixed and filled into a new vial. For each ml of food 2.5 µl Pioglitazone (14 µM final concentration, 2 µg/µl stock solution, diluted in DMSO; Sigma-Aldrich, St. Louis, USA) were added. The equivalent amount of DMSO served as control. Flies were starved in an empty vial for at least six hours to ensure synchronized feeding when set to Pioglitazone. Food was renewed after three days and fly midguts were dissected after five days.
Immunohistochemistry
Request a detailed protocolGuts were dissected in PBS and transferred into 4% PFA immediately after dissection and staining was performed on an orbital shaker. After 45 min of fixation guts were washed once with PBS for 10 min. Antibodies were diluted in 0.5% PBT + 5% normal goat serum. The incubation with primary antibodies (1:250 anti-Dlg-1 [mouse; Developmental studies Hybridoma Bank (DSHB)]; 1:50 anti-Pros [mouse; DSHB]; 1:2000 anti-pH3 [rabbit; Merck Millipore, 06–570]; 1:50 anti-EcR common Ag10.2 [mouse; DSHB]; 1:500 anti-HA High Affinity 3F10 [rat; Merck, Sigma-Aldrich]; 1:1500 anti-ß-Galactosidase preabsorbed [rabbit; Cappel Research Products]) was performed at 4°C over night. After washing with PBS guts were incubated with secondary antibodies (1:500 Goat anti-MouseAlexa647 [Invitrogen]; 1:500 Goat anti-RatAlexa647 [Invitrogen]; 1:500 Goat anti-RabbitAlexa647 [Invitrogen]) and DAPI (1:1000; 100 µg/ml stock solution in 0.18 M Tris pH 7.4; DAPI No. 18860, Serva, Heidelberg) for at least 3 hr at RT. Guts were washed a last time with PBS prior to mounting in Fluoromount-G Mounting Medium (Electron Microscopy Sciences).
X-Gal staining of Drosophila midguts
Request a detailed protocolGuts were dissected in PBS and transferred into 4% PFA immediately after dissection. After 20 min of fixation, guts were washed three times with 0.3% PBT. Stainingbuffer (0.15M NaCl; 1 mM MgCl2; 10 mM Na-phosphate buffer pH 7.2; 3.3 mM F3Fe(CN)6, 3.3 mM K4Fe(CN)6; 0.3% Triton X-100) was heated to 65°C and 3% X-Gal added. The guts were stained for at least 1 hr at 37°C until a dark blue staining became visible. Guts were washed two times in 0.3% PBT prior to mounting in Fluoromount-G Mounting Medium (Electron Microscopy Sciences). Stained midguts were imaged using an Axiophot2 microscope (Carl Zeiss) equipped with an AxioCam MRc (Carl Zeiss).
OilRedO staining of Drosophila midguts
Request a detailed protocolGuts were dissected in PBS and transferred into 4% PFA immediately after dissection. After 45 min of fixation, guts were washed in consecutive applications of 1xPBS, double-distilled H2O, and 60% isopropanol. Guts were stained in a 6:4 dilution of OilRedO (Sigma-Aldrich, 0.1% stock solution diluted in isopropanol) in dH2O for 20 min, then washed in 60% isopropanol and dH2O. After mounting in Fluoromount-G Mounting Medium (Electron Microscopy Sciences, emsdiasum) PMG were imaged using an Axiophot2 microscope (Carl Zeiss) equipped with an AxioCam MRc (Carl Zeiss). OilRedO staining intensity was analyzed using Fiji. RGB channels were split and the green channel was subtracted from the red channel to eliminate background signal. A fixed threshold was set, and guts were manually outlined as a ROI. The mean intensity of the resulting signal within the ROI was measured.
Image acquisition
Request a detailed protocolPosterior midguts were imaged using an LSM 880 Airyscan confocal microscope (Carl Zeiss) using ‘Plan-Apochromat 10x/0.45 M27’, ‘Plan-Apochromat 20x/0.8 M27’ and ‘C-Apochromat 40x/1.20 W Corr M27’ objectives. Image resolution was set to at least 2048 × 2048 pixels. At least three focal planes (1 µm distance) were combined into a Z-stack to image one cell layer and to compensate for gut curvature.
For determining whole midgut length, an Axio Zoom.V16 (Carl Zeiss) was employed with DAPI filter and 1x/0.25-objective.
