The hepcidin regulator erythroferrone is a new member of the erythropoiesis-iron-bone circuitry
- Division of Hematology Oncology, Tisch Cancer Institute, Icahn School of Medicine at Mount Sinai, United States
- The Mount Sinai Bone Program, Departments of Medicine and Pharmacological Sciences, and Center for Translational Medicine and Pharmacology, Icahn School of Medicine at Mount Sinai, United States
- Center for Iron Disorders, University of California, Los Angeles (UCLA), United States
- Department of Pediatrics, Division of Hematology, and Penn Center for Musculoskeletal Disorders, Children’s Hospital of Philadelphia (CHOP), University of Pennsylvania, Perelman School of Medicine, United States
- Intrinsic Lifesciences, LLC, United States
- Department of Pediatrics, Saint Louis University School of Medicine, United States
Abstract
Background:
Erythroblast erythroferrone (ERFE) secretion inhibits hepcidin expression by sequestering several bone morphogenetic protein (BMP) family members to increase iron availability for erythropoiesis.
Methods:
To address whether ERFE functions also in bone and whether the mechanism of ERFE action in bone involves BMPs, we utilize the Erfe-/- mouse model as well as β–thalassemic (Hbbth3/+) mice with systemic loss of ERFE expression. In additional, we employ comprehensive skeletal phenotyping analyses as well as functional assays in vitro to address mechanistically the function of ERFE in bone.
Results:
We report that ERFE expression in osteoblasts is higher compared with erythroblasts, is independent of erythropoietin, and functional in suppressing hepatocyte hepcidin expression. Erfe-/- mice display low–bone–mass arising from increased bone resorption despite a concomitant increase in bone formation. Consistently, Erfe-/- osteoblasts exhibit enhanced mineralization, Sost and Rankl expression, and BMP–mediated signaling ex vivo. The ERFE effect on osteoclasts is mediated through increased osteoblastic RANKL and sclerostin expression, increasing osteoclastogenesis in Erfe-/- mice. Importantly, Erfe loss in Hbbth3/+mice, a disease model with increased ERFE expression, triggers profound osteoclastic bone resorption and bone loss.
Conclusions:
Together, ERFE exerts an osteoprotective effect by modulating BMP signaling in osteoblasts, decreasing RANKL production to limit osteoclastogenesis, and prevents excessive bone loss during expanded erythropoiesis in β–thalassemia.
Funding:
YZG acknowledges the support of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01 DK107670 to YZG and DK095112 to RF, SR, and YZG). MZ acknowledges the support of the National Institute on Aging (U19 AG60917) and NIDDK (R01 DK113627). TY acknowledges the support of the National Institute on Aging (R01 AG71870). SR acknowledges the support of NIDDK (R01 DK090554) and Commonwealth Universal Research Enhancement (CURE) Program Pennsylvania.
Introduction
It has become increasingly clear that both erythropoiesis and skeletal homeostasis are susceptible to changes in iron metabolism, especially during stress or ineffective erythropoiesis. Diseases of ineffective erythropoiesis, such as β-thalassemia, one of the most common forms of inherited anemia worldwide (Weatherall et al., 2010), are thus associated with bone loss, primarily at cortical sites (Haidar et al., 2011; Vogiatzi et al., 2006). β-thalassemia results from β-globin gene mutations that cause ineffective erythropoiesis, splenomegaly, and anemia (Weatherall, 1998; Pootrakul et al., 1988; Rund et al., 2005; Centis et al., 2000). Patients with homozygous mutations have either red blood cell (RBC) transfusion-dependent β-thalassemia major (TDT) or a relatively milder anemia, namely non-transfusion-dependent β-thalassemia intermedia (NTDT).
Both TDT and NTDT generally present with anemia and iron overload, requiring iron chelation therapy. Surprisingly, however, TDT patients show more marked decrements in bone mineral density (BMD) compared with NTDT, despite chronic RBC transfusion that suppresses expanded and ineffective erythropoiesis. Optimization of RBC transfusion has reduced the frequency of overt bone disease, such as frontal bossing, maxillary hyperplasia, and limb deformities, and importantly, has enabled prolonged survival (Kwiatkowski et al., 2012). Nonetheless, growth patterns have not significantly improved (Wallace et al., 2009), and low bone mass remains a frequent, significant, and poorly understood complication even in optimally treated patients. As such, β-thalassemia-induced bone disease has warranted formal guidelines for management (Thalassemia Clinical Research Network et al., 2015).
Proposed mechanisms of bone loss in β-thalassemia include direct effects of abnormal erythroid proliferation (Schnitzler and Mesquita, 1998; Gurevitch and Slavin, 2006), increased circulating erythropoietin (Epo) (Singbrant et al., 2011), iron toxicity (Weinberg, 2006), oxidative stress (Basu et al., 2001), inflammation (Lacativa and Farias, 2010), and changes in bone marrow adiposity (Schwartz, 2015). Strong negative correlations between BMD and systemic iron concentrations (Imel et al., 2016) and the profound bone loss noted in patients with hereditary hemochromatosis (Guggenbuhl et al., 2006) underscore the premise that BMD and iron homeostasis may be associated causally. However, mice lacking the transferrin receptor, TFR2, display increased rather than decreased bone mass and mineralization despite iron overload (Rauner et al., 2019). These latter findings prompted us to take a fresh look at the mechanisms underpinning bone loss in diseases of iron dysregulation.
Erythroferrone (ERFE), a protein secreted by bone marrow erythroblasts, is a potent negative regulator of hepcidin (Kautz et al., 2014); hepcidin inhibits iron absorption and recycling (Nemeth et al., 2005). Thus, hepcidin suppression by increased ERFE enables an increase in iron availability during stress erythropoiesis. Very recently, ERFE has been shown to bind and sequester certain members of the bone morphogenetic protein (BMP) family, prominently BMP2, BMP6, and the BMP2/6 heterodimer (Arezes et al., 2018; Wang et al., 2020). BMPs stimulate bone formation by osteoblasts during skeletal development, modeling, and ongoing remodeling (Hogan, 1996). We thus hypothesized that, by modifying BMP availability, ERFE may be a key player in the newly discovered erythropoiesis-iron-bone circuitry. As a result, ERFE may also be an important link between altered iron metabolism, abnormal erythropoiesis, and bone loss in β-thalassemia.
The known mechanism of ERFE action—BMP sequestration—predicts that ERFE loss, by enhancing BMP availability, may stimulate osteoblastic bone formation (Salazar et al., 2016). Alternatively, recent literature shows that loss of BMP signaling increases bone mass through direct osteoclast inhibition and Wnt activation (Broege et al., 2013; Jensen et al., 2009; Kamiya et al., 2016; Kamiya et al., 2008; Baud’huin et al., 2012; Gooding et al., 2019), predicting that ERFE loss would lead to decreased bone mass. Here, we demonstrate that global deletion of Erfe in mice results in a low-bone-mass phenotype, which is phenocopied in β-thalassemic mice lacking ERFE (i.e. Hbbth3/+;Erfe-/- mice). Despite the osteopenic phenotype, we found that ERFE loss stimulated mineralization in cell culture. The net loss of bone in Erfe-/- mice in the face of a pro-osteoblastic action could therefore only be attributed to a parallel increase in bone resorption, which we found was the case in both Erfe-/- and Hbbth3/+;Erfe-/- mice. Furthermore, the increase in osteoclastogenesis was osteoblast-mediated exerted via an increased expression of Sost and Tnfsf11, the gene encoding RANKL. Together, our data provide compelling evidence that ERFE loss induces BMP-mediated osteoblast differentiation, but upregulates Sost and Tnfsf11 to increase osteoclastogenesis with net bone loss. Therefore, high ERFE levels in β-thalassemia are osteoprotective and prevent the bone loss when erythropoiesis is expanded.
Materials and methods
Mouse lines
Request a detailed protocolC57BL/6 and β-thalassemic (Hbbth3/+) mice (Yang et al., 1995) were originally purchased from Jackson Laboratories. Erfe-/- mice were a generous gift from Tomas Ganz (UCLA) (Kautz et al., 2014). Progeny of Erfe-/-mice crossed with Hbbth3/+ yielded Hbbth3/+;Erfe-/- mice. The mice have been backcrossed onto a C57BL/6 background for more than 11 generations. All mice had ad libitum access to food and water and were bred and housed in the animal facility under AAALAC guidelines. Experimental protocols were approved by the Institutional Animal Care and Use Committee at Icahn School of Medicine at Mount Sinai.
