Intracellular glycosyl hydrolase PslG shapes bacterial cell fate, signaling, and the biofilm development of Pseudomonas aeruginosa
- Frontiers Science Center for Synthetic Biology and Key Laboratory of Systems Bioengineering (Ministry of Education), School of Chemical Engineering and Technology, Tianjin University, China
- State Key Laboratory of Microbial Resources, Institute of Microbiology, Chinese Academy of Sciences, China
- University of Chinese Academy of Sciences, China
Abstract
Biofilm formation is one of most important causes leading to persistent infections. Exopolysaccharides are usually a main component of biofilm matrix. Genes encoding glycosyl hydrolases are often found in gene clusters that are involved in the exopolysaccharide synthesis. It remains elusive about the functions of intracellular glycosyl hydrolase and why a polysaccharide synthesis gene cluster requires a glycosyl hydrolase-encoding gene. Here, we systematically studied the physiologically relevant role of intracellular PslG, a glycosyl hydrolase whose encoding gene is co-transcribed with 15 psl genes, which is responsible for the synthesis of exopolysaccharide PSL, a key biofilm matrix polysaccharide in opportunistic pathogen Pseudomonas aeruginosa. We showed that lack of PslG or its hydrolytic activity in this opportunistic pathogen enhances the signaling function of PSL, changes the relative level of cyclic-di-GMP within daughter cells during cell division and shapes the localization of PSL on bacterial periphery, thus results in long chains of bacterial cells, fast-forming biofilm microcolonies. Our results reveal the important roles of intracellular PslG on the cell fate and biofilm development.
Editor's evaluation
One of the major findings related to the production of long chain polysaccharide polymers by bacteria is why there often is a hydrolytic enzyme encoded within the gene cluster encoding the biosynthetic proteins. At a basic level, these hydrolases cleave the chains to prevent toxic accumulation of intracellular polysaccharides, but it has now been discovered that a glycosyl hydrolase acting on the Pseudomonas aeruginosa PSL polysaccharide affects other important responses of this Gram-negative bacterium. The PslG hydrolase regulated cell length, PSL cell surface localization and signaling via cyclic-di-GMP, overall controlling accumulation of the cells into a protective biofilm.
https://doi.org/10.7554/eLife.72778.sa0Introduction
Structured, surfaced-associated communities of microorganism known as biofilms are important life forms of bacteria prevailing in nature, industrial, and clinical settings (Costerton et al., 1995; Stoodley et al., 2002). In general, biofilm development involves four specific stages: attachment, microcolony formation, matured microcolonies, and dispersal. Bacteria within biofilms are embedded in an extracellular matrix that protects bacterial cells from antibiotics, host defenses, and environmental stresses. Even though the components of biofilm matrix differ from species to species, it generally composes of exopolysaccharides, proteins, and nucleic acids (Stoodley et al., 2002; Flemming and Wingender, 2010). Exopolysaccharides are critical biofilm matrix components for many bacteria, which often promote attachment to surfaces and other cells, act as a scaffold to help maintain biofilm structure, and provide protection (Stewart and Costerton, 2001; Häussler and Parsek, 2010; Stewart and Costerton, 2001). Gene encoding glycosyl hydrolase is often found in gene clusters that are involved in the synthesis of exopolysaccharide (Franklin et al., 2011). Beyond of exopolysaccharide degradation, little is known about whether these genes affect bacterial physiology and biofilm development.
Pseudomonas aeruginosa is an opportunistic human pathogen that can cause life-threatening infections in cystic fibrosis (CF) patients and immune-compromised individuals (Govan and Deretic, 1996; Lyczak et al., 2000; Ramsey and Wozniak, 2005). P. aeruginosa can produce at least three different types of exopolysaccharides: alginate, PEL, and PSL (also named as Pel/Psl polysaccharide previously). Alginate is not expressed at high levels in the majority of non-CF isolates (Colvin et al., 2012), whereas PSL is expressed by most P. aeruginosa natural and clinical isolates (Colvin et al., 2012; Ma et al., 2012; Jennings et al., 2021). PEL is significant for biofilm formation when PSL cannot be synthesized (Jennings et al., 2015). In P. aeruginosa PAO1, PSL is a primary scaffold matrix component that can form a fibre-like matrix to enmesh bacteria within a biofilm (Colvin et al., 2012; Ma et al., 2009; Wang et al., 2013). PSL has shown multiple functions in the biofilm formation of PAO1. For example, PSL can act as a ‘molecular glue’ to promote bacteria cell-cell and cell-surface interactions (Ma et al., 2006; Ma et al., 2009). PSL trails on a surface guide bacteria exploration and microcolony formation (Zhao et al., 2013). Moreover, PSL can also work as a barrier to protect bacteria from antibiotics and phagocytic cells (Mishra et al., 2012; Billings et al., 2013; Tseng et al., 2013). Interestingly, PSL can function as a signal to stimulate biofilm formation through affecting intracellular signal molecule cyclic-di-GMP (c-di-GMP) (Irie et al., 2012).
PSL is synthesized by psl operon, containing 15 co-transcribed genes (pslABCDEFGHIJKLMNO) (Byrd et al., 2009). PslG is a glycosyl hydrolase that encoded by pslG wthin psl operon. PslG has been shown to degrade PSL in vitro or within biofilm matrix which is released from dead bacteria (Yu et al., 2015; Zhao et al., 2018), hence it can inhibit biofilm formation and disrupt a formed biofilm at a nanomolar concentration (Yu et al., 2015; Baker et al., 2016). PslG was first thought to be essential for PSL synthesis, since deletion of pslG gene led to a loss of PSL production (Byrd et al., 2009). Later studies found that deletion of pslG in the earlier work has a polar effect on the expression of pslH and thus resulted in the loss of PSL production, and absence of pslG itself did not result in a complete loss of PSL production per se, but led to a less production of PSL and reduced bacterial initial attachment compared with PAO1 (Baker et al., 2015; Wu et al., 2019). PslG is localized mainly at the inner membrane and some in the periplasm. PslA, PslD, and PslE help PslG anchoring in the inner membrane, which is critical for PslG to be involved in the biosynthesis of PSL (Wu et al., 2019). In addition, the glycoside hydrolytic activity of PslG is also important for PSL production and the key amino acid residues for this activity are E165 and E276 (Wu et al., 2019; Yu et al., 2015).
C-di-GMP is an important second messenger controlling a wide range of cellular processes in many bacteria, such as motility, cell differentiation, biofilm formation and production of virulence factors (Römling et al., 2013). Reports have shown that c-di-GMP is asymmetrically distributed among daughter cells upon bacterial cell division and the asymmetric division on surfaces produces specialized cell types, a spreader for dissemination and a striker for local tissue damage (Christen et al., 2010; Laventie et al., 2019). It has not been investigated whether an intracellular glycoside hydrolase would affect the c-di-GMP level.
In this work, aiming to study the effect of PslG on the cell fate and biofilm development of P. aeruginosa at the single cell level, we systematically studied the pslG in-frame deletion mutants by employing a high-throughput bacterial tracking technique (Zhao et al., 2013). The morphology and motility behavior of bacterial cells in the course of biofilm development were analyzed at the single-cell level. Using pCdrA::gfp reporter, the c-di-GMP level of each cell was also monitored. Microscopically, the attachment behavior of cells on the microtiter surfaces and the pellicles formed at the air-liquid interface were also characterized. Our data suggest that lacking of pslG impacts cell morphology, the signaling function of PSL, the c-di-GMP distribution and bacteria distribution within a biofilm. Based on our results together with those in literature, a model is proposed to understand the role of pslG in the biofilm development.
Results
ΔpslG strains cannot form rings on microtiter dish wells even when PSL production is induced to the wild-type level
Our previous study showed that the pslG in-frame deletion mutant (was named as ΔpslG2 by Wu et al., 2019 hereafter termed as ΔpslG) decreased the production of PSL and bacterial initial attachment on the microtiter surface (Wu et al., 2019). To know whether the attachment defect of ΔpslG mutant is due to PSL reduction, we replaced the promoter of psl operon by PBAD promoter in ΔpslG background, resulting in a PAO1-derived Psl-inducible ΔpslG strain (named as PBAD-pslΔpslG), whose PSL production can be induced by the concentration of arabinose. When induced with 0.5% arabinose, PBAD-pslΔpslG strain produced similar amount of PSL as that of PAO1 (Figure 1B). However, it was not able to form a ring at the air-liquid interface as that seen in either PAO1 or PBAD-psl strain (referred as WFPA801 previously, Ma et al., 2006) under 0.5% arabinose induction (Figure 1B, rings are indicated by an arrow). The ring formation can be recovered by either PBAD-pslG inserted in chromosome attB site (PBAD-psl ΔpslG attB::pslG in Figure 1B) or pslG knocked into the ΔpslG mutants at the original location of pslG (PBAD-pslΔpslG::pslG in Figure 1B). Whereas PslGE165Q, E276Q (two key hydrolytic active sites of PslG were mutated) cannot complement this phenotype (Figure 1B). These results indicate that PslG and its glycoside hydrolytic activity are important for the ring formation at the air-liquid interface.
Figure 1 with 1 supplement see all
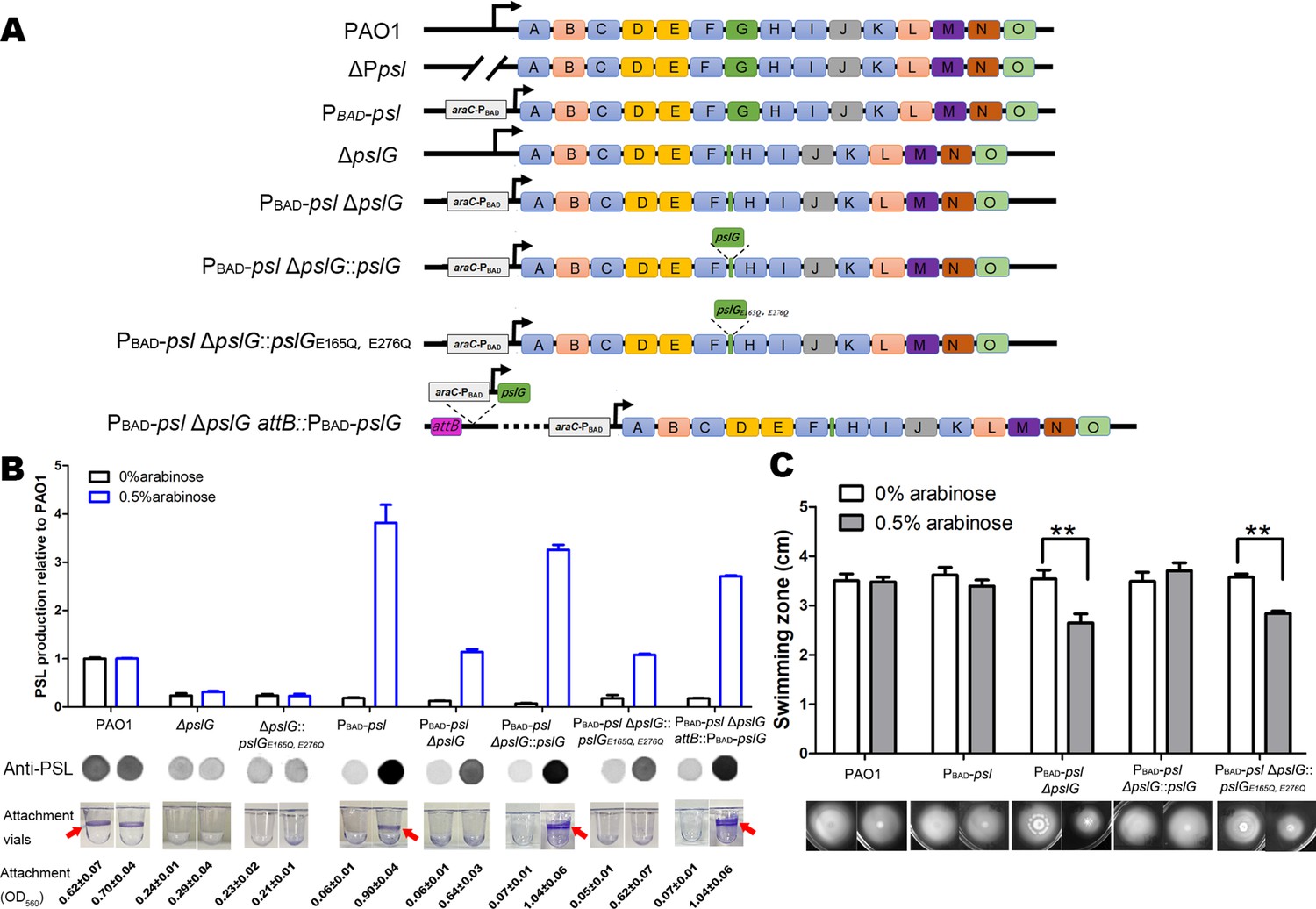
Inducing PSL production in ΔpslG background cannot recover its defects on bacterial initial attachment and yet affects swimming motility.
