Cardiac pathologies in mouse loss of imprinting models are due to misexpression of H19 long noncoding RNA
- Division of Intramural Research, Eunice Kennedy Shriver National Institute of Child Health and Human Development, National Institutes of Health, United States
- Pathology Core, National Heart Lung and Blood Institute, National Institutes of Health, United States
- Laboratory of Vascular and Matrix Genetics, National Heart Lung and Blood Institute, National Institutes of Health, United States
- Murine Phenotyping Core, National Heart Lung and Blood Institute, National Institutes of Health, United States
Abstract
Maternal loss of imprinting (LOI) at the H19/IGF2 locus results in biallelic IGF2 and reduced H19 expression and is associated with Beckwith–-Wiedemann syndrome (BWS). We use mouse models for LOI to understand the relative importance of Igf2 and H19 mis-expression in BWS phenotypes. Here we focus on cardiovascular phenotypes and show that neonatal cardiomegaly is exclusively dependent on increased Igf2. Circulating IGF2 binds cardiomyocyte receptors to hyperactivate mTOR signaling, resulting in cellular hyperplasia and hypertrophy. These Igf2-dependent phenotypes are transient: cardiac size returns to normal once Igf2 expression is suppressed postnatally. However, reduced H19 expression is sufficient to cause progressive heart pathologies including fibrosis and reduced ventricular function. In the heart, H19 expression is primarily in endothelial cells (ECs) and regulates EC differentiation both in vivo and in vitro. Finally, we establish novel mouse models to show that cardiac phenotypes depend on H19 lncRNA interactions with Mirlet7 microRNAs.
Introduction
There are 100–200 imprinted genes in mammals. These genes are organized into discrete clusters where monoallelic expression is dependent on a shared regulatory element known as the Imprinting Control Region (ICR) (Barlow and Bartolomei, 2014). Imprinted genes are frequently involved in human disease and developmental disorders (Eggermann et al., 2015; Feinberg and Tycko, 2004; Horsthemke, 2014; Kalish et al., 2014; Peters, 2014). Sometimes, these diseases are due to inactivating point mutations of the only transcriptionally active allele. Alternatively, imprinting diseases are caused by disruption of ICR function, leading to mis-expression of all genes in the cluster.
One imprinted cluster is the IGF2/H19 locus on human chromosome 11p15.5. Imprinting in this >100 kb region is determined by the H19ICR, located just upstream of the H19 promoter (Kaffer et al., 2000; Thorvaldsen et al., 1998). As described in Figure 1A, the H19ICR organizes the locus such that transcription of the IGF2 (Insulin-like Growth Factor 2) and H19 genes are restricted to the paternal and maternal chromosomes, respectively (Ideraabdullah et al., 2008; Murrell, 2011; Yoon et al., 2007). (Note that in medical genetics, the H19ICR is also known as Imprinting Center one or IC1).
Figure 1
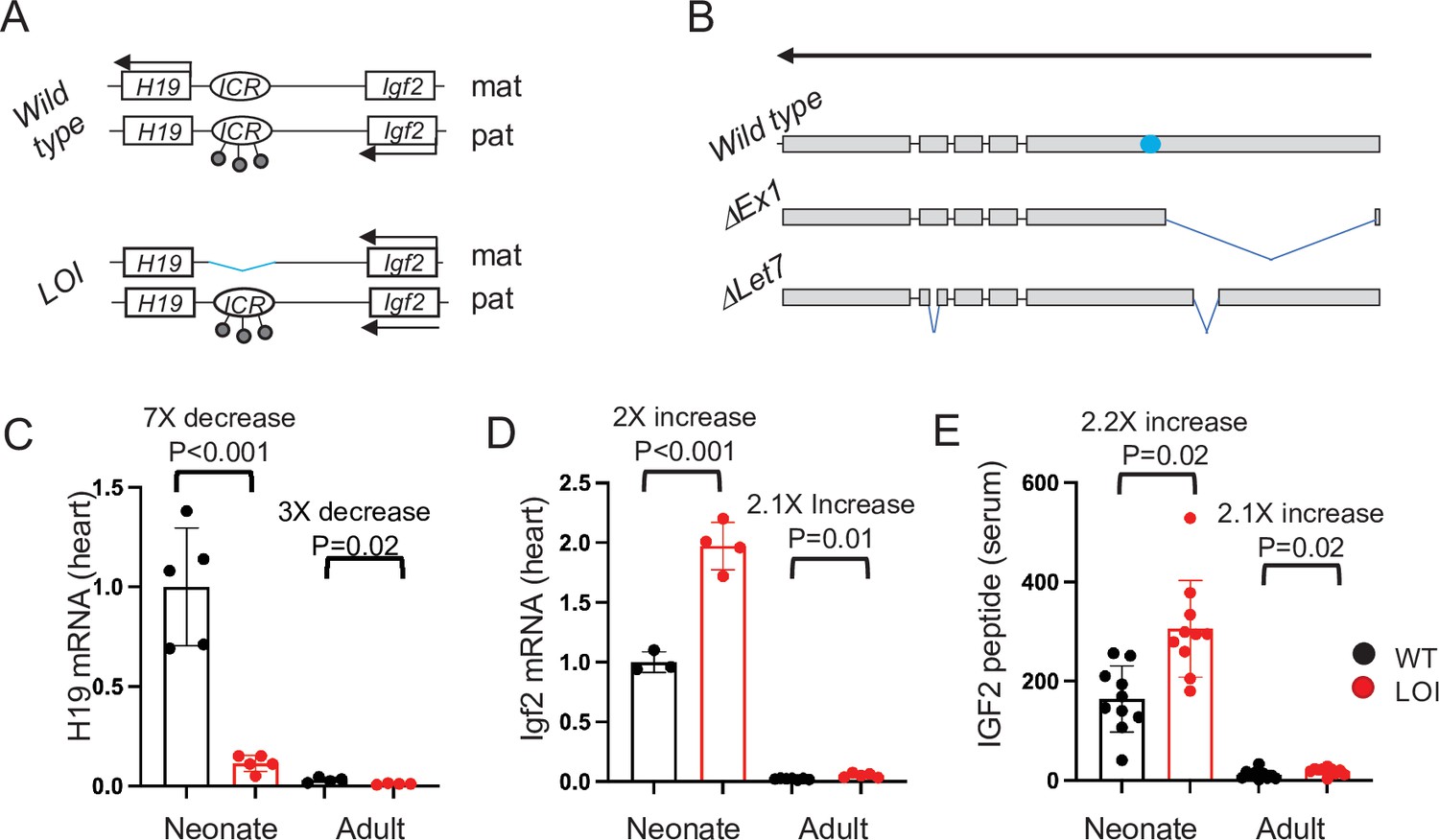
The H19/Igf2 locus.
(A) Schematic of maternal (mat) and paternal (pat) chromosomes in wild-type and in loss of imprinting (LOI) mice. Gene expression is indicated by horizontal arrows. In wild-type mice, the paternal copy of the imprinting control region (ICR) is inactivated by DNA methylation (filled lollipops). In LOI patients, inappropriate inactivation of the maternal ICR typically occurs due to microdeletion or to inappropriate DNA methylation. In the mouse LOI model, the maternal ICR is inactivated by deletion. (B) Schematic of wild-type, ΔEx1, and ΔLet7 H19 alleles. H19 exons 1–5 are shown as filled rectangles. ΔEx1 is a 700 bp deletion at the 5’ end of exon 1. ΔLet7 was constructed for this study by simultaneous deletion of Mirlet7 binding sites in H19 exons 1 and 4. The blue oval identifies coding sequences for Mir675-3p and -5 p. Arrowheads show the direction of transcription. (C–E). Maternal loss of imprinting results in reduced H19 lncRNA and 2× doses of Igf2. (C, D) Hearts were isolated from wild type (WT) or from H19ΔICR/H19+ (LOI) littermates at postnatal day 2 or at 2 months. RNAs were extracted, analyzed by qRT-PCR, normalized to GAPDH, and then normalized to levels observed in wild-type neonates. Despite the dramatic postnatal repression, H19 expression in adults remains substantial as H19 lncRNA is in the top 10-percentile of all RNAs. (C) IGF2 peptide levels in serum were measured by ELISA. Statistical significance was evaluated with Student’s t-test type 2.
-
Figure 1—source data 1
Maternal Loss of Imprinting (LOI) results in decreased Igf2 and increased H19 expression.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig1-data1-v2.xlsx
IGF2 encodes a peptide hormone that binds to and activates the insulin receptor (InsR) and insulin-like growth factor one receptor (Igf1R) kinases to promote cell growth and proliferation in many cell types including cardiomyocytes (Bergman et al., 2013; Geng et al., 2017; Li et al., 2011; Wang et al., 2019). In contrast, the functional product of the H19 gene is a 2.3 kb long non-coding RNA whose biochemical functions remain controversial (Brannan et al., 1990; Gabory et al., 2010). Reported roles for the H19 lncRNA include (1) acting as the precursor for microRNAs (Mir675-3p and Mir675-5p) (Cai and Cullen, 2007; Keniry et al., 2012), (2) regulating the bioavailability of Mirlet7 (let-7) microRNAs (Gao et al., 2014; Geng et al., 2018; Kallen et al., 2013; Li et al., 2015), (3) interacting with p53 protein to reduce its function (Hadji et al., 2016; Park et al., 2017; Peng et al., 2017; Yang et al., 2012; Zhang et al., 2019; Zhang et al., 2017), and (4) regulating DNA methylation to thereby modulate gene expression (Zhou et al., 2019; Zhou et al., 2015).
In humans, disruption of the maternally inherited H19ICR results in biallelic IGF2 along with reduced H19 expression and is associated with the developmental disorder, Beckwith–Wiedemann syndrome (BWS) (Jacob et al., 2013). BWS is a fetal overgrowth disorder but the specific manifestations of overgrowth vary between patients. Cardiomegaly is a common newborn presentation but typically resolves without treatment. Cardiomyopathies are rarer and include ventricular dilation, valve/septal defects, fibrotic and rhabdomyoma tumors, and vascular abnormalities (Cohen, 2005; Descartes et al., 2008; Drut et al., 2006; Elliott et al., 1994; Greenwood et al., 1977; Knopp et al., 2015; Longardt et al., 2014; Ryan et al., 1989; Satgé et al., 2005). BWS incidence correlates with artificial reproductive technologies (ART) (DeBaun et al., 2003; Gicquel et al., 2003; Halliday et al., 2004; Hattori et al., 2019; Johnson et al., 2018; Maher et al., 2003; Mussa et al., 2017), and among BWS patients, the frequency of heart defects is higher in those born via ART (Tenorio et al., 2016).
We have generated a mouse model that recapitulates the molecular loss of imprinting (LOI) phenotypes of BWS (Figure 1A; Srivastava et al., 2000). That is, deletion of the H19ICR on the maternal chromosome results in biallelic Igf2 and reduced levels of H19. In this study, we show that the LOI mouse model displays cardiovascular defects seen in BWS patients. Genetic and developmental analyses indicate that mis-expression of Igf2 and H19 act independently on distinct cell types to cause the cardiac phenotypes. During fetal development, increased circulating IGF2 activates AKT/mTOR pathways in cardiomyocytes resulting in cellular hypertrophy and hyperplasia. This neonatal hypertrophy is transient, non-pathologic, and unaffected by the presence or absence of a functional H19 gene. However, loss of H19 lncRNA results in cardiac fibrosis and hypertrophy and a progressive cardiac pathology in adult animals. In both neonatal and adult hearts, H19 lncRNA expression is restricted to endothelial cells (ECs). In vivo, loss of H19 results in high incidence of ECs that co-express endothelial and mesenchymal markers. Similarly, primary cardiac endothelial cells can be driven toward a mesenchymal phenotype by manipulating H19 expression levels. Thus, this research identifies a novel developmental role for the H19 lncRNA in regulating cardiac endothelial cells. In fact, this role for H19 in restricting endothelial cell transitions in the heart is unexpected given previous analyses of H19 function in vitro in transformed cell lines. Finally, we describe structure–function analyses in two novel mouse models (Figure 1B) and show that H19 lncRNA acts by regulating Mirlet7 bioavailability.
