A critical residue in the α1M2–M3 linker regulating mammalian GABAA receptor pore gating by diazepam
- University of Texas at Austin, Department of Neuroscience, United States
Abstract
Benzodiazepines (BZDs) are a class of widely prescribed psychotropic drugs that modulate activity of GABAA receptors (GABAARs), neurotransmitter-gated ion channels critical for synaptic transmission. However, the physical basis of this modulation is poorly understood. We explore the role of an important gating domain, the α1M2–M3 linker, in linkage between the BZD site and pore gate. To probe energetics of this coupling without complication from bound agonist, we use a gain of function mutant (α1L9'Tβ2γ2L) directly activated by BZDs. We identify a specific residue whose mutation (α1V279A) more than doubles the energetic contribution of the BZD positive modulator diazepam (DZ) to pore opening and also enhances DZ potentiation of GABA-evoked currents in a wild-type background. In contrast, other linker mutations have little effect on DZ efficiency, but generally impair unliganded pore opening. Our observations reveal an important residue regulating BZD-pore linkage, thereby shedding new light on the molecular mechanism of these drugs.
Introduction
Benzodiazepines (BZDs) (e.g. Valium, Xanax) are one of the most widely prescribed psychotropic drugs today. An estimated nearly 100 million adults in the United States are prescribed a BZD annually (Agarwal and Landon, 2019; Bachhuber et al., 2016; Olfson et al., 2015). Their anxiolytic and sedative properties are used as therapies for conditions including anxiety, panic, insomnia, seizures, muscle spasms, pain, and alcohol withdrawal (Möhler et al., 2002). Although largely effective, BZDs have undesirable effects, including tolerance, addiction, dependence, and withdrawal symptoms, and are often co-abused with alcohol and opioids (Fluyau et al., 2018; Schmitz, 2016; Jones and McAninch, 2015; Jones et al., 2010). Novel therapies with reduced risks are imperative for safer long-term treatment options.
The therapeutic effects of BZDs are conferred upon binding to and modulating the activity of GABAARs, which are the primary inhibitory neurotransmitter-gated ion channels in the central nervous system (Smart and Stephenson, 2019). GABAARs are part of the Cys-loop superfamily of pentameric ligand-gated ion channels (pLGICs) including homologous glycine (Gly), nicotinic acetylcholine (nACh), serotonin type 3 (5-HT3), and zinc-activated receptors as well as prokaryotic homologs (Pless and Sivilotti, 2018; Nemecz et al., 2016; Tasneem et al., 2004; Connolly and Wafford, 2004). Each pentameric GABAAR is comprised of subtype-specific combinations of five homologous but nonidentical subunits (α1-6, β1-3, γ1-3, δ, ε, π, θ, ρ1-3) that together form a central chloride conducting pore (Figure 1; Olsen and Sieghart, 2008). The most prominent subtype at synapses is comprised of α1, βn, and γ2 subunits (Sieghart and Sperk, 2002). GABAARs provide a critical balance with excitatory signaling for normal neural function, and not surprisingly, their dysfunction is related to disorders such as epilepsy, autism spectrum disorder, intellectual disability, schizophrenia, and neurodevelopmental disorders such as fragile X syndrome (Hernandez and Macdonald, 2019; Gao et al., 2018; Mahdavi et al., 2018; Braat and Kooy, 2015; Vien et al., 2015; Rudolph and Möhler, 2014; Limon et al., 2012; Ramamoorthi and Lin, 2011; Solís-Añez et al., 2007). Although pharmacological manipulation of GABAARs is a powerful approach to tuning neural signaling, the rational design of novel therapies is challenged by a lack of understanding of the molecular mechanism by which drugs such as BZDs modulate channel behavior.
Figure 1 with 1 supplement see all
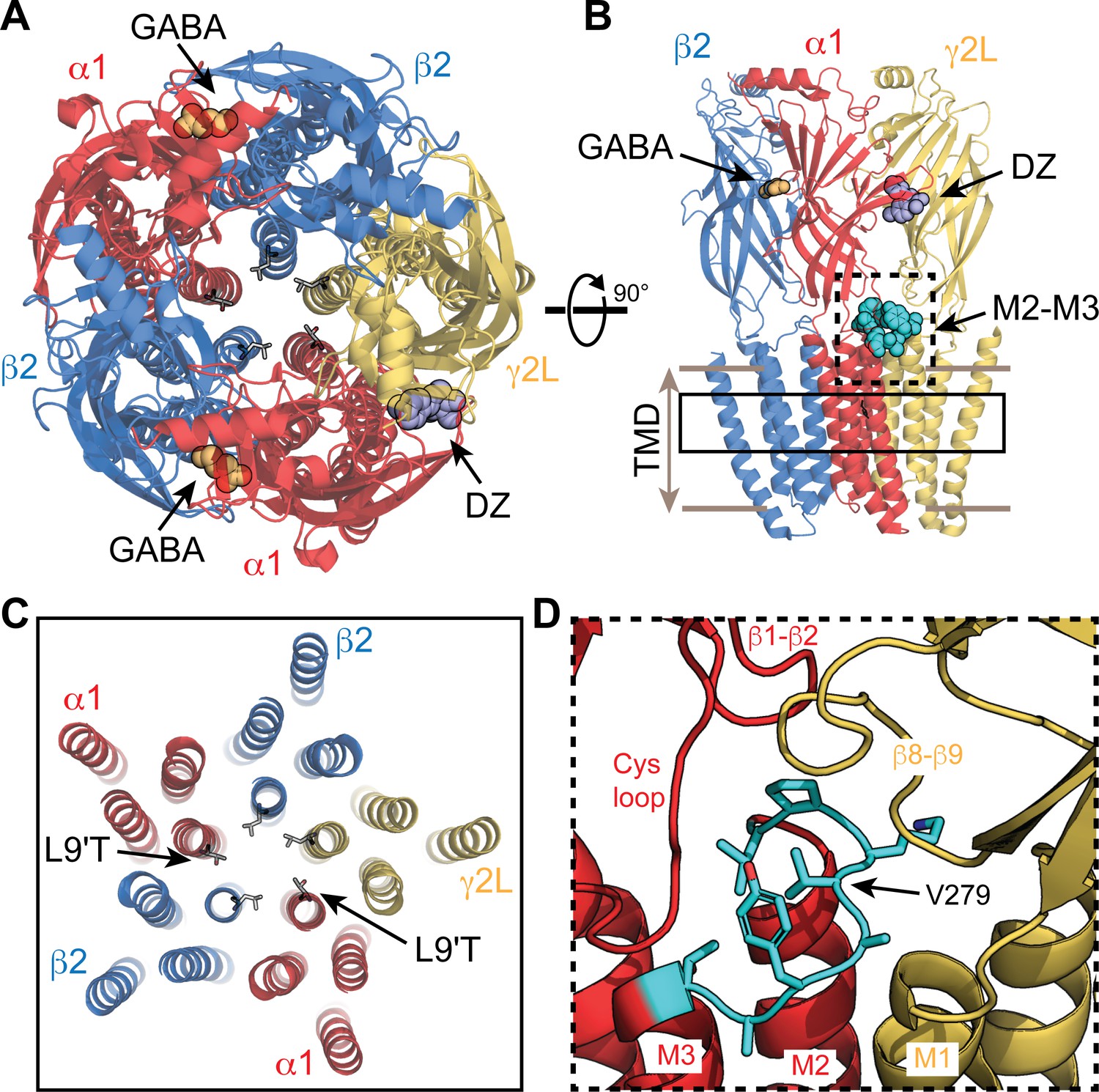
Visual representation of an α1β2γ2 GABAA receptor from cryo-EM map PDB 6X3X.
(A,B) View from the extracellular space (A) and parallel to the membrane plane (B). Bound GABA and DZ are shown as gold and lavender spheres, respectively. The 9' pore residue from each subunit is shown in stick representation, all leucine except for the mutation α1L9'T, which was generated in PyMol as a visualization aid. One of the two α1M2–M3 linkers is shown as cyan spheres in (B). (C) Same view as in (A) for a slice through the transmembrane domains indicated by the solid box in (B). (D) Detail for the dashed box in (B). The α1M2–M3 linker (L276-T283; rat numbering) is colored cyan with side chains shown in stick representation.
In conjunction with structural models of mostly homomeric pLGICs, recent cryo-EM models of heteromeric GABAARs with bound neurotransmitter and BZD provide an important conceptual aid to understand the mechanism of action of these drugs (Kim et al., 2020; Masiulis et al., 2019; Laverty et al., 2019; Phulera et al., 2018; Zhu et al., 2018). Consistent with earlier functional studies (Sigel and Ernst, 2018; Sigel and Lüscher, 2011; Sigel, 2002; Wagner and Czajkowski, 2001; Kucken et al., 2000; McKernan et al., 1998; Pritchett et al., 1989; Gibbs et al., 1985), they show that BZDs are bound at a specific recognition site in the extracellular domain between α and γ subunits (Figure 1). However, these structures have yet to clarify the mechanism by which BZDs modulate channel activity.
Kinetic models of interacting domains and thermodynamic linkage analysis can be used to estimate the energy of interaction between binding sites and the pore gate from bulk measures of ensemble average activity (Goldschen-Ohm et al., 2014; Chowdhury and Chanda, 2013; Wyman and Gill, 1990). Coupled with mutagenesis or other perturbations, these interactions can be probed to elucidate their physical basis. However, these approaches are challenged for BZDs because they do not evoke robust channel opening by themselves, but instead modulate responses to an agonist such as GABA. Thus, typical experimental measures reflect channels with both agonist and BZD bound. This makes it difficult to dissect whether the drug has altered either agonist binding or channel gating as the two processes are intimately coupled (Colquhoun, 1998). This challenge has contributed to differing conclusions postulating that BZDs alter either agonist binding (Goldschen-Ohm et al., 2010; Perrais and Ropert, 1999; Mellor and Randall, 1997; Rogers et al., 1994; Vicini et al., 1987), pore gating (Li et al., 2013; Downing et al., 2005; Campo-Soria et al., 2006; Rüsch and Forman, 2005), or an intermediate preactivation step (Goldschen-Ohm et al., 2014; Gielen et al., 2012). The ability of BZDs to potentiate current responses to saturating concentrations of partial agonists, and also to directly gate gain of function mutants, implies that the drug’s effect is not due to changes in binding alone. A combination of effects on both binding and gating as predicted by changes in intermediate gating steps energetically coupled with both binding sites and the pore gate is plausible (Goldschen-Ohm et al., 2014). However, the molecular identity of any such intermediate states remains unclear.
