Loss of N1-methylation of G37 in tRNA induces ribosome stalling and reprograms gene expression
- Department of Biochemistry and Molecular Biology, Thomas Jefferson University, United States
- Department of Molecular Biology and Genetics, Johns Hopkins University School of Medicine, United States
Abstract
N1-methylation of G37 is required for a subset of tRNAs to maintain the translational reading-frame. While loss of m1G37 increases ribosomal +1 frameshifting, whether it incurs additional translational defects is unknown. Here, we address this question by applying ribosome profiling to gain a genome-wide view of the effects of m1G37 deficiency on protein synthesis. Using E coli as a model, we show that m1G37 deficiency induces ribosome stalling at codons that are normally translated by m1G37-containing tRNAs. Stalling occurs during decoding of affected codons at the ribosomal A site, indicating a distinct mechanism than that of +1 frameshifting, which occurs after the affected codons leave the A site. Enzyme- and cell-based assays show that m1G37 deficiency reduces tRNA aminoacylation and in some cases peptide-bond formation. We observe changes of gene expression in m1G37 deficiency similar to those in the stringent response that is typically induced by deficiency of amino acids. This work demonstrates a previously unrecognized function of m1G37 that emphasizes its role throughout the entire elongation cycle of protein synthesis, providing new insight into its essentiality for bacterial growth and survival.
Introduction
N1-methylation of G37 in tRNA, generating m1G37 on the 3'-side of the anticodon, is a post-transcriptional modification that is essential for life (Björk et al., 2001). It has been specifically associated across evolution with all isoacceptors of tRNAPro, which are species that share the same prolyl specificity of aminoacylation but differ in the primary sequence and in the anticodon triplet and yet collectively decode the Pro CCN codons (N = A, C, G, and U). Similarly, m1G37 is conserved in the CCG isoacceptor of tRNAArg(tRNAArg(CCG)), with the anticodon CCG for pairing with the CGG codon, and it is conserved in isoacceptors tRNALeu(GAG), tRNALeu(UAG), and tRNALeu(CAG) for pairing with CUU and CUC codons (CU[C/U]) and CU[A/G] codons, respectively (Björk et al., 2001; Li et al., 1997). Additionally, m1G37 may also be present in other tRNAs in higher eukaryotes. The m1G37 methylation of tRNA is required for cell growth and viability; elimination of the enzyme responsible for the methylation causes cell death in yeast and in bacteria (Björk et al., 2007; Björk et al., 2001; Masuda et al., 2019). The established function of m1G37 is to maintain the translational reading-frame during protein synthesis (Björk et al., 1989; Hagervall et al., 1993; Qian et al., 1998). Loss of m1G37 elevates frequencies of ribosomal +1 frameshifting in kinetic assays with reconstituted E. coli ribosomes (Gamper et al., 2015a), and in cell-based assays in E. coli and Salmonella (Gamper et al., 2021; Gamper et al., 2015a). Unlike ribosomal miscoding, +1 frameshifting is almost always deleterious, altering the translational reading-frame, inducing premature termination of protein synthesis, and ultimately cell death.
Recent work has shed light on how loss of m1G37 induces ribosomal +1 frameshifting. While non-methylated tRNAs were thought to pair incorrectly in the ribosomal A site (the aminoacyl-tRNA [aa-tRNA] binding site) during decoding (Roth, 1981), they were found to occupy the correct reading-frame in X-ray crystal structures and in kinetic assays (Gamper et al., 2021; Maehigashi et al., 2014). Maintenance of the correct reading-frame in the A site is consistent with the strict ribosomal A site structure that selects for accurate anticodon-codon pairing during decoding (Ogle et al., 2001; Ogle and Ramakrishnan, 2005). Even genetically isolated high-efficiency +1--frameshifting tRNAs, which usually contain an extra nucleotide inserted to the anticodon loop (Atkins and Björk, 2009), were found to occupy the correct reading-frame in the A site (Dunham et al., 2007; Fagan et al., 2014; Gamper et al., 2021; Maehigashi et al., 2014). These genetically isolated +1-frameshifting tRNAs, however, were found to occupy the triplet +1-frame in the ribosomal P site (the peptidyl-tRNA binding site) (Hong et al., 2018).
All existing evidence points to tRNA +1 frameshifting occurring after decoding at the A site. A frameshift-prone tRNA, containing the natural anticodon loop but lacking m1G37, is shown in kinetic assays to undergo +1 frameshifting during translocation from the A site to the P site, or during occupancy in the P site next to an empty A site (Gamper et al., 2015a). A genetically isolated high-efficiency +1-frameshifting tRNA, containing an expanded anticodon loop, is also shown in kinetic assays to undergo +1 frameshifting during translocation (Gamper et al., 2021). The notion of +1 frameshifting during translocation is supported by a recent cryo-EM structural analysis of a canonical tRNA translating a frameshift-prone mRNA sequence (Demo et al., 2021). Similarly, the notion of +1 frameshifting within the P site is supported by X-ray crystal structures of a P site-bound frameshift-prone tRNA with a natural anticodon loop (Hoffer et al., 2020). In these latter structures, while m1G37 stabilizes the tRNA on an mRNA codon that is prone to frameshifting, loss of m1G37 destabilizes the tRNA-ribosome interaction and induces +1 frameshifting within large conformational changes of the ribosome.
Here, we seek to determine whether m1G37 plays additional roles beyond maintaining the ribosome translational reading-frame. An earlier study suggested that loss of m1G37 delayed the tRNA anticodon-codon pairing interaction during decoding (Li et al., 1997), raising the possibility of a role at the ribosomal A site. To test this possibility more broadly, we employ the approach of ribosome profiling to determine ribosome positions during translation of the entire transcriptome of a cell (Ingolia et al., 2009) and to monitor how ribosome density changes upon loss of m1G37. Using E. coli as a model, we show that deficiency of m1G37 induces global ribosome stalling, most notably at Pro codons CCN, the Arg codon CGG, and the Leu codon CUA, all of which are translated by tRNAs that are normally methylated with m1G37. Stalling is most prominent when the affected codons are being decoded at the ribosomal A site, indicating a distinct mechanism than that of +1 frameshifting. Enzyme- and cell-based assays show that m1G37 deficiency reduces aminoacylation of the affected tRNAs, including all isoacceptors of tRNAPro and the isoacceptor tRNAArg(CCG), and that additionally it reduces rates of peptide-bond formation for some of these tRNAs. Most significantly, stalling induces programmatic changes in gene expression that are consistent with changes occurring during the bacterial stringent response under environmental stress of nutrient starvation. These findings support a model in which m1G37 deficiency reduces levels of aa-tRNAs at the ribosomal A site and prevents peptide-bond formation in some cases, leading to ribosome stalling. Binding of uncharged tRNAs to the A site would then induce programmatic changes of gene expression similar to those occurring during the bacterial stringent response through activation of the ppGpp synthase RelA (Gourse et al., 2018). The importance of m1G37 for the ribosomal activity at the A site, together with its already demonstrated importance in maintaining the translational reading-frame from the A site to the P site and within the P site (Gamper et al., 2021; Gamper et al., 2015a), establishes the involvement of the methylation throughout the entire elongation cycle of protein synthesis. This sustained involvement of m1G37 during protein synthesis underscores its indispensable role in bacterial viability and survival.
Results
E. coli strains with conditional m1G37 deficiency
Because m1G37 is essential for cell viability, a simple knock-out of the gene responsible for its biosynthesis cannot be made. Previous studies of cellular functions of m1G37 relied on temperature-sensitive variants of the gene responsible for m1G37 biosynthesis whose protein product became inactivated at elevated temperatures (Björk and Nilsson, 2003; Masuda et al., 2013). Because elevated temperatures induce changes in gene expression, we took a different approach to conditionally deplete m1G37 to study its role in protein synthesis. Interestingly, while m1G37 is conserved in evolution, the genes responsible for its biosynthesis are distinct – being trmD in bacteria and trm5 in archaea and eukaryotes (Christian et al., 2004). The protein products of trmD and trm5 are fundamentally different from each other in structure and mechanism (Christian and Hou, 2007; Christian et al., 2010a; Christian et al., 2010b; Christian et al., 2016; Lahoud et al., 2011; Sakaguchi et al., 2014). We recently constructed conditional m1G37-deficient strains of E. coli and Salmonella, in which the trmD locus is deleted from the chromosome and cell viability of the trmD-knock-out (trmD-KO) strain is maintained by a plasmid-borne human trm5 that is under arabinose (Ara)-controlled expression (Gamper et al., 2015a; Masuda et al., 2019). Upon induction with Ara, expression of human trm5 is sufficient to supply m1G37-tRNAs to support bacterial viability (trmD-KO (trm5+)) (Christian et al., 2004), whereas upon replacement of Ara with glucose (Glc), expression of human trm5 is arrested and the human enzyme is degraded inside bacterial cells (trmD-KO (trm5–)) (Christian et al., 2013). As a control, a trmD-wild-type (trmD-WT) strain was created, where trmD remains on the chromosome and expression of the plasmid-borne trm5 in the presence of Ara (trmD-WT (trm5+)), or its repression in the presence of Glc (trmD-WT (trm5–)), did not affect cell viability.
To avoid the possibility of artifacts by studying only one conditional m1G37-deficient strain, we developed a second conditional m1G37-deficient strain to compare data and to strengthen conclusions. In this second conditional strain, we chose E. coli as a model and extended trmD at the chromosomal locus with a degron sequence, adding YALAA to the C-terminus of the TrmD protein (the trmD-deg strain) to allow rapid degradation by the protease ClpXP. We controlled TrmD degradation by inducing the expression of clpXP from a plasmid using Ara, or by repressing the expression using Glc (Carr et al., 2012). Because over-expression of the plasmid-borne clpXP could also target proteins without the degron tag, we generated a control strain (trmD-cont) for comparison. In this trmD-cont strain, we added the coding sequence for the degron tag after the stop codon of trmD to maintain the same gene length as in trmD-deg, but without expression of the tag (Figure 1—figure supplement 1A and B).
Overnight cultures of trmD-deg and trmD-cont strains were grown in LB + Glc and were spotted onto an LB + Ara plate to turn on expression of clpXP. Analysis of a serial dilution of each strain confirmed that trmD-deg cells rapidly lost viability, whereas trmD-cont cells retained viability (Figure 1A). In liquid culture, in which each strain was freshly diluted into LB + Ara at OD600 0.1 and grown to 0.3–0.4, followed by a second round of dilution and re-growth, we observed that the re-growth of the trmD-deg strain was retarded in the third round, whereas that of the trmD-cont strain was robust (Figure 1B). Although the growth defect of the trmD-deg strain only manifested in the third cycle of dilution, the level of TrmD protein drastically decreased in 30 min after the first dilution into fresh LB + Ara (as in Figure 1B), whereas that in the trmD-cont strain remained stable up to 90 min (Figure 1C) and longer (Figure 1—figure supplement 1C). We speculate that it takes time for cultures to express clpXP to a threshold level, after which TrmD is rapidly degraded. A similar pattern of drastic reduction of the target protein following clpXP induction was reported previously (Carr et al., 2012). We also speculate that the clpXP expression plasmid, which has the strongest expression strength among a library of plasmids varying in Shine-Dalgarno sequences (Materials and methods), could also contribute to the drastic degradation of TrmD.
Figure 1 with 1 supplement see all
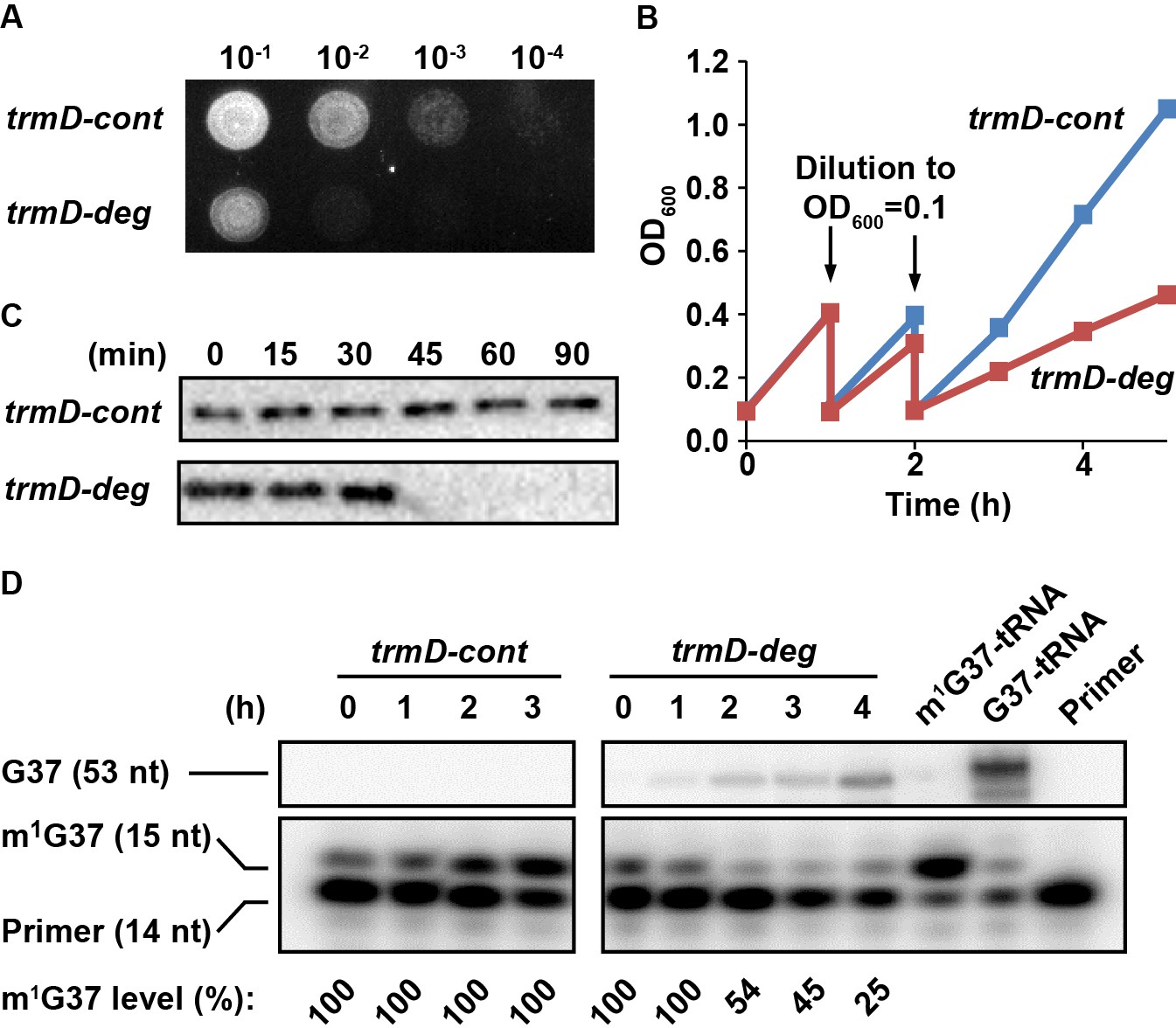
Conditional m1G37 deficiency of the E. coli trmD-deg strain.