Results
Defective structure and function in hearts from mice with H19/Igf2 maternal LOI
Wild-type and LOI mice were generated by crossing H19ΔICR/H19+ with wild-type C57Bl/6 J males. (See Figure 1A for a description of the H19ΔICR allele). In mice (as in humans), maternal LOI results in biallelic (2×) expression of Igf2 and reduced levels of H19 RNA (Figure 1C–E). Hearts isolated from P1 LOI mice display cellular hyperplasia and cellular hypertrophy. Hyperplasia is indicated by increased staining for Ki-67 (a marker for cell proliferation) in tissue sections (Figure 2A,C) and by increased levels of Ki-67 and of cyclins E1 and D1 in protein extracts (Figure 2D, Figure 2—figure supplement 1A). Cellular hypertrophy is demonstrated by measuring surface areas of primary cardiomyocytes isolated from wild-type and LOI neonates (Figure 2E,F). Apart from their increased size, neonatal LOI hearts do not display any obvious pathologies. For example, we did not see increased fibrosis or expression of protein markers associated with heart disease. Furthermore, by 2 months of age, we were unable to distinguish LOI mice by cardiomegaly as measured by heart weight/tibia length ratios: WT = 6.7 ± 0.3 mg/mm (N = 8), LOI = 6.8 ± 0.3 mg/mm (N = 13), p=0.79 (Student’s t-test).
Figure 2 with 4 supplements see all
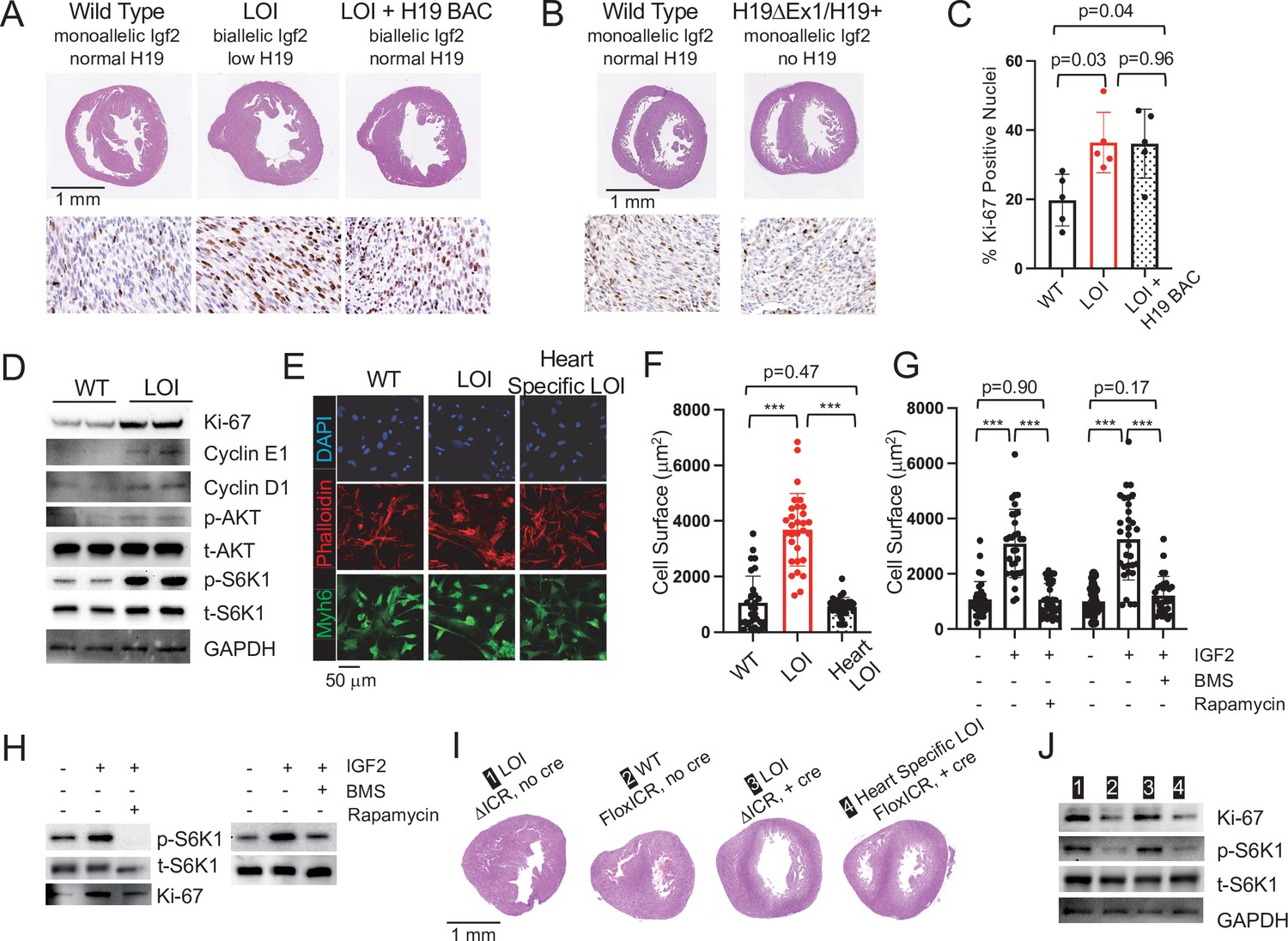
Cardiac hypertrophy in neonatal LOI mice is mediated by circulating IGF2 activation of AKT/mTOR signaling in cardiomyocytes and is independent of H19 gene function.
(A, B) Heart morphology in wild-type, LOI, and LOI+ H19 BAC littermates (A) or in wild-type and H19-deficient littermates (B) Top panels, transverse sections were taken from fixed hearts at 200 mm from the apex. Bottom panels, Ki-67 (brown stain) is a marker for cell proliferation. LOI, H19ΔICR /H19+; LOI+ BAC, H19ΔICR/H19+ that also carry a 140 kb Bacterial Artificial Chromosome transgene that restores normal H19 expression (Figure 2—figure supplement 2). Notice the thickened walls, misshaped right ventricles, and high levels of Ki-67 expression in LOI and in LOI+ BAC transgenic neonates. (C) Quantitation of Ki-67 expression as assayed in panel A. (N = 5). (D) Immunoblot analyses of heart extracts prepared from wild-type and LOI littermates. LOI hearts show increased levels of proliferation markers, Ki-67, Cyclin EI, and Cyclin D1 and also increased levels of phosphorylated AKT and S6K1 (a target of mTORC1). See Figure 2—figure supplement 1A for quantifed results. (E, F) Cardiomyocyte cellular hypertrophy in LOI animals is cell non-autonomous. Primary cardiomyocyte cultures were prepared from wild type, LOI, and from littermates carrying an ICR deletion only in cardiomyocytes (see below). Cells were cultured overnight, stained for MYH6 (to identify cardiomyocytes) and Phalloidin (to facilitate measurement of surface areas). For each culture (N = 5 per genotype), at least 30 cells were measured. (G, H) Exogenous IGF2 peptide induces cellular hypertrophy in wild-type cardiomyocytes through mTOR pathways. Primary cardiomyocytes were prepared from wild-type neonates and cultured overnight with IGF2 before measurement of cell surface area (G) or preparation of protein extracts for immunoblotting. (H) The effect of increased IGF2 is prevented by treatment with BMS 754807 or with Rapamycin. BMS inhibits IgfR1 and Ins2 receptor kinases (Carboni et al., 2009). Rapamcyin blocks a subset of mTOR activities (Li et al., 2014). See Figure 2—figure supplement 1B for quantified results. (I, J) LOI phenotypes in cardiomyocytes are cell non-autonomous. H19ICRflox/H19ΔICR females were crossed with males carrying the Myh6-Cre transgene to generate four kinds of pups: H19ΔICR/H19+ (#1) and H19ΔICR/H19+ Myh6 Cre (#3) will display LOI in all cell types; H19ICRflox/H19+ (#2) will display wild-type expression patterns for Igf2 and H19; and H19ICRflox/H19+ Myh6 Cre mice will show LOI only in cardiomyocytes. Hearts were analyzed for cellular hypertrophy (E), megacardia and hyperplasia (I), and protein expression (J). See Figure 2—figure supplement 1C for quantified results of protein expression. In all assays, H19ICRflox/H19+ Myh6 Cre mice were highly similar to their wild-type littermates and distinct from the congenital LOI littermates. *p<0.05; ***p<0.001 (Student’s t-test). LOI, loss of imprinting (H19ΔICR/H19+).
-
Figure 2—source data 1
Analyses of LOI phenotype in neonatal mice.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig2-data1-v2.xlsx
We continued to monitor cardiovascular phenotypes in LOI and wild-type mice until 19 months of age. By 6 months, LOI mice displayed cardiac hypertrophy as measured by a 28% increase in heart weight/tibia length ratios (wild type = 10.0 ± 1.7 mg/mm, N = 8; LOI = 12.8 ± 0.2 mg/mm, N = 10; p=0.005). Transverse sections revealed increased fiber diameter in LOI hearts (wild type = 10.2 ± 0.7 μm; LOI = 14.4 ± 0.8 μm; p=0.007) (Figure 3A). Cardiac hypertrophy is often a poor prognostic sign and is associated with most forms of heart failure (Heinzel et al., 2015; Vakili et al., 2001). However, hypertrophy can also be physiologic (McMullen and Jennings, 2007; Shimizu and Minamino, 2016). The hypertrophy in LOI mice might be considered pathologic based on increased levels of ANP, Myh7, cleaved Caspase-3, cleaved Caspase-7, and cleaved PARP proteins as well as decreased levels of Serca2 protein in all LOI mice by 1 year of age (Figure 3B, Figure 3—figure supplement 1; Mitra et al., 2013; van Empel et al., 2005). Finally, both interstitial and perivascular fibrosis are prominent in LOI animals by 6 months of age (Figure 3C,D).