Skeletal phenotyping
Request a detailed protocolSkeletal phenotyping was conducted on 6-week- and/or 5-month-old male mice, unless otherwise noted. Mice were injected with calcein (15 mg/kg, Sigma C0875) and xylenol orange (90 mg/kg, Sigma 52097), at days 8 and 2, respectively, prior to sacrifice. Briefly, for histomorphometry, the left femur, both tibias, and L1-L3 were fixed in neutral buffered formalin (10%, v/v) for 48 hr at 4°C; transferred to sucrose (30%, w/v) at 4°C overnight; and embedded and sectioned at –25°C (5–6 µm thick sections, 10X) (Dyment et al., 2016). Unstained sections were analyzed by fluorescence microscopy (Leica Upright DM5500) to determine the mineralizing surface and interlabel distance using image J. Von Kossa staining of sections was used to quantify fractional bone volume (BV/TV) and trabecular thickness (Tb.Th). Tartrate-resistant acid phosphatase (ACP5) staining (Sigma 387A) was used to identify osteoclasts, counterstained with aniline blue using Olympus Stereoscope MVX10 (1X). Images were analyzed by TrapHisto and OsteoidHisto (van 't Hof et al., 2017). On the day of sacrifice, BMD was also measured in intact mice (Shi et al., 2016). Frozen bone sections were incubated for 4 min at room temperature in Alkaline Phosphatase Substrate Solution ImmPACT Vector Red (Vector Laboratories). After washing with buffer, the sections were counterstained with hematoxylin (Vector Laboratories) and mounted with VectaMount AQ Mounting Medium (Vector Laboratories). Sections were visualized using Olympus BH-2 Microscope and images obtained with OMAX A35180U3 Camera were analyzed by ImageJ.
Isolation and culture of bone marrow cells
Request a detailed protocolFor osteoblast cultures, fresh bone marrow cells were seeded in 12-well plates (0.6 × 106 cells per well) under differentiating conditions (αMEM, 10% FBS, 1% penicillin/streptomycin, 1 M β-glycerol phosphate, and 0.5 M ascorbic acid) for 21 days to induce the formation of mature, mineralizing osteoblast colonies, Cfu-ob, as before (Maridas et al., 2018). Cultured osteoblasts were treated with BMP2 (50 ng/ml) or BMP6 (50 ng/ml) for 30 min on day 3 of culture to assess effects on BMP-mediated signaling, as previously (Rauner et al., 2019). For osteoclast cultures, bone marrow hematopoietic stem cells (non–adherent) from wild type and Erfe-/- were seeded in six-well plates (106 cells per well) in the presence of αMEM, 10% FBS, 1% penicillin/streptomycin and M-CSF (25 ng/mL, PeproTech) for 48 hr, followed by the addition of RANK-L (50 ng/mL, PeproTech) for 5 days. In experiments testing Epo responsiveness, 20 U/ml of Epo was added for the duration of the differentiation process, for 21 and 5 days in osteoblast and osteoclast cultures, respectively.
Erythroblasts were isolated from bone marrow and purified using CD45 beads, as previously (Li et al., 2017) with minor modifications. Briefly, mouse femur was flushed, single-cell suspensions incubated with anti-CD45 magnetic beads (Mylteni), and erythroid lineage-enriched cells that flowed through the column were collected. Erythroid-enriched cells were incubated with anti-mouse TER119-phycoerythrin Cy7 (PE-Cy7) (BioLegend) and CD44-allophycocyanin (APC) (Tonbo, Biosciences). Non-erythroid and necrotic cells were identified and excluded from analyses using anti-CD45 (BD Pharmigen), anti-CD11b, and anti-Gr1 (APC-Cy7) (Tonbo, Biosciences) antibodies. Erythroid precursors were selected by gating and analyzed using TER119, CD44, and forward scatter as previously described (Li et al., 2017). Samples were analyzed on either FACSCanto I or LSRFortessa flow cytometer (BD Biosciences). To determine levels of Erfe mRNA expression in Epo–stimulated conditions, erythroblasts were cultured in the presence or absence of 20 U/ml Epo for 15 hr as described (Kautz et al., 2014).
Primary hepatocyte culture
Request a detailed protocolHepatocytes were isolated by perfusion with collagenase and liver digestion, as described previously (Merlin et al., 2017). Briefly, 0.025% (w/v) collagenase type IV (Gibco) and 5 mM CaCl2 was added to Leffert perfusion buffer containing 10 mM HEPES, 3 mM KCl, 130 mM NaCl, 1 mM NaH2PO4.H2O, and 10 mM D-glucose (Sigma). Live cells were purified by Percoll (Sigma) and plated in six-well plates (0.25 × 106 cells per well) in William’s Medium E (Sigma) supplemented with antibiotics and 5% fetal bovine serum (FBS) for 2 hr to allow the hepatocytes to attach. Cells were starved overnight with William’s Medium E lacking FBS, and were then treated for 6 hr with conditioned or control media from wild type or Erfe-/- osteoblast and osteoclast cultures (day 6 and day 5, respectively) in the presence of 50% (v/v) William’s Medium E and 5% FBS.
Quantitative PCR
Request a detailed protocolRNA was purified from osteoblasts, osteoclasts, erythroblasts, and hepatocytes using PureLink RNA (Sigma) and analyzed with SuperScript III Platinum SYBR Green One-Step (Invitrogen). As previously described (Koide et al., 2017; Dumas et al., 2008), ΔΔCT values were used to calculate fold increases relative to β-actin, α-tubulin, and RLP4. Primers are listed in Table 1.
Table 1
Primers used in the presented studies.
| Gene | Forward (sense) | Reverse (antisense) |
|---|---|---|
| Actb | TTCTTTGCAGCTCCTTCGTT | ATGGAGGGGAATACAGCCC |
| Tubb | CTGGAGCAGTTTGACGACAC | TGCCTTTGTGCACTGGTATG |
| Hamp | TGAGCAGCACCACCTATCTC | ACTGGGAATTGTTACAGCATTT |
| Acp5 | ACCTGTGCTTCCTCCAGGAT | TCTCAGGGTGGGAGTGGG |
| Ctsk | CCATATGTGGGCCAGGATG | TCAGGGCTTTCTCGTTCCC |
| Runx2 | GTGGCCACTTACCACAGAGC | GTTCTGAGGCGGGACACC |
| Alpl | ACACCTTGACTGTGGTTACTGCTGA | CCTTGTAGCCAGGCCCGTTA |
| Osx | TGAGGAAGAAGCCCATTCAC | GTGGTCGCTTCTGGTAAAGC |
| Col1a1 | CCTGGCAAAGACGGACTCAAC | GCTGAAGTCATAACCGCCACTG |
| Tnsf11 | CAGCCATTTGCACACCTCAC | GTCTGTAGGTACGCTTCCCG |
| Opg | ACAGTTTGCCTGGGACCAAA | CAGGCTCTCCATCAAGGCAA |
| Dmp1 | GGGCTGTGTTGTGCAAGACA | GGTGCACACCTGACCTTCTTTAA |
| Fam132b | ATGGGGCTGGAGAACAGC | TGGCATTGTCCAAGAAGACA |
Western immunoblotting and ELISA
Request a detailed protocolFor western immunoblotting, differentiated cells at day three were lysed in ice cold SDS page lysis buffer (2% SDS, 50 mM Tris-HCl, pH 7.4, 10 mM EDTA) with protease and phosphatase inhibitors. 20 µg of heat–denatured protein was loaded onto a 10% gel, run, and transferred onto a 0.4 µm nitrocellulose membrane (Thermo Scientific). After blocking with 5% BSA in Tris–buffered saline with 1% Tween-20 (TBS-T), the membranes were incubated with primary antibodies to signaling proteins (Table 2) overnight at 4°C, washed, and incubated with the corresponding HRP–conjugated secondary antibodies at room temperature. Proteins were visualized using the ImageQuant LAS 4010 and quantified using Image J. Osteoblast supernatants from wild type and Erfe-/- mice were collected and centrifuged for 10 min at 10,000 x g, and BMP2 (Abnova) and RANKL (R and D) concentrations were measured by ELISAs. Serum BMP2 concentration was determined using mouse BMP2 ELISA (abnova, KA0542), per manufacturers instructions. ERFE concentration in conditioned media was determined as described (Kautz et al., 2015) with the substitution of DELFIA europium–conjugated streptavidin for horseradish-peroxidase-conjugated streptavidin. Fluorescence was measured by CLARIOstar plate reader.