(A) Growth of trmD-deg and trmD-cont G78 cells on an LB plate. An overnight culture of trmD-deg and trmD-cont cells in LB + Glc was serially diluted, spotted on an LB + Ara plate to turn on clpXP expression, and incubated at 37°C overnight. (B) Representative growth of trmD-deg and trmD-cont G78 cells in a liquid LB culture. An overnight culture of trmD-deg and trmD-cont G78 cells in LB + Glc was grown in LB for 1–2 hr, then diluted into LB + Ara at OD600 of 0.1 and grown to OD600 of 0.3 at 37°C. The cycle of dilution and re-growth was repeated three times to observe a growth defect of the trmD-deg strain. (C) Western blot analysis of TrmD protein. An overnight culture of trmD-deg and trmD-cont cells in LB + Glc was diluted to LB + Ara at T = 0, and was sampled at the indicated time points. Cell lysates were separated on a 12% SDS-PAGE and TrmD protein was detected by primary antibody against E. coli TrmD and a secondary antibody against rabbit IgG. (D) Primer-extension inhibition analysis. An overnight culture of trmD-deg and trmD-cont cells in LB + Glc was diluted to LB + Ara at T = 0 and the fresh culture was taken through three cycles of dilution and re-growth. Total RNA was extracted over the time course, probed with a 5'-[32P]-labeled DNA primer targeting E. coli tRNALeu/CAG, and analyzed by a 12% PAGE/7 M urea gel and phosphorimaging. Primer extension would terminate in the control at 1 nt downstream of m1G37, generating a 15 nt fragment, whereas primer extension would continue to the 5'-end in TrmD deficiency, generating a 53 nt fragment. The fraction of m1G37 in each sample is calculated based on all primer-extension products as shown in the source file, not including the primer. Because the same amount of cell culture was used for extraction of RNA, an increased primer-extension stop at the 15 nt band relative to the primer position was observed in trmD-cont cells, reflecting an increased cell density 0–3 hr and increased synthesis of m1G37-tRNA. In all samples collected for trmD-cont cells, no synthesis of the read-through 53 nt band was observed, indicating 100% methylation. At the time of cell harvest, the m1G37 level was 100% for trmD-cont cells at T = 3 hr, but was below 25% for trmD-deg cells at T = 4 hr.
To measure cellular levels of m1G37 during this series of dilution and re-growth, we designed a 5'-[32P]-labeled primer of 14 nucleotides (14 nt) complementary to E. coli tRNALeu(CAG). The presence of m1G37 would inhibit primer extension, producing a 15 nt product, whereas the absence of m1G37 would permit primer extension to the 5'-end, producing a 53 nt product. The level of m1G37 was calculated as the fraction of the 15 nt product in the sum of all of the primer-extension products of each reaction. The primer was not included in the calculation due to its molar excess in these reactions. Analysis of total RNA samples, collected after the first dilution into fresh LB + Ara in the time course (as in Figure 1B), showed that the 15 nt product progressively decreased in trmD-deg cells, indicating gradual loss of m1G37, but that it remained stable in trmD-cont cells (Figure 1D). At the time of cell harvesting, m1G37 in trmD-deg cells was at or below 25% (T = 4.2 hr), whereas that in trmD-cont cells was 100% (T = 3 hr). Collectively, these results demonstrate the ability of the degron approach to control the stability of TrmD and to produce conditional m1G37 deficiency in E. coli cells.
Codon-specific ribosome stalling in m1G37 deficiency
We performed ribosome profiling and RNA-seq analyses on two biological replicates of the trmD-deg and trmD-cont strains, which were cultured in the presence of clpXP expression with three cycles of dilution and re-growth and were harvested at OD600 of 0.3. We also obtained a third set of samples using trmD-KO (trm5–) and trmD-WT (trm5–) strains, which were cultured in the absence of trm5 expression in three cycles of dilution and re-growth and were harvested at OD600 of 0.3. Given that m1G37 is associated with a specific set of E. coli tRNAs, we looked in the ribosome profiling data for local differences in the A site occupancy for each codon in m1G37 deficiency. If loss of m1G37 impaired tRNA decoding, we expected that the ribosome would linger on affected codons to accumulate higher levels of density. To quantify these codon-specific effects, we defined a pause score for each codon as the ribosome density at the first nt of the codon normalized by the average ribosome density on the gene where that codon occurs. We computed the pause score for all of the 61 sense codons by averaging the pause score at thousands of instances of each codon across the entire transcriptome. Strikingly, we observed significant increases in the pause score for a set of codons when each was positioned at the ribosomal A site during decoding (Figure 2). Most notably, ribosome density at Pro codons was dramatically increased in m1G37 deficiency, showing an increase in the pause score from 1.4 in trmD-cont cells to 3.5 in trmD-deg cells (Figure 2A, left). Similarly, the pause score at Pro codons increased from 1.2 in trmD-WT (trm5–) cells to 2.5 in trmD-KO (trm5–) cells (Figure 2A, right). The increase in the pause score for all Pro codons indicates that m1G37 deficiency affected decoding by some or all of the isoacceptors of tRNAPro, leading to strong ribosome pausing during elongation.
Figure 2
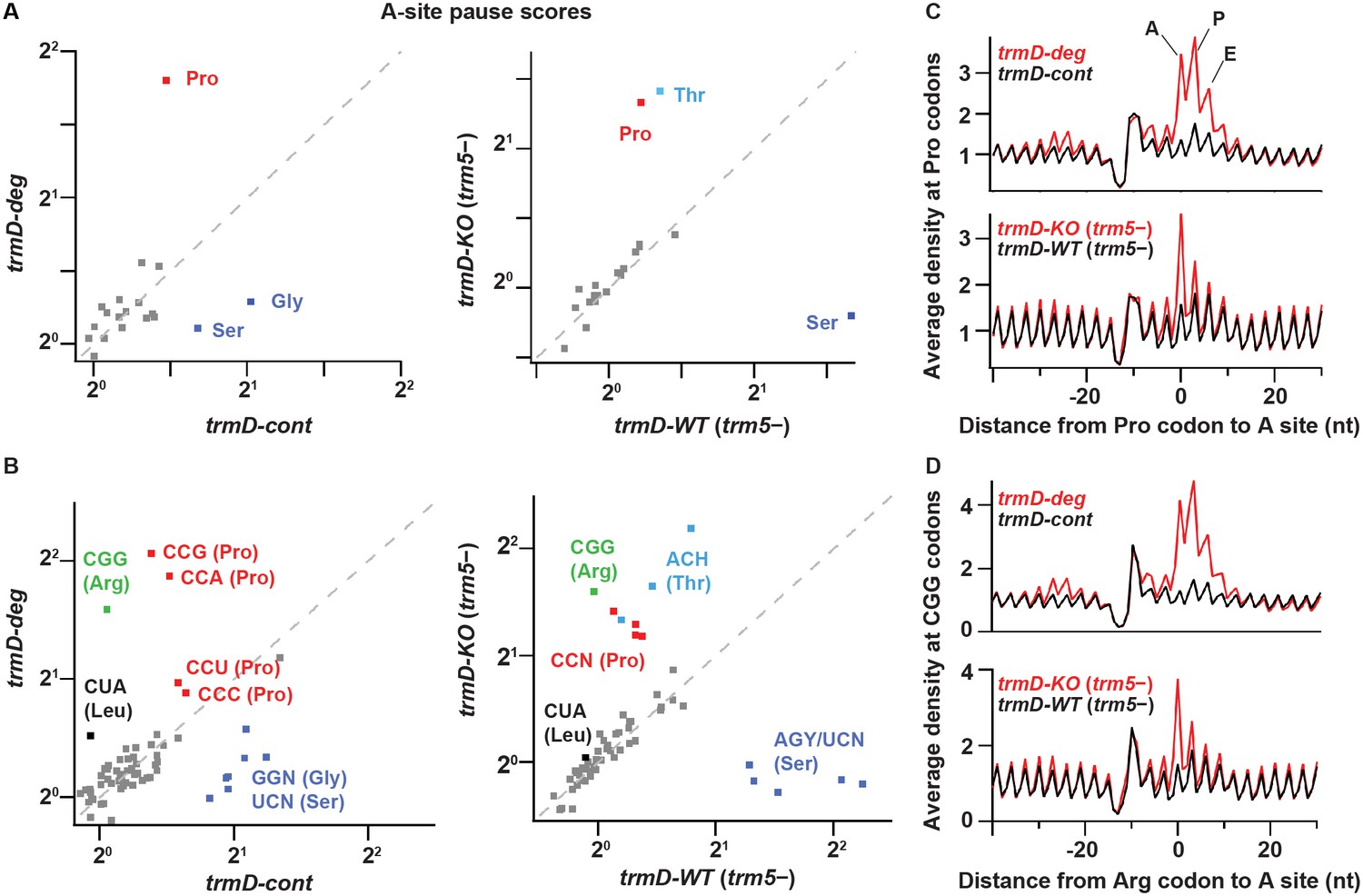
TrmD depletion leads to ribosome pausing at CCN (Pro) and CGG (Arg) codons.
(A) Pause scores for codons positioned in the ribosomal A site. The codons are grouped by amino acid. Left panel: data from biological replicates of trmD-deg and trmD-cont strains upon ClpXP induction. Each biological replicate is an independent culture of cells. Right panel: data from trmD-KO (trm5–) and trmD-WT (trm5–) strains upon turning off trm5. (B) The same as above but showing the 61 sense codons individually. (C) Plots of average ribosome density aligned at Pro codons for trmD-deg strain (top) and trmD-KO (trm5–) strain (bottom) in red with their respective control strains in black. The peaks corresponding to the ribosomal A, P, and E sites are labeled. (D) Plots of average ribosome density aligned at CGG codons as above. Note that trmD-deg samples were obtained with the traditional ribosome profiling lysis buffer, whereas trmD-KO (trm5–) samples were obtained with a high Mg2+ lysis buffer that more effectively arrests translation after cell lysis, revealing these pauses at higher resolution.
We also observed differences in ribosome pausing at other codons (Figure 2A), which likely resulted from artifacts of the ribosome profiling method and not from translation differences due to m1G37 deficiency. There were stronger pauses at Ser and Gly codons in the control samples of trmD-cont than in the trmD-deg samples, and stronger pauses at Ser codons in the control samples of trmD-WT (trm5–) than in the trmD-KO (trm5–) samples. These pauses were likely due to artifacts of harvesting bacterial cultures. We previously observed pauses at Ser and Gly codons in cells harvested by filtration and showed that filtration reduced aminoacylation of the tRNA cognate to these codons (Mohammad et al., 2019). These Ser and Gly pauses, however, are less obvious in strains where protein synthesis is defective (e.g., due to loss of the elongation factor EFP) (Woolstenhulme et al., 2015), which may explain their lower pause scores in the trmD-deg and trmD-KO (trm5–) samples than in the control samples. We have also observed variable pausing at Thr codons under different harvesting conditions in unpublished work, although those pauses have not been as well characterized. Given that all of the samples described here were harvested by filtration, we attributed the pauses at Ser, Gly, and Thr codons (labeled in blue in Figure 2A) to artifacts of cell growth and harvesting and did not consider them further.
At the codon level, we observed higher pause scores for all four Pro codons (CCN, in red, Figure 2B) in m1G37-deficient cells relative to control cells, as shown in trmD-deg vs. trmD-cont strains (left) and in trmD-KO (trm5–) vs. trmD-WT (trm5–) strains (right). In both replicates of the trmD-deg strain, Pro codons CCG and CCA had significantly higher pause scores than CCC and CCU. In contrast, all four CCN codons showed similar pause scores in the trmD-KO (trm5–) strain. While the reason for these differences between the two strains is not clear, the implication is that ribosomes paused at all four Pro codons in both strains. Also, we observed elevated pause scores on the CGG codon (in green) in m1G37 deficiency, increasing from 1.0 in control cells to 3.0 in trmD-deg cells and in trmD-KO (trm5–) cells. Notably, CGG was the only Arg codon where pausing was observed upon loss of m1G37, whereas the other Arg codons CG[A/C/U], AGA, and AGG were not affected. Additionally, we observed a small increase of the pause score at the Leu codon CUA in m1G37 deficiency at 1.4 relative to the pause score of 1.0 in control samples (Figure 2B). Together, these data showed significant pauses at all Pro codons CCN, the Arg CGG codon, and to a lesser extent the Leu CUA codon. All of the paused codons are translated by tRNAs that are substrates of m1G37 methylation, although pauses at Leu codons CU[C/G/U], which are translated by tRNAs that should also be methylated with m1G37, were not observed.
The increase in the average pause scores in m1G37 deficiency indicates accumulation of ribosome density when the affected codon is positioned in the A site. To explore the possibility of other effects on translation, we looked more broadly at the average ribosome density aligned to a specific codon of interest. For trmD-deg vs. trmD-cont samples, we observed strong increases of ribosome density in the A, P, and E sites at Pro codons, and at the Arg CGG codons in m1G37 deficiency, with the highest peak in the ribosomal P site (Figure 2C–D, top). Although these findings could be interpreted to mean that m1G37 deficiency in tRNAs for these codons led to pausing when each tRNA was in the P or E site, we recognized that the trmD-deg and trmD-cont samples were obtained using the traditional lysis buffer with chloramphenicol to arrest translation in the lysate. We have since discovered that ribosomes continue to translate a few codons in the lysate under these conditions (Mohammad et al., 2019), suggesting that the several peaks observed in m1G37 deficiency likely arose from a strong pause at the A site that was blurred by ongoing translation in the lysate during sample preparation. In contrast, the trmD-KO (trm5–) and trmD-WT (trm5–) samples were prepared with a buffer containing 150 mM MgCl2, which immediately arrests ribosomes without the need for antibiotics (Mohammad et al., 2019). In these samples, we observed that pauses at Pro codons and at Arg CGG codons (Figure 2C–D, bottom) were the strongest when each codon was positioned at the A site, without an increased density in the P and E sites. We conclude that pauses are primarily in the A site, due to defects in decoding associated with m1G37 deficiency.
Interestingly, we observed increased ribosome density ~25 nt upstream of affected codons in trmD-deg relative to trmD-cont samples (Figure 2C–D, top). This distance is roughly equivalent to the footprint length of a single ribosome, indicating that the increased density was due to collision of an upstream ribosome with a paused ribosome that was struggling to decode an affected codon. The collision of two ribosomes suggests that the pausing of the downstream ribosome at the affected codon is significantly prolonged. These findings are consistent with our prior observation of pausing and formation of disomes upon treatment of cells with mupirocin, an antibiotic that blocks aminoacylation of tRNAIle (Mohammad et al., 2019).
Reduced aminoacylation and A-site peptide-bond formation of m1G37-deficient tRNAs
The observed ribosome pausing at specific codons in m1G37 deficiency raised two possibilities for the tRNAs translating these codons. First, m1G37 deficiency may reduce aminoacylation of these tRNAs by the respective aminoacyl-tRNA synthetases (aaRSs), preventing them from forming a ternary complex (TC) with EF-Tu-GTP and entering the ribosomal A site. Second, m1G37 deficiency may prevent these tRNAs from peptide-bond formation at the A site, leading to ribosome stalling. We addressed these two possibilities, which are not mutually exclusive, with all three isoacceptors of tRNAPro and the tRNAArg(CCG) isoacceptor. To isolate the effect of m1G37, we prepared tRNAs as T7 RNA polymerase (RNAP) transcripts lacking m1G37 or any other post-transcriptional modification (the G37-state) and compared their activity to transcripts that were subsequently modified with m1G37 by TrmD in vitro (the m1G37-state). We confirmed that the level of methylation in the m1G37-state was high, reaching nearly 100% for the m1G37-state of E. coli tRNAPro(UGG) and nearly 70% for the m1G37-state of E. coli tRNAArg(CCG) (Figure 3—figure supplement 1A). In addition, we also purified some of these tRNAs from cells (the native-state) containing the full complement of natural post-transcriptional modifications.