Figure 3 with 4 supplements see all
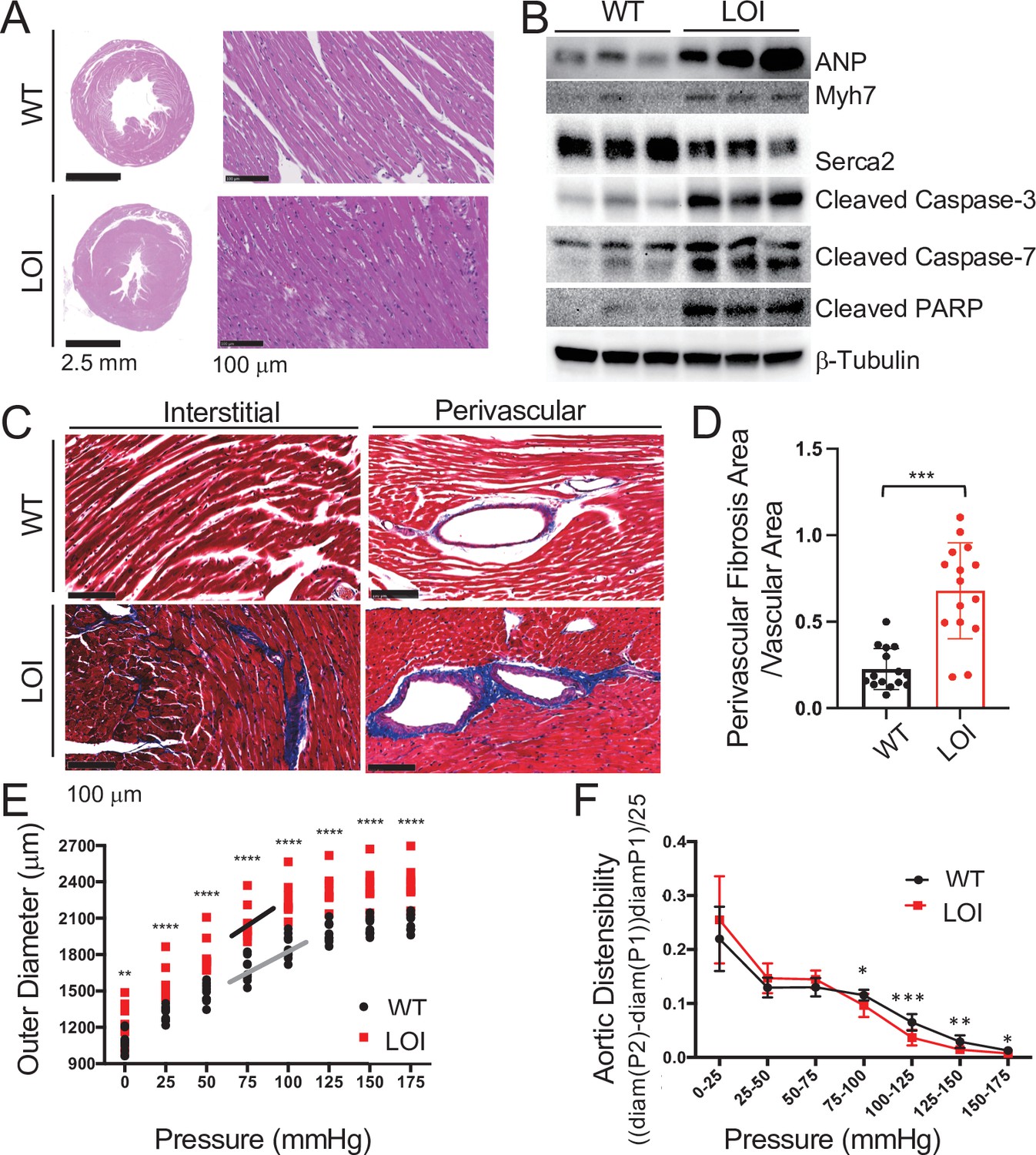
Cardiomyopathies in adult LOI mice.
(A) Transverse sections were collected midway along the longitudinal axis from hearts collected from 6-month-old wild-type (WT) and LOI mice and stained with hematoxylin and eosin. (B) Immunoblot analyses of whole heart extracts prepared from 1 year WT and LOI mice. Note the altered expression of ANP (Atrial Natriuretic Peptide), Myh7 (Myosin Heavy Chain 7), Serca2 (Sarco/endoplasmic reticulum Ca++ ATPase), Cleaved Caspase-3, and Cleaved Poly ADP Ribose Polymerase (PARP). β-Tubulin is a loading control. See Figure 3—figure supplement 1 for quantified data. (C, D) Masson’s trichrome staining of sections described in (A). Red, muscle fibers; blue, collagen. Sections from five wild-type and five LOI animals were used to calculate fibrosis. Statistical significance was evaluated with Student’s t-test type 2. (E, F) Ascending aortas were isolated from 10 wild-type and 8 LOI mice at age 20 months and pressure-diameter curves generated. (E) Increased diameters across a wide range of applied pressures. (F) Increased segmental distensibility across physiologically relevant pressures. Data were analyzed by two-way repeated measure ANOVA. Analyses of carotid arteris are described in Figure 3—figure supplement 4.
-
Figure 3—source data 1
Analyses of cardiomyopathies in adult LOI mice.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig3-data1-v2.xlsx
Table 1 summarizes echocardiography phenotypes from 13-month-old mice. Left ventricles (LV) from LOI mice are dilated (as measured by increased LV volumes at both systole and diastole), mildly hypertrophic (as measured by increased wall thickness, LVAW diastole and LVPW diastole), and show diminished function (as measured by reduced ejection fractions, % EF). LOI mice showed large increases in velocity and turbulence of blood flow from the LV outflow tract. Finally, major vessel lumen diameters (measured at the aortic arch and the first brachial arch) were >30% larger in LOI mice.
Table 1
Echocardiograph of wild-type (WT) and loss of imprinting (LOI) mice at 13 and at 16 months.
| Phenotype | 13 months | 16 months | ||||||
|---|---|---|---|---|---|---|---|---|
| Mean ± SEM | p-value | % Change | Mean ± SEM | p-value | % Change | |||
| Wt (N = 11) | Loi (N = 10) | Wt (N = 11) | Loi (N = 9) | |||||
| Heart rate (bpm) | 504 ± 12 | 499 ± 11 | 0.77 | -1 | 529 ± 20 | 540 ± 16 | 0.77 | 2 |
| LV volume systole (μl) | 24.1 ± 1.5 | 39.2 ± 4.7 | 0.01 | 63 | 28.3 ± 1.4 | 46.6 ± 1.6 | <0.001 | 65 |
| LV volume diastole (μl) | 67.9 ± 3.0 | 79.8 ± 4.6 | 0.05 | 17 | 74.2 ± 2.8 | 91.3 ± 3.3 | <0.001 | 23 |
| LV EF (%) | 64.7 ± 1.2 | 51.9 ± 3.8 | <0.01 | –19 | 62.5 ± 0.9 | 48.6 ± 1.7 | <0.001 | –22 |
| LVAW systole (mm) | 1.40 ± 0.01 | 1.44 ± 0.02 | 0.09 | 3 | 1.39 ± 0.01 | 1.52 ± 0.04 | 0.01 | 8 |
| LVAW diastole (mm) | 0.88 ± 0.01 | 1.01 ± 0.03 | <0.01 | 15 | 0.91 ± 0.01 | 1.08 ± 0.04 | < 0.001 | 20 |
| LVPW systole (mm) | 1.35 ± 0.01 | 1.39 ± 0.02 | 0.17 | 3 | 1.34 ± 0.01 | 1.42 ± 0.03 | 0.4 | 6 |
| LVPW diastole (mm) | 0.87 ± 0.02 | 0.98 ± 0.04 | 0.02 | 12 | 0.88 ± 0.02 | 1.08 ± 0.04 | <0.001 | 19 |
| LVOT mean gradient | 2.4 ± 0.2 | 6.8 ± 1.8 | 0.04 | 185 | 2.3 ± 0.2 | 4.9 ± 1.1 | 0.04 | 115 |
| LVOT mean velocity | 768 ± 30 | 1211 ± 162 | 0.02 | 58 | 750 ± 37 | 1066 ± 112 | 0.02 | 42 |
| LVOT peak gradient | 5.8 ± 0.3 | 15.7 ± 3.7 | 0.03 | 170 | 5.5 ± 0.5 | 13.3 ± 3.3 | 0.04 | 143 |
| LVOT peak velocity | 1201 ± 35 | 1870 ± 215 | 0.01 | 56 | 1158 ± 52 | 1732 ± 200 | 0.02 | 50 |
| Aorta systole (mm) | 1.65 ± 0.04 | 2.14 ± 0.10 | <0.01 | 30 | 1.70 ± 0.04 | 2.25 ± 0.12 | 0.002 | 32 |
| Aorta diastole (mm) | 1.44 ± 0.04 | 1.86 ± 0.07 | 0.001 | 30 | 1.48 ± 0.04 | 2.07 ± 0.13 | 0.002 | 40 |
| First brachial arch (mm) | 0.78 ± 0.03 | 1.06 ± 0.07 | 0.003 | 36 | 0.78 ± 0.02 | 1.09 ± 0.08 | 0.004 | 40 |
-
Table 1—source data 1
Analyses of heart function in wild type and LOI adult mice.
- https://cdn.elifesciences.org/articles/67250/elife-67250-table1-data1-v2.xlsx
Scatterplots of echocardiography data from 13 month animals show that most LOI phenotypes are heterogeneous and are not normally distributed (Figure 3—figure supplement 2). Rather phenotypes for volume, mass, ejection fraction, and outflow tract velocity and turbulence are all bimodal: 6–7 animals display mild phenotypes, and 3–4 animals display extreme pathologies (Figure 3—figure supplement 2A–D). The only exception to this pattern is seen in arterial diameter phenotypes. In this case, the variance among LOI animals is low (like their WT cohorts) and all the LOI animals display a pathologic phenotype (Figure 3—figure supplement 2E,F).
Supplementary file 1 summarizes correlations between the various phenotypes identified by echocardiography. Cardiac function as measured by ejection fraction is inversely correlated with LV volume (RR = 0.81). However, function correlates only moderately with wall thickness (RR = 0.53) and not at all with outflow tract defects (RR = 0.02), or with aortic diameter (RR < 0.01). Thus, LOI associated phenotypes are not uniformly penetrant. Rather, each mouse presents a distinct array of defects. The only invariant is that all LOI mice have arterial diameters larger than their wild-type counterparts.
The right-hand columns in Table 1 summarize echocardiography results from the same mice at 16 months of age. We observed the same ventricular abnormalities: reduced ejection fraction, increased chamber size, and increased wall thickness. However, on scatter plots, we see that wild-type and mutant animals now show non-overlapping phenotypes, consistent with the idea that ventricular failure is progressing in LOI mice (Figure 3—figure supplement 2). Note that the LOI mouse with the poorest function at 13 months (25 % EF) died prior to this second analysis.
Finally, in vivo analyses at 19 months identified significant pathological reductions in both systolic blood pressure (WT = 105 ± 2, LOI = 93 ± 3, p=0.01) and pulse pressure (WT = 37 ± 1, LOI = 28 ± 1, p<0.001) in mutant mice (Figure 3—figure supplement 3). These data confirm that H19/Igf2 LOI has a substantial effect on cardiovascular function.
As described above, increased artery diameter is a phenotype where by 13 months, LOI and WT mice sorted into phenotypically distinct cohorts. This suggested that abnormal blood vessel structure might be a relatively primary defect. We focused additional attention to this phenotype and measured outer diameters of isolated ascending aorta and carotid arteries in response to applied pressures on a pressure myograph (Figure 3E, Figure 3—figure supplement 4A). Arteries from LOI mice are larger in diameter across all applied pressures. Moreover, across normal physiological pressure ranges (75–125 mmHg) arteries from mutant mice are more sensitive to changes in pressure and lumens reach their maximum diameter at lower pressures. They are appropriately distensible at low (elastic) pressures but are stiffer than WT vessels over higher pressure intervals, including most physiologic pressures (Figure 3F, Figure 3—figure supplement 4B).
In sum, H19/Igf2 LOI in mice results in transient neonatal cardiomegaly and then a progressive cardiomyopathy. Note that results shown in Figure 3 and in Table 1 describe comparisons of age-matched male mice. Adult LOI females consistently showed relatively weak phenotypes and p-values were not significant (data now shown). However, neonatal hypertrophy and hyperplasia occurs in both male and female pups. This apparent paradox was the first clue that the relationship between the neonatal hypertrophy and the adult disease phenotypes was not straightforward.
