Distinct architectural requirements for the parS centromeric sequence of the pSM19035 plasmid partition machinery
- Laboratory of Molecular Biology, National Institute of Diabetes and Digestive and Kidney Diseases, National Institutes of Health, United States
- Departamento de Biotecnología Microbiana, Centro Nacional de Biotecnología, Consejo Superior de Investigaciones Científicas, Spain
Abstract
Three-component ParABS partition systems ensure stable inheritance of many bacterial chromosomes and low-copy-number plasmids. ParA localizes to the nucleoid through its ATP-dependent nonspecific DNA-binding activity, whereas centromere-like parS-DNA and ParB form partition complexes that activate ParA-ATPase to drive the system dynamics. The essential parS sequence arrangements vary among ParABS systems, reflecting the architectural diversity of their partition complexes. Here, we focus on the pSM19035 plasmid partition system that uses a ParBpSM of the ribbon-helix-helix (RHH) family. We show that parSpSM with four or more contiguous ParBpSM-binding sequence repeats is required to assemble a stable ParApSM-ParBpSM complex and efficiently activate the ParApSM-ATPase, stimulating complex disassembly. Disruption of the contiguity of the parSpSM sequence array destabilizes the ParApSM-ParBpSM complex and prevents efficient ATPase activation. Our findings reveal the unique architecture of the pSM19035 partition complex and how it interacts with nucleoid-bound ParApSM-ATP.
Editor's evaluation
The work by Volante et al. studied a plasmid partition system, in which the authors discovered that four or more contiguous ParS sequence repeats are required to assemble a stable partitioning ParAB complex and activate the ParA ATPase. The work reveals a plasmid partitioning mechanism in which the mechanic property of DNA and its interaction with the partition complex may drive the directional movement of the plasmid.
https://doi.org/10.7554/eLife.79480.sa0Introduction
Faithful chromosome segregation is essential for the proliferation of bacterial cells, and low-copy-number plasmids also need a robust partition mechanism for their stable inheritance. However, prokaryotes do not possess the mitotic machinery of eukaryotes: instead, alternative active DNA partition systems have evolved, among which ParABS systems (also called class I partition systems) are the most widespread. Basic ParABS systems consist of three components, a partition ATPase (ParA), a ‘centromere’-binding protein (ParB), and a cis-acting centromere-like DNA site (parS).
ATP-activated ParA dimers bind nonspecific DNA (nsDNA) and localize to the nucleoid in vivo (Ebersbach and Gerdes, 2001; Ebersbach and Gerdes, 2004; Pratto et al., 2008; Ringgaard et al., 2009). The parS sites, often composed of multiple tandem repeats of binding consensus sequences for ParBs, demark the DNA-cargos that are translocated and positioned into the two halves of the cell before cell division by recruiting ParB molecules to assemble partition complexes (PCs). ParB proteins fall into two structurally unrelated groups, dimeric helix-turn-helix (HTH), and dimeric ribbon-helix-helix (RHH) DNA-binding proteins. HTH-ParBs have been shown to bind not only site-specifically to their cognate parS sequences but also to spread many kilobase pairs into the DNA neighboring the parS sites (Murray et al., 2006; Graham et al., 2014; Soh et al., 2019; Rodionov et al., 1999; Lynch and Wang, 1995; Breier and Grossman, 2007; Jalal et al., 2020; Osorio-Valeriano et al., 2019; Sanchez et al., 2015). Therefore, they form large PCs containing many ParB molecules bound to condensed DNA around parS, and ParB spreading activity is essential for their partition function (Rodionov et al., 1999; Debaugny et al., 2018). In contrast, RHH-ParB does not spread beyond parS site judged by the absence of ParB-mediated silencing of parS-proximal gene expression (J. C. Alonso, unpublished observation), unlike HTH-ParBs (Lynch and Wang, 1995; Rodionov et al., 1999), but like HTH-ParBs, they interact with their cognate ParA proteins via their N-terminus (Radnedge et al., 1998; Figge et al., 2003; Barillà et al., 2007). RHH-ParB proteins also control the expression of the proteins involved in the partition system and plasmid copy number control by binding parS sites, which overlaps promoters of their genes (de la Hoz et al., 2000).
