Functional interrogation of HOXA9 regulome in MLLr leukemia via reporter-based CRISPR/Cas9 screen
- Division of Hematology/Oncology, University of Alabama at Birmingham, United States
- O’Neal Comprehensive Cancer Center, University of Alabama at Birmingham, United States
- Department of Tumor Cell Biology, St. Jude Children’s Research Hospital, United States
- Cancer Biology Program/Comprehensive Cancer Center, St. Jude Children’s Research Hospital, United States
- Department of Pharmaceutical Sciences, St. Jude Children’s Research Hospital, United States
- Department of Computational Biology, St. Jude Children’s Research Hospital, United States
- Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, United States
- Department of Biomedical Engineering School of Engineering, University of Alabama at Birmingham, United States
Abstract
Aberrant HOXA9 expression is a hallmark of most aggressive acute leukemias, notably those with KMT2A (MLL) gene rearrangements. HOXA9 overexpression not only predicts poor diagnosis and outcome but also plays a critical role in leukemia transformation and maintenance. However, our current understanding of HOXA9 regulation in leukemia is limited, hindering development of therapeutic strategies. Here, we generated the HOXA9-mCherry knock-in reporter cell lines to dissect HOXA9 regulation. By utilizing the reporter and CRISPR/Cas9 screens, we identified transcription factors controlling HOXA9 expression, including a novel regulator, USF2, whose depletion significantly down-regulated HOXA9 expression and impaired MLLr leukemia cell proliferation. Ectopic expression of Hoxa9 rescued impaired leukemia cell proliferation upon USF2 loss. Cut and Run analysis revealed the direct occupancy of USF2 at HOXA9 promoter in MLLr leukemia cells. Collectively, the HOXA9 reporter facilitated the functional interrogation of the HOXA9 regulome and has advanced our understanding of the molecular regulation network in HOXA9-driven leukemia.
Introduction
Dysregulation of the homeobox (HOX)-containing transcription factor HOXA9 is a prominent feature in most aggressive acute leukemias (Collins and Hess, 2016a; Alharbi et al., 2013). During normal hematopoiesis, HOXA9 plays a critical role in hematopoietic stem cell expansion and is epigenetically silenced during lineage differentiation (Alharbi et al., 2013). In certain leukemia subtypes, this regulatory switch fails and HOXA9 is maintained at high levels to promote leukemogenesis. However, the mechanisms governing HOXA9 expression remain to be fully understood. HOXA9 overexpression is commonly observed in over 70% of human acute myeloid leukemia (AML) cases and ~10% of acute lymphoblastic leukemia (ALL) cases (Jambon et al., 2019). Notably, the high expression of HOXA9 is sharply correlated with poor prognosis and outcome in human leukemia (Golub et al., 1999; Baccelli et al., 2019). An accumulating body of evidence indicates that HOXA9 dysregulation is both sufficient and necessary for leukemic transformation (Collins and Hess, 2016a; Alharbi et al., 2013). Forced expression of HOXA9 enforces self-renewal, impairs myeloid differentiation of murine marrow progenitors, and ultimately leads to late onset of leukemia transformation (Bach et al., 2010), which is accelerated by co-expression with interacting partner protein MEIS1 (Kroon, 1998). Conversely, knocking down HOXA9 expression results in leukemic cell differentiation and apoptosis (Ayton and Cleary, 2003; Zeisig et al., 2004). Thus, excessive HOXA9 expression has emerged as a critical mechanism of leukemia transformation in many hematopoietic malignancies.
Consistent with the broad overexpression pattern of HOXA9 in many leukemia cases, a wide variety of genetic alterations in leukemia contribute to HOXA9 dysregulation including MLL gene rearrangements (MLLr), NPM1 mutations, NUP98-fusions, EZH2 loss-of-function mutations, ASXL1 mutations, MOZ fusions and other chromosome alterations (Collins and Hess, 2016a; Jambon et al., 2019; De Braekeleer et al., 2014; Collins and Hess, 2016b). Additionally, our recent work shows that DNMT3A hotspot mutations may also contribute to HOXA9 overexpression by preventing DNA methylation at its regulatory regions (Lu et al., 2016). Given that genomic variation of HOXA9 including NUP98-HOXA9 fusion and gene amplification accounted for less than 2% of HOXA9 overexpression in AML cases (Xu et al., 2016; Gough et al., 2011; Nakamura et al., 1996), uncovering the upstream epigenetic and transcriptional regulators of HOXA9 in leukemia could advance the design of novel therapeutic interventions. For example, because MLLr proteins recruit the histone methyltransferase DOT1L to the HOXA locus promoting hyper-methylation at histone H3 lysine 79 and subsequent high HOXA9 transcription (Krivtsov et al., 2008), selective DOT1L inhibitors have been exploited to inhibit leukemia development and HOXA9 expression in MLLr leukemias and are now in clinical trials (Chen et al., 2015; Stein and Tallman, 2015). However, DOT1L inhibitors usually act slowly and their effects remain sub-optimal. To date, most known HOXA9 regulator proteins are epigenetic modifiers, and little is known about which DNA-binding transcription factors are involved in directly regulating HOXA9 expression in acute leukemia (Godfrey et al., 2017; Daigle et al., 2011; Yu et al., 2012; Shi et al., 2012).
Previous studies have also advocated that the organization of chromatin domains at the HOXA gene cluster contributes to high HOXA9 expression in cancer cells (Luo et al., 2018; Xu et al., 2014). Specifically, CCCTC-binding factor CTCF may potentiate HOXA9 expression through direct binding at the conserved motif between HOXA7 and HOXA9 (CBS7/9) to establish necessary chromatin looping interaction networks in MLLr AML MOLM13 cells (Luo et al., 2018; Luo et al., 2019). In contrast, Ghasemi et al. reported that HOXA gene expression was maintained in the CTCF-binding site deletion mutants derived from AML OCI-AML3 cells, suggesting that transcriptional activity at the HOXA locus in NPM1-mutant AML cells does not require long-range CTCF-mediated chromatin interactions (Ghasemi et al., 2020). These data also suggest that CTCF may play a cell-type-dependent role on HOXA9 regulation. However, whether loss of CTCF has a direct effect on HOXA9 expression remains to be studied. Lastly, although the clinical significance of HOXA9 has been recognized for more than two decades, it is technically difficult to systematically discover regulators of HOXA9 in acute leukemia owing to the lack of an endogenous reporter to dictate HOXA9 expression.
In this work, we sought to establish an endogenous reporter system enabling real-time monitoring of HOXA9 expression in conjunction with high-throughput CRISPR/Cas9 screening in a human B-ALL MLLr t(4;11) cell line SEM and a AML MLLr t(6;11) cell line OCI-AML2 equipped with an endogenous HOXA9P2A-mCherry reporter allele. The HOXA9P2A-mCherry reporter allele authentically recapitulated endogenous transcription of the HOXA9 gene and did not affect endogenous transcription of other adjacent HOXA genes. To gain a global understanding of the transcription factors regulating HOXA9 expression, we performed a CRISPR/Cas9 loss-of-function screen specifically targeting 1639 human transcription factors. Our screening robustly re-identified expected targets such as KMT2A, DOT1L and HOXA9 itself. Surprisingly, the CRISPR screen and global depletion of CTCF via siRNA and degron-associated protein degradation all demonstrated that HOXA9 does not downregulate upon CTCF loss. More importantly, we identified novel functional regulators of HOXA9 including Upstream Transcription Factor 2 (USF2). USF2 depletion selectively downregulated HOXA9 expression in MLLr leukemia cells and impaired cell growth, which could be rescued by ectopic expression of HOXA9 and its partner MEIS1. Thus, our HOXA9P2A-mCherry reporter lines are robust tools for discovery of novel HOXA9 regulators.
Results
Establishment and characterization of the HOXA9P2A-mCherry reporter human MLLr leukemia cell line
As shown by many previous studies, HOXA9 overexpression was observed in refractory MLL-rearranged ALL and AML patients (Gu et al., 2019; Haferlach et al., 2010; Kohlmann et al., 2008; Figure 1—figure supplement 1A–C). Therefore, we utilized our previously reported high-efficiency knock-in strategy, ‘CHASE knock-in’ (Hyle et al., 2019), to deliver the P2A-mCherry cassette upstream of the HOXA9 stop codon in a patient-derived human B-ALL cell line, SEM, which has a typical B-ALL signature along with a t(4;11) translocation and maintains one single allele expression of the HOXA gene cluster (Figure 1—figure supplement 1D). Because the P2A-mediated ribosome skipping disrupts the synthesis of the glycyl-prolyl peptide bond at the C-terminus of the P2A peptide, translation leads to dissociation of the P2A peptide and its immediate downstream mCherry protein (Kim et al., 2011). Therefore, the knock-in allele would produce a functional HOXA9 protein under control of the endogenous promoter and intrinsic cis-regulatory elements while delivering a separate mCherry protein. In brief, we constructed the knock-in vector containing a P2A-mCherry cassette flanked with 5’ and 3’ HOXA9 homology arms (HAs) of approximately 800-bps, which were cloned from SEM cells. A single guide RNA (sgRNA) and a protospacer adjacent motif (PAM) sequence targeting the genomic sequence 5’ of the HOXA9 stop codon was inserted into the 5’ end of the 5’ HA and 3’ end of the 3’ HA (Figure 1A). When the HA/knock-in cassette was co-electroporated with an all-in-one vector expressing wild-type Cas9 and the same HOXA9 sgRNA, the HA/knock-in cassette was released from the donor vector with two nuclease cleavages and delivered to the target genomic region where double-strand breaks occurred. Successful knock-in cells were enriched by flow cytometry sorting for mCherry (Figure 1B) and characterized via genotyping PCR and Sanger sequencing (Figure 1C). To examine the possibility of random integration of the P2A-mCherry cassette, fluorescence in situ hybridization (FISH) was performed with a P2A-mCherry DNA probe (red) and a FITC-labeled fosmid DNA probe targeting the HOXA9 locus (green). On-target knock-in cells displayed co-localization of red and green fluorescence without random integration signals in the rest of genome (Figure 1D and Figure 1—figure supplement 2A–D). The bulk knock-in population from SEM cells, hereafter called HOXA9P2A-mCherry, was used as a reporter cell line for the entire study. Similarly, a HOXA9P2A-mCherry allele was delivered to a human MLLr AML cell line OCI-AML2 (Figure 1—figure supplement 2E). Many knock-in studies reported the exogenous DNA fragment may affect normal endogenous gene expression in a complex chromatin niche (Liu et al., 2019; Zu et al., 2013). Therefore, to test whether the inserted P2A-mCherry segment would affect the gene expression pattern of HOXA9 and its neighboring HOXA cluster genes, Q-PCR analysis was conducted on both wild-type (WT) and HOXA9P2A-mCherry knock-in (KI) cells. RNA-seq data collected from SEM cells in our previous studies suggested that HOXA7, HOXA9 and HOXA10 were the only highly expressed HOXA genes in MLLr leukemia SEM cells (Hyle et al., 2019; Figure 1E), and that these patterns were indistinguishable between WT and KI populations, indicating the P2A-mCherry knock-in did not alter the gene expression landscape at the HOXA cluster (Figure 1F).