Hypertrophy and hyperplasia in neonatal LOI mice is dependent on hyperactivation of mTOR/AKT signaling by increased dosage of IGF2 peptide
To understand the specific roles for mis-expression of Igf2 and of H19 in neonatal cardiomegaly, we performed two genetic analyses. First, we rescued H19 expression in an LOI background by introducing a 140 kb H19 Bacterial Artificial Chromosome (H19 BAC) (Kaffer et al., 2001; Kaffer et al., 2000; Figure 2—figure supplement 2) but still saw cardiomyocyte hypertrophy and hyperplasia in neonates (Figure 2A,C). Second, we tested the effect of removing H19 in a background where Igf2 remains monoallelic by comparing H19ΔEx1/H19+ (Figure 1B) with wild-type littermates. (Reduced expression of H19 lncRNA in H19ΔEx1/H19+ is shown in Figure 2—figure supplement 3A.) Loss of H19 lncRNA does not result in neonatal cardiomyocyte hypertrophy or hyperplasia (Figure 2B). Altogether, we conclude that loss of H19 lncRNA does not contribute to neonatal hypergrowth. Rather, this neonatal hypertrophy is dependent only upon biallelic (2× dosage) Igf2 transcription.
IGF2 peptide works by binding and activating InsR and IgfR kinases and mTOR/AKT signaling is a known downstream target of these receptor kinases (Bergman et al., 2013). In addition, studies document the role of AKT/mTOR signaling in cardiomyocyte cell division and hypertrophy (Geng et al., 2017; Li et al., 2011; Sciarretta et al., 2014; Shen et al., 2020; Wang et al., 2019). Consistent with a critical role for AKT/mTOR signaling in LOI-dependent neonatal hypertrophy, hearts from LOI neonates show increased levels of phosphorylated AKT and of phosphorylated S6K1, a downstream marker for mTORC1 activity (Figure 2D, Figure 2—figure supplement 1A). Moreover, the LOI cellular hypertrophy and pAKT hyperactivation phenotypes can be phenocopied by treatment of wild-type primary cardiomyocytes with IGF2 peptide. However, IGF2 action is blocked by BMS-754807, a specific inhibitor of the receptor kinase, or by treatment with rapamycin, an mTOR signaling pathway inhibitor (Figure 2G and H; Figure 2—figure supplement 1B).
Igf2 is widely expressed in the embryo. In fact, expression of Igf2 is low in the heart relative to other tissues, especially liver and skeletal muscle (Figure 2—figure supplement 4), which are believed to be the major source of circulating IGF2 peptide. Within the heart, IGF2 originates in both cardiomyocytes but also in endothelial cells (Shen et al., 2020). To assess the role of biallelic expression of Igf2 in the cardiomyocytes themselves, we crossed H19ΔICR/H19ICRflox females with H19+/H19+ males carrying the Myh6-Cre transgene. H19ICRflox is an allele where the H19ICR is flanked with loxP sites, so that cre recombination results in deletion of the ICR (Srivastava et al., 2000). We used PCR analyses to demonstrate that the transgene drives efficient ICR deletion in the heart but not in other tissues tested (skeletal muscle, liver, kidney, brain, thymus, spleen, and lung). Our cross generated wild-type mice (H19ICRflox/H19+) and two kinds of LOI controls (H19ΔICR/H19+;+ Myh6 Cre and H19ΔICR/H19+) that we compared with experimental mice that had cardiomyocyte-specific LOI (H19ICRflox/H19+;+ Myh6 Cre). Cardiomyocyte-specific ICR deletion does not cause hypertrophy. Rather, H19ICRFlox/H19+ Myh6 Cre mice were indistinguishable from their wild-type littermates (Figure 2E,F,I,J; Figure 2—figure supplement 2C).
Note that our data demonstrate that the effects of LOI on neonatal cardiomyocytes are cell non-autonomous but do not rule out a cell autonomous role in other cell types, including cardiac endothelium.
Cardiac disease in adults is dependent only on loss of H19 lncRNA expression. Biallelic Igf2 and the resultant hypertrophy in neonatal hearts are not relevant to the adult LOI phenotype.
While the H19 BAC transgene does not prevent neonatal cardiomegaly, it does successfully prevent adult pathologies. That is, hearts from 6 month LOI mice carrying the H19 transgene are not enlarged as determined by heart weight/tibia length ratios (LOI = 12.9 ± 0.6 mg/mm, N = 3; LOI+ H19 BAC transgene = 11.5 ± 0.7, N = 3; p<0.05), are not fibrotic (Figure 4A,B), and do not express cardiomyopathy markers (Figure 4C; Figure 4—figure supplement 1A). Thus, loss of H19 is necessary to induce LOI cardiomyopathies.
Figure 4 with 1 supplement see all
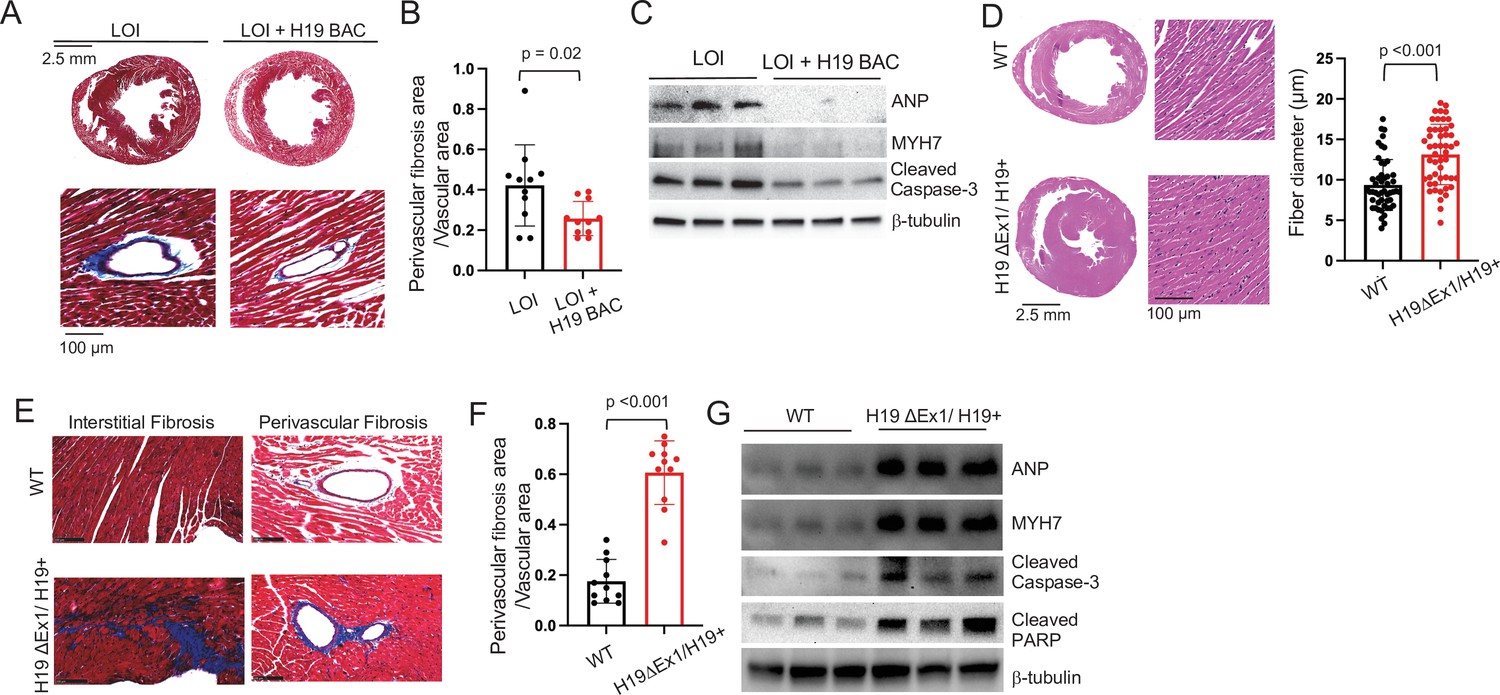
LOI pathologies in adult mice are H19-dependent.
(A–C) An H19 transgene rescues pathologies in LOI mice. Phenotypes of LOI mice or their LOI littermates that also carry an H19 Bacterial Artificial Chromosome transgene that restores wild-type levels of H19 RNA (LOI+ H19 BAC). (D–G) H19 deletion is sufficient to cause cardiac pathologies. Phenotypes in wild-type (WT) mice and in littermates carrying the H19ΔEx1 deletion. For histology (A, B, E, F) hearts were isolated from 6 month old animals and transverse sections collected midway along the longitudinal axes before staining with hematoxylin and eosin (D) or with Masson’s trichrome (A, B, E, F) Statistical significance was analyzed using Student’s t-test type 2. For immunoblotting (C, G), hearts were isolated from 1 year animals and investigated for ANP, Myh7, Serca2, Cleaved Caspase-3, and Cleaved PARP. β-Tubulin is a loading control. See Figure 4—figure supplement 1 for quantified data.
-
Figure 4—source data 1
Pathologies in LOI mice are caused by decreased function of the H19 lncRNA.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig4-data1-v2.xlsx
We next investigated whether loss of H19 is sufficient to induce pathologies and also investigated exactly which H19 RNA was important. The H19 gene encodes a 2.3 kb lncRNA which is exported to the cytoplasm but also is the precursor for microRNAs, Mir675-5p and Mir675-3p (Cai and Cullen, 2007). Since LOI mice show reduced levels of both the lncRNA and of Mir675-3p and -5 p and because the H19 BAC transgene restores expression of both the lncRNA and the Mir675 microRNAs, these models were not helpful in determining which RNA species prevents cardiac pathology. The H19ΔEx1 allele is a 700 bp deletion of the 5’ end of exon one that leaves bases encoding the Mir675-3p and Mir675-5p intact (Figure 1B). This ΔEx1 deletion does not prevent H19 transcription but rather, reduces H19 lncRNA levels by destabilizing the truncated transcript (Srivastava et al., 2003), raising the possibility that the ΔEx1 mutation might affect only the lncRNA. In fact, we show here that levels of Mir675-5p and -3 p are unaltered in H19ΔEx1/H19+ (Figure 2—figure supplement 3). Yet, 6-month-old H19ΔEx1/H19+ mice display LOI cardiac pathologies including hypertrophy (Figure 4D), fibrosis (Figure 4E,F), and expression of disease markers (Figure 4G, Figure 4—figure supplement 1B). Thus, we conclude that loss of H19 lncRNA is sufficient to induce cardiomyopathy in adult mice.
In addition to establishing the critical importance of H19 lncRNA, these genetic experiments also uncouple neonatal hypertrophy and adult pathology: neonatal LOI+ H19 BAC mice show hypertrophy but do not develop adult pathologies, while neonatal H19ΔEx1/H19+ have normal-sized hearts but do develop pathologies. Thus, neonatal cardiomegaly is not a risk factor for adult pathologies.
H19 lncRNA regulates the frequency of endothelial to mesenchymal transition in mice and in isolated primary endothelial cell cultures
H19 expression is not uniform throughout the heart but rather restricted to endothelial cells (ECs) (Figure 5A,B). In fetal and neonatal hearts H19 is expressed in all endothelial cells including microvasculature. In adults, H19 expression is patchy and patterns vary between mice but are always restricted to endocardium and endothelial cells lining major coronary vessels (Figure 5A). Localization was confirmed in vasculature by co-staining for both endothelial and smooth muscle markers. For example, in coronary vessels, H19 RNA expression exclusively overlapped with endothelial specific marker von-Willebrand’s factor (Figure 5B).
Figure 5 with 1 supplement see all

H19 influences gene expression and cell fate in cardiac endothelial cells.
