Identifying molecular features that are associated with biological function of intrinsically disordered protein regions
- Department of Cell and Systems Biology, University of Toronto, Canada
- Program in Molecular Medicine, Hospital for Sick Children, Canada
- Department of Biochemistry, University of Toronto, Canada
Figures
Figure 1 with 1 supplement

Schematic of the FAIDR statistical model.
Example inputs (function, molecular features) and outputs (FAIDR probability, association with function) are shown. Given a set of features (e.g., molecular features comprising evolutionary signatures, as shown) for one or more IDRs in a given protein and functional annotation information (e.g., whether or not the protein is annotated with mRNA binding function or nucleolus function, as shown), FAIDR outputs the probability that the IDR is associated with the given function as well as the strength and direction of the association with function for each molecular feature. Molecular features comprising the evolutionary signatures are represented by a heatmap scaling from blue (decreased mean or variance of the feature compared to our null expectation of IDR evolution) to yellow (increased mean or variance of the feature compared to our null expectation of IDR evolution). The association with function is represented by a heatmap scaling from blue (negative association) to yellow (positive association). For example, for nucleolus function (as shown), mean CKII consensus sites are strongly positively associated with this function (indicated with yellow), whereas for mRNA binding (also shown), mean threonine content is negatively associated with this function (indicated with blue).
Figure 1—figure supplement 1
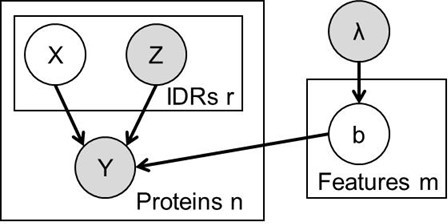
The FAIDR probabilistic model.
Unfilled circles represent hidden variables, and gray circles represent known variables. Functions or phenotypes are treated as binary variables, where Y = 1 if the protein is associated with that function or phenotype and Y = 0 if the protein is not. For the ith protein, we have a set of features, Zi for each of ri disordered regions. We use