Aminoacylation of tRNAPro with Pro was performed with purified E. coli ProRS under steady-state multi-turnover conditions (Zhang et al., 2006). The initial rate as a function of the concentration of each tRNA was measured and the data were fit to the Michaelis-Menten equation to derive kinetic parameters Km (tRNA) and kcat. For all three isoacceptors of tRNAPro, the catalytic efficiency kcat/Km was decreased from the m1G37-state to the G37-state by 3- to 10-fold (Figure 3A and Figure 3—source data 1). The reduction in kcat/Km was driven by an increase in Km for all three tRNAs, indicating that m1G37 may be important for binding of each tRNA to ProRS (Figure 3—source data 1A). This apparent binding defect makes sense structurally because m1G37 is immediately downstream of the two conserved anticodon nucleotides G35-G36, which are the major determinants of tRNAPro binding to ProRS (Cusack et al., 1998; Yaremchuk et al., 2000; Yaremchuk et al., 2001). The largest decrease in kcat/Km (10-fold) upon loss of m1G37 was observed for the UGG isoacceptor. Unique among the isoacceptors of tRNAPro, the UGG isoacceptor is required for cell growth and survival (Nasvall et al., 2004) and is most critically dependent on m1G37 for maintaining the translational reading-frame (Gamper et al., 2015a). The critical role that m1G37 plays in the UGG isoacceptor was further highlighted by the finding that aminoacylation reaction with the m1G37-state tRNA has the same kcat/Km values as the native-state tRNA (Figure 3—source data 1A), indicating that all other post-transcriptional modifications played little or no role in aminoacylation.
Figure 3 with 1 supplement see all
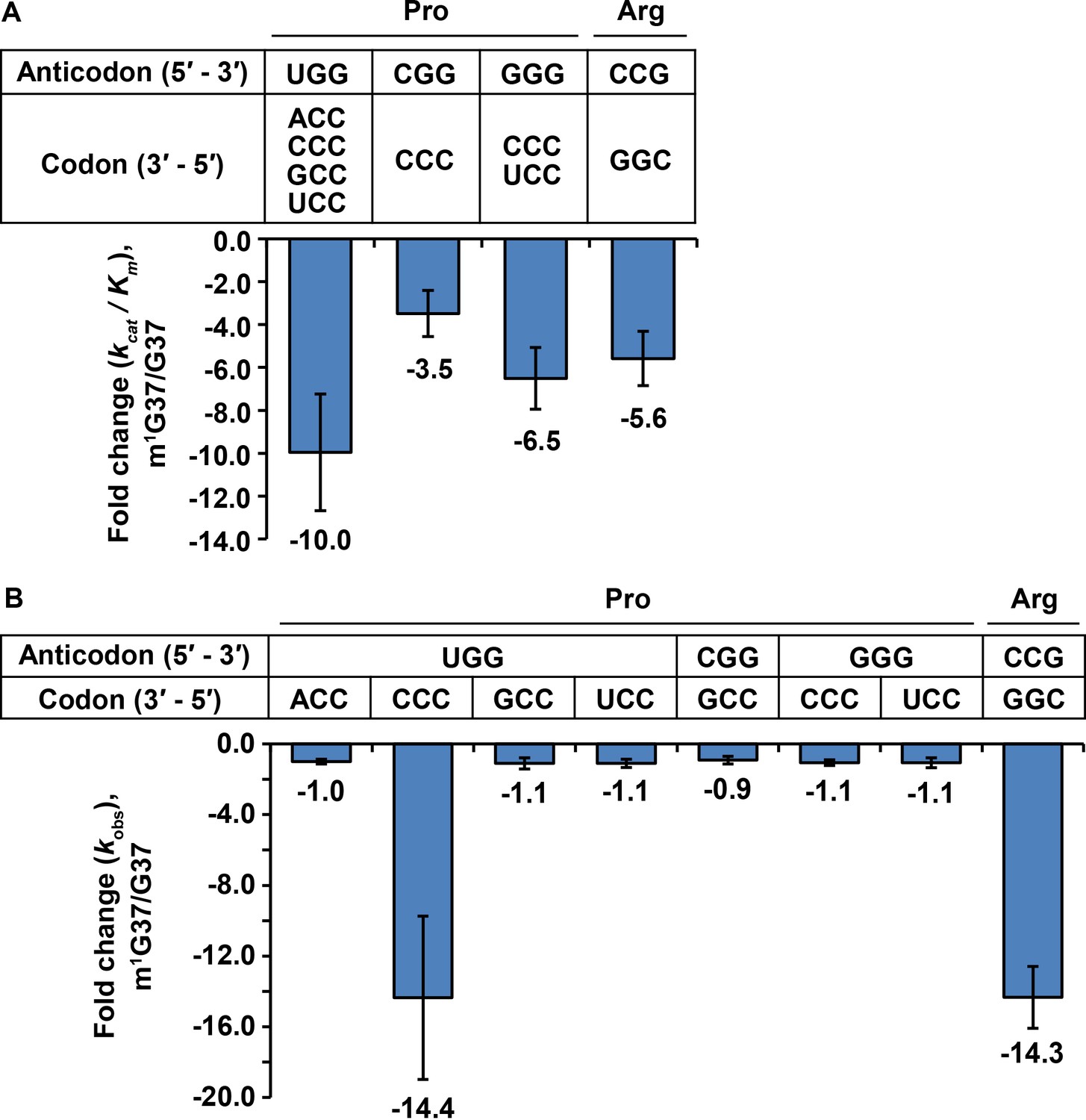
Kinetic parameters showing the loss of tRNA aminoacylation and peptide-bond formation at the A site in m1G37 deficiency.
(A) Fold-change in the loss of kcat/Km of tRNA aminoacylation from the m1G37-state to the G37-state. Bar graphs are shown for the UGG, the CGG, and the GGG isoacceptors of E. coli tRNAPro, and for the CCG isoacceptor of E. coli tRNAArg, with the fold-change value displayed at the bottom of each bar. The bars are SD of three independent replicates (n = 3), and the data are presented as the mean ± SD for each sample. (B) Fold-change in the loss of kobs from the m1G37-state to the G37-state in overall reaction at the A site leading to peptide-bond formation. Bar graphs are shown for the UGG, CGG, and GGG isoacceptors of E. coli tRNAPro, and for the CCG isoacceptor of E. coli tRNAArg. Each tRNA is paired to the cognate codon, and the fold-change value is displayed at the bottom of each bar as the mean ± SD of three independent replicates (n = 3).
Aminoacylation of the tRNAArg(CCG) isoacceptor with Arg was performed with purified E. coli ArgRS under steady-state multi-turnover conditions. The kcat/Km of aminoacylation was decreased from the m1G37-state to the G37-sate by 5.6-fold (Figure 3A and Figure 3—source data 1A). This decrease was largely driven by a loss in kcat (Figure 3—source data 1A), indicating that m1G37 contributed to catalysis, which is a different effect than that observed for tRNAPro, where loss of m1G37 likely reduced tRNA binding to ProRS. Additionally, we observed a loss of 14.3-fold in kcat/Km from the native-state to the G37-state (Figure 3—source data 1A), greater than the 5.6-fold loss due to m1G37 alone, indicating that other post-transcriptional modifications played a role in aminoacylation of this tRNAArg, which also contrasts the observation that m1G37 alone is sufficient for rapid aminoacylation of tRNAPro. Together, these results show that m1G37 is required for efficient aminoacylation of all three isoacceptors of tRNAPro and the tRNAArg(CCG) isoacceptor, and that loss of m1G37 reduces aminoacylation in all cases, although by apparently different mechanisms.
To determine whether m1G37 deficiency reduced peptide-bond formation with affected tRNA in the A site, we used our E. coli in vitro translation system of purified components and supplemented it with requisite tRNAs and translation factors to perform a series of ensemble rapid kinetic studies (Gamper et al., 2021; Gamper et al., 2015a; Gamper et al., 2015b). We assayed for the synthesis of a peptide bond between the [35S]-fMet moiety of the P site [35S]-fMet-tRNAfMet of a 70S initiation complex (70S IC) and the aminoacyl moiety of a TC delivered to the A site. This assay monitored all of the reactions at the A site, including the initial binding of the TC to the A site, EF-Tu-catalyzed GTP hydrolysis, accommodation of the aa-tRNA to the A site, and peptidyl transfer. Each TC carried a fully aminoacylated tRNA in the G37-state or in the m1G37-state, in excess of the 70S IC, to allow evaluation of the activity of peptide-bond formation. The TC was kept at a limiting concentration (0.2 μM), relative to the previously reported Kd (3–4 μM) (Cochella and Green, 2005), such that the dipeptide-bond formation catalyzed by a limiting 70S IC (0.1 μM) proceeded linearly over time, as shown for tRNAPro(UGG) against the CCA codon (Figure 3—figure supplements 1B and C) and for tRNAArg(CCG) against the CGG codon (Figure 3—figure supplement 1D and E). The slope of each linear production (kobs) under these limiting conditions represented the catalytic efficiency kcat/Km of the overall reaction of peptide-bond formation at the A site, which is a composite term that could be limited by any of the intermediate steps.
For tRNAPro, the UGG isoacceptor was assayed at all four Pro codons CCN, the CGG isoacceptor was assayed at the codon CCG, and the GGG isoacceptor was assayed at the codons CC[C/U] (Figure 3B and Figure 3—source data 1B). The results of these assays showed that, of all of the tested anticodon-codon pairs, loss of m1G37 only had a significant effect on kobs with the UGG isoacceptor at the CCC codon, decreasing kobs by 14.4-fold from the m1G37-state to the G37-state (Figure 3B and Figure 3—source data 1B). Thus, in contrast to aminoacylation, where all isoacceptors of tRNAPro were affected by loss of m1G37, loss of m1G37 only affected the overall reaction of dipeptide formation for one isoacceptor at one codon. Likewise, the CCG isoacceptor of tRNAArg was assayed at the cognate codon CGG, showing a significant decrease of kobs by 14.3-fold from the m1G37-state to the G37-state (Figure 3B and Figure 3—source data 1B). Thus, for the single isoacceptor of tRNAArg, loss of m1G37 reduced the activity of both aminoacylation and the overall reaction of dipeptide formation at the A site.
Loss of aminoacylation in m1G37-deficient cells
Following up on the results of kinetic studies in vitro, we investigated whether m1G37 deficiency led to loss of aminoacylation of affected tRNAs in vivo. Total RNA was extracted from E. coli trmD-KO (trm5+) and trmD-KO (trm5–) cells using an acid buffer (pH 4.5) and run on an acid-urea gel to preserve the levels of charged aa-tRNA vs. uncharged tRNA at the time of harvest. Northern blots with probes against tRNAPro(UGG) and tRNAArg(CCG) showed that both had reduced aa-tRNA levels, decreasing from 84% to 68% and from 74% to 62% respectively in m1G37 deficiency (Figure 4A, left and middle panels). We confirmed that the loss of aminoacylation of these two tRNAs was specific to m1G37 deficiency. Acid-urea gel analysis of E. coli tRNATyr(QUA) (Q: queuosine), which contains ms2i6A37 (2-methyl-thio-N6-isopentenyl adenosine) and is not methylated by TrmD, showed that its aa-tRNA level was similar (78% vs. 81%) whether m1G37 was abundant or deficient (Figure 4A, right panel).
Figure 4
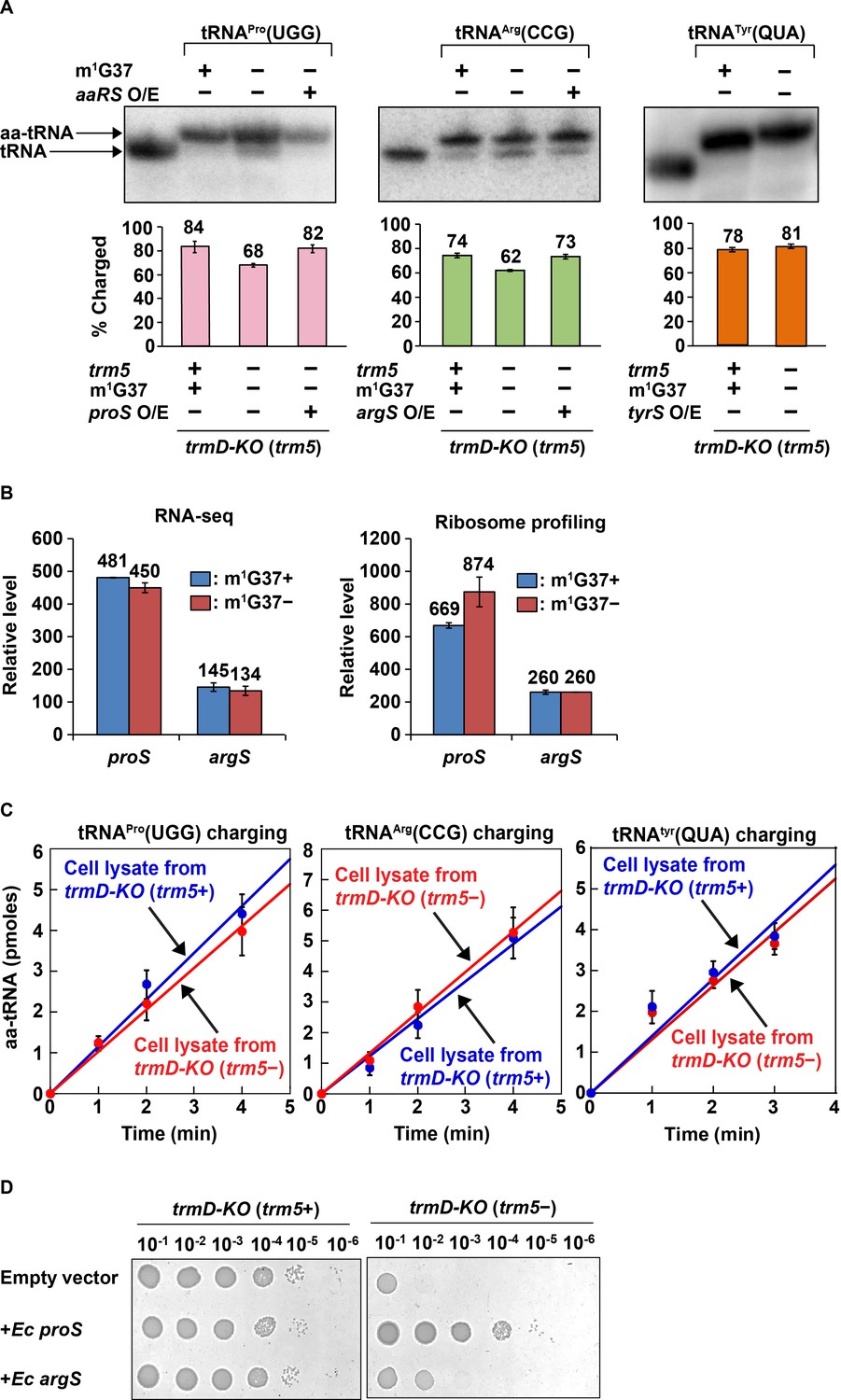
Acid-urea gel analysis of loss of tRNA aminoacylation in m1G37-deficient E. coli cells.