Quantification of proliferation and intensity measurements
Request a detailed protocolMaximum intensity projections were calculated from Z-stack images of PMG by Fiji (ImageJ 1.51 n, Wayne Rasband, National Institutes of Health, USA). Total cell number and RFP-positive cell count of ReDDM (Antonello et al., 2015a) guts were analyzed semi-automatically by a self-written macro for Fiji whereas GFP-positive cells were counted manually (macro available from the authors).
For fluorescence or OilRedO intensity measurements, intestines were scanned with fixed laser/exposure time settings and measured in Fiji. The region of interest was selected manually, and mean intensity of the area was determined. This way, relative EcR and ß-Galactosidase protein levels were measured in antibody-stainings, relative SREBP activity was analyzed in PMG cells expressing mCD8::GFP under the control of Srebp-GAL4, and amount of triglycerides was analyzed by OilRedO stainings.
Jaspar
Request a detailed protocolThe open-access webtool JASPAR (Khan et al., 2018) was utilized to predict transcription factor binding sites within the 5’-UTR of EcI. It was specifically scanned for binding motifs related to Ecdysone and Juvenile Hormone signaling.
RNA isolation and cDNA synthesis
Request a detailed protocolThe R5 regions or ovaries of at least three female flies were dissected and transferred into a droplet of RNAlater Solution (Invitrogen by Thermo Fisher Scientific) on ice. The tissue was homogenized in 100 µl peqGOLD TriFast (VWR Life Science) and total RNA was isolated as specified by the manufacturer. The following cDNA synthesis was performed with 250 ng of total RNA and the SuperScript IV Reverse Transcriptase (Invitrogen by Thermo Fisher Scientific) using a 1:1 mixture of oligo-dT primers and random hexamers directly upon RNA isolation. Prior to Real-time qPCR, cDNA samples were diluted 1:2 in dH2O.
Verification of gene expression in the adult midgut by PCR
Request a detailed protocolTo verify gene expression in the adult Drosophila midgut, PCRs were performed with primer pairs specific for ovo and the Svb isoform, or isoform-specific primer pairs for EcR spanning at least one exon-exon boundary. PCRs were performed with Q5 High-Fidelity DNA Polymerase (NEB) for at least 30 cycles. Reaction products were loaded on an agarose gel (1.5%) and separated by electrophoresis to verify lengths of PCR products.
| Primer | Forward (5’−3’) | Reverse (5’−3’) |
|---|---|---|
| EcR.A | GGGGTCTAAGAAACATTTTGAGG | CCATTTGCAGCTGCAGCCGACGT |
| EcR.B1 | GCACGTACGAAGCCCGATCGCGT | CCGGACTCGTTGCCGCAGAGCC |
| EcR.B2 | GCACGTACGAAGCCCGATCGCGT | CTCTTCCCTCTGTTCACGCCC |
| ovo | CGCAGAGCCAAGATGTACGTG | GATAGTGGACCTCCGGCT |
| SvbRep | ACAGTAAGTTGCGAGCCGG | TGTTTTGGGGTGTCCTTTCGTG |
Real-time qPCR
Request a detailed protocolExpression levels of Ecdysone signaling pathway-associated genes were determined in VF and MF. Eclosed OregonR or EcRE-LacZ flies were aged for 4d before mating. After 72 hr of mating, RNA was isolated and cDNA synthetized before running qPCRs. After an enzyme activation step (20 s 95°C), 40 cycles of denaturation (2 s 95°C), primer annealing (20 s 58°C) and elongation (20 s 72°C) were run. Primers were designed to anneal at 59°C. Reaction was set up with KAPA SYBR FAST Universal (Roche) in a total volume of 10 µl. All qPCR results were normalized to the house-keeping gene rp49.