Table 2
Antibodies used in the presented studies.
| Antibody | Company | # catalog | Dilution | BSA/Milk (5%) | Rabbit/mouse |
|---|---|---|---|---|---|
| PRIMARY antibodies | |||||
| pSMAD 1/5/8 | Cell signaling | 9511 | 1:1000 | BSA | Rabbit |
| pSMAD 1/5/8 (monoclonal) | Cell signaling | 9516 | 1:1000 | BSA | Rabbit |
| SMAD 1 | Cell signaling | 6944S | 1:1000 | BSA | Rabbit |
| p-ERK (monoclonal) | Cell signaling | 4376 | 1:1000 | BSA | Rabbit |
| ERK | Cell signaling | 4695 | 1:1000 | BSA | Rabbit |
| pp38 | Cell signaling | 4511 | 1:1000 | BSA | Rabbit |
| p38 | Cell signaling | 8690 | 1:1000 | BSA | Rabbit |
| Beta-actin | ThermoFisher | MA515452 | 1:1000 | BSA | Mouse |
| SECONDARY antibodies | |||||
| Rabbit | Cell signaling | 7074 | 1:5000 | BSA | |
| Mouse | GE Healthcare | NXA931V | 1:20000 | BSA | |
Complete blood counts
Request a detailed protocolPeripheral blood (100 µL from each mouse) was collected from the retro-orbital vein in EDTA-coated tubes and analyzed by IDEXX Procyte Hematology Analyzer.
Statistical analyses
Request a detailed protocolData are reported as means ± SEM. Unpaired Student’s t-test was used to determine if differences between groups were significant at p<0.05.
Results
To understand if ERFE has a role in regulating skeletal integrity in health, we first studied the effect of ERFE loss on BMD and bone remodeling in adult Erfe-/- mice, as well as in compound mutant mice in which the Erfe gene was deleted on a β-thalassemia Hbbth3/+ background. Compared with wild-type littermates, both 6-week-old and 5-month-old male Erfe-/- mice showed significant reductions in whole body BMD, and BMD at mainly cortical (femur and tibia) sites (Figure 1A and B). However, in contrast to young mice, the older Erfe-/- mice did not show a difference in lumbar spine BMD compared with wild-type littermates. Interestingly, unlike hypogonadal bone loss, which is predominantly trabecular, the sustained reduction in femur and tibia BMD is consistent with prominent cortical loss seen in patients with β-thalassemia (Haidar et al., 2011; Vogiatzi et al., 2006).
Figure 1 with 1 supplement see all
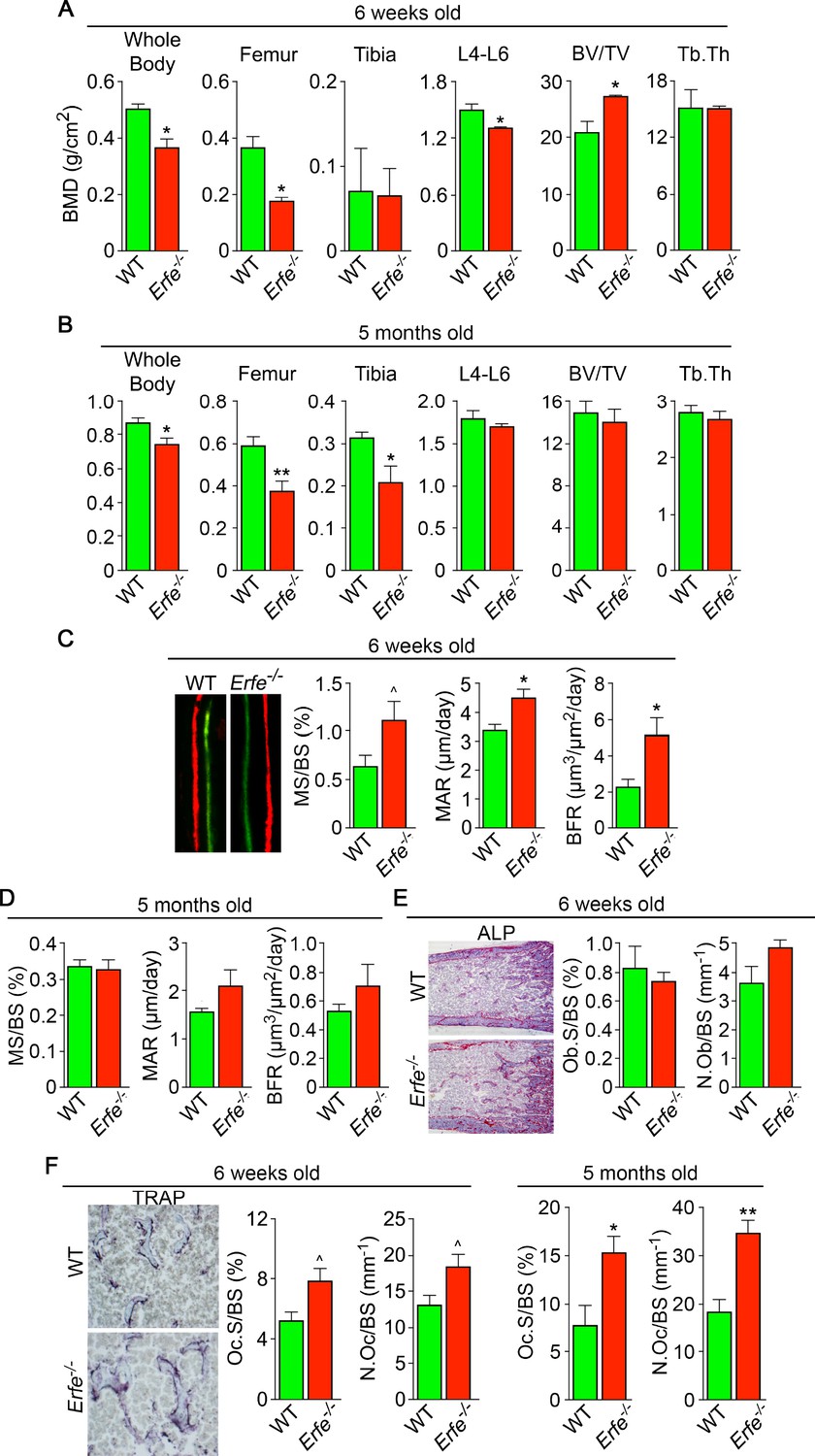
ERFE loss results in high turnover osteoporosis.
Bone mineral density (BMD) measured in whole body, femur, tibia, and lumbar spine (L4–L6) along with bone volume (BV/TV) and trabecular thickness (Tb.Th) in growing (6-week-old) (A) and mature (5-month-old) (B) Erfe-/- and wild-type (WT) littermates. Dynamic histomorphometry following two i.p. injections of calcein (green) and xylenol orange (red) given at days 8 and 2, respectively. Representative dual labels from the epiphysis are shown, together with measured and derived parameters, namely mineralizing surface (MS) as a function of bone surface (BS), mineral apposition rate (MAR) and bone formation rate (BFR) in 6-week-old (C) and 5-month-old (D) mice. (E) Alkaline phosphatase staining (magenta) in sections of femura demonstrates no differences in osteoblast surface (Ob.S) and number (N.Ob) as a function of BS in 6-week-old Erfe-/- and WT mice. (F) TRAP staining at the epiphysis showing both osteoclast surface (Oc.S) and number (N.Oc) as a function of BS. Statistics: Mean ± SEM; unpaired two-tailed Student’s t-test; *p<0.05, **p<0.01, ^0.05 < p < 0.1, N = 3–6 mice per group.