(A) Northern blots of acid-urea gels showing the fractional distribution of charged (aa-tRNA) vs. uncharged (tRNA) in total RNA prepared from trmD-KO cells complemented by the Ara-controlled trm5. Three E. coli tRNAs were probed: tRNAPro(UGG) (left panel), tRNAArg(CCG) (middle panel), and tRNATyr(QUA) (right panel), each under m1G37-abundant (+) and m1G37-deficient (–) conditions. Of the three, the m1G37-dependent tRNAPro(UGG) and tRNAArg(CCG) were further probed in m1G37-deficient cells with over-expression (O/E) of proS and argS, respectively. In contrast, the m1G37-independent tRNATyr(QUA) served as a control and was not probed for O/E of tyrS. The % of aa-tRNA in each case is calculated from the sum of the charged aminoacyl-tRNA (aa-tRNA) and the uncharged tRNA and is shown in the bar graph below for three biological replicates (n = 3). (B) Relative levels of expression of E. coli proS and argS in RNA-seq and ribosome-profiling analysis in m1G37+ and m1G37– conditions of two biological replicates. (C) Aminoacylation activity in E. coli cell lysates prepared in m1G37+ and m1G37– conditions. Three enzymes, encoded by proS (left panel), argS (middle panel), and tryS (right panel), were each tested against the cognate tRNA (i.e., m1G37-state tRNAPro(UGG), m1G37-state tRNAArg(CCG), and A37-state tRNATyr(QUA), respectively). Data are the average of three biological replicates (n = 3). Aminoacylation was performed under substrate-limiting conditions to obtain a linear line of product formation over time. (D) Viability of E. coli trmD-KO (trm5+) and trmD-KO (trm5–) cells harboring an empty vector, or the vector over-expressing E. coli proS or argS, was evaluated by spotting a serial dilution of cells onto an M9 plate with or without Ara for creating trmD-KO (trm5+) and trmD-KO (trm5–) conditions.
The loss of aminoacylation of tRNAPro(UGG) and tRNAArg(CCG) is not due to the loss of the corresponding charging enzymes encoded by proS and argS, respectively. RNA-seq and ribosome profiling data showed that the expression level of these two enzymes was unaffected by m1G37 deficiency (Figure 4B). Moreover, cell lysates in m1G37-abundant (trm5+) and m1G37-deficient (trm5–) conditions exhibited a similar aminoacylation activity when assayed with the methylated m1G37-tRNAPro(UGG) or m1G37-tRNAArg(CCG) (Figure 4C, left and middle panels), indicating that the enzymatic activity of proS and argS was similar between the two growth conditions. As expected, aminoacylation of the control tRNATyr(QUA) was also similar between the two cell lysates (Figure 4C, right panel), validating that equivalent amounts of cell lysates were added to the different reactions.
Additional data showed that the lost aminoacylation of tRNAPro(UGG) and tRNAArg(CGG) in m1G37 deficiency was recovered by over-expression of proS and argS in trmD-KO (trm5–) cells (Figure 4A, left and middle panels, the third lane of each). This recovery indicates that the poor aminoacylation kinetics of m1G37-deficient tRNA can be overcome by increasing the corresponding aaRS enzyme. We asked whether these higher levels of aminoacylation would restore cell viability by improving the translation activity of m1G37-deficient tRNAs. Indeed, while control cells in m1G37-abundant conditions (trmD-KO (trm5+)) grew robustly, with or without over-expression of a plasmid-borne proS or argS, m1G37-deficient cells (trmD-KO (trm5–)) grew poorly with an empty plasmid, but improved viability upon over-expression of proS or argS (Figure 4D). Notably, while the loss of aminoacylation in m1G37 deficiency was similar to tRNAPro(UGG) and tRNAArg(CCG) in cell lysates (Figure 4A, left and middle panels), the recovery of cell viability upon over-expression of proS approached that of the positive control (trmD-KO (trm5+)), whereas that of over-expression of argS was much weaker (Figure 4D). This indicates that it is the loss of aminoacylation of tRNAPro species that is the driver for limiting cell growth under m1G37 deficiency.
Taken together, these findings show that m1G37 deficiency reduces the aminoacylation level of all tRNAPro species and tRNAArg(CCG) in vitro and in vivo. This loss of aminoacylation levels compromises cell viability but can be overcome by over-expression of proS and to a lesser extent over-expression of argS.
Changes in gene expression by m1G37 deficiency
We next asked what changes in gene expression took place in m1G37 deficiency. Using the DESeq algorithm (Love et al., 2014) to analyze the RNA-seq data, we identified genes whose steady-state levels of mRNAs were altered with statistical significance by loss of m1G37. We found 220 genes with more than 2-fold higher expression (p < 0.01) in the trmD-deg strain (shown in red, Figure 5A). Conversely, we identified 166 genes that were repressed by more than 2-fold (p < 0.01, colored in blue, Figure 5A). For both the up- and down-regulated genes, we identified several pathways that are affected and whose genes are enriched at statistically significant levels (Figure 5C). We also observed changes at the translational level. Using the Xtail algorithm (Xiao et al., 2016) to identify changes in ribosome occupancy (RO = Ribo-seq density/RNA-seq density), we found 71 genes with higher levels of apparent ribosome occupancy (1.7- to 6.5-fold increase) in the trmD-deg strain relative to the trmD-cont strain (p < 0.01, Figure 5B). Because the magnitude of transcriptional changes was greater than that of translational changes, yielding clearer results on which pathways are affected, we focused on the transcriptional changes arising from m1G37 deficiency.
Figure 5
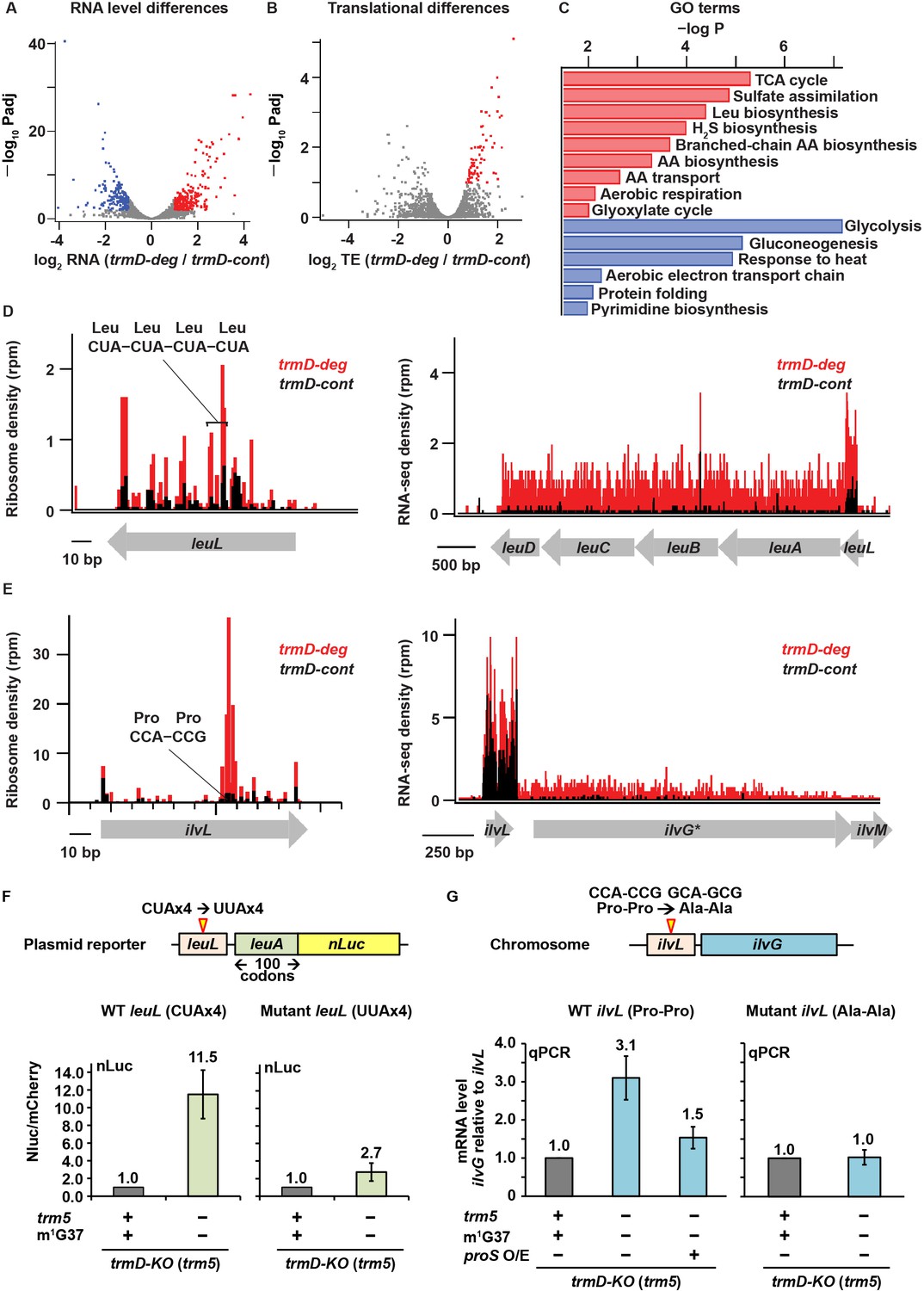
Changes in gene expression arising from m1G37 deficiency.
(A) Volcano plot showing differences in steady-state RNA levels (from RNA-seq data analyzed by DESeq) using samples of two replicates of trmD-deg and trmD-cont strains upon ClpXP induction. Genes that are more than 2-fold higher in the mutant with p < 0.01 are colored red (n = 220); those that are more than 2-fold lower with p < 0.01 are colored blue (n = 166). (B) Volcano plot showing differences in ribosome occupancy (Ribo-seq/RNA-seq analyzed by Xtail) using samples from two replicates of trmD-deg and trmD-cont strains upon ClpXP induction. Of these, 95 genes that are up-regulated (p < 0.1) are colored red. (C) Gene ontology categories for genes enriched in the up-regulated genes (red) and down-regulated genes (blue) from (A). (D) Gene model showing ribosome reads (in rpm) and RNA-seq density (in rpm, reads per million mapped reads) at the leuL leader sequence upstream of the leuABCD operon. (E) Gene model showing ribosome reads (in rpm) and RNA-seq density (in rpm) at the ilvL leader sequence upstream of the ilvGMEDA operon. (F) A plasmid reporter construct that demonstrates the codon-specific effect in the leuL leader sequence on expression of the downstream leuA gene. The plasmid reporter encodes the genomic sequence of leuL, followed by the first 100 codons of leuA, in the native sequence or with substitution of the four consecutive CUA codons with four UUA codons. The 100 codons of leuA are then fused to the nano-luciferase (nLuc) gene in-frame. Expression of nLuc, normalized by co-expression of mCherry in a separate plasmid within the same cell, is shown for m1G37+ and m1G37– conditions for the average of three biological replicates (n = 3). (G) Analysis of the CCA-CCG (Pro-Pro) codons in the genomic locus of ilvL as the determinant of regulation of gene expression of the downstream ilvG gene. A variant Escherichia coli strain was created that changed the CCA-CCG (Pro-Pro) codons to GCA-GCG (Ala-Ala) codons at the natural genomic locus, using CRISPR/Cas editing. Expression of ilvG was monitored by qPCR analysis in m1G37+ and m1G37– conditions and in m1G37– condition with over-expression of proS as shown as the average of three biological replicates (n = 3).
Many of the genes with higher RNA levels upon TrmD depletion are involved in amino acid biosynthesis and transport (Figure 5C). For example, induction of the Leu operon (leuA, leuB, leuC, and leuD) was particularly strong (~16-fold). This Leu operon is regulated by transcriptional attenuation depending on the efficiency of translation of the upstream leuL leader sequence (Wessler and Calvo, 1981; Wohlgemuth et al., 2013). As with leader sequences of other amino acid biosynthesis operons (Kolter and Yanofsky, 1982), translation of leuL serves as a sensitive genetic switch that has evolved to sense levels of aa-tRNAs and to respond to amino acid starvation by up-regulating the downstream biosynthetic pathways (Wohlgemuth et al., 2013). We observed ribosomal pausing in leuL at the four consecutive Leu CUA codons in m1G37 deficiency (Figure 5D), consistent with our observation of ribosome pausing at CUA codons (Figure 2B), suggesting that the pausing would prevent transcriptional termination and allow transcription elongation into the downstream Leu biosynthetic genes, resulting in elevated RNA levels. In agreement with this transcriptional attenuation mechanism, we observed in the control trmD-cont strain efficient transcriptional termination downstream of leuL, where the average RNA-seq density (adjusted for length) decreased 29-fold from the leuL leader sequence to the first gene in the operon (leuA), whereas we observed in the trmD-deg strain only a 4-fold decrease (Figure 5D). These results support the notion that ribosome pausing at the four consecutive Leu CUA codons in leuL during m1G37 deficiency reduced transcriptional termination of the downstream leuA gene (Figure 5D).
Another example is the ilvL leader sequence upstream of the ilvGMEDA operon encoding genes for biosynthesis of branched chain amino acids Ile, Leu, and Val. Ribosome pausing in ilvL would prevent transcriptional termination and allow expression of the downstream biosynthetic genes (Nargang et al., 1980). Although this genetic switch has evolved to sense changes in levels of aa-tRNAs associated with the amino acids produced by the downstream genes, we instead observed strong ribosome pauses at two consecutive Pro codons CCA-CCG in m1G37 deficiency (Figure 5E). These strong pauses within the ilvL leader sequence could trigger anti-termination, explaining the higher RNA-seq levels of the downstream gene ilvG. Indeed, while we observed in the control strain a 45-fold decrease in the RNA-seq density from the region around ilvL to the first half of the downstream ilvG gene, we observed in the trmD-deg strain only a 9-fold decrease in the RNA-seq density (Figure 5E). Notably, the ilvG gene in strains derived from E. coli K12 MG1655, such as the one used here, has a mutation that induces a frameshift midway through the gene, suggesting that it may be a pseudogene, although the ilvG gene appears intact in other E. coli strains.
We confirmed that the changes of expression of these two operons were due to pauses at specific codons induced by m1G37 deficiency in cognate tRNAs. To examine the effect of the leuL leader sequence on expression of leuA, we generated a plasmid reporter construct containing leuL and the first 100 codons of leuA fused in-frame to the nano-luciferase (nLuc) gene (Figure 5F). Upon expression of the WT reporter construct, m1G37 deficiency in trmD-KO (trm5–) cells elevated the nLuc readout by 11.5-fold relative to control cells, consistent with the notion that ribosome stalling at the four consecutive Leu CUA codons in leuL prevents transcriptional termination and allows transcription of the downstream leuA-nLuc fusion. In contrast, a smaller activation during m1G37 deficiency (only 2.7-fold) was observed with a second nLuc reporter where the four consecutive Leu CUA codons were changed to Leu UUA codons, which do not require m1G37 for translation.