Here we examined linkage between the BZD site and pore gate in isolation from any effect of the drug on agonist binding using a spontaneously active gain of function mutant (α1L9'Tβ2γ2L) that is directly gated by BZDs alone (Scheller and Forman, 2002; Chang and Weiss, 1999). In the α1L9'T background, we serially mutated each residue in the α1M2–M3 linker to alanine and assessed the effect on modulation of the channel pore by the BZD positive modulator DZ (Valium) in the absence of agonist. The M2–M3 linker is a loop following the pore lining M2 helix that together with several other important interfacial loops defines the region connecting extracellular ligand binding and transmembrane domains known to be crucially involved in the gating process of pLGICs (Figure 1). Structural models show that agonist binding is associated with an outward displacement of the M2–M3 linker from the central pore axis (Nemecz et al., 2016). Mutations in the linker generally impair gating by agonists (Kash et al., 2003; O'Shea and Harrison, 2000), and some are associated with genetic diseases such as epilepsy in GABAARs (Hales et al., 2006; Hernandez and Macdonald, 2019) or hyperekplexia in GlyRs (Bode and Lynch, 2014). However, the role of the M2–M3 linker in drug modulation by BZDs is less understood.
We show that alanine substitutions throughout the α1M2–M3 linker generally impair unliganded pore gating, whereas they have little effect on the efficiency by which chemical energy from DZ binding is transmitted to the pore gate. The notable exception is α1V279A, which more than doubles DZ’s energetic contribution to pore opening, whereas larger side chains at this site do not. In a wild-type background, α1V279A enhances DZ potentiation of currents evoked by even saturating GABA, consistent with a direct effect on the pore closed/open equilibrium. Our observations identify an important residue in the α1M2–M3 linker regulating the efficiency of BZD modulation in GABAARs.
Results
Alanine substitutions in the α1M2–M3 linker generally impair unliganded gating
We use the gain of function mutation α1L9'T that confers spontaneous channel gating in the absence of agonist that can be further modulated by BZDs (Scheller and Forman, 2002; Chang and Weiss, 1999). The primary purpose for this mutation is to enable macroscopic current responses to report on pore gating by a BZD in the absence of agonist. Initially, we examine unliganded and GABA-evoked gating and in later sections turn to BZDs. In the α1L9'Tβ2γ2L (α1L9'T or L9'T) gain of function background, we assessed the effects of alanine substitutions in the α1M2–M3 linker on unliganded and GABA-evoked channel activity. We restricted the scan primarily to the flexible loop between helical regions associated with M2 and M3 from α1L276-T283 (Figure 1; Figure 1—figure supplement 1). Two of these positions, α1A280 and α1A282, are natively alanine and were not mutated. Briefly, Xenopus laevis oocytes were co-injected with mRNA for α, β, and γ subunits in a 1:1:10 ratio, and current responses to microfluidic application of ligands were recorded in two-electrode voltage clamp. Each recording consisted of a series of 10 second pulses of GABA at various concentrations bookended by 10 second pulses of 1 mM picrotoxin (PTX) (Figure 2). Current block by the pore blocker PTX was used to assess the amount of spontaneous activity and to correct for drift or rundown over the course of the experiment (see Figure 2—figure supplement 1). In comparison, wild-type receptors have little to no PTX-sensitive spontaneous activity (Figure 2—figure supplement 3). Normalized concentration–response relationships for the additional GABA-evoked current above the spontaneous current baseline in the L9'T background are shown in Figure 2. As compared to α1L9'T, all of the alanine mutations exhibited an increase in the EC50 and/or steepness of the GABA concentration–response relationship (Figure 2; Figure 2—figure supplement 2). The increased EC50 is generally consistent with previous reports of α1M2–M3 linker mutations in a wild-type background (O'Shea and Harrison, 2000) and implies that side chain interactions in this region are important for channel gating by agonists. The steepness of the concentration–response relationship for α1L9'T was fairly shallow, with a hill slope slightly below one. This could reflect an increased efficiency of mono-liganded gating in the gain of function background. In such a case, the steeper hill slopes conferred by α1L276A, α1V279A, and α1Y281A may be due to a reduction in the efficiency of mono- vs di-liganded gating, although this was not verified.
Figure 2 with 3 supplements see all
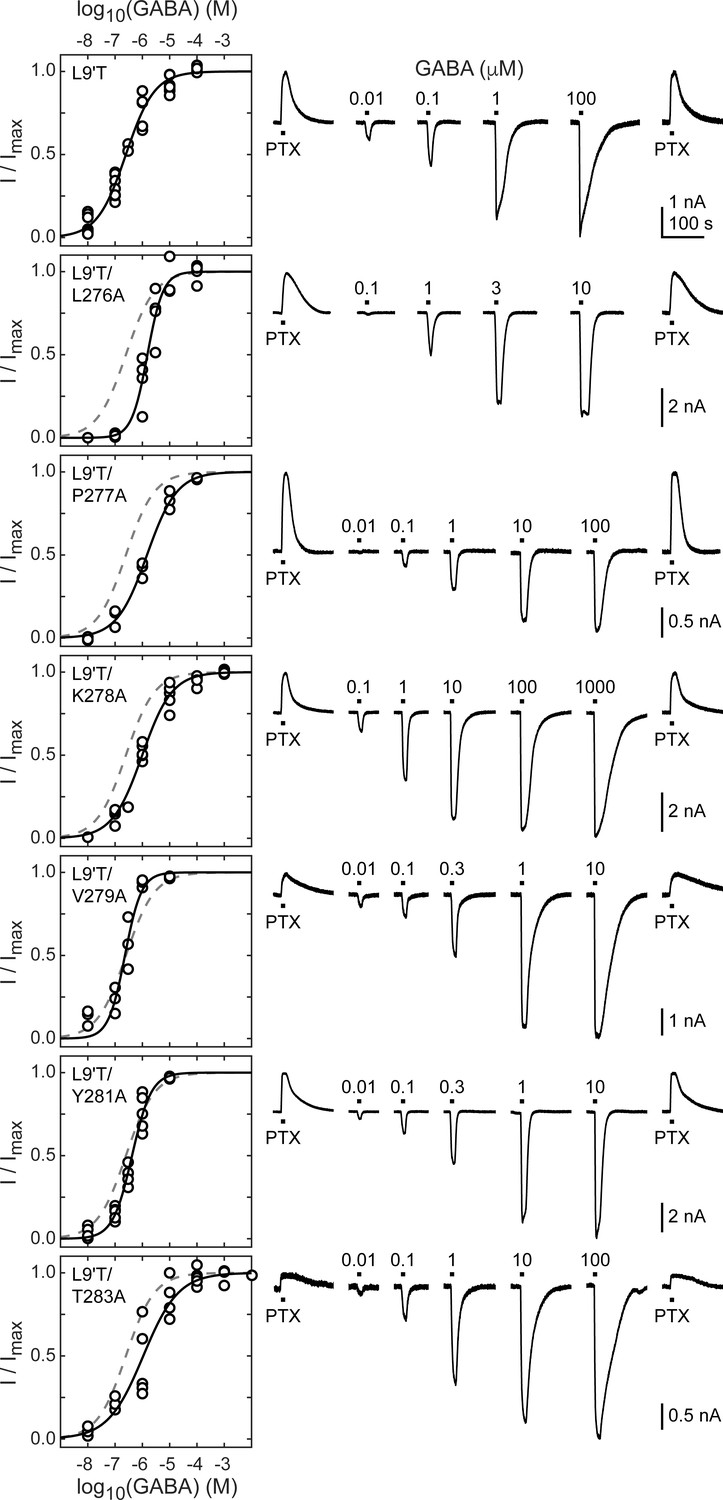
Spontaneous PTX-sensitive and GABA-evoked currents for α1M2–M3 linker alanine substitutions in the gain of function α1L9'Tβ2γ2L background.
(Left) Summary of normalized GABA concentration–response relationships for GABA-evoked currents with the zero current baseline set to the level of spontaneous activity. Solid line is a fit of the pooled data across oocytes to Equation 1, and the dashed line is the fit for the L9'T background. Fit parameters are EC50, hill slope (# oocytes): L9'T = 0.25 μM, 0.83 (7); L9'T/L276A = 1.53 μM, 1.51 (4); L9'T/P277A = 1.48 μM, 0.80 (3); L9'T/K278A = 0.99 μM, 0.79 (5); L9'T/V279A = 0.23 μM, 1.44 (4); L9'T/Y281A = 0.42 μM, 1.23 (5); L9'T/T283A = 1.08 μM, 0.67 (5). Parameters for fits to individual oocytes are summarized in Figure 2—figure supplement 2. (Right) Example currents in response to 10 second pulses of the pore blocker PTX (1 mM) or GABA (concentration in micromolar indicated above each pulse). Responses to GABA were bookended by application of PTX to assess the amount of spontaneous current and to normalize any drift or rundown during the experiment (see Figure 2—figure supplement 1).
The level of unliganded channel activity was determined from the ratio of the spontaneous current amplitude as assessed by block with PTX (IPTX) to the maximal current amplitude evoked with saturating GABA (IGABA-max) (Figure 3A). The unliganded open probability was estimated as the product of the ratio IPTX/IGABA-max and the open probability in saturating GABA (Po-GABA-max). Under the assumption that Po-GABA-max is similar for all constructs, all α1M2–M3 linker mutations except for α1P277A reduced the unliganded open probability by approximately two-fold (Figure 3B, Table 1). Po-GABA-max is ~0.85 in wild-type channels (Keramidas and Harrison, 2010), and likely to be closer to 1.0 in a gain of function background such as α1L9'T that is further known to desensitize much more slowly than wild-type (Scheller and Forman, 2002). This assumption was verified for single L9'T/V279A receptors (Figure 7—figure supplement 1). Thus, any reduction in Po-GABA-max for the linker mutations should only further reduce their estimated unliganded open probability. In summary, these data show that alanine mutations in the α1M2–M3 linker generally impair channel gating by increasing GABA EC50 and/or reducing unliganded pore opening. This suggests that α1M2–M3 linker side chain interactions play an important role in the closed vs. open pore equilibrium even in the absence of agonist.