Bone resorption and bone formation are tightly coupled to maintain bone mass during each remodeling cycle (Zaidi, 2007). Bone is lost when either both processes are increased―with resorption exceeding formation, as in hypogonadism―or when there is uncoupling in which formation decreases while resorption rises, as in glucocorticoid excess (Zaidi, 2007). To differentiate between relative increases and uncoupling, we measured both formation and resorption in intact bone. Dynamic histomorphometry performed after the sequential injections of calcein and xylenol orange, which yielded dual fluorescent labels, allowed us to derive parameters of bone formation. We observed that mineralizing surface (MS), mineral apposition rate (MAR) and bone formation rates (BFR) were all increased in young Erfe-/- mice, consistent with the pro-osteoblastic (anabolic) action of ERFE deficiency (see below) (Figure 1C and D). No differences in MS, MAR, and BFR were noted in 5-month-old mice (Figure 1D). We also analyzed alkaline phosphatase stained sections of femurs to find no difference in osteoblast surfaces (Ob.S) or osteoblast number (N.Ob) per bone surface (BS) in 5–week–old Erfe-/- relative to wild type littermates (Figure 1E).
Finally, to study whether an increase in osteoclastic bone resorption caused the notable reduction in BMD in Erfe-/- mice, we measured TRAP-positive osteoclast surfaces (Oc.S) and number (N.Oc) per bone surface (BS). Both Oc.S/BS and N.Oc/BS were increased significantly in Erfe-/- compared with wild type bones in older mice, and to a lesser extent, in younger mice (Figure 1F). Thus, the overall low-bone-mass phenotype in Erfe-/- mice primarily resulted from a relative increase in osteoclastic bone resorption over osteoblastic bone formation, suggesting that ERFE has a function in preventing skeletal loss. To confirm that decreased BMD in Erfe-/- mice did not result from changes in erythropoiesis, we measured circulating red blood cells (RBCs) and reticulocytes, and bone marrow erythroblasts. We also measured spleen weight given the ubiquity of compensatory erythropoiesis that results in splenomegaly. Our results show no differences between 6-week-old wild type and Erfe-/- mice (Figure 1—figure supplement 1), consistent with what has been previously reported in Erfe-/- mice (Kautz et al., 2014).
To probe the mechanism of action of ERFE on osteoblastic bone formation and osteoclastic bone resorption, we first asked which cells in bone marrow produce ERFE, and whether secreted ERFE was functional. Intriguingly, time course studies in differentiating osteoblasts revealed that Erfe expression was 10- and twofold higher at 3 and 21 days of culture, respectively, compared with cultured erythroblasts––the only previously known source of ERFE in bone marrow (Figure 2A). To confirm that cultured cells were of the osteoblastic lineage, we evaluated Alp expression, a known marker of osteoblast lineage, and demonstrate increased Alp expression as early as day 3 in culture (Figure 2—figure supplement 1). Furthermore, Erfe expression in mature osteoclasts was similar to cultured erythroblasts, with little expression in immature osteoclasts (Figure 2B). Likewise, conditioned media from osteoblast cultures revealed increased ERFE concentration at 3 days with no differences in conditioned media from osteoclast cultures (Figure 2C).
Figure 2 with 1 supplement see all
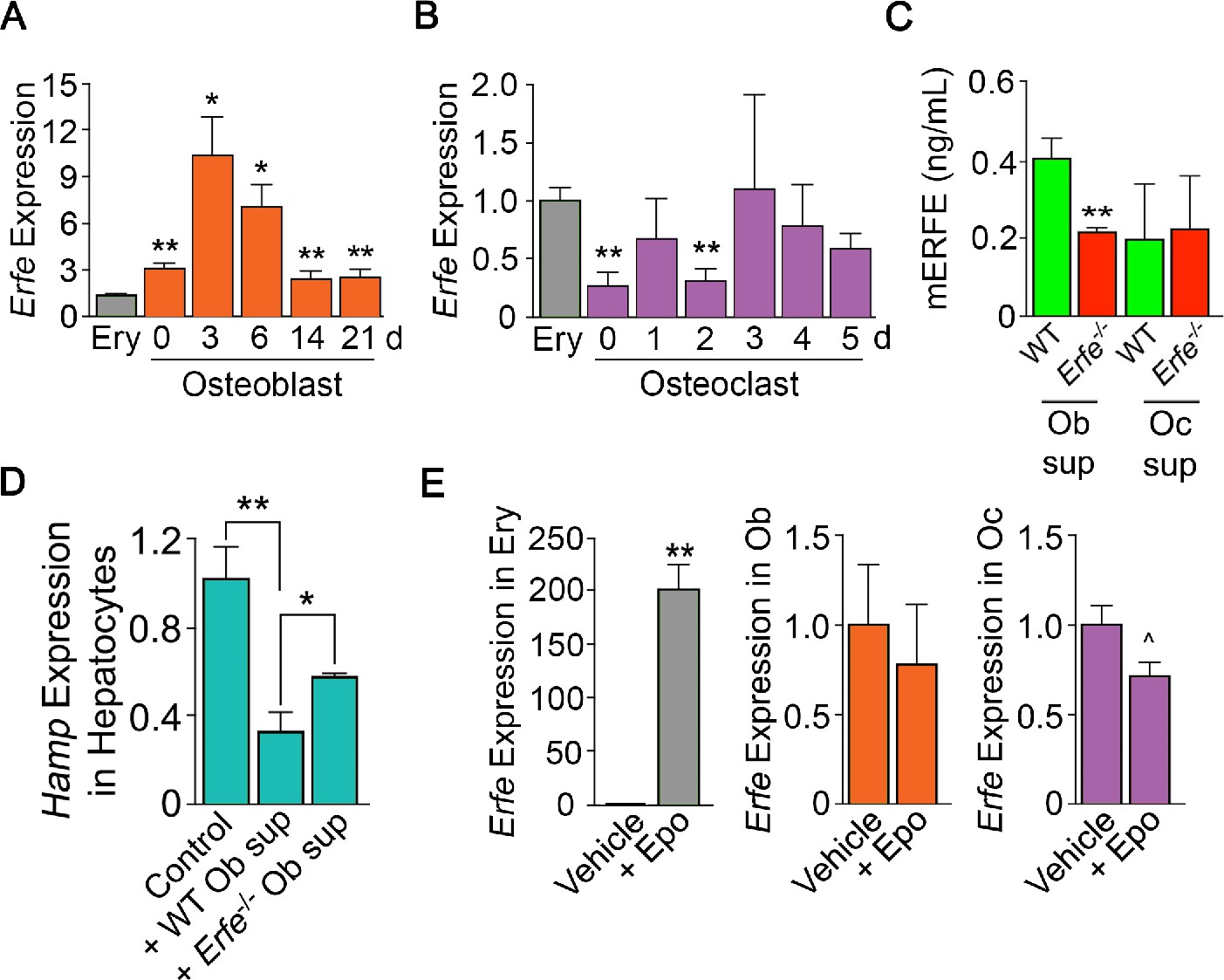
ERFE is expressed at higher levels in osteoblasts than in erythroblasts.
(A) Quantitative PCR showing high levels of Erfe expression in osteoblasts from wild type mice cultured under differentiating conditions. Notably, at 3 days of culture, there was a 10-fold greater expression in osteoblasts relative to bone-marrow-derived wild-type cultured erythroblasts. (B) Quantitative PCR showing comparable levels of Erfe expression in osteoclasts at 3–5 days of culture relative to bone-marrow-derived wild-type cultured erythroblasts from wild-type mice cultured under differentiating conditions. (C) Increased supernatant murine ERFE (mERFE) concentration in day 3 osteoblast cultures and no difference in day 5 osteoclast cultures from wild type relative to Erfe-/- mice (detection limit of 0.2 ng/ml mERFE). (D) Hepcidin (HAMP) expression is suppressed in primary wild-type hepatocytes in response to conditioned media from wild-type relative to Erfe-/- osteoblast cultures (day 6), confirming functionality of osteoblast-derived ERFE. Control hepatocytes were exposed to osteoblast culture media. (E) Unlike in erythroblasts, Erfe expression in cultured wild-type osteoblasts and osteoclasts does not respond to erythropoietin (Epo). Statistics: Mean ± SEM; unpaired two-tailed Student’s t-test; *p<0.05, **p<0.01, ^0.05 < P < 0.1, N = 3 wells per group.
