A regulatory pathway that selectively up-regulates elongasome function in the absence of class A PBPs
- Department of Microbiology, Cornell University, United States
Abstract
Bacteria surround themselves with peptidoglycan, an adaptable enclosure that contributes to cell shape and stability. Peptidoglycan assembly relies on penicillin-binding proteins (PBPs) acting in concert with SEDS-family transglycosylases RodA and FtsW, which support cell elongation and division respectively. In Bacillus subtilis, cells lacking all four PBPs with transglycosylase activity (aPBPs) are viable. Here, we show that the alternative sigma factor σI is essential in the absence of aPBPs. Defects in aPBP-dependent wall synthesis are compensated by σI-dependent upregulation of an MreB homolog, MreBH, which localizes the LytE autolysin to the RodA-containing elongasome complex. Suppressor analysis reveals that cells unable to activate this σI stress response acquire gain-of-function mutations in the essential histidine kinase WalK, which also elevates expression of sigI, mreBH and lytE. These results reveal compensatory mechanisms that balance the directional peptidoglycan synthesis arising from the elongasome complex with the more diffusive action of aPBPs.
Introduction
Nearly all bacterial cells are surrounded by a peptidoglycan (PG) cell wall that provides a protective barrier, helps resist cell swelling and lysis under hypoosmotic conditions, and contributes to cell shape determination (Egan et al., 2020; Zhao et al., 2017). PG functions as a large, covalently linked macromolecular enclosure and is actively remodeled to allow cell growth and division. The basic processes of PG synthesis are broadly conserved, and the detailed pathways are well documented. PG synthesis initiates with the diversion of sugars from central metabolism to form the two amino-sugars, N-acetylglucosamine (NAG) and N-acetylmuramic acid (NAM), and the incorporation of amino acids to form the stem peptide (Barreteau et al., 2008). The ultimate product of these cytosolic reactions is lipid II, a disaccharide pentapeptide precursor unit linked to an undecaprenyl pyrophosphate carrier lipid (van Heijenoort, 2007). Lipid II is flipped across the membrane (Sham et al., 2014; Meeske et al., 2015) where it interacts with two key enzymatic activities to assemble the PG layer: a transglycosylase (TG) function joins the disaccharide unit to form long, linear chains of alternating NAG-NAM residues, and a transpeptidase (TP) activity crosslinks a subset of the pentapeptide side chains to link the glycan strands together. Crucially, insertion of new glycan strands requires endopeptidases that can cleave existing crosslinks to facilitate cell wall expansion (Singh et al., 2012; Hashimoto et al., 2012; Do et al., 2020).
Most bacteria require PG for survival, except under very specific conditions (Claessen and Errington, 2019). This, combined with the absence of PG in eukaryotes, makes PG synthesis and stability an excellent target for antibiotics. One class of PG-targeting antibiotics, the beta-lactams, account for more than 60% of the global market (Klein et al., 2018). Beta-lactam antibiotics interfere with PG synthesis by covalently modifying penicillin-binding proteins (PBPs), named for their affinity for the first widely used member of this drug family. All PBPs have TP activity, and beta-lactams mimic the substrate of the transpeptidation reaction (Tipper and Strominger, 1965). Many PBPs also have TG activity, and these bifunctional PBPs are designated class A PBPs, or aPBPs (McPherson and Popham, 2003). Other PBPs, designated bPBPs, only have TP activity, and must work in coordination with enzymes that provide TG activity (Wei et al., 2003; Taguchi et al., 2019; Rohs et al., 2018; Özbaykal et al., 2020).
While the basic outline of PG assembly has been understood for decades, the last few years have seen major strides in our understanding of how PG synthesis is coordinated in time and space (Zhao et al., 2017; Egan et al., 2020). Moreover, PG synthesis can be regulated as a function of cell growth, division, nutritional status, and in response to externally imposed stresses such as the action of antibiotics (Delhaye et al., 2019; Typas et al., 2012; Helmann, 2016). B. subtilis has been a leading model system for understanding PG synthesis in rod-shaped, Gram-positive bacteria. Seminal work in this system established, for example, that the sites of PG synthesis during cell elongation seem to be correlated with cytoskeletal filaments assembled from MreB and its paralogs, MreBH and Mbl (Kawai et al., 2009). This synthesis occurs in arcs that are perpendicular to the long access of the cell and is driven by a putative complex known as the elongasome (Garner et al., 2011). Cell division, in contrast, occurs at mid-cell during vegetative growth and is directed by a different cytoskeletal filament, FtsZ, in a complex called the divisome (Mahone and Goley, 2020). In early models, it was suggested that the major aPBP, PBP1 (encoded by the ponA gene), shuttled between the elongasome and divisome to provide the needed TG and TP activities (Claessen et al., 2008). However, bPBPs clearly also play important roles in synthesis (Wei et al., 2003). The composition and dynamic nature of these complementary systems has been subject of intensive study.
A key finding that challenged our understanding of PG synthesis in B. subtilis was the observation that a strain lacking all four known aPBPs was viable and still synthesized an apparently normal PG layer (McPherson and Popham, 2003). This implied that there must be another protein with TG activity and, unlike aPBP-associated TG activity, this activity was insensitive to inhibition by moenomycin (MOE). MOE, like many PG synthesis inhibitors, activates the σM stress response (Mascher et al., 2007). Moreover, sigM null mutants are highly MOE sensitive (Mascher et al., 2007), which suggested that the missing TG might be part of the σM regulon. Indeed, the elongasome-associated TG has been identified as the SEDS family protein RodA (Meeske et al., 2016; Emami et al., 2017), a known member of the σM regulon (Eiamphungporn and Helmann, 2008; Helmann, 2016). A RodA paralog, FtsW, provides TG activity in the context of the divisome (Taguchi et al., 2019; Liu et al., 2018).
Our current understanding of PG synthesis during cell elongation in B. subtilis suggests that the bulk of synthesis is provided by the elongasome, with RodA serving as TG and PBP2a and PbpH, and perhaps also aPBPs, serving as TP (Emami et al., 2017; Meeske et al., 2016). This action is directional, largely oriented perpendicular to the long cell axis, and is balanced by a more diffusive activity of aPBPs (Dion et al., 2019; Vigouroux et al., 2020). Cells that rely exclusively on the elongasome for growth are longer and thinner, whereas those that rely predominantly on aPBPs tend to be wider and shorter (Dion et al., 2019). Many PG synthesis inhibitors activate the σM regulon, and this leads to elevated expression of many key PG biosynthetic enzymes (MurB, Amj, BcrC), elongasome components (MreB, RodA, MreCD), and the major aPBP (PBP1) (Eiamphungporn and Helmann, 2008; Helmann, 2016). However, some antibiotics may act selectively on the aPBPs or the elongasome, and it is less clear how cells might act to balance these two biosynthetic activities.
Here, we sought to define pathways important for fitness in cells that rely exclusively on the elongasome for cell elongation. We demonstrate that cells lacking aPBPs, or even just PBP1 (ponA), require a regulatory pathway that selectively increases expression of elongasome-associated proteins. Specifically, ΔponA mutant cells are unable to grow in the absence of σI, which induces transcription of genes encoding MreBH and an associated autolysin, LytE. Factors that facilitate σI activity, including the RasP intramembrane peptidase and its regulator EcsAB, are therefore also essential under these conditions. Further support for the importance of MreBH and LytE derives from analysis of a suppressor mutation that activates the WalKR two-component system, and thereby also restores viability to a ΔrasPΔponA double mutant by up-regulating these same elongasome components. These results suggest that the σI stress response acting in concert with the WalKR system helps to maintain balanced activity of the elongasome and the aPBPs during cell elongation.
Results
The EcsAB-RasP pathway is essential in the absence of class A PBPs
Bacteria often use overlapping or redundant systems to sustain essential pathways such as PG synthesis. To identify genes with significant roles in elongasome activity in B. subtilis, we constructed a strain (designated Δ4) lacking all four class A PBPs (aPBPs), and which therefore relies solely on the elongasome for PG synthesis during cell elongation (McPherson and Popham, 2003). A Tn-Seq approach was employed to identify genes essential in the Δ4 strain but not in the wild-type (WT) background. We identified the ecsAB operon as having numerous mariner transposon insertions in WT, but very few in the Δ4 strain (Figure 1—figure supplement 1). We verified conditional essentiality of ecsA by determining the plating efficiency of a clean, unmarked deletion mutant (ΔecsA) in a ponA depletion background in the presence and absence of the genes encoding the other 3 aPBPs (pbpD, pbpF, pbpG). Interestingly, ecsA was not only essential in the Δ4 background but also with depletion of ponA alone (Figure 1A). Mutations that impair PG synthesis can often be rescued by growth on plates amended with 20 mM MgSO4, which leads to decreased activity of autolysins and thereby helps restore balance between PG synthesis and degradation pathways (Formstone and Errington, 2005). Indeed, an ΔecsAΔponA mutant was viable when streaked on high Mg plates, and growth was Mg-dependent (Figure 1B).