(A): A schematic of the psl operon in PAO1 and its corresponding mutants used in this study. Genes pslA-O are shown in boxes (not to scale). Angled lines represent the extent of deleted sequence, and black arrows indicate transcriptional start sites (not to scale). (B): The Psl production of tested strains inducing with 0% or 0.5% arabinose. The amount of Psl was determined by immune-dot blotting and normalized to the level of PAO1. A representative of dot blotting as well as corresponding microtiter dish wells and their crystal violet (CV) reading (OD560) posted CV staining in attachment assay were shown under each corresponding column. Arrows indicate the ring at air-liquid interface. (C): The swimming motility of tested strains inducing with 0% or 0.5% arabinose. The corresponding image of swimming zone was shown under each column. Statistical significances were measured using student’s t-test (**, p < 0. 01).
-
Figure 1—source data 1
Figure 1B, C source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig1-data1-v2.xlsx
Flagella and Type IV pili (T4P) are also important for the initial attachment of P. aeruginosa (O’Toole and Kolter, 1998; Bruzaud et al., 2015). We then tested the flagellum-driven swimming motility and T4P-mediated twitching motility of PBAD-pslΔpslG strain to evaluate the function of flagella and T4P. Without arabinose induction, PBAD-pslΔpslG strain exhibited similar swimming ability and twitching motility as did PAO1 and PBAD-psl (Figure 1C and Figure 1—figure supplement 1). However, with 0.5% arabinose induction, PBAD-pslΔpslG strain (having a wild type level of PSL production, Figure 1B) showed reduced swimming zone compared to that of either PAO1 or PBAD-psl (Figure 1C). T4P-mediated twitching motility of PBAD-pslΔpslG was not affected under conditions with or without arabinose (Figure 1—figure supplement 1C). These results demonstrate that pslG deletion does not affect the function of flagella or T4P directly, yet inducing PSL production in PBAD-pslΔpslG attenuates the swimming motility with no effect on bacterial growth (Figure 1—figure supplement 1D), which might impact its attachment phenotype. Taken together, our results show that increasing PSL production in PBAD-pslΔpslG could not rescue its defect on attachment, suggesting that ΔpslG might have multiple effects on bacterial physiology.
ΔpslG impacts the bacterial distribution and maximum thickness of pellicles
We then investigated the effect of ΔpslG on the biofilms formed at the air-liquid interface, termed as pellicles, by using confocal laser scanning microscopy. The total pellicle biomass of ΔpslG is similar to that of PAO1 after 24 hr growth, although ΔpslG produced much less PSL and had defect on initial attachment (Figures 1, 2A and C). However, ΔpslG has significant higher maximum thickness than that of PAO1 (Figure 2B). In addition, there are less bacteria in each section image of ΔpslG pellicles compared to that of PAO1 (Figure 2D, left and middle panel). The PSL matrix in ΔpslG pellicles shows weaker fluorescent intensities than that of PAO1 (Figure 2D, middle and right panels), which is consistent with their corresponding PSL production. In spite of that, the fibre-like PSL can be detected in the pellicles of ΔpslG, which have a radial pattern as previously described for PAO1 pellicles (Figure 2D, middle panels) (Wang et al., 2013). These results suggest that the pslG deletion might impact bacterial distribution within biofilms.
Figure 2
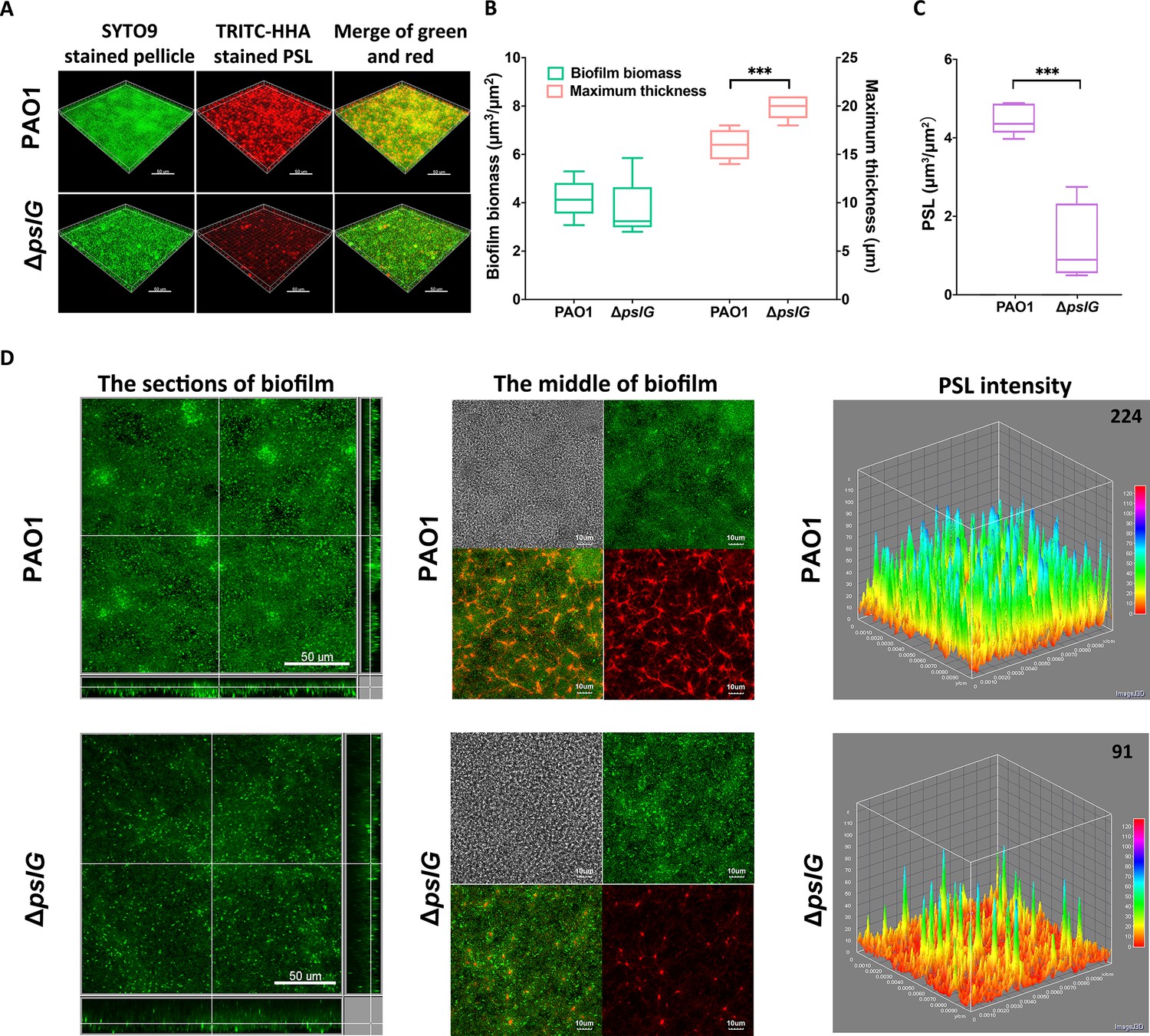
Comparison of pellicles formed by PAO1 and ΔpslG mutant.
(A): Three-dimensional images of 24 hr air-liquid interface biofilms (pellicles) formed by PAO1 and ΔpslG. (B): Biofilm biomass and maximum thickness of PAO1 or ΔpslG strain. (C): PSL in pellicles of PAO1 and ΔpslG. (D): Typical section images of pellicles formed by PAO1 and ΔpslG. Left panel, section images showed the top-down view (square) and side view (rectangle) of corresponding pellicles. Middle panel, section images at the middle of corresponding pellicles. The distribution of bacteria (green), the fibre-like PSL matrix (red) and corresponding DIC images (grey) were shown. Right panel, PSL fluorescence intensity in corresponding biofilm images shown in the middle panel (the average intensity of PSL in per μm3 biofilm is shown in the upper right corner). Green, SYTO9 stained bacteria, Red, TRITC-HHA stained PSL. Statistical significances were measured using student’s t-test (***, p < 0.001 when compared to PAO1). Scale bar: 50 μm for A and the left panel in (D); 10 μm for the middle panel in D.