To examine the effect of the ilvL leader sequence on expression of ilvG, we altered the two Pro codons CCA-CCG in ilvL to two Ala codons GCA-GCG by genome editing using CRISPR and followed the expression of the downstream ilvG gene with qPCR (Figure 5G). With the wild-type ilvL sequence, ilvG was up-regulated 3.1-fold in m1G37-deficient trmD-KO (trm5–) cells relative to m1G37-abundant trmD-KO (trm5+) cells (left panel, bar 2). In contrast, no increase was observed for the mutant ilvL sequence (right panel, bar 2). These results support the notion that ribosome stalling at the Pro codons in ilvL activates expression of the downstream ilvG in m1G37 deficiency. Furthermore, given our observation that over-expression of proS restored aminoacylation and cell viability in m1G37 deficiency (Figure 4D), we asked if it might also minimize pausing and induction of this genetic switch. We found that over-expression of proS in trmD-KO (trm5–) cells with the wild-type ilvL leader sequence reduced the activation of ilvG from 3.1-fold to 1.5-fold (left panel, bars 2 and 3). This result is consistent with the notion that elevated proS expression restores aminoacylation levels of tRNAPro in cells, even in m1G37 deficiency, relieving ribosome stalling and repressing the downstream ilvG gene.
In addition to Leu, Ile, and Val, the biosynthetic pathways for Trp, His, and Cys were highly up-regulated in the trmD-deg strain (Figure 5C), even though there is no evidence of ribosome stalling at the 5'-leader of the relevant operons. The up-regulation of these pathways suggests that they are activated by a genome-wide response due to ribosome stalling at Pro CCN, Arg CGG, and Leu CUA codons (Figure 2B). The increase in expression of the Cys biosynthesis pathway is correlated with the enrichment in gene ontology (GO) terms for genes involved in sulfate assimilation and hydrogen sulfide biosynthesis (Figure 5C) and is consistent with the notion that cysteine is synthesized from serine in bacteria by incorporation of sulfide to O-acetylserine (Kredich and Tomkins, 1966).
Changes of gene expression in m1G37 deficiency consistent with the stringent response
The high levels of expression of amino acid biosynthesis genes in m1G37 deficiency was reminiscent of the bacterial stringent response, which is triggered in nutrient starvation by uncharged tRNAs binding to the ribosomal A site, activating synthesis of ppGpp catalyzed by the RelA protein (Gourse et al., 2018). In the stringent response, ppGpp binds to two sites on RNA polymerase (RNAP) and re-programs the transcriptional landscape (Gourse et al., 2018; Ross et al., 2016; Ross et al., 2013), shutting down gene expression of rRNAs and ribosome proteins, while up-regulating amino acid biosynthesis genes to respond to amino acid starvation. Our observation that m1G37 deficiency reduced aminoacylation of all isoacceptors of tRNAPro and the tRNAArg(CCG) isoacceptor raised the possibility that these effects would induce the programmatic changes in gene expression similar to those in the stringent response.
To test this possibility, we compared the changes in RNA levels in m1G37 deficiency with the changes in RNA levels upon induction of the stringent response. We used the dataset for the stringent response obtained by Gourse and co-workers that compared changes in RNA levels between an E. coli strain containing the ppGpp-binding sites in RNAP and a variant lacking the binding sites upon over-expression of a relA mutant that was constitutively activated to synthesize ppGpp (Sanchez-Vazquez et al., 2019). This dataset focused on E. coli genes that exhibited transcriptional up- and down-regulation in response to direct binding of ppGpp to RNAP, rather than indirect effects from starvation-induced changes in metabolism. After 5 min of induction of the relA mutant, the dataset identified a set of 401 down-regulated genes (by >2-fold), a set of 321 up-regulated genes (by >2-fold), and a set of 3036 genes with less than 2-fold changes (Sanchez-Vazquez et al., 2019). Using these same sets of genes, we found in our RNA-seq data that genes that were down-regulated upon relA over-expression were also down-regulated in m1G37 deficiency (log2(median) = –0.36; t-test p = 8.9 × 10–15) (Figure 6A). Likewise, genes that were up-regulated in relA over-expression were also up-regulated in m1G37 deficiency (Figure 6A), although the extent of up-regulation in m1G37 deficiency was smaller (log2(median) = 0.29; t-test p = 2.3 × 10–11). Additionally, the non-responsive genes showing less than 2-fold changes upon relA over-expression also showed little change in m1G37 deficiency (log2(median) ~ 0.04). Together, these results show that genes that are strongly induced or repressed by the stringent response are affected in a similar way by m1G37 deficiency, arguing that m1G37 deficiency induces changes in gene expression similar to those of the stringent response.
Figure 6
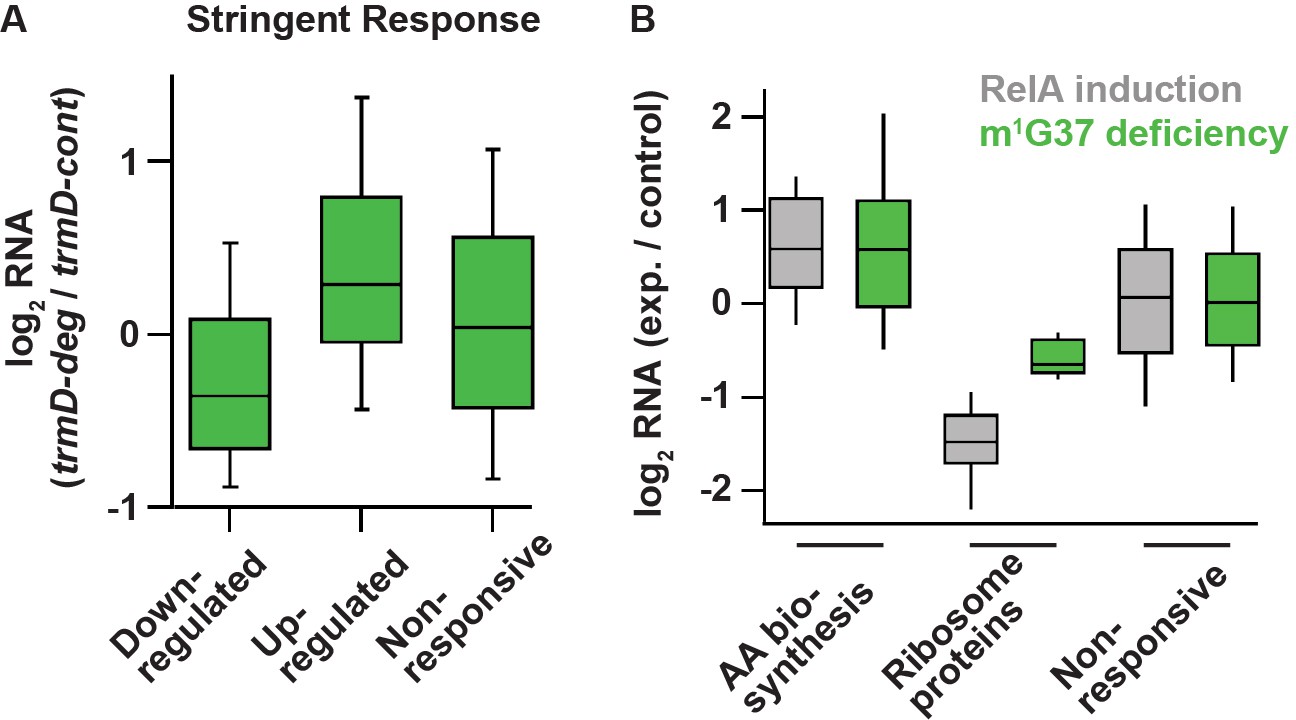
Deficiency of m1G37 induces gene expression similar to that of the bacterial stringent response.
(A) A set of genes published previously by Gourse et al. (Sanchez-Vazquez et al., 2019), whose steady-state RNA levels changed more than 2-fold upon induction of the stringent response by RelA over-expression (down n = 401 genes, up n = 321 genes, other = 3036 genes). Using these same sets of genes, the ratio of RNA levels in trmD-deg and trmD-cont strains upon ClpXP induction are shown. (B) Changes in expression in genes involved in amino acid biosynthesis (n = 89), genes encoding ribosomal proteins (n = 47), and other genes.
Following up on this observation, we next asked to what extent two characteristic pathways known to be regulated by the stringent response were also affected by m1G37 deficiency. In nutrient starvation, bacterial cells down-regulate ribosome biosynthesis due to the reduced demand for protein production, but up-regulate amino acid biosynthesis (Gourse et al., 2018). As expected, the published RNA-seq data upon relA over-expression showed that 89 genes associated with amino acid biosynthesis in the EcoCyc database were more highly expressed than other genes (log2(median) = 0.59, Mann-Whitney p = 4.8 × 10–12). Intriguingly, we found that these genes were also up-regulated to a similar extent in m1G37 deficiency (log2(median) = 0.58, Mann-Whitney p = 1.2 × 10–8) (Figure 6B), consistent with the enrichment of these genes in our GO annotation (Figure 5C). Likewise, 47 ribosome protein genes were down-regulated upon relA over-expression (log2(median) = –1.48, Mann-Whitney p = 1.6 × 10–24) and they were also down-regulated in m1G37 deficiency, although to a lesser extent (log2(median) = –0.64, Mann-Whitney p = 1.1 × 10–12) (Figure 6B). As a control, we also found that the genes that were not induced in these sets (i.e., not ribosome proteins and not involved in amino acid biosynthesis) were less responsive to relA over-expression, and that they were also less responsive in m1G37 deficiency (Figure 6B). Collectively, the parallel changes in steady-state RNA levels between m1G37 deficiency and relA over-expression were remarkable, suggesting that loss of m1G37 triggers a response similar to that of the relA-dependent stringent response.
Metabolic changes
Some of the most striking changes in gene expression in m1G37 deficiency occurred in central metabolic pathways: glycolysis, the citric acid (the tricarboxylic acid [TCA]) cycle, and fatty acid oxidation. For example, 10 genes associated with the TCA cycle were significantly up-regulated in the trmD-deg strain, making this pathway the most enriched in the GO terms (Figure 5C). Genes in the glyoxylate cycle are also up-regulated. Notably, because the glyoxylate cycle bypasses the decarboxylation reactions and instead replenishes TCA cycle intermediates, it is reasonable that these changes may be to build up metabolic intermediates for amino acid biosynthesis. The two-carbon units entering the TCA cycle likely arise primarily from fatty acid oxidation, whose enzymes are highly up-regulated in m1G37 deficiency, as opposed to the glycolysis pathway, whose genes are strongly repressed (Figure 5C). Thus, it appears that m1G37 deficiency shifts metabolism away from consuming Glc toward consuming fatty acids and that cells prioritize building up metabolic intermediates to support amino acid biogenesis.
Discussion
Here, we provide a genome-wide view of the effect of m1G37 deficiency on protein synthesis upon depletion of TrmD in E. coli. This genome-wide view is important, because TrmD is ranked as a high-priority antibacterial target (White and Kell, 2004), due to its fundamental distinction from Trm5, and its conservation throughout the bacterial domain, essentiality for bacterial life, and possession of a small molecule-binding site for drug targeting. The small molecule-binding site in TrmD is unique, consisting of a protein topological knot-fold that holds the methyl donor S-adenosyl methionine in an unusual shape (Ahn et al., 2003; Elkins et al., 2003; Ito et al., 2015), which is different from that in Trm5 and in most other methyl transferases (Goto-Ito et al., 2009), indicating the possibility to explore novel chemical space and diversity of drugs targeting TrmD. Additionally, we have shown that TrmD deficiency can disrupt the double-membrane cell-envelope structure of Gram-negative bacteria, thus facilitating permeability of multiple drugs into cells, preventing efflux of these drugs, and accelerating bactericidal action (Masuda et al., 2019). This multitude of advantages of targeting TrmD emphasizes that a better understanding of the genome-wide function of its m1G37 product will help develop a successful antibacterial strategy.
Using ribosome profiling, we show that m1G37 deficiency causes ribosome stalling at codons specific for tRNAs that are methylated by TrmD, including strong pauses at all Pro CCN codons and the Arg CGG codon and weak pauses at the Leu CUA codon. Stalling is primarily observed when the affected codons are in the ribosomal A site, indicating a distinct mechanistic feature than that of +1 frameshifting, which occurs in m1G37 deficiency after decoding at the A site and during tRNA translocation to the P site and occupancy within the P site (Gamper et al., 2021; Gamper et al., 2015a). The importance of m1G37 at the ribosomal A site thus expands the spectrum of its biology, which can be summarized in a model that explains its indispensability throughout the entire elongation cycle of protein synthesis (Figure 7). In this model, m1G37 deficiency induces ribosome stalling in the A site, triggering changes in gene expression and possibly affecting mRNA stability and co-translational protein folding. Ribosome stalling can be prolonged if a subset of the affected tRNAs is unable to perform peptide-bond formation in the A site, leading to ribosome collisions that increase the frequency of frameshifting (Smith et al., 2019). Even if some of the m1G37-deficient tRNAs manage to enter the A site and participate in peptide-bond formation, they would induce +1 frameshifting during translocation to the P site and destabilize interaction with the ribosomal P site (Hoffer et al., 2020), leading to premature fall-off from the ribosome and termination of protein synthesis. In contrast to starvation that can be alleviated by re-supply of nutrients, this series of defects associated with m1G37 deficiency cannot be readily resolved, thus underscoring the essentiality of m1G37 in protein synthesis that is coupled to cell growth and survival.