To determine whether osteoblast-derived ERFE was functional, we established a bioassay based on the known inhibitory action of ERFE on hepcidin (Hamp) expression. For this, wild-type hepatocytes were exposed to supernatants from differentiating wild type or Erfe-/- osteoblasts. Hamp expression was suppressed with Erfe-/- osteoblast supernatants, but importantly, this suppression was significantly greater with wild type supernatants (Figure 2D). No Hamp suppression was evident with wild type or Erfe-/- osteoclast supernatants. This latter suggests that osteoblast– but not osteoclast-derived ERFE is functional. However, as Erfe-/- supernatants also suppressed Hamp expression, other yet unknown osteoblast-derived factors likely function in hepcidin regulation. Finally, unlike in erythroblasts, Erfe expression in mature osteoblasts or osteoclasts was not responsive to Epo (Figure 2E).
Given that osteoblasts secrete ERFE that is known to inhibit hepcidin (Kautz et al., 2014) by sequestering BMPs (Arezes et al., 2018; Wang et al., 2020; Arezes et al., 2020) that are skeletal anabolics (Hogan, 1996), we measured serum BMP2 concentration to find elevated BMP2 levels in Erfe-/- relative to wild-type mice (Figure 3A). Given the specific importance of BMP2 in bone remodeling (Salazar et al., 2016), these results are consistent with the previously demonstrated sequestration of BMP2, along with BMP6, by ERFE (Wang et al., 2020)—namely, loss of ERFE led to decreased BMP sequestration. We thus hypothesized that ERFE functions in modulating bone formation by sequestering BMPs and tested whether the loss of ERFE facilitates BMP2-mediated signaling in the osteoblast in vitro. We found that the concentration of BMP2 was higher in supernatants from cultured Erfe-/- osteoblasts compared with wild type cultures (Figure 3B). Consistent with this difference, BMP2-activated signaling pathways, namely phosphorylated Smad1/5/8 and ERK1/2, but not phosphorylated p38, were enhanced in Erfe-/- compared with wild-type osteoblasts (Figure 3C).
Figure 3 with 1 supplement see all
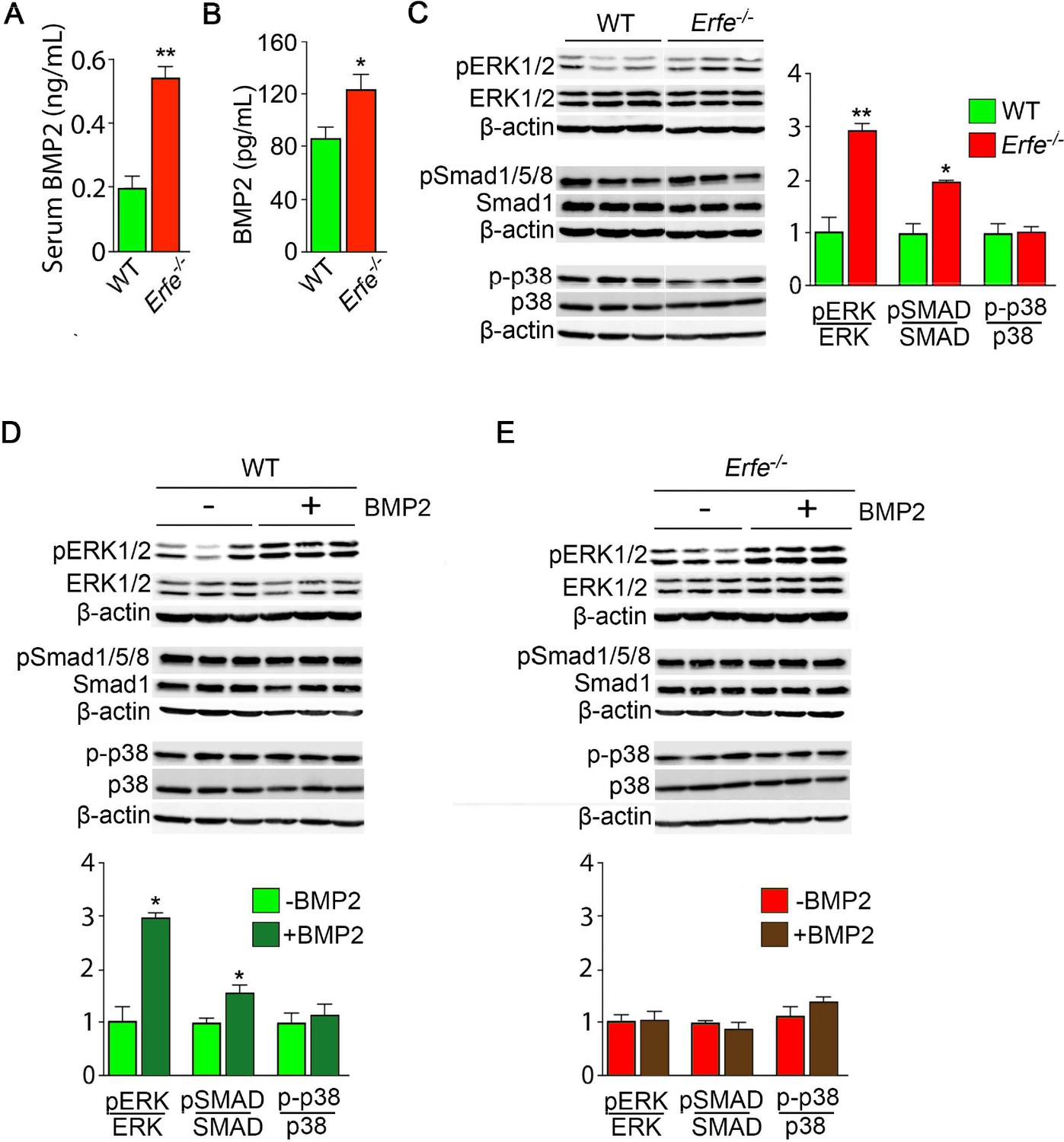
ERFE function on bone involves BMP-2 sequestration.
(A) BMP2 ELISA demonstrates elevated BMP2 concentration in serum samples from Erfe-/- relative to WT mice (N = 4 per group). In the 3-day cultures, there was an increase in BMP2 concentration in culture supernatants from Erfe-/- relative to WT osteoblasts (N = 6 per group) (B). (C) Similarly, signaling via the known BMP receptor pathways, namely ERK1/2 and Smad1/5/8, without changes in p38/MAPK, increase in Erfe-/- relative to WT osteoblasts; western blots with quantification shown. Finally, pSmad1/5/8 and pERK1/2 signaling is further induced by BMP2 (50 ng/ml) only in WT (D) but not in Erfe-/- (E) osteoblasts. Statistics: Mean ± SEM; unpaired two-tailed Student’s t-test; *p<0.05, **p<0.01.
To further understand how ERFE impacts BMP2-mediated signaling, we evaluated the effect of BMP2 on wild type and Erfe-/- osteoblasts in vitro. Treatment with BMP2 (50 ng/ml) in osteoblast cultures showed that pSmad1/5/8 and pERK signaling was not further induced in Erfe-/- relative to wild-type osteoblasts (Figure 3D and E). In all, the data establish that increased BMP2 in Erfe-/- mice leads to maximal induction of BMP signaling that remains unaffected by the further addition of BMP2. To test whether an ERFE effect on bone is BMP2 specific, we also repeated these experiments using BMP6, demonstrating results similar to the effects of BMP2 on BMP signaling pathways in wild type and Erfe-/- osteoblasts in vitro (Figure 3—figure supplement 1). These findings support the hypothesis that ERFE functions in bone by sequestering BMPs, thus, attenuating downstream signaling.
We studied whether the stimulation of bone formation in Erfe-/- mice was due to a cell–autonomous action of ERFE on osteoblasts. For this, we compared the ability of wild type and Erfe-/- bone marrow stromal cells ex vivo to differentiate into mature mineralizing colony forming units–osteoblastoid (Cfu-ob). Stromal cells from 5-month-old Erfe-/- mice showed enhanced von Kossa staining of mineralizing Cfu-ob colonies (Figure 4A). This mineralizing phenotype was associated with enhanced expression of the osteoblast transcription factors Runx2 and Sp7, and downstream genes Sost and Tnfsf11 (Yang et al., 2010; Pérez-Campo et al., 2016), increased supernatant RANKL levels, and suppressed expression of Opg (Figure 4B and C). Enhanced RANKL profoundly increases osteoclastogenesis, as noted below, while sclerostin, encoded by the Sost gene, reduces the production of OPG, hence further increasing osteoclast formation.