Figure 3
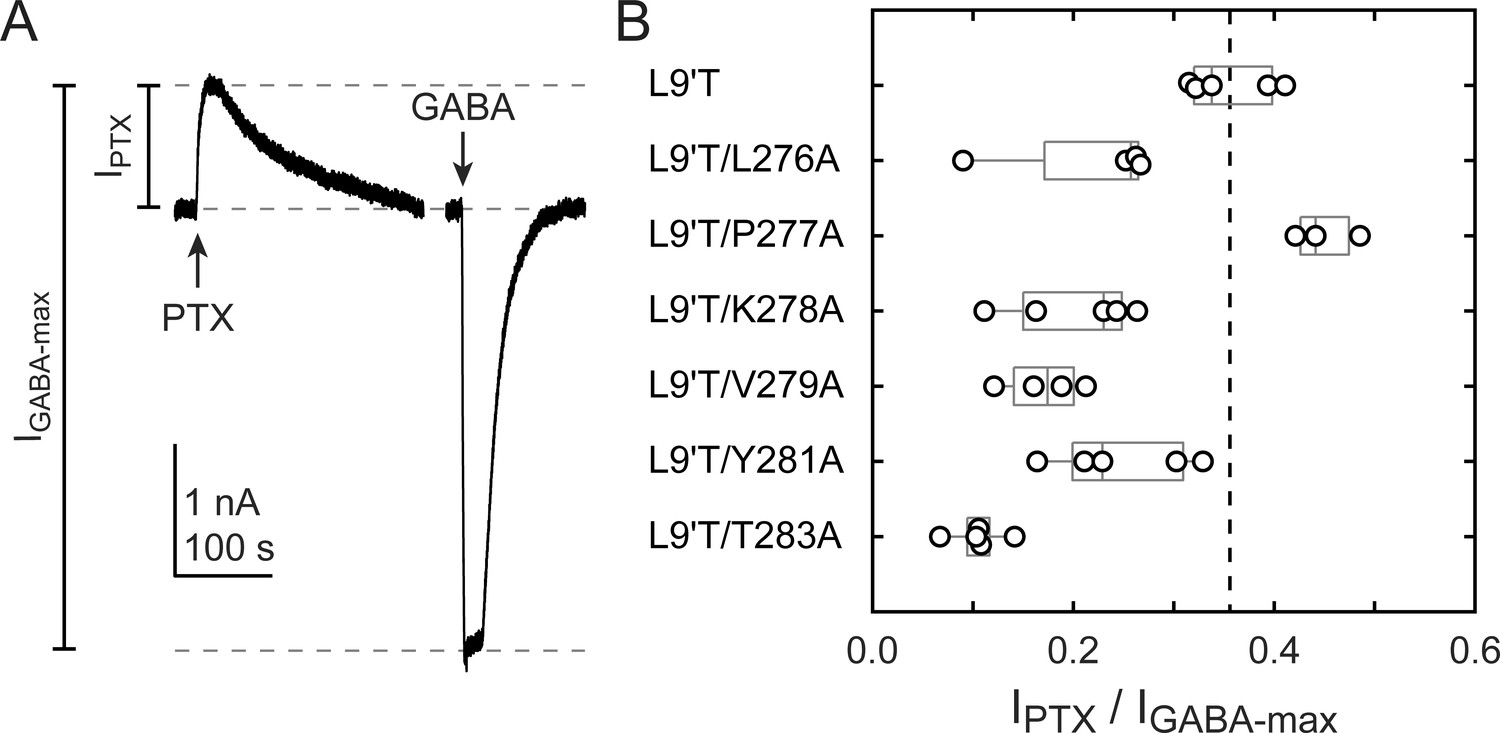
Ratio of PTX-sensitive to maximal GABA-evoked current amplitude.
(A) Example currents from α1L9'T/V279Aβ2γ2L receptors elicited by 10 second pulses of either 1 mM PTX or 10 μM GABA. (B) Summary of the ratio of PTX-sensitive to maximal GABA-evoked current amplitude for individual oocytes. Gray box plots indicate the median and 25th and 75th percentiles. The vertical dashed line is the mean for L9'T. These ratios were estimated as being approximately proportional to the unliganded open probability by a factor that is the open probability in saturating GABA.
All but one alanine substitution in the α1M2–M3 linker have little effect on activation by DZ relative to unliganded activity
BZD positive modulators alone evoke channel opening with extremely low probability (Campo-Soria et al., 2006), thus necessitating coapplication with an agonist to obtain robust currents from wild-type receptors (Germann et al., 2019). However, dissecting the effects of BZDs on either agonist binding or channel gating is severely challenged in the presence of an agonist because the two processes are intimately coupled (Colquhoun, 1998). To isolate effects on gating apart from any effects on agonist binding, we examined the effect of alanine substitutions in the α1M2–M3 linker on DZ-evoked currents in the background of the gain of function mutation α1L9'T in the absence of agonist. Concentration–response relationships for currents evoked by 10 second pulses of DZ were bookended by 10 second pulses of 1 mM PTX to assess spontaneous activity (Figure 4). Drift or rundown was corrected as described for GABA-evoked currents (Figure 2—figure supplement 1). Normalized concentration–response relationships for the additional DZ-elicited current above the spontaneous current baseline were fit to Equation 1. Alanine substitutions in the α1M2–M3 linker have no obvious effect on DZ concentration–response relationships with the exception of α1L276A that confers a right shift and reduction in steepness (Figure 4; Figure 4—figure supplement 1). Reports for the EC50 of DZ potentiation in wild-type receptors are similar to the DZ EC50 for α1L9'T receptors reported here and in previous studies (Li et al., 2013; Campo-Soria et al., 2006; Rüsch and Forman, 2005; Walters et al., 2000).
Figure 4 with 1 supplement see all
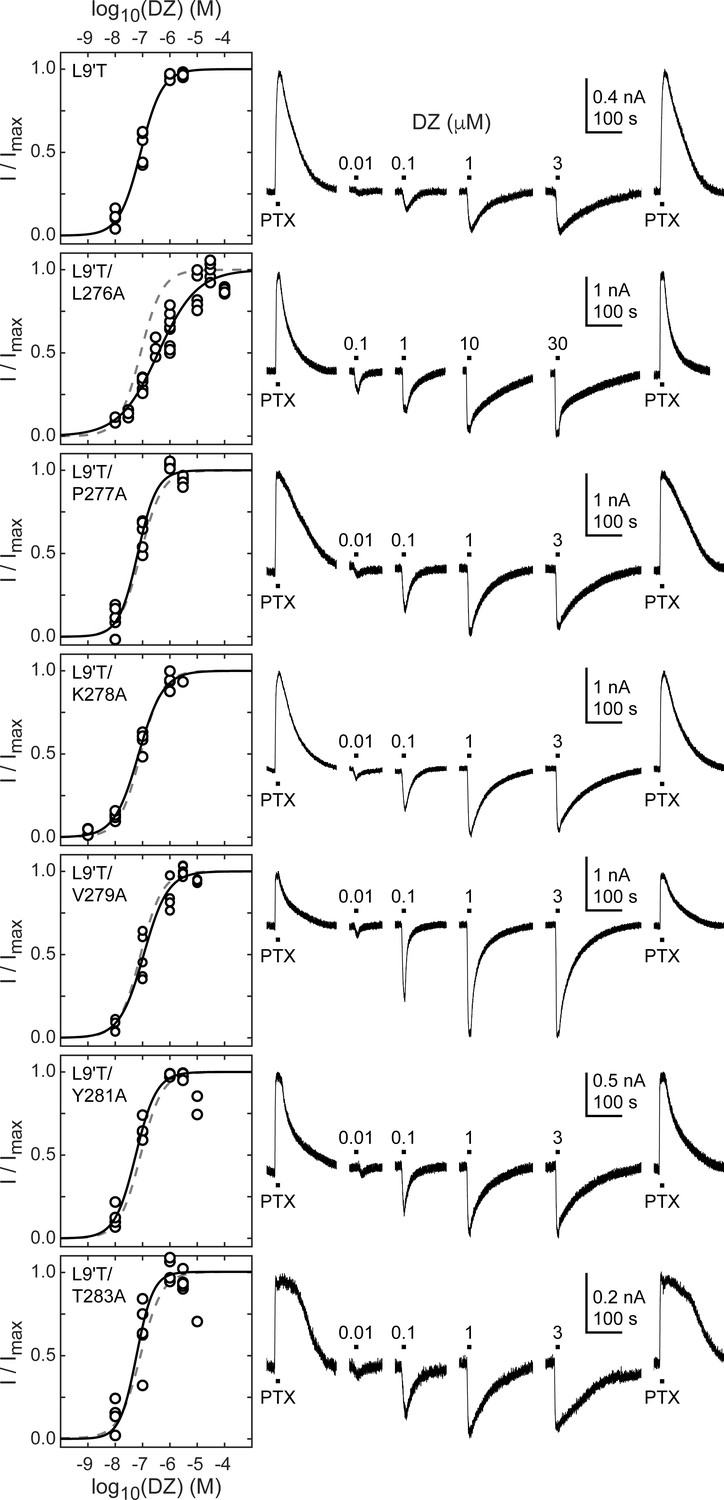
Spontaneous PTX-sensitive and DZ-evoked currents for α1M2–M3 linker alanine substitutions in the gain of function α1L9'Tβ2γ2L background.
(Left) Summary of normalized DZ concentration–response relationships for DZ-evoked currents with the zero current baseline set to the level of spontaneous activity. Solid line is a fit of the pooled data across oocytes to Equation 1, and the dashed line is the fit for the L9'T background. Reduced peak responses to 10 μM DZ for L9'T/Y281A and L9'T/T283A were omitted from the fits. Fit parameters are EC50, hill slope (# oocytes): L9'T = 85 nM, 1.10 (5); L9'T/L276A = 380 nM, 0.61 (8); L9'T/P277A = 67 nM, 1.24 (5); L9'T/K278A = 72 nM, 0.97 (4); L9'T/V279A = 110 nM, 0.98 (5); L9'T/Y281A = 56 nM, 1.14 (4); L9'T/T283A = 60 nM, 1.42 (6). Parameters for fits to individual oocytes are summarized in Figure 4—figure supplement 1. (Right) Example currents in response to 10 second pulses of the pore blocker PTX (1 mM) or DZ (concentration in micromolar indicated above each pulse). Responses to DZ were bookended by application of PTX to assess the amount of spontaneous current and to normalize any drift or rundown during the experiment (see Figure 2—figure supplement 1).
The ratio of the maximal DZ-evoked current amplitude (IDZ-max) to PTX-sensitive current amplitude (IPTX) is a measure for how well DZ activates the channel relative to its spontaneous unliganded activity. This ratio is largely independent of alanine substitution in the α1M2–M3 linker with the notable exception of α1V279A (Figure 5, Table 1). Thus, apart from α1V279A, DZ-evoked currents are predictably proportional to the amount of unliganded activity. This suggests that α1V279 has a unique role in DZ modulation, whereas side chains of other linker residues are involved little or not at all.