a hidden variable, Xi, to represent the IDR that is responsible for the function, so that if Xij = 1, the jth IDR in the ith protein is the one responsible for the function. Throughout we used an exponential prior, λ = 0.2 on the coefficients, b (equivalent to a constant L1 penalty) that we found to limit overfitting for our functions with small numbers of positives.
Figure 2 with 1 supplement
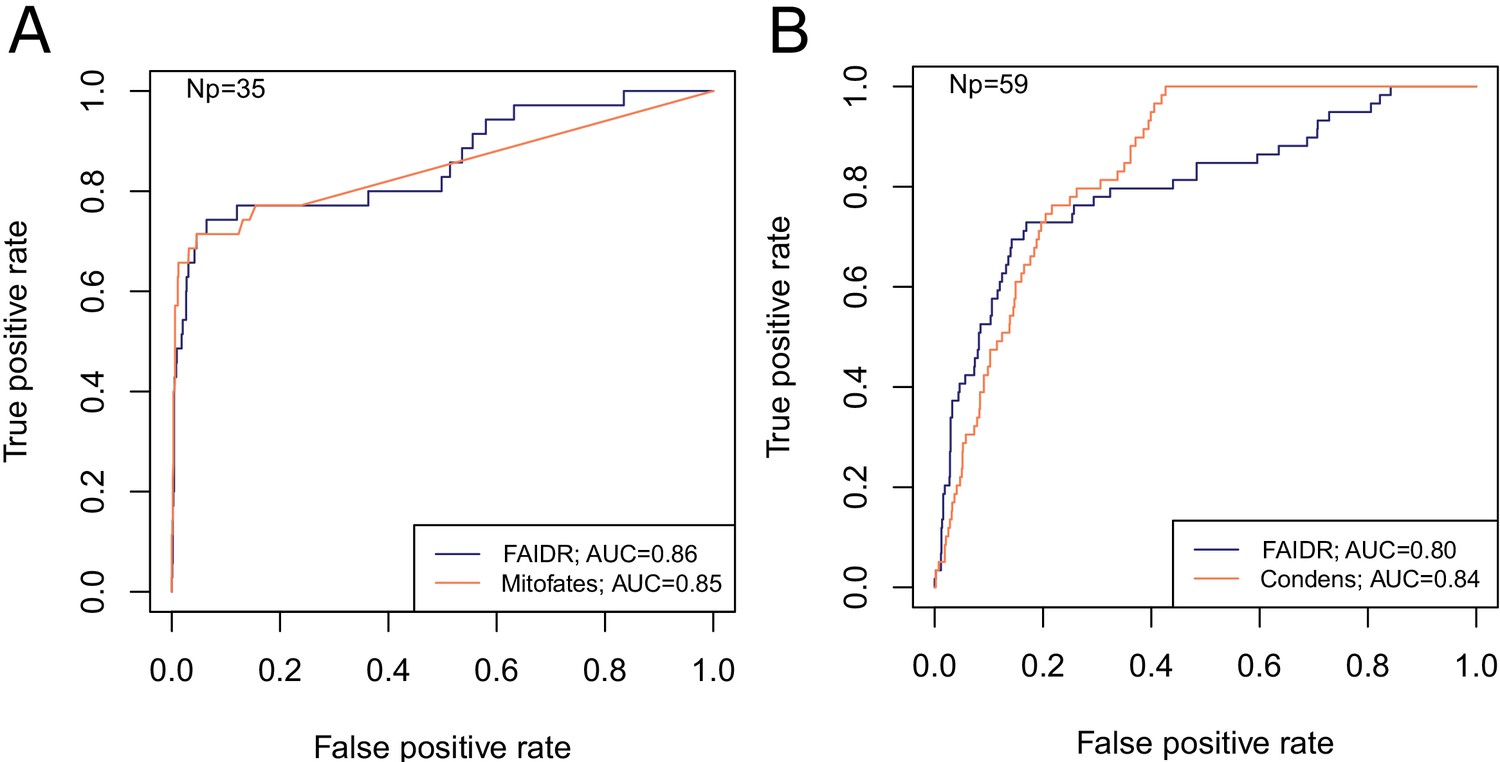
FAIDR trained on evolutionary signatures of IDRs can predict unseen data comparably to state-of-the-art, specific predictors for mitochondrial targeting signals (Mitofates [Fukasawa et al., 2015]) and Cdc28 substrates (Condens [Lai et al., 2012]).
Receiver operating curves (ROC) on a held-out 20% sample are shown for FAIDR trained on evolutionary signatures (blue) versus Mitofates (A) and Condens (B) (orange). Area under curve (AUC) is indicated in legend on bottom right for each method. The number of positive IDRs (Np) in the held-out 20% is indicated in the top left of each plot.
-
Figure 2—source data 1
Data for ROC curves including IDR coordinates, FAIDR probabilities for held out IDRs and ground truth labels from experimental data mapped to each IDR.
idr_name_protein_coordinates.txt defines IDR names as amino acid coordinates in each protein. heldout_idr_probability_cdc28_mito.csv is the FAIDR probability for IDRs that were not included in the training set (‘held-out IDRs’). total_idr_expt_db_overlap_lai_2009_cdc28.csv is the binary summary of whether or not each IDR is phosphorylated by Cdc28 according to experimental data mapped from protein coordinates to each IDR (data compiled by Lai et al., 2012). total_idr_expt_db_overlap_vogtle_2009_mitochondrial_nterm.csv is the binary summary of whether or not each IDR is a mitochondrial N-terminal targeting signal according to experimental data mapped from protein coordinates to each IDR (based on a proteome-wide experimental study by Vögtle et al., 2009).
- https://cdn.elifesciences.org/articles/60220/elife-60220-fig2-data1-v2.zip
Figure 2—figure supplement 1

Predicting diverse protein function and phenotype using FAIDR.