-
Figure 2—source data 1
Figure 2B, C source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig2-data1-v2.xlsx
Single-cell tracking analysis indicates that loss of PslG or its glycoside hydrolytic activity promotes microcolony formation in flow-cell systems
To further understand the effect of ΔpslG, by employing bacterial tracking techniques, we observed ΔpslG cell behavior at the single-cell level. Figure 3A shows the surface coverage obtained by all tracked bacterial trajectories for a specific time period during microcolony formation. Red color indicates the surface area that has been visited by bacteria, while black color indicates a ‘fresh’ surface area that has never been visited. Bacterial cells are shown in blue. Under the same total bacterial visits (marked as N in Figure 3A), the difference in the surface coverage between ΔpslG and PAO1 is not obvious at N~10,000. As N increases, the surface coverage of ΔpslG is clearly less than that of PAO1 (Figure 3A). At N~100,000, ΔpslG has a surface coverage of 52% ± 6% while PAO1 has 81% ± 10%. Compared with PAO1, the less efficiency in covering the surface leads to a more non-uniform bacterial visit distribution for ΔpslG (Figure 3B). Correspondingly, by fitting the distribution of bacterial visits with a power law, different power law exponents were obtained (see one example in Figure 3C). The averaged power law exponents of three repeats are –2.9 ± 0.1 for PAO1 and –2.5 ± 0.1 for ΔpslG. Such differences in bacterial visits distribution resulted that the time required to observe a visible microcolony (defined as clusters of more than 30 cells in this study, marked by dash lines in Figure 3D) in the field of view is shorter for ΔpslG (5.9 ± 1.7 hr) than that for PAO1 (8.7 ± 2.4 hr). In addition, after 10 hr of growth, compared with PAO1, ΔpslG formed more microcolonies in the field of view (Figure 3E). The microcolony formation phenotype of ΔpslG mutants can be reverted back to WT-like when they are complemented with PslG by knocking pslG into the ΔpslG mutants at the original location of pslG, namely ΔpslG::pslG. As illustrated in Figure 3, at N~100,000, the surface coverage of ΔpslG::pslG reaches to 70% ± 7% and the power law exponent of the bacterial visit distribution is –3.2 ± 0.13 (averaged over three repeats), both are closer to those of PAO1 than those of ΔpslG. The number of microcolonies formed by ΔpslG::pslG in the field of view after 10 hr of growth is also similar to that of PAO1 and less than that of ΔpslG. The fast microcolony formation phenotype of ΔpslG mutant is also able to be complemented by PslG that is expressed from plasmid (Figure 3—figure supplement 1). However, ΔpslG::pslGE165Q,E276Q, in which the key glycoside hydrolytic activity sites are mutated cannot recover WT-like phenotype (Figure 3 and Figure 3—figure supplement 1), suggesting the importance of PslG hydrolytic activities. Complementation tests were also performed in strains from PBAD-psl background, and similar trends were observed (Figure 3—figure supplement 2). PEL has been shown to play a role in bacterial aggregation (Jennings et al., 2021), thus we also tested PEL-negative strains in wild type (ΔPpel) or the ΔpslG background (ΔpslGΔPpel). The phenotype of ΔpslGΔPpel was similar to that of ΔpslG, suggesting little contribution of PEL on the fast microcolony formation of ΔpslG (Figure 3 and Figure 3—figure supplement 3). Tracking GFP-tagged bacteria in a flow cell also shows that the PAO1 biofilms tend to spread on surfaces (Figure 3F and Video 1) while ΔpslG cells tend to accumulate and form microcolonies with strong fluorescence intensity (Figure 3F and Video 2). This is consistent with the results of bacterial visit distribution map shown in Figure 3B. Taken together, these results indicate that loss of pslG promotes microcolonies formation by changing the surface exploration of bacteria during microcolony formation, a phenomenon that has been reported mostly for strains producing a high level of PSL (Zhao et al., 2013).
Figure 3 with 3 supplements see all
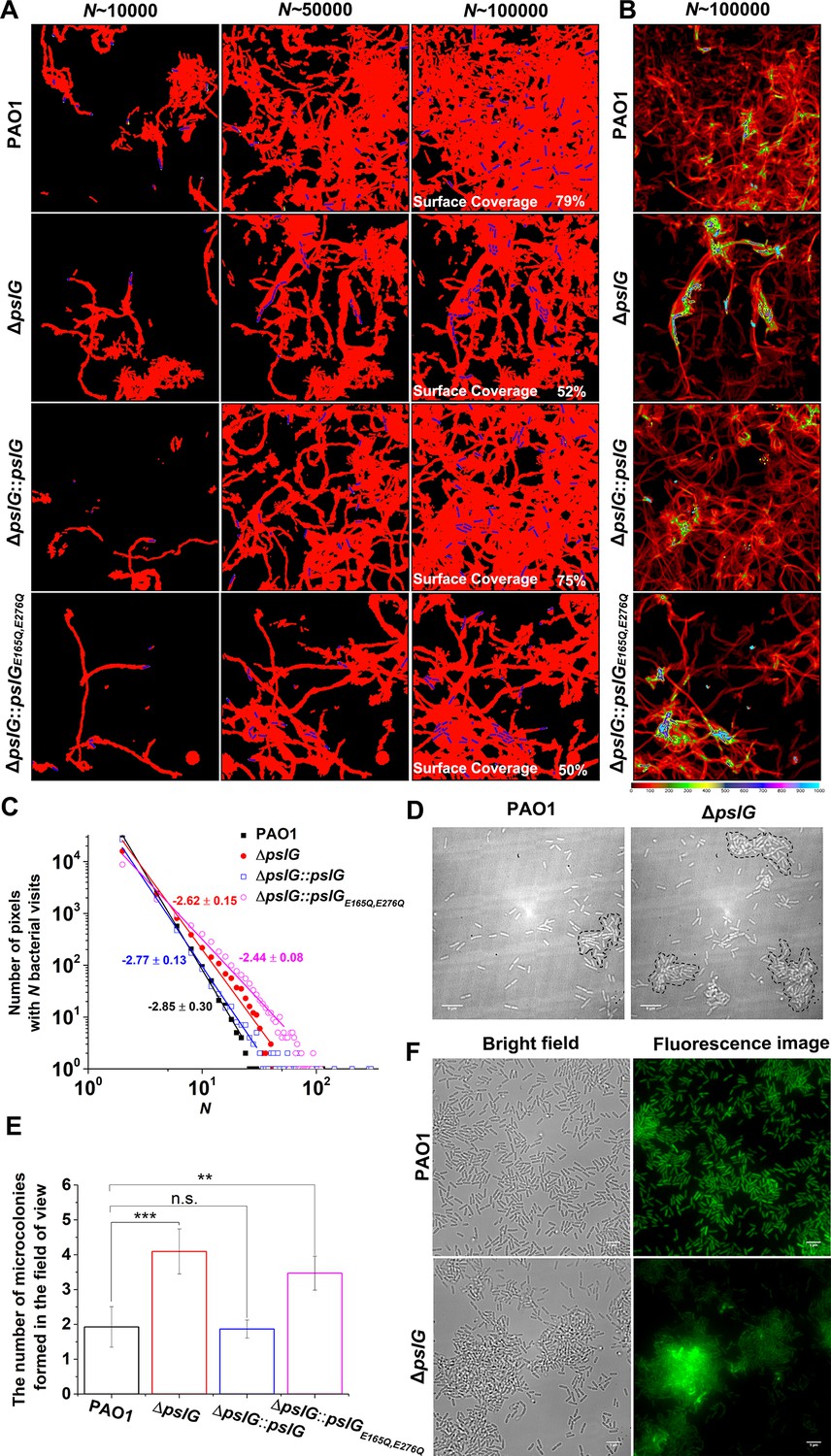
Effects of pslG on the formation of microcolonies in flow-cell systems.
(A): Surface coverage maps at a total of 10,000, 50,000, and 100,000 bacterial visits for PAO1, ΔpslG, ΔpslG::pslG, and ΔpslG::pslGE165Q,E276Q cells. Red color indicates the surface area that has been visited or contaminated, while black color indicates a ‘fresh’ surface area. Bacteria in the current frame are shown in blue. The surface coverage in the figure is the result of an experiment. (B): The intensity map of bacterial visits at N ~ 100,000. The color scale of black to cyan represents bacterial visits of 0–1,000. (C): The graph displays one measurement result for the visit frequency distributions of PAO1, ΔpslG, ΔpslG::pslG and ΔpslG::pslGE165Q,E276Q at N ~ 100,000. The slope in the figure is the fitting result of an experiment. (D): Examples of microcolonies (enclosed by dash lines) formed by PAO1 and ΔpslG cells, respectively, cultured in a flow cell for about 8 hr. (E): The number of microcolonies in the field of view formed by PAO1, ΔpslG, ΔpslG::pslG and ΔpslG::pslGE165Q,E276Q at 10 hr after inoculation in a flow cell. The number (N) of frames analyzed are 14, 43, 52, and 51 for PAO1, ΔpslG, ΔpslG::pslG, and ΔpslG::pslGE165Q,E276Q, respectively. Error bars represent standard deviations of the means. Statistical significances were measured using one-way ANOVE. n.s., not significant; *p < 0.05; **p < 0.001; ***p < 0.0001. F. Snapshots taken at 10 hr after inoculation in a flow cell, showing the microcolonies formed by ΔpslG and PAO1. Bacteria were tagged by GFP. Fluorescence images and corresponding bright-field images were shown. Scale bar, 5 μm.
-
Figure 3—source data 1
Figure 3C source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig3-data1-v2.xlsx
-
Figure 3—source data 2
Figure 3E source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig3-data2-v2.xlsx
Video 1
An example video of tracking the biofilm formation of Gfp-tagged PAO1 cells.
The video was taken at a frame interval of 10 min for 10 hr and was played back at 5 fps. Scale bar, 5 μm.
Video 2
An example video of tracking the biofilm formation of Gfp-tagged ΔpslG cells.
The video was taken at a frame interval of 10 min for 10 hr and was played back at 5 fps. Scale bar, 5 μm.
T4P-driven motility is not the main factor in promoting the microcolony formation of ΔpslG strains in flow-cell systems
T4P-driven motilities on surface, such as walking and crawling, affect microcolonies formation in flow-cell systems (Conrad et al., 2011). To investigate why ΔpslG promotes microcolonies formation, we calculated the twitching speed of bacterial cells. To minimize the effect due to possible different production of PSL, we compared the measurements between PAO1 and PBAD-pslΔpslG under 0.5% arabinose, under which both strains show relatively similar production of PSL (Figure 1). The results show a slightly reduced average speed and a higher crawling percentage of PBAD-pslΔpslG cells compared with PAO1 (Figure 1—figure supplement 1A, B). But such differences are not statistically significant (P = 0.29), indicating that the twitching motility may not be the main factor in promoting the microcolony formation.
ΔpslG shapes the localization of PSL on bacterial periphery, leading to long chains of bacterial cells that are connected by PSL
During bacterial tracking in flow cells, we frequently observed long chains of bacterial cells in the ΔpslG strain or PBAD-pslΔpslG strain when PSL production was induced with arabinose (Figure 4A), which typically started to appear 1~2 hr after inoculation of bacteria into a flow cell under tested conditions and could reach to about 50% of cell population at a later time (Figure 4B). Such long chains of cells were not observed in strains that have intact pslG or when PSL production was not induced in PBAD-pslΔpslG (Figure 4—figure supplement 1). In a typical cell division process, two daughter cells will be disconnected from each other when the formation of septum is completed. However, in pslG deletion mutants, the two daughter cells could not separate into physically disconnected progenies, leading to a cell chain (See one example of PBAD-pslΔpslG in Video 3). The length of chains varied. Among all the observed chains, the chains consisting of 4 cells were observed most frequently, which is true both in ΔpslG and in PBAD-pslΔpslG strains (Figure 4C). These bacterial cell chains can grow as cells continue to divide, yet some can also be broken by bending of chains (Video 3), suggesting that the bacterial chains are not connected by septum. In addition, bacterial cell chains were also observed in liquid culture of ΔpslG mutants (data not shown), indicating that bacterial adhering on surfaces is not required for the formation of bacterial cell chains.