| Primer | Forward (5’−3’) | Reverse (5’−3’) |
|---|---|---|
| Eip74EF-A | AGAAACTTCGAGGCAATAGGGT | TGTGCGGCCTCATCTCAAG |
| Eip74EF-B | TGGCCATCCCACAACGC | GGGCGGAAATGAACCTGTTG |
| Eip75B-A | CCTGTGCCAGAAGTTCGATGA | AAGAATCCATCGGCATCTTCGT |
| Eip75B-B | CGTCTAGCTCGATTCCTGATCTA | CGGAAGAATCCCTTGCAACC |
| Eip75B-C | CTGTGGTTCCGGCGGATT | TCGAATTCTATGTTGAGTTCTGGTT |
| EcI | TGCAGTGCCGCTCTCAACTGTACC | TCACAGTAACCGTTGACCGCCTCC |
| EcR.A | GTGTTCGGTGAAAAACGCAA | TCCTAGCAACTGAGCTTTTGTAGAC |
| EcR.B1 | TTAACGGTTGTTCGCTCGCA | AGTGCGGGAAACAATCAGAGCAT |
| EcR.B2 | GTTAACGGTTGTTCGCTCGC | TGCGGGAAACAATCAGAGCATA |
| Kr-h1(A) | ACAATTTTATGATTCAGCCACAACC | GTTAGTGGAGGCGGAACCTG |
| Kr-h1(B) | AAATCTTGGGCACCCAAACAA | GTTGTGGCTGAATCTTTCGC |
| Lac-Z | ATCAGGATATGTGGCGGATGAGCG | AGTACAGCGCGGCTGAAATCATC |
| rp49 | TGGTTTCCGGCAAGCTTCAA | TGTTGTCGATACCCTTGGGC |
20-HE isolation and titer determination
Request a detailed protocol20-HE titers were determined in VF and MF of the same age. Eclosed OregonR or heterozygous ovoD1 mutant flies were aged for 3d before mating. After 24 hr or 48 hr at least 20 adult female flies were pierced in the thorax with a needle and put into a 150 µl-PCR tube that was punctured in its very bottom. Hemolymph was harvested by centrifugation (5.000x g 5 min RT) and collected in a 1.5 ml-reaction tube. Total weight of flies was determined before and after centrifugation. Typical yields of hemolymph were around 0.6–1 mg. The isolated hemolymph was mixed with 500 µl MeOH, centrifuged (12.000x g 20 min 4°C) and the supernatant was transferred into a new 1.5 ml-reaction tube. MeOH was evaporated at 30°C in a vacuum centrifuge (Eppendorf concentrator plus) and 20HE was resuspended in 100 µl EIA buffer and stored at −20°C until usage.
Ecdysone levels were determined using the 20-Hydroxyecdysone Enzyme Immunoassay kit according to manufacturer’s instructions (#A05120.96 wells; Bertin Bioreagent). 20HE titer was normalized to hemolymph yield.
Statistical analysis
Request a detailed protocolFigures of quantifications were assembled, and statistics were run in GraphPad Prism 6.01. For single comparisons, data sets were analyzed by two-sided unpaired t-test. For multiple comparisons, data sets were analyzed by one-way ANOVA and Tukey’s post-hoc test. Significant differences are displayed as * for p≤0.05, ** for p≤0.01, *** for p≤0.001 and **** for p≤0.0001.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files. Source data files have been provided in a separate Excel File.
References
-
A view through a chromatin loop: insights into the ecdysone activation of early genes in DrosophilaNucleic Acids Research 42:10409–10424.https://doi.org/10.1093/nar/gku754
-
Ecdysteroids in adult males and females of Drosophila melanogasterJournal of Insect Physiology 30:823–830.https://doi.org/10.1016/0022-1910(84)90019-2
-
Genetic analysis of three dominant Female-Sterile mutations located on the X chromosome of Drosophila melanogasterGenetics 105:309–325.
-
Recent use of hormone replacement therapy and the prevalence of colorectal adenomasCancer Epidemiology Biomarkers Prevention 7:227–230.
-
Control and biochemical nature of the ecdysteroidogenic pathwayAnnual Review of Entomology 47:883–916.https://doi.org/10.1146/annurev.ento.47.091201.145302
-
Adaptation of the maternal intestine during lactationJournal of Mammary Gland Biology and Neoplasia 2:243–252.https://doi.org/10.1023/a:1026332304435
-
Ecdysteroid titers in mated and unmated Drosophila melanogaster femalesJournal of Insect Physiology 45:571–577.https://doi.org/10.1016/S0022-1910(99)00038-4
-
Gender disparities in metastatic colorectal Cancer survivalClinical Cancer Research 15:6391–6397.https://doi.org/10.1158/1078-0432.CCR-09-0877
-
Control of endodermal endocrine development by Hes-1Nature Genetics 24:36–44.https://doi.org/10.1038/71657
-
Steroid regulated programmed cell death during Drosophila metamorphosisDevelopment 124:4673–4683.