Figure 1 with 1 supplement see all
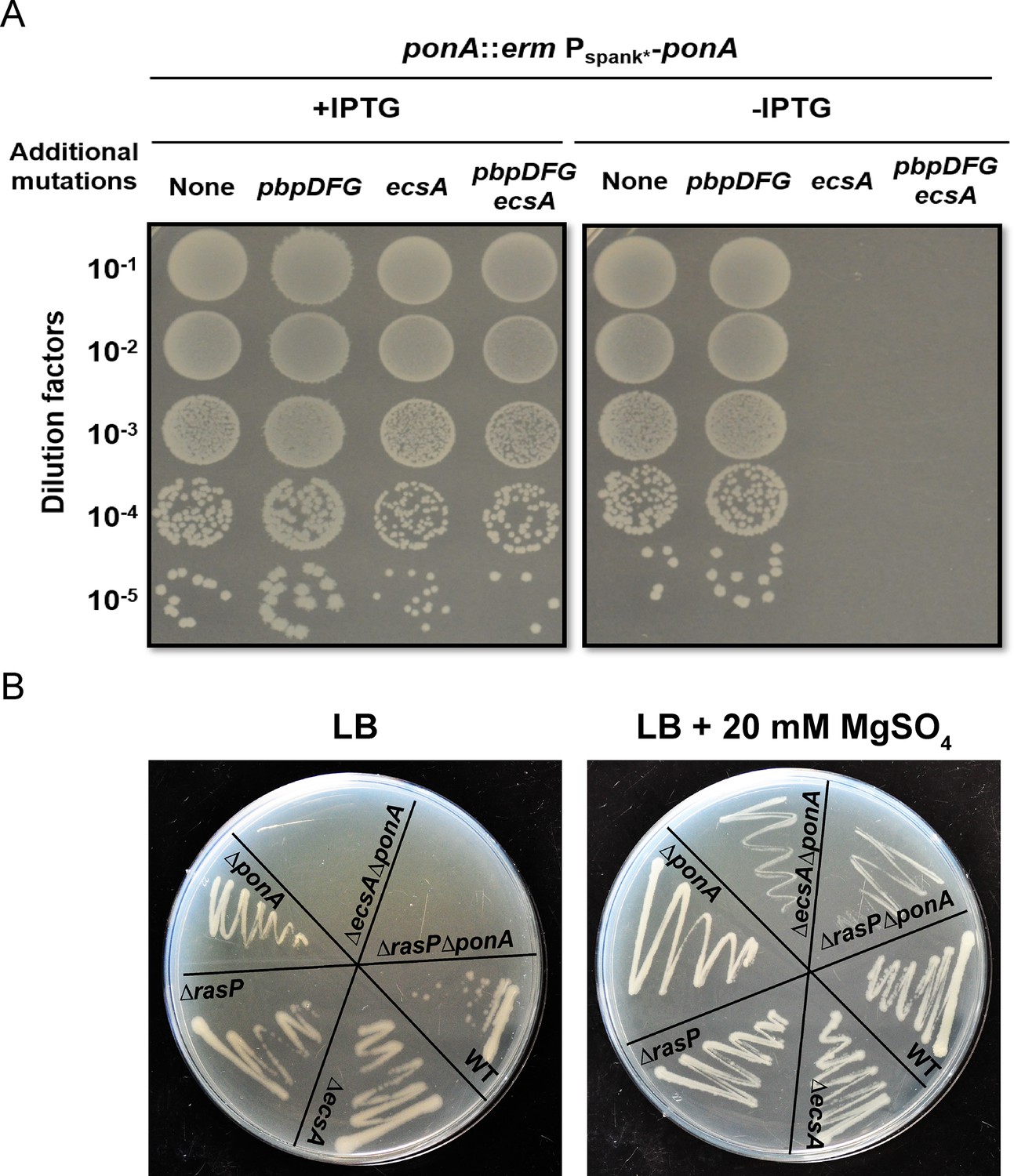
The ecsA and ponA genes are synthetic lethal in LB medium.
(A) Plating efficiency of ecsA deletion mutants. Right panel: spot dilutions were used to assess the effect of an ecsA null mutation on growth in a ponA depletion background (-IPTG) with and without additional mutations in pbpD, pbpF, pbpG (to mimic the Δ4 A PBP background). Left panel: ponA was induced (+IPTG) from the Pspank* promoter. (B) Growth of ΔecsA, ΔrasP, ΔponA and the double mutants ΔecsAΔponA and ΔrasPΔponA on LB agar plates with and without supplementation with 20 mM MgSO4.
EcsA has been designated as part of an ABC-type transporter involved in the expression and secretion of proteins (Leskelä et al., 1999). Deletion of ecsA has a profound effect on the intramembrane protease RasP, with similar phenotypes noted for the ecsA and rasP deletion mutants (Heinrich et al., 2008). Consequently, we tested whether the essential role of EcsA in the ΔponA strain was due to RasP. Indeed, viability of ΔrasPΔponA, like ΔecsAΔponA, depended on high Mg concentrations (Figure 1B). The above data highlight the importance of the EcsAB-RasP pathway in maintaining viability in the absence of aPBPs.
Mutants defective in the EcsAB-RasP pathway are sensitive to antibiotics that inhibit aPBPs
Upregulation of elongasome activity is known to alleviate aPBP defects (Meeske et al., 2016). Based on the observed conditional essentiality, we hypothesized that the EcsAB-RasP pathway might functionally compensate for the absence of aPBPs. As a first test of this hypothesis, we measured sensitivity to moenomycin (MOE), a specific inhibitor of aPBP-associated TG activity (Van Heijenoort et al., 1978; Chen et al., 2019). Indeed, ecsA and rasP mutants were MOE sensitive with a four-fold decrease in minimum inhibitory concentration (MIC) relative to WT (Table 1). This was not due to a general growth defect: ecsA and rasP single mutants grew as well as WT in the absence of MOE, albeit with some lysis in stationary phase (Figure 2A), consistent with previous observations (Heinrich et al., 2008). This antibiotic sensitivity could be complemented by ectopic expression of ecsAB or rasP, respectively (Figure 2—figure supplement 1). Moreover, ΔecsAΔrasP had a similar MOE sensitivity as ΔrasP (Figure 2A), suggesting that the synthetic lethality of ecsA with ponA is mediated through its known downstream effect on the activity of RasP (Heinrich et al., 2008). In contrast to MOE, the ΔrasP and ΔponA mutants had a similar sensitivity as WT when tested for sensitivity to antibiotics that act on substrates common to both the elongasome and aPBP-dependent pathways of PG synthesis. For example, both nisin (Wiedemann et al., 2001) and vancomycin (Watanakunakorn, 1984) bind the common lipid II intermediate (Figure 2—figure supplement 1). Together, these results suggest that the EcsAB-RasP pathway is critical when aPBPs are compromised, but not as a general response to inhibition of PG synthesis.
Figure 2 with 2 supplements see all
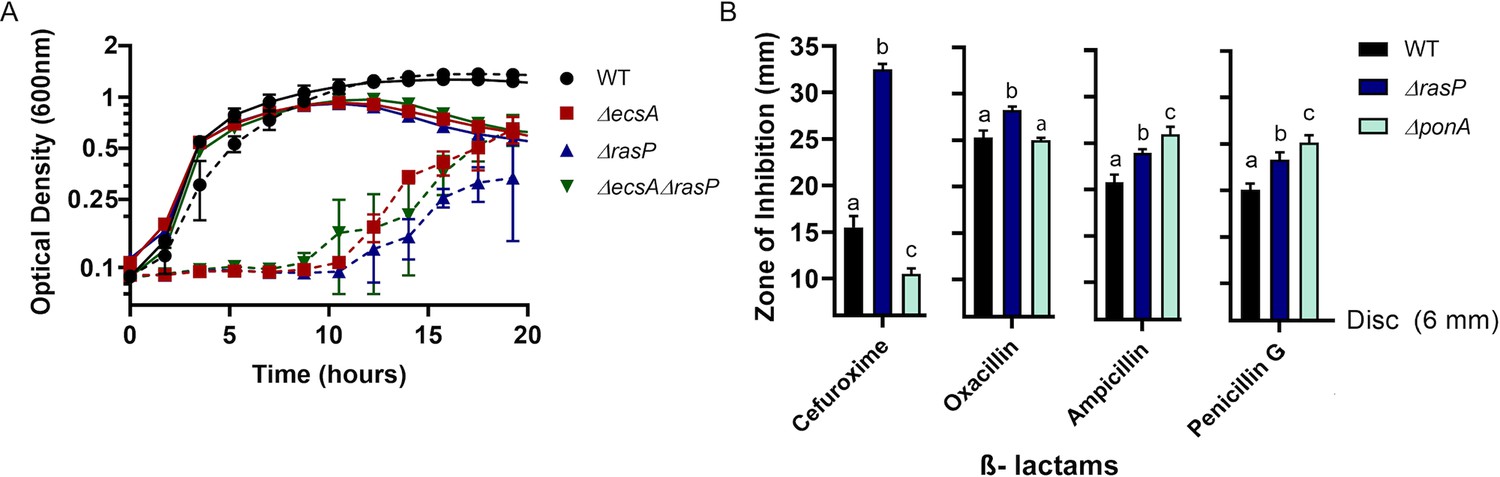
The EcsAB-RasP pathway is important for intrinsic antibiotic resistance.
(A) Growth kinetics of WT, ΔecsA, ΔrasP and the ΔecsAΔrasP double mutant in liquid LB medium with (dotted lines) and without (continuous lines) 0.4 µg/mL moenomycin (MOE). (B) β-lactam sensitivity of ΔrasP and ΔponA strains determined by disc diffusion assay using cefuroxime (CEF) (10 µg), oxacillin (3 µg), ampicillin (15 µg), and penicillin G (20 units). No comparison was done between antibiotic groups. P-value cutoff of <0.001 was used.
-
Figure 2—source data 1
Data of growth kinetics and zone of inhibition.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig2-data1-v1.xlsx
Table 1
Minimum inhibitory concentration (MIC) of various strains for moenomycin in µg/mL.
| Strains | Moenomycin MIC (µg/mL) |
|---|---|
| WT | 1.6 |
| ΔecsA | 0.4 |
| ΔrasP | 0.4 |
| ΔponA | >1.6 |
| ΔsigW | 1.6 |
| ΔsigV | 1.6 |
| ΔsigI | 0.4 |
| Δ25ftsL | 1.6 |
We next sought to test antibiotics that, unlike MOE, inhibit aPBPs at their TP active site. We reasoned that a stress response important for elongasome activity should also provide resistance to antibiotics that inhibit aPBPs, assuming they do not also interfere with the bPBPs essential for the elongasome. We tested 4 β-lactams (cefuroxime, oxacillin, ampicillin and penicillin G) for their inhibition profiles against ΔrasP and ΔponA strains. Oxacillin and cefuroxime (CEF) were previously suggested to preferentially inhibit aPBPs (Sassine et al., 2017; Sharifzadeh et al., 2020), whereas penicillin G preferentially inhibits bPBPs (Sassine et al., 2017). Consistently, oxacillin and CEF had highest activity against ∆rasP, whereas penicillin G and ampicillin had the highest activity against ∆ponA, which encodes the major aPBP, PBP1 (Figure 2B). These results support the idea that the EcsAB-RasP pathway functionally compensates either for the absence of aPBPs or for their chemical inhibition at either the TG (MOE) or TP (CEF) active sites.
Interestingly, the ∆ponA mutant was actually more CEF resistant than WT. Thus, PBP1 inactivated by CEF may be deleterious to the cell. This is suggestive of futile cycling, a process in which inactivation of the TP active site leads to an ongoing generation and degradation of uncrosslinked PG strands driven by the aPBP-associated TG (Cho et al., 2014; Waxman et al., 1980). To explore this idea further, we treated WT cells with sub-inhibitory concentrations of two drugs simultaneously, MOE and CEF, that inhibit the two different active sites of the aPBP proteins. If CEF results in futile cycling, we reasoned that MOE might antagonize this effect. In contrast, MOE and CEF together resulted in synergistic inhibition (Figure 2—figure supplement 2). This is consistent with the same target drug synergy model, as previously described for E. coli protein synthesis inhibitors (Yilancioglu, 2019) and drugs used to treat human diseases (Jia et al., 2009), but does not support the hypothesis of CEF-dependent futile cycling.