Figure 5
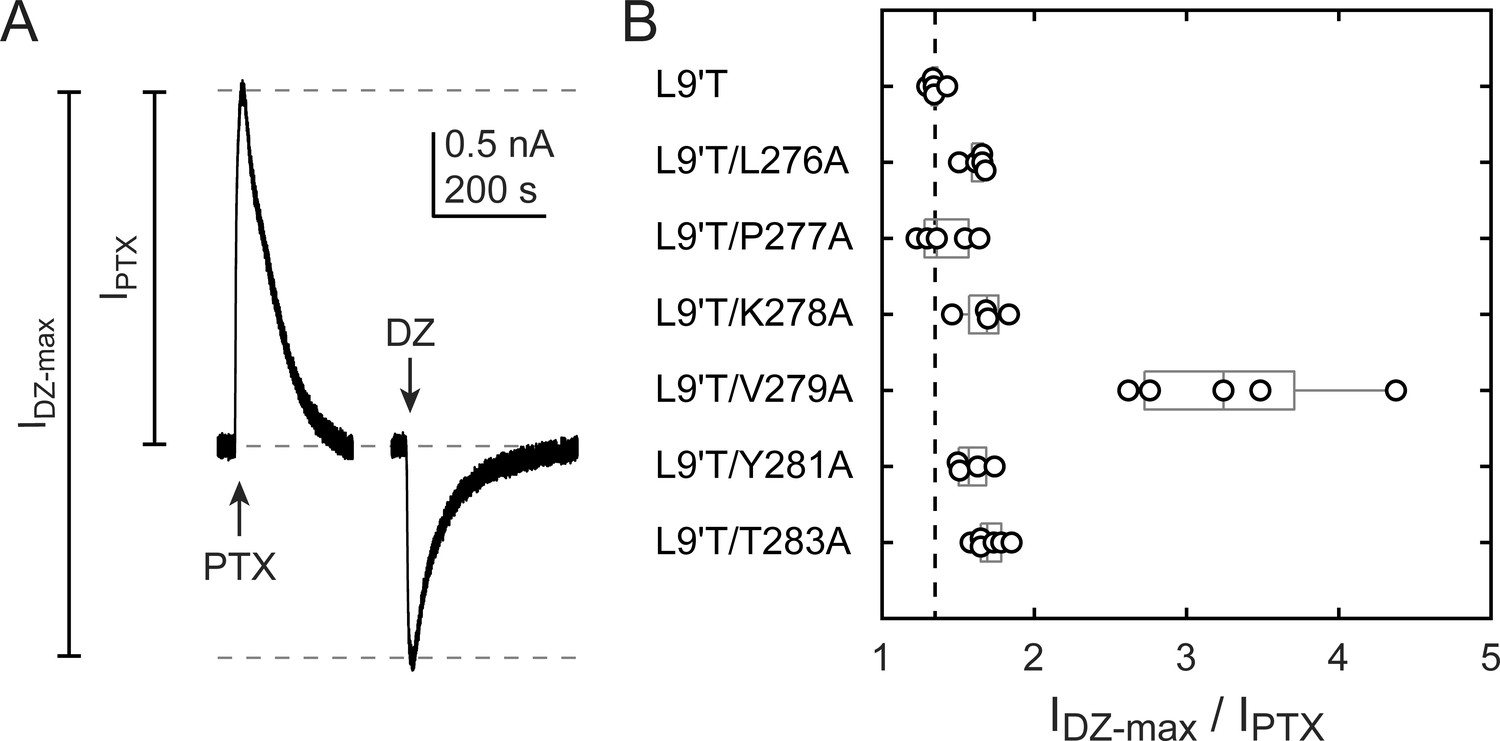
Ratio of maximal DZ-evoked to PTX-sensitive current amplitude.
(A) Example currents from α1L9'T/K278Aβ2γ2L receptors elicited by 10 second pulses of either 1 mM PTX or 1 μM DZ. (B) Summary of the ratio of maximal DZ-evoked to PTX-sensitive current amplitude for individual oocytes. Gray box plots indicate the median and 25th and 75th percentiles. The vertical dashed line is the mean for L9'T. These ratios were used as estimates for the approximate fold-change in open probability upon DZ binding.
Dependence of DZ gating on charge and volume at α1V279
The M2–M3 linker has both high sequence and structural similarity in pLGICs. The valine at position 279 in the GABAAR α1 subunit is located near the center of the linker and is nearly always an aliphatic residue in different GABAAR subunits as well as in subunits of other pLGICs, with the exception of AChR, where it is a threonine. Even where sequences differ, structural models of the M2–M3 linker for multiple pLGICs are highly similar. To explore the side chain properties relevant to DZ gating, we examined the effect of introducing a charged aspartate or bulky tryptophan at position 279 in the α1L9'T background. We recorded unliganded PTX-sensitive and GABA- or DZ-evoked currents as described above and compared their relative current amplitudes and concentration–response relationships (Figure 6). The negative charge introduced by α1V279D severely inhibits unliganded gating, exhibiting very little PTX-sensitive current in relation to robust currents evoked with saturating GABA. In contrast, addition of the bulky side chain α1V279W only slightly impairs unliganded gating to a similar degree as substitution with alanine. These data suggest that side chain volume at this position is less critical for unliganded gating, whereas introduction of a negative charge nearly abolishes spontaneous activity despite the α1L9'T pore mutation. Consistent with impaired pore gating, α1V279D increases the GABA EC50 by ~100-fold.
Figure 6
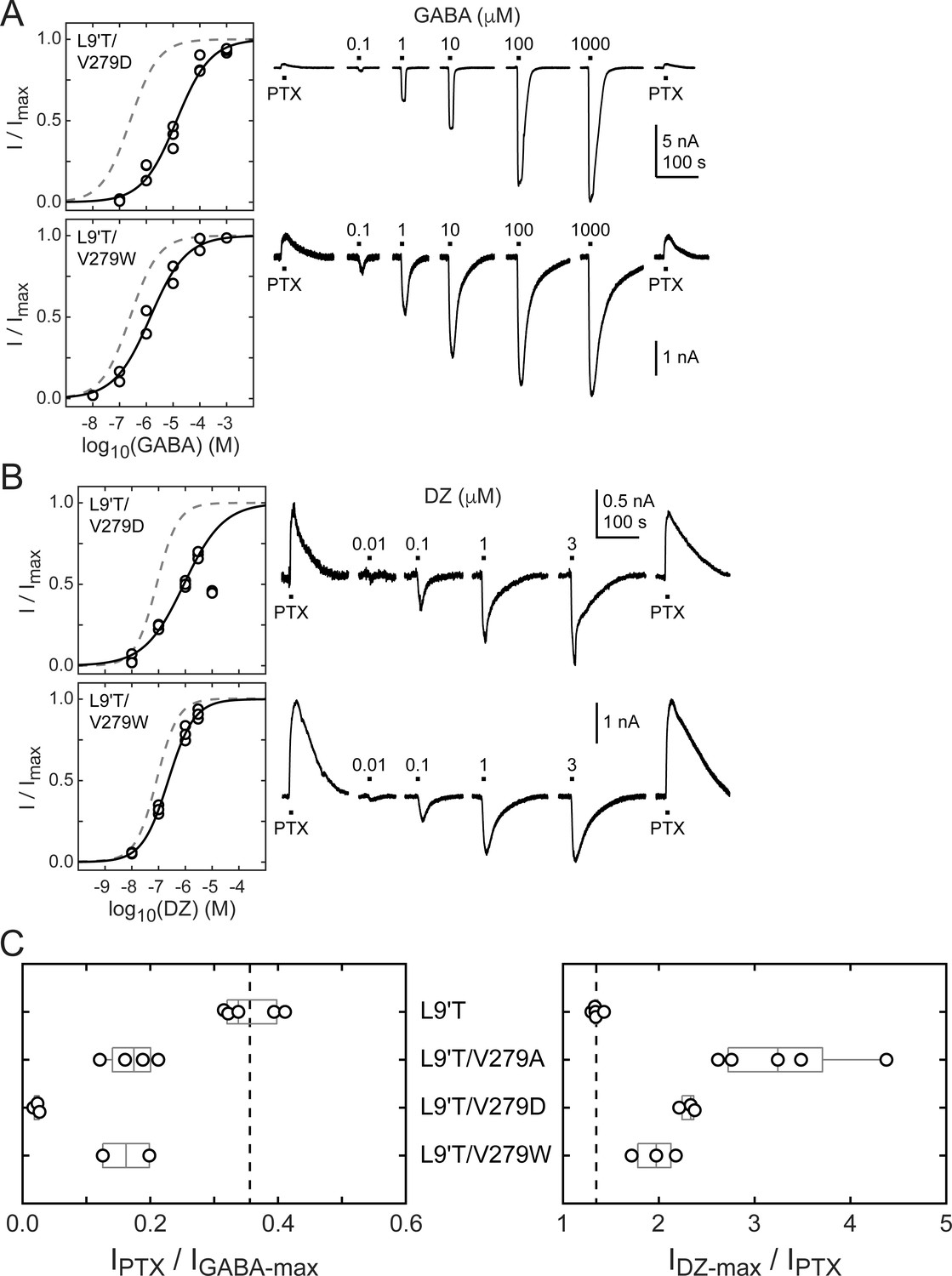
Spontaneous PTX-sensitive and GABA- or DZ-evoked currents for α1V279D and α1V279W in the gain of function α1L9'Tβ2γ2L background.
(A) Summary of normalized GABA concentration–response relationships for GABA-evoked currents with the zero current baseline set to the level of spontaneous activity (left). Solid line is a fit of the combined data across oocytes to Equation 1, and the dashed line is the fit for the L9'T background. Fit parameters are EC50, hill slope (# oocytes): L9'T/V279D = 13 μM, 0.68 (3); L9'T/V279W = 1.4 μM, 0.65 (2). Example currents in response to 10 second pulses of the pore blocker PTX (1 mM) or GABA (concentration in micromolar indicated above each pulse) (right). (B) Same as in (A) except for DZ-evoked currents. Responses from L9'T/V279D to 10 μM DZ were excluded from the fit. Fit parameters are EC50, hill slope (# oocytes): L9'T/V279D = 0.88 μM, 0.58 (3); L9'T/V279W = 0.23 μM, 0.90 (3). (C) Summary of the ratios of PTX-sensitive and either maximal GABA- or DZ-evoked current amplitudes for individual oocytes. Gray box plots indicate the median and 25th and 75th percentiles. The vertical dashed line is the mean for L9'T. These ratios were used as estimates for the approximate fold-change in open probability upon DZ binding.
The ratio of DZ-evoked to PTX-sensitive current amplitude is only slightly increased by α1V279W, whereas it is further increased by α1V279D, albeit not to the extent of α1V279A (Figure 6C). However, we were unable to reach saturation for DZ-evoked currents from α1V279D due to a reduction in peak current amplitude at higher DZ concentrations consistent with occupation of a secondary lower affinity inhibitory site (Figure 6B). Thus, our observations for α1V279D reflect a lower limit on channel activity evoked by DZ binding. Even if α1V279D has an appreciable effect on the ratio of DZ-evoked to unliganded current, the overall effect of this mutation on channel function is likely to be dominated by its inhibition of pore gating. In contrast, α1V279W has similar effects to alanine substitutions at other linker residues. These data suggest that DZ gating is specifically enhanced by a reduction of side chain volume at position 279.
The mutation α1V279A increases DZ’s energetic contribution to pore gating
To estimate the amount of chemical energy from DZ binding that is transmitted to the pore gate, we employed a simple model of channel gating between closed (C) and open (O) pore states in both unliganded and DZ-bound conditions (Figure 7A). The model assumes that gating can be approximated with a single closed and open states in each liganded condition. The free energy differences for pore gating in unliganded (ΔG0) and DZ-bound (ΔG1) conditions were calculated according to Equations 2–3 under the assumption that Po-GABA-max ~ 1. A comparison of ΔG0 versus ΔG1 shows that DZ binding confers a uniform ΔΔGDZ = ΔG1− ΔG0 = −0.4 kcal/mol to the pore gating equilibrium independent of α1M2–M3 linker mutation with the notable exception f α1V279A that more than doubles ΔΔGDZ (Figure 7B,C and Table 1). Importantly, this result is largely independent of our assumption for Po-GABA-max (see bold vs. light gray symbols in Figure 7B). Nonetheless, we verified that single L9'T/V279A channels in saturating GABA open with high probability to a similar maximal conductance level as wild-type and L9'T channels (Figure 7—figure supplement 1). These observations suggest that coupling between the BZD site and pore gate is relatively independent of individual α1M2–M3 linker side chain interactions except for α1V279, which natively hinders DZ gating as compared to its substitution with alanine. Strikingly, introduction of a bulky tryptophan or charged aspartate at position 279 have much smaller effects on ΔΔGDZ, consistent with the idea that the small side chain volume of alanine is the most relevant factor for increased DZ efficiency.