Figure 7
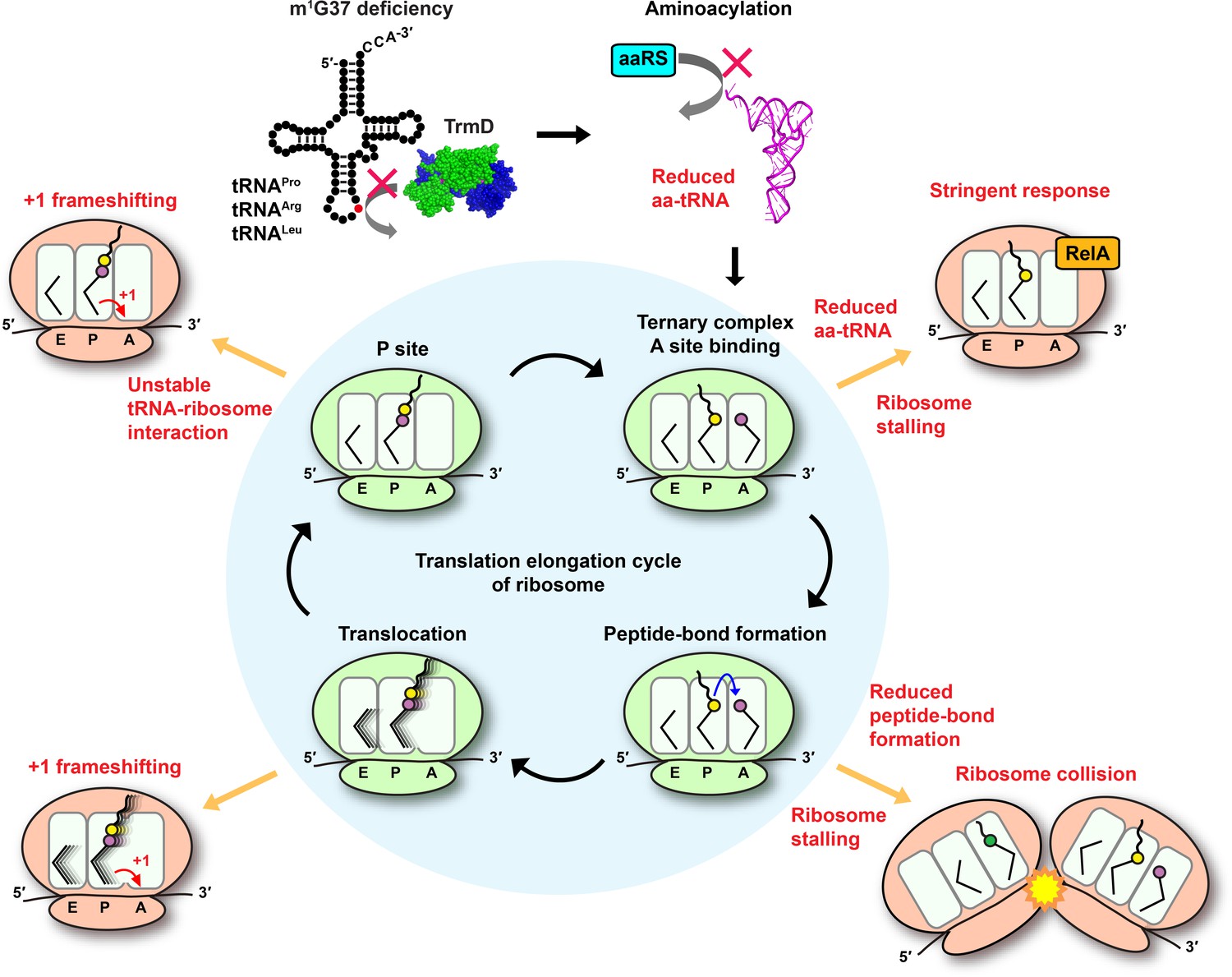
Deficiency of m1G37 impairs the elongation cycle of protein synthesis in bacteria.
A cloverleaf structure of tRNA, showing the position of m1G37 in red. Loss of m1G37 reduces aminoacylation, lowering levels of aa-tRNAs at the ribosome A site, stalling the ribosome, and activating programmatic changes in gene expression similar to those induced by the bacterial stringent response upon RelA sensing of uncharged tRNA and synthesis of ppGpp. Ribosome stalling at the A site is prolonged if the m1G37-deficient aa-tRNA is delayed in peptide-bond formation, leading to ribosome collision. Even if the m1G37-deficient tRNA manages to enter the A site and participates in peptide-bond formation, it would promote +1 frameshifting during translocation from the A site to the P site and during occupancy within the P site, while also destabilizing the P site structure and possibly falling off from the P site to prematurely terminate protein synthesis. Accumulation of these defects ultimately collapses the entire elongation cycle and leads to cell death.
An important finding of this work is that ribosome stalling in m1G37 deficiency is primarily driven by loss of aminoacylation of affected tRNAs, which is shown in enzyme-based kinetic assays for all isoacceptors examined, and in cell-based acid-urea assays for tRNAPro(UGG) and tRNAArg(CCG), two major TrmD-dependent species in bacteria. While the loss of aminoacylation in m1G37 deficiency is not due to the loss of ProRS or ArgRS in cells, it can be restored by over-expression of either enzyme, which improves cell viability. These results clearly demonstrate that the loss of aminoacylation in m1G37 deficiency, and consequently the accumulation of uncharged tRNAs, is the underlying basis of ribosome stalling that induces significant changes in gene expression. Direct changes include up-regulation of the leuABCD and ilvGMEDA operons. Indirect changes include hundreds of genes that are activated and similar to those in the bacterial stringent response – the central bacterial response to nutrient starvation upon RelA sensing of uncharged tRNAs binding to the A site of translating ribosomes. These changes certainly make sense given that we see higher levels of deacylated tRNAs accumulating in m1G37 deficiency and that the binding of deacylated tRNA to the A site is known to induce (p)ppGpp synthesis by RelA on the ribosome. We did not test the role of RelA in the indirect changes in gene expression in m1G37 deficiency, because the trmD-deg and trmD-KO (trm5–) cells are already substantially compromised in cell viability and the deletion of relA is expected to further reduce viability. Another confounding factor is that E. coli contains two genes responsible for ppGpp synthesis: relA and spoT, which have overlapping functions. Upon relA deletion, the protein product of spoT can still synthesize ppGpp in response to some of the metabolic changes that are observed here. Nonetheless, the changes in gene expression in trmD deficiency largely parallel with those observed during the stringent response, consistent with the notion that the loss of aminoacylation in m1G37 deficiency activates the stringent response.
The significance of this work is that it demonstrates a stress response in bacteria similar to the stress response in eukaryotes that is activated by changes of the post-transcriptional modification state of tRNA. In yeast, loss of the 5-methoxycarbonylmethyl (mcm5) and/or the 2-thio (s2) groups from the normal mcm5s2U34-state tRNA activates the GCN4-mediated pathway (Zinshteyn and Gilbert, 2013). This GCN4 pathway in yeast is translationally regulated in response to a variety of insults, most notably to amino acid starvation (Hinnebusch, 2005). In yeast, loss of the threonyl-carbamoyl group from the normal t6A37-state tRNA also activates the GCN4 response (Thiaville et al., 2016). In Drosophila, deficiency of the t6A37-state in a specific tRNA activates the target of rapamycin response (Rojas-Benitez et al., 2015), which reprograms gene expression in response to amino acid deficiency. In Neurospora, loss of the inosine (I) group from the normal I34-state tRNA activates the CPC-1 (cross-pathway-control-1) response, which is the ortholog of the yeast GCN4 response (Lyu et al., 2020). Mechanistically, loss of the s2 group from the mcm5s2U34-state has been shown to reduce binding and accommodation of tRNA to the ribosomal A site and translocation to the P site (Ranjan and Rodnina, 2017). While we had previously shown that loss of the s2 group from the related cmnm5s2U34 (5-carboxy-amino-methyl-2-thio-uridine 34)-state in E. coli tRNAGln also reduced tRNA aminoacylation and the subsequent binding and accommodation to the A site (Rodriguez-Hernandez et al., 2013), we did not investigate the cellular response due to the deficiency in bacteria. Thus, this work on m1G37 deficiency provides a previously unrecognized parallel between E. coli and eukaryotes in a stress response that activates amino acid biosynthesis upon loss of a post-transcriptional modification of tRNA. The parallel suggests the evolution of a conserved cellular priority that activates amino acid biosynthesis in response to nutrient deficiency or to tRNA modification deficiency, both of which compromise active protein synthesis and threaten cell viability.
The molecular insight obtained from this study is important for antibacterial therapeutics. The observation that over-expression of proS most effectively rescues cell viability in m1G37 deficiency (Figure 4D) indicates that aminoacylation of tRNAPro is a major determinant of cell viability. Notably, loss of aminoacylation of tRNAPro is uniformly manifested at the Km (tRNA) step in m1G37 deficiency (Figure 3—source data 1A), suggesting that the immediate proximity of the methylation to the aminoacylation-determinant anticodon G36 nucleotide is important for the bacterial ProRS. In contrast, human ProRS has diverged in tRNA recognition by shifting the emphasis toward the G35 nucleotide, which is more distant from m1G37 (Burke et al., 2001). This shift suggests the possibility that the human enzyme may better accommodate m1G37-deficient tRNAs for aminoacylation, and that the eukaryotic response to m1G37 deficiency is executed in a step other than aminoacylation for decoding at the A site. These possibilities remain to be tested in future experiments.
In summary, we show here that m1G37 is required for the ribosomal activity at the A site and that, when combined with previous studies, we emphasize the importance of m1G37 for the entire elongation cycle of protein synthesis. This new insight suggests the possibility that drug targeting of TrmD can be used in combination with classic antibiotics that target the elongation cycle of protein synthesis. For example, targeting TrmD can be explored in combination of antibiotics that target the A site (e.g., tetracycline [Tet], negamycin), those that target the translocation reaction (e.g., viomycin, spectinomycin), and those that target the P site (e.g., streptogramin A, bactobolin A, and retapamulin) (Vázquez-Laslop and Mankin, 2018). This combinatorial approach may produce a synergistic and accelerated bactericidal effect that can combat the rapid emergence of multi-drug resistance of pathogens.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Escherichia coli) | G78 trmD-deg | This paper | TrmD is fused with a C-terminal YALAA degron tag | |
| Strain, strain background (Escherichia coli) | G78 trmD-cont | This paper | Control strain without a degron tag | |
| Strain, strain background (Escherichia coli) | MG1655 trmD-KO | Masuda et al., 2019 | ||
| Strain, strain background (Escherichia coli) | MG1655 trmD-KO ilvL Ala-Ala mutant | This paper | CRISPR mutant possessing GCA-GCG codons replacing CCA-CCG codons on ilvL gene | |
| Strain, strain background (Escherichia coli) | JM109 trmD-KO | Demo et al., 2021 | ||
| Strain, strain background (Escherichia coli) | BL21(DE3) trmD-KO | Gamper et al., 2015a | ||
| Strain, strain background (Escherichia coli) | BL21(DE3) | CGSC | CGSC#: 12504 | |
| Antibody | anti-TrmD (Rabbit polyclonal) | Li and Björk, 1999 doi: 10.1017/S1355838299980834 | WB (1:10000) | |
| Antibody | anti-rabbit IgG (Goat polyclonal) | Sigma-Aldrich | Cat#: A0545 | WB(1:160000) |
| Peptide, recombinant protein | Proteinase K | Promega | Cat#: V302B | |
| Peptide, recombinant protein | Micrococcal nuclease | Roche | Cat#: 10107921001 | |
| Peptide, recombinant protein | Superscript III reverse transcriptase | Invitrogen | Cat#: 18080–044 | |
| Peptide, recombinant protein | RQ1 RNase-free DNase | Promega | Cat#: M6101 | |
| Commercial assay or kit | TruSeq Stranded Total RNA kit | Illumina | Cat#: 20020598 | |
| Commercial assay or kit | SuperSignal West Pico Chemiluminescent Substrate | Thermo Fisher Scientific | Cat#: 34080 | |
| Commercial assay or kit | NEBuilder HiFi DNA Assembly Cloning Kit | New England Biolabs | Cat#: E5520S | |
| Commercial assay or kit | Nano-Glo Luciferase Assay System | Promega | Cat#: N1110 | |
| Commercial assay or kit | TRIzol | Invitrogen | Cat#: 15596026 | |
| Commercial assay or kit | Direct-zol RNA MiniPrep Kits | Zymo Research | Cat#: R2051 | |
| Commercial assay or kit | RevertAid First Strand cDNA synthesis Kit | Thermo Fisher Scientific | Cat#: K1622 | |
| Commercial assay or kit | SYBR Green I Master | Roche | Cat#: 04707516001 | |
| Software, algorithm | Skewer | Jiang et al., 2014 | ||
| Software, algorithm | bowtie | Langmead et al., 2009 | ||
| Software, algorithm | DESeq | Love et al., 2014 | ||
| Software, algorithm | Xtail | Xiao et al., 2016 | ||
| Software, algorithm | DAVID | Huang et al., 2007 | ||
| Software, algorithm | Pathway Tools | Karp et al., 2021 | ||
| Software, algorithm | ImageQuant | GE Healthcare | ||
| Software, algorithm | Kaleidagraph | Synergy software | ||
| Software, algorithm | ImageJ | NIH | ||
| Other | MOPS EZ Rich Defined | Teknova | Cat#: M2105 | Growth medium |
Construction of trmD-deg and trmD-cont strains
Request a detailed protocolE. coli trmD-deg strain was constructed as described (Carr et al., 2012). The degron tag was amplified from the template DNA (provided by Dr Sean Moore), which encodes a FLAG tag, a His6 tag, and the degron sequence YALAA followed by the promoter and the coding sequence of the Tet resistance gene. This region was amplified with primers homologous to the 3'-end and flanking sequence of the chromosomal trmD locus. The PCR product was purified and electroporated into competent cells of the recombinogenic E. coli strain SM1405 expressing λRed recombinase (Datsenko and Wanner, 2000). Recombination was confirmed by PCR analysis of colonies on plates containing Tet using primers homologous to the 3'-end of the chromosomal trmD locus. The P1 lysate of the confirmed trmD-deg strain was used for transduction into the recipient E. coli G78 strain whose chromosomal clpX was deleted (Carr et al., 2012). A resultant P1 transductant was selected on plates with Tet, confirmed by PCR, and transformed with a library of the plasmid pclpPX harboring a cassette encoding clpP and clpX and random mutations at the promoter and SD region that control expression of the cassette (Carr et al., 2012). Transformants were screened on LB + Ara (0.2%) plates to turn on expression of the cassette and to identify the clone with the highest efficiency of the degron activity as indicated by Western blots (Figure 1C). E. coli trmD-cont strain was created similarly, except that the degron tag coding for the YALAA sequence was placed after the stop codon of trmD. The P1 transductant harboring trmD-cont in the G78 strain was transformed with the pclpPX plasmid that showed the highest degron activity, generating the trmD-cont control strain. The primer sequences are shown below (5' to 3'):
Forward recombination:
CGGAACACGCACAACAGCAACATAAACATGATGGGATGGCGGGTGGCTCCGACTACAAGG
Reverse recombination:
ATAATTTAATCTCTTATCCTGGGTAAACTGATATCTCGGGGGCTTAGGTCGAGGTGGCCC
Forward confirmation at the trmD locus:
ATGTGGATTGGCATAATTAGCCTGTTTCC
Reverse confirmation at the trmD locus:
GAATTCCGGTTACGAATAGCGATAACCACGCC
Growth conditions
Request a detailed protocolE. coli G78 strains of trmD-deg and trmD-cont were grown in LB + Glc at 37°C overnight, inoculated into 20 mL of fresh LB + Ara at 1:100, and grown for 2 hr at 37°C into the start of the log phase. This growth cycle was repeated three times. In the first cycle, cells were inoculated into 20 mL of LB + Ara (0.2%) and 10 mM Ser at OD600 of 0.1 and grown for 1 hr at 37°C to OD600 of ~0.4, at which point TrmD protein was no longer detected by Western blots. In the second cycle, cells were inoculated into 100 mL of LB + Ara (0.2%) and 10 mM Ser at OD600 of 0.1 and grown for 1 hr at 37°C to OD600 of ~0.4. In the third cycle, cells were inoculated into 500 mL at OD600 of 0.1 and grown for 2–3 hr at 37°C to OD600 of ~0.3. Cells of 200–300 mL were harvested by rapid filtration (see below). Strains of trmD-KO (trm5–) and trmD-WT (trm5–) were grown in LB + Ara (0.2%) at 37°C overnight and were then taken through three cycles of growth in MOPS Medium (EZ Rich Defined, Teknova) containing Glc as the only carbon source. In the first cycle, cells in the overnight culture were inoculated into 10 mL of MOPS at OD600 of 0.1 and grown for 4 hr at 37°C to turn off the Ara-dependent expression of trm5 and to deplete m1G37-tRNAs. In the second cycle, cells were inoculated into 25 mL of MOPS + Glc and grown for 3 hr, and in the third cycle, cells were inoculated into 300 mL of MOPS + Glc and grown 2–4 hr at 37°C until OD600 of ~0.3. Cells of 200–300 mL were harvested by rapid filtration (see below).