Figure 1 with 2 supplements see all
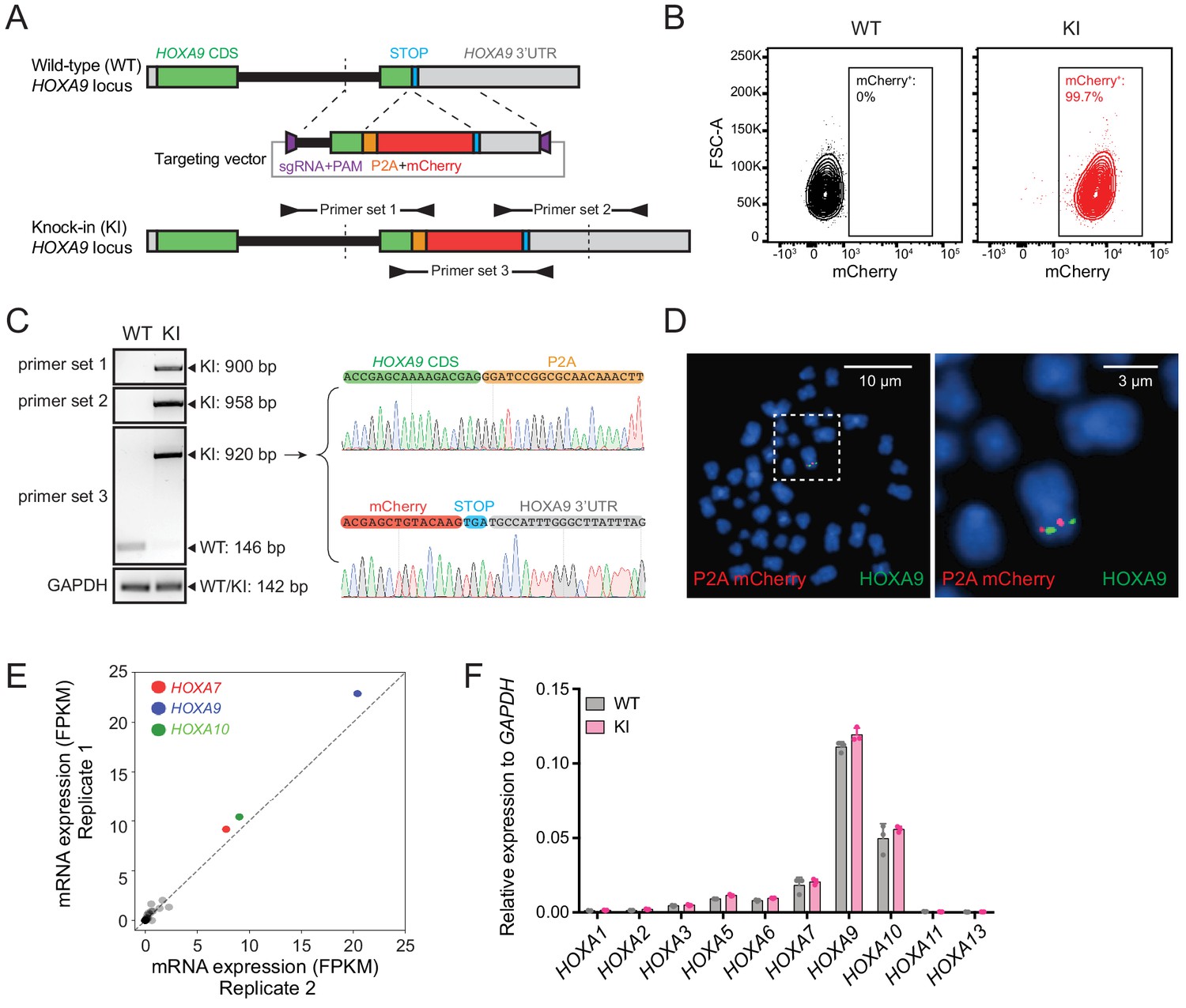
Establishment and characterization of the HOXA9P2A-mCherry reporter human MLLr leukemia cell line.
(A) Schematic diagram of the knock-in design and genotyping PCR primer design for the HOXA9P2A-mCherry reporter allele. (B) Flow cytometry analysis of HOXA9P2A-mCherry reporter cells. Wild-type SEM cells were used as negative controls. (C) Genotyping PCR products from the 5′ and 3′ knock-in boundaries were sequenced to verify the seamless knock-in of the mCherry reporter gene to the endogenous locus. (D) Fluorescence in situ hybridization of the P2A-mCherry knock-in cassette in HOXA9P2A-mCherry reporter cells. The P2A-mCherry DNA was labeled with a red-dUTP by nick translation, and a HOXA9 BAC clone was labeled with a green-dUTP. The cells were then stained with 4,6-diamidino-2-phenylindole (DAPI) to visualize the nuclei. A representative metaphase cell image is shown for the pattern of hybridization (pairing of red and green signals). (E) RNA-seq data of all HOXA cluster genes were illustrated as log2 (normalized numbers of FPKM) from two replicate samples of SEM cells. HOXA7, HOXA9, and HOXA10 were highlighted by color code. (F) Q-PCR analysis confirmed the unaffected HOXA cluster gene transcription between HOXA9P2A-mCherry reporter (KI) and WT SEM cells. Data shown are means ± SEM from replicate independent experiments. *p<0.05 of two-tailed Student’s t test.
The HOXA9P2A-mCherry reporter allele recapitulates endogenous transcription of HOXA9 in MLLr cells
To evaluate whether the HOXA9P2A-mCherry reporter allele would faithfully respond to the transcriptional regulation of the cellular HOXA9 promoter, we genetically perturbed or pharmaceutically inhibited HOXA9’s upstream regulators. Previous studies have shown that DOT1L and ENL positively regulate HOXA9 expression in MLLr leukemia via direct occupancy on HOXA9’s promoter (Zeisig et al., 2004; Chen et al., 2015). Therefore, two sgRNAs targeting the coding region of DOT1L (sgDOT1L) and ENL (sgENL) were infected into the HOXA9P2A-mCherry cells expressing Cas9. Flow cytometry and Q-PCR analysis each revealed that mCherry and HOXA9 expression were both downregulated by sgRNAs targeting DOT1L or ENL (Figure 2A–D), and that the mCherry expression correlated well with the expression of HOXA9 (Figure 2E). Additionally, a DOT1L-selective inhibitor, SGC0946 (Yu et al., 2012), was supplemented at different dosages for 6 days to the HOXA9P2A-mCherry cells in culture resulting in a dosage-dependent reduction of mCherry fluorescence intensity measured by fluorescence imaging (Figure 2F–G) and flow cytometry (Figure 2H). Again, Q-PCR analysis of the DMSO- and SGC0946-treated cells showed that mRNA expression of mCherry was significantly correlated with that of HOXA9 (Pearson’s r = 0.90, p<0.001) (Figure 2I). Similarly, the HOXA9P2A-mCherry knock-in OCI-AML2 reporter line was also comprehensively characterized (Figure 2—figure supplement 1A–G). Taken together, these data confirm that the newly established HOXA9P2A-mCherry alleles were authentically controlled by the endogenous HOXA9 promoter and its local chromatin niche.
Figure 2 with 1 supplement see all
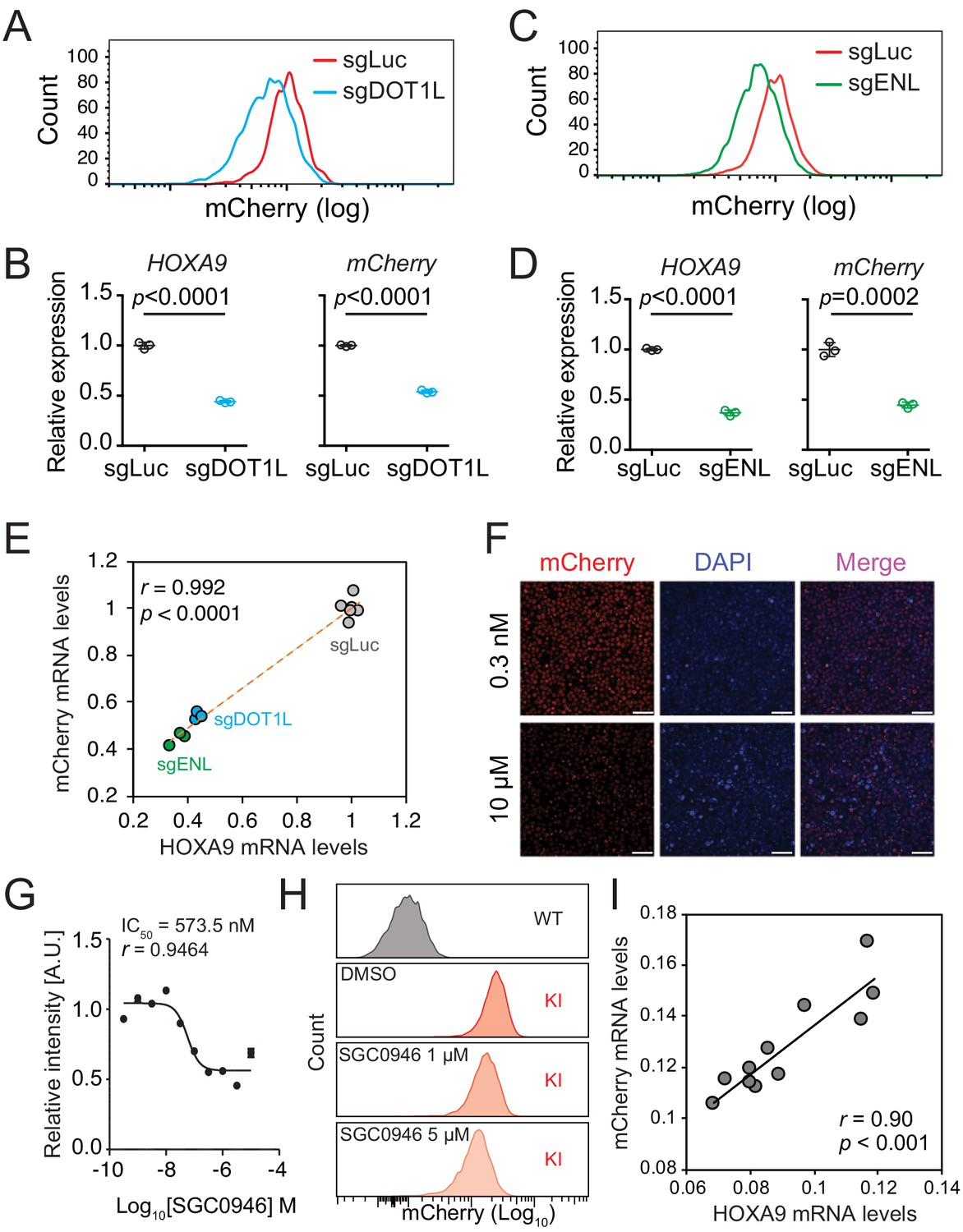
The HOXA9P2A-mCherry reporter allele recapitulates endogenous transcription of HOXA9 in MLLr SEM cells.
(A) Flow cytometry analysis of the HOXA9P2A-mCherry cells targeted with luciferase-sgRNA (sgLuc) and DOT1L-sgRNA (sgDOT1L). (B) Q-PCR analysis of the HOXA9P2A-mCherry cells targeted with sgLuc and sgDOT1L by using specific primers targeting the mRNA sequences of mCherry and HOXA9. Three biological replicates were performed. Data shown are means ± SEM from replicate independent experiments. The p-value was calculated by performing a two-tailed t-test. (C) Flow cytometry analysis of the HOXA9P2A-mCherry cells targeted with luciferase-sgRNA (sgLuc) and ENL-sgRNA (sgENL). (D) Q-PCR analysis of the HOXA9P2A-mCherry cells targeted with sgLuc and sgENL by using specific primers targeting the mRNA sequence of mCherry and HOXA9. Three biological replicates were performed. The p-value was calculated by performing a two-tailed t-test. (E) The correlation of transcription reduction in mCherry and HOXA9 in response to CRISPR–mediated targeting was calculated by Pearson’s correlation test. (F) Fluorescence imaging was performed on the HOXA9P2A-mCherry cells treated with various dosages of DOT1L inhibitor SGC0946 for six days. Representative images were shown for comparison between 0.3 nM and 10 µM dosages. For each dosage treatment, four replicates were conducted (scale bar 50 μm). (G) Fluorescence curve was generated according to mCherry intensity in response to dosage-dependent treatment of drug for 6 days. About 20,000 cells were split in each of the 384-well at the starting time point. (H) Flow cytometry analysis of the HOXA9P2A-mCherry cells treated with DMSO and various dosages of the DOT1L inhibitor SGC0946. (I) Q-PCR analysis of the HOXA9P2A-mCherry cells with or without the 6-day treatment of the DOT1L inhibitor SGC0946 by using specific primers targeting the mRNA sequences of mCherry and HOXA9. The correlation of transcription reduction in mCherry and HOXA9 in response to inhibitor–mediated transcription repression was calculated by performing Pearson’s correlation test.