Figure 4 with 1 supplement see all
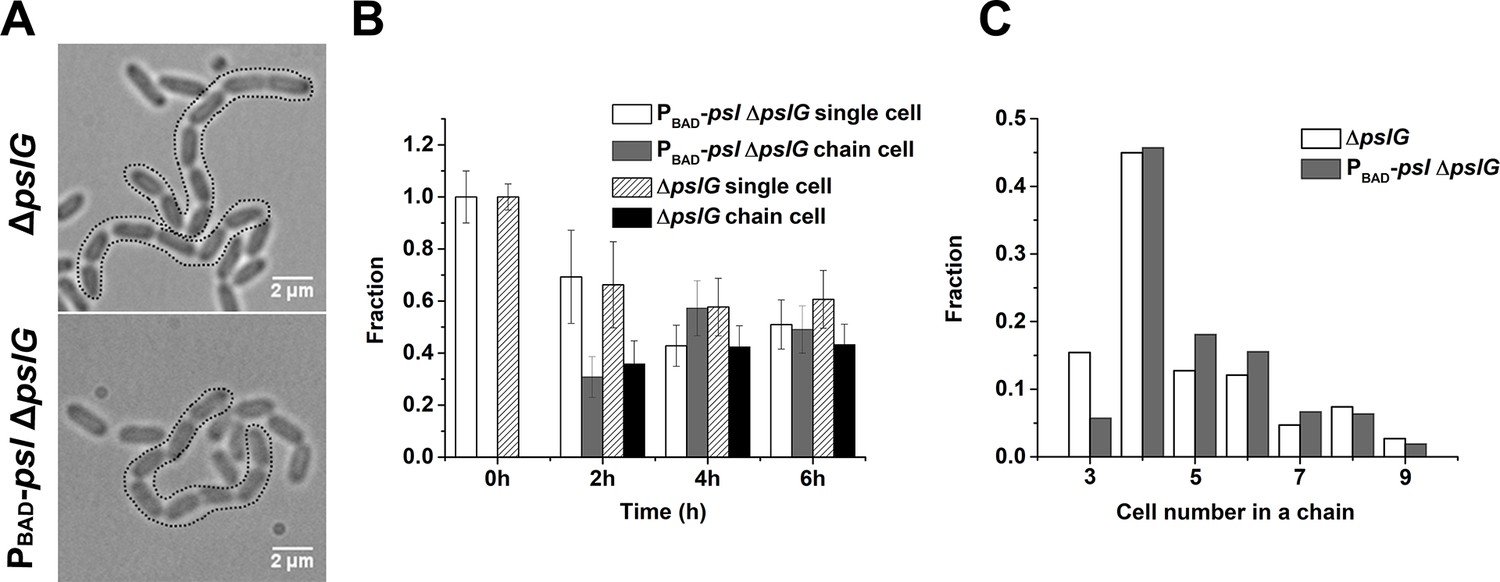
Characterization of long bacterial chains of ΔpslG strains.
(A): Examples of long bacterial chains (indicated by black dotted outlines) formed by ΔpslG and PBAD-pslΔpslG cells. (B): The faction of single isolated bacterial cell and cell in chains at different time points after inoculation in a flow cell. The number of analyzed picture in each strain is n = 88 (about 3200 cells) for PBAD-pslΔpslG and n = 87 (about 3500 cells) for ΔpslG. (C): The number distribution of cells consisted in a chain. The number of analyzed cells is n = 301 for PBAD-pslΔpslG and n = 322 for ΔpslG. Scale bar, 2 μm.
-
Figure 4—source data 1
Figure 4B source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Figure 4C source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig4-data2-v2.xlsx
Video 3
An example video of the formation of PBAD-pslΔpslG long cell-chain.
The video was taken at a frame interval of 5 min for 3.5 hr and was played back at 5 fps. Scale bar, 5 μm.
To investigate whether PSL has any contribution on the formation of such long bacterial chains, we stained PSL in the biofilm formed in flow cells by FITC-HHA (green fluorescence dyes FITC labeled lectin HHA). The PSL of ΔpslG strains is tightly associated with bacteria compared to PAO1 and PBAD-psl with arabinose (Figure 5A and Figure 5—figure supplement 1). We also used cell membrane stain FM4-64 to help locate cell periphery and septum. Strikingly, strong PSL signal is often found around septa in ΔpslG strains and strains with catalytic site mutation (PBAD-pslΔpslG::pslGE165Q, E276Q or ΔpslG::pslGE165Q, E276Q), which barely observed in strains with wild type pslG (such as PAO1 and PBAD-psl) under the same growth conditions (Figure 5A and Figure 5—figure supplement 1).
Figure 5 with 3 supplements see all
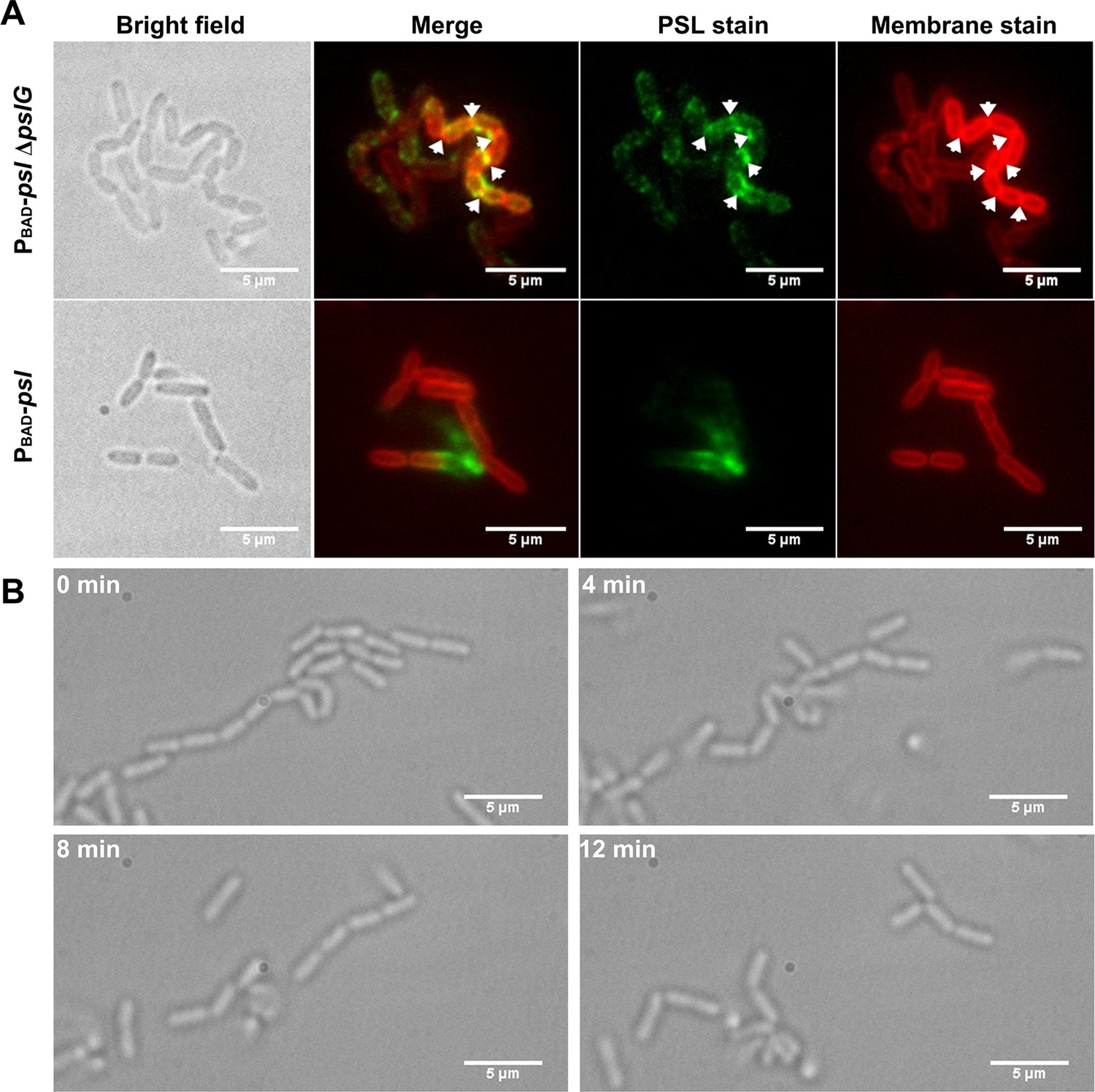
Cells in bacterial chains are connected by PSL and can be disassembled by PslG supplied exogenously.
(A): Fluorescence staining of long bacterial chains formed by PBAD-pslΔpslG cells. Staining of control samples of PBAD-psl is also shown. Green shows PSL stained by FITC-HHA, and red shows the bacterial cell membranes stained by FM4-64. Arrows in the PSL stain image indicate the bright spotted PSL locations, and arrows in the membrane stain image show the septum locations. (B): Time–lapse images show the break-up process of long bacterial chains of PBAD-pslΔpslG when PslG was supplied. Scale bar, 5 μm.
From the PSL staining results, we speculate that PSL might help to connect bacterial cells together to form long bacterial chains. To test this hypothesis, we first tested whether the PBAD-pslΔpslG could form long bacterial chains when PSL is not produced. Under a culture condition without arabinose, the transcription of psl operon in PBAD-pslΔpslG is not induced, no bacterial chains were observed as shown in Figure 4—figure supplement 1, suggesting that PSL production is required for the formation of long bacterial chains. Next, we treated the long bacterial chains with purified PslG, which has been shown to be able to disaggregate microcolonies and matured biofilms by hydrolyzing PSL (Yu et al., 2015). At 4 min after addition of exogenous PslG, the long chains of bacterial cells were seen clearly to start to be broken up, and they were completely disconnected into single cells after 12 min of PslG treatment (Figure 5B) (Videos 4 and 5). Bacterial chains can be separated by exogenous PslG within a few minutes further confirmed that the bacterial chains were not connected by septum. These results together with the fact that PSL is a ‘sticky’ exopolysaccharide suggest that bacterial cells are frequently connected by PSL in ΔpslG mutant strains, leading to the long bacterial cell chains.
Video 4
An example video of the degradation of PBAD-pslΔpslG long cell-chain.
The video was taken at a frame interval of 1 min for 0.5 hr and was played back at 5 fps. Scale bar, 5 μm.
Video 5
An example video of the degradation of ΔpslG long cell-chain.
The video was taken at a frame interval of 1 s for 10 min and was played back at 5 fps. Scale bar, 5 μm.