Average area under the receiver operating curve (AUC), computed using ROCR (Sing et al., 2009) on the held-out 20% in fivefold cross-validation is shown, computed using either the 82 molecular features on the Saccharomyces cerevisiae IDR sequence alone (unfilled bars) or from evolutionary signatures (filled bars, see text). Error bars represent the standard deviation. Functional annotations are from GO (obtained from SGD [Cherry et al., 2012]), except gene deletion phenotypes (inviable, respiratory growth, RNA accumulation, from SGD), Cdc28 substrates (yeastKID, Sharifpoor et al., 2011), mRNPs (Mitchell et al., 2013), and reversible heat aggregation (Wallace et al., 2015).
Figure 3
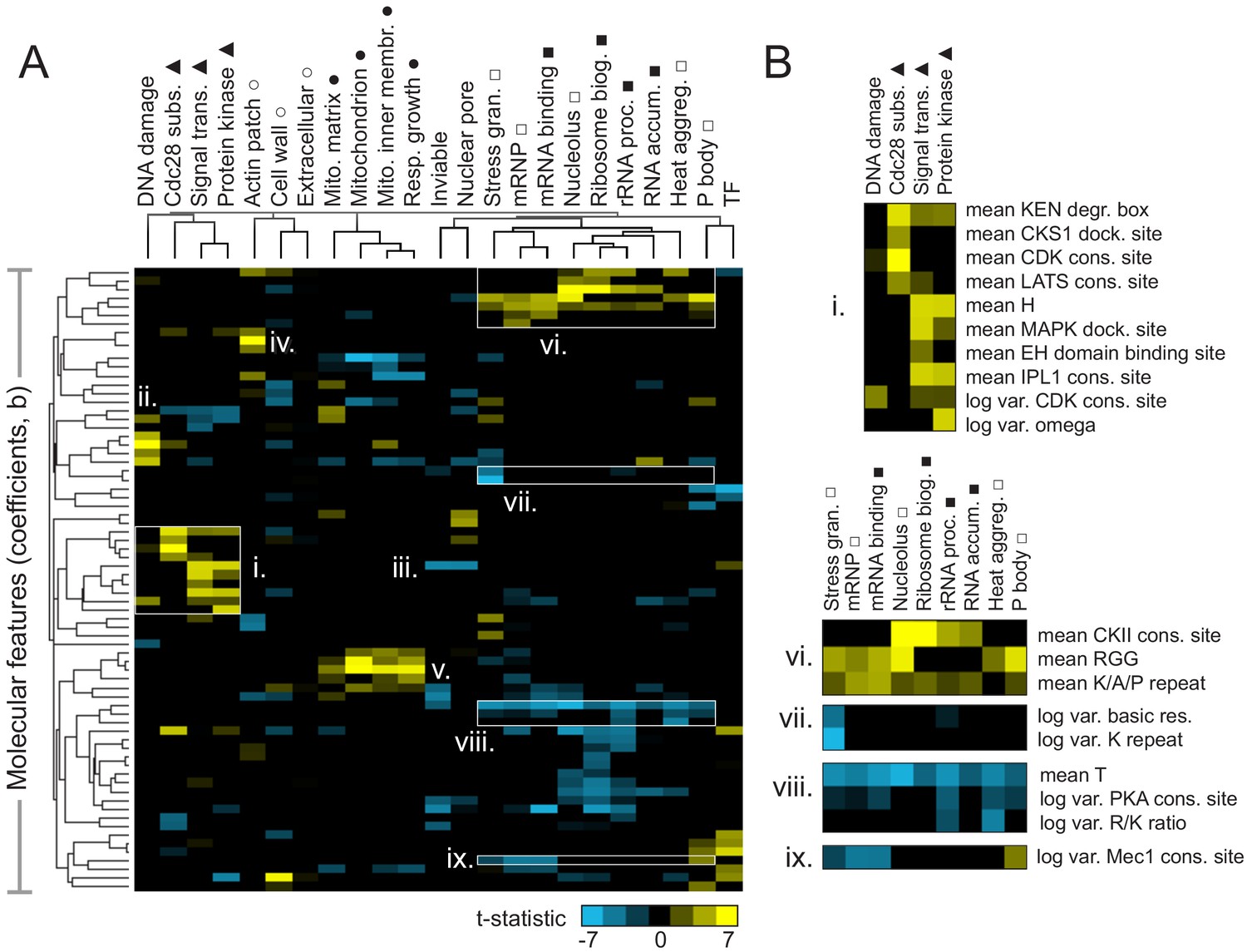
Molecular features of IDRs are associated with specific functions.