Pooled CRISPR/Cas9 screening identified a novel transcription factor, USF2, that regulates HOXA9 expression
Although a few regulators of HOXA9 in MLLr leukemia have been previously identified (Zeisig et al., 2004; Collins and Hess, 2016b; Collins et al., 2014; Li et al., 2013a; Sun et al., 2013; Li et al., 2013b; Ogawara et al., 2015; de Bock et al., 2018; Lynch et al., 2019), to date a comprehensive CRISPR/Cas9 screen to unbiasedly identify novel upstream regulatory factors of HOXA9 has not been feasible owing to the lack of a reliable reporter cell line. Therefore, we combined the HOXA9P2A-mCherry reporter line and an in-house CRISPR-Cas9 sgRNA library targeting 1639 human transcription factors to identify novel regulatory effectors (Lambert et al., 2018). In this library, seven sgRNAs spanning multiple coding exons were designed per transcription factor, seven sgRNAs targeting DOT1L were included as a positive control, and an additional 100 non-targeting sgRNAs were included as negative controls. Two paralleled screens were performed on the same HOXA9P2A-mCherry reporter line stably expressing Cas9 and the lentiviral sgRNA library at a low M.O.I. (less than 0.3). Cells were selected with antibiotics, enriched, and fractionated by flow cytometric sorting for the top 10% (mCherryHigh) and bottom 10% (mCherryLow) mCherry populations, followed by genomic DNA extraction, PCR, and deep sequencing to identify differentially represented sgRNAs (Figure 3A). The differentially represented sgRNAs were calculated by DEseq2 analysis and combined for MAGeCK testing at the gene level (Li et al., 2014). The positive control genes HOXA9 and DOT1L were identified among the top hits between mCherryHigh and mCherryLow populations, suggesting that the screening was successful (Figure 3B). To mitigate the possibility that key upstream regulators of HOXA9 could be missed due to a survival disadvantage, we conducted an independent CRISPR/Cas9 TF screen in HOXA9P2A-mCherry reporter SEM cells with ectopically expressed HOXA9 together with its functional partner MEIS1. In this regard, exogenously expressed HOXA9 could rescue the potential cell loss due to decreased HOXA9 expression in SEM cells, while the level of endogenous HOXA9 is still monitored by the mCherry reporter. As a result, our CRISPR screening using the HOXA9/MEIS1 pre-rescued reporter line has identified more well-known regulators of HOXA9, which are also considered survival essential genes. Among the top 10 hits from this screen, DOT1L and HOXA9 were enriched. KMT2A, the translocation partner of MLL-AF4 in SEM cells, was identified in the HOXA9-MEIS1 rescue TF screen but not the original screen without ectopic expression of HOXA9. Notably, the MYST acetyltransferase HBO1 (also known as KAT7 or MYST2) and several members of the HBO1 protein complex, which were recently shown as critical regulators of leukemia stem cell maintenance, were also identified among the top hits (MacPherson et al., 2020; Au et al., 2020). Most importantly, USF2 was enriched among the top hits in both screens (Figure 3B), suggesting USF2 is likely a positive regulator with less survival essentiality compared with KMT2A. Consistent with the significant enrichment of these three candidates at the gene level, DEseq2 analysis (Love et al., 2014) and sgRNA enrichment plotting both suggested that most of the sgRNAs against these genes were differentially represented (Figure 3C–F and Figure 3—figure supplement 1A–F). Importantly, all the non-targeting control sgRNAs were similarly distributed across mCherryHigh and mCherryLow populations, indicating that the sorting-based screen did not bias the enrichment.
Figure 3 with 3 supplements see all
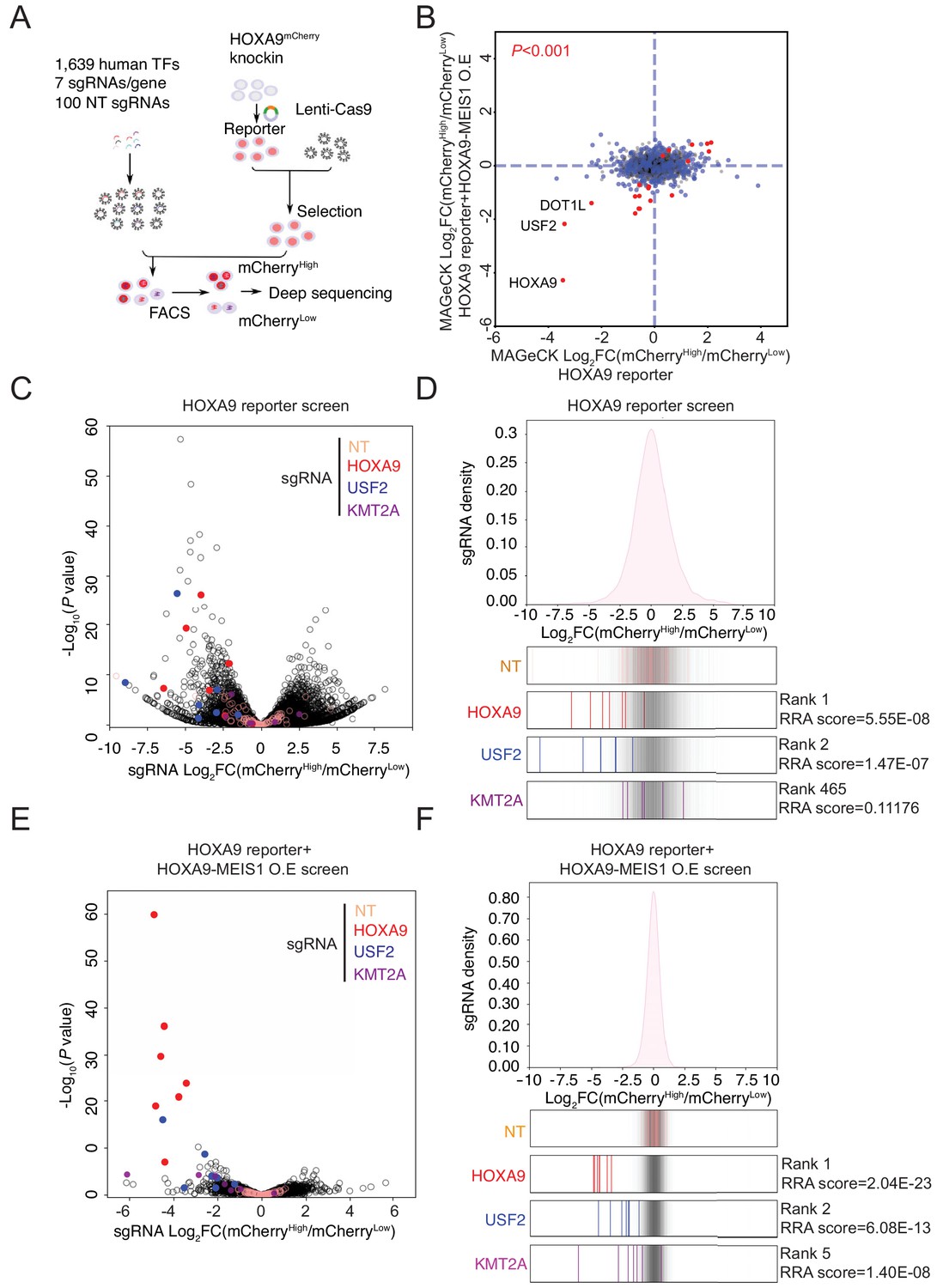
Pooled CRISPR/Cas9 screening identified a novel transcription factor, USF2, regulating HOXA9.
(A) Schematic diagram of a working model of loss-of-function CRISPR screening targeting 1639 human transcription factors. (B) The enrichment score of seven sgRNAs against each transcription factor was combined by the MAGeCK algorithm. Positive regulators of HOXA9 were compared between parental reporter strain and HOXA9-MEIS1 overexpressed screens. Overlapped top hits including HOXA9, USF2 and DOT1L were highlighted. (C) The overall distribution of all sgRNAs from the parental SEM HOXA9 reporter screening was shown based on the p-value and the DEseq2 score calculated by Log2[Fold Change (mCherryHigh/mCherryLow)]. NT, HOXA9, USF2 and KMT2A sgRNAs were highlighted by different color code. (D) The ratio for all sgRNAs targeting HOXA9, USF2, and KMT2A, are shown between mCherryHigh and mCherryLow sorted population. NT sgRNAs were overlaid on a gray gradient depicting the overall distribution. NT: 100 sgRNAs. Transcription factors: seven sgRNAs/each. RRA score of each gene was collected from MAGeCK analysis. (E) The overall distribution of all sgRNAs from the HOXA9-MEIS1 overexpressing SEM HOXA9 reporter screening was shown based on the p-value and the DEseq2 score calculated by Log2[Fold Change (mCherryHigh/mCherryLow)]. NT, HOXA9, USF2 and KMT2A sgRNAs were highlighted by different color code. (F) The ratio for all sgRNAs targeting HOXA9, USF2, and KMT2A, are shown between mCherryHigh and mCherryLow sorted population. NT sgRNAs were overlaid on a gray gradient depicting the overall distribution. NT: 100 sgRNAs. Transcription factors: seven sgRNAs/each. RRA score of each gene was collected from MAGeCK analysis.
Interestingly, the most-characterized looping factors, CTCF and YY1, were not enriched in the HOXA9P2A-mCherry reporter screen (Figure 3B). CTCF was reported to be essential for HOXA9 expression by occupying the boundary sequence between HOXA7 and HOXA9 (CBS7/9) in MLLr AML cell line MOLM13 (Luo et al., 2018). CRISPR-mediated deletion of the core sequence CTCF-binding motif in CBS7/9 significantly decreased HOXA9 expression and tumor progression (Luo et al., 2018; Luo et al., 2019). Given that CTCF is generally essential for cell survival, it is possible that cells targeted by CTCF sgRNAs in the HOXA9P2A-mCherry reporter and TF screen quickly dropped out of the population and were unable to be enriched as a regulator of HOXA9. To mitigate the challenge, we utilized a previously described auxin-inducible degron (AID) cellular system (Hyle et al., 2019; Morawska and Ulrich, 2013; Natsume et al., 2016; Nora et al., 2017) to acutely deplete the CTCF protein in SEM cells and evaluate the immediate transcriptional response of HOXA9 (Figure 3—figure supplement 2A). Upon acute depletion of CTCF via auxin (IAA) treatment in three CTCFAID bi-allelic knock-in clones, the protein expression of a previously identified vulnerable gene as positive control, MYC, was significantly inhibited (Figure 3—figure supplement 2B). Moreover, a Cut and Run assay using CTCF antibody for chromatin immunoprecipitation confirmed loss of CTCF occupancy throughout the HOXA9 locus, including CBS7/9 (Figure 3—figure supplement 2C). However, RNA-seq data (Figure 3—figure supplement 2D–E) and Q-PCR analysis (Figure 3—figure supplement 2F–G) collected from these three clones further confirmed the observation that loss of CTCF occupancy did not correlate with a decrease in HOXA7 or HOXA9 expression at the mRNA level. Instead, long-term depletion of CTCF by auxin for 48 hr slightly increased the transcription of HOXA7 and HOXA9. Upon washout of auxin from culture medium for an additional 48 hr, both HOXA7 and HOXA9 expression were restored to levels indistinguishable from those of the parental untreated cells (Figure 3—figure supplement 2D–G). Additionally, siRNA-mediated knock-down of CTCF in SEM cells did not change the transcription level of HOXA7 or HOXA9 (Figure 3—figure supplement 2H–J). However, suppressing CTCF in human colorectal cancer cell line HCT116 notably reduced HOXA7 and HOXA9 expression (Figure 3—figure supplement 3A–E), consistent with the finding in MLLr AML cell line MOLM13 (Luo et al., 2018). Collectively, these data further confirmed the results of our CRISPR screening that CTCF is not a key regulator of HOXA9 in MLLr B-ALL SEM and likely plays a role in regulating HOXA9 transcription in a cell-type-specific manner.