EcsAB-RasP functions through σI to sustain cell wall synthesis in the absence of aPBPs
RasP functions as an intramembrane protease for the activation of multiple stress response pathways, and our results suggest it may be important for PG synthesis when aPBPs are missing or inhibited. RasP proteolytically inactivates the anti-sigma factors RsiW (regulator of σW) (Schöbel et al., 2004), RsiV (regulator of σV) (Hastie et al., 2013) and RsgI (regulator of σI) (Liu et al., 2017). In the absence of RasP, these σ factors can not be activated. RasP also cleaves FtsL, a cell division protein (Bramkamp et al., 2006). To determine which of these RasP targets may contribute to elongasome activity, we took advantage of the fact that MOE and CEF selectively inactivate aPBPs. Therefore, MOE and CEF resistance provides a readout of elongasome function. We tested mutants lacking each of the three RasP-dependent sigma factors or containing Δ25FtsL, coding for a functional, but truncated FtsL (deleted in amino acids 2–26) variant that is not subject to cleavage by RasP (Bramkamp et al., 2006). The ΔecsA and ΔrasP mutants were 4-fold more sensitive to MOE than WT (0.4 vs. 1.6 µg/mL), whereas for ΔponA the (MIC) was >1.6 µg/mL (Table 1; Figure 3—figure supplement 1). The MIC was unaffected by deletion of sigW or sigV or by the non-cleavable FtsL (1.6 µg/mL). However, the ΔsigI mutant was significantly more sensitive to MOE with the MIC being 0.4 µg/mL, similar to ΔrasP. This suggests that σI is required for optimal function of the MOE-insensitive elongasome.
Similar results were observed when CEF sensitivity was monitored (Figure 3A). Of the known RasP targets, σI contributes the most to CEF resistance. Moreover, the ΔsigWΔsigI mutant phenocopies the ΔrasP mutant, suggesting that activation of σI and σW largely accounts for the role of RasP in CEF resistance. In addition, the sensitivity of the ΔecsA and ΔrasP mutants was not further increased by mutation of sigW or sigI (Figure 3—figure supplement 2), indicative of them being in the same pathway. Finally, deletion of rsgI, encoding the σI anti-sigma factor, led to a significant decrease in CEF sensitivity of the ΔecsA and ΔrasP mutants. ΔrsgI was more sensitive to CEF compared to WT, which may be due to increased activity of σI and its associated autolysins. In contrast, deletion of rsiW, encoding the σW anti-sigma factor, led to a much less pronounced effect (Figure 3—figure supplement 2). Thus, σI plays a dominant role in intrinsic CEF resistance, and as expected this activity relies on the RasP-dependent degradation of the RsgI anti-sigma factor.
Figure 3 with 2 supplements see all
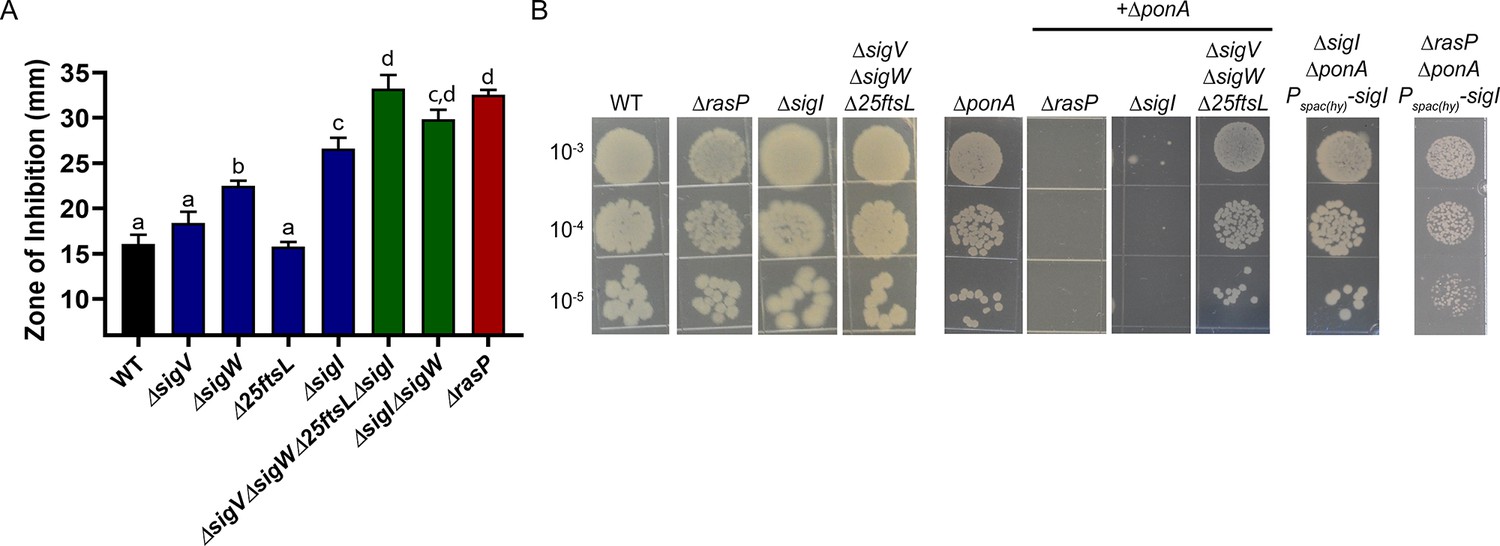
The EcsAB-RasP pathway functions largely through sigI.
(A) CEF (10 µg) sensitivity (disc diffusion assay) for WT, ΔrasP, ΔsigV, ΔsigW, Δ25ftsL, ΔsigI, ΔsigWΔsigI and ΔsigVΔsigWΔ25ftsLΔsigI strains. P-value cut-off of <0.0001 was used. (B) Plating efficiency of ΔrasP, ΔsigI and ΔsigVΔsigWΔ25ftsL strains in WT and ΔponA deletion background. This assay was done by plating 10 µL of mid-log phase cultures (grown in LB with 20 mM MgSO4) on LB agar plates (no Mg supplementation). The plating efficiency of ΔsigIΔponA double mutant was also evaluated after ectopic expression of sigI from the leaky promoter Pspac(hy).
-
Figure 3—source data 1
Data of zone of inhibition.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig3-data1-v1.xlsx
The importance of σI in the absence of aPBPs was confirmed by determining the plating efficiency of ΔsigIΔponA double mutant (Figure 3B). The double mutant could survive with high Mg2+, but was unable to grow on LB. This synthetic lethality of the ΔsigIΔponA and ΔrasPΔponA strains was suppressed by ectopically expressing the sigI gene from the leaky promoter Pspac(hy). Thus, decreased σI activity can fully explain the ∆rasP antibiotic sensitivity phenotypes, and we therefore conclude that one or more members of the σI regulon must facilitate growth under conditions of impaired aPBP activity.
σI supports elongasome function by regulating MreBH and LytE
Next, we sought to identify the σI-dependent genes important for survival in the absence of aPBPs. Of the genes directly regulated by σI (Ramaniuk et al., 2018), five (mreBH, lytE, gsiB, fabI and bcrC) have known or likely roles related to cell envelope functions. GsiB is a general stress response protein (Michna et al., 2016) and FabI is involved in fatty acid synthesis (Heath et al., 2000). BcrC functions in undecaprenylpyrophosphate recycling (Bernard et al., 2005; Zhao et al., 2016; Radeck et al., 2017b), and MreBH and LytE are both elongasome-associated proteins. MreBH, one of three MreB-family proteins that associate with the elongasome, sequesters and directs the LytE endopeptidase to the sites of insertion of new peptidoglycan (Carballido-López et al., 2006). To further define the role of σI in sustaining viability during aPBP inhibition, we conducted CEF/MOE sensitivity assays using single mutants of σI-controlled genes. The mreBH, lytE and bcrC single mutants exhibited slightly higher sensitivity for both CEF and MOE (Figure 4—figure supplement 1), however, they did not entirely phenocopy the sigI phenotype. The ΔmreBHΔlytE double mutant exhibited the same level of CEF and MOE sensitivity as both the rasP and sigI mutants (Figure 4A–B). Thus, these results suggest that the EcsAB-RasP-σI pathway primarily acts through MreBH and LytE to control elongasome function.
Figure 4 with 1 supplement see all
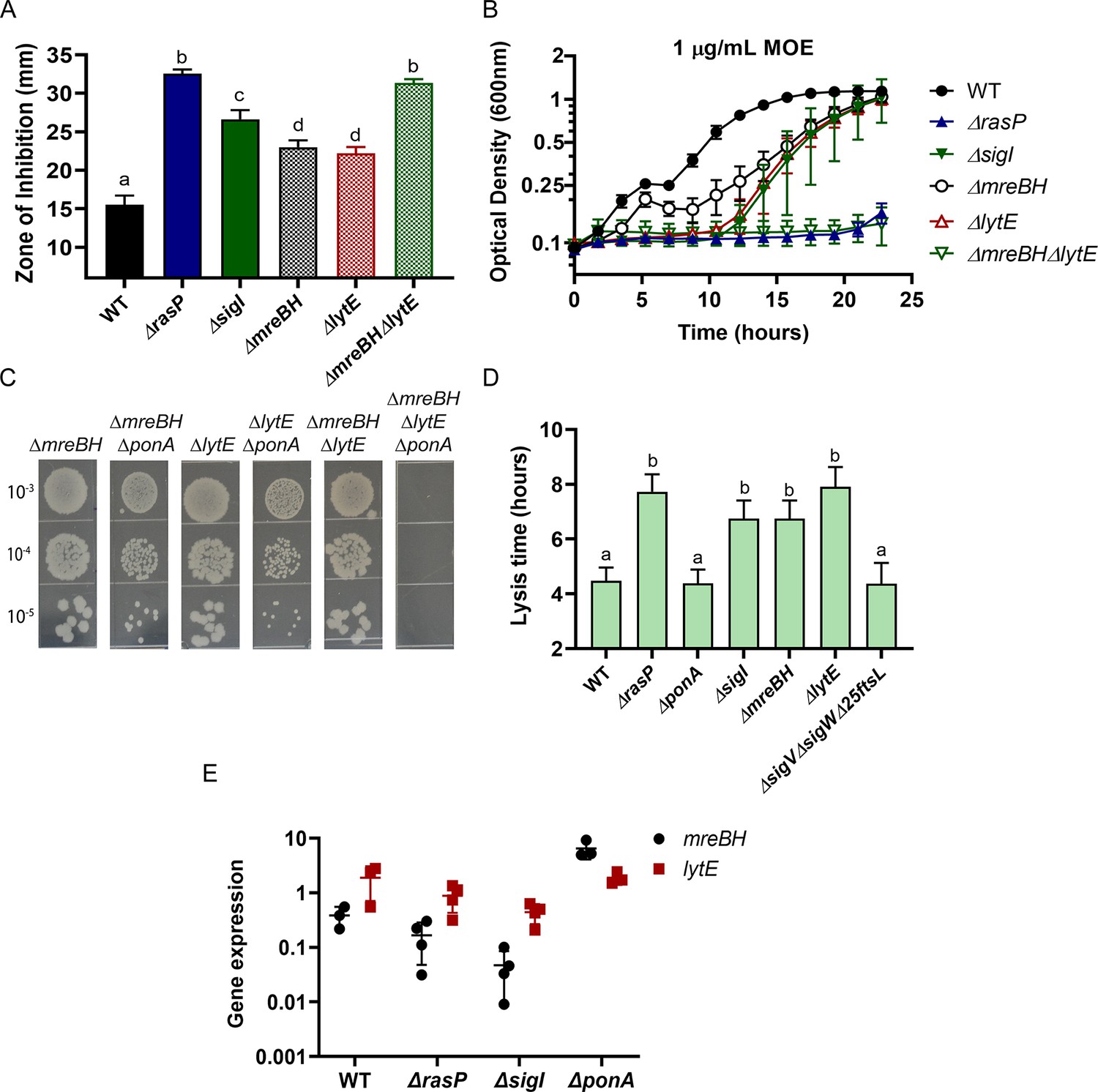
σI functions by increasing expression of mreBH and lytE.