(A) Hierarchical clustering of t-statistics obtained from regression of 23 functions and phenotypes on evolutionary signatures (means and variances of 82 molecular features). Symbols beside function/phenotype names indicate related functions/phenotypes. Indicated subgroups (i–ix) are referred to and described in the main text. (B) Examples of positive and negatively predictive molecular features for signaling (i) and PMLOs (vi-ix).
-
Figure 3—source data 1
Raw T-statistics used for cluster analysis and display.
The remaining molecular features (n = 73) and the associated functions/phenotypes (n = 23) were then hierarchically clustered using weighted uncentered correlation distance (‘cluster’ and ‘calculate weights’ checked off with cutoff = 0.1 and exponent = 1) on Cluster 3.0, using average linkage. The clustered t-statistics underlying Figure 3 (tstat_table_atleast1_over_3_v2.cdt) can be viewed/explored using Java Treeview.
- https://cdn.elifesciences.org/articles/60220/elife-60220-fig3-data1-v2.zip
Figure 4 with 1 supplement
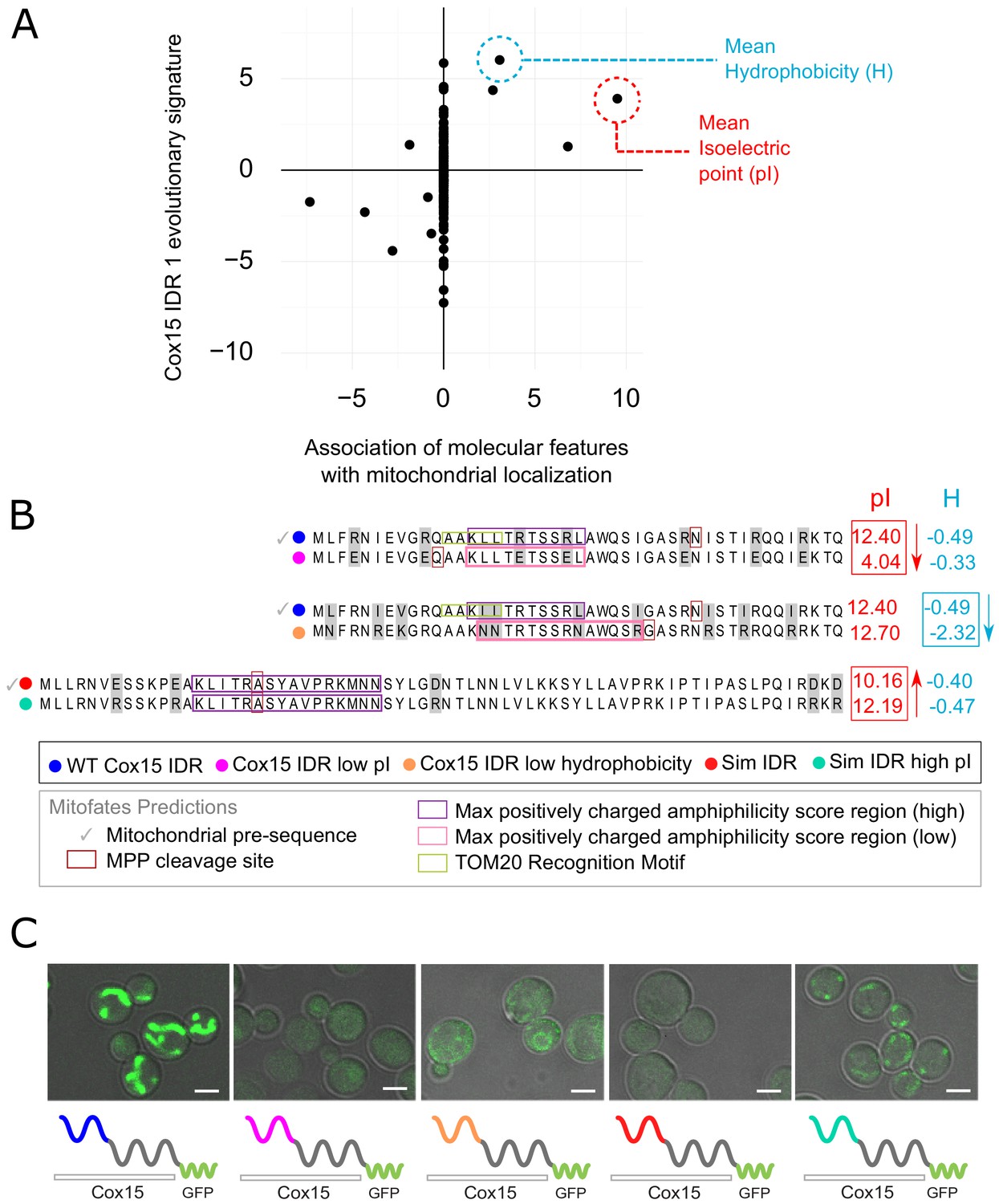
Molecular features predicted to be associated with ‘mitochondrion’ GO annotation affect mitochondrial localization phenotype.