(A) In situ staining for H19 (brown) in hearts from wild-type P1 neonates or 6 month adults. (B) Combined in situ and immunohistochemistry for hearts isolated from wild-type and H19ΔEx1/H19+ littermates at 6 months shows expression of H19 is concentrated in endothelial cells. Sections were stained for H19 lncRNA and then with antibodies to the endothelial marker, vWF (von Willebrand factor), or to the smooth muscle marker, α−SMA (alpha smooth muscle actin). (C) MA blot showing differences in expression of polyadenylated RNAs in H19-deficient endothelial cells. Endothelial cells were isolated from wild-type (N = 4) and H19ΔEx1/H19+ (N = 3) P2 neonatal hearts based on CD31 expression. RNAs were isolated and polyadenylated transcripts were quantitated. Genes marked in red are significantly differentially expressed at FDR < 0.05. Gene ontology analyses are described in Figure 5—figure supplement 1. (D) Transient transfection of H19ΔEx1/H19+ cardiac endothelial cells with an H19-expression vector rescues expression of key EndMT genes. Cardiac endothelial cells were isolated from wild-type and H19-deficient P2 hearts as described in (D) and transfected with empty expression vector (pcDNA3) or with pcDNA3 carrying mouse H19 gDNA (pcDNA3-H19). After + hours in culture, RNA was extracted and cDNAs synthesized and analyzed for H19, SM22α, Snail, or Slug. For each gene, cDNA levels were normalized to GAPDH and then to the levels seen in wild-type cells transfected with pcDNA3 only. (E, F). Increased frequency of EndMT transitioning cells in H19-deficient mice. (E) Hearts from wild type and H19ΔEx1/H19+ were isolated at e14.5, P1, and at 6 months. Sections were probed for endothelial cell markers (CD31 or IB4) and for mesenchymal markers (αSMA or SM22α) to identify cells co-expressing these genes. (F) Frequencies of cells co-expressing endothelial and mesenchymal markers in adult and P1 hearts. The role of H19 was determined by three independent comparisons: wild type vs. Η19ΔEx1/H19+, LOI vs LOI+ H19 BAC, wild type vs LOI. (D, F). Statistical significance was evaluated using Student’s t-test type 2.
-
Figure 5—source data 1
H19 regulates gene expression and cell fate in cardiac endothelial cells.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig5-data1-v2.xlsx
To identify a possible function for H19 RNA, we performed transcriptomic analyses comparing RNAs isolated from wild-type and H19-deficient P1 hearts. Using whole heart extracts, we did not identify significant differences in gene expression. We next compared RNAs isolated from purified ECs. Hearts were dissociated into single cells using enzyme digestion and mechanical agitation and then endothelial cells were isolated based on expression of CD31 antigen. About 30,000 cells per neonatal heart were isolated to >95% purity. RNA sequencing identified 228 differentially expressed genes (DEGs) with adjusted p-values of <0.1, including 111 upregulated and 117 downregulated transcripts (Figure 5C). GO analysis for biological, cellular, and molecular pathways gives evidence for a change in cellular identity (Figure 5—figure supplement 1). Specifically, enriched biological pathways included positive regulation of mesenchymal cell proliferation, positive regulation of endothelial cell migration, and cell adhesion (n = 36, p-adj = 0.003). Cellular pathways showed enrichment for genes coding for extracellular matrix (n = 42, p-adj = 1.83E-10). Enriched molecular function categories include extracellular matrix binding and TGFβ binding (n = 7, p-adj = 0.0001; n = 5, p-adj = 0.1), as well as other pathways that are especially active during endothelial to mesenchymal transition (EndMT). EndMT is not an identifiable GO term, however, we conducted a PubMed search of the 188 DEGs described in the PubMed literature database and noted that 63 DEGs were implicated in EndMT as either players in driving the transition or as markers. Some examples include Transforming growth factor beta receptor 3 (Tgfbr3, up 1.5×, padj = 0.01), Collagen Type XIII α1 chain (Col13a1, up 2.0×, padj = 4.2E-09), Bone Morphogenic Protein 6 (Bmp6, down 0.6×, padj = 7.0E-05), Latent Transforming Growth Factor Binding Protein 4 (Ltbp4, down 0.5×, padj = 0.008), Connective Tissue Growth Factor (Ctgf, down 0.7×, padj = 0.06), Slit Guidance Ligand 2, (Slit2, up 1.6×, padj = 2.5E-05), and α2 macroglobin (A2m, down 0.6×, 5.6E-05). Due to the results of the GO term analysis as well as the PubMed search, we speculated that H19 might play a role in regulating EndMT.
To directly test the role of H19 in regulating EC gene biology, we isolated primary ECs from wild-type and H19-deficient P2 littermates and transfected with an H19 expression vector or with an empty control vector and then assayed gene expression after 24 hr. H19 expression reduces expression of a mesenchymal cell marker (SM22α) and of genes encoding transcription factors critical for EndMT (Snail and Slug) (Figure 5D).
EndMT is an essential part of the normal development of many tissues/organs including heart. For example, EndMT is critical in cardiac valve development (Kisanuki et al., 2001; Markwald et al., 1977). Studies also report that EndMT contributes to cardiac diseases including cardiac fibrosis, valve calcification, and endocardial elastofibrosis (Evrard et al., 2016; Goumans et al., 2008; Piera-Velazquez et al., 2011; Zeisberg et al., 2007). During the actual EC transition, cells will transiently express endothelial markers (like CD31or IB4) simultaneously with mesenchymal markers (like aSMA or SM22a). To understand the impact of H19 deficiency on EC transition in vivo, we fixed and sectioned hearts isolated at several developmental stages from H19ΔEx1/H19+ and their wild-type littermates mice and looked for co-staining of these endothelial and mesenchymal markers. At each stage, we focused on the regions of the heart where H19 expressing cells were particularly abundant, assuming that this is where a phenotype would be most readily observed. In e14.5 embryos, we looked at endocardium, epicardium, valves, and blood vessels. In P1 embryos, we looked at endocardium, valves, and blood vessels. In adult hearts, we looked at endocardium. Comparable sections for wild-type and mutant mice were identified by a cardiac pathologist blinded to genotype before we stained for EC and mesenchymal markers. In each stage. we noted significant changes in co-staining frequency indicating that the likelihood of EC cell transition is increased in the absence of H19 (Figure 5E,F). We confirmed that these results through independent analyses that compared LOI mice with their wild-type littermates (H19ΔICR/H19+ vs. H19+/H19+) and that compared LOI mice with LOI littermates that also carried the H19 BAC transgene H19ΔICR/H19+ vs H19ΔICR/H19+ H19 BAC Transgene (Figure 5F).
Mirlet7 binding sites on the H19 lncRNA are essential for normal cardiac physiology
H19 lncRNA is known to physically bind Mirlet7g (let-7g) and Mirlet7i (let-7i) in exon one and Mirlet7e (let-7e) in exon four in mice (Kallen et al., 2013). One proposed mechanism for H19 lncRNA function is that it regulates Mirlet7 microRNAs via these interactions to modulate their biological activities. Mirlet7 miRNAs are known to play a role in cardiovascular diseases including cardiac hypertrophy, cardiac fibrosis, dilated cardiomyopathy, and myocardial infarction (Bao et al., 2013).
To test the role of H19’s Mirlet7 binding in preventing cardiomyopathy, we used CRISPR/Cas9 genome editing to delete Mirlet7 binding sites in the H19 gene (Figures 1A and 6A). Mice carrying this mutation (H19ΔLet7/ H19ΔLet7) express H19 at wild-type levels (Figure 6B), which shows that the deletions do not disrupt lncRNA expression or stability. We used sequence-specific biotinylated oligonucleotides to purify H19 lncRNA from extracts prepared from wild-type and from H19Δlet7/H19Δlet7 cells and assayed for co-purification of Mirlet7 microRNAs. These experiments provided direct biochemical evidence that H19 and Mirlet7 RNAs do physically associate and also that these interactions are lost in our H19Δlet7 deletion model (Figure 6—figure supplement 1).
Figure 6 with 2 supplements see all

H19’s Mirlet7 binding domains are essential for normal function.
(A) The H19ΔLet7 allele was generated by deleting 25 and 48 bp sequences within exons 1 and 4 to eliminate binding sites for Mirlet7g, Mirlet7i, and Mirlet7e miRNAs. (B) The H19ΔLet7 allele is expressed at wild-type levels. RNAs were isolated from hearts from H19ΔLet7/H19+ and quantitated by qRT-PCR, normalizing first to GAPDH and then to the levels of H19 observed in H19+/H19+. Similarly, Igf2 is expressed at equivalent levels in wild-type and mutant mice. Thus phenotypes associated with the H19Δlet7 allele should be ascribed to changes in H19 function and not to changes in H19 levels or in Igf2 expression. See Figure 6—figure supplement 1 for experiments demonstrating that the H19Δlet7 lncRNA cannot interact with Mirlet7 miRNAs. (C) Transverse sections were collected midway along the longitudinal axis from hearts collected from 12-month-old wild-type (N = 4) and mutant (N = 3) littermates and stained with hematoxylin and eosin. (D) Fiber diameters were quantitated using three sections per mouse. (E, F). Masson’s trichrome staining of sections described in panel (C) Red, muscle fibers; blue, collagen. Sections from three wild-type and four mutant littermates were used to calculate fibrosis. (G) Immunoblot analyses of whole heart extracts prepared from 12 month WT (N = 3) and mutant littermates (N = 3). Altered expression of ANP, Myh7, Cleaved Caspase-3, Cleaved Caspase-7, and Cleaved PARP. β-tubulin is a loading control. Quantified data is presented in Figure 6—figure supplement 2. Statistical significance was evaluated with Student’s t-test type 2.
-
Figure 6—source data 1
Mirlet7 binding sites on the H19lncRNA are essential to prevent cardiomyopathy.
- https://cdn.elifesciences.org/articles/67250/elife-67250-fig6-data1-v2.xlsx
One year old H19ΔLet7/H19ΔLet7 mice displayed cardiomegaly as measured by a 32% increase in heart weight/tibia length ratios (wild type = 7.5 ± 1.7 mg/mm, N = 4; H19ΔLet7/H19+ = 9.9 ± 2.2 mg/mm, N = 5; p=0.007). Transverse sections also suggested hypertrophy (Figure 6C), which was quantified as increased fiber diameter (Figure 6D). The cardiac hypertrophy in H19ΔLet7/H19ΔLet7 mice was accompanied by increased interstitial and perivascular fibrosis (Figure 6E,F). The pathologic nature of the observed cardiac myopathies in these mutant mice was confirmed by the increased levels of cardiomyopathy markers (Figure 6G, Figure 6—figure supplement 2). Thus our results support a role for Mirlet7 miRNA binding to H19 lncRNA in preventing cardiomyopathies.
Discussion
BWS is an overgrowth disorder with significant patient to patient variation in disease symptoms (Jacob et al., 2013). An explanation for some of this variability is that independent molecular mechanisms for BWS exist (Weksberg et al., 2010). More than 50% of BWS cases are associated with epigenetic lesions that disrupt expression of CDKN1C, an imprinted gene closely linked to IGF2/H19 but under control of its own ICR (IC2). (More rarely, BWS cases are associated with pathogenic lesions in the CDKN1C peptide coding sequences.) About 5% of BWS cases are associated with disrupted imprinting at the IGF2/H19 locus. About 20% of cases are associated with paternal uniparental disomy of the entire region (potentially affecting both CDKN1C and IGF2/H19) (IC1 and IC2), and another 20% of cases are of unknown origin.