(A) CEF (10 µg) sensitivity (disc diffusion assay) of ΔmreBH, ΔlytE and ΔmreBHΔlytE strains. Significance was determined with a P-value cut-off of <0.0001. (B) Growth kinetics of the mutants in LB medium with 1 µg/mL MOE. (C) Plating efficiency of the ΔmreBH, ΔlytE, and ΔmreBHΔlytE mutants alone and in combination with ΔponA. (D) The autolytic potential of the cells (WT, ΔponA, ΔrasP, ΔsigI, ΔmreBH, ΔlytE and ΔsigVΔsigWΔ25ftsL) measured by the time taken to reach 50% of initial cell density on treatment with sodium azide. P-value cut-off of <0.0001 was used. (E) Gene expression values (2-Δct) of mreBH and lytE normalized to gyrA plotted on log10 scale for WT, ΔrasP, ΔsigI and ΔponA strains.
-
Figure 4—source data 1
Data of zone of inhibition, MOE growth kinetics, lysis time and gene expression.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig4-data1-v1.xlsx
To further validate the importance of MreBH and LytE, we created deletion mutants in the ΔponA background (Figure 4C). A ΔmreBHΔponA double mutant could be constructed only when the cells were initially plated on LB supplemented with high Mg2+. Once constructed, however, this mutant and the ΔlytEΔponA double mutant did not exhibit a plating defect on LB. In contrast, the triple mutant of ΔmreBHΔlytEΔponA was synthetic lethal and could not be plated on LB agar without Mg2+ supplementation. These data suggest an additive role for MreBH and LytE in the effective functioning of the elongasome, likely due to the ability of LytE to retain some function in the absence of MreBH, and MreBH having functional roles beyond localization of LytE.
B. subtilis has two partially redundant D,L-endopeptidases, LytE and CwlO, which are collectively essential for cell viability (Hashimoto et al., 2012). The involvement of σI in the expression of lytE has already been established since both ΔlytEΔcwlO and ΔsigIΔcwlO are synthetic lethal (Salzberg et al., 2013). Consistently, ΔrasPΔcwlO was also synthetic lethal (Figure 4—figure supplement 1). To confirm that LytE activity was reduced in the rasP and sigI mutants we evaluated the autolytic potential of the cells. Cells were treated with sodium azide, which disrupts membrane potential and activates autolysins (Jolliffe et al., 1981; Wang et al., 2014). By monitoring the time taken for a 50% reduction in optical density, we found that the ΔlytE mutant had a lower rate of autolysis (Figure 4D). Similar to ΔlytE, we observed that ΔrasP, ΔsigI and ΔmreBH also had lower autolytic potential, consistent with a role in affecting LytE expression or activity.
Next, we evaluated the expression levels of mreBH and lytE in ΔrasP, ΔsigI and ΔponA mutants (Figure 4E). In the ΔponA mutant, mreBH was significantly upregulated, whereas lytE was unchanged. In ΔsigI, both mreBH and lytE expression was significantly lower. This suggests that ΔponA cells require higher levels of MreBH to direct the autolytic activity of LytE to support optimal elongasome function, and that activation of σI mediates increased mreBH expression. As a result, the reduced expression of mreBH in ΔrasP and ΔsigI strains likely contributes to the synthetic lethality with ΔponA.
Balance in the MreBH-LytE activity is essential for optimal elongasome function
We complemented the conditional essentiality of mreBH and lytE by ectopically expressing each of these genes individually as well as in combination in different mutant backgrounds. These strains were used to evaluate the relative importance of each gene upon inhibition of PBP1 by monitoring their CEF resistance. Although ectopic expression of mreBH complements the CEF sensitivity of ΔmreBH, it is unable to restore CEF resistance to the ΔmreBHΔlytE double mutant (Figure 5A). However, when both mreBH and lytE were ectopically expressed, the strain was significantly more CEF resistant than WT (Figure 5A). Similarly, induction of mreBH modestly increased CEF resistance of ΔrasP (Figure 5B), but not a ΔrasPΔlytE double mutant. Similar results were obtained in cells where pbpD, pbpF and pbpG were deleted (data not shown) indicating no indirect effect of MreBH on these aPBPs. In ΔsigI, however, mreBH expression alone had no significant impact on CEF resistance, perhaps due to reduced availability of LytE. Thus, increasing MreBH levels likely functions to increase elongasome activity by facilitating the localized action of LytE. Conversely, the Pspac(hy)lytE overexpression construct could not be introduced into the ΔrasP and ΔsigI mutants. We speculate that high LytE, in cells that have reduced expression of mreBH, leads to delocalized and unregulated autolysin activity. Collectively, these results further support a model in which a major role of MreBH is in directing LytE to sites of ongoing, elongasome-dependent PG synthesis.
Figure 5

MreBH and LytE function cooperatively to increase elongasome function.
(A) CEF (10 µg) sensitivity (disc diffusion assay) of the ΔmreBH, ΔlytE, and ΔmreBHΔlytE strains with and complementation by ectopic expression of genes from the leaky promoter, Pspac(hy), or (for the ΔmreBHΔlytE strain) expression of mreBH from a xylose inducible promoter (Pxyl) and lytE from the Pspac(hy). P-value cut-off of <0.0001 was used. (B) CEF sensitivity (as for panel A) for ΔrasP and ΔsigI mutants with ectopic expression of mreBH from Pspac(hy) in the presence and absence of lytE. P-value cut-off of <0.0001 was used. Cell length (C) and width (D) of WT, ΔponA, ΔrasP, ΔsigI, ΔmreBHΔlytE, and ΔmreBH and ΔlytE strains was determined using at least 100 cells for each strain. P-value cut-off of <0.005 was used.
-
Figure 5—source data 1
Data of zone of inhibition and cell size measurements.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig5-data1-v1.xlsx
The elongasome is critical for the maintenance of rod-shape, as judged by the spherical morphology of conditional mutants that are depleted for either the RodA transglycosylase or the two class B PBPs that provide transpeptidase activity (Boylan and Mendelson, 1969; Wei et al., 2003). The maintenance of rod shape is also affected by the balance between the directional motion of the elongasome and the random diffusive motion of PBP1 (Dion et al., 2019). Any imbalance in the activities of the two systems can lead to change in cell morphology. Overexpression of MreB or other elongasome proteins leads to cells that are longer and thinner, whereas overexpression of PBP1 leads to shorter and wider cells (Dion et al., 2019). Thus, we hypothesized that the effects of the σI regulatory system (acting through mreBH and lytE) on elongasome function would be revealed by monitoring cell morphology. We imaged WT, ΔrasP, ΔsigI, ΔmreBH, ΔlytE, ΔmreBHΔlytE and ΔponA cells and quantified the cell length and width using MicrobeJ (Ducret et al., 2016). Indeed, ΔrasP, ΔsigI and ΔmreBHΔlytE mutants were significantly shorter (Figure 5C) and wider (Figure 5D) compared to the WT, which indicates that these cells were primarily utilizing PBP1 for PG synthesis. ΔmreBH and ΔlytE mutants individually also had lower elongasome activity. In contrast, the ΔponA mutant formed significantly thinner cells due to PG synthesis being contributed mainly by the elongasome. These data support the conclusion that the rasP, sigI and mreBH-lytE genes all support elongasome function.
Suppressor analysis confirms the importance of mreBH and lytE in cells dependent on elongasome
Next, we took advantage of the ΔrasPΔponA synthetic lethality to isolate suppressors that grow on LB agar plates. Using whole-genome resequencing, we identified three strains with point mutations in walK (Ala241Asp, Ser385Leu, Asp274Ala). WalK is the sensor kinase of the essential two-component system WalKR, which regulates cell wall metabolism (Takada and Yoshikawa, 2018). WalR has binding sites upstream of sigI, mreBH and lytE and activates expression of these genes under heat stress (Huang et al., 2013). In addition to their regulation by σI, sigI and lytE also have σA-dependent promoters. WalR may function in conjunction with the σA holoenzyme, which together with σI controls lytE expression (Tseng et al., 2011). Taking into account the importance of WalKR in the expression of sigI, mreBH and lytE, we characterized one of the suppressor mutants of WalK, wherein aspartate 274 is changed to alanine (D274A).
Residue 274 lies in the cytoplasmic Per-Arnt-Sim (PAS) domain of WalK (Figure 6A). PAS domains have been linked to signal sensing (Taylor and Zhulin, 1999) and may be involved in protein dimerization (Huang et al., 1993). Recently, the cytoplasmic PAS domain of S. aureus WalK was found to bind zinc at a site including D274. Moreover, mutation in this binding site, which is highly conserved in WalK orthologs (Monk et al., 2019), led to increased kinase activity. We therefore hypothesized that the WalKD274A suppressor (denoted as WalK*) led to increased activity of the WalKR two-component system. We used CRISPR mutagenesis to introduce the walK* allele into WT cells and then confirmed that this allele suppressed the synthetic lethality of ΔrasPΔponA (Figure 6B).