(A) Feature vector of Z-scores (evolutionary signature) of the Cox15 N-terminal IDR (Cox15 IDR 1) (y-axis) is plotted against predictive features (t-statistics) for mitochondrial function as determined by FAIDR (x-axis). Two of the top features associated with mitochondrial localization are mean isoelectric point (pI), circled in red, and mean hydrophobicity (H), circled in blue. (B) Amino acid sequences for each Cox15 IDR variant. Wild-type and simulated Cox15 IDR sequences are compared to the same sequence with mutations altering isoelectric point (pI) and hydrophobicity. Sequences with variable residues (gray) are visualized with Jalview (Waterhouse et al., 2009). Predictions from Mitofates (Fukasawa et al., 2015), a predictor of mitochondrial targeting sequences, are also shown: prediction of whether or not the given sequence is a mitochondrial pre-sequence (check mark), as well as the predicted location of the cleavage site in the wild-type and mutated sequences (red box), the regions predicted to have a high (purple box) or low (pink box) max positively charged amphiphilicity score, and region of the TOM20 (receptor) recognition motif (green box). (C) Micrographs showing the mitochondrial localization phenotype for different budding yeast strains that differ in their Cox15 N-terminal IDRs. Green shows GFP-tagged Cox15 localization. Left to right: wild-type IDR, Cox15 IDR with low pI, Cox15 IDR with low hydrophobicity, simulated IDR, simulated IDR with high pI. Scale bar represents 1 µm.