USF2 is required to maintain HOXA9 expression in MLLr leukemia
Aside from the positive controls confirmed from the CRISPR/Cas9 transcription factor screen in HOXA9P2A-mCherry cells, the top-ranked candidate among positive regulators was USF2. To further validate the CRISPR screen result and investigate the regulatory effect of USF2 on HOXA9 expression, we individually delivered four lentiviral sgRNAs targeting USF2 exons 1, 2, 7, and 9 into the HOXA9P2A-mCherry reporter line stably expressing Cas9. Similar to the results seen in sgENL targeted cells, USF2 knock-down significantly decreased the mCherry fluorescence in a time-dependent manner compared to that of luciferase sgRNA-targeted control (sgLuc) (Figure 4A and Figure 4—figure supplement 1). Q-PCR and immunoblotting analysis further confirmed the concordant downregulation of both HOXA9 and mCherry (Figure 4B–C). Collectively, these data suggest that USF2 positively controls HOXA9 expression in the MLLr B-ALL SEM cell line. USF2 was reported to generally bind to a symmetrical DNA sequence (E-box motif) (5’CACGTG3’) in a variety of cellular promoters (Henrion et al., 1995). Publicly available ChIP-seq data collected from human ES cells suggested that USF2 can directly bind to the conserved E-box element at both HOXA7 and HOXA9 promoters (Cheng et al., 2014). A Cut and Run assay was performed in control sgLuc and sgUSF2 targeted SEM cells to study genome-wide USF2 occupancy. In control SEM cells, USF2 bound to HOXA1, HOXA-AS3, HOXA7, and HOXA9 in HOXA cluster. Upon USF2 depletion, binding occupancy at these regions was significantly reduced (Figure 4D), further supporting the specificity of the USF2 binding identified by the Cut and Run assay. Taken together, these data suggest that USF2 could regulate HOXA9 expression as well as other HOXA genes through interactions with its regulatory elements at the HOXA cluster gene loci.
Figure 4 with 1 supplement see all
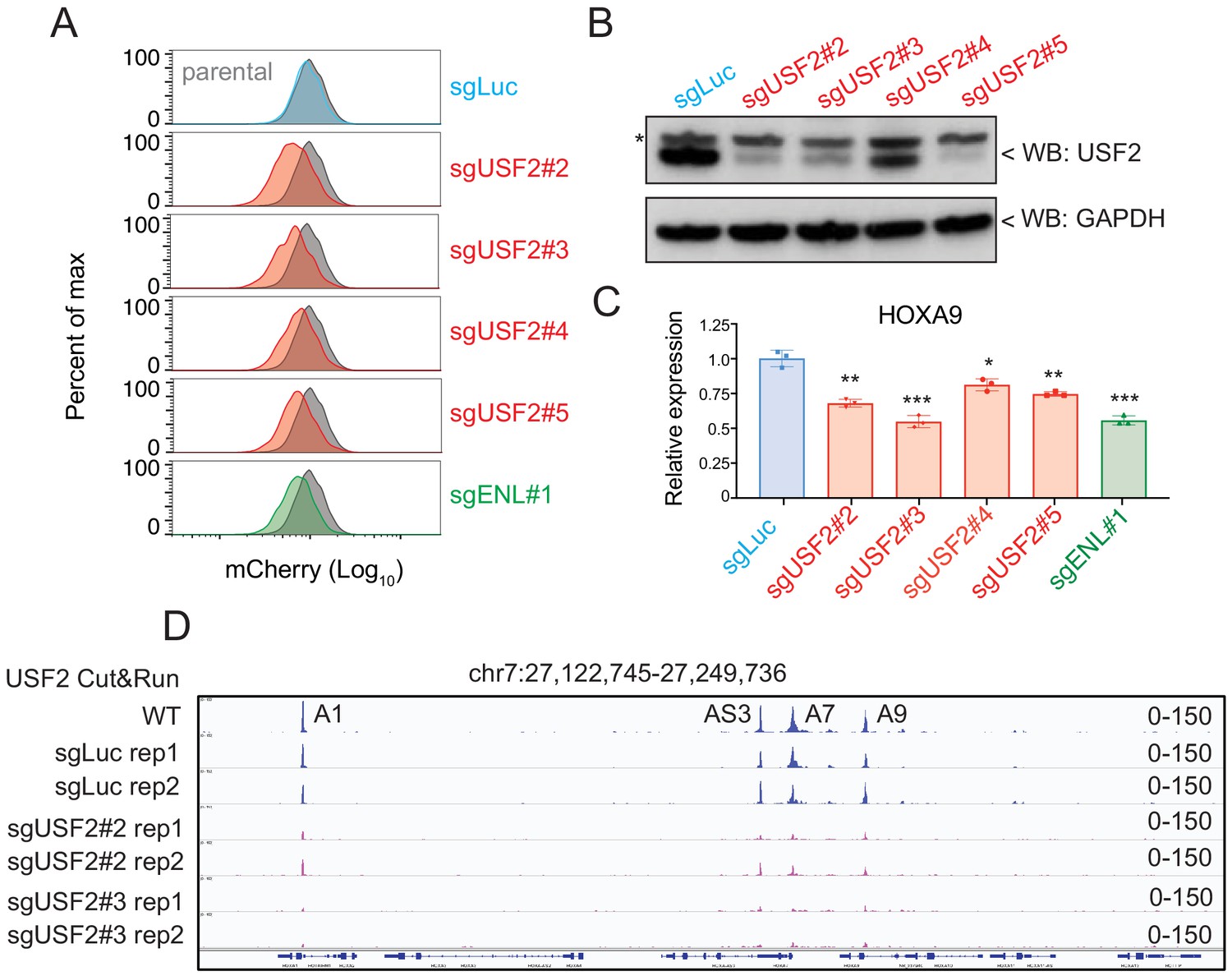
USF2 is required to maintain HOXA9 expression in MLLr leukemia.
(A) Flow cytometry analysis was performed at day 8 on the HOXA9P2A-mCherry cells targeted with lentiviral Cas9 and four sgRNAs against USF2. The sgENL-targeted cells were used as positive controls while sgLuc targeted cells were used as negative controls. (B) Q-PCR analysis was conducted on the USF2-targeted cells to monitor the reduction of HOXA9. The sgENL targeted cells were used as positive controls while sgLuc-targeted cells were used as negative controls. Data shown are means ± SEM from three independent experiments. *p<0.05, **p<0.01, ***p<0.001, two-tailed Student’s t test. (C) Immunoblotting of USF2 in USF2 sgRNAs targeted cells. '*' denoted non-specific bands. (D) USF2 occupancy changes in sgLuc and sgUSF2-targeted SEM cells were characterized in HOXA9 locus (A1, HOXA1; AS3, HOXA-AS3; A7, HOXA7; A9, HOXA9).
USF2 is an essential gene in MLLr B-ALL by controlling HOXA9 expression
To unbiasedly evaluate the survival dependency of USF2 in SEM cells, we conducted a dropout CRISPR/Cas9 screen by targeting 1639 transcription factors. SEM cells infected with the pooled library of sgRNAs were collected at day 0 and day 12 to sequence for sgRNA distribution (Figure 5A). In accordance with prior genome-wide CRISPR screens and functional studies in B-ALL, many survival dependent genes were identified in the top 50 genes in our screen including PAX5, DOT1L, ZFP64, YY1, MEF2C, MYC, and KMT2A (Gu et al., 2019; Hyle et al., 2019; Pridans et al., 2008; Lu et al., 2018). USF2 was ranked as the top 24th essential gene in MLLr SEM cells (Figure 5B). Taken together, these findings suggest that the USF2/HOXA9 axis might play a role in supporting MLLr B-ALL cell proliferation. To evaluate the importance of the USF2/HOXA9 axis in MLLr B-ALL progression, we sought to investigate the knockout phenotype of USF2 in MLLr B-ALL cells. A competition-based proliferation assay was performed by infecting SEMCas9 cells with a lentiviral-mCherry-sgRNAs against the HOXA9 promoter at ~50% targeting efficiency (Figure 5C). The proportion of mCherry+ cells were monitored over a 12-day time course (days 3, 6, 9, and 12) to investigate the proliferation disadvantage of HOXA9 knock-down cells (Figure 5D). Next, the same assay was performed by infecting SEMCas9 cells with three individual lentiviral-mCherry-sgRNAs against USF2 (sgRNA-2,–3 and 5) at ~50% infection efficiency. As a result, the proliferation-arrested phenotype was observed in all three sgRNA targeted cells but not in cells targeted with sgLuc (Figure 5E). Importantly, in SEM cells constitutively expressing ectopic retroviral mouse Hoxa9 (SEMHOXA9), USF2 knock-down had little effect on cell growth (Figure 5F), suggesting that HOXA9 is a functional and essential downstream gene of USF2 in USF2-mediated leukemia propagation.
Figure 5 with 3 supplements see all
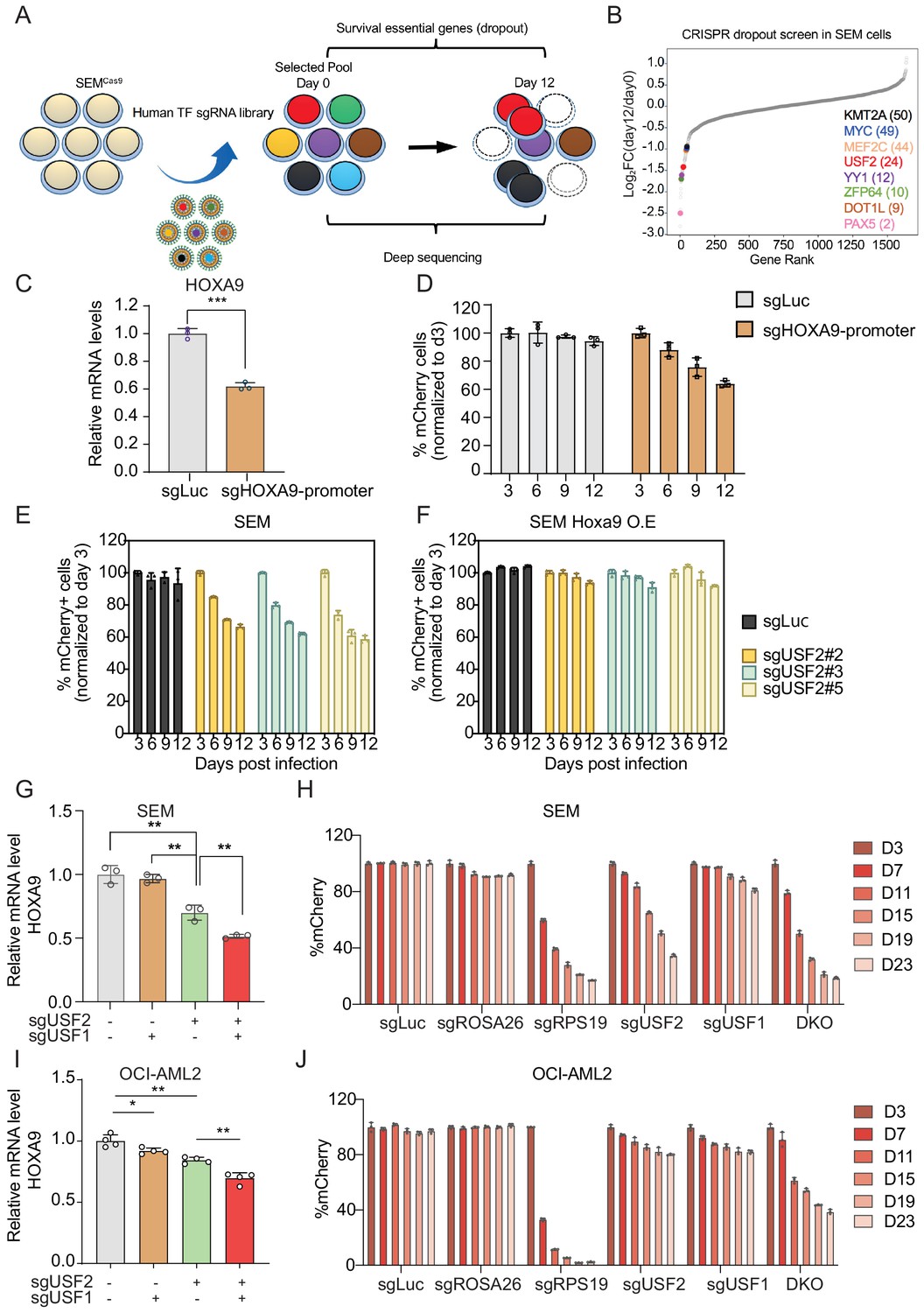
USF1 and USF2 synergistically regulate HOXA9 expression in MLLr leukemia.