This long-cell-chain phenotype can be complemented by wild type PslG, but not hydrolytic activity sites mutation PslGE165Q, E276Q (Figure 5—figure supplement 2), suggesting that the hydrolytic activity of PslG plays a key role on the formation of long bacterial cell chains. To check whether exopolysaccharide PEL and alginate play roles on the observed phenotype induced by pslG deletion, we also tested PAO1-derived alginate-negative strain ΔalgD as well as PEL-negative strains ΔPpel and ΔpslGΔPpel. The phenotype of either ΔPpel or ΔalgD is similar to that of PAO1, whereas ΔpslGΔPpel exhibits a similar phenotype as ΔpslG strains, for bacterial chains formation, microcolonies formation, and initial attachment (Figure 5—figure supplement 3, Figure 3—figure supplement 3, Figure 1—figure supplement 1C). These results indicate that PEL and alginate may have little effect on the observed phenotype induced by pslG deletion in this study. This data also imply that the long bacterial chains might be a contributor for ΔpslG strains to promote microcolony formation.
Lack of PslG or its hydrolytic activity has effects on the fate and C-di-GMP distribution of daughter cells during cell division
C-di-GMP is a critical intracellular signal molecule that affects a variety of cell activities including cell motility, cell fate after division and biofilm formation. We then monitored the c-di-GMP level of cells for each cell division event by employing pCdrA::gfp as a reporter (Irie et al., 2012). Cells with high c-di-GMP levels show strong fluorescence intensity as previously described (Irie et al., 2012). We focused on the first bacterial division events after cells attached on the surface and analyzed the fluorescence intensity (corresponding to c-di-GMP levels) in two daughter cells right after division (Figure 6A). By comparing the fluorescence intensity of each daughter cell relative to its mother cell, the division events can be classified into three types: none of the daughter cells becomes bright (none-bright), one daughter cell becomes bright (one-bright) and both of daughter cells become bright (two-bright). The results show that none-bright type is observed most frequently in all tested strains, which has an occurrence probability of ~ 60% for PAO1, ~ 58% for ΔpslG, and ~ 52% for PBAD-pslΔpslG. Interestingly, both ΔpslG mutants show a relatively higher probability of two-bright type (~17% for PBAD-pslΔpslG strain, ~ 16% for ΔpslG strain) than that of PAO1 (~11%) (Figure 6B and Figure 6—figure supplement 1). In addition, the catalytic site mutated strains show a phenotype similar to ΔpslG strains, have a relatively higher probability of two-bright type (~19% for both ΔpslG::pslGE165Q, E276Q and PBAD-pslΔpslG::pslGE165Q, E276Q strain) (Figure 6B). Thus, compared with PAO1, ΔpslG cells and bacterial cells with the catalytic site mutated PslG would have a higher probability to have both daughter cells with high c-di-GMP levels. During asymmetric divisions, daughter cells with high c-di-GMP levels keep staying on the surface and daughter cells with low c-di-GMP levels tend to move away (Christen et al., 2010; Laventie et al., 2019). Therefore, both daughter cells with high c-di-GMP levels might enhance bacterial stay on the surface and alter their movement pattern. In addition, a high level of c-di-GMP enhances PSL production. An earlier work has shown that cells form a microcolony through a PSL-based rich-get-richer mechanism (Zhao et al., 2013), during which founder cells can be very important. The cells of two-bright cases have high c-di-GMP levels and thus can act as founder cells to promote microcolony formation. Altogether, the slight change in c-di-GMP levels of daughter cells in pslG mutants can be likely one of reasons to promote the formation of microcolony and long bacterial chains.
Figure 6 with 1 supplement see all
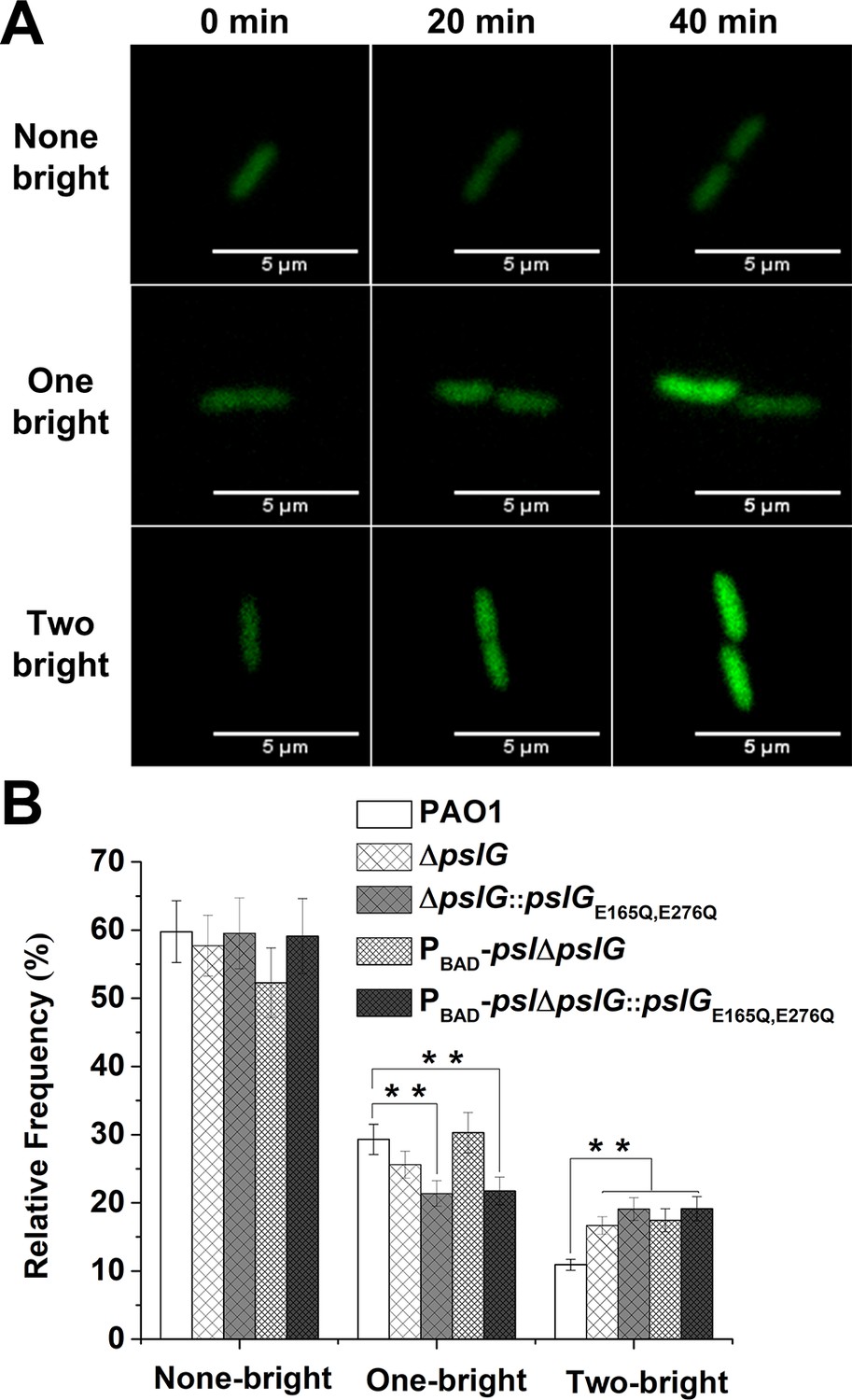
Lacking of PslG or its hydrolytic activity has effects on the fate and c-di-GMP distribution of daughter cells.
(A): Three types of cell division based on fluorescence intensity changes of daughter cells relative to that of their mother cell: none of daughter cells becomes bright (none-bright), one daughter cell becomes bright (one-bright), and both daughter cells become bright (two-bright). Examples given are PAO1 cells. The fluorescence is from pCdrA::gfp, which acts as a reporter for the c-di-GMP level of cells. (B): The measured probability of three types of division in PAO1, ΔpslG, PBAD-pslΔpslG, ΔpslG::pslGE165Q,E276Q, and PBAD-pslΔpslG::pslGE165Q,E276Q. The total number of analyzed division events from more than three repeats is n = 174 for PAO1, n = 168 for ΔpslG, n = 109 for PBAD-pslΔpslG, n = 131 for ΔpslG::pslGE165Q,E276Q, and n = 115 for PBAD-pslΔpslG::pslGE165Q,E276Q. Statistical significances were measured using one-way ANOVA. n.s., not significant; **p < 0. 01. Scale bar, 5 μm.
-
Figure 6—source data 1
Figure 6B source data.
- https://cdn.elifesciences.org/articles/72778/elife-72778-fig6-data1-v2.xlsx
PSL produced by pslG mutants has a stronger signaling function
As shown in Figure 5, PSL is often localized around septa in ΔpslG strains. Since PSL can have a signaling function by stimulating intracellular c-di-GMP production (Irie et al., 2012), we speculated that PSL produced from ΔpslG might have different signaling properties. To test the signaling functions of PSL, we set up a co-culture system, which contained a PSL donor strain and a reporter strain. PAO1 harboring the plasmid pCdrA::gfp was utilized as an intracellular c-di-GMP reporter strain while PAO1, ΔPpsl, ΔpslG, and ΔpslG:: pslGE165Q,E276Q strains as PSL donors respectively, in which ΔPpsl (named as WFPA800 previously) was used as a negative control because it does not produce PSL due to deletion of the promoter of psl operon (Figure 1A). As shown in Figure 7, PAO1 that produces wild type level of PSL can induce a stronger GFP fluorescence signal in the reporter stain compared to ΔPpsl (Figure 7A,B). Strikingly, ΔpslG strain and the hydrolytic activity sites mutant (ΔpslG:: pslGE165Q,E276Q) stimulated a higher fluorescence signal than that of PAO1 (Figure 7B) although both of which produce less PSL (about 30% of PAO1 level, Figure 1A), suggesting that PSL synthesized from these two pslG mutants has a stronger signaling effect on stimulating the intracellular c-di-GMP production than that of wild type. We then examined the impact of exogenous PslG on the fluorescence signal of PAO1/pCdrA::gfp. After 10 hr of PslG treatment, the fluorescence intensity of PAO1/pCdrA::gfp was significantly reduced compared to the non-treatment control and it had a dose-dependent manner ( Figure 7C, D). This further suggests that the hydrolysis of PslG might reduce the signaling property of PSL.