There are conflicting studies regarding the correlation of artificial reproductive technology (ART) in the development of BWS. Doornbos et al., 2007 suggested no direct effect on the increase of imprinted diseases after correcting for the increased fertility problems of the parents. However, more recent studies have examined the effect of ART on epigenetic changes (Odom and Segars, 2010) and suggest a 6- to 10-fold increased risk for BWS specifically because of the increased chance that IGF2/H19 imprinting is disrupted (Hattori et al., 2019; Johnson et al., 2018; Mussa et al., 2017). Interestingly, BWS patients associated with ART are more likely to show cardiac problems (Tenorio et al., 2016), suggesting a role for IGF2/H19 expression in normal heart development and function.
In this study, we characterize a mouse model for Igf2/H19 LOI. This model deletes the Imprinting Control Region upstream of the H19 promoter and recapitulates the molecular phenotype of BWS patients: biallelic (i.e. 2× dosage) IGF2 and reduced H19 RNA. Here we show that this model not only phenocopies the transient cardiomegaly observed in neonates but also displays cardiovascular dysfunctions that are only rarely observed in patients.
To elucidate the molecular and developmental etiology of these cardiovascular phenotypes, we characterized two additional mouse models that independently altered expression of Igf2 and of H19. These genetic analyses demonstrated that overexpression of Igf2 and loss of H19 play distinct roles in driving BWS cardiac phenotypes. In neonates, increased levels of circulating IGF2 results in hyperactivation of mTOR signaling in cardiomyocytes and thus leads to cardiomyocyte hyperplasia and cellular hypertrophy but the resultant cardiomegaly in mice is transient. As in humans, expression of Igf2 in mice is strongly downregulated after birth and organ sizes return toward normal. Loss of H19, however, results in progressive cardiac pathology. Aged H19 deficient mice show increased fibrosis, expression of markers indicative of cardiac failure, abnormal echocardiography phenotypes, low blood pressure, and aberrant vasculature. Thus, in the mouse LOI model, disease phenotypes are not restricted to fetal and neonatal stages. It will be interesting and important to assess whether this is true in other mammals.
In hearts, H19 expression is restricted to endothelial cells. To understand the significance of H19 expression we isolated cardiac ECs from wild-type and mutant neonates. Transcriptome analyses showed altered expression of genes associated with endothelial to mesenchymal transition, suggesting that H19 might help regulate EC cell fate. Supporting this idea, we saw that forcing expression of H19 in primary ECs prevents activation of mesenchymal gene expression patterns. Finally, we saw that H19-deficient mice show significant increases in the frequency of EC cells simultaneously expressing mesenchymal markers.
The ability of some ECs to transition of mesenchymal cells is necessary for normal development and thus can be assumed to be an essential property of ECs. The phenotypes of H19-deficient mice do not suggest that H19 lncRNA is the single key molecule regulating EC cell fate. Even in LOI mice, EndMT is almost always occurring only when developmentally appropriate. Rather, our data indicate that H19 RNA levels play a role in modulating the fate decision, so that cells lacking H19 are modestly but measurably more likely to switch toward a transitional state where both EC and mesenchymal markers are expressed. It is interesting to note one commonality of key pathways disrupted by loss of H19 lncRNA is that they share regulation by TGFβ signaling, suggesting that the observed 50 % reduction in expression of TGFβ receptors might be a key phenotype in H19-deficient ECs (Goumans et al., 2008).
Our findings extend earlier studies showing patterns of H19 expression in development and in response to injury suggesting a role for H19 in vascular physiology and pathology (Jiang et al., 2016; Kim et al., 1994). Moreover, Voellenkle et al., 2016 recently described a role for lncRNAs including H19 in the physiology of umbilical vein endothelial cells.
Our results also agree with in vitro studies that demonstrated an important role for H19 in regulating EMT in cancer cells (Li et al., 2019; Ma et al., 2014; Matouk et al., 2016; Matouk et al., 2014; Wu et al., 2019; Zhang et al., 2018). In these previous analyses, H19 function was determined by transfecting cancer cells with H19-expression vectors and analyzing cell motility and gene expression. However, in contrast to our findings that activation of mesenchymal expression is associated with loss of H19, these in vitro analyses find EMT is induced by increasing H19 RNA. This discrepancy emphasizes the useful role for genetic animal models in addressing developmental disorders where phenotypes are coming from cumulative changes in multiple cell types and over long periods of time. In animal models, observed phenotypes are due to the cumulative effect of the mutation in many cell types and over developmental time. In vitro studies of H19 have focused on the effect of acute changes in levels of H19 in a single cell type.
Our analyses cannot address the fate of cells that co-express EC and mesenchymal markers. Do they all proceed toward full EndMT, do they return toward EC fates, or do they teeter in between? These questions can be addressed in future experiments using conditional H19 deletion alleles and cell fate markers.
H19 can be a very abundant transcript. In neonatal ECs, H19 lncRNA represents about 1% of all polyadenylated RNA. Yet its biochemical functions remain unclear. Various studies support the idea that H19 functions as a microRNA precursor (Cai and Cullen, 2007; Dey et al., 2014; Keniry et al., 2012), as a p53 protein inhibitor (Hadji et al., 2016; Park et al., 2017; Yang et al., 2012; Zhang et al., 2017), as a regulator of DNA methylation (Zhou et al., 2019; Zhou et al., 2015), and as a modulator of Mirlet7 microRNA functions (Gao et al., 2014; Geng et al., 2018; Kallen et al., 2013; Peng et al., 2017; Zhang et al., 2019; Zhang et al., 2017). It is possible that H19 functions vary from cell type to cell type (Raveh et al., 2015). Alternatively, these functions might co-exist in a single cell but analyses to date have only looked at H19 function from single perspectives and have missed its ability to perform in multiple pathways. To address this issue, we have begun to generate mutant H19 alleles that disrupt specific functions. Here we show that H19ΔEx1/H19+ have 100× reduced levels of lncRNA but almost normal levels of Mir675-3p and -5 p and still show cardiac pathology. Thus, the pathologies in LOI mice depend on the loss of H19 lncRNA. To then address how the lncRNA might function, we generated mice carrying an H19 allele missing Mirlet7 binding sites. These mice show cardiac pathologies including extreme fibrosis. We find the fibrosis phenotype in H19ΔLet7 mice to be especially interesting. We speculate that intensity relative to that seen in an H19 null is most consistent with the idea that H19 lncRNA has multiple roles in the cell and by disrupting only one role we have altered some balance, so that the animal is worse off than having no H19 at all.
The strong phenotype in H19ΔLet7 mice is consistent with several previous studies that emphasize the importance of H19 lncRNA interactions with Mirlet7 miRNAs but it is also paradoxical in that multiple studies of Mirlet7 function in hearts indicate that Mirlet7 functions as an anti-fibrotic factor. That is, reduced Mirlet7 is a risk factor for fibrosis and fibrosis induces Mirlet7, presumably as a corrective measure (Bao et al., 2013; Elliot et al., 2019; Sun et al., 2019; Wang et al., 2015). The increased fibrosis in H19ΔLet7 mice suggests that the simple model (that H19 binds to and reduces Mirlet7 bioavailability) is not correct or, more likely, that complex developmental interactions play critical roles that determine phenotypes in ways that are not yet understood. Either way, our results confirm the importance of animal models and the need for even more sophisticated conditional deletions.
H19 and Igf2 are generally thought of as fetal genes since their expression is so strongly repressed after birth. This fact might suggest that the adult phenotypes in H19-deficient mice are downstream effects of the loss of H19 in the developing heart. However, as already mentioned, at peak expression, H19 levels are extraordinarily high. Thus, even after 100-fold developmentally regulated decrease, H19 remains one of the top 100 genes in terms of RNA levels. For this reason, conditional ablation models will be needed to determine exactly when H19 expression is important.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Mus musculus) | H19 | GeneBank | NR_130973.1 | |
| Strain, strain background (Mus musculus) | C57BL/6 J (B6) | Jackson Labs | Jax: 000664 | |
| Strain, strain background (Mus musculus) | B6.FVB-Tg (Myh6-cre)2,182Mds/J | Jackson Labs | Jax: 011038 | |
| Strain, strain background (Mus musculus) | H19ΔICR | Previous study from this lab | PMID:10817754 | Maintained by B6 backcross |
| Strain, strain background (mouse) | H19 BAC Transgene | Previous study from this lab | PMID:10921905 | Maintained by B6 backcross |
| Strain, strain background (mouse) | H19ΔEx1 | Previous study from this lab | PMID:12270940 | Maintained by B6 backcross |
| Strain, strain background (mouse) | H19ΔLet7 | This study | See Materials and methods | Maintained by B6 backcross |
| Cell line (Rattus norvegicucs) | H9c2 cells: Rat myocardium (embryo) | ATCC CRL-1446 | RRID:CVCL_0286 | |
| Transfected construct (vector control) | pcDNA3.1+ | Invitrogen | V790-20 | |
| Biological sample (Mus musculus) | Primary myoblasts | This study | See Materials and methods | Isolated from neonates |
| Biological sample (Mus musculus) | Primary cardiac endothelial cells | This study | See Materials and methods | Freshly isolated from neonates |
| Biological sample (Mus musculus) | Primary cardiomycytes | This study | See Materials and methods | Freshly isolated from neonates |
| Antibody | See Supplementary file 3 for list of 23 primary and six secondary antibodies | |||
| Recombinant DNA reagent | pcDNA3-H19 | This study | Mouse EcoRI-SalI fragment cloned into Nhe-XhoI of pcDNA3 | |
| Sequence-based reagent | See Supplementary file 2 for list of oligonucleotides used for PCR | |||
| Peptide, recombinant protein | Human Recombinant IGF2 | Preprotech | Catalog # 100–12 | |
| Commercial assay or kit | Mouse IGF2 ELISA Kit | Abcam | ab100696 | |
| Commercial assay or kit | H19 in situ probe | Advanced Cell Diagnostics | ACD 423751 | |
| Commercial assay or kit | H19 in situ probe | Advanced Cell Diagnostics | ACD 322360 | |
| Chemical compound, drug | BMS 754807 | Active Biochem | A-1013 | |
| Chemical compound, drug | MEK1/2 Inhibitor PD98059 | Cell Signaling Technology | #9900 | |
| Software, algorithm | DESeq2 | RRID:SCR_015687 | ||
| Software, algorithm | Bioconductor | RRID:SCR_006442 | ||
| Software, algorithm | Feature Counts | RRID:SCR_012919 | ||
| Software, algorithm | Debian | RRID:SCR_006638 | ||
| Software, algorithm | STAR | RRID:SCR_004463 |
Animal studies
Request a detailed protocolAll mice were bred and housed in accordance with National Institutes of Health and United States Public Health Service policies. Animal research was performed only after protocols were approved by the National Institute of Child Health and Human Development Animal Care and Use Committee.
H19ΔICR/H19+ (Srivastava et al., 2000) and wild-type littermates or H19ΔEx1/H19+ (Srivastava et al., 2003) and wild-type littermates were generated by backcrossing heterozygous females with C57BL/6 J males (Jackson Labs 000664). For tissue-specific LOI, we crossed H19ΔICR/H19ICRflox females (Srivastava et al., 2000) with males hemizygous for the Myh6-Cre transgene (Jackson Labs 011038) (Agah et al., 1997). The H19 BAC transgene was generated as described (Kaffer et al., 2001; Kaffer et al., 2000) and used to generate H19ΔICR/H19+ BAC+ females for backcrossing with C57BL/6 J males.