(A) Flow diagram of dropout CRISPR screening procedure. (B) Gene ranking of all transcription factors from dropout screening was illustrated. The enrichment score of seven sgRNAs against each transcription factor was combined by the MAGeCK algorithm. (C) Q-PCR was conducted to monitor HOXA9 expression upon CRISPR targeting on its promoter. (D) Competitive proliferation assay was conducted by infecting SEMCas9 cells with Lentiviral-mCherry-sgRNAs against HOXA9 promoter at about 50% efficiency. The mCherry% was quantified every three days by flow cytometry to evaluate the growth disadvantage. (E) Competitive proliferation assay was conducted by infecting SEMCas9 cells with Lentiviral-mCherry-sgRNAs against luciferase (sgLuc) and USF2 (sgUSF2#2, 2#3 and 2#5) at about 50% efficiency. The mCherry% was quantified every 3 days by flow cytometry to evaluate the growth disadvantage. (F) Rescued competitive proliferation assay was conducted by infecting SEM cells overexpressing ectopic Hoxa9 with Lentiviral-mCherry-sgRNAs against luciferase (sgLuc) and USF2 (sgUSF2#2, 2#3 and 2#5) at about 50% efficiency. The mCherry% was quantified every 3 days by flow cytometry to evaluate the growth disadvantage. (G) Q-PCR analysis was conducted on the sgUSF2, sgUSF1 and sgUSF1/2-targeted SEM cells to monitor the reduction of HOXA9. Data shown are means ± SEM from three independent experiments. **p<0.01, two-tailed Student’s t test. (H) Competitive proliferation assay was conducted by infecting SEMCas9 cells with Lentiviral-mCherry-sgLuc, sgUSF1, sgUSF2, and sgUSF1/2 (DKO) at about 50% efficiency. The mCherry% was quantified at days 3, 7, 11, 15, 19, and 23 by flow cytometry to evaluate the growth disadvantage. A guide RNA targeting the survival essential gene RPS19 was included as a positive control for Cas9 activity. Guide RNAs targeting Luciferase gene (sgLuc) and the human ROSA26 gene (sgROSA26) were included as a negative control. (I) Q-PCR analysis was conducted on the sgUSF2, sgUSF1 and sgUSF1/2 targeted OCI-AML2 cells to monitor the reduction of HOXA9. Data shown are means ± SEM from three independent experiments. *p<0.05, **p<0.01, two-tailed Student’s t test. (J) Competitive proliferation assay was conducted by infecting OCI-AML2Cas9 cells with Lentiviral-mCherry-sgLuc, sgUSF1, sgUSF2, and sgUSF1/2 (DKO) at about 50% efficiency. The mCherry% was quantified at days 3, 7, 11, 15, 19, and 23 by flow cytometry to evaluate the growth disadvantage. A guide RNA targeting the survival essential gene RPS19 was included as a positive control for Cas9 activity. Guide RNAs targeting Luciferase gene (sgLuc) and the human ROSA26 gene (sgROSA26) were included as negative controls.
USF1 and USF2 synergistically regulate HOXA9 expression in MLLr leukemia
Previously, other studies identified the USF2 homolog protein USF1 shares a similar protein structure with USF2 (49, 53). USF1 and USF2 bind to the same type of E-box elements and are also able to form homo- or heterodimers (Kumari and Usdin, 2001; Wang and Sul, 1995; Prasad and Singh, 2008; Spohrer et al., 2017) suggesting that these two proteins may function in synergy to regulate HOXA9. Interestingly, in our HOXA9-reporter-based CRISPR screen, USF1 was also among the top 50 positive regulator genes identified (49th) (Supplementary file 2). To test whether USF1 and USF2 have redundant roles in regulating HOXA9 expression, we co-delivered sgRNAs against USF2 (sgUSF2) and USF1 (sgUSF1) to the SEM HOXA9P2A-mCherry reporter line stably expressing Cas9. Notably, both the flow cytometry and Q-PCR analysis confirmed a significant decrease in HOXA9 expression with double inactivation of USF1 and USF2 compared with inactivation of USF2 alone (Figure 5G and Figure 5—figure supplement 1A), which was also supported by a synergetic effect in the competitive proliferation assay (Figure 5H). To further evaluate whether USF2 and USF1 could regulate HOXA9 expression in other MLLr leukemias, sgUSF2 and sgUSF1 alone or in combination were delivered into the human MLLr AML cell line OCI-AML2 which carried the MLL-AF6 translocation. Similar to observations in SEM cells, USF1 or USF2 CRISPR targeting resulted in notably suppressed HOXA9 expression (Figure 5I). In addition, USF1 and USF2 synergistically regulate HOXA9 expression and leukemia survival in OCI-AML2 (Figure 5J and Figure 5—figure supplement 1B-E). In NOMO-1 MLLr AML cells, USF2 downregulation also notably decreased expression of HOXA9 (Figure 5—figure supplement 1C) comparable to levels observed in SEM cells. Interestingly, in human MLLr AML cell line MOLM13, individual knockout of USF1 or USF2 did not affect HOXA9 expression nor cell survival. However, USF1 and USF2 double-knockout cells demonstrated suppressed HOXA9 expression and reduced survival (Figure 5—figure supplement 1F-G). Taken together, the data suggests that loss of one USF family member gene may lead to varying degrees of compensatory regulation of HOXA9 by the untargeted USF gene, whereas loss of both USF genes results in a more robust abrogation of HOXA9 expression.
To examine if USF2 regulation of HOXA9 expression was unique to MLLr leukemias, we used two sgRNAs, sgUSF2#2 and sgUSF2#3, to knockdown USF2 expression in two human non-MLLr leukemia cell lines, OCI-AML3 and U937, which both express HOXA9. Upon complete USF2 depletion, HOXA9 expression remained unchanged, suggesting the USF2/HOXA9 axis may function in a MLLr-dependent manner (Figure 5—figure supplement 2A-D). Lastly, a transcriptome analysis from the to-date largest human B-ALL transcriptome cohort (N = 1988 patients) (Gu et al., 2019) identified USF2 expression to be significantly correlated with HOXA9 in MLLr-subtype patients (N = 136 patients) (Figure 5—figure supplement 3A-D) highlighting that the USF2 and HOXA9 regulation axis could have clinical relevance for patients in this specific subtype.
Discussion
HOX genes are a cluster of genes strictly regulated in development by various transcription and epigenetic modulators. Dysregulation of HOX genes has been frequently linked to human diseases, particularly cancer. Here, we focus on HOXA9, the aberrant expression of which is one of the most significant features in the most aggressive human leukemias. The HOXA9P2A-mCherry knock-in MLLr cell line derived in this study fully recapitulated transcriptional regulation of the endogenous gene. Previously, Godmin, et al. derived two mouse strains by delivering the in-frame GFP cassette to two different murine Hox genes, Hoxa1 and Hoxc13, to visualize the proteins during mouse embryogenesis (Godwin et al., 1998). Although this previous study certainly added to the repertoire of research tools available to investigate HOXA-related gene expression and gene function, our HOXA9 reporter cell line provides a unique intrinsic cellular model with which to study transcriptional regulation of human HOXA9 directly. Additionally, the CHASE-knock-in protocol developed to generate the HOXA9 reporter is user-friendly, highly efficient, robust to reproduce and could be easily adapted to a wide variety of HOXA9-driven human leukemia cell models and other HOXA9-expressing cancer types.
In mammalian cells, each chromosome is hierarchically organized into hundreds of megabase-sized TADs (ENCODE Project Consortium, 2012; Ji et al., 2016; Rowley et al., 2017; Rowley and Corces, 2018), each of which is insulated by the boundary elements. Within the TAD scaffold, promoter/enhancer physical contacts intricately regulate gene expression (Pombo and Dillon, 2015). Intra-TAD chromatin interactions can be facilitated by a pair of CTCF-binding sites engaged in contact with each other when they are in a convergent linear orientation (Rao et al., 2014; Vietri Rudan et al., 2015). The HOXA9 cluster is located on the TAD boundary, providing an opportunity to interact with neighboring genomic elements. However, because of the low resolution of publicly available Hi-C data and the lack of DpnI restriction enzyme sites within the HOXA gene cluster that are necessary to generate high-quality 3C libraries, the impact of chromatin interaction regulation of HOXA9 remains unclear. Using a chromosome conformation capture-based PCR assay and CRISPR-mediated deletion of a minimal CTCF-binding motif between HOXA7 and HOXA9 (CBS7/9), Luo and colleagues proposed that the CTCF boundary was crucial for higher order chromatin organization by showing the depletion of CBS7/9 disrupted chromatin interactions and significantly reduced HOXA9 transcription in MLLr AML MOLM13 cells with t(9;11) (Luo et al., 2018; Luo et al., 2019). In our study, the loss-of-function results from auxin-inducible degradation of CTCF, siRNA-mediated CTCF knock-down, and the unbiased transcription factor screening suggested that CTCF is not required to maintain HOXA9 expression in SEM cells with MLLr with t(4;11). We speculate that the discrepancy could be due to the following reasons. Although both cell lines carried the MLLr translocation as a driver oncogenic mutation, MOLM13 and SEM were classified as AML and B-ALL, respectively. Besides the lineage difference, SEM cells are also less sensitive to many well-known pharmaceutical inhibitors including JQ1 and DOT1L inhibitor. Therefore, we hypothesized that other as yet to be identified looping factors might be involved in the transcriptional regulation of the HOXA9 locus in MLLr SEM cells, and that CTCF regulates HOXA9 expression in a cell-type-specific context.
By performing unbiased CRISPR screens designed to target 1639 known human transcription factors in a HOXA9P2A-mCherry reporter cell line, we identified USF2 as a novel regulator of HOXA9. In addition, two known HOXA9 regulators, HOXA9 and DOT1L, were identified among the top hits supporting the reliable sensitivity of both the reporter system and the CRISPR screening strategy. USF2 is a ubiquitously expressed basic helix-loop-helix-leucine-zip transcription factor that generally recognizes E-box DNA motifs (Henrion et al., 1995; Groenen et al., 1996; Luo and Sawadogo, 1996). USF1 and USF2 usually form homo- or heterodimers to modulate gene expression (Kumari and Usdin, 2001). Interestingly, USF1 was also enriched in our CRISPR screening. Moreover, the function of USF2 in controlling leukemia progression has not been reported. Although our study identified the regulatory function of USF1/USF2 on HOXA9 maintenance and leukemia cell survival in MLLr B-ALL and AML cell lines, other HOXA9-independent functions of USF1/2 cannot be excluded and requires further studies.