Figure 7 with 1 supplement see all
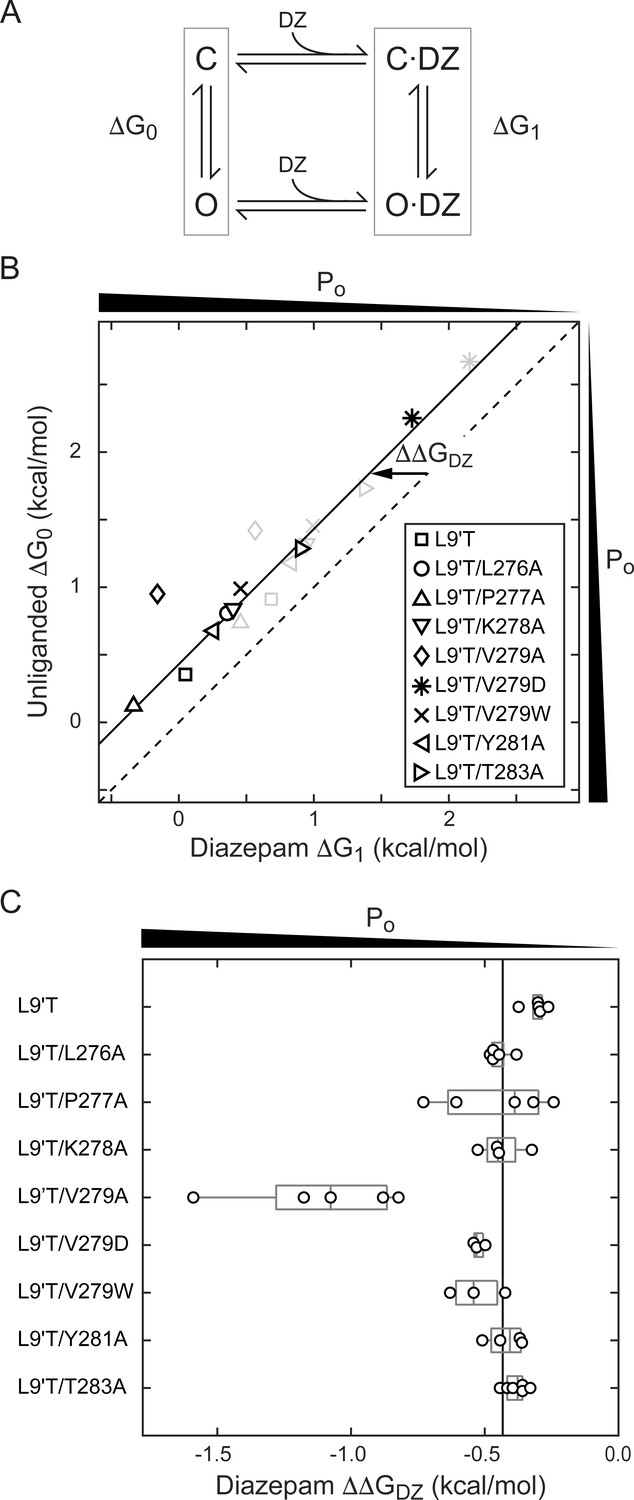
A critical residue in the α1M2–M3 linker regulating DZ’s energetic contribution to pore gating.
(A) A simple model approximating channel gating between closed (C) and open (O) pore states in both unliganded and DZ-bound conditions. (B) Relationship between gating free energy in unliganded (ΔG0) and DZ-bound (ΔG1) conditions for α1L9'Tβ2γ2L receptors and α1M2–M3 linker mutations assuming Po–GABA–max = 1 (bold symbols) or Po–GABA–max = 0.5 (light gray symbols) (Equations 2–3). ΔG values are the change in energy from closed to open states, such that negative values favor opening. To illustrate this, we indicate the direction of increasing open probability (Po) along each axis. Points are the mean across oocytes. The dashed line of symmetry reflects ΔG0 = ΔG1 where DZ would have no effect on pore gating. The solid line is a fit to ΔG1 = ΔG0 + ΔΔGDZ for all of the data points except the outlier α1V279A given Po-GABA-max = 1. The good description of the data suggests that DZ’s energetic contribution to pore gating is the same for all of the constructs on this line, estimated as ΔΔGDZ = −0.4 kcal/mol. In contrast, α1V279A more than doubles the energy that DZ binding transmits to the pore gate. A comparison of the bold and light gray symbols shows that reducing Po–GABA–max to 0.5 primarily shifts the data points along the fitted line with only minor changes in ΔΔGDZ, indicating that our assumption for the value of Po–GABA–max is not critical for interpretation of ΔΔGDZ. Nonetheless, we verified that single L9'T/V279A receptors open with high probability (Figure 7—figure supplement 1). (C) Summary of ΔΔGDZ for individual oocytes. Negative values of ΔΔGDZ increase channel open probability. Gray box plots indicate the median and 25th and 75th percentiles. The vertical line is the position of linear fit in (B) corresponding to −0.4 kcal/mol.
The mutation α1V279A enhances DZ potentiation of GABA-evoked currents in a wild-type background
If gating in the α1L9'T background is relevant to neurotransmitter-driven gating in native receptors, then the mutation α1V279A should both impair gating by GABA and enhance DZ potentiation of GABA-evoked currents in a wild-type background. To test this, we compared GABA-evoked current responses in α1β2γ2L (wild-type) and α1V279Aβ2γ2L (V279A) receptors, as well as the ability of 1 μM DZ to potentiate these responses. First, GABA EC50 is increased by α1V279A, consistent with impaired unliganded gating (Figure 8A). This effect is similar, although slightly larger than previously observed in α2β1γ2S receptors (O'Shea and Harrison, 2000). Second, DZ potentiates GABA-evoked peak current responses to a greater extent in V279A than in wild-type receptors, consistent with enhanced coupling between the BZD site and pore gate in the mutant (Figure 8B,C). In contrast to wild-type, DZ also potentiates V279A currents evoked with a saturating concentration of GABA. Together, these data suggest that the additional DZ modulation conferred by α1V279A is both independent of agonist association and additive to DZ potentiation in wild-type receptors.
Figure 8 with 1 supplement see all
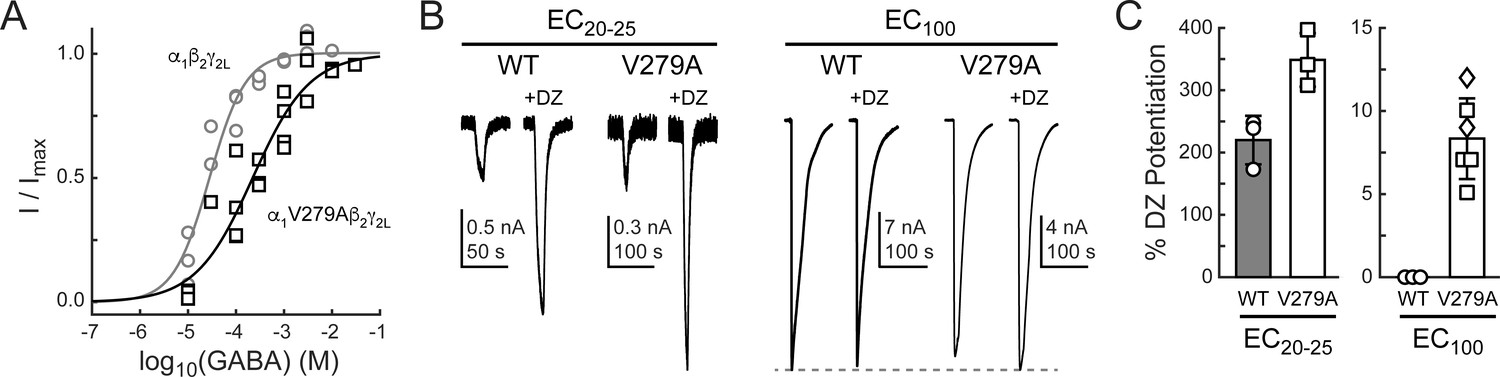
The mutation α1V279A enhances DZ potentiation of GABA-evoked current amplitudes in a wild-type (WT) α1β2γ2L background.
(A) Normalized GABA concentration–response relationships for WT (circles, three oocytes) and V279A (squares, four oocytes). Solid lines are fits to Equation 1 for all oocytes combined. Fit parameters are EC50, hill slope (# oocytes): WT = 28 μM, 1.2 (3); V279A = 222 μM, 0.7 (4). (B) Potentiation of current amplitudes evoked by 10 second pulses of either subsaturating EC20-25 (WT: 10 μM, V279A: 30 μM) or saturating EC100 (WT: 3 mM, V279A: 3–30 mM) GABA by 1 μM DZ. (C) Summary of potentiation as shown in (B) for individual oocytes. For V279A EC100, squares indicate 3 or 10 mM GABA and diamonds 30 mM GABA.
In addition to the BZD positive modulator DZ, we examined the effect of the α1V279A mutation on the BZD negative modulator FG-7142 (Im et al., 1995). Responses to ~EC20-25 GABA were reduced in the presence of FG-7142 in both WT and V279A receptors, but to a slightly lesser extent in V279A (Figure 8—figure supplement 1). Distinct mechanisms for BZD positive and negative modulation could explain the observed reduction in negative modulation as compared to enhancement of positive modulation conferred by α1V279A. Alternatively, our observations are also consistent with the idea that α1V279A stabilizes an open channel configuration specifically in the BZD-bound complex and that this stabilization works in concert with positive modulators but hinders negative modulators.
α1L9'T and α1V279A have independent and additive effects on the pore-gating equilibrium
To further explore the idea that α1V279A confers its effects primarily by altering pore gating in DZ-bound receptors, we asked whether a simple Monod–Wyman–Changeux (MWC) model of receptor behavior can account for our observations (Figure 9A). This model has been widely used to describe pseudo-steady-state behavior in GABAA receptors (Steinbach and Akk, 2019; Rüsch and Forman, 2005; Chang and Weiss, 1999). For comparison with model simulations, we estimated open probability (Po) as a function of GABA or DZ concentration (black curves in Figure 9B) from fits of Equation 1 to the data shown in Figures 2, 4, and 8 where the minimum and maximum Po were scaled according to our observed PTX-sensitive and GABA- or DZ-evoked current amplitudes. In the α1L9'T background, the maximum GABA-evoked Po was allowed to vary during optimization, and the minimum unliganded Po was scaled relative to the maximum by the observed mean ratio of IPTX/IGABA-max (Figure 3). The maximum DZ-evoked Po was scaled relative to the minimum unliganded Po by the observed mean ratio of IDZ-max/IPTX (Figure 5). The maximal Po was set to 0.85 for α1β2γ2L receptors (Keramidas and Harrison, 2010) and allowed to vary for α1V279Aβ2γ2L receptors.