Western blots
Request a detailed protocolE. coli G78 strains of trmD-deg and trmD-cont were grown overnight in LB and then diluted 1:100 into fresh LB + Ara (0.2%). Cells were grown at 37°C and were lysed 0, 15, 30, 45, 60, and 90 min after inoculation. Cell lysate of 15 μg of protein was separated on 12% SDS-PAGE and levels of TrmD were probed by an anti-TrmD primary antibody (provided by Dr Glenn Bjork) at a dilution of 1:10,000 and a secondary anti-rabbit IgG antibody (Sigma-Aldrich) at a dilution of 1:160,000. Signals were detected by the SuperSignal West Pico Chemiluminescent Substrate (Thermo Fisher Scientific in the Chemi-Doc XRS+ System [Bio-Rad]).
Primer-extension assays
Request a detailed protocolPrimer-extension analyses of E. coli tRNALeu(CAG) were performed as described (Masuda et al., 2019) on trmD-deg and trmD-cont lysates generated from the same volume of cells collected at the indicated time points after switching to LB + Ara. A DNA primer targeting nucleotides 40–54 of the tRNA was chemically synthesized, [32P]-labeled at the 5'-end by T4 polynucleotide kinase, annealed to the tRNA, and extended by Superscript III reverse transcriptase (Invitrogen) at 200 U/µL with 6 μM each dNTP in 50 mM Tris-HCl, pH 8.3, 3 mM MgCl2, 75 mM KCl, and 1 mM dithiothreitol (DTT) at 55°C for 30 min. The reaction was quenched with 10 mM EDTA at 70°C for 15 min. cDNA products were separated by 12% PAGE/7 M urea and visualized by phosphorimaging. The extension product of the read-through cDNA is 53 nt in length, whereas the extension inhibition product is 15 nt in length.
Cell harvesting and lysis
Request a detailed protocolWe used two different cell harvesting strategies to arrest ribosomes and block translation after cell lysis. The strategy of cell harvesting can influence the quality of ribosome profiling data (Mohammad et al., 2019). Cultures of trmD-deg and trmD-cont were harvested by rapid filtration on a Kontes 99 mm filtration apparatus and 0.45 µm nitrocellulose filter (Whatman) and the cells were flash-frozen in liquid nitrogen. Cells were lysed in lysis buffer (20 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, 5 mM CaCl2, 100 U/mL RNase-free DNase I, and 1 mM chloramphenicol). Due to sequence-specific inhibition by chloramphenicol (Marks et al., 2016; Mohammad et al., 2016; Nakahigashi et al., 2014; Orelle et al., 2013), this cell harvesting strategy was more prone to artifacts at the codon level, creating apparent pauses when the smaller amino acids Ala, Gly, and Ser were at the penultimate position in nascent polypeptide chains. Furthermore, chloramphenicol does not fully arrest translation in cell lysates, blurring the signal (Mohammad et al., 2019). In contrast, cultures of trmD-KO (trm5–) and trmD-WT (trm5–), while also harvested by rapid filtration and flash-freezing, were lysed in a buffer lacking chloramphenicol but containing high concentrations of MgCl2 (20 mM Tris-HCl, pH 8.0, 150 mM MgCl2, 100 mM NH4Cl, 5 mM CaCl2, 0.4% Triton X-100, 0.1% NP-40, and 100 U/mL RNase-free DNase I). In this lysis buffer, the high MgCl2 inhibits translation by preventing ribosomal conformational changes required for elongation (Mohammad et al., 2019). After lysis, the samples in the high MgCl2 buffer were centrifuged over a sucrose cushion to collect polysomes, which were resuspended in the standard lysis buffer without the high salt to allow micrococcal nuclease (MNase) digestion and isolation of ribosome-protected mRNA fragments for deep sequencing analysis.
Library preparation
Request a detailed protocolA 10–54% sucrose density gradient was prepared in the Gradient Master 108 (Biocomp) in the gradient buffer (20 mM Tris-HCl, pH 8.0, 10 mM MgCl2, 100 mM NH4Cl, and 2 mM DTT). A cell lysate of 5–20 AU was loaded onto the sucrose density gradient and centrifuged in an SW41 rotor at 35,000 rpm for 2.5 hr at 4°C. Fractionation was performed on a Piston Gradient Fractionator (Biocomp). Libraries for ribosome profiling and RNA-seq were prepared as described (Mohammad et al., 2016; Woolstenhulme et al., 2015) on RNA fragments 15–45 nt in length, with the exception of RNA-seq libraries for the trmD-KO (trm5–) and trmD-WT (trm5–) samples, which were prepared using the TruSeq Stranded Total RNA kit (Illumina). The libraries were analyzed on a high-sensitivity BioAnalyzer (Agilent) and sequenced on the HiSeq2500 Illumina instrument.
Analysis of sequencing data
Request a detailed protocolThe adaptor sequence CTGTAGGCACCATCAATAGATCGGAAGAGCACACGTCTGAA-CTCCAGTCA was removed by Skewer version 0.2.2 (Jiang et al., 2014). After reads mapping onto tRNA and rRNA were removed, the remaining reads were aligned to E. coli MG1655 genome NC_000913.2 using bowtie version 1.1.2, requiring unique mapping sites and allowing two mismatches (Langmead et al., 2009). The position of the ribosome was assigned using the 3'-end of reads. Reads 10–40 nt in length were included in analyses unless otherwise specified for all samples except for RNA-seq samples from trmD-KO (trm5–) and trmD-WT (trm5–) prepared using the TruSeq kit, which were 50 nt in length.
For calculating pause scores, only reads 24–40 nt in length were used in trmD-KO (trm5–) and trmD-WT (trm5–) ribosome profiling samples because there was uncertainty about the position of the ribosome on reads shorter than 24 nt due to the different lysis buffer. In calculating pause scores, we only included genes with more than 0.1 reads per codon on average. The first seven and last seven codons in each ORF were ignored. For each instance of a codon of interest, we calculated a pause score by taking the density at the first nt only in the A site codon and dividing it by the mean density on the ORF. The average pause scores reported (Figure 2) represent the mean of the scores for all instances (typically thousands) of the codon of interest.
For analyses of changes in gene expression, we calculated for each gene the density of the RNA-seq or Ribo-seq in units of RPKM and then used the DESeq and Xtail packages in R with two replicates of the trmD-deg and trmD-cont samples to compute the log2 fold change in expression and the –log(padj) value shown in Figure 5. Enrichment for GO terms was determined using the functional annotation tools in DAVID at david.ncifcrf.gov (Huang et al., 2007). RNA-seq data were mapped onto metabolic pathways using the Pathway Collage tool at ecocyc.org (Karp et al., 2021; Keseler et al., 2017).
Aminoacylation of tRNA in vitro
Request a detailed protocolEach tRNA was aminoacylated with the cognate amino acid by the cognate aaRS enzyme that had been over-expressed in BL21 (DE3) and purified via binding to a Ni-NTA resin and elution with imidazole (Zhang et al., 2006). Each tRNA was heat-denatured at 85°C for 3 min and re-annealed at 37°C for 15 min. Aminoacylation in steady-state conditions was performed at 37°C in a 30 μL reaction of 0.25–20 μM tRNA, 5 nM aaRS, and 20 μM [3H]-amino acid (Perkin Elmer, 7.5 Ci/mmol) in the aminoacylation buffer of 20 mM KCl, 10 mM MgCl2, 4 mM DTT, 0.2 mg/mL bovine serum albumin, 2 mM ATP (pH 8.0), and 50 mM Tris-HCl, pH 7.5 (Liu et al., 2011). Reaction aliquots of 5 µL were removed at different time intervals and precipitated with 5% (w/v) trichloroacetic acid (TCA) on filter pads for 10 min twice. Filter pads were washed with 95% ethanol twice, with ether once, air-dried, and measured for radioactivity in Tri-Carb 4910 TR scintillation counter (Beckman). Counts were converted to pmoles using the specific activity of the [3H]-amino acid after correcting for signal quenching by filter pads. Data corresponding to the initial rate of aminoacylation as a function of the tRNA substrate concentration were fit to the Michaelis-Menten equation to derive the Km (tRNA), kcat (catalytic turnover of the enzyme), and kcat/Km (tRNA) (the catalytic efficiency of aminoacylation).
Peptide-bond formation assays in vitro
Request a detailed protocolPeptide-bond formation assays were performed on mRNAs that varied in the second codon position (shown as XXX below) but maintained all other nucleotides, including the SD sequence (underlined) and the AUG start codon (bold face).
5'-GGGAAGGAGGUAAAAAUG-XXX-CGU-UCU-AAG-(CAC)7-3'
Each reaction was carried out in 50 mM Tris-HCl, pH 7.5, 70 mM NH4Cl, 30 mM KCl, 3.5 mM MgCl2, 1 mM DTT, and 0.5 mM spermidine at 20°C as described (Gamper et al., 2015a; Liu et al., 2011). E. coli 70S ICs were formed by incubating 70S ribosomes, mRNA, [35S]-fMet-tRNAfMet, and initiation factors (IFs) 1, 2, and 3 with GTP for 25 min at 37°C in the reaction buffer. Each E. coli tRNA (4 μM) was aminoacylated with the cognate amino acid by the cognate aaRS (1 μM) to rapidly achieve plateau level of aminoacylation as confirmed by enzymatic assays. To form a TC, the aminoacylation reaction was incubated with activated EF-Tu-GTP, which was formed separately for 15 min at 37°C, and incubated in an ice bath for 15 min. To monitor peptide-bond formation, 70S ICs templated with an mRNA were mixed with a TC in an RQF-3 Kintek chemical quench apparatus. Final concentrations in each reaction (30 μL) were 0.1 µM for the 70S IC; 1.0 µM for mRNA; 0.5 µM each for IFs 1, 2, and 3; 0.25 µM for [35S]-fMet-tRNAfMet; 1.0 µM for EF-Tu for each aa-tRNA; 0.2 µM each for the aa-tRNAs; and 1 mM for GTP. Reactions were conducted at 20°C and were quenched by adding 60 μL of KOH to 0.5 M. After a brief incubation at 37°C in the quench (90 μL), aliquots of 1.8 µL were spotted onto a cellulose-backed plastic thin layer chromatography sheet and electrophoresed at 1000 V in PYRAC buffer (62 mM pyridine, 3.48 M acetic acid, pH 2.7) until the marker dye bromophenol blue (BPB) reached the water-oil interface at the anode. The position of the origin was adjusted to maximize separation of the expected oligopeptide products. The separation of unreacted [35S]-fMet from each of the [35S]-fMet-peptide products was visualized by phosphor-imaging and quantified by ImageQuant (GE Healthcare) and kinetic plots were fitted using Kaleidagraph (Synergy software).
Acid-urea gel and Northern analysis
View detailed protocolTotal RNA was extracted from cells in an acidic condition (pH 4.5) to maintain aa-tRNA levels (Parker et al., 2020). Cell cultures at the appropriate OD were mixed at a 1:1 vol ratio with 10% TCA and incubated on ice for 10 min. Cells were spun down at 8000 rpm (=7600× g) for 5 min at 4°C, resuspended in the extraction buffer (0.3 M NaOAc, pH 4.5, and 10 mM EDTA), and mixed with an equal volume of ice-cold phenol-chloroform-isoamyl alcohol (83:17:3.4), pH 5.0. The mixture was vortexed for five cycles of 1 min vortex and 1 min rest on ice, and spun at 12,000 rpm (=13,500× g) for 10 min at 4°C. Samples with proS over-expression were extracted one more time with phenol-chloroform-isoamyl alcohol (83:17:3.4) pH 5.0 for five cycles of 1 min vortex and 1 min rest on ice. The aqueous phase was separated by spinning at 12,000 rpm (=13,500× g) for 10 min at 4°C, and was mixed with an equal volume of cold isopropanol and incubated at −20°C for 30 min for precipitation. The RNA-containing pellet was collected after centrifugation at 14,000 rpm for 20 min at 4°C, washed with 70% ethanol in 30% 10 mM NaOAc, pH 4.5, dried, and dissolved in the gel-loading buffer (10 mM NaOAc, pH 4.5, and 1 mM EDTA). The RNA of proS over-expression samples dissolved in the gel-loading buffer was incubated with Proteinase K (V302B, Promega) at the final protease concentration of 0.1 mg/mL on ice for 5 min. Due to the high lability of the prolyl linkage in Pro-tRNAPro to hydrolysis (Peacock et al., 2014), total RNA was immediately separated on an acid-urea gel. Assuming that tRNA accounts for 10% of total RNA, we made calculation to load 700 ng of total tRNA of each sample, mixed with two volumes of the acid-urea loading buffer (0.1 M NaOAc, pH 5.0, 9 M urea, 0.05% BPB, and 0.05% xylene cyanol [XC]). An intermediate size of acid-urea gel (14 × 17 cm) was used to resolve Pro-tRNAPro from uncharged tRNAPro, whereas a Bio-Rad standard size mini gel was used to resolve Arg-tRNAArg and Tyr-tRNATyr from uncharged tRNA. Each acid-urea gel was made with 6.5% polyacrylamide, 7 M urea, and 0.1 M NaOAc, pH 5.0, and run at 250 V for 3 hr 45 min at 4°C for the intermediate size gel and at 100 V for 2 hr 30 min for the mini gel. Following electrophoresis, the region between BPB and XC was excised and washed in 1× Tris-borate pH 8.0 and EDTA (TBE) buffer. Gels were transferred to a wetted nitrocellulose membrane in 1× TBE using Trans-Blot Turbo Transfer System (1704150, Bio-Rad) at constant 25 V for 20 min. The electroblotted membranes were briefly air-dried before crosslinking the RNA on the membrane using an ‘optimal crosslink’ in a UV crossliker (FB-UVXL-1000, Thermo Fisher Scientific). The membranes were pre-incubated in a hybridization buffer (0.9 M NaCl, 90 mM Tris-HCl, pH 7.5, 6 mM EDTA, 0.3% SDS, and 1% dry milk) at 37°C for 1 hr. Three DNA oligonucleotide probes, one targeting positions 18–36 of E. coli tRNAPro(UGG) (5'-CCAAACCAGTTGCGCTACCA-3'), one targeting positions 19–36 of E. coli tRNAArg(CCG) (5'-CGGAGGGCAGCGCTCTAT-3'), and the third targeting positions 18–36 of E. coli tRNATyr(QUA) (5'-TACAGTCTGCTCCCTTTGGC-3') were each [32P]-labeled at the 5′-end. These probes (each at 106 cpm) were incubated with the pre-washed membrane in the hybridization buffer for 12 hr while shaking. The probes were washed off by a 2× SSC buffer (0.3 M NaCl, 30 mM Na-citrate) at 37°C two times, each time with mild shaking for 10 min. The membranes were then dried and exposed to an imaging plate overnight. Imaging analysis was performed with a phosphor-imager (Typhoon IP, GE Healthcare) and the bands were quantified using ImageJ software (NIH). The charged fraction was calculated as the area encompassing the charged band divided by the sum of the charged and uncharged bands.