Figure 4
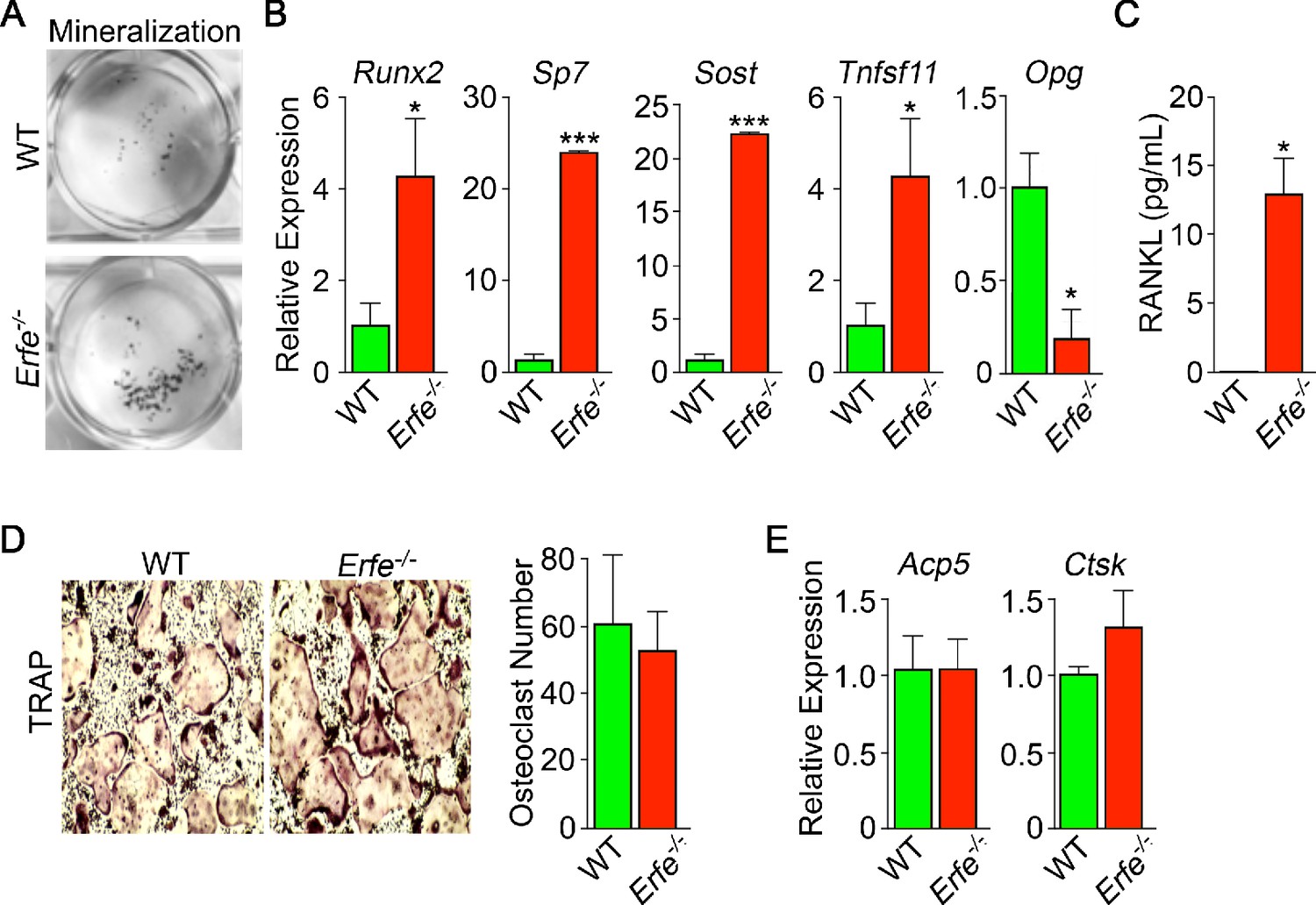
Mechanism of action of ERFE on bone involves interplay between osteoblastic RANKL and sclerostin.
Osteoblasts from 5-month-old wild type and Erfe-/- mouse bone marrow cultured under differentiating conditions for 5 or 21 days. Loss of ERFE resulted in accelerated mineralization, noted by an increase in Von Kossa-stained nodules (A). Consistent with the cellular phenotype is the upregulation in Erfe-/- osteoblasts of Runx2, Sp7, Sost, and Tnfsf11 expression and suppression of Opg expression (quantitative PCR on 21-day cultures) (B) as well as increased secreted RANKL (ELISA on 3 day cultures) (C). In vitro osteoclastogenesis assays show that ERFE loss does not alter osteoclast number, as measured by TRAP staining (D), or the expression of osteoclast genes, namely Acp5 or Ctsk (E). Statistics: Mean ± SEM; unpaired two-tailed Student’s t-test; *p<0.05, **p<0.01; wells per group – three for A-C.
Given that Erfe is expressed in osteoclasts, and that Erfe-/- mice display a pro–resorptive phenotype, we questioned whether ERFE directly affected the osteoclast, or whether the action resulted via a primary osteoblastic effect. Erfe-/- bone marrow cell cultures derived from 5-month-old mice showed no difference in TRAP-positive osteoclast number compared to wild-type cultures (Figure 4D). Consistent with this, the program of osteoclast gene expression remained unchanged in these 5-day cultures (Figure 4E). The data collectively suggest that the absence of ERFE results in the de-sequestration of BMP2, stimulates the osteoblast to upregulate RANKL and sclerostin, and thus enhances osteoclastic bone resorption indirectly.
Finally, we explored whether ERFE mediates osteoprotection in Hbbth3/+ mice, a model of human NTDT given that Erfe is upregulated in Hbbth3/+ marrow erythroblasts (Vogiatzi et al., 2006; Kautz et al., 2014; Li et al., 2017; Kautz et al., 2015; Vogiatzi et al., 2010). We crossed Hbbth3/+ mice with Erfe-/- mice to generate Hbbth3/+;Erfe-/- compound mutants. Whole body and site-specific measurements at mainly cortical sites, namely femur and tibia, and vertebral trabecular (L4-L6) bone showed striking reductions in BMD in 5-month-old Hbbth3/+;Erfe-/- mutants compared with Hbbth3/wt mice, most notably in cortical bone (Figure 5A). The trabecular bone loss was consistent with reduced histomorphometrically determined fractional bone volume (BV/TV) and trabecular thickness (Tb.Th) in the femoral epiphyses (Figure 5B). There was a trend toward increases in MAR (Figure 5C), but a significant increase in TRAP-positive N.Oc and Oc.S in Hbbth3/+;Erfe-/- bones compared with wild-type controls (Figure 5D)—changes expected to produce reduction in bone mass. These findings document ERFE-mediated skeletal protection in β-thalassemia.
Figure 5 with 1 supplement see all

ERFE loss in β-thalassemia mice causes profound bone loss.
(A) Bone mineral density (BMD) measured in whole body, femur, tibia, and lumbar spine (L4–L6) in 5-month-old β-thalassemia mice (Hbbth3/+ mice) and compound Hbbth3/+;Erfe-/- mutants. (B) Representative section of femoral epiphyses stained with Von Kossa, and quantitative estimates of bone volume (BV/TV) and trabecular thickness (Tb.Th). (C) Dynamic histomorphometry following two i.p. injections of calcein (green) and xylenol orange (red) given at days 8 and 2, respectively. Shown are measured and derived parameters, namely mineralizing surface (MS), mineral apposition rate (MAR) and bone formation rate (BFR). (D) Representative image of TRAP (ACP5) staining of femoral epiphysis, also showing both osteoclast surface (Oc.S) and number (N.Oc), expressed as a function of bone surface (BS). Statistics: Mean ± SEM; unpaired two-tailed Student’s t-test; *p<0.05, **p<0.01; N = 4–5 mice per group.