Figure 6
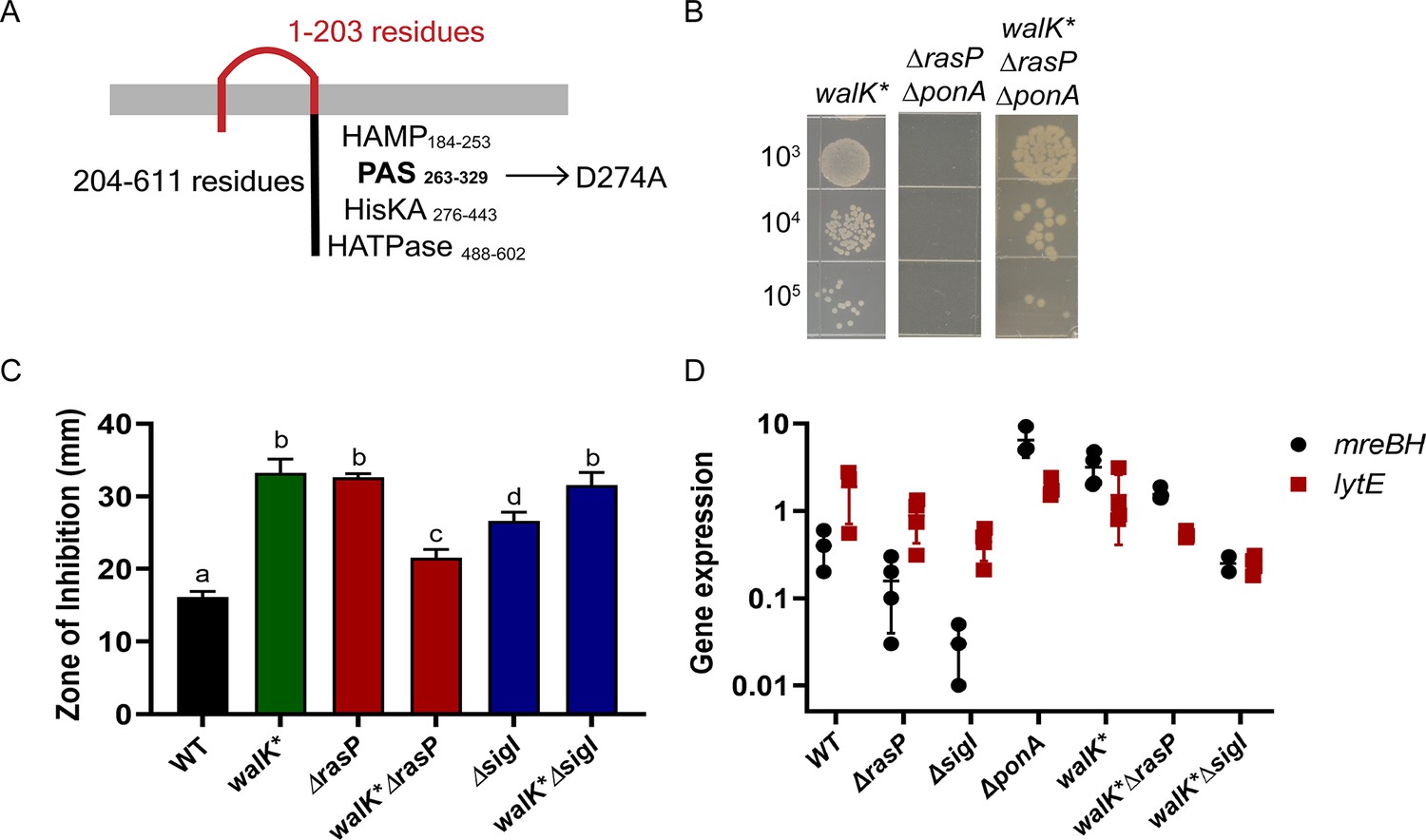
A walK* suppressor mutation elevates mreBH transcription.
(A) The D274 residue of WalK is part of a PAS-domain associated Zn-binding motif. (B) A walK* mutation rescues growth of the ΔrasPΔponA strain as monitored by a spot dilution assay. (C) CEF (10 µg) resistance (disc diffusion assay) of ΔrasP and ΔsigI and the respective double mutants of walK*ΔrasP and walK*sigI. A P-value cut-off of <0.0001 was used. (D) The effect of walK* on the expression profile of mreBH and lytE genes, alone and in combination with ΔrasP and ΔsigI. The gene expression values (2-Δct) were normalized with the house-keeping gene gyrA and then plotted on a log10 scale.
-
Figure 6—source data 1
Data of zone of inhibition and gene expression.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig6-data1-v1.xlsx
We next aimed to test the effect of WalK* on gene expression and cell wall homeostasis. The sigI and lytE genes can be expressed through their σA promoter after activation by WalR (Salzberg et al., 2013; Tseng et al., 2011). However, mreBH lacks an annotated σA promoter, implying that the expression of mreBH may rely on WalR activation of the σI holoenzyme. To test this hypothesis, we measured CEF sensitivity of walK*ΔrasP and walK*ΔsigI strains (Figure 6C). Although walK* increased CEF resistance of the ΔrasP strain, it could not rescue the ΔsigI strain. This supports the idea that WalR may act in conjunction with σI to activate transcription of mreBH, and thereby augment elongasome activity. Increased activation of WalK* can lead to increased expression of not only lytE, but also cwlO (Takada and Yoshikawa, 2018). This could lead to elevated autolysin levels that might account for the higher CEF sensitivity of walK* alone compared to WT.
We further quantified the mRNA levels of mreBH and lytE in the walK* strain and in the walK*ΔrasP and walK*ΔsigI strains (Figure 6D). The walK* allele led to increased expression of both mreBH and lytE. Moreover, these levels were similar to that observed in the ΔponA background, suggesting that deletion of ponA leads to a compensatory increase in mreBH and lytE mediated by the WalKR. However, they were lower for the walK*ΔsigI strain. These data suggest that walK* leads to increased activation of WalR, which then leads to increased transcription of sigI and thereby of mreBH and lytE. This ultimately leads to the survival of the ΔrasPΔponA strain. These data also validate the importance of RasP and σI in the regulation of MreBH and LytE and their significant impact on elongasome activity, especially in the ΔponA background.
Additive role of σI and σM in regulating the elongasome activity
While our results suggest a critical role for σI in aPBP-elongasome homeostasis through its regulation of MreBH and LytE, previous studies have indicated that the extracytoplasmic (ECF) sigma factor σM also plays a significant role in B. subtilis cell wall homeostasis. σM regulates the expression of rodA, mreB, mreC and mreD (core components of the elongasome), as well as ponA and other genes involved in PG synthesis (Eiamphungporn and Helmann, 2008; Luo and Helmann, 2012). To determine the relative contribution of σM to cell survival during aPBP inhibition, we used PM* mutations that selectively inactivate σM-dependent promoters of genes encoding elongasome components. We constructed the PM*rodA and PM*ponA strains that are unable to upregulate rodA and ponA, respectively, and a PM*maf strain that cannot upregulate the mreBCD genes located downstream of the intragenic PM inside maf (Eiamphungporn and Helmann, 2008). We also constructed the double mutant PM*rodA PM*maf strain. The CEF sensitivity of PM*rodA and PM*rodA-PM*maf was similar to that of the sigM mutant (Figure 7A). Neither PM*maf nor PM*ponA were CEF sensitive. Thus, under conditions where CEF has inhibited PBP1, σM helps restore peptidoglycan synthesis primarily by increasing the expression of rodA to increase elongasome activity. In contrast, the double mutants of ΔecsAΔsigM, ΔrasPΔsigM and ΔsigIΔsigM revealed an additive effect with respect to CEF sensitivity (Figure 7B). Thus, the role of the elongasome in PG synthesis can be regulated through two-independent pathways: the EcsAB-RasP-σI pathway acts by regulating MreBH and LytE, and the σM pathway acts through RodA.
Figure 7
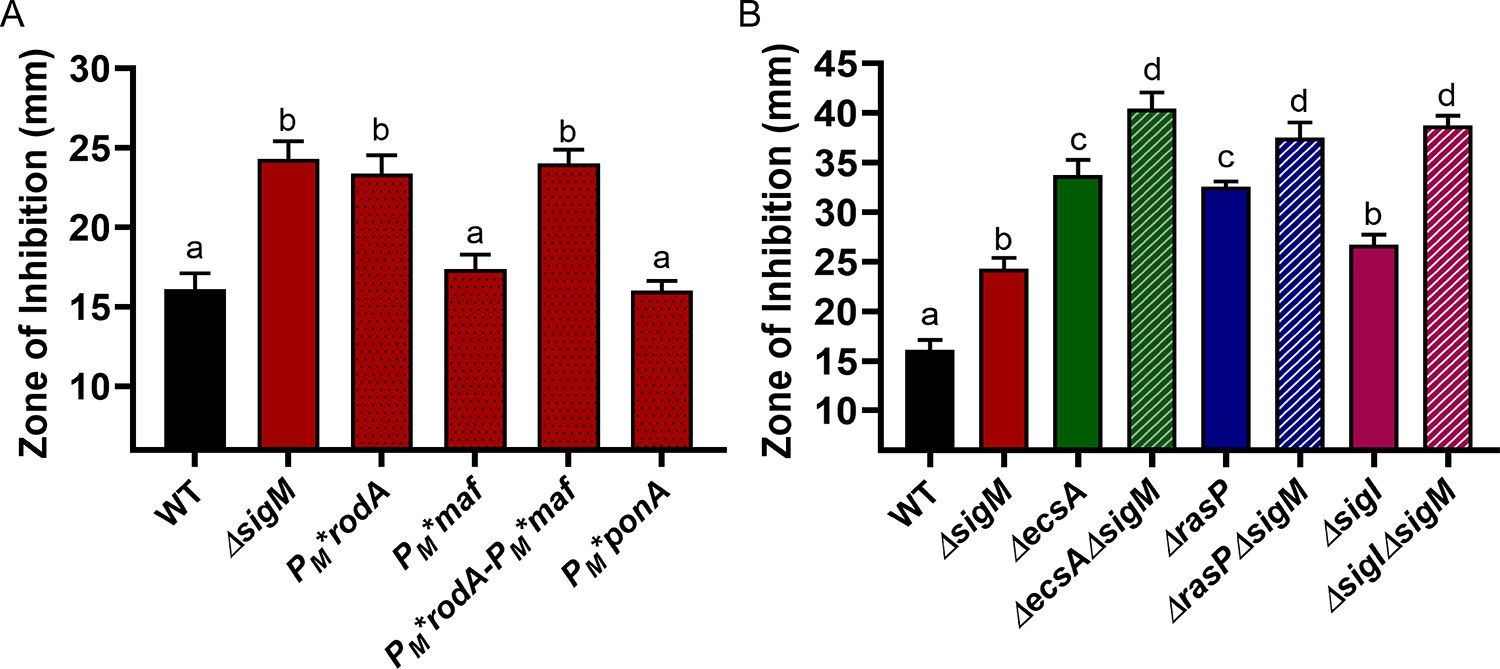
σM contributes additively with σI to CEF resistance by increasing expression of rodA.
CEF (10 µg) sensitivity (disc diffusion assay) for (A) WT, ΔsigM and promoter mutants of PM*rodA, PM*maf (which controls expression of mreBCD), PM*rodA-PM*maf and PM*ponA and (B) WT and ΔsigM mutants, alone and in combination with ΔecsA, ΔrasP and ΔsigI. P-value cut-off of <0.0001 was used for both the graphs.
-
Figure 7—source data 1
Data of zone of inhibition.