Figure 7
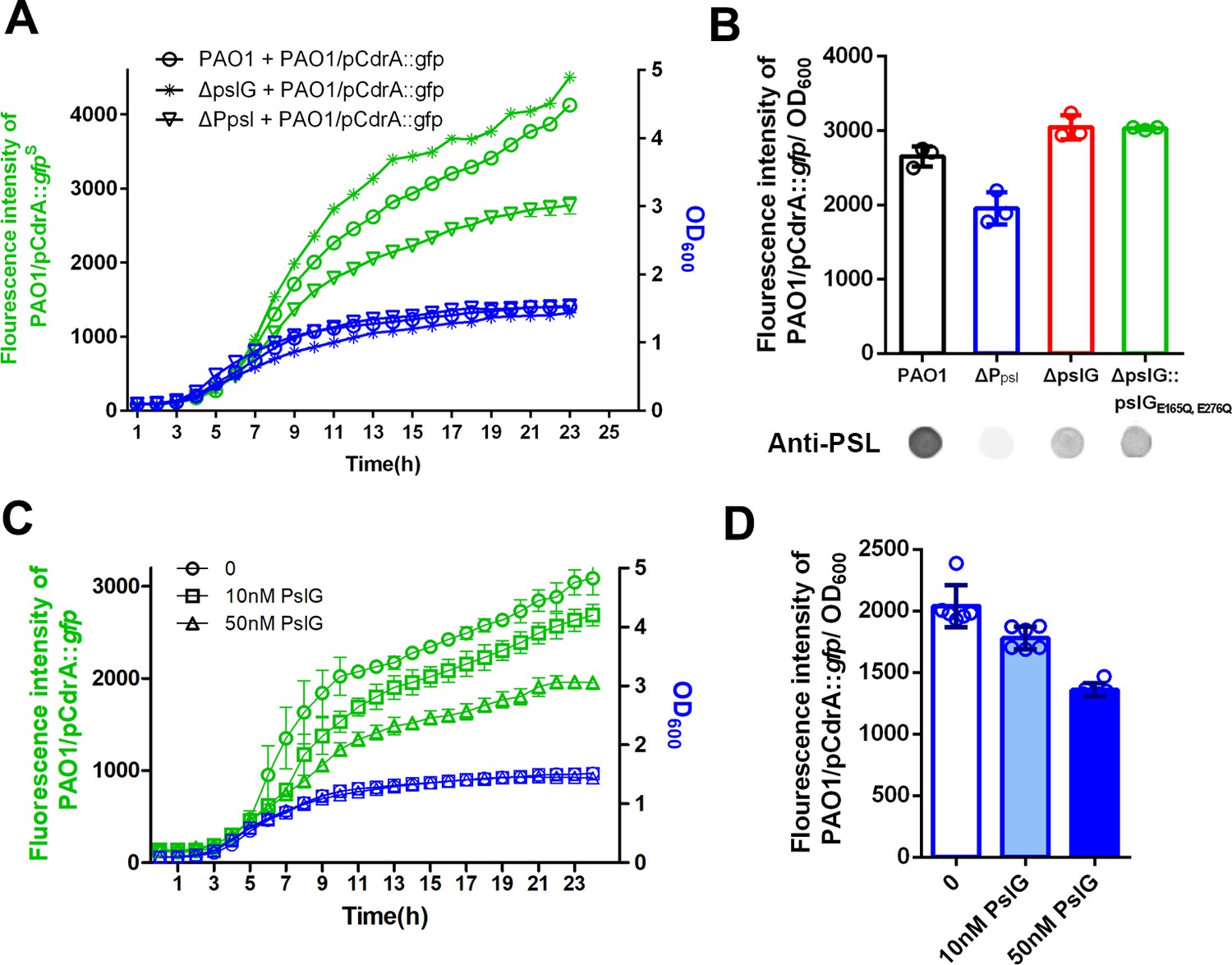
PSL produced by pslG mutant has a stronger signaling function.
(A): A coculture system using pCdrA::gfp as a reporter plasmid to evaluate the intracellular c-di-GMP. The reporter strain (PAO1/pCdrA::gfp) was mixed with PSL provider strain (PAO1, ΔpslG, or ΔPpsl: a PSL-negative mutant) at a ratio of 1: 1, and the GFP fluorescence value and OD600 in each co-culture system was recorded once per hour for 24 hr. (B): The fluorescence intensity per OD600 of PAO1/pCdrA::gfp after 24 hr co-culture with PSL provider strain PAO1, ΔPpsl, ΔpslG, or ΔpslG::pslGE165Q,E276Q. PSL production of each donor strain was shown under corresponding column. Statistical significances were measured using student’s t-test (**, p < 0. 01 when compared to PAO1). (C): The fluorescence intensity and corresponding OD600 of PAO1/pCdrA::gfp during 24 hr of PslG treatment. (D): The fluorescence intensity per OD600 of PAO1/pCdrA::gfp post 24 hr of treatment with different concentration of PslG.
Taken together, our results show that lack of intracellular PslG shapes the localization of PSL on bacterial periphery, enhances the signaling function of PSL, which might further affect the c-di-GMP distribution in daughter cells during cell division, leads to changing of bacterial surface exploration, daughter cells being connected together after cell division and thus the formation of bacterial chains and microcolonies. Strikingly, all these phenotypes depend on the hydrolytic activity of PslG and PSL production, implying that PslG, as a glycosyl hydrolase, can modulate the signaling property of PSL by its hydrolytic activity to affect P. aeruginosa biofilm development.
Discussion
PslG has been shown to be an efficient PSL degrader in vitro and in biofilm matrix, yet its physiologically relevant role within a bacterium or biofilm is ill-defined. In this work, by systematically studying the pslG knock-out mutants both at a single cell and community level, the effects of pslG on the bacterial physiology and surface behavior have been illustrated comprehensively. Based on our results, we propose a model for the role of pslG in the biofilm development of P. aeruginosa as the following (Figure 8).
Figure 8
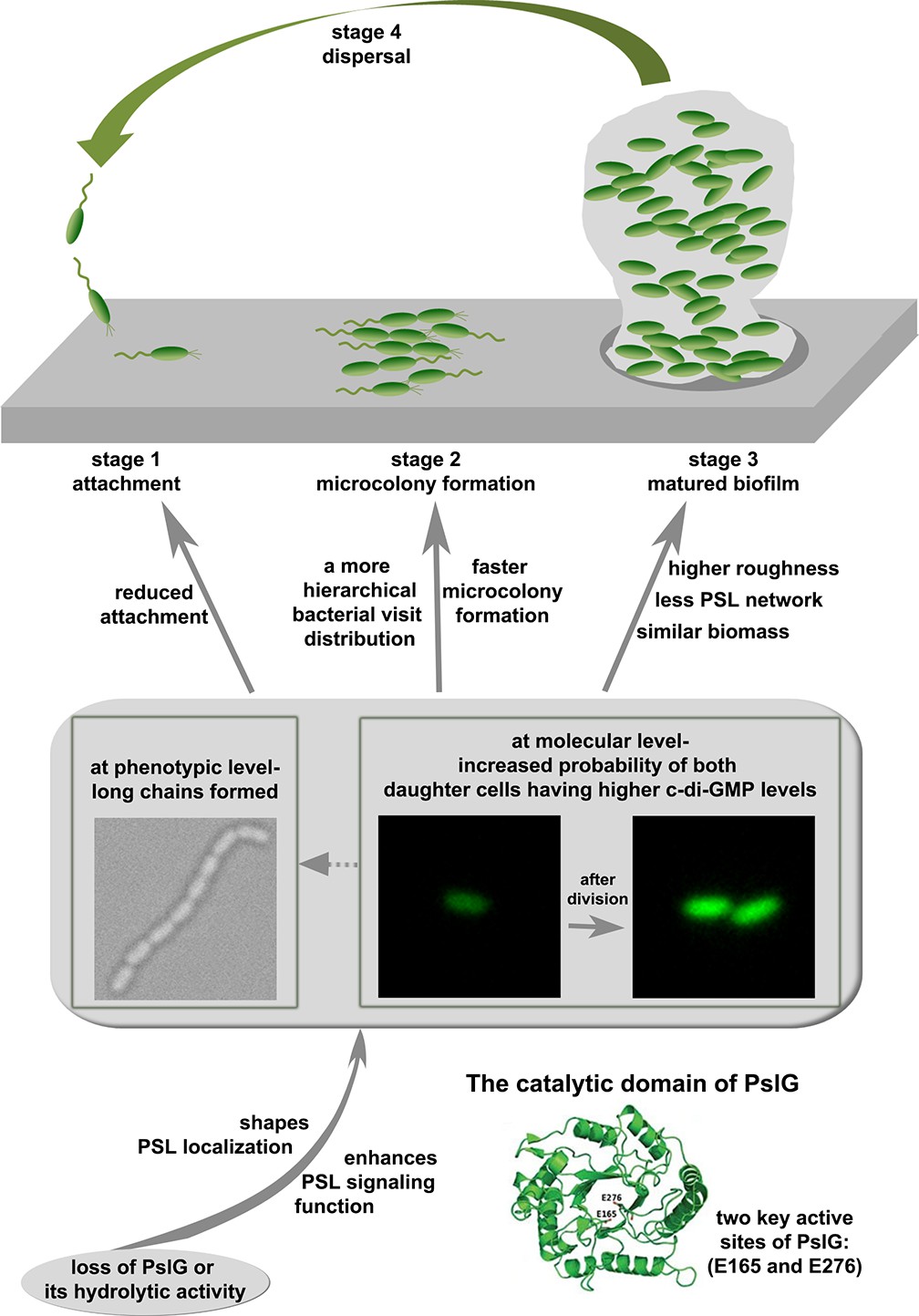
A schematic to show the effects of pslG in the biofilm development of P. aeruginosa.
The structural analysis of PslG has shown that it has a catalytic domain for the hydrolysis of PSL, and is typically considered to modify or hydrolyze the PSL polymer before PSL are secreted out of the bacterial cell (Yu et al., 2015; Baker et al., 2015). Earlier studies have shown that PslG is localized mainly on the inner membrane and a little in the periplasm (Wu et al., 2019; Baker et al., 2015). It has been proposed that PslG on membrane hydrolyzes PSL polymer during biosynthesis to help the release of PSL at right time (Wu et al., 2019) and the portion in periplasm is to degrade any PSL accumulated in the periplasm space (Baker et al., 2015). In this study, our results revealed that it is very important for P. aeruginosa and its communities to have a glycosyl hydrolase encoding gene, pslG within the polysaccharide synthesis gene cluster. Loss of PslG or its catalytic activity affects the properties and functions of PSL, including its localization and signaling. The consequence of these changes has two aspects. One is that phenotypically long chains of cells are formed. The long chains are not observed in strains with intact pslG. Such long chains seem not induced by the adherence on a surface as they can also be observed in liquid cultures. Rather, there are multiple lines of evidences to support that PSL is the main factor for the formation of long chains. Firstly, by adding PslG externally into cell cultures, long chains were observed to disassemble into single cells, presumably due to the hydrolysis of PSL by PslG as shown in literature (Yu et al., 2015; Baker et al., 2015; Zhang et al., 2018). Secondly, such long chains are not observed in PBAD-pslΔpslG when PSL production is not induced (under 0% arabinose). Thirdly, the fluorescence staining experiments also show that PSL are often localized around septa of cells, where daughter cells are supposed to be disconnected after division.
The other aspect as a consequence of the change in PSL due to the loss of PslG is that, at the molecular level, the probability of both daughter cells having a higher level of c-di-GMP than that of their mother cell in a division event is increased. The increased level of c-di-GMP then would result in reduced cell motility and promote cells to transit to biofilm style (Römling et al., 2013). This may also help cells to form long chains by reducing the breakage of chains due to reduced cell motility and increased PSL production.