In summary, we revealed that candidate transcription factors identified from the CRISPR/Cas9 screen including USF2 and USF1, regulate HOXA9 thereby providing a more comprehensive understanding about how the HOXA9 locus is regulated in human cancer cells. Given the well-recognized role of HOXA9 in hematopoietic malignancies, we anticipate the HOXA9 reporter cells will advance many lines of investigation including drug screening and the identification of concordant epigenetic modifiers/transcription factors that are required for activation and maintenance of HOXA9 expression in leukemia progression. Collectively, these efforts would clarify the molecular mechanisms underlying aberrant HOXA9 activation in leukemias, thus providing the foundation to develop clinically relevant therapies to target the expression and/or function of HOXA9 in leukemia patients.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Cell line (Homo sapiens) | SEM | DSMZ | ACC546 | CVCL_0095 |
| Cell line (Homo sapiens) | NOMO-1 | DSMZ | ACC542 | CVCL_1609 |
| Cell line (Homo sapiens) | OCI-AML2 | DSMZ | ACC99 | CVCL_1619 |
| Cell line (Homo sapiens) | OCI-AML3 | DSMZ | ACC582 | CVCL_1844 |
| Cell line (Homo sapiens) | MOLM13 | DSMZ | ACC554 | CVCL_2119 |
| Cell line (Homo sapiens) | U937 | ATCC | CRL-1593.2 | CVCL_0007 |
| Cell line (Homo sapiens) | 293T | ATCC | CRL-3216 | CVCL_0063 |
| Cell line (Homo sapiens) | SEM-HOXA9P2A-mCherry | This eLife study | Reporter derived from SEM cells via knock-in | Cell line is available upon request to Dr. Chunliang Li |
| Cell line (Homo sapiens) | OCI-AML2-HOXA9P2A-mCherry | This eLife study | Reporter derived from OCI-AML2 cells via knock-in | Cell line is available upon request to Dr. Chunliang Li |
| Antibody | Anti-USF2 (Rabbit polyclonal) | Novus | NBP1-92649 | IP, IB (1:2,000) AB_11007053 |
| Antibody | Anti-USF1 | Proteintech | 22327–1-AP | IB (1: 2,000) AB_2060867 |
| Antibody | Anti-CTCF (Rabbit polyclonal) | Abcam | ab70303 | IB (1:1,000) AB_1209546 |
| Antibody | Anti-MYC (Rabbit polyclonal) | Cell Signaling Technology | 9402 | IB (1:1000) AB_2151827 |
| Antibody | Anti-GAPDH | Thermo Fisher Scientific | AM4300 | IB (1:10,000) AB_437392 |
| Antibody | Anti-Vinculin | Proteintech | 26520–1-AP | IB (1:2,000) AB_2868558 |
| Sequence-based reagent | U6-Forward sequencing primer | This paper | sgRNA sequencing primer | 5’GAGGGCCTATTTCCCATGAT3’ |
| Sequence-based reagent | sgRNA sequence | This paper | sgRNA targeting HOXA9 on C-terminus | 5’AAAGACGAGTGATGCCATTT3’ |
| Sequence-based reagent | HOXA9 5’HA cloning primer F | This paper | HOXA9 knockin reporter cloning | 5’GGCCGATTCCTTCCACTTCT3’ |
| Sequence-based reagent | HOXA9 5’HA cloning primer R | This paper | HOXA9 knockin reporter cloning | 5’TCACTCGTCTTTTGCTCGGT3’ |
| Sequence-based reagent | HOXA9 3’HA cloning primer F | This paper | HOXA9 knockin reporter cloning | 5’ACCGAGCAAAAGACGAGTGA3’ |
| Sequence-based reagent | HOXA9 3’HA cloning primer R | This paper | HOXA9 knockin reporter cloning | 5’CACTGTTCGTCTGGTGCAAA3’. |
| Sequence-based reagent | Infusion cloning F | This paper | HOXA9 knockin reporter cloning | 5’AAGACCGAGCAAAAGACGAGGGATCCGGCGCAACAAACTT3’ |
| Sequence-based reagent | Infusion cloning R | This paper | HOXA9 knockin reporter cloning | 5’AATAAGCCCAAATGGCATCACTTGTACAGCTCGTCCATGC3‘ |
| Sequence-based reagent | Infusion cloning of mCherry F | This paper | HOXA9 knockin reporter cloning | 5’AAAGACGAGTGATGCCATTTGGGATGAGGCTGCGGGCGAC3’ |
| Sequence-based reagent | Infusion cloning of mCherry R | This paper | HOXA9 knockin reporter cloning | 5’AAAGACGAGTGATGCCATTTGGGTATATATACAATAGACAAGACAGGAC3’ |
| Sequence-based reagent | DOT1L-sgRNA | This paper | sgRNA sequence | 5’TCAGCTTCGAGAGCATGCAG3’ |
| Sequence-based reagent | ENL-sgRNA | This paper | sgRNA sequence | 5’TCACCTGGACGGTGCACTGG3’ |
| Sequence-based reagent | USF2-sgRNA#2 | This paper | sgRNA sequence | 5’AGAAGAGCCCAGCACAACGA3’ |
| Sequence-based reagent | USF2-sgRNA#3 | This paper | sgRNA sequence | 5’TGTTTTCCGCAGTGGAGCGG3’ |
| Sequence-based reagent | USF2-sgRNA#4 | This paper | sgRNA sequence | 5’CCGGGGATCTTACCTGGCGG3’ |
| Sequence-based reagent | USF2-sgRNA#5 | This paper | sgRNA sequence | 5’CAGCCACGACAAGGGACCCG3’ |
| Sequence-based reagent | USF1-sgRNA | This paper | sgRNA sequence | 5’CTATACTTACTTCCCCAGCA3’ |
| Sequence-based reagent | Luciferase-sgRNA | This paper | sgRNA sequence | 5’CCCGGCGCCATTCTATCCGC3’ |
| Sequence-based reagent | ROSA26-sgRNA | This paper | sgRNA sequence | 5’ACCTACCACACTAGCCCGA3’ |
| Sequence-based reagent | RPS19-sgRNA | This paper | sgRNA sequence | 5’GTAGAACCAGTTCTCATCGT3’ |
| Sequence-based reagent | HOXA9-promoter sgRNA | This paper | sgRNA sequence | 5’GATTTCATGTAACAACTTGG3’ |
| Sequence-based reagent | CTCF-F | This study | Q-PCR primer | 5’TTTGTCTGTTCTAAGTGTGGGAAA3’ |
| Sequence-based reagent | CTCF-R | This study | Q-PCR primer | 5’TTAGAGCGCATCTTTCTTTTTCTT3’ |
| Sequence-based reagent | GAPDH-F | This study | Q-PCR primer | 5’AGGGCTGCTTTTAACTCTGGT3’ |
| Sequence-based reagent | GAPDH-R | This study | Q-PCR primer | 5’CCCCACTTGATTTTGGAGGGA3’ |
| Sequence-based reagent | ACTB-F | This study | Q-PCR primer | GAGCACAGAGCCTCGCCTTT |
| Sequence-based reagent | ACTB-R | This study | Q-PCR primer | GAGCGCGGCGATATCATCA |
| Sequence-based reagent | HOXA1-F | This study | Q-PCR primer | 5’CCAGCCACCAAGAAGCCTGT3’ |
| Sequence-based reagent | HOXA1-R | This study | Q-PCR primer | 5’CCAGTTCCGTGAGCTGCTTG3’ |
| Sequence-based reagent | HOXA2-F | This study | Q-PCR primer | 5’ACAGCGAAGGGAAATGTAAAAGC3’ |
| Sequence-based reagent | HOXA2-R | This study | Q-PCR primer | 5’GGGCCCCAGAGACGCTAA3’ |
| Sequence-based reagent | HOXA3-F | This study | Q-PCR primer | 5’TGCAAAAAGCGACCTACTACGA3’ |
| Sequence-based reagent | HOXA3-R | This study | Q-PCR primer | 5’CGTCGGCGCCCAAAG3’ |
| Sequence-based reagent | HOXA4-F | This study | Q-PCR primer | 5’CGTGGTGTACCCCTGGATGA3’ |
| Sequence-based reagent | HOXA4-R | This study | Q-PCR primer | 5’AAGACCTGCTGCCGGGTGTA3’ |
| Sequence-based reagent | HOXA5-F | This study | Q-PCR primer | 5’TCTACCCCTGGATGCGCAAG3’ |
| Sequence-based reagent | HOXA5-R | This study | Q-PCR primer | 5’AATCCTCCTTCTGCGGGTCA3’ |
| Sequence-based reagent | HOXA6-F | This study | Q-PCR primer | 5’TGGATGCAGCGGATGAACTC3’ |
| Sequence-based reagent | HOXA6-R | This study | Q-PCR primer | 5’CCGTGTCAGGTAGCGGTTGA3’ |
| Sequence-based reagent | HOXA7-F | This study | Q-PCR primer | 5’TCTGCAGTGACCTCGCCAAA3’ |
| Sequence-based reagent | HOXA7-R | This study | Q-PCR primer | 5’AGCGTCTGGTAGCGCGTGTA3’ |
| Sequence-based reagent | HOXA9-F | This study | Q-PCR primer | 5’AAAAACAACCCAGCGAAGGC3’ |
| Sequence-based reagent | HOXA9-R | This study | Q-PCR primer | 5’ACCGCTTTTTCCGAGTGGAG3’ |
| Sequence-based reagent | HOXA10-F | This study | Q-PCR primer | 5’CCTTCCGAGAGCAGCAAAGC3’ |
| Sequence-based reagent | HOXA10-R | This study | Q-PCR primer | 5’CAGCGCTTCTTCCGACCACT3’ |
| Sequence-based reagent | HOXA11-F | This study | Q-PCR primer | 5’ACAGGCTTTCGACCAGTTTTTC3’ |
| Sequence-based reagent | HOXA11-R | This study | Q-PCR primer | 5’CCTTCTCGGCGCTCTTGTC3’ |
| Sequence-based reagent | HOXA13-F | This study | Q-PCR primer | 5’ACTCTGCCCGACGTGGTCTC3’ |
| Sequence-based reagent | HOXA13-R | This study | Q-PCR primer | 5’TTCGTGGCGTATTCCCGTTC3’ |
| Sequence-based reagent | mCherry-F | This study | Q-PCR primer | 5’CACTACGACGCTGAGGTCAA3’ |
| Sequence-based reagent | mCherry-R | This study | Q-PCR primer | 5’TAGTCCTCGTTGTGGGAGGT3’ |
| Sequence-based reagent | siRNA: nontargeting control | Thermo Fisher | siRNA oligo | Silencer Select |
| Sequence-based reagent | siRNA: CTCF | Thermo Fisher | siRNA oligo | Silencer Select |
| Sequence-based reagent | DNA oligo pool | CustomArray | sgRNA synthesis | |
| Commercial assay or kit | In-Fusion HD Cloning | Clontech | 638909 | |
| Commercial assay or kit | Polybrene | EMD Millipore | TR-1003-G | |
| Commercial assay or kit | DAPI | Sigma | D9542-10MG | |
| Commercial assay or kit | Lonza nucleofector Kit | Lonza | VCA-1003 | |
| Commercial assay or kit | Q5 High-Fidelity DNA Polymerase | New England Biolabs | M0491L | |
| Commercial assay or kit | CloneAMP HiFI PCR Premix | Clontech | 639298 | |
| Commercial assay or kit | Quick-DNA Miniprep Kit | Zymo | D3025 | |
| Commercial assay or kit | NEB Next UltraII DNA Library Prep Kit | NEB | E7645S | |
| Commercial assay or kit | ZymoPURE II Plasmid Midiprep Kit | Zymo | D4201 | |
| Commercial assay or kit | TRIzol | Thermo Fisher Scientific | 15596026 | |
| Commercial assay or kit | High-Capacity cDNA Reverse Transcription Kit | Applied Biosystems | 4374966 | |
| Commercial assay or kit | FAST SYBR Green Master Mix | Applied Biosystems | 4385612 | |
| Recombinant DNA reagent | pSpCas9(BB)−2A-GFP | Addgene | 48138 | PX458 |
| Recombinant DNA reagent | TOPO-cloning vector | Thermo Fisher Scientific | 450641 | |
| Recombinant DNA reagent | Lenti-Cas9-Blast plasmid | Addgene | 83480 | |
| Recombinant DNA reagent | Lenti-Guide-Puro plasmid | Addgene | 52963 | |
| Recombinant DNA reagent | LRCherry2.1 | Addgene | 108099 | |
| Recombinant DNA reagent | LRNeo-2.1 vector | This study | Subclone from LRCherry2.1 | |
| Recombinant DNA reagent | Lenti-Guide-Puro-IRES-CFP plasmid | This study | Subclone from Lenti-Guide-Puro | |
| Recombinant DNA reagent | HOXA9-MEIS1 OE | This study | Subclone from mouse cDNA | |
| Recombinant DNA reagent | mHoxa9 OE | This study | Subclone from mouse cDNA | |
| Chemical compound, drug | Puromycin | InvivoGen | ant-pr-1 | |
| Chemical compound, drug | Neomycin | GeminiBio | 400–121P | |
| Chemical compound, drug | Blasticidine | Gibco | A1113903 | |
| Chemical compound, drug | SGC0946 | MedChemExpress | HY-15650 | DOT1L inhibitor |