- https://cdn.elifesciences.org/articles/57902/elife-57902-fig7-data1-v1.xlsx
Discussion
Peptidoglycan (PG) is a defining feature of bacteria. This cellular enclosure must provide stability, yet at the same time be highly dynamic and adaptable. During growth, PG is continuously remodeled, which involves the action of autolysins, hydrolytic enzymes that cleave links within and between the glycan strands (Vollmer et al., 2008; Egan et al., 2020). These hydrolases are essential for the insertion of new glycan strands into the existing structure (Hashimoto et al., 2012; Singh et al., 2012). Cell shape maintenance requires that the sites of new PG synthesis be spatially regulated, often in response to the activity of cytoskeletal filaments such as the MreB (Domínguez-Escobar et al., 2011) and FtsZ proteins (Mahone and Goley, 2020).
B. subtilis, a genetically tractable model organism, has provided an important system for investigating the pathways of PG synthesis in rod-shaped, Gram positive bacteria. During cell elongation, a multiprotein complex designated the elongasome is the primary biosynthetic machine for inserting new glycan strands. In B. subtilis, there are three MreB paralogs (MreB, Mbl and MreBH), which colocalize to form elongasome-associated cytoskeletal filaments along the cell periphery (Carballido-López et al., 2006; Garner et al., 2011). Cells lacking all three paralogs lose their rod shape and become spheres which ultimately lyse (Kawai et al., 2009). Whereas MreB and Mbl are critical for the circumferential motion of the elongasome, the role of MreBH is less clear, and seems related to its ability to recruit LytE (Carballido-López et al., 2006). PG synthesis by the elongasome relies on the activity of RodA as TG, with bPBPs providing TP activity (Figure 8A). A separate complex, the divisome, builds the cross-walls prior to cell separation (Mahone and Goley, 2020).
Figure 8
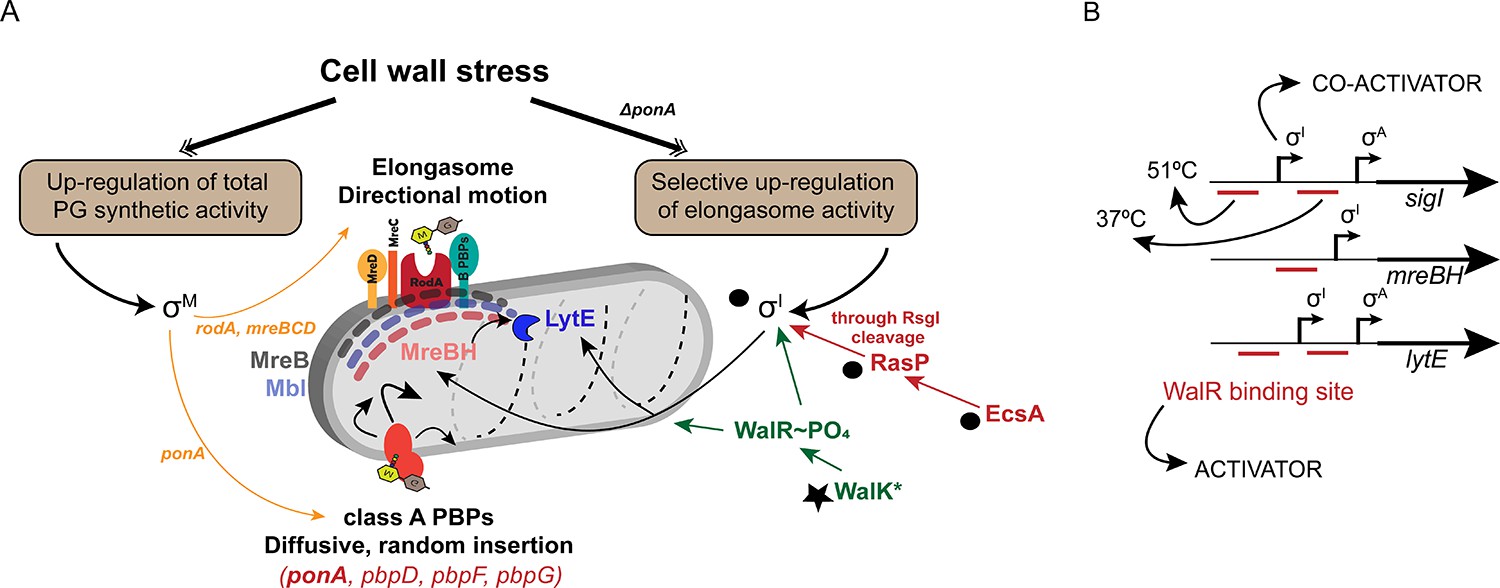
σI co-ordinates with WalKR to regulate elongasome function, and complements the σM dependent stress response.
(A) PG synthesis potential is dictated by the activity of the elongasome in collaboration with aPBPs. Cell wall stress activates σM (left), which up-regulates both pathways. In the absence of aPBPs, cells up-regulate elongasome activity through σI, which increases expression of genes (mreBH and lytE) important for elongasome function. Synthetic lethal relationships are shown here between deletion of ponA and genes in the σI pathway (black circles). Bypass of synthetic lethality can be compensated by a gain of function mutation in walK (star). (B) The promoter regions of sigI, mreBH and lytE are shown, depicting the binding sites of WalR and σI as annotated before (Huang et al., 2013). σI and WalR act as activators for the expression of sigI and lytE from the σA promoter. The downstream WalR binding site is important for expression of sigI and lytE at 37°C whereas the upstream binding site is crucial for the heat induction of these genes at 51°C.
Because of its unique chemical composition, PG synthesis requires numerous highly conserved enzymes, which thereby present attractive targets for antibiotics (Bugg et al., 2011). Inhibitors of PG synthesis may result in spheroplast formation, cell lysis, or morphological defects, depending on the antibiotic target and the organism (Cross et al., 2019; Emami et al., 2017). Many of our most familiar antibiotics are natural products of soil bacteria, including Bacillus spp. (Kaspar et al., 2019; Stein, 2005) and many actinobacteria (Mahajan, 2012). Like other soil bacteria, B. subtilis has substantial intrinsic resistance to many antibiotics (Kingston et al., 2013; Radeck et al., 2017a; Helmann, 2016). We have explored these intrinsic resistance mechanisms by analysis of cell envelope stress responses, including those controlled by alternative sigma factors (Helmann, 2016). For example, σV is induced by and provides resistance to lysozyme by covalently modifying PG (Guariglia-Oropeza and Helmann, 2011), whereas σW is induced by and provides resistance to membrane-active bacteriocins (Butcher and Helmann, 2006; Kingston et al., 2011).
The σM response is selectively induced by stresses during PG synthesis and contributes to resistance to a wide-variety of PG synthesis inhibitors, including MOE, CEF, and bacitracin (Helmann, 2016; Mascher et al., 2007). The σM regulon serves to both upregulate PG synthetic capacity, and to compensate for stresses resulting from PG inhibition. The former includes the up-regulation of elongasome components (Figure 8A) and PG biosynthetic enzymes (PBP1, Ddl, MurB, MurF, BcrC, Amj) (Eiamphungporn and Helmann, 2008). The latter includes the large regulon controlled by the Spx transcription factor that protects cells against antibiotic-associated oxidative stress (Rojas-Tapias and Helmann, 2018). Finally, it has recently been shown that induction of a σM-regulated ppGpp synthase, YwaC, increases the number of persister cells following antibiotic exposure (Fung et al., 2020).
Here, we identify a major role for another alternative sigma factor, σI, in conferring intrinsic resistance to important cell wall antibiotics, MOE and CEF. Induction of σI, which requires the EcsAB-RasP regulatory pathway (Liu et al., 2017), selectively elevates elongasome function by increasing the expression of the MreB paralog, MreBH, and the associated autolytic endopeptidase LytE (Carballido-López et al., 2006). This stress response is critical in cells lacking PBP1, as judged by the synthetic lethality of ΔsigI ΔponA mutants (Figure 3B). This stress response functions in coordination with both the σM stress response (Figure 7A), which increases elongasome function by upregulation of the RodA TG (Meeske et al., 2016; Emami et al., 2017), and the essential WalKR two-component system (Figures 6 and 8). Although σI was previously linked to heat-stress (Zuber et al., 2001), virulence in B. anthracis (Kim and Wilson, 2016), and control of autolysin synthesis (Salzberg et al., 2013), our results reveal new insights into its role in cell envelope stress.
This study also highlights the complex regulation of the mreBH and lytE genes. WalR, σI and σA binding sites have been previously annotated in the promoters of sigI, mreBH and lytE (Figure 8B). The WalK (D274A) gain of function mutant suppresses the lethal phenotype of ΔrasPΔponA by induction of mreBH and lytE (Figure 6). However, induction was not significant in the σI mutant. We conclude that co-activation by WalR and σI is required for induction of these two genes. The signals sensed by WalK are unclear, but it was recently suggested that peptidoglycan cleavage products generated by LytE and CwlO can be sensed by WalK to balance the activity of these proteins (Dobihal et al., 2019). Moreover, it was previously observed that sigI activation enhances the growth of mbl mutants (Schirner and Errington, 2009), which we suggest was likely due to increasing elongasome activity through mreBH and lytE.
Collectively, our results reveal that WalKR and σI act in coordination to maintain optimal elongasome activity, and these pathways complement the general PG stress response activated by σM (Figure 8). One general theme that has emerged is that PG synthesis involves multiple, functionally overlapping systems, often with one being inducible by antibiotic inhibition of the other. For example, the inducible UPP phosphatase BcrC complements the activity of UppP (Radeck et al., 2017b; Zhao et al., 2016), and the σM-regulated Amj functions as a second lipid II flippase that is critical when MurJ is inhibited (Chamakura et al., 2017; Meeske et al., 2015). Similarly, inhibition of aPBPs by MOE leads to an essential, compensatory induction of RodA (Meeske et al., 2016; Emami et al., 2017). Here, it is shown that this single σM-regulated target gene can largely account for the CEF sensitivity of sigM mutants (Figure 7). This increase in RodA, together with the induction of MreBH and LytE, serves to boost the biosynthetic potential of the elongasome. These results reveal mechanisms that allow diverse PG biosynthetic complexes to coordinate their activities, in both time and space. The highly orchestrated processes that direct and coordinate PG synthesis are important both for intrinsic antibiotic resistance, as explored here and are ultimately responsible for the enormous diversity of bacterial morphologies (Caccamo and Brun, 2018).