As it is widely known that PSL plays a very important role in the biofilms of P. aeruginosa, then both aforementioned aspects would have an effect on the biofilm development at different stages. At the attachment stage, reduced swimming motility, which can be caused by the higher level of c-di-GMP, can contribute to the reduced surface attachment observed on microtiter surfaces. Long chains of cells may also contribute to the reduced swimming motility. However, under our experimental setup, we cannot measure the swimming of long chains in liquid cultures when they move toward to the air-liquid interface. After cells attach on a surface, both the formation of long chains and the increased level of c-di-GMP in two daughter cells will help both daughter cells to stay on the surface, and result in a more hierarchical bacterial visit distribution (Figure 3), which then lead to an earlier formation of microcolony. As cells continue to grow and proliferate, such differences in cell behavior between ΔpslG and PAO1 finally result in matured biofilms with different structures as shown in pellicles formed at the air-liquid interface, where pellicles of ΔpslG are rougher than those of PAO1 although their biomasses are similar. We note that the aforementioned effect of pslG on the biofilm development is also dependent on the synthesis of PSL.
Thus, by revealing the important roles of PslG on bacterial physiology and the biofilm development, our results further expand our understandings of PslG functions. To the best of our knowledge, the ability of a glycosyl hydrolase to affect bacterial c-di-GMP levels has not been reported until this work. It would be also very interesting to see in the future whether other glycosyl hydrolases in different bacterial species can have similar functions.
Similarly, glycosyl hydrolase has also been found to be encoded in gene clusters that are involved in the synthesis of other exopolysaccharides. For example, algL, a gene within the alginate synthesis operon of P. aeruginosa, also encodes a lyase to degrade alginate (Franklin et al., 2011). An early work showed that ΔalgL in a CF isolate mucoid strain FRD1 results in cell death, due to accumulation of alginate in the periplasm (Jain and Ohman, 2005). Interestingly, a later work showed that ΔalgL in a PAO1-derived mucoid strain PDO300 does not cause cell death, yet increases alginate production (Wang et al., 2016). A recent study further revealed that the function of AlgL in PAO1 is to clear the periplasmic space of accumulated alginate during polymer biosynthesis, while lack of AlgL might inhibit bacterial growth under certain conditions (Gheorghita et al., 2022). pelA is another example of encoding a hydrolase in the PEL synthesis operon (Franklin et al., 2011), which has been shown to inhibit biofilm formation as that of PslG (Baker et al., 2016). It would be interesting to investigate whether algL or pelA affects bacterial intracellular c-di-GMP levels.
In summary, in this study, we have provided a comprehensive analysis on the effect of pslG on the biofilm development of P. aeruginosa. Our results indicate that although pslG is not essential for synthesis of PSL, it plays an important role in regulating the proper functions of PSL, and loss of pslG or its hydrolytic activity results in malfunction of PSL, which then cause changes in both morphology and surface behavior of bacterial cells through PSL-mediated interactions. This work shed light on better understanding the role of PslG and would be helpful in developing new ways for biofilm control through pslG-based PSL regulation.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Pseudomonas aeruginosa) | PAO1 | Holloway, 1955 | Prototroph. | |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG | Wu et al., 2019 | In-frame deletion of pslG. | Ma’s lab, strain No. P539. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔPpsl | Ma et al., 2006 | PSL-negative, the promoter of psl operon deletion mutant. | Previous name is WFPA80. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-psl | Ma et al., 2006 | PSL-inducible strain, the promoter of psl operon is replaced by araC-PBAD. | Previous name is WFPA801 |
| Strain, strain background (Pseudomonas aeruginosa) | ΔPpel | Ma et al., 2012 | PEL-negative, the promoter of pel operon deletion mutant. | Previous name is WFPA830. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-pel | Ma et al., 2012 | PEL-inducible strain, the promoter of pel operon is replaced by araC-PBAD. | Previous name is WFPA831. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-psl ΔpslG | This study | In-frame deletion of pslG in PBAD-psl background. | Ma’s lab, strain No. P977. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG ΔPpel | This study | The promoter of pel operon deletion strain in ΔpslG background. | Ma’s lab, strain No. P1717. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-psl ΔpslG ΔPpel | This study | The promoter of pel operon deletion strain in PBAD-psl ΔpslG background. | Ma’s lab, strain No. P1711. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔalgD | Whitchurch et al., 2002 | Alginate-negative, the algD:: tet deletion mutant of PAO1. | Previous name is WFPA1. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG::pslG | Wu et al., 2019 | pslG was inserted into pslG deletion mutant at chromosome pslG locus. | Ma’s lab, strain No. P963. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG::pslGE165Q, E276Q | Wu et al., 2019 | pslG was replaced by the active sites mutated pslG (E165Q + E276 Q). | Ma’s lab, strain No. P964. |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG attB::PBAD-pslG | Wu et al., 2019 | PBAD-pslG was inserted into pslG deletion mutant at chromosome attB site. | Ma’s lab, strain No. P1716. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-psl ΔpslG::pslG | This study | pslG was inserted into PBAD-pslΔpslG strain at chromosome pslG locus. | Ma’s lab, strain No. P967. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-pslΔpslG::pslGE165Q,E276Q | Wu et al., 2019 | pslG was replaced by the active sites mutated pslG (E165Q, E276Q) in PBAD-psl strain. | Ma’s lab, strain No. P966. |
| Strain, strain background (Pseudomonas aeruginosa) | PBAD-psl ΔpslG attB::PBAD-pslG | This study | PBAD-pslG was inserted into PBAD-psl ΔpslG strain at chromosome attB site. | Ma’s lab, strain No. P1715. |
| Strain, strain background (Pseudomonas aeruginosa) | PAO1/pCdrA::gfp | Rybtke et al., 2012 | PAO1 strain carrying plasmid pCdrA::gfp. AmpR, GmR. | |
| Strain, strain background (Pseudomonas aeruginosa) | PAO1/pHERD20T | Wu et al., 2019 | PAO1 strain carrying plasmid pHERD20T | |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG /pHERD20T | Wu et al., 2019 | ΔpslG strain carrying plasmid pHERD20T | |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG /pG | Yu et al., 2015 | ΔpslG strain carrying plasmid pHERD20T-pslG. AmpR | |
| Strain, strain background (Pseudomonas aeruginosa) | ΔpslG /pGDM | Wu et al., 2019 | ΔpslG strain carrying plasmid pHERD20T-pslGDM, pHERD20T with double active sites mutated pslG(E165Q + E276 Q), AmpR | |
| Recombinant DNA reagent | pCdrA::gfp (plasmid) | Rybtke et al., 2012 | pUCP22Not-RNase III-gfp (ASV)-T0-T1, a cyclic di-GMP level reporter consisting of the cyclic di-GMP-responsive cdrA promoter fused to gfp gene, AmpR, GmR | |
| Recombinant DNA reagent | pHERD20T (plasmid) | Qiu et al., 2008 | pUCP20T Plac replaced with 1.3 kb AflII-EcoRI fragment of araC-PBAD cassette. AmpR | |
| Recombinant DNA reagent | pG(plasmid) | Yu et al., 2015 | pHERD20T-pslG. AmpR | |
| Recombinant DNA reagent | pGDM (plasmid) | Wu et al., 2019 | pHERD20T with double active sites mutated pslG (E165Q + E276 Q), AmpR | |
| Recombinant DNA reagent | pEX18Gm (plasmid) | Hoang et al., 1998 | Allelic exchange vector, GmR | |
| Recombinant DNA reagent | pSW196 (plasmid) | Baynham et al., 2006 | Modified from mini-CTX with pBAD30-based vector, for inserting an arabinose-inducible gene at the neutral attB site. TcR | |
| Recombinant DNA reagent | pFLP2 (plasmid) | Hoang et al., 1998 | Source of Flp recombinase, AmpR | |
| Antibody | anti-ePsl (Rabbit polyclonal) | Byrd et al., 2009 | Exopolysaccharide Psl specific antibody. | IF(1:1667) |
| Other | TRITC-HHA | EY-lab, INC | Fluorescent labeled lectin HHA. | |
| Other | FITC-HHA | EY-lab, INC | Fluorescent labeled lectin HHA. | |
| Other | Syto9 | Invitrogen, Molecular probes | Green-fluorescent nucleic acid stain. | |
| Other | FM4-64 | Invitrogen,Molecular probes | Lipophilic Styryl Dye. |
Bacterial strains and growth conditions
Request a detailed protocolAll P. aeruginosa stains used in study were listed in the key resources table. P. aeruginosa stains were grown at 37 °C in LB without sodium chloride (LBNS) or Jensen’s, a chemically defined media (Jensen et al., 1980). Biofilms of P. aeruginosa were grown in Jensen’s medium at 30 °C. L-arabinose (Sigma) was used as inducer for genes transcribed from PBAD promoter in P. aeruginosa. Antibiotics for P. aeruginosa were added at the following concentrations: gentamicin 30 μg/mL; ampicillin 100 μg/mL; carbenicillin 300 μg/mL. For Pseudomonas selection media, irgasan at 25 μg/mL was used.
The psl-inducible strains PBAD-pslΔpslG was constructed in accordance with PBAD-psl (the promoter of psl operon in PAO1 was replaced by araC-PBAD-psl) (Ma et al., 2006). Briefly, plasmid pMA9 (Ma et al., 2006) was transferred into pslG deletion mutant by conjugation (Wu et al., 2019). All deletion mutants were constructed by the similar in-frame deletion strategy (Ma et al., 2006). For single recombination mutant selection, LBNS plates with 30 μg/mL gentamycin and 25 μg/mL irgasan were used; for double recombination mutant selection, LBNS plates containing 10% sucrose were used. Gene insertion at the attB site of P. aeruginosa was performed as described previously (Hoang et al., 2000).
Bacterial attachment on microtiter dish
Request a detailed protocolThe assay was done as described previously with modifications (Ma et al., 2006; O’Toole, 2011). Overnight culture was 1/100 diluted into Jensen’s media (with or without arabinose) and incubated at 37 °C with shaking until the OD600 reached 0.5. The 100 μL of such culture was inoculated into 96-well PVC microtiter dish (BD Falcon), and incubated at 30 °C for 30 min. Then the planktonic and loosely adherent bacteria cells were washed off by rinsing the plate in water. The remaining surface-attached cells were stained by 0.1% crystal violet, solubilized in 30% acetic acid, and finally the value of OD560 was measured.
Motility assay
Request a detailed protocolSwimming motility assay was performed as preciously described (Zhao et al., 2018). Briefly, strains were grown overnight on LBNS plates. Single colony was stab-inoculated with a sterile toothpick on the surface of Jensen’s plates (0.3% BD Bacto Agar). Plates were incubated upright at 37 °C overnight. Swimming zones were measured accordingly. For the twitching motility assay, the strains were stab inoculated with a sterile toothpick into the bottom of thin Jensen’s plates, cultivated at 30 °C for 2–3 days, and the twitching motility zones were visualized at the agar plate interface (Wang et al., 2013).
PSL dot-blotting
Request a detailed protocolStrains were incubated in Jensen’s medium with shaking at 30 °C for 24 hr. Cells of an OD600 of 4 were collected by centrifugation to extract crude bacterial surface-bound exopolysaccharides. Pellet was re-suspended in 100 μL of 0.5 M EDTA, and boiled at 100 °C for 5 min. After centrifugation at 13,000 g for 10 min, the supernatant fraction was treated with 0.5 mg/mL proteinase K at 60 °C for 1 hr and proteinase K was then inactivated at 80 °C for 30 min. PSL immunoblotting was performed as previously described using PSL antibody (Byrd et al., 2009). ImageJ software was used to quantify the immunoblot data. The protein concentration of each sample culture was measured by a BCA protein assay kit (Thermo) to ensure the same amount of cell lysate was used in each experiment.