The H19ΔLet7 allele was generated using CRISPR/Cas9 gene editing of RI mouse embryonic stem cells (ESCs). In step 1, we used gRNAs 5’-CACCGAGGGTTGCCAGTAAAGACTG-3’ and 5’-CACCGCTGCCTCCAGGGAGGTGAT-3’ to delete 25 bp (AGACTGAGGCCGCTGCCTCCAGGGAGGTGAT) in exon 1. In step 2, we used gRNAs: 5’-CACCGCTTCTTGATTCAGAACGAGA-3’ and 5’-CACCGACCACTACACTACCTGCCTC-3’ to delete 48 bp (CGTTCTGAATCAAGAAGATGCTGCAATCAGAACCACTACACTACCTGC) in exon 4. Positive clones were identified by PCR screens and then confirmed by sequencing 686 bp spanning the exon 1 deletion and 1086 bp spanning the exon four deletion. Founder mice were obtained by injecting mutated ESCs into C57BL/6 J blastocyts and then backcrossed twice with C57BL/6 J females.
Genotypes were determined by PCR analyses of gDNAs extracted from ear punch biopsies (Supplementary file 2).
Electrocardiography measurements
Request a detailed protocolTransthoracic echocardiography was performed using a high-frequency linear array ultrasound system (Vevo 2100, VisualSonics) and the MS-400 Transducer (VisualSonics) with a center operating frequency of 30 MHz, broadband frequency of 18–38 MHz, axial resolution of 50 mm, and footprint of 20 × 5 mm. M-mode images of the left ventricle were collected from the parasternal short-axis view at the midpapillary muscles at a 90° clockwise rotation of the imaging probe from the parasternal long-axis view. Form the M-mode images, the left ventricle systolic and diastolic posterior and anterior wall thicknesses and end-systolic and -diastolic internal left ventricle chamber dimensions were measured using the leading-edge method. Left ventricular functional values of fractional shortening and ejection fraction were calculated from the wall thicknesses and chamber dimension measurements using system software. Mice were imaged in the supine position while placed on heated platform after light anesthesia using isoflurane delivered by nose cone.
Blood pressure measurements
Request a detailed protocolAfter sedation with isoflurane, a pressure catheter (1.0-Fr, model SPR1000, Millar Instruments, Houston, TX) was inserted into the right carotid and advanced to the ascending aorta. After 5 min acclimation, pressures were recorded using Chart 5 software (AD Instruments, Colorado Springs, CO) (Knutsen et al., 2018).
Arterial pressure-diameter testing
Request a detailed protocolAscending aortas (from the root to just distal to the innominate branch point) and left carotid arteries (from the transverse aorta to 6 mm up the common carotid) were dissected and mounted on a pressure arteriograph (Danish Myotechnology, Copenhagen, Denmark) in balanced physiological saline (130 mM NaCl, 4.7 mM KCl, 1.6 mM CaCl2, 1.18 mM MgSO4·7H2O, 1.17 mM KH2PO4, 14.8 mM NaHCO3, 5.5 mM dextrose, and 0.026 mM EDTA, pH 7.4) at 37 °C. Vessels were transilluminated under a microscope connected to a charge-coupled device camera and computerized measurement system (Myoview, Danish Myotechnology) to allow continuous recording of vessel diameters. Prior to data capture vessels were pressurized and stretched to in vivo length (Wagenseil et al., 2005). Intravascular pressure was increased from 0 to 175 mmHg in 25 mmHg steps. At each step, the outer diameter (OD) of the vessel was measured and manually recorded. Segmental distensibility was calculated from the pressure diameter curves as follows: distensibility (SD25) over a 25 mmHg interval = [ODHigher Pressure (H) − ODLower Pressure(L)]/OD(L)/25 (Knutsen et al., 2018).
Histological analyses
Request a detailed protocolHearts from adult mice were fixed by Langendorff perfusion or by transcardiac perfusion using 4 % paraformaldehyde (PFA) and embedded in paraffin. Fetal and neonatal hearts were isolated and then fixed by submersion in 4 % PFA before embedding. From embedded hearts, we obtained 5 mm transverse sections for analysis. Masson’s Trichrome (Sigma Aldrich, HT15, St. Louis Missouri) and Picosirius Red (Sigma Aldrich, 365548) staining were according to supplier’s instructions. Fiber diameter index was quantitated using Hamamatsu-NDP software.
Immunofluorescence and immunohistochemistry
Request a detailed protocolPrimary myocytes and H19C2 cells were fixed with 4% PFA, permeabilized with 0.5% Triton, and blocked with 10% normal serum before incubation with antibodies. Paraffin sections were deparaffinized and rehydrated according to standard protocols. Antigen retrieval was applied using citrate buffer (Abcam, 1b93679, Cambridge, MA) for 20 min and then maintained at a sub-boiling temperature for 10 min. Sections were treated with serum-free blocking solution (DAKO, X0909, Santa Clara, CA) and all antibodies (Supplementary file 3) diluted in antibody diluent solution (DAKO, S0809). Secondary staining was performed for 30 min at RT. Samples were imaged with a Carl Zeiss 880 laser scanning microscope using a 40× oil immersion objective. Images were composed and edited in ZEN&LSM image software provided by Carl Zeiss or Illustrator 6.0 (Adobe).
RNA in situ hybridization
Request a detailed protocolSingle color probes for H19 were purchased from Advanced Cell Diagnostics (ACD 423751, Newark, CA). RNA in-situ hybridization was performed on paraffin sections using the 2.5 HD Brown Detection Kit (ACD 322310). For dual staining with antibodies, we used H19-RD chromagen kit (ACD 322360).
Immunoblotting
Request a detailed protocolCell extracts and tissue extracts were prepared using M-PER mammalian protein extraction buffer (Thermo Fisher 78501, Waltham, MA) or T-PER tissue protein extraction buffer (Thermo Fisher 78510), respectively. Protein concentrations were assayed using a BCA Protein Assay Kit (Pierce 23227, Waltham, MA). Proteins were fractionated by electrophoresis on 12% or on 4%–20% SDS−PAGE gels and then transferred to nitrocellulose. Antibodies (Supplementary file 3) were diluted in antibody enhancer buffer (Pierce 46644).
Cell culture
Request a detailed protocolPrimary cardiomyocytes were isolated form P1 pups using the Pierce Primary Cardiomyocyte Isolation Kit (Thermo Fisher 88281). H19c2 (Branco et al., 2015) cells were purchased from ATCC (CXRL-1446). Cells were grown at 37°C in 5% CO2 in DMEM + 10% FBS. Cardiomyocyte identity was confirmed by cell morphology and by immunohistochemistry for cardiomyoyte-specific markers including Myh6. Cell surface index was quantitated using Carl Zeiss-LSM software (n = 50 for each of three independent experiments).
To prepare primary endothelial cells, neonatal hearts were isolated and dissociated into single cells using Miltenyi Biotec Neonatal Heart Dissociation Kit (130-098-373, Gaithersburg, MD) but omitting the Red Cell Lysis step. Endothelial cells were purified based on CD31 expression (Miltenyi Biotec Neonatal Cardiac Endothelial Cell Isolation Kit, 130-104-183).
Primary myoblasts were generated and grown as described (Park et al., 2017). Cell identify is confirmed by expression of myoblast-specific markers (including MyoG), and by determining that upon serum depletion, >90% cells will differentate into elongated, multinucleate myotubes.
Cells are negative for mycoplasma contamination.
Quantitative real-time PCR for RNA samples
Request a detailed protocolConventional RNAs were prepared from three to five independent biological samples, analyzed using a Thermo Fisher NANODROP 2000c to evaluate purity and yield, and then stored at –70°C. cDNA samples were prepared with and without reverse transcriptase using oligo-dT primers (Roche, 04 887 352 001). cDNAs were analyzed using SYBR Green (Roche, 04 887 352 001) on the Roche Light Cycler 480 II (45 cycles with annealing at 60°C) using primers described in Supplementary file 2. For each primer pair, we established standard curves to evaluate slope, y-intercepts, and PCR efficiency and to determine the dynamic range of the assay. Assay specificity was determined by melting point analyses and gel electrophoresis.
For microRNA analyses, we used mirVanaTM miRNA Isolation Kit and TaqMan MicroRNA Assays Thermo Fisher, 4437975; Assay ID 001973 (U6), 001940 (Mir675-5p), 001941 (Mir675-3p).
RNA pull-down
Request a detailed protocolPrimary myoblast cell cultures were generated and grown as described (Park et al., 2017). Cell RNA pull-down assay was performed essentially as previously described (Lee et al., 2020). Briefly, cells were fixed for 10 minutes at room temperature with 2.5% formaldehyde in PBS before quenching with 0.125 M glycine for 5 min. Cells were rinsed, resuspended in NET-2 buffer (50 mM Tris–HCl, pH 7.4, 150–300 mM NaCl, 0.05% NP-40, 0.05% deoxycholic acid, 1 mM PMSF, 2 mM benzamidine), and sonicated. Cell lysates were incubated with biotin-labeled probes synthesized by Eurofins Genomics and then pulled down with streptavidin beads (Sigma-Aldrich). Precipitated RNA and miRNA were subsequently subjected to RT-qPCR. The sequences of biotin-labeled h19 probes used in this study were as follows: CTCAGTCTTTACTGGCAACC-BIOTIN, TGTAAAATCCCTCTGGAGTC-BIOTIN, CTCCCTAGAAACTCATTCAT-BIOTIN, CTTCGAGACACCGATCACTG-BIOTIN, ATGTCATGTCTTTCTGTCAC-BIOTIN, TTGACACCATCTGTTCTTTC-BIOTIN, AAGAGGTTTACACACTCGCT-BIOTIN. The sequence of biotin-labeled control probe was CCTACGCCACCAATTTCGT-BIOTIN.
ELISA
Request a detailed protocolIGF2 secreted peptide was assayed with the Mouse IGF2 ELISA KIT (Abcam, ab100696) on 10 independent samples.
RNA sequencing and analyses
Request a detailed protocolFor analyses in adult animals, RNAs were isolated from 6 month H19ΔICR/H19ΔICR and H19+/H19+ littermates (two per genotype) using RNeasy Plus Mini Kit (Qiagen). Samples with RNA Integrity numbers > 9 were Ribosomal RNA depleted using RiboZero Gold Kit (Illumina). Libraries were prepared using an RNA Sample Prep V2 Kit (Illumina) and sequenced (Illumina HiSeq2500) to generate paired-end 101 bp reads that were aligned to the mouse genome version mm10 using STAR v2.5.3a (Dobin et al., 2013). Differential expression analyses were performed using DESeq2 (Love et al., 2014).
For analyses in neonates, RNAs were isolated from purified cardiac endothelial cells isolated from H19ΔEx1/H19+ (N = 3) and H19+/H19+ (N = 4) littermates. Libraries were generated from samples with RNA Integrity Numbers > 9 and were sequenced and analyzed as described above.
Data availability
Sequencing data are deposited in the NCBI Gene Expression Omnibus (GEO) under series accession number GSE111418.
-
NCBI Gene Expression OmnibusID GSE111418. Mis-expression of Igf2 and H19 work independently on distinct cell types to cause cardiomyopathy in a Beckwtih Wiedemann mouse model.