Figure 9 with 1 supplement see all
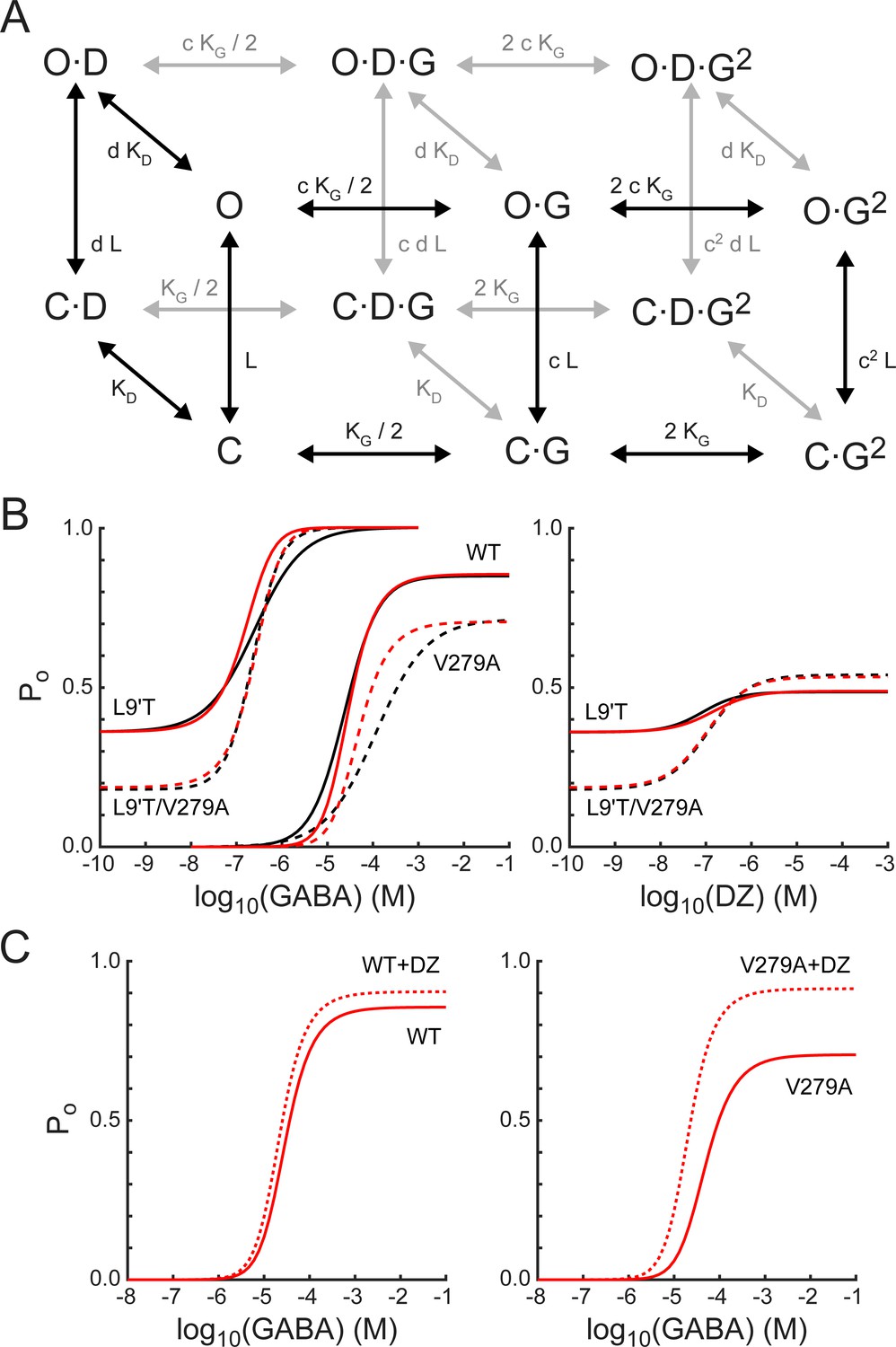
A simple MWC model of channel gating largely accounts for the observed effects of α1L9'T and α1V279A via independent and additive effects on the pore-gating equilibrium.
(A) The model depicts channel gating between closed (C) and open (O) states with independent binding of two GABA (G) and one DZ (D) molecule. L is the ratio of closed to open state probabilities in the absence of ligand, KG and KD are the respective dissociation constants for GABA or DZ, and c and d are the respective factors by which GABA or DZ binding influence channel opening. The probability to be in an open state is given by Equation 4. (B) Estimated open probability (Po) from the data in Figures 2–5 (black) overlaid with model simulations (Equation 4, red). See main text for a detailed description. Model parameters are: LL9'T = 1.8; LL9'T/V279A = 4.4; LWT = 18000; KG = 53 μM; KD = 180 nM; c = 0.0031; d = 0.59; dV279A = 0.20. (C) The model’s prediction for potentiation of WT and V279A GABA-evoked responses by 1μM DZ.
-
Figure 9—source code 1
MWC simulation code.
- https://cdn.elifesciences.org/articles/64400/elife-64400-fig9-code1-v2.zip
Table 1
Summary of peak current ratios and ΔΔGDZ for mutations in the L9'T background.
Data are mean ± standard deviation (# oocytes), and individual data points are shown in Figures 3B, 5B, 6C, and 7C.
| IPTX /IGABA-max | IDZ-max /IPTX | ΔΔGDZ (kcal/mol) | |
|---|---|---|---|
| L9'T | 0.36 ± 0.04 (5) | 1.35 ± 0.05 (5) | −0.31 ± 0.04 (5) |
| L9'T/L276A | 0.22 ± 0.09 (4) | 1.62 ± 0.07 (5) | −0.45 ± 0.04 (5) |
| L9'T/P277A | 0.45 ± 0.03 (3) | 1.41 ± 0.17 (5) | −0.46 ± 0.20 (5) |
| L9'T/K278A | 0.20 ± 0.06 (5) | 1.67 ± 0.15 (4) | −0.44 ± 0.08 (4) |
| L9'T/V279A | 0.17 ± 0.04 (4) | 3.30 ± 0.70 (5) | −1.11 ± 0.30 (5) |
| L9'T/V279D | 0.02 ± 0.005 (3) | 2.31 ± 0.08 (3) | −0.52 ± 0.02 (3) |
| L9'T/V279W | 0.16 ± 0.05 (2) | 1.96 ± 0.23 (3) | −0.53 ± 0.10 (3) |
| L9'T/Y281A | 0.25 ± 0.07 (5) | 1.59 ± 0.11 (4) | −0.42 ± 0.07 (4) |
| L9'T/T283A | 0.11 ± 0.03 (5) | 1.71 ± 0.10 (6) | −0.38 ± 0.04 (6) |
The model assumes independent GABA and DZ binding. Model parameters were optimized by globally minimizing the sum of squared residuals between simulated (Equation 4) and estimated Po as a function of GABA or DZ concentration for α1L9'Tβ2γ2L, α1L9'T/V279Aβ2γ2L, and α1β2γ2L receptors (Figure 9B). Responses to α1V279Aβ2γ2L receptors were not considered during optimization and treated as a model prediction. The mutations α1L9'T and α1V279A were allowed to perturb the parameter L with the assumption that their effects are independent in the L9'T/V279A double mutant. For wild-type receptors, L was fixed according to its approximate relation with EC50 as (Steinbach and Akk, 2019; Akk et al., 2018; Scheller and Forman, 2002; Chang and Weiss, 1999). The value of d was assumed to be the same for L9'T and wild-type receptors and allowed to vary in the presence of the α1V279A mutation. The resulting optimized model parameters (Figure 9 caption) were highly constrained by the data (i.e. estimated Po), and the model does a fairly good job of describing our observed responses to either GABA or DZ, differing somewhat in the steepness of their concentration dependence (Figure 9B). Furthermore, the model qualitatively accounts for observations from α1V279Aβ2γ2L receptors even though they were not considered during optimization. The model’s predicted maximum Po approached 1.0 in the α1L9'T background, consistent with a gain of function phenotype. In contrast, maximal Po was reduced in α1V279Aβ2γ2L as compared to wild-type receptors, as expected for a reduction in unliganded activity. These results suggest that α1L9'T and α1V279A have independent and additive effects on the gating equilibrium, and that for α1V279A, this effect depends on DZ occupancy.
Although responses to GABA or DZ alone are largely explained by this model, DZ potentiation of GABA-evoked responses in a wild-type background is either underestimated or overestimated for subsaturating or saturating GABA concentrations, respectively (Figure 9C). Previous applications of this model to BZD modulation in gain of function backgrounds predict a larger left shift of the Po curve in the presence of drug more similar to that observed, in part due to a smaller value of d (Campo-Soria et al., 2006; Downing et al., 2005; Rüsch and Forman, 2005). However, reducing d also enhances the predicted potentiation of responses to saturating GABA, contrary to that observed for wild-type receptors. Thus, the model is either too simplistic or its assumptions too strict to describe all aspects of DZ potentiation in wild-type channels that both rapidly desensitize and may involve drug modulation of agonist binding and/or intermediate gating steps (Goldschen-Ohm et al., 2014; Gielen et al., 2012). Nonetheless, the model does predict an increase in DZ potentiation of responses evoked by both subsaturating and saturating GABA for α1V279A, qualitatively similar to that observed. For responses to saturating GABA in V279A, 42% of the predicted potentiation is accounted for by the lower Po-GABA-max as compared to WT, and the remainder reflects the mutations enhancement of DZ gating.
Effects of alanine mutations on gating by GABA
To explore the effects of the mutations on gating by GABA, we fit estimated open probability versus GABA concentration relationships using the model in Figure 9A (front face only in the absence of DZ). Estimated open probabilities from fits of Equation 1 to the data in Figure 2 were scaled according to the assumption that Po-GABA-max = 1 as described above. We first asked whether all of the mutations could be explained solely by differences in their intrinsic closed–open equilibrium (parameter L). Figure 9—figure supplement 1A shows model predictions where all constructs share identical affinities for GABA (i.e. parameters KG and c). The overall rough qualitative agreement with the data suggests that differences in unliganded open probability are likely to account for much of the observed effects. For comparison, we next constrained L based on the estimated unliganded Po as L = (1 − Po)/Po and allowed GABA affinity to differ across constructs. The resulting model fits were slightly improved to a nearly equivalent extent regardless of whether the relative affinity for closed versus open states (parameter c) was held constant (Figure 9—figure supplement 1B) or allowed to vary (Figure 9—figure supplement 1C). Thus, the simplest conclusion is that mutations with right-shifted EC50s reduce GABA affinity for the closed state, although we cannot rule out compensatory changes in affinity for closed and open states.