Figure 4—figure supplement 1
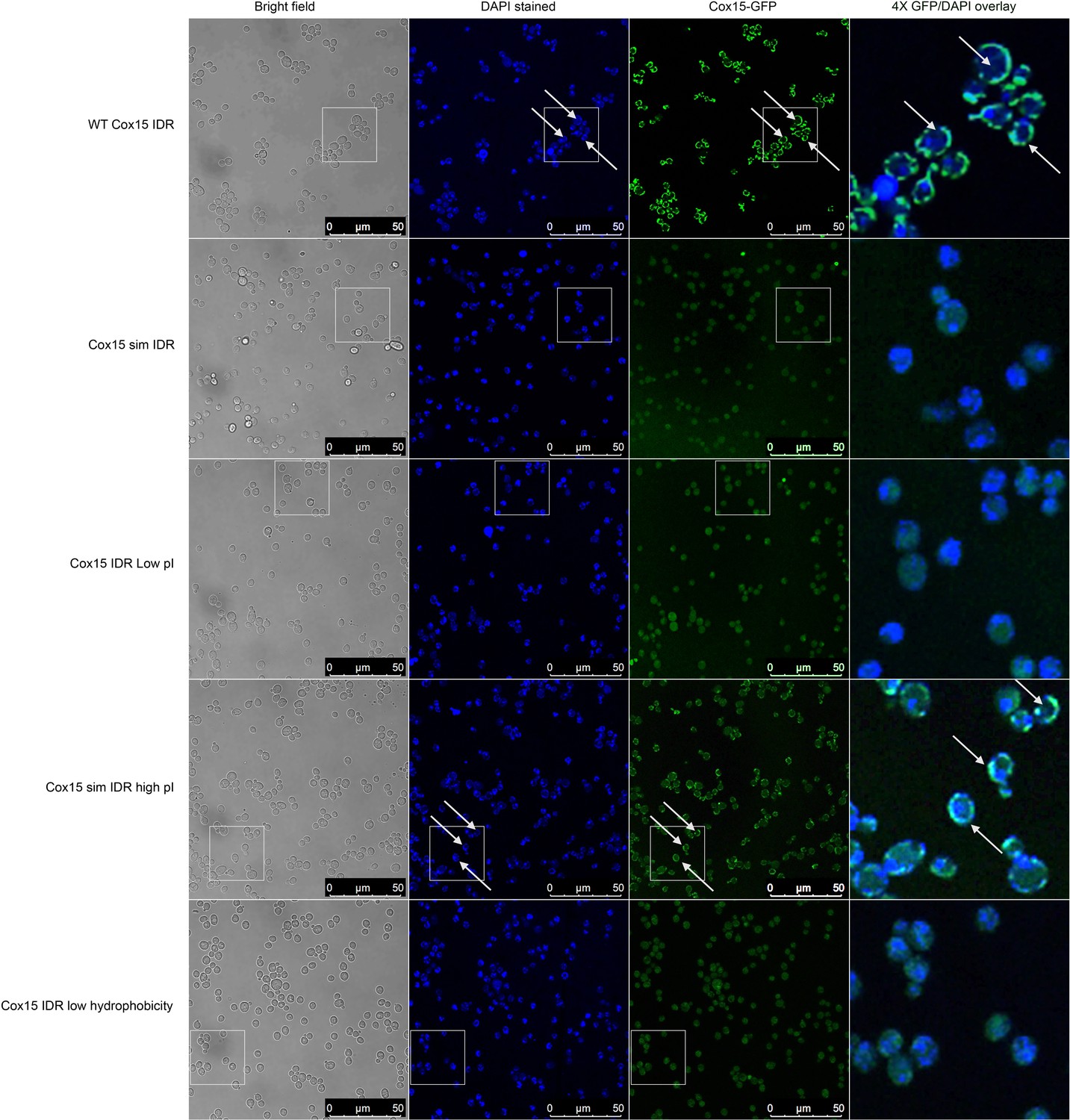
Micrographs showing (columns, left to right) bright-field, DAPI, Cox15-GFP, and 4× zoomed Cox15-GFP/DAPI overlay of different budding yeast strains with different Cox15 IDR genotypes (labels on left side of image).
DAPI stains DNA in the nucleus and mitochondria. Cox15 WT and Cox15 sim IDR high pI strain images indicate that Cox15-GFP is localized to mitochondria in these strains, while Cox15-GFP is not localized to the mitochondria in Cox15 sim IDR, Cox15 IDR low pI, and Cox15 IDR low hydrophobicity. White boxes indicate 4× zoomed in areas that are shown in the right-most column. White arrows indicate areas where there is overlap of non-nuclear DAPI and Cox15-GFP (i.e., mitochondrial localization).