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Bacillus subtilis, strain 168) | WT | Lab stock | B. subtilis 168 | (see Materials and methods) |
| Recombinant DNA reagent | This study | E. coli with pMarA1 | (see Materials and methods) | |
| Recombinant DNA reagent | HB20725 | This study | 168 pMarA1 | (see Materials and methods) |
| Recombinant DNA reagent | HB20738 | This study | pbpDFG null; ponA::erm;pMarA | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Δ4 Class A PBP | This study | ponA::erm; pbpDFG::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ponA::erm Pspank*-ponA | This study | ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | pbpDFG ponA::erm Pspank*-ponA | This study | pbpDFG::null; ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ecsA ponA::erm Pspank*-ponA | This study | ecsA::null; ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | pbpDFG ecsA-ponA::erm Pspank*-ponA | This study | ecsA::null;pbpDFG::null; ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ytxG ponA::erm Pspank*-ponA | This study | ytxG::null; ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | pbpDFG ytxG ponA::erm Pspank*-ponA | This study | ytxG::null;pbpDFG::null;ycgO::Pspank*-ponA; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsA | This study | ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasP | This study | rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔponA | This study | ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔponA | This study | ecsA::null;ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔponA | This study | rasP::null;ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔrasP | This study | ecsA::null;rasP::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsA Pspac(hy)-ecsA | This study | amyE::Pspac(hy)-ecsA; ecsA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsA Pspac(hy)-ecsAecsB | This study | amyE::Pspac(hy)-ecsAB; ecsA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasP Pspac(hy)-rasP | This study | amyE::Pspac(hy)-rasP; rasP::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigW | This study | sigW::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigV | This study | sigV::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigI | This study | sigI::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Δ25ftsL | This study | Made using CRISPR to remove the 2-26th AAs of FtsL so it is no longer a target of RasP | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigVΔsigW Δ25ftsLΔsigI | This study | sigV::null;sigW::null; Δ25ftsL;sigI::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigIΔsigW | This study | sigI::null;sigW::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigVΔsigW Δ25ftsL | This study | sigV::null;sigW::null; Δ25ftsL | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigIΔponA Pspac(hy)-sigI | This study | sigI::null; amyE::Pspac(hy)-sigI; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔsigI | This study | sigI::null;ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔsigW | This study | sigW::null;ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔsigI | This study | sigI::null;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔsigW | This study | sigW::null;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrsgI | This study | rsgI::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrsiW | This study | rsiW::mls | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔrsgI | This study | rsgI::null;ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔrsiW | This study | rsiW::mls;ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔrsgI | This study | rsgI::null;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔrsiW | This study | rsiW::mls;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigM | This study | sigM::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔecsAΔsigM | This study | sigM::null;ecsA::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔsigM | This study | sigM::null;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigIΔsigM | This study | sigM::null;sigI::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Pm*rodA | Zhao et al., 2019 | WT 168 transformed with CRISPR plasmid to remove Pm of rodA | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Pm* maf | Zhao et al., 2019 | WT 168 transformed wth pMUTIN to introduce maf-Pm*(TGTT) | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Pm*rodA Pm*murG | This study | Pm*murG transformed with CRISPR plasmid to remove Pm of ProdA | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | Pm*ponA | This study | WT168 transformed with CRISPR plasmid to remove Pm of ponA | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBH | This study | mreBH::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔlytE | This study | lytE::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔgsiB | This study | gsiB::spec | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔfabI | This study | fabI::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔbcrC | This study | bcrC::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBHΔlytE | This study | mreBH::null;lytE::null | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBHΔponA | This study | mreBH::null;ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔlytEΔponA | This study | lytE::null;ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBHΔlytE ΔponA | This study | mreBH::null;lytE::null; ponA::erm | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBH Pspac(hy)-mreBH | This study | mreBH::null; amyE::Pspac(hy)-mreBH | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔlytE Pspac(hy)-lytE | This study | lytE::null; amyE::Pspac(hy)-lytE | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBHΔlytE Pxyl-mreBH | This study | mreBH::null;lytE::null; lacA::Pxyl-mreBH | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔmreBHΔlytE Pxyl-mreBH Pspac(hy)-lytE | This study | lytE::null; amyE::Pspac(hy)-lytE; lacA::Pxyl-mreBH; mreBH::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔmreBH Pspac(hy)-mreBH | This study | mreBH::null; amyE::Pspac(hy)-mreBH; rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔrasPΔmreBH ΔlytE Pspac(hy)-mreBH | This study | mreBH::null;lytE::null; amyE::Pspac(hy)-mreBH; rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigIΔmreBH Pspac(hy)-mreBH | This study | mreBH::null; amyE::Pspac(hy)-mreBH; sigI::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | ΔsigIΔmreBHΔlytE Pspac(hy)-mreBH | This study | mreBH::null;lytE::null; amyE::Pspac(hy)-mreBH; sigI::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | walK* | This study | WalKD274A, constructed using CRISPR | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | walK*ΔrasP | This study | WalKD274A;rasP::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | walK*ΔsigI | This study | WalKD274A;sigI::kan | (see Materials and methods) |
| Strain, strain background (Bacillus subtilis, strain 168) | walK*ΔrasPΔponA | This study | WalKD274A;rasP::kan; ponA::erm | (see Materials and methods) |
| Recombinant DNA reagent | pMarA | Le Breton et al., 2006 | a plasmid harboring the mariner-Himar1 transposase | |
| Recombinant DNA reagent | pMarA1 | Modified pMarA to introduce MmeI sites | ||
| Recombinant DNA reagent | pDR244 | BGSC (ECE274) | To remove the kan/erm cassette from BKE strains | |
| Recombinant DNA reagent | pAM012 | Meeske et al., 2015 | For Pspank*-ponA constructs | |
| Recombinant DNA reagent | pPL82 | For Pspac(hy) constructs at amyE locus | ||
| Recombinant DNA reagent | pBS2EXylRPxylA | BGSC (ECE741) | For Pxyl constructs at lacA locus |
Bacterial strains, plasmids and growth conditions
Request a detailed protocolAll stains were grown in lysogeny broth (LB) medium at 37°C. Liquid cultures were aerated on an orbital shaker at 300 rpm. Glycerol stocks were streaked on LB agar plates and incubated overnight at 37°C. Conditionally synthetic lethal strains were grown in LB medium with 20 mM MgSO4.
Bacterial strains used in this study have been listed in the Key Resources Table. For all deletion mutants, primary strains were ordered from the BKK/BKE collection available at the Bacillus Genetic Stock Centre (BGSC) (Koo et al., 2017). These gene deletions with the antibiotic cassette (kanamycin or erythromycin) were then transformed into our WT 168 strain using natural competence induced in modified competence (MC) medium. rasP, ecsA and ponA deletion strains had very low natural competence. Thus, other mutations were introduced using SPP1 phage transduction as described (Kearns et al., 2005). The null mutants were constructed using pDR244, which removes the resistance cassette leading to clean in-frame deletions (Koo et al., 2017). The resulting gene deletions (designated Δ) were confirmed with check primers listed in Supplementary file 1.
Genes were ectopically expressed at amyE under promoter Pspac(hy) using pPL82 plasmid (Quisel et al., 2001). MreBH was also expressed at the lacA locus under xylose inducible promoter Pxyl using plasmid pECE741 (Popp et al., 2017). The respective genes were amplified from genomic DNA using primers listed in Supplementary file 1. The required restriction enzyme sites (and if required a ribosome binding site (RBS)) were incorporated in the primers used for gene amplification. CRISPR-Cas9 mutagenesis was carried out using pJOE8999 plasmid as described before (Altenbuchner, 2016). The primers used to construct the repair fragment and guide RNAs are in Supplementary file 1. The whole sequence of the genes was confirmed by Sanger sequencing (Biotechnology Resources core facility at Cornell University).
Transposon mutagenesis
Request a detailed protocolThe transposon-sequencing (Tn-Seq) was performed using modified pMarA (Le Breton et al., 2006). pMarA is a plasmid harboring the mariner-Himar1 transposase gene and a temperature-sensitive replicon to select for transposition events. Two MmeI sites were introduced flanking the BstXI and PstI sites to generate plasmid pMarA1 (HE8334). The plasmid was transformed into WT Bacillus subtilis and ΔpbpDFG ponA::erm mutant at 28°C selecting for KanR on LB plates supplemented with 10 mM MgSO4 (final concentration) to generate strain HB20725 and HB20738, respectively. Liquid cultures of HB20725 and HB20738 harboring plasmid-borne transposons were grown at 28°C in liquid LB medium with 10 mM MgSO4 to mid-exponential phase (OD600 ~0.4), diluted and spread on LB plates containing kanamycin and 10 mM MgSO4. Plates were incubated overnight at 48°C to select for transposition events, and the ones with distinct single colonies (not too crowded, and about 500 colonies per plate) were pooled together. Two hundred and forty plates with a total of >100,000 independent colonies were pooled together for each strain, and their genomic DNA was isolated. For each strain, 10 µg of genomic DNA was digested using MmeI, purified and ligated with sequencing adaptors. Illumina sequencing was performed and DNA adjacent to the transposon insertion sites were matched to Bacillus subtilis reference genome NC_000964.3 using CLC workbench version 8.5.1. Matching results were visualized using CLC workbench, and quantified using Tn-seq Explorer software (Solaimanpour et al., 2015). For visualization of transposon insertions, IGV genome browser was used (Robinson et al., 2011).
Plating efficiency
Request a detailed protocolFor plating efficiency (spot dilution) assays, the cultures were grown in LB medium with 20 mM MgSO4 to ~0.4 OD600. 1 mL of culture was centrifuged at 5000 rpm for 5 min and resuspended in LB medium (without MgSO4). 10-fold serial dilutions were done in LB medium and 10 µL was plated/spotted on LB agar plates, allowed to air-dry for 10–15 min, and incubated overnight at 37°C.
Growth kinetics and MIC determinations
Request a detailed protocolCultures were grown in LB medium to ~0.4 OD600. 1 µL of this culture was inoculated in each well containing 200 µL of LB media with the required drug concentration. Honeycomb 100-well plates were used for the assay. The increase in the OD600 of the culture was monitored real-time using Bioscreen C growth curve analyzer (Growth curves USA). Readings were taken at every 15 min interval up to 24 hr under constant shaking conditions at 37°C. For MIC determination, two-fold increase in the drug concentration was screened ranging from (0.2 to 1.6 µg/mL). The minimum concentration which inhibited the growth (less than 0.2 OD600) up to at least 10 hr of incubation was considered as the MIC for the strain.
Disc diffusion assays
Request a detailed protocolAntibiotic sensitivity was screened by determining the zone of inhibition using a disc diffusion assay. Cultures were allowed the grow up to ~0.4 OD600. 100 µL of this culture was added to 4 mL of top agar (0.75% agar) kept at 50°C to prevent it from solidifying. This was poured on to 15 mL LB agar plates (1.5% agar). The top agar was allowed to air-dry for 30 min. A Whatmann paper filter disc of 6 mm was then put on the top agar. The required amount of drug was added on the disc immediately. The plates were incubated overnight at 37°C and the diameter of the clear zone of inhibition was measured. For all histograms, the zone of inhibition (Y-axis) starts from 6 mm which is the disc diameter. For strains having the inducible promoter Pxyl, both the top agar and LB agar plates were made with 0.1% xylose.