To confirm that decreased BMD in Hbbth3/+;Erfe-/- mice did not result from further expanded erythropoiesis, we measured circulating RBCs and reticulocytes, bone marrow erythroblasts, and spleen weight. Our results demonstrate a mildly decreased RBC count and hemoglobin, but no differences in spleen weight or bone marrow erythroblasts between 6-week-old Hbbth3/+ and Hbbth3/+;Erfe-/- mice (Figure 5—figure supplement 1). This is consistent with what has been previously reported in Hbbth3/+;Erfe-/- mice (Kautz et al., 2015).
Discussion
To date, the only known function of ERFE was on hepatocellular hepcidin expression exerted through the sequestration of BMPs (Arezes et al., 2018; Wang et al., 2020). Using genetically–modified mice and in vitro assays, we identify a new role for ERFE in skeletal protection. First, we show that Erfe expression is higher in osteoblasts comapred with erthyroblasts. Second, we find that ERFE is a potent down-regulator of BMP2–mediated signaling and RANK-L production by osteoblasts. Third, ERFE loss in vivo enhances bone formation, while also stimulating resorption by inducing the expression of osteoblastic Tnfsf11 and Sost. The net effect of these opposing changes is bone loss in both young and old mice (Figure 6). Fourth, although also produced by osteoclasts, ERFE displays no cell-autonomous actions on osteoclast function. Taken together, and consistent with prior inferences (Broege et al., 2013; Jensen et al., 2009; Kamiya et al., 2016; Kamiya et al., 2008; Baud’huin et al., 2012; Gooding et al., 2019), ERFE functions to protect the skeleton by negatively regulating BMP signaling in osteoblasts, with indirect inhibitory effects on osteoclastic bone resorption.
Figure 6
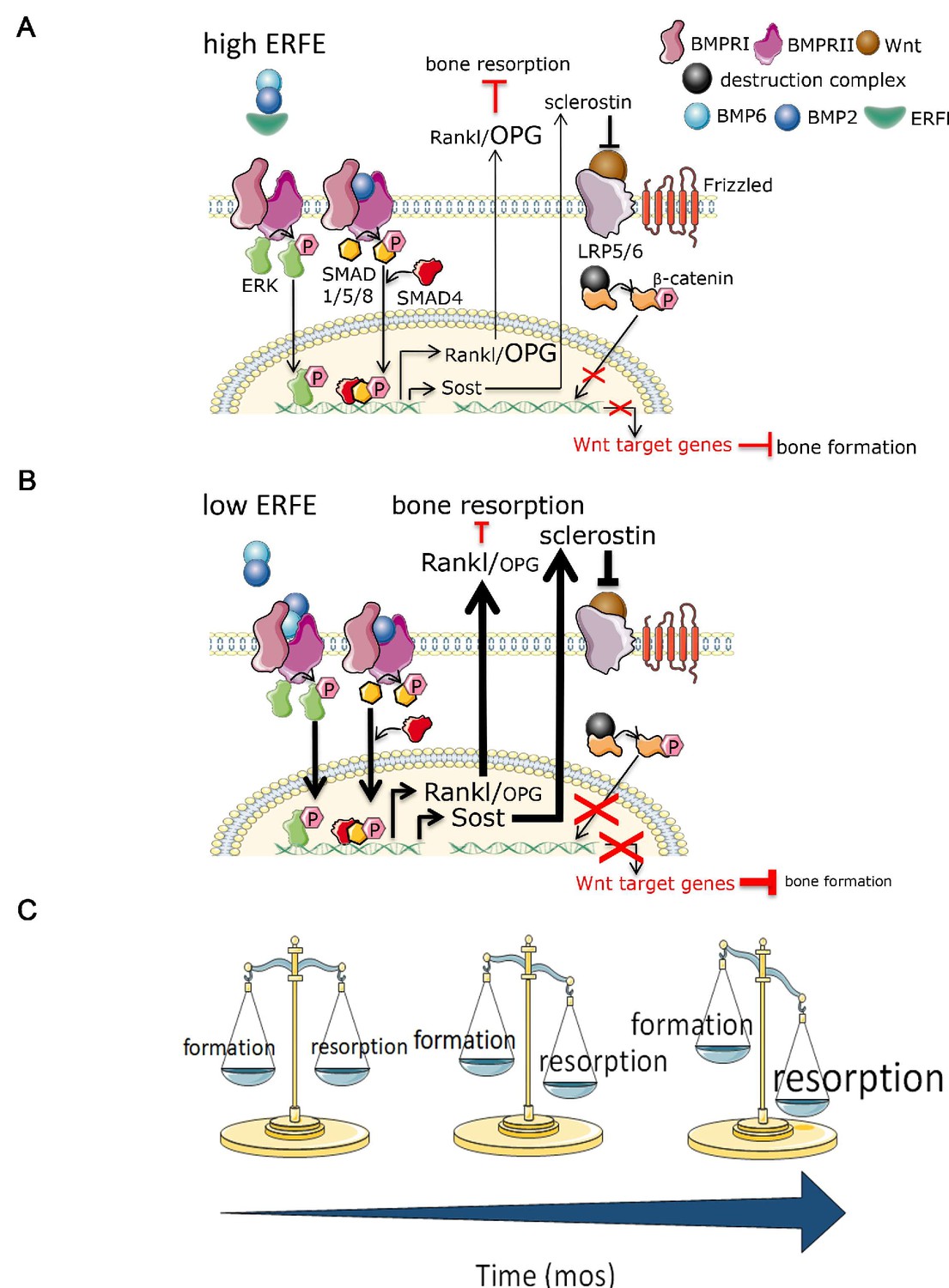
Putative osteoprotective function of ERFE in health and in β-thalassemia.
In conditions of elevated ERFE (A), sich as β-thalassemia, more BMP2 and BMP6 is sequestered, decreasing signaling through the BMP/Smad and ERK pathways. This would result in decreased Sost and Rankl expression to decrease osteoclastogenesis and bone resorption. In contrast, when ERFE is low (B), increased BMP2, and possibly BMP6, leads to increased osteoclastogenesis with consequent decrease in bone formation. (C) Together, ERFE loss leads to a greater degree of progressively increased bone resorption relative to bone formation with age. ERFE = erythroferrone; BMP = bone morphogenetic protein; BMPR = BMP receptor; SOST = Sclerostin; RANKL = receptor activator of nuclear factor kappa-B ligand; OPG = osteoprotegrin; LRP = lipoprotein receptor-related protein; Wnt = wingless type MMTV integration site family.
Bone is a highly dynamic and purposefully organized tissue which undergoes constant remodeling in response to changing metabolic and mechanical needs. Bone remodeling is a process in which bone resorption by osteoclasts is balanced by synthesis of new bone by osteoblasts, which then undergo terminal differentiation to become mechanosensory osteocytes. Multiple local cytokines and systemic hormones regulate the delicate balance of bone resorption and bone formation, enabling bone cells to communicate among themselves as well as with other cells in the bone marrow. For example, osteocytes secrete Sclerostin, encoded by the SOST gene, which, in turn, inhibits further osteoblast differentiation. Osteoblasts and osteocytes also secrete RANKL and OPG. Osteoclasts express RANK, the RANKL receptor, and binding stimulates the differentiation of osteoclast precursors into mature osteoclasts; OPG sequesters RANKL to prevent unrestricted osteoclast differentiation. SOST optimizes the relative proportion of RANKL and OPG to induce bone resorption. Our findings demonstrate that while ERFE loss leads to increased bone mineralization in vitro and bone formation increases as expected with age, the composite effect in vivo results in greater enhancement of osteoclastogenesis relative to osteoblastogenesis in Erfe-/- relative to wild-type mice, consequently decreasing BMD (Figure 6).
We intentionally compared growing (6-week-old) and mature (5-month-old) mice to assess the potentially distinct or cumulative effect of ERFE loss on bone growth and/or remodeling, respectively. Our results demonstrate that ERFE loss leads to impaired BMD in both cohorts of mice. However, while MS/BS, MAR, BFR, and BV/TV are increased in 6-week-old Erfe-/- relative to wild-type mice, no differences are evident between 5-month-old Erfe-/- and wild-type mice. These findings strongly suggest that over time, while both bone formation and resorption increase relative to younger mice, the balance between them favors bone resorption (Figure 6C). This interpretation is further corroborated by a more profound increase in osteoclast surface and osteoclast number in 5-month-old Erfe-/- relative to wild type than in 6-week-old mice.