| Software, algorithm | Fluorescene Imaging | Perkin Elmer | Columbus Image Data Storage and Analysis system | |
| Software, algorithm | MAGeCK | https://sourceforge.net/ p/mageck/wiki/Home/ | PMID:25476604 | |
| Software, algorithm | MACS2 | https://github.com/macs3-project/MACS; Zhang et al., 2008 | ||
| Software, algorithm | Cutadapt | https://cutadapt.readthedocs.io/en/v1.9.1/installation.html | 1.9.1 | |
| Software, algorithm | BWA | https://github.com/lh3/bwa/releases; Li, 2013 | 0.7.17-r1188 | |
| Software, algorithm | Samtools | http://www.htslib.org/ | Htslib 1.6 | |
| Software, algorithm | IGV | http://software.broadinstitute.org/software/igv/ | IGV2.3.97 | |
| Software, algorithm | ChIPseeker | https://guangchuangyu.github.io/software/ChIPseeker/; Yu et al., 2015 | ||
| Software, algorithm | TRANSFAC | http://gene-regulation.com/pub/databases.html | ||
| Software, algorithm | JASPAR | http://jaspar.genereg.net/ | 8th release (2020) | |
| Software, algorithm | FIMO | http://meme-suite.org/doc/fimo.html | ||
| Software, algorithm | DESeq2 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html | ||
| Software, algorithm | GraphPad Prism | version 8.0 | ||
| Software, algorithm | Flowjo | version 10.0 | ||
| Software, algorithm | Bowtie | http://bowtie-bio.sourceforge.net/index.shtml | ||
| Software, algorithm | BamCoverage | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
Cell culture
Request a detailed protocolSEM cells (ACC-546, DSMZ), OCI-AML2 (ACC-99, DSMZ), Cas9-expressing OCI-AML3 (originally from ACC-582, a kind gift from Dr. Christopher Vakoc) and MOLM13 (ACC-554, DSMZ) were maintained in RPMI-1640 medium (Lonza) containing 10% fetal bovine serum (FBS) (HyClone), and 1% penicillin/streptomycin (Thermo Fisher Scientific) at 37°C, 5% CO2 atmosphere and 95% humidity. Basal medium for culturing 293 T cells is DMEM (HyClone). All passages of cells used in this study were mycoplasma-free. Cell identity was confirmed by STR analysis.
Vector construction
Request a detailed protocolA pair of oligomers containing a 20 bp sgRNA (5’-AAAGACGAGTGATGCCATTT-3’) sequence targeting the surrounding genomic segment of HOXA9 stop codon was synthesized (Thermo Fisher Scientific) and cloned into the all-in-one vector, pSpCas9(BB)−2A-GFP (Addgene #48138) between BsmBI sites. Correct clones were screened and confirmed by Sanger sequencing with the U6-Forward sequencing primer (5’-GAGGGCCTATTTCCCATGAT-3’). To construct a CHASE-knock-in donor vector delivering a P2A-mCherry DNA segment to the endogenous HOXA9 locus, a two-step cloning protocol was used. The ~800 bp 5′ and 3’ homology arm (HA) flanking the endogenous sgRNA target was amplified from SEM cells. The 5’ HA PCR primer sequences are 5’-GGCCGATTCCTTCCACTTCT-3’ and 5’-TCACTCGTCTTTTGCTCGGT-3’, and the 3’ HA PCR primer sequences are 5’-ACCGAGCAAAAGACGAGTGA-3’ and 5’-CACTGTTCGTCTGGTGCAAA-3’. The P2A-mCherry DNA fragment was amplified from p16INK4A-P2A-mCherry knock-in donor vector (Zhang et al., 2019) using a pair of primers containing overlapping sequences of 5’ HA or 3’ HA for in-fusion cloning (forward primer: 5’-AAGACCGAGCAAAAGACGAGGGATCCGGCGCAACAAACTT-3’; reverse primer: 5’- AATAAGCCCAAATGGCATCACTTGTACAGCTCGTCCATGC-3‘). The 5’ HA-P2A-mCherry-3’ HA in-fusion cloning product was further supplemented with 23 bp target sgRNA and PAM sequences at both 5’ and 3’ ends through PCR amplification using primers 5’-AAAGACGAGTGATGCCATTTGGGATGAGGCTGCGGGCGAC-3’ and 5’-AAAGACGAGTGATGCCATTTGGGTATATATACAATAGACAAGACAGGAC-3’. The cloning PCR reactions were performed using Q5 High-Fidelity DNA Polymerase (New England Biolabs # M0491L), and the cycling parameters were as follows for all cloning: 98°C for 30 s, followed by 98°C for 15 s, 72°C for 20 s, and 72°C for 30 s per kb for 40 cycles. The final PCR product was conducted into TOPO cloning vector (Thermo Fisher Scientific #450641). Sanger sequencing was performed to ensure that the knock-in DNA was cloned in-frame with the HAs. The Lenti-Cas9-Blast plasmid (Addgene #83480) and the Lenti-Guide-Puro plasmid (Addgene #52963) were purchased from Addgene. For candidate validation of CRISPR screen, sgRNA sequences against DOT1L (5’-TCAGCTTCGAGAGCATGCAG-3’), ENL (5’-TCACCTGGACGGTGCACTGG-3’), USF2 (#2: 5’-AGAAGAGCCCAGCACAACGA-3’, #3: 5’-TGTTTTCCGCAGTGGAGCGG-3’, #4: 5’-CCGGGGATCTTACCTGGCGG-3’, and #5: 5’-CAGCCACGACAAGGGACCCG-3’) were cloned into an in-house-made Lenti-Guide-Puro-IRES-CFP vector. The sgRNA sequence against USF1 (3#, 5’-CTATACTTACTTCCCCAGCA-3’) was cloned into an in-house-made LRNeo-2.1 vector in which the mCherry-expressing cassette of LRCherry2.1 (Addgene #108099) was replaced by Neomycin. For competitive proliferation assay, sgRNAs against Luciferase (Luc)(5’-CCCGGCGCCATTCTATCCGC-3’) and USF2 (#2, #3 and #5 as above) were cloned into mCherry-expressing LRCherry2.1 (Addgene #108099) vector.
Generation of a HOXA9P2A-mCherry reporter allele
Request a detailed protocolSEM and OCI-AML2 were electroporated by using the Nucleofector-2b device (Lonza) with the V-kit and program X-001. For HOXA9P2A-mCherry knock-in delivery, 2.5 μg of the donor plasmid and 2.5 μg of the CRISPR/Cas9-HOXA9-C-terminus-sgRNA all-in-one plasmid were used for 5 million SEM cells. Twenty-four hours after transfection, cells were sorted for the GFP fluorescent marker linked to Cas9 expression vector to enrich the transfected cell population. After the sorted cells recovered in culture for up to 3 weeks, a second sort was performed to select cells for successful knock-in by sorting for cells expressing the knock-in mCherry fluorescent marker. Two weeks later, a third sort was repeated based on the selection mCherry-expressing cells.
Characterization of successful knock-in events by PCR and Sanger sequencing
Request a detailed protocolDNA from single-cell-derived bacterial or cell colonies was extracted with a Quick-DNA Miniprep Kit (Zymo #D3025). Combinatorial primer sets designed to recognize the 5′ and 3′ knock-in boundaries were used with the following PCR cycling conditions: 98°C for two mins, followed by 40 cycles of 98°C for 30 s and 68°C for 60 s. The sequences for genotyping primers are provided in Supplementary file 1. After electrophoresis, the bands that were at the expected size were cut out, purified, and sequenced with two specific primers (Supplementary file 1).
CRISPR library construction and screening
View detailed protocolA set of ~11,000 sgRNA oligos that target 1639 human transcription factors were designed for array-based oligonucleotide synthesis (CustomArray). Unique binding of each sgRNA was verified by sequence blast against the whole human genome. In the sgRNA pooled library, seven gRNAs against each of the 1639 human transcription factors were obtained from validated sgRNA libraries published previously (Wang et al., 2015; Doench et al., 2016; Sanjana et al., 2014; Ma et al., 2015; Tzelepis et al., 2016; Hart et al., 2015; Hart et al., 2017; Smith et al., 2008; Park et al., 2017). The synthesized oligo pool was amplified by PCR and cloned into LentiGuide-Puro backbone (#52963) by in-fusion assembly (Clontech #638909). The HOXA9P2A-mCherry reporter cell line was overexpressed with lentiviral Cas9 followed by infection of pooled sgRNA library at low M.O.I (~0.3). Infected cells were selected by blasticidine and puromycin and later sorted for mCherryHigh and mCherryLow populations between days 10–12. The sgRNA sequences were recovered by genomic PCR analysis and deep sequencing using MiSeq for single-end 150 bp read length (Illumina). The primer sequences used for cloning and sequencing are listed in Supplementary file 1. The sgRNA sequences are described in Supplementary file 2. High-titer lentivirus stocks were generated in 293 T cells as previously described (Vo et al., 2017).
Data analysis of CRISPR screening
Request a detailed protocolThe raw FASTQ data were de-barcoded and mapped to the original reference sgRNA library. The differentially enriched sgRNAs were defined by comparing normalized counts between sorted cells in the top 10% and those in the bottom 10% of mCherry-expressing bulk populations. Two independent replicate screenings were performed with the HOXA9P2A-mCherry reporter cell line stably expressing Cas9. Normalized counts for each sgRNA were extracted and used to identify differentially enriched sgRNA by DESeq2 (Love et al., 2014). The combined analysis of seven sgRNAs against each human transcription factor was conducted by using the MAGeCK algorithm (Li et al., 2014). Detailed screening results were included in Supplementary file 2.
Fluorescence imaging and analysis
Request a detailed protocol0.1% of DMSO (vehicle control) or 10 doses of SGC0946 with a half log scale (0.3 nM-10 μM) were first dispensed into 384-well plates (in quadruplicate, four wells per dose). Suspension-cultured SEM cells were immediately plated into the 384-well plate (20,000 cells / well). Six days after drug treatment, the cells were fixed with 4% paraformaldehyde for 10 mins at room temperature, followed by Hoechst staining for 15 mins at room temperature. Fluorescence images (Hoechst and mCherry) were taken by a CellVoyager 8000 high content imager (Yokogawa). The acquired images were processed by using the Columbus Image Data Storage and Analysis system (Perkin Elmer) to count the number of positive cells and measure fluorescent intensity. To determine the changes of mCherry intensity in SEM expressing HOXA9P2A-mCherry, we measured average mCherry intensity of four fields per well and normalized to vehicle (0.1% DMSO) treated control. Wild-type SEMs with no fluorescence were included as negative controls.