Figure 5 with 1 supplement
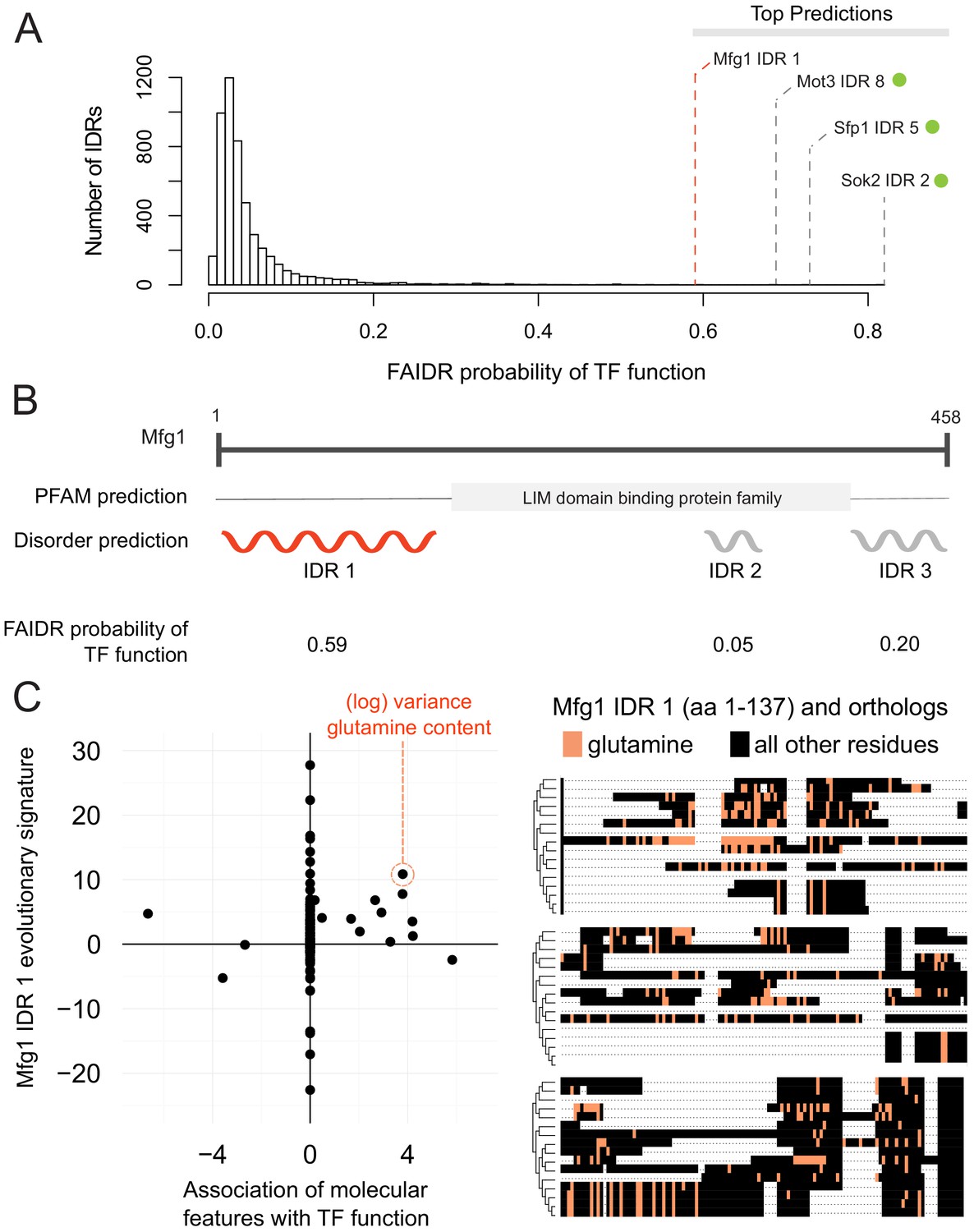
Identification of a specific IDR sequence associated with transcription factor activity in an uncharacterized protein.