Autolytic potential
Request a detailed protocol200 µL of cells (~0.4 OD600) were added in each well of a 100-well honeycomb plate. To this, 0.05 M of sodium azide (from 5 M stock) was added. Immediately, the real-time monitoring of the decrease in OD600 was started with Bioscreen C. Readings were taken every 15 min for up to 24 hr. The time at which 50% of the cells had lysed was noted for each mutant. The time taken (in hours) was plotted as lysis time for each strain. Sodium azide stock was prepared fresh before every experiment.
Real-time PCR
Request a detailed protocolGene expression for mreBH and lytE was determined by real-time PCR using primers in Table S2. RNA was purified from 1.5 mL of ~0.4 OD600 cells using the RNeasy Kit from Qiagen as per the manufacturer’s instructions. 2 µg of RNA was used to prepare 20 µL of cDNA to achieve a final concentration of 100 ng/µL using High capacity cDNA reverse transcription kit from Applied Biosystems. The gene expression levels were measured using 100 ng of cDNA using 0.5 µM of gene specific primers and 1X SYBR green (Bio-Rad) in CFX connect real-time system from Bio-Rad. gyrA was used a house-keeping gene. Gene expression values (2-Δct) were plotted after normalization with gyrA.
Cell size measurements
Request a detailed protocolA very thin agar pad was prepared on slides from 0.8% agarose. 10 µL of cells (~0.4 OD600) were spotted and allowed to air dry for 10 min before putting on a cover slip. Cells were imaged using Olympus BX61. Images were captured using Cooke Sensicam camera system under 100X magnification with immersion oil. The images were then analyzed for their length and width using MicrobeJ (Ducret et al., 2016), a plugin for imageJ (Schneider et al., 2012).
Suppressor analysis
Request a detailed protocolSpontaneous suppressors were picked from LB agar plates for ΔecsAΔponA and ΔrasPΔponA. 12 suppressors were selected from each background and their chromosomal DNA extracted using Qiagen DNA extraction kit. DNA was sequenced using the Illumina platform at the Biotechnology Resources core facility at Cornell University. The results were trimmed, mapped and aligned with the ΔecsAΔponA and ΔrasPΔponA backgrounds using CLC genomics workbench.
Statistical analysis
Request a detailed protocolAll the experiments were performed with a minimum of 3 biological replicates. For microscopy images, at least 100 cells per strain were quantified for their cell length and width. One-way ANOVA was used to calculate the statistical significance. Tukey’s comparison test was used to determine significance between all the strains. P-value cut-offs have been mentioned in the figure legends. Different letters represent data which are significantly different. Same letter represents mean values which are not statistically different. Significance between two strains was determined using student’s t-test.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Editing of the Bacillus subtilis genome by the CRISPR-Cas9 systemApplied and Environmental Microbiology 82:5421–5427.https://doi.org/10.1128/AEM.01453-16
-
Cytoplasmic steps of peptidoglycan biosynthesisFEMS Microbiology Reviews 32:168–207.https://doi.org/10.1111/j.1574-6976.2008.00104.x
-
BcrC from Bacillus subtilis Acts as an Undecaprenyl Pyrophosphate Phosphatase in Bacitracin ResistanceJournal of Biological Chemistry 280:28852–28857.https://doi.org/10.1074/jbc.M413750200
-
Initial characterization of a temperature-sensitive rod--mutant of Bacillus subtilisJournal of Bacteriology 100:1316–1321.https://doi.org/10.1128/JB.100.3.1316-1321.1969
-
Bacterial cell wall assembly: still an attractive antibacterial targetTrends in Biotechnology 29:167–173.https://doi.org/10.1016/j.tibtech.2010.12.006
-
The molecular basis of noncanonical bacterial morphologyTrends in Microbiology 26:191–208.https://doi.org/10.1016/j.tim.2017.09.012
-
A viral protein antibiotic inhibits lipid II flippase activityNature Microbiology 2:1480–1484.https://doi.org/10.1038/s41564-017-0023-4
-
Targeting the bacterial transglycosylase: antibiotic development from a structural perspectiveACS Infectious Diseases 5:1493–1504.https://doi.org/10.1021/acsinfecdis.9b00118
-
Cell wall deficiency as a coping strategy for stressTrends in Microbiology 27:1025–1033.https://doi.org/10.1016/j.tim.2019.07.008
-
Spheroplast-Mediated carbapenem tolerance in Gram-Negative pathogensAntimicrobial Agents and Chemotherapy 63:19.https://doi.org/10.1128/AAC.00756-19
-
A fly on the wall: how stress response systems can sense and respond to damage to peptidoglycanFrontiers in Cellular and Infection Microbiology 9:380.https://doi.org/10.3389/fcimb.2019.00380
-
Uncovering the activities, biological roles, and regulation of bacterial cell wall hydrolases and tailoring enzymesJournal of Biological Chemistry 295:3347–3361.https://doi.org/10.1074/jbc.REV119.010155
-
Regulation of peptidoglycan synthesis and remodellingNature Reviews Microbiology 18:446–460.https://doi.org/10.1038/s41579-020-0366-3
-
The fractional inhibitory concentration (FIC) index as a measure of synergyJournal of Antimicrobial Chemotherapy 11:427–433.https://doi.org/10.1093/jac/11.5.427
-
The enoyl-[acyl-carrier-protein] reductases FabI and FabL from Bacillus subtilisJournal of Biological Chemistry 275:40128–40133.https://doi.org/10.1074/jbc.M005611200
-
Bacillus subtilis extracytoplasmic function (ECF) sigma factors and defense of the cell envelopeCurrent Opinion in Microbiology 30:122–132.https://doi.org/10.1016/j.mib.2016.02.002
-
Mechanisms of drug combinations: interaction and network perspectivesNature Reviews Drug Discovery 8:111–128.https://doi.org/10.1038/nrd2683
-
Bioactive secondary metabolites from Bacillus subtilis: A Comprehensive ReviewJournal of Natural Products 82:2038–2053.https://doi.org/10.1021/acs.jnatprod.9b00110
-
A master regulator for biofilm formation by Bacillus subtilisMolecular Microbiology 55:739–749.https://doi.org/10.1111/j.1365-2958.2004.04440.x
-
In vivo random mutagenesis of Bacillus subtilis by use of TnYLB-1, a mariner-based transposonApplied and Environmental Microbiology 72:327–333.https://doi.org/10.1128/AEM.72.1.327-333.2006
-
Antibacterial agents from actinomycetes - A reviewFrontiers in Bioscience E4:240–253.https://doi.org/10.2741/e373
-
Bacterial cell division at a glanceJournal of Cell Science 133:jcs237057.https://doi.org/10.1242/jcs.237057
-
Regulatory overlap and functional redundancy among Bacillus subtilis extracytoplasmic function sigma factorsJournal of Bacteriology 189:6919–6927.https://doi.org/10.1128/JB.00904-07
-
Peptidoglycan synthesis in the absence of class A penicillin-binding proteins in Bacillus subtilisJournal of Bacteriology 185:1423–1431.https://doi.org/10.1128/JB.185.4.1423-1431.2003
-
SubtiWiki 2.0--an integrated database for the model organism Bacillus subtilisNucleic Acids Research 44:D654–D662.https://doi.org/10.1093/nar/gkv1006
-
Synergy, antagonism, and what the chequerboard puts between themJournal of Antimicrobial Chemotherapy 52:1.https://doi.org/10.1093/jac/dkg301
-
In vivo effects of sporulation kinases on mutant Spo0A proteins in Bacillus subtilisJournal of Bacteriology 183:6573–6578.https://doi.org/10.1128/JB.183.22.6573-6578.2001
-
Functional redundancy of division specific penicillin-binding proteins in Bacillus subtilisMolecular Microbiology 106:304–318.https://doi.org/10.1111/mmi.13765
-
The cell wall regulator {sigma}I specifically suppresses the lethal phenotype of mbl mutants in Bacillus subtilisJournal of Bacteriology 191:1404–1413.https://doi.org/10.1128/JB.01497-08
-
NIH image to ImageJ: 25 years of image analysisNature Methods 9:671–675.https://doi.org/10.1038/nmeth.2089
-
Bacillus subtilis antibiotics: structures, syntheses and specific functionsMolecular Microbiology 56:845–857.https://doi.org/10.1111/j.1365-2958.2005.04587.x
-
Essentiality and function of WalK/WalR two-component system: the past, present, and future of researchBioscience, Biotechnology, and Biochemistry 82:741–751.https://doi.org/10.1080/09168451.2018.1444466
-
PAS domains: internal sensors of oxygen, redox potential, and lightMicrobiology and Molecular Biology Reviews 63:479–506.https://doi.org/10.1128/MMBR.63.2.479-506.1999
-
From the regulation of peptidoglycan synthesis to bacterial growth and morphologyNature Reviews Microbiology 10:123–136.https://doi.org/10.1038/nrmicro2677
-
Lipid intermediates in the biosynthesis of bacterial peptidoglycanMicrobiology and Molecular Biology Reviews 71:620–635.https://doi.org/10.1128/MMBR.00016-07
-
Bacterial peptidoglycan (murein) hydrolasesFEMS Microbiology Reviews 32:259–286.https://doi.org/10.1111/j.1574-6976.2007.00099.x
-
Mode of action and in-vitro activity of vancomycinJournal of Antimicrobial Chemotherapy 14:7–18.https://doi.org/10.1093/jac/14.suppl_D.7
-
Linear, uncross-linked peptidoglycan secreted by penicillin-treated Bacillus subtilis Isolation and characterization as a substrate for penicillin-sensitive D-alanine carboxypeptidasesJournal of Biological Chemistry 255:11577–11587.
-
Depletion of undecaprenyl pyrophosphate phosphatases disrupts cell envelope biogenesis in Bacillus subtilisJournal of Bacteriology 198:2925–2935.https://doi.org/10.1128/JB.00507-16
-
Don't let sleeping dogmas lie: new views of peptidoglycan synthesis and its regulationMolecular Microbiology 106:847–860.https://doi.org/10.1111/mmi.13853
-
Putative sigma factor SigI (YkoZ) of Bacillus subtilis is induced by heat shockJournal of Bacteriology 183:1472–1475.https://doi.org/10.1128/JB.183.4.1472-1475.2001
Article and author information
Author details
Funding
National Institutes of Health (R35GM122461)
- John D Helmann
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Research reported in this publication was supported by the National Institutes of Health under award number R35GM122461 to JDH. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
We thank Ahmed Gaballa, Gumpanat Mahipant, Daniel Roistacher, Anna Weaver, Ivano Pezzotta, Jessica Willdigg, Chloe Murrell and Annette Choi for their contributions to the early stages of this project. We also thank Alex Meeske and David Rudner for plasmid pAM012 that contains the Pspank* promoter.
Copyright
© 2020, Patel et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,834
- views
-
- 250
- downloads
-
- 47
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 47
- citations for umbrella DOI https://doi.org/10.7554/eLife.57902
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A regulatory pathway that selectively up-regulates elongasome function in the absence of class A PBPs
eLife 9:e57902.
https://doi.org/10.7554/eLife.57902




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}