ParA–ParB interaction leads to activation of the ParA-ATPase, which is most efficient in the presence of nsDNA and parS DNA (Ah-Seng et al., 2009; Chu et al., 2019; Pratto et al., 2008; Taylor et al., 2021) and leads to dissociation of ParA from nsDNA. Interaction dynamics between the nucleoid-bound ParA and ParB in the PC prior to ATP hydrolysis and ParA dissociation determine the dynamics of the PC relative to the nucleoid. The common results of most systems in vivo appear to be the establishment of equidistant distribution of two or more PCs along the nucleoid(s) so that at cell division each daughter cell inherits at least one copy of the plasmid DNA (Sengupta et al., 2010; Ringgaard et al., 2009; Lioy et al., 2015; McLeod et al., 2017).
With biochemical findings and observations from live-cell imaging approaches accumulating in the field, combined with experiments using reconstituted cell-free reaction systems, a diffusion-ratchet mechanism of the ParABS partition was proposed. Here, the driving force for the DNA-cargo motion is generated by a propagating nucleoid-bound ParA distribution gradient (Vecchiarelli et al., 2010; Hwang et al., 2013; Vecchiarelli et al., 2013; Vecchiarelli et al., 2014; Sugawara and Kaneko, 2011). Additional models related to the diffusion-ratchet mechanism have also been proposed based on high-resolution imaging observations (Lim et al., 2014; Le Gall et al., 2016). While findings supporting diffusion-ratchet-type models for ParABS systems accumulate, many molecular details required to put the model on quantitatively solid ground are lacking. Large number of HTH-ParB dimers load onto a PC by ParB ‘spreading’ to facilitate partitioning. Resulting high concentration of ParB at the PC assures near saturation ParB-binding to the local nucleoid-bound ParA molecules, leading to efficient sensing of the local ParA distribution gradient by the PC. Combined with relatively short lifetime of the ParA/ParB-mediated cargo-nucleoid bridges and sufficient ParA-ATP reloading rate to the nucleoid would maintain enough cargo-nucleoid bridges needed to significantly suppress thermal diffusion of the cargo without completely blocking the motion of the cargo. Proper balance among these parameters is needed for efficient plasmid partition by diffusion-ratchet mechanism. Insufficient free diffusion suppression results in random diffusion of the cargo, and oversuppression by too many or too stable bridges blocks the cargo motion altogether (Hu et al., 2015; Hu et al., 2017; Taylor et al., 2021). However, it is unclear how a system with RHH-ParBs, which cannot spread ParB beyond the parS sites and thus can load only limited number of ParB dimers to a PC, can fit into the diffusion-ratchet paradigm. Nevertheless, live-cell imaging studies of systems that use RHH-ParB proteins showed an oscillating dynamic ParA distribution pattern on the nucleoid and PC chasing the receding tail of the ParA distribution (Ringgaard et al., 2009; Lioy et al., 2015; McLeod et al., 2017) similar to observations with the F-plasmid partition system and other related systems involving HTH-ParB proteins (Hatano and Niki, 2010; Schofield et al., 2010) for which there is accumulating evidence supporting the diffusion-ratchet mechanism. Therefore, we suspect systems with RHH-ParBs also operate via a diffusion-ratchet-type mechanism. A better understanding of how the PCs are organized for this group and a quantitative understanding of the interaction dynamics of these PCs with nucleoid-bound ParA molecules are critical for advancing our mechanistic understanding of these systems.