Cut and Run assay
Request a detailed protocolCut and Run assay was conducted following the protocol described previously (Skene and Henikoff, 2017). In brief, three million cells were collected for each sample. The USF2 antibody (NBP1-92649, Novus) was used at a 1:100 dilution. Library construction was performed using the NEBNext UltraII DNA Library Prep Kit from NEB (E7645S). Indexed samples were run using the Illumina Next-seq 300-cycle kit. Cut and Run raw reads were mapped to genome hg19. by bowtie 2.3.4 with default parameter. The mapping file were converse to. bw file by bamCoverage (Langmead and Salzberg, 2012; Ramírez et al., 2014).
Flow cytometry
Request a detailed protocolSuspension-cultured SEM and OCI-AML2 cells were collected by centrifugation at 800Xg, filtered through a 70 µm filter, and analyzed for mCherry on a BD FACS Aria III flow cytometer with a negative control. The 4,6-diamidino-2-phenylindole (DAPI) staining was conducted prior to sorting to exclude dead cells.
Inhibitor treatment
Request a detailed protocolSEM and OCI-AML2 cells were seeded at a density of 1 × 105 cells/mL in medium supplemented with DMSO vehicle or different doses (from 0.5 µM to 15 μM) of the DOT1L inhibitor SGC0946 (MedChemExpress #HY-15650). Medium was replaced every three days, and fresh inhibitor was added. At day-6 post-treatment, cells were collected for flow cytometry analysis and RNA extraction.
Fluorescence in situ hybridization
Request a detailed protocolAn ~800 bp purified P2A-mCherry DNA fragment was labeled with a red-dUTP (AF594, Molecular Probes) by nick translation, and a HOXA9 BAC clone (CH17-412I12/7p15.2) was labeled with a green-dUTP (AF488, Molecular Probes). Both of labeled probes were combined with sheared human DNA and independently hybridized to fix the interphase and metaphase nuclei derived from each sample by using routine cytogenetic methods in a solution containing 50% formamide, 10% dextran sulfate, and 2XSSC. The cells were then stained with DAPI and analyzed.
Quantitative real-time PCR
Request a detailed protocolTotal RNA was collected by using TRIzol (Thermo Fisher Scientific #15596026) or Direct-zol RNA Miniprep Kit (Zymo #R2052). Reverse transcription was performed by using a High-Capacity cDNA Reverse Transcriptase Kit (Applied Biosystems #4374966). Real-time PCR was performed by using FAST SYBR Green Master Mix (Applied Biosystems #4385612) in accordance with the manufacturer’s instructions. Relative gene expression was determined by using the ΔΔ-CT method (Schmittgen and Livak, 2008). All Q-PCR primers used in this study are listed in Supplementary file 1.
Competitive proliferation assay
Request a detailed protocolFor evaluating the impact of USF2 sgRNAs on leukemia expansion, cell cultures were lentivirally transduced with individual USF2 sgRNAs in mCherry expressing vector, followed by measurement of the mCherry-positive percentage at various days post-infection using flow cytometry. The rate of mCherry-positive percentage was normalized to that of Day 3 and declined over time, which was used to infer a defect in cell accumulation conferred by a given sgRNA targeting USF2 relative to the uninfected cells in the same culture.
Immunoblotting
Request a detailed protocolCells lysate was prepared by using RIPA buffer followed with SDS-PAGE (Thermo Fisher Scientific) and transferred to a PVDF membrane according to the manufacturer’s protocols (Bio-Rad) at constant 100 V for 1 hr. After blocking incubation with 5% non-fat milk in TBS-T (10 mM Tris, pH 8.0, 150 mM NaCl, 0.5% Tween-20) for 1 hr at room temperature, the membrane was incubated with antibodies against GAPDH (Thermo Fisher Scientific, AM4300, 1:10,000), MYC (Cell Signaling Technology, #9402, 1:1,000), USF2 (Novus, NBP1-92649, 1:2,000), USF1 (Proteintech, 22327–1-AP, 1:2,000), GAPDH (Thermo Fisher Scientific, AM4300, 1:10,000), Vinculin (Proteintech, 26520–1-AP, 1:2,000) and CTCF (abcam, ab70303, 1:1,000) at 4°C for 12 hr with gentle shaking. Membranes were washed three times for 30 min and incubated with a 1:2000 dilution of horseradish peroxidase-conjugated anti-mouse or anti-rabbit antibodies for 2 hr at room temperature. Blots were washed with TBS-T three times for 30 min and developed with the ECL system (Amersham Biosciences) according to the manufacturer’s protocol.
Statistics
All values are shown as the mean ± SEM. Statistical analyses were performed with GraphPad Prism software, version 8.0. p-Values were calculated by performing a two-tailed t-test.
Data availability
All plasmids created in this study will be deposited to Addgene. Raw data collected from Cut&Run were deposited at NCBI GEO (GSE140664). Raw data collected from CRISPR screening were included in Supplementary File 2. Publicly available dataset used in this study were cited accordingly including Figures 1E and S5D: GSE120781; Figure 1-supplement 1A-C: GSE13159; Figure 3-supplement 2C: GSE126619, GSE74812, GSE89485; Figure 3-supplement 3A: ENCODE (HCT116); Figure 5-supplement 3A-C: European Genome-phenome Archive (EGA) under accession number EGAS00001003266, EGAS00001000654, EGAS00001001952, EGAS00001001923, EGAS00001002217 and EGAS00001000447.
-
NCBI Gene Expression OmnibusID GSE140664. Functional Interrogation of HOXA9 Regulome in Leukemia via Endogenous Reporter-based CRISPR/Cas9 screen.
-
NCBI Gene Expression OmnibusID GSE120781. Acute deletion of CTCF disrupted enhancer-promoter regulation of MYC in human cancer cells.
-
NCBI Gene Expression OmnibusID GSE13159. Microarray Innovations in LEukemia (MILE) study: Stage 1 data.
-
NCBI Gene Expression OmnibusID GSE126619. Acute deletion of CTCF disrupted enhancer-promoter regulation of MYC in human cancer cells.
-
NCBI Gene Expression OmnibusID GSE74812. MLL-rearranged acute lymphoblastic leukemias upregulate BCL-2 through H3K79 methylation and are highly sensitive to the BCL-2 specific antagonist ABT-199.
-
NCBI Gene Expression OmnibusID GSE89485. Therapeutic targeting MLL degradation pathways in MLL-rearranged leukemia.
-
European Genome-phenome ArchiveID EGAS00001003266. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
-
European Genome-phenome ArchiveID EGAS00001000654. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
-
European Genome-phenome ArchiveID EGAS00001001952. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
-
European Genome-phenome ArchiveID EGAS00001001923. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
-
European Genome-phenome ArchiveID EGAS00001002217. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
-
European Genome-phenome ArchiveID EGAS00001000447. PAX5-driven Subtypes of B-cell Acute Lymphoblastic Leukemia.
References
-
Transformation of myeloid progenitors by MLL oncoproteins is dependent on Hoxa7 and Hoxa9Genes & Development 17:2298–2307.https://doi.org/10.1101/gad.1111603
-
Deregulation of the HOXA9/MEIS1 Axis in acute leukemiaCurrent Opinion in Hematology 23:354–361.https://doi.org/10.1097/MOH.0000000000000245
-
Hox gene dysregulation in acute myeloid leukemiaFuture Oncology 10:475–495.https://doi.org/10.2217/fon.13.195
-
Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9Nature Biotechnology 34:184–191.https://doi.org/10.1038/nbt.3437
-
MLL-AF4 binds directly to a BCL-2 specific enhancer and modulates H3K27 acetylationExperimental Hematology 47:64–75.https://doi.org/10.1016/j.exphem.2016.11.003
-
Evaluation and design of Genome-Wide CRISPR/SpCas9 knockout screensG3: Genes, Genomes, Genetics 7:2719–2727.https://doi.org/10.1534/g3.117.041277
-
Acute depletion of CTCF directly affects MYC regulation through loss of enhancer-promoter loopingNucleic Acids Research 47:6699–6713.https://doi.org/10.1093/nar/gkz462
-
Interaction of the transcription factors USF1, USF2, and alpha -Pal/Nrf-1 with the FMR1 promoter implications for fragile X mental retardation syndromeThe Journal of Biological Chemistry 276:4357–4364.https://doi.org/10.1074/jbc.M009629200
-
Fast gapped-read alignment with bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
A SALL4/MLL/HOXA9 pathway in murine and human myeloid leukemogenesisJournal of Clinical Investigation 123:4195–4207.https://doi.org/10.1172/JCI62891
-
Three-dimensional genome architecture: players and mechanismsNature Reviews Molecular Cell Biology 16:245–257.https://doi.org/10.1038/nrm3965
-
Interaction of USF1/USF2 and alpha-Pal/Nrf1 to Fmr-1 promoter increases in mouse brain during agingBiochemical and Biophysical Research Communications 376:347–351.https://doi.org/10.1016/j.bbrc.2008.08.155
-
Identification of Pax5 target genes in early B cell differentiationThe Journal of Immunology 180:1719–1728.https://doi.org/10.4049/jimmunol.180.3.1719
-
deepTools: a flexible platform for exploring deep-sequencing dataNucleic Acids Research 42:W187–W191.https://doi.org/10.1093/nar/gku365
-
Organizational principles of 3D genome architectureNature Reviews Genetics 19:789–800.https://doi.org/10.1038/s41576-018-0060-8
-
Improved vectors and genome-wide libraries for CRISPR screeningNature Methods 11:783–784.https://doi.org/10.1038/nmeth.3047
-
Analyzing real-time PCR data by the comparative C(T) methodNature Protocols 3:1101–1108.https://doi.org/10.1038/nprot.2008.73
-
Mixed lineage rearranged leukaemia: pathogenesis and targeting DOT1LCurrent Opinion in Hematology 22:92–96.https://doi.org/10.1097/MOH.0000000000000123
-
Upstream stimulatory factors bind to insulin response sequence of the fatty acid synthase promoter USF1 is regulatedJournal of Biological Chemistry 270:28716–28722.https://doi.org/10.1074/jbc.270.48.28716
-
CTCF controls HOXA cluster silencing and mediates PRC2-repressive higher-order chromatin structure in NT2/D1 cellsMolecular and Cellular Biology 34:3867–3879.https://doi.org/10.1128/MCB.00567-14
-
Hoxa9 and Meis1 are key targets for MLL-ENL-mediated cellular immortalizationMolecular and Cellular Biology 24:617–628.https://doi.org/10.1128/MCB.24.2.617-628.2004
-
Model-based analysis of ChIP-Seq (MACS)Genome Biology 9:R137.https://doi.org/10.1186/gb-2008-9-9-r137
Article and author information
Author details
Funding
Leukemia Research Foundation
- Rui Lu
Concern Foundation
- Rui Lu
American Cancer Society (IRG15-59-IRG)
- Rui Lu
Young Supporters Board of the O’Neal Comprehensive Cancer Center
- Rui Lu
National Cancer Institute (P30CA021765-37)
- Chunliang Li
National Heart, Lung, and Blood Institute (R01HL153220)
- Yang Zhou
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We gratefully acknowledge the staffs of the Hartwell Sequencing, Cytogenetics, Flow Cytometry and Cell Sorting Shared Resource facility within the Comprehensive Cancer Center of St. Jude Children’s Research Hospital. We thank Li and Lu laboratory members for critical comments and discussion. We thank Dr. Cherise Guess for helping with scientific editing.
Copyright
© 2020, Zhang et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,956
- views
-
- 922
- downloads
-
- 39
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 39
- citations for umbrella DOI https://doi.org/10.7554/eLife.57858
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Functional interrogation of HOXA9 regulome in MLLr leukemia via reporter-based CRISPR/Cas9 screen
eLife 9:e57858.
https://doi.org/10.7554/eLife.57858




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}