We show that supernatants from wild-type osteoblast cultures suppress Hamp expression, suggesting that BMP sequestration is likely a common mechanism that underpins both the hepatocellular and skeletal actions of ERFE. Recombinant ERFE binds certain bone-active BMPs, namely BMP2, BMP6, and the BMP2/6 heterodimer (Arezes et al., 2018; Wang et al., 2020), of which BMP2 is most relevant to adult bone formation (Salazar et al., 2016). We posit that ERFE is a negative regulator of osteoblastic bone formation, and that the absence of ERFE increases bioavailable BMP to promote osteoblast differentiation. Our results indeed demonstrate that BMP2 levels are elevated in supernatants from cultured Erfe-/- osteoblasts, with enhanced downstream signals, notably phosphorylated Smad1/5/8 and ERK1/2. In all, the findings provide strong support for BMP sequestration as the mechanism of action of ERFE on bone.
We have also used the β-thalassemia mouse, Hbbth3/+, as a relevant disease model to study a role for ERFE in β-thalassemia, a condition with known elevations in ERFE. The results presented reflect evidence derived from analysis of male mice. We anticipate that similar differences would be expected in female mice. Chronic erythroid expansion in β-thalassemia is associated with a thinning of cortical bone resulting in bone loss (Haidar et al., 2011; Vogiatzi et al., 2006). It is therefore surprising that patients with the more severe forms of β-thalassemia, namely TDT, in whom RBC transfusions lead to suppression of endogenous erythropoiesis, exhibit significantly greater decrements in BMD than NTDT patients (Vogiatzi et al., 2010). We have previously shown that ERFE is suppressed post-transfusion in TDT patients, and is significantly higher in NTDT patients (Ganz et al., 2017). Our finding of a marked reduction of bone mass in Hbbth3/+ mice with genetically deleted Erfe (or Hbbth3/+;Erfe-/- mice) compared with Hbbth3/+ mice provides strong evidence for a protective function of ERFE in preventing further worsening of the bone loss phenotype in β-thalassemia.
Taken together, our findings uncover ERFE as a novel regulator of bone mass via its modulation of BMP signaling in osteoblasts. In addition, because RBC transfusion suppresses erythropoiesis and thus decreases ERFE in both mice (Kautz et al., 2015) and patients (Ganz et al., 2017) with β-thalassemia, a relative decrement of ERFE may explain the more severe bone disease in TDT than in NTDT patients. As a consequence, our findings identify ERFE as a promising new therapeutic target for hematologic diseases associated with bone loss, such as β-thalassemia.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Association between Oxidative Stress and Bone Mineral DensityBiochemical and Biophysical Research Communications 288:275–279.https://doi.org/10.1006/bbrc.2001.5747
-
Bone Morphogenetic Proteins Signal Via SMAD and Mitogen-activated Protein (MAP) Kinase Pathways at Distinct Times during OsteoclastogenesisJournal of Biological Chemistry 288:37230–37240.https://doi.org/10.1074/jbc.M113.496950
-
High-Throughput, Multi-Image cryohistology of mineralized tissuesJournal of Visualized Experiments 115:54468.https://doi.org/10.3791/54468
-
Texture analysis of X-ray radiographs of iliac bone is correlated with bone micro-CTOsteoporosis International 17:447–454.https://doi.org/10.1007/s00198-005-0007-8
-
The hematological etiology of osteoporosisMedical Hypotheses 67:729–735.https://doi.org/10.1016/j.mehy.2006.03.051
-
Bone morphogenetic proteins: multifunctional regulators of vertebrate developmentGenes & Development 10:1580–1594.https://doi.org/10.1101/gad.10.13.1580
-
Bone Morphogenic Protein 2 Activates Protein Kinase D to Regulate Histone Deacetylase 7 Localization and Repression of Runx2Journal of Biological Chemistry 284:2225–2234.https://doi.org/10.1074/jbc.M800586200
-
Bone Formation Is Coupled to Resorption Via Suppression of Sclerostin Expression by OsteoclastsJournal of Bone and Mineral Research 32:2074–2086.https://doi.org/10.1002/jbmr.3175
-
Osteoporosis and inflammationArquivos Brasileiros De Endocrinologia & Metabologia 54:123–132.https://doi.org/10.1590/S0004-27302010000200007
-
Isolation, culture, and differentiation of bone marrow stromal cells and osteoclast progenitors from miceJournal of Visualized Experiments 131:e56750.https://doi.org/10.3791/56750
-
A Novel Platform for Immune Tolerance Induction in Hemophilia A MiceMolecular Therapy 25:1815–1830.https://doi.org/10.1016/j.ymthe.2017.04.029
-
Osterix and RUNX2 are Transcriptional Regulators of Sclerostin in Human BoneCalcified Tissue International 99:302–309.https://doi.org/10.1007/s00223-016-0144-4
-
BMP signalling in skeletal development, disease and repairNature Reviews Endocrinology 12:203–221.https://doi.org/10.1038/nrendo.2016.12
-
Bone Marrow Composition and Bone Microarchitecture and Turnover in Blacks and WhitesJournal of Bone and Mineral Research 13:1300–1307.https://doi.org/10.1359/jbmr.1998.13.8.1300
-
Marrow fat and bone: review of clinical findingsFrontiers in Endocrinology 6:40.https://doi.org/10.3389/fendo.2015.00040
-
Guidelines for the standard monitoring of patients with thalassemia: report of the thalassemia longitudinal cohortJournal of Pediatric Hematology/Oncology 37:e162–e169.https://doi.org/10.1097/MPH.0000000000000307
-
4 Pathophysiology of thalassaemiaBaillière's Clinical Haematology 11:127–146.https://doi.org/10.1016/S0950-3536(98)80072-3
-
The Population Genetics and Dynamics of the ThalassemiasHematology/Oncology Clinics of North America 24:1021–1031.https://doi.org/10.1016/j.hoc.2010.08.010
-
Sclerostin is a direct target of osteoblast-specific transcription factor osterixBiochemical and Biophysical Research Communications 400:684–688.https://doi.org/10.1016/j.bbrc.2010.08.128
-
Skeletal remodeling in health and diseaseNature Medicine 13:791–801.https://doi.org/10.1038/nm1593
Article and author information
Author details
Funding
National Institute of Diabetes and Digestive and Kidney Diseases (DK107670)
- Yelena Ginzburg
National Institute of Diabetes and Digestive and Kidney Diseases (DK095112)
- Robert Fleming
- Stefano Rivella
- Yelena Ginzburg
National Institute of Diabetes and Digestive and Kidney Diseases (DK113627)
- Mone Zaidi
National Institute on Aging (AG60917)
- Mone Zaidi
National Institute of Diabetes and Digestive and Kidney Diseases (DK09055)
- Stefano Rivella
National Institute on Aging (AG71870)
- Tony Yuen
National Institute of Diabetes and Digestive and Kidney Diseases (DK090554)
- Stefano Rivella
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We sincerely appreciate Tomas Ganz and Chun-Ling (‘Grace’) Jung (UCLA), as well as Martina Rauner (University of Dresden) for many stimulating and helpful discussions and Ronald Hoffman (Icahn School of Medicine at Mount Sinai) for continued mentoring and advocacy. YZG acknowledges the support of the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) (R01 DK107670 to YZG and DK095112 to RF, SR, and YZG). MZ acknowledges the support of the National Institute on Aging (U19 AG60917) and NIDDK (R01 DK113627). TY acknowledges the support of the National Institute on Aging (R01 AG71870). SR acknowledges the support of NIDDK (R01 DK090554) and Commonwealth Universal Research Enhancement (CURE) Program Pennsylvania.
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (#16-0143) of the Icahn School of Medicine.
Copyright
© 2021, Castro-Mollo et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,497
- views
-
- 314
- downloads
-
- 31
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 31
- citations for umbrella DOI https://doi.org/10.7554/eLife.68217
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The hepcidin regulator erythroferrone is a new member of the erythropoiesis-iron-bone circuitry
eLife 10:e68217.
https://doi.org/10.7554/eLife.68217




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}