In this study, we focused on the pSM19035 partition system of Streptococcus pyogenes. This plasmid harbors a ParABS system composed of ParApSM (also called Delta), an RHH ParBpSM (also called Omega), and six parSpSM sites, each comprising 7–10 consecutive non-palindromic 7-bp-long sequence repeats (5′-WATCACW-3′, symbolized by →) that overlap the promoter regions of copS, δ (coding ParApSM) and ω (coding ParBpSM) genes. Each ParBpSM dimer binds one copy of the 7 bp parSpSM consensus sequence. However, the affinity for a single repeat is low, while two dimers bind with high affinity to two direct (→→) or inverted (→←) repeats forming dimers of dimers (de la Hoz et al., 2004; Weihofen et al., 2006; Welfle et al., 2005). Within the structures of these ParBpSM-parSpSM complexes, DNA does not show significant curvature, and although the protein dimers bound to each DNA sequence repeat slightly deviates from dyad symmetry due to the bound DNA sequence asymmetry, full-size parSpSM-ParBpSM complexes could be modeled as nearly straight DNA wrapped by left-handed spiral arrangement of ParBpSM dimers (Weihofen et al., 2006). Atomic force microscopy images of the complex involving seven parSpSM consensus sequence repeats supported its straight arrangement and the lack of spreading (Pratto et al., 2009). ParApSM, unlike most other ParAs, forms dimers in solution in the absence of ATP (Pratto et al., 2008). Like other ParAs, it also undergoes a conformational transition upon binding ATP that increases its affinity for nsDNA (Soberón et al., 2011; Pratto et al., 2008). In the ATP-bound form, ParApSM has been shown to bind nsDNA forming limited size patches containing several ParApSM dimers at random location, instead of individual dimers independently distributed on the nsDNA (Pratto et al., 2009). In the presence of parSpSM DNA and ParBpSM, several parSpSM-ParBpSM mini-filaments and ParApSM-nsDNA patches appeared to bind together to form large protein-DNA complexes bridging multiple DNA molecules (Pratto et al., 2008; Pratto et al., 2009; Soberón et al., 2011; Lioy et al., 2015). Interactions among these components fueled by ATP hydrolysis are thought to drive dynamic oscillations of the nucleoid-bound ParApSM in vivo, which resembles those observed for the TP228 ParABS system (Lioy et al., 2015; McLeod et al., 2017).
Despite accumulating information summarized above, how the observed inter-molecular interactions coordinate the in vivo system dynamics resulting in robust plasmid partitioning remains a mystery. To approach this puzzle, here we studied functional requirements of the parSpSM sequence-structure necessary for ParBpSM-mediated activation of the ParApSM ATPase. We found a minimum of four contiguous repeats of the parSpSM heptad consensus sequence without a gap is necessary for full activity. Kinetics of the nsDNA-bound parSpSM-ParBpSM-ParApSM complex formation and disassembly indicated the presence of a complex multistep process involved in ATPase activation.
Results
ParApSM ATPase is synergistically activated by nsDNA, ParBpSM, and parSpSM-DNA
To define the requirements for stimulation of the ParApSM ATPase activity by ParBpSM, the steady-state ParApSM ATP turnover rate was measured with varying concentrations of ParBpSM in the presence or absence of different duplex DNA cofactors. The turnover rate of ParApSM ATPase alone is very low at 37°C (0.9 ± 0.1 ATP/ParA-dimer/h, N = 3; Figure 1A; no DNA). In the presence of a saturating concentration of double-stranded DNA (40 μg/ml pBR322 plasmid DNA plus 23–38 μg/ml double-stranded oligonucleotide with or without parSpSM sequence), to which ATP-ParApSM dimers can bind to support ATPase activation by ParBpSM (see below), no significant rate change was observed (1.0 ± 0.1 h–1, N = 46, Figure 1A, pool of all measurements at [ParBpSM] = 0). Next, effects of the parSpSM-DNA in addition to 40 μg/ml nsDNA and ParBpSM were examined. ParApSM ATP hydrolysis was stimulated up to ~20-fold (kcat = 20.5 ± 2.9 h–1, N = 7) in the presence of ParBpSM and oligonucleotide duplex