Flow cell assembly, sterilization, and washing of the system
Request a detailed protocolFlow cells made of polycarbonate were purchased from the Department of Systems Biology, Technical University of Denmark. Each flow cell has three identical rectangle channels (40 × 4 × 1 mm3) and was assembled by attaching a cover glass as substratum as previously described (Sternberg and Tolker-Nielsen, 2006). The assembled flow cell was connected to a syringe through a 0.22 μm filter (Millipore) using silicon tubing. Then the whole system was sterilized overnight with 3% H2O2 at 3 mL/hr using a syringe pump (Harvard Apparatus). After sterilization, autoclaved, deionized water was used to wash the whole system overnight. Before inoculation of bacteria into the flow cell, the system was flushed for 5 min at a flow rate of 30 mL/hr by Jensen’s medium using a syringe pump (Harvard Apparatus). Then the medium flow was stopped and 1 mL of a diluted bacteria culture (OD600 ~ 0.01) were injected directly into the channel of the flow cell using a 1 mL syringe equipped with a needle. A 5-min incubation period was allowed after inoculation to let cells attaching to the surface, which was then followed by a medium flow with a large flow rate of 30 mL/hr for 5 min to wash out floating cells. After that the flow rate was set to 3 mL/hr, and image recording was started. In this work, the flow cell experiments were conducted at 30 ℃.
Biofilms and image acquisition
Request a detailed protocolPellicles (air-liquid interface biofilms) were grown in glass chambers (Chambered # 1.5 German Coverglass System, Nunc) with a glass coverslip at the bottom of each chamber as described previously (Wang et al., 2013). 1/100 dilution of a saturated (overnight) culture in Jensen’s media for P. aeruginosa was inoculated into the chamber, and incubated at 30 °C for 24 hr. The PSL was stained with lectin TRITC-HHA (EY lab, INC) at 100 μg/mL for 2 hr in the dark. Then bacteria were strained with SYTO9 (5 μM final concentration, Molecular Probes, Invitrogen) for 15 min. Fluorescent images were obtained using a FV1000 CLSM (Olympus, Japan). The excitation/emission parameters for TRITC-HHA and SYTO9 were 554 nm/570 nm and 480 nm/500 nm, respectively. CLSM-captured images were analyzed using COMSTAT software (Heydorn et al., 2000).
For flow cell experiments, the flow was stopped before staining. PSL was stained with lectin FITC-HHA (EY lab, INC) at 100 μg/mL for 20 min in the dark, and then the flow was running for a short time to flush out the non-binding dye. Subsequently, bacteria were tagged by Gfp or stained by cell membrane stain FM4-64 (10 μM final concentration, Molecular Probes) for 2 min in the dark (without flow). Next, the flow was resumed to flush out the dye and ready for examination under microscope. Images were captured using an EMCCD camera (Andor iXon) on a Leica DMi8 microscope equipped with Zero Drift autofocus system. The image size is 66.5 μm × 66.5 μm (1,024 × 1,024 pixels). The images were recorded with a 100 × oil objective (plus 2 × magnifier).
Detection of PSL signaling function on stimulating bacterial intracellular C-di-GMP
Request a detailed protocolThe c-di-GMP levels were determined using pCdrA::gfp as a reporter as described previously (Rybtke et al., 2012). The growth curve and green fluorescent signal of PAO1 /pCdrA::gfp were measured via recording the OD600 values and the corresponding GFP fluorescence (Ex/Em 488/520) by a Synergy H4 hybrid reader (BioTek). The promoter activity was calibrated as the relative fluorescence divided by the OD600. In co-culture system, PAO1 harboring plasmid pCdrA::gfp was the reporter strain to indicate the level of intracellular c-di-GMP. PAO1, PBAD-psl, ΔpslG, and ΔpslG::pslGE165Q,E276Q strains were PSL donor strains, respectively. In PslG treatment assay, PslG was added into cultures when inoculating PAO1 /pCdrA::gfp, the OD600 values and the corresponding GFP fluorescence were tracked for 24 hr.
Single-cell tracking image analysis
Request a detailed protocolImages were processed and analyzed in the same way as described in reference (Zhang et al., 2018). Simply, 16-bit greyscale images were first converted to binary images for the detection of bacteria with a standard image processing algorithm. Geometry information of cells such as center position, size and aspect ratio etc. were then collected. Bacterial trajectories were obtained by connecting cell positions in all frames of a time series, from which bacterial motion can be measured and analyzed. Specifically, the twitching speed of each tracked cell at frame n was calculated by the displacement of the cell between nth and (n + 1)th frames divided by the corresponding time interval.
For quantitatively comparing the fluorescence intensity of cells containing pCdrA::gfp reporter between mother cell and daughter cells, first the fluorescence intensity of each cell I was measured by the averaged fluorescence intensity value within the area enclosed by the cell envelope. The mother cell was measured when it irreversibly attached to the surface (typically 40~50 min before the division completed), and the daughter cells were measured right after the division (i.e. the two daughter cells are completely separated. In practice, the daughter cells were measured within 10 min right after the division completed due to the 10-min time interval for the fluorescent image recording). Then the ratio of fluorescence intensity between each daughter cell (Idau) and its mother cell (Imot) was calculated, γ = Idau/Imot. The relative standard deviation was estimated by . Here, refers to standard deviation of γ. Similar for and . We define a daughter cell to be fluorescent brighter than its mother cell if , here is the averaged value of the relative standard deviation for all analyzed division events.
A cluster is an aggregation of multiple cells. We used a minimum distance criterion to judge whether a cell belonged to a cluster or not. If the minimum distance between any point of the scrutinized cell body and any point of any cell body of the cluster, is smaller than 0.5 μm (i.e. about one width of a bacterial cell), then the scrutinized cell is considered to belong to the cluster, otherwise not.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting file. Source Data files have been provided for Figure 1,2, 3,4,6, and 7.
References
-
Flagella but not type IV pili are involved in the initial adhesion of Pseudomonas aeruginosa PAO1 to hydrophobic or superhydrophobic surfacesColloids and Surfaces. B, Biointerfaces 131:59–66.https://doi.org/10.1016/j.colsurfb.2015.04.036
-
Asymmetrical distribution of the second messenger c-di-GMP upon bacterial cell divisionScience (New York, N.Y.) 328:1295–1297.https://doi.org/10.1126/science.1188658
-
Flagella and pili-mediated near-surface single-cell motility mechanisms in P. aeruginosaBiophysical Journal 100:1608–1616.https://doi.org/10.1016/j.bpj.2011.02.020
-
Microbial biofilmsAnnual Review of Microbiology 49:711–745.https://doi.org/10.1146/annurev.mi.49.100195.003431
-
The Pseudomonas aeruginosa homeostasis enzyme AlgL clears the periplasmic space of accumulated alginate during polymer biosynthesisThe Journal of Biological Chemistry 298:101560.https://doi.org/10.1016/j.jbc.2021.101560
-
Biofilms 2009: new perspectives at the heart of surface-associated microbial communitiesJournal of Bacteriology 192:2941–2949.https://doi.org/10.1128/JB.00332-10
-
Quantification of biofilm structures by the novel computer program COMSTATMicrobiology (Reading, England) 146 (Pt 10):2395–2407.https://doi.org/10.1099/00221287-146-10-2395
-
Genetic recombination in Pseudomonas aeruginosaJournal of General Microbiology 13:572–581.https://doi.org/10.1099/00221287-13-3-572
-
Role of an alginate lyase for alginate transport in mucoid Pseudomonas aeruginosaInfection and Immunity 73:6429–6436.https://doi.org/10.1128/IAI.73.10.6429-6436.2005
-
Nutritional factors controlling exocellular protease production by Pseudomonas aeruginosaJournal of Bacteriology 144:844–847.https://doi.org/10.1128/jb.144.2.844-847.1980
-
Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunistMicrobes and Infection 2:1051–1060.https://doi.org/10.1016/s1286-4579(00)01259-4
-
Microtiter dish biofilm formation assayJournal of Visualized Experiments 30:2437.https://doi.org/10.3791/2437
-
PBAD-based shuttle vectors for functional analysis of toxic and highly regulated genes in Pseudomonas and Burkholderia spp and other bacteriaApplied and Environmental Microbiology 74:7422–7426.https://doi.org/10.1128/AEM.01369-08
-
Cyclic di-GMP: the first 25 years of a universal bacterial second messengerMicrobiology and Molecular Biology Reviews 77:1–52.https://doi.org/10.1128/MMBR.00043-12
-
Fluorescence-based reporter for gauging cyclic di-GMP levels in Pseudomonas aeruginosaApplied and Environmental Microbiology 78:5060–5069.https://doi.org/10.1128/AEM.00414-12
-
Growing and analyzing biofilms in flow cellsCurrent Protocols in Microbiology Chapter 1:Unit 1B.2.https://doi.org/10.1002/9780471729259.mc01b02s00
-
Antibiotic resistance of bacteria in biofilmsLancet (London, England) 358:135–138.https://doi.org/10.1016/s0140-6736(01)05321-1
-
Biofilms as complex differentiated communitiesAnnual Review of Microbiology 56:187–209.https://doi.org/10.1146/annurev.micro.56.012302.160705
-
The extracellular matrix protects Pseudomonas aeruginosa biofilms by limiting the penetration of tobramycinEnvironmental Microbiology 15:2865–2878.https://doi.org/10.1111/1462-2920.12155
-
Effects of PslG on the Surface Movement of Pseudomonas aeruginosaApplied and Environmental Microbiology 84:e00219-18.https://doi.org/10.1128/AEM.00219-18
Article and author information
Author details
Funding
National Key Research and Development Program of China (2018YFA0902102)
- Kun Zhao
National Natural Science Foundation of China (91951204)
- Luyan Ma
National Natural Science Foundation of China (32070033)
- Di Wang
National Natural Science Foundation of China (21621004)
- Luyan Ma
National Key Research and Development Program of China (2019YFA0905501)
- Di Wang
National Key Research and Development Program of China (2019YFC804104)
- Luyan Ma
National Key Research and Development Program of China (2021YFA0909500)
- Luyan Ma
PetroChina Company Limited
- Huijun Wu
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work is supported by the National Key R&D Program of China (2018YFA0902102, 2021YFA0909500, 2019YFC804104, and 2019YFA0905501), the National Natural Science Foundation of China (91951204, 21621004, 32070033), and Research on basic science and technology of the strategic reserve fund projects of PetroChina Company Limited (No. 2020D-5008). The funders had no role in the study design, data collection and interpretation, or the decision to submit the work for publication.
Copyright
© 2022, Zhang et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,424
- views
-
- 300
- downloads
-
- 13
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 13
- citations for umbrella DOI https://doi.org/10.7554/eLife.72778
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Intracellular glycosyl hydrolase PslG shapes bacterial cell fate, signaling, and the biofilm development of Pseudomonas aeruginosa
eLife 11:e72778.
https://doi.org/10.7554/eLife.72778




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}