Analysis of aaRS activity in cell lysates
Request a detailed protocolHarvested cells were resuspended in a lysis buffer (50 mM Tris-HCl pH 8.0 and 150 mM NaCl) and were disrupted by a Bioruptor Pico device (Diagenode) through 12 cycles of sonication and intermittent rest at 10 and 45 s, respectively. The generated cell lysate was cleared of debris by ultracentrifugation through MTX-150 with the S100-AT4 rotor (Thermo Fisher Scientific) at 500,000× g for 2 hr at 4°C, and was concentrated through Amicon Ultra-4 Centrifugal Filter Units (Millipore) to ~0.5 mL. The concentrated lysate was washed with 4 mL of the binding buffer (10 mM Tris-HCl pH 7.5, 30 mM KCl, 10 mM MgCl2, and 1 mM DTT) and concentrated again through the same Amicon unit to 1.6 mL. To prepare for post-DEAE fraction of the lysate, two aliquots of 300 µL DEAE sepharose were washed with 1 mL water by spinning at 3000× g for 2 min, and two washes with 1 mL of the binding buffer each. The 1.6 mL concentrated cell lysate was divided into two, and each was mixed with the washed DEAE sepharose and incubated at 4°C with rotation for 1 hr. The slurry was transferred to a gravity column and washed with 10 volumes of 1.2 mL of the binding buffer. The aaRS-containing fraction was eluted by 4 mL of the elution buffer (10 mM Tris-HCl pH 7.5, 400 mM KCl, 10 mM MgCl2, and 1 mM DTT), concentrated by two steps of Amicon Ultra-0.5 mL centrifugal filtering down to 100 µL, exchanged with the dialysis buffer (20 mM Tris-HCl pH 7.5, 130 mM KCl, 10 mM MgCl2, and 1 mM DTT), and followed by dialysis in a Slide-A-Lyzer MINI Dialysis Device, 10 k MWCO (Thermo Fisher Scientific) to remove small molecules. This post-DEAE fraction was determined for protein concentration by the Bradford assay. Molar concentration of proteins in the post-DEAE cell lysate was estimated based on an average molecular weight of E. coli proteins at 40 kDa.
Transcripts of E. coli tRNAPro(UGG), tRNAArg(CCG), and tRNATyr(QUA) were made by in vitro transcription with T7 RNAP to generate the G37-state of each. Transcripts of E. coli tRNAPro(UGG) and tRNAArg(CCG) were subsequently methylated with m1G37 by purified recombinant E. coli TrmD (Christian et al., 2004) to generate the m1G37-state of each. Intracellular concentration of ProRS, ArgRS, and TyrRS in the post-DEAE lysate was estimated assuming each representing 1% of total proteins to allow calculation of the molar concentration of each enzyme. The final concentration of each enzyme was used at 20 nM ProRS, 20 nM ArgRS, and 7 nM TyrRS in the post-DEAE fraction for aminoacylation of 200 nM tRNA in order to produce linear synthesis of aa-tRNA over time Figure 4C. Aminoacylation was performed with 20 µM [3H]-proline, [3H]-arginine, or [3H]-tyrosine and quantified as above (see section ‘Aminoacylation of tRNA in vitro’).
To investigate the effect of over-expression of aaRSs, E. coli trmD-KO strain in BL21(DE3) maintained by human trm5 (Gamper et al., 2015a; Masuda et al., 2019) was transformed with plasmid pET22b expressing E. coli proS, argS, or an empty vector. Overnight cultures in MOPS medium with 0.15% Ara + 0.05% Glc were inoculated 1:100 to fresh MOPS medium with 0.15% Ara + 0.05% Glc to induce the m1G37+ condition, or to MOPS with 0.2% Glc to induce the m1G37– condition, each harboring 0.025 mM isopropyl-β-D-thiogalactoside (IPTG) to induce expression of the plasmid-borne gene. After two cycles of growth and dilution at 37°C, cells were harvested at OD600 = 0.3–0.5 for isolation of total RNA and for preparation of cell lysate. To assay viability on a plate, overnight cultures in LB medium supplemented with 0.2% Ara were inoculated to fresh LB without Ara and grown for 3 hr to deplete pre-existing Trm5 protein and m1G37-tRNAs. After depletion, cells were made by a 10-fold serial dilution, spotted on M9 plates containing 0.025 mM IPTG, and 0.2% Ara or 0.2% Glc as the only carbon source, and grown at 37°C for overnight.
Reporter assay
Request a detailed protocolThe leuL-leuA genes from the leu operon was cloned from the E. coli MG1655 genomic DNA into pKK223-3 by NEBuilder HiFi DNA Assembly Cloning Kit (NEB) and the first 300 bp portion of leuA was translationally fused with the nLuc reporter gene. The four consecutive Leu codons CUA in leuL were mutated into the synonymous UUA codons by inverse PCR and self-ligation. The WT (CUA) and mutant (UUA) plasmids were transformed into E. coli JM109 trmD-KO cells maintained by human trm5 (Demo et al., 2021) together with pZS2R-mCherry plasmid for constitutive expression of mCherry for normalization. Cells were grown overnight in MOPS medium in the m1G37+ condition and inoculated 1:100 to fresh MOPS medium containing 0.1 mM IPTG and grown in m1G37+ and m1G37− conditions for 6 hr at 37°C. The nLuc readout was measured using Nano-Glo Luciferase Assay System (Promega), followed by normalization by the mCherry fluorescence level.
qPCR analysis
Request a detailed protocolTo test the effect of the Pro-Pro CCA-CCG codon motif on gene expression of ilvL, mutations were introduced to change the motif to Ala-Ala GCA-GCG on the E. coli chromosome of trmD-KO/MG1655 strain, using CRISPR-Cas9 mutagenesis (Reisch and Prather, 2017). WT and CRISPR-edited mutant cells were grown and harvested for total RNA extraction using TRIzol (Invitrogen) and Direct-zol RNA MiniPrep Kits (Zymo Research) per manufacturer’s instructions. It was processed to remove genomic DNA using RQ1 RNase-free DNase (M6101, Promega), followed by cDNA synthesis using RevertAid First Strand cDNA synthesis Kit (K1622, Thermo Fisher Scientific) per manufacturer’s instructions. qPCR was carried out using LightCycler 96 Instrument. For each targeted gene, 18 µL of qPCR master mix containing 10 µL of SYBR Green I Master (04707516001, Roche), 7.6 µL of nuclease-free PCR grade water, 0.2 pmoles of gene-specific forward primer, and 0.2 pmoles of gene-specific reverse primer was combined with 2 µL of cDNA template into each well of a 96-well plate (LightCycler 480 Multiwell Plate 96, white, 04729692001, Roche). qPCR amplification was performed using the following cycling conditions: preincubation for 10 min at 95°C, followed by 45 cycles of three steps of amplification with 10 s at 95°C, 10 s at 48°C, and 10 s at 72°C. The transcript level for each gene was determined using the standard curve generated from the control template by plotting the Cq values (y-axis) against the log initial concentration (x-axis). Results were confirmed by normalization with idnT mRNA. Primers used are shown below (5' to 3'):
Forward primer for ilvL: ATGACAGCCCTTCTACG
Reverse primer for ilvL: CTAAGCCTTTCCTCGTC
Forward primer for ilvG: ATGAATGGCGCACAGTGG
Reverse primer for ilvG: CCTGCTCATGTCGGCAT
Data availability
Request a detailed protocolSequencing data have been deposited in raw FASTQ files at the SRA and processed WIG files at the GEO under accession code GSE165592. Custom Python scripts used to analyze the ribosome profiling and RNA-seq data is freely available at https://github.com/greenlabjhmi/2021_TrmD (copy archived at swh:1:rev:034808ee3fd651b0cf94d091b52aff7c6955ef1c) (Buskirk, 2021).
Data availability
Sequencing data have been deposited in raw FASTQ files at the SRA and processed WIG files at the GEO under accession code GSE165592. Custom Python scripts used to analyze the ribosome profiling and RNA-seq data is freely available at https://github.com/greenlabjhmi/2021_TrmD (copy archived at https://archive.softwareheritage.org/swh:1:rev:034808ee3fd651b0cf94d091b52aff7c6955ef1c).
-
NCBI Gene Expression OmnibusID GSE165592. Loss of N1-methylation of G37 in tRNA induces ribosome stalling and reprograms gene expression.
References
-
A gripping tale of ribosomal frameshifting: extragenic suppressors of frameshift mutations spotlight P-site realignmentMicrobiology and Molecular Biology Reviews 73:178–210.https://doi.org/10.1128/MMBR.00010-08
-
A primordial tRNA modification required for the evolution of life?The EMBO Journal 20:231–239.https://doi.org/10.1093/emboj/20.1.231
-
Divergent adaptation of tRNA recognition by Methanococcus jannaschii prolyl-tRNA synthetaseJournal of Biological Chemistry 276:20286–20291.https://doi.org/10.1074/jbc.M100456200
-
Rapid depletion of target proteins allows identification of coincident physiological responsesJournal of Bacteriology 194:5932–5940.https://doi.org/10.1128/JB.00913-12
-
Distinct origins of tRNA(m1G37) methyltransferaseJournal of Molecular Biology 339:707–719.https://doi.org/10.1016/j.jmb.2004.04.025
-
Control of catalytic cycle by a pair of analogous tRNA modification enzymesJournal of Molecular Biology 400:204–217.https://doi.org/10.1016/j.jmb.2010.05.003
-
Methyl transfer by substrate signaling from a knotted protein foldNature Structural & Molecular Biology 23:941–948.https://doi.org/10.1038/nsmb.3282
-
Distinct determinants of tRNA recognition by the TrmD and Trm5 methyl transferasesJournal of Molecular Biology 373:623–632.https://doi.org/10.1016/j.jmb.2007.08.010
-
Insights into catalysis by a knotted TrmD tRNA methyltransferaseJournal of Molecular Biology 333:931–949.https://doi.org/10.1016/j.jmb.2003.09.011
-
Maintenance of protein synthesis reading frame by EF-P and m(1)G37-tRNANature Communications 6:7226.https://doi.org/10.1038/ncomms8226
-
The UGG isoacceptor of tRNAPro is naturally prone to frameshiftsInternational Journal of Molecular Sciences 16:14866–14883.https://doi.org/10.3390/ijms160714866
-
Tertiary structure checkpoint at Anticodon loop modification in tRNA functional maturationNature Structural & Molecular Biology 16:1109–1115.https://doi.org/10.1038/nsmb.1653
-
Transcriptional responses to ppGpp and DksAAnnual Review of Microbiology 72:163–184.https://doi.org/10.1146/annurev-micro-090817-062444
-
Deficiency of 1-methylguanosine in tRNA from Salmonella typhimurium induces frameshifting by quadruplet translocationJournal of Molecular Biology 232:756–765.https://doi.org/10.1006/jmbi.1993.1429
-
Translational regulation of GCN4 and the general amino acid control of yeastAnnual Review of Microbiology 59:407–450.https://doi.org/10.1146/annurev.micro.59.031805.133833
-
Pathway tools version 23.0 update: software for pathway/genome informatics and systems biologyBriefings in Bioinformatics 22:109–126.https://doi.org/10.1093/bib/bbz104
-
The EcoCyc database: reflecting new knowledge about Escherichia coli K-12Nucleic Acids Research 45:D543–D550.https://doi.org/10.1093/nar/gkw1003
-
Attenuation in amino acid biosynthetic operonsAnnual Review of Genetics 16:113–134.https://doi.org/10.1146/annurev.ge.16.120182.000553
-
The enzymic synthesis of L-cysteine in Escherichia coli and Salmonella typhimuriumJournal of Biological Chemistry 241:4955–4965.https://doi.org/10.1016/S0021-9258(18)99657-2
-
The temperature sensitivity of a mutation in the essential tRNA modification enzyme tRNA methyltransferase D (TrmD)Journal of Biological Chemistry 288:28987–28996.https://doi.org/10.1074/jbc.M113.485797
-
Structural insights into translational fidelityAnnual Review of Biochemistry 74:129–177.https://doi.org/10.1146/annurev.biochem.74.061903.155440
-
Tools for characterizing bacterial protein synthesis inhibitorsAntimicrobial Agents and Chemotherapy 57:5994–6004.https://doi.org/10.1128/AAC.01673-13
-
Thio-Modification of tRNA at the wobble position as regulator of the kinetics of decoding and translocation on the ribosomeJournal of the American Chemical Society 139:5857–5864.https://doi.org/10.1021/jacs.7b00727
-
Scarless Cas9 assisted recombineering (no-SCAR) in Escherichia coli, an Easy-to-Use system for genome editingCurrent Protocols in Molecular Biology 117:31–38.https://doi.org/10.1002/cpmb.29
-
Structural and mechanistic basis for enhanced translational efficiency by 2-thiouridine at the tRNA anticodon wobble positionJournal of Molecular Biology 425:3888–3906.https://doi.org/10.1016/j.jmb.2013.05.018
-
The levels of a universally conserved tRNA modification regulate cell growthJournal of Biological Chemistry 290:18699–18707.https://doi.org/10.1074/jbc.M115.665406
-
A divalent metal ion-dependent N(1)-methyl transfer to G37-tRNAChemistry & Biology 21:1351–1360.https://doi.org/10.1016/j.chembiol.2014.07.023
-
Context-Specific action of ribosomal antibioticsAnnual Review of Microbiology 72:185–207.https://doi.org/10.1146/annurev-micro-090817-062329
-
Control of leu operon expression in Escherichia coli by a transcription attenuation mechanismJournal of Molecular Biology 149:579–597.https://doi.org/10.1016/0022-2836(81)90348-X
-
Comparative genomic assessment of novel broad-spectrum targets for antibacterial drugsComparative and Functional Genomics 5:304–327.https://doi.org/10.1002/cfg.411
-
Translational sensitivity of the Escherichia coli genome to fluctuating tRNA availabilityNucleic Acids Research 41:8021–8033.https://doi.org/10.1093/nar/gkt602
-
Genome-wide assessment of differential translations with ribosome profiling dataNature Communications 7:11194.https://doi.org/10.1038/ncomms11194
-
Distinct kinetic mechanisms of the two classes of Aminoacyl-tRNA synthetasesJournal of Molecular Biology 361:300–311.https://doi.org/10.1016/j.jmb.2006.06.015
Article and author information
Author details
Funding
National Institute of General Medical Sciences (GM134931)
- Ya-Ming Hou
National Institute of General Medical Sciences (GM110113)
- Allen R Buskirk
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank Chris Woolstenhulme and Ryuma Matsubara for preliminary experiments, Sean Moore and Ana Carr for materials to generate the trmD-deg strain, and Glenn Bjork for anti-TrmD antibodies. This study was funded by a JSPS fellowship to IM, NIH grants GM134931 to YMH, and GM110113 to ARB. The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Copyright
© 2021, Masuda et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,142
- views
-
- 337
- downloads
-
- 41
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 41
- citations for umbrella DOI https://doi.org/10.7554/eLife.70619
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Loss of N1-methylation of G37 in tRNA induces ribosome stalling and reprograms gene expression
eLife 10:e70619.
https://doi.org/10.7554/eLife.70619




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}