Discussion
The main conclusions of this study are as follows: First, in the α1L9'T gain of function background alanine substitutions in the α1M2–M3 linker generally impair unliganded pore opening, indicating that side chain interactions with the linker are important for gating even in the absence of bound agonist. Second, the same mutations have no effect on the amount of chemical energy from DZ binding transmitted to the pore gate, except for α1V279A which more than doubles DZ’s energetic contribution to pore gating. Thus, α1V279 plays a crucial role to natively hinder drug modulation as compared to its substitution with alanine. Third, introduction of a bulky tryptophan or charged aspartate at position 279 is less favorable than the smaller alanine, suggesting that DZ modulation is inhibited by side chain interactions at the center of the linker. Fourth, α1V279D severely impairs unliganded gating. Fifth, α1V279A similarly enhances DZ potentiation of GABA-evoked currents in a wild-type background. Sixth, the effects of α1V279A in both α1L9'T and wild-type backgrounds are explained by specific changes in the pore gating equilibrium and its coupling to DZ binding at the BZD site.
The use of gain of function mutations to study modulatory or weakly activating ligands is well appreciated (Germann et al., 2019; Akk et al., 2018; Campo-Soria et al., 2006; Downing et al., 2005; Rüsch and Forman, 2005; Findlay et al., 2001). Single-channel gating dynamics for combinations of gain of function mutations in AChR suggest that the unliganded and agonist-bound gating mechanisms are similar if not identical and also that they are similarly affected by mutations (Purohit and Auerbach, 2009). Consistent with this idea, MWC models of pseudo steady-state GABAAR function have been largely successful in describing the effects of gain of function mutations with changes to the pore gating equilibrium independent of agonist binding (Steinbach and Akk, 2019; Downing et al., 2005; Campo-Soria et al., 2006; Rüsch and Forman, 2005; Chang and Weiss, 1999). The model reported here in Figure 9 supports this conclusion.
Although the effects of α1V279A can also be explained by specific changes to the pore-gating equilibrium, they are dependent on DZ occupation of the BZD site. From the model in Figure 9A, we compute an efficiency for GABA of , similar to that reported previously (Nayak et al., 2019). In contrast, DZ efficiency is . Whereas α1V279A decreases unliganded pore opening, it also increases DZ efficiency () to the extent that DZ binding overcomes its intrinsic inhibitory effect and results in similar or even enhanced activation as compared to wild type (Figure 9). Interestingly, an M2–M3 linker mutation in the γ2 subunit was similarly observed to impair activation by GABA and enhance modulation by the general anesthetic propofol (O'Shea et al., 2009).
Our estimates for the energetic effect of DZ binding on pore gating (−0.4 kcal/mol) are similar to prior estimates from direct gating in L9'S/T backgrounds and from kinetic models (Goldschen-Ohm et al., 2014; Gielen et al., 2012; Downing et al., 2005; Rüsch and Forman, 2005). This is approximately 10-fold less than the energy derived from binding each molecule of GABA (Goldschen-Ohm et al., 2014; Maksay, 1994; Jackson, 1992). However, it is sufficient to produce a relevant change in open probability in channels with small free energy differences between closed and open states, such as conferred by single bound agonists, partial agonists, other allosteric modulators, or gain of function mutations. From a clinical perspective, such small perturbations are likely to be more easily tolerated in patients. Given that the M2–M3 linker is associated with early movements during AChR activation (Purohit et al., 2013), it is possible that biasing the linker toward its active conformation in the α1L9'T background could limit our ability to observe its full range of motion. In this case, our observed effects of linker mutations on DZ modulation may underestimate their effects on the full activation pathway.
The interface between the extracellular agonist- and BZD-binding domains and the transmembrane helices is formed largely by several loops including the M2–M3 linker, Cys loop, β1–β2 loop and β8–β9 linker, and pre-M1 segment from neighboring subunits (Figure 1). Mutagenesis suggests that interactions between these domains are important for channel gating by agonists (Kash et al., 2003; O'Shea and Harrison, 2000) thought to involve an outward radial displacement of the M2–M3 linker (Nemecz et al., 2016). Rate versus free energy linear relationships in AChR suggest that the M2–M3 linker moves early during channel activation, similar to rearrangements at agonist binding sites (Purohit et al., 2013). Consistent with these observations, mutant cycle analysis indicates strong long-range coupling between residues in the M2–M3 linker and agonist binding sites (Gupta et al., 2017). Given the homology between the BZD site at the α/γ interface and agonist sites at β/α interfaces, interactions between the M2–M3 linker and BZD site are reasonable, although they need not be very strong given the relatively small energy DZ contributes to pore gating. As with agonist sites, such interactions could be mediated by global backbone conformational fluctuations or via distinct structural components, or both. Either way, side chain interactions at position 279 in the α1 subunit play an important role in coupling between the BZD site and pore gate.
In structural models, the M2–M3 linker adopts a C-shaped conformation with α1V279 oriented inwards toward the center of the arc (Figure 1D; Kim et al., 2020; Laverty et al., 2019; Masiulis et al., 2019; Phulera et al., 2018; Zhu et al., 2018). We hypothesize that the side chain at position 279 is centrally involved in steric interactions between linker residues near the top of the M2 and M3 helices. Removing this obstruction (α1V279A) would allow the linker to become more compact and bring the M2 and M3 helices closer together. In contrast, similar or larger side chains (α1V279D/W) would sterically inhibit such a conformational change. In this case, we speculate that the closer proximity of the M3 helix could impair unliganded pore gating by hindering radial expansion of the pore lining M2 helix. Conversely, a more compact transmembrane domain may also enhance coupling with the BZD site (Kim et al., 2020). Valine and threonine residues in GlyR and AchR aligning with V279 (Figure 1—figure supplement 1) adopt a similar conformation (Kumar et al., 2020; Nemecz et al., 2016; Du et al., 2015), suggesting that this residue has a conserved role in other pLGICs.
Alternatively, removing most of the central side chain may simply alter linker flexibility. Linker flexibility has been suggested to be inversely correlated with gating, where stabilizing interactions between α1R19' (located just below α1V279) and the linker backbone may promote coupling between extracellular and intracellular domains (Masiulis et al., 2019). Thus, the strong inhibition of unliganded opening conferred by α1V279D could reflect competition for α1R19', thereby weakening its interaction with the linker backbone and increasing linker flexibility. However, this does not provide a simple explanation for the opposing effects of α1V279A on unliganded versus DZ-bound gating. Regardless, it is important to keep in mind that the energetic changes that we observe for DZ gating are on the order of a hydrogen bond or two, and thus can be accounted for by relatively subtle changes.
The β8-β9 linker and pre-M1 in the neighboring γ2 subunit come in close proximity to the α1M2–M3 linker and are known to be important for BZD modulation, with the β8-β9 linker also contributing to the BZD-binding site (Hanson and Czajkowski, 2011; Hanson and Czajkowski, 2008). In AChR strong coupling with the adjacent β8-β9 linker and pre-M1 segment of the neighboring subunit occurs primarily via the threonine aligning with α1V279 in GABAAR and its neighboring serine (Gupta et al., 2017). A reorientation of the linker due to removal of steric interactions near its center could alter coupling to the BZD site via interactions with the γ2 β8-β9 linker and/or pre-M1. For example, a compression of the ends of the M2–M3 linker would cause the middle of the linker to be squeezed outward toward the neighboring γ2 subunit, potentially enhancing intersubunit coupling. Comparison of GlyR structures in apo or antagonist-bound versus agonist-bound conformations indicates that intersubunit interactions between the M2–M3 linker in the vicinity of the valine aligning with V279 and the following serine, and the top of the M1 helix in the adjacent subunit are weakened during channel activation (Du et al., 2015; Kumar et al., 2020). Thus, a stronger intersubunit coupling in this area could potentially both reduce unliganded opening and enhance coupling with rearrangements of the γ2 subunit upon BZD binding. However, other regions including the β4-β5 linker in the channel’s outer vestibule also affect BZD modulation of agonist-evoked currents (Pflanz et al., 2018; Venkatachalan and Czajkowski, 2012). Thus, BZD modulation likely involves larger domain fluctuations in addition to any specific molecular pathways involving α1V279.
Recent structures of αβγ receptors show DZ bound not only at the classical α/γ site in the extracellular domain, but also in the transmembrane domain below the M2–M3 linker at β/α and γ/β intersubunit interfaces (Kim et al., 2020; Masiulis et al., 2019). Thus, it is intriguing to speculate that enhanced DZ gating as conferred by α1V279A may involve occupation of a transmembrane site. However, these structures were solved in the presence of 100–200 μM DZ, and the relevance of these transmembrane sites to high affinity binding of ~1 μM DZ at the classical site is unclear (Walters et al., 2000). Also, DZ binding in the transmembrane domain was not observed at α/γ or α/β interfaces. Therefore, we favor the interpretation that our observed effects reflect altered functional coupling with the classical site.
Our observations reveal a critical residue V279 in the α1M2–M3 linker, which regulates energetic coupling between the BZD site and the pore gate, whereas other linker side chains contribute little or not at all. These data shed new light on the molecular basis for GABAAR modulation by one of the most widely prescribed classes of psychotropic drugs.
Materials and methods
Mutagenesis and expression in oocytes
Request a detailed protocolDNA for wild-type GABAAR rat α1, β2, and γ2L subunits were a gift from Dr. Cynthia Czajkowski. Single alanine substitutions were introduced throughout the α1M2–M3 linker from L276-T283 in addition to the gain of function α1L9'T pore mutation (rat α1L263T) (QuikChange II, Qiagen). Mutations V279D and V279W were introduced similarly. Each construct was verified by forward and reverse sequencing of the entire gene. mRNA for each construct was generated (mMessage mMachine T7, Ambion) for expression in X. laevis oocytes (EcoCyte Bioscience, Austin, TX). Oocytes were injected with 27–54 ng of total mRNA for α, β, and γ subunits in a 1:1:10 ratio (Boileau et al., 2002) (Nanoject, Drummond Scientific). Oocytes were incubated in ND96 (in mM: 96 NaCl, 2 KCl, 1 MgCl2, 1.8 CaCl2, 5 HEPES, pH 7.2) with 100 mg/ml gentamicin at 18°C.
Two-electrode voltage clamp recording and analysis
Request a detailed protocolCurrents from expressed channels 1–3 days post-injection were recorded in two-electrode voltage clamp (Dagan TEV-200, HEKA ITC and Patchmaster software). Oocytes were held at −80 mV and perfused continuously with buffer (ND96) or buffer containing PTX, GABA, or DZ. PTX was diluted from a 1 M stock solution in DMSO. DZ was diluted from a 10 mM stock solution in DMSO. Fresh PTX and DZ stock solutions were tested several times with no change in results. GABA was dissolved directly from powder. A microfluidic pump (Elveflow OB1 MK3+) and rotary valve (Elveflow MUX Distributor) provided consistent and repeatable perfusion and solution exchange across experiments, which limited solution exchange variability to primarily differences between oocytes only. Ten second pulses of PTX, GABA, or DZ were followed by 3–6 min in buffer to allow currents to return to baseline. Recorded currents were analyzed with custom scripts in MATLAB (Mathworks). Recordings of concentration–response relationships were bookended by pulses of PTX to correct for any drift or rundown during the experiment. Briefly, currents were baseline subtracted to correct for drift and then scaled by a linear fit to the peak of each PTX response to correct for rundown (Figure 2—figure supplement 1). The amount of current rundown was variable across oocytes, with no clear relation to specific constructs. Concentration–response relationships were fit to the Hill equation:
(1)
where is the peak current response, is ligand concentration, is the concentration eliciting a half-maximal response, and is the Hill slope.