References
-
Let-7 in cardiovascular diseases, heart development and cardiovascular differentiation from stem cellsInternational Journal of Molecular Sciences 14:23086–23102.https://doi.org/10.3390/ijms141123086
-
Genomic imprinting in mammalsCold Spring Harbor Perspectives in Biology 6:a018382.https://doi.org/10.1101/cshperspect.a018382
-
The product of the h19 gene may function as an RNAMolecular and Cellular Biology 10:28–36.https://doi.org/10.1128/mcb.10.1.28-36.1990
-
BMS-754807, a small molecule inhibitor of insulin-like growth factor-1R/IRMolecular Cancer Therapeutics 8:3341–3349.https://doi.org/10.1158/1535-7163.MCT-09-0499
-
Beckwith-wiedemann syndrome: Historical, clinicopathological, and etiopathogenetic perspectivesPediatric and Developmental Pathology 8:287–304.https://doi.org/10.1007/s10024-005-1154-9
-
Association of in vitro fertilization with Beckwith-wiedemann syndrome and epigenetic alterations of lit1 and h19American Journal of Human Genetics 72:156–160.https://doi.org/10.1086/346031
-
Constitutional H19 hypermethylation in a patient with isolated cardiac tumorAmerican Journal of Medical Genetics. Part A 146A:2126–2129.https://doi.org/10.1002/ajmg.a.32421
-
Star: Ultrafast Universal RNA-SEQ alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Vascular malformation and choroid plexus adrenal heterotopia: New findings in Beckwith-wiedemann syndrome?Fetal and Pediatric Pathology 25:191–197.https://doi.org/10.1080/15513810601015704
-
MICRORNA let-7 downregulates ligand-independent estrogen receptor-mediated male-predominant pulmonary fibrosisAmerican Journal of Respiratory and Critical Care Medicine 200:1246–1257.https://doi.org/10.1164/rccm.201903-0508OC
-
The H19/let-7 double-negative feedback loop contributes to glucose metabolism in muscle cellsNucleic Acids Research 42:13799–13811.https://doi.org/10.1093/nar/gku1160
-
In vitro fertilization may increase the risk of Beckwith-wiedemann syndrome related to the abnormal imprinting of the KCN1OT geneAmerican Journal of Human Genetics 72:1338–1341.https://doi.org/10.1086/374824
-
Transforming growth factor beta-induced endothelial-to-mesenchymal transition: A switch to cardiac fibrosis?Trends in Cardiovascular Medicine 18:293–298.https://doi.org/10.1016/j.tcm.2009.01.001
-
Cardiovascular abnormalities in the Beckwith-wiedemann syndromeAmerican Journal of Diseases of Children 131:293–294.https://doi.org/10.1001/archpedi.1977.02120160047007
-
Beckwith-Wiedemann syndrome and IVF: a case-control studyAmerican Journal of Human Genetics 75:526–528.https://doi.org/10.1086/423902
-
Association of four imprinting disorders and ARTClinical Epigenetics 11:21.https://doi.org/10.1186/s13148-019-0623-3
-
Myocardial hypertrophy and its role in heart failure with preserved ejection fractionJournal of Applied Physiology 119:1233–1242.https://doi.org/10.1152/japplphysiol.00374.2015
-
In brief: Genomic imprinting and imprinting diseasesThe Journal of Pathology 232:485–487.https://doi.org/10.1002/path.4326
-
Genomic imprinting mechanisms in mammalsMutation Research 647:77–85.https://doi.org/10.1016/j.mrfmmm.2008.08.008
-
Overrepresentation of pregnancies conceived by artificial reproductive technology in prenatally identified fetuses with Beckwith-Wiedemann syndromeJournal of Assisted Reproduction and Genetics 35:985–992.https://doi.org/10.1007/s10815-018-1228-z
-
A transcriptional insulator at the imprinted H19/Igf2 locusGenes & Development 14:1908–1919.
-
Regulatory mechanisms at the mouse Igf2/H19 locusMolecular and Cellular Biology 21:8189–8196.https://doi.org/10.1128/MCB.21.23.8189-8196.2001
-
Epigenetics and imprinting in human diseaseThe International Journal of Developmental Biology 58:291–298.https://doi.org/10.1387/ijdb.140077mb
-
The imprinted H19 lncRNA antagonizes let-7 microRNAsMolecular Cell 52:101–112.https://doi.org/10.1016/j.molcel.2013.08.027
-
The H19 lincrna is a developmental reservoir of mir-675 that suppresses growth and igf1rNature Cell Biology 14:659–665.https://doi.org/10.1038/ncb2521
-
H19, a developmentally regulated gene, is reexpressed in rat vascular smooth muscle cells after injuryThe Journal of Clinical Investigation 93:355–360.https://doi.org/10.1172/JCI116967
-
Tie2-cre transgenic mice: A new model for endothelial cell-lineage analysis in vivoDevelopmental Biology 230:230–242.https://doi.org/10.1006/dbio.2000.0106
-
twenty-one years to the right diagnosis - clinical overlap of Simpson-Golabi-Behmel and Beckwith-Wiedemann syndromeAmerican Journal of Medical Genetics. Part A 167A:151–155.https://doi.org/10.1002/ajmg.a.36825
-
Minoxidil improves vascular compliance, restores cerebral blood flow, and alters extracellular matrix gene expression in a model of chronic vascular stiffnessAmerican Journal of Physiology. Heart and Circulatory Physiology 315:H18–H32.https://doi.org/10.1152/ajpheart.00683.2017
-
A novel long noncoding RNA Linc-ASEN represses cellular senescence through multileveled reduction of p21 expressionCell Death and Differentiation 27:1844–1861.https://doi.org/10.1038/s41418-019-0467-6
-
Rapamycin: one drug, many effectsCell Metabolism 19:373–379.https://doi.org/10.1016/j.cmet.2014.01.001
-
H19 derived microRNA-675 regulates cell proliferation and migration through CDK6 in gliomaAmerican Journal of Translational Research 7:1747–1764.
-
LNCRNA H19 promotes the development of hepatitis b related hepatocellular carcinoma through regulating microrna-22 via EMT pathwayEuropean Review for Medical and Pharmacological Sciences 23:5392–5401.https://doi.org/10.26355/eurrev_201906_18208
-
Fetal intracardiac rhabdomyoma in Beckwith-wiedemann syndromeJournal of Clinical Ultrasound 42:569–573.https://doi.org/10.1002/jcu.22164
-
Beckwith-Wiedemann syndrome and assisted reproduction technology (ARTJournal of Medical Genetics 40:62–64.https://doi.org/10.1136/jmg.40.1.62
-
Structural development of endocardial cushionsThe American Journal of Anatomy 148:85–119.https://doi.org/10.1002/aja.1001480108
-
Oncofetal H19 RNA promotes tumor metastasisBiochimica et Biophysica Acta 1843:1414–1426.https://doi.org/10.1016/j.bbamcr.2014.03.023
-
Differences between pathological and physiological cardiac hypertrophy: Novel therapeutic strategies to treat heart failureClinical and Experimental Pharmacology & Physiology 34:255–262.https://doi.org/10.1111/j.1440-1681.2007.04585.x
-
Setting up and maintaining differential insulators and boundaries for genomic imprintingBiochemistry and Cell Biology = Biochimie et Biologie Cellulaire 89:469–478.https://doi.org/10.1139/o11-043
-
Imprinting disorders and assisted reproductive technologyCurrent Opinion in Endocrinology, Diabetes, and Obesity 17:517–522.https://doi.org/10.1097/MED.0b013e32834040a3
-
The role of genomic imprinting in biology and disease: An expanding viewNature Reviews. Genetics 15:517–530.https://doi.org/10.1038/nrg3766
-
Role of endothelial-mesenchymal transition (endomt) in the pathogenesis of fibrotic disordersThe American Journal of Pathology 179:1074–1080.https://doi.org/10.1016/j.ajpath.2011.06.001
-
Reversible obstructive hypertrophic cardiomyopathy in the Beckwith-wiedemann syndromePediatric Cardiology 10:225–228.https://doi.org/10.1007/BF02083298
-
A third case of cardiac neoplasm in a fetus with Beckwith-wiedemann syndrome: Epicardial angiofibromaFetal Diagnosis and Therapy 20:44–47.https://doi.org/10.1159/000081368
-
Mammalian target of rapamycin signaling in cardiac physiology and diseaseCirculation Research 114:549–564.https://doi.org/10.1161/CIRCRESAHA.114.302022
-
Physiological and pathological cardiac hypertrophyJournal of Molecular and Cellular Cardiology 97:245–262.https://doi.org/10.1016/j.yjmcc.2016.06.001
-
H19 and igf2 monoallelic expression is regulated in two distinct ways by a shared cis acting regulatory region upstream of H19Genes & Development 14:1186–1195.
-
Imprint control element-mediated secondary methylation imprints at the IGF2/H19 locusThe Journal of Biological Chemistry 278:5977–5983.https://doi.org/10.1074/jbc.M208437200
-
Exosomal MIRNA let-7 from menstrual blood-derived endometrial stem cells alleviates pulmonary fibrosis through regulating mitochondrial dna damageOxidative Medicine and Cellular Longevity 2019:4506303.https://doi.org/10.1155/2019/4506303
-
Clinical and molecular analyses of Beckwith-wiedemann syndrome: Comparison between spontaneous conception and assisted reproduction techniquesAmerican Journal of Medical Genetics. Part A 170:2740–2749.https://doi.org/10.1002/ajmg.a.37852
-
Prognostic implications of left ventricular hypertrophyAmerican Heart Journal 141:334–341.https://doi.org/10.1067/mhj.2001.113218
-
Myocyte apoptosis in heart failureCardiovascular Research 67:21–29.https://doi.org/10.1016/j.cardiores.2005.04.012
-
Effects of elastin haploinsufficiency on the mechanical behavior of mouse arteriesAmerican Journal of Physiology. Heart and Circulatory Physiology 289:H1209–H1217.https://doi.org/10.1152/ajpheart.00046.2005
-
Beckwith-wiedemann syndromeEuropean Journal of Human Genetics 18:8–14.https://doi.org/10.1038/ejhg.2009.106
-
Long non-coding RNA H19 mediates ovarian cancer cell cisplatin-resistance and migration during EMTInternational Journal of Clinical and Experimental Pathology 12:2506–2515.
-
Analysis of the H19ICR insulatorMolecular and Cellular Biology 27:3499–3510.https://doi.org/10.1128/MCB.02170-06
-
Endothelial-to-mesenchymal transition contributes to cardiac fibrosisNature Medicine 13:952–961.https://doi.org/10.1038/nm1613
-
The interplay of Lncrna-h19 and its binding partners in physiological process and gastric carcinogenesisInternational Journal of Molecular Sciences 18:450.https://doi.org/10.3390/ijms18020450
-
LNCRNA H19 promotes epithelial-mesenchymal transition (EMT) by targeting mir-484 in human lung cancer cellsJournal of Cellular Biochemistry 119:4447–4457.https://doi.org/10.1002/jcb.26537
Article and author information
Author details
Funding
Eunice Kennedy Shriver National Institute of Child Health and Human Development (ZIAHD001804)
- Karl Pfeifer
National Heart, Lung, and Blood Institute (ZIA HL006247)
- Beth A Kozel
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Victoria Biggs and Jeanne Yimdjo for animal husbandry. This work was supported by the Division of Intramural Research of the Eunice Kennedy Shriver National Institute of Child Health and Human Development.
Ethics
This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All animals were handled according to approved institutional animal care and use committee (IACUC) protocols (050 and 063) of the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Surgery was performed under Avertin anesthesia and every effort was made to minimize suffering.
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 1,414
- views
-
- 196
- downloads
-
- 14
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 14
- citations for umbrella DOI https://doi.org/10.7554/eLife.67250
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cardiac pathologies in mouse loss of imprinting models are due to misexpression of H19 long noncoding RNA
eLife 10:e67250.
https://doi.org/10.7554/eLife.67250




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}