(A) Histogram of probabilities for association of IDRs with sequence-specific DNA binding (transcription factor or ‘TF’) function. Top predictions are indicated. From the top predictions, IDRs from known transcription factors are indicated with a green dot. Mfg1 is an uncharacterized protein. (B) Mfg1 protein coordinates, with all known domain annotations (via PFAM), disorder prediction (via DISOPRED3), and FAIDR probability for association with sequence-specific DNA binding indicated. (C) Feature vector (evolutionary signature) for Mfg1 IDR 1 (y-axis) is plotted against predictive features for transcription factor/sequence-specific DNA binding function as determined by FAIDR (x-axis). Log variance in glutamine (Q) residues is highlighted as one of the top features (orange circle) associated with TF function. Glutamine residues (Q) are indicated (in orange) in the IDR of Mfg1 and orthologs obtained from the YGOB (Byrne and Wolfe, 2005). All other (non-glutamine) residues are indicated in black. Multiple sequence alignment of orthologs is visualized using Jalview (Waterhouse et al., 2009). Species in alignment (from bottom to top) are as follows: Saccharomyces cerevisiae, Saccharomyces mikatae, Saccharomyces kudriavzevii, Saccharomyces uvarum, Candida glabrata, Kazachstania africana, Kazachstania naganishii, Naumovozyma castellii, Naumovozyma dairenensis, Zygosaccharomyces rouxii, Torulaspora delbrueckii, Eremothecium (Ashbya) gossypii, Eremothecium (Ashbya) cymbalariae, Lachancea kluyveri, Lachancea thermotolerans, and Lachancea waltii.
Figure 5—figure supplement 1

Example of modular IDR function predicted by FAIDR.
(A) FAIDR probabilities for the IDRs in Ebp2. (B) and (C) Feature vectors (evolutionary signatures) for Ebp2 IDRs (y-axis) plotted against predictive features for nucleolus (B) and mRNA binding (C) (x-axis) as determined by FAIDR. Top predictive features for nucleolus (B) and mRNA binding (C) are highlighted in red. (D) and (E) Alignments of IDRs from Ebp2, where CKII consensus phosphorylation sites ([ST].[DE][DE]) and (lack of) threonine (T) are indicated by black highlighting, respectively. Arrows above the sequence indicate residues reported to be phosphorylated (from SGD [Cherry et al., 2012]) in Saccharomyces cerevisiae (top protein sequence in each case). Shade of blue represents percent identity in the multiple sequence alignment (darker blue is higher percent identity). Ebp2 is annotated in GO (The Gene Ontology Consortium, 2019) to be localized to the nucleolus and to bind mRNA. (D) and (E) The first IDR reveals an abundance of CKII consensus phosphorylation sites, while the second IDR reveals reduction in threonine (T). Remarkably, these predictions are consistent with the report that a C-terminal truncation mutant of Ebp2 including the second IDR (Ionescu et al., 2004) leads to defects that are distinct from the nucleolar rRNA-related functions. This example illustrates how different IDRs in multi-IDR proteins can be associated with different functions: the IDRs contain different molecular features.
Additional files
-
Supplementary file 1
Table of function and phenotype abbreviations used in this article, corresponding function and phenotype full names, references, and function and phenotype name used in data frames/source data (i.e., name with no spaces).
- https://cdn.elifesciences.org/articles/60220/elife-60220-supp1-v2.zip
-
Transparent reporting form
- https://cdn.elifesciences.org/articles/60220/elife-60220-transrepform-v2.docx
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Identifying molecular features that are associated with biological function of intrinsically disordered protein regions
eLife 10:e60220.
https://doi.org/10.7554/eLife.60220




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}