Single-channel recording
Request a detailed protocolHEK293T cells were transfected with DNA for rat α1L9'T/V279A, β2 and γ2L subunits in a 1:1:1 ratio. Single-channel currents were recorded 16–32 hr post-transfection from excised inside-out patches clamped at −80 mV. Currents were acquired at 20 kHz and low-pass filtered at 2 kHz (Axopatch 200A, HEKA ITC, and PatchMaster software). Extracellular (pipet) solution was (in mM): 145 NaCl, 2.5 KCl, 2 CaCl2, 1 MgCl2, 10 HEPES, 4 Dextrose, pH 7.3 with NaOH. Intracellular (bath) solution was (in mM): 140 KCl, 10 EGTA, 10 HEPES, 2 MgATP, pH 7.3 with KOH. GABA was dissolved in the extracellular solution.
DZ-gating model
Request a detailed protocolFor the model in Figure 7A, the free energy difference for unliganded (ΔG0) and DZ-bound (ΔG1) gating was estimated as follows:
(2)
(3)
where , , and are as shown in Figures 3 and 5, k is the Boltzmann constant, T is temperature, and is the open probability in saturating GABA, which was assumed to be approximately 1. Importantly, even if this assumption is incorrect, the effect on DZ’s energetic contribution to pore gating should be minimal and our overall conclusion unchanged (see Figure 7B). Also, this assumption was verified for single L9'T/V279A receptors (Figure 7—figure supplement 1).
MWC model
Request a detailed protocolFor the model in Figure 9A, the probability to be in an open (O) state is given by:
(4)
where L is the ratio of closed (C) to open (O) state probabilities in the absence of ligand, KG and KD are the respective dissociation constants for GABA or DZ, and c and d are the respective factors by which GABA or DZ binding influence pore opening.
Data availability
Source code for the modeling depicted in Figure 9 has been provided.
References
-
Increasing benzodiazepine prescriptions and overdose mortality in the United States, 1996-2013American Journal of Public Health 106:686–688.https://doi.org/10.2105/AJPH.2016.303061
-
Mechanism of action of benzodiazepines on GABAA receptorsBritish Journal of Pharmacology 148:984–990.https://doi.org/10.1038/sj.bjp.0706796
-
Free-energy relationships in ion channels activated by voltage and ligandJournal of General Physiology 141:11–28.https://doi.org/10.1085/jgp.201210860
-
The Cys-loop superfamily of ligand-gated ion channels: the impact of receptor structure on functionBiochemical Society Transactions 32:529–534.https://doi.org/10.1042/bst0320529
-
Challenges of the pharmacological management of benzodiazepine withdrawal, dependence, and discontinuationTherapeutic Advances in Psychopharmacology 8:147–168.https://doi.org/10.1177/2045125317753340
-
Impaired GABA neural circuits are critical for fragile X syndromeNeural Plasticity 2018:1–7.https://doi.org/10.1155/2018/8423420
-
Benzodiazepine receptor photoaffinity labeling: correlation of function with bindingEuropean Journal of Pharmacology 110:171–180.https://doi.org/10.1016/0014-2999(85)90209-2
-
Benzodiazepines modulate GABAA receptors by regulating the preactivation step after GABA bindingJournal of Neuroscience 32:5707–5715.https://doi.org/10.1523/JNEUROSCI.5663-11.2012
-
A nonequilibrium binary elements-based kinetic model for benzodiazepine regulation of GABAA receptorsJournal of General Physiology 144:27–39.https://doi.org/10.1085/jgp.201411183
-
A mechanism for acetylcholine receptor gating based on structure, coupling, phi, and flipJournal of General Physiology 149:85–103.https://doi.org/10.1085/jgp.201611673
-
An asymmetric contribution to γ-Aminobutyric type A receptor function of a conserved lysine within TM2–3 of α1, β2, and γ2 subunitsJournal of Biological Chemistry 281:17034–17043.https://doi.org/10.1074/jbc.M603599200
-
Structural mechanisms underlying benzodiazepine modulation of the GABAA receptorJournal of Neuroscience 28:3490–3499.https://doi.org/10.1523/JNEUROSCI.5727-07.2008
-
Disulphide trapping of the GABAA receptor reveals the importance of the coupling interface in the action of benzodiazepinesBritish Journal of Pharmacology 162:673–687.https://doi.org/10.1111/j.1476-5381.2010.01073.x
-
Interaction of β-carboline inverse agonists for the benzodiazepine site with another site on GABAA receptorsBritish Journal of Pharmacology 114:1040–1044.https://doi.org/10.1111/j.1476-5381.1995.tb13310.x
-
Alcohol involvement in opioid pain reliever and benzodiazepine drug abuse-related emergency department visits and drug-related deaths - United statesMMWR. Morbidity and Mortality Weekly Report 63:881–885.
-
Emergency department visits and overdose deaths from combined use of opioids and benzodiazepinesAmerican Journal of Preventive Medicine 49:493–501.https://doi.org/10.1016/j.amepre.2015.03.040
-
The activation mechanism of α1β2γ2S and α3β3γ2S GABAA receptorsJournal of General Physiology 135:59–75.https://doi.org/10.1085/jgp.200910317
-
Identification of benzodiazepine binding site residues in the gamma2 subunit of the gamma-aminobutyric acid(A) receptorMolecular Pharmacology 57:932–939.
-
Meta-Analysis of the association between GABA receptor polymorphisms and autism spectrum disorder (ASD)Journal of Molecular Neuroscience 65:1–9.https://doi.org/10.1007/s12031-018-1073-7
-
Photoaffinity labeling of the benzodiazepine binding site of alpha1beta3gamma2 gamma-aminobutyric acidA receptors with flunitrazepam identifies a subset of ligands that interact directly with His102 of the alpha subunit and predicts orientation of these within the benzodiazepine pharmacophoreMolecular Pharmacology 54:33–43.https://doi.org/10.1124/mol.54.1.33
-
Frequency-dependent actions of benzodiazepines on GABAA receptors in cultured murine cerebellar granule cellsThe Journal of Physiology 503 ( Pt 2:353–369.https://doi.org/10.1111/j.1469-7793.1997.353bh.x
-
A new benzodiazepine pharmacologyJournal of Pharmacology and Experimental Therapeutics 300:2–8.https://doi.org/10.1124/jpet.300.1.2
-
Efficiency measures the conversion of agonist binding energy into receptor conformational changeJournal of General Physiology 151:465–477.https://doi.org/10.1085/jgp.201812215
-
Arg-274 and Leu-277 of the γ-Aminobutyric Acid Type A Receptor α2 Subunit Define Agonist Efficacy and PotencyJournal of Biological Chemistry 275:22764–22768.https://doi.org/10.1074/jbc.M001299200
-
Benzodiazepine use in the United StatesJAMA Psychiatry 72:136–142.https://doi.org/10.1001/jamapsychiatry.2014.1763
-
Subtypes of gamma-aminobutyric acid(A) receptors: classification on the basis of subunit composition, pharmacology, and functionPharmacological Reviews 60:243–260.
-
An intersubunit electrostatic interaction in the GABAA receptor facilitates its responses to benzodiazepinesJournal of Biological Chemistry 293:8264–8274.https://doi.org/10.1074/jbc.RA118.002128
-
Functional anatomy of an allosteric proteinNature Communications 4:2984.https://doi.org/10.1038/ncomms3984
-
The contribution of GABAergic dysfunction to neurodevelopmental disordersTrends in Molecular Medicine 17:452–462.https://doi.org/10.1016/j.molmed.2011.03.003
-
GABAA receptor subtypes: therapeutic potential in down syndrome, affective disorders, schizophrenia, and autismAnnual Review of Pharmacology and Toxicology 54:483–507.https://doi.org/10.1146/annurev-pharmtox-011613-135947
-
Coupled and uncoupled gating and desensitization effects by pore domain mutations in GABA(A) receptorsThe Journal of Neuroscience 22:8411–8421.
-
Benzodiazepine use, misuse, and abuse: a reviewMental Health Clinician 6:120–126.https://doi.org/10.9740/mhc.2016.05.120
-
Subunit composition, distribution and function of GABA(A) receptor subtypesCurrent Topics in Medicinal Chemistry 2:795–816.https://doi.org/10.2174/1568026023393507
-
Mapping of the benzodiazepine recognition site on GABA(A) receptorsCurrent Topics in Medicinal Chemistry 2:833–839.https://doi.org/10.2174/1568026023393444
-
The benzodiazepine binding sites of GABAA ReceptorsTrends in Pharmacological Sciences 39:659–671.https://doi.org/10.1016/j.tips.2018.03.006
-
A closer look at the high affinity benzodiazepine binding site on GABAA receptorsCurrent Topics in Medicinal Chemistry 11:241–246.https://doi.org/10.2174/156802611794863562
-
A half century of γ-aminobutyric acidBrain and Neuroscience Advances 3:239821281985824.https://doi.org/10.1177/2398212819858249
-
Autism, chromosome 15 and the GABAergic dysfunction hypothesisInvestigacion Clinica 48:529–541.
-
Actions of benzodiazepine and beta-carboline derivatives on gamma-aminobutyric acid-activated Cl- channels recorded from membrane patches of neonatal rat cortical neurons in cultureThe Journal of Pharmacology and Experimental Therapeutics 243:1195–1201.
-
Benzodiazepines act on GABAA receptors via two distinct and separable mechanismsNature Neuroscience 3:1274–1281.https://doi.org/10.1038/81800
-
BookBinding and Linkage: Functional Chemistry of Biological MacromoleculesUniversity Science Books.https://doi.org/10.1016/0014-5793(91)81192-B
Article and author information
Author details
Marcel P Goldschen-Ohm

Funding
University of Texas at Austin (Department of Neuroscience Startup)
- Marcel P Goldschen-Ohm
University of Texas at Austin (STARS)
- Marcel P Goldschen-Ohm
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Dr. Cynthia Czajkowski for gifting DNA for wild-type GABAAR subunits, and Drs. Richard Aldrich and Eric Senning for helpful discussions and shared use of equipment.
Copyright
© 2021, Nors et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,539
- views
-
- 192
- downloads
-
- 20
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 20
- citations for umbrella DOI https://doi.org/10.7554/eLife.64400
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
A critical residue in the α1M2–M3 linker regulating mammalian GABAA receptor pore gating by diazepam
eLife 10:e64400.
https://doi.org/10.7554/eLife.64400




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}