-
The juvenile hormone signaling pathway in insect developmentAnnual Review of Entomology 58:181–204.https://doi.org/10.1146/annurev-ento-120811-153700
-
PPAR gamma activation is neuroprotective in a Drosophila model of ALS based on TDP-43Human Molecular Genetics 24:1741–1754.https://doi.org/10.1093/hmg/ddu587
-
Ecdysone coordinates the timing and amounts of E74A and E74B transcription in DrosophilaGenes & Development 5:1067–1079.https://doi.org/10.1101/gad.5.6.1067
-
Nuclear receptors — a perspective from DrosophilaNature Reviews Genetics 6:311–323.https://doi.org/10.1038/nrg1581
-
Reproductive and post-reproductive life history of wild-caught Drosophila melanogaster under laboratory conditionsJournal of Evolutionary Biology 26:1508–1520.https://doi.org/10.1111/jeb.12155
-
Steroid regulation of postembryonic development and reproduction in DrosophilaTrends in Endocrinology & Metabolism 11:276–280.https://doi.org/10.1016/S1043-2760(00)00282-4
-
Fatty acid auxotrophy in Drosophila larvae lacking SREBPCell Metabolism 3:439–448.https://doi.org/10.1016/j.cmet.2006.04.011
-
Prognostic significance of TWEAK expression in colorectal Cancer and effect of its inhibition on invasionAnnals of Surgical Oncology 19 Suppl 3:385–394.https://doi.org/10.1245/s10434-011-1825-x
-
Intestinal-specific PPARgamma deficiency enhances tumorigenesis in ApcMin/+ miceInternational Journal of Cancer 119:2339–2346.https://doi.org/10.1002/ijc.22115
-
The ovo locus is required for sex-specific germ line maintenance in DrosophilaGenes & Development 1:913–923.https://doi.org/10.1101/gad.1.9.913
-
Niche appropriation by Drosophila intestinal stem cell tumoursNature Cell Biology 17:1182–1192.https://doi.org/10.1038/ncb3214
-
Brush border spectrin is required for early endosome recycling in DrosophilaJournal of Cell Science 119:1361–1370.https://doi.org/10.1242/jcs.02839
-
Connecting metabolism and reproduction: roles of central energy sensors and key molecular mediatorsMolecular and Cellular Endocrinology 397:4–14.https://doi.org/10.1016/j.mce.2014.09.027
-
Peroxisome proliferator-activated receptor γ-dependent regulation of lipolytic nodes and metabolic flexibilityMolecular and Cellular Biology 32:1555–1565.https://doi.org/10.1128/MCB.06154-11
-
Ecdysone receptor expression and activity in adult Drosophila melanogasterJournal of Insect Physiology 57:899–907.https://doi.org/10.1016/j.jinsphys.2011.03.027
-
Regulation of the endocycle/gene amplification switch by notch and ecdysone signalingJournal of Cell Biology 182:885–896.https://doi.org/10.1083/jcb.200802084
-
Endocrine insights into the evolution of metamorphosis in insectsAnnual Review of Entomology 47:467–500.https://doi.org/10.1146/annurev.ento.47.091201.145230
-
Placental peroxisome proliferator-activated receptor-gamma is up-regulated by pregnancy serumThe Journal of Clinical Endocrinology and Metabolism 85:3808–3814.https://doi.org/10.1210/jcem.85.10.6847
-
Thiazolidinedione, a peroxisome proliferator-activated receptor-gamma ligand, inhibits growth and metastasis of HT-29 human Colon cancer cells through differentiation-promoting effectsInternational Journal of Oncology 25:631–639.
Article and author information
Author details
Funding
Deutsche Forschungsgemeinschaft (RE 34532-1)
- Tobias Reiff
Wilhelm Sander-Stiftung (2018.145.1)
- Lisa Zipper
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Maria Dominguez, Thomas Klein, Oren Schuldiner, Francois Schweisguth, Naoki Yamanaka, the Bloomington Drosophila Stock Center (NIHP400D018537), the Transgenic RNAi Project (TRiP) at Harvard Medical School (NIK/NIGMS R01-GM084947) and the Vienna Drosophila Resource Center (VDRC, http://www.vdrc.at) for providing transgenic fly stocks. We thank the Center for Advanced Imaging (CAi) at HHU for providing microscopy services. TR thanks Maria Dominguez in whose lab he initiated this project and Thomas Klein for being a very supportive host. We also thank Zeus Antonello, Nahuel Villegas, Hendrik Pannen and Thomas Klein for comments on the manuscript. The project is funded by a Deutsche Forschungsgesellschaft (DFG-Sachbeihilfe RE 34532–1) grant. LZ is supported by the Wilhelm Sander-Stiftung (2018.145.1).
Copyright
© 2020, Zipper et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,656
- views
-
- 574
- downloads
-
- 96
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 96
- citations for umbrella DOI https://doi.org/10.7554/eLife.55795
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Ecdysone steroid hormone remote controls intestinal stem cell fate decisions via the PPARγ-homolog Eip75B in Drosophila
eLife 9:e55795.
https://doi.org/10.7554/eLife.55795




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}