DNA containing one of the native arrangements containing seven parSpSM heptad-sequence-repeats (7R-parSpSM, →→←→→←←) (Figure 1A). The stimulation approached saturation around 2 μM ParBpSM at varying ParApSM concentrations (Figure 1—figure supplement 1A). In contrast, when the parSpSM DNA fragment was replaced with one having a scrambled sequence, ParBpSM stimulated ParApSM ATPase activity only to 2.4 ± 1.1 h–1 even in the presence of 8 μM ParBpSM that should have allowed non-parSpSM DNA binding (N = 3*) (Figure 1A, scram). Similarly, a low level of ParApSM ATPase stimulation was observed with ParBpSM1-27 peptide, which lacked the DNA-binding and dimerization domains (Figure 1—figure supplement 1B). These results demonstrated that full ParApSM-ATPase stimulation by ParBpSM requires specific parSpSM interactions. Even the low parSpSM-independent ATPase stimulation was not detected in the absence of DNA (Figure 1A, no DNA). Similarly, the low-level ATPase stimulation by non-parSpSM-binding ParBpSM1-27 peptide was not detected without DNA (Figure 1—figure supplement 1B). We conclude ParApSM-nsDNA binding is required for the ATPase activation by ParBpSM, as reported for ParAF ATPase activation by ParBF (Taylor et al., 2021). Consistently, in the absence of nsDNA, excess ParBpSM relative to the concentration of 7R-parSpSM in the reaction inhibited ATPase stimulation, presumably competing with ParApSM for parSpSM DNA binding (Figure 1—figure supplement 1C).
Figure 1 with 3 supplements see all

ParApSM ATPase activation by ParBpSM, parSpSM, and nsDNA.
(A) Efficient activation of ParApSM ATPase by ParBpSM exhibits critical dependency on parSpSM heptad-sequence-repeat number. The ATPase reactions contained ParApSM (2 μM), pBR322 DNA (60 μM in bp, unless noted otherwise), ParBpSM (at the concentration indicated), and parSpSM duplex substrates (4.4 μM of the 7 bp consensus sequence repeats) or equal amount of a scrambled sequence duplex (scram). (B) Comparison of parSpSM containing four contiguous repeats with different heptad orientation arrangements. (C) parSpSM containing different heptad arrangements of three contiguous repeats and two contiguous triple heptad repeats with a gap fails to fully activate the ParApSM ATPase. (D) 7R-parSpSM concentration dependence of ParApSM ATPase activity. Reaction mixtures contained ParApSM (2 μM), ParBpSM (2 μM), pBR322 DNA (60 μM in bp), and increasing concentration of 7R-parSpSM duplex (duplex fragment concentration shown, the ratio of parSpSM heptad repeat sequence to ParBpSM dimers are indicated on top). (E) The numbers and arrangement of the heptad repeats of the parSpSM fragments used in this study (also see Figure 1—figure supplement 2). Data points represent means and standard errors of mean (SEM) of N repeated experiments (N* represents repeats for majority of data points, see Figure 1—source data 1 for details). Curves were fitted after subtraction of the background in the absence of ParApSM to an equation v-v0 = (vmax[B]n)/(KAn + [B]n). The maximum turnover rates (vmax) cited in the text represent mean ± 95% confidence intervals (symmetrized to the larger estimated errors from the mean for simplicity).
Formation of short clusters of ParApSM molecules bound to nsDNA might be taken as a hint for cytoskeletal filament treadmilling models for this class of partition systems, rather than diffusion-ratchet model. However, the low maximum ATP turnover rate (~0.3 /min) observed would be too slow for a treadmilling filament model. Combined with low abundance of ParApSM molecules inside a cell insufficient to form axial protein filaments, the treadmilling model is highly unlikely to fit this system.
Efficient ParApSM-ATPase stimulation requires ParBpSM bound to parSpSM-DNA with at least four contiguous heptad-sequence-repeats
We asked whether entire 7R-parSpSM is required for the full stimulation of ParApSM ATPase by ParBpSM. A series of deletions of parSpSM heptad-sequence-repeats were made and their ATPase stimulation activities were tested (Figure 1E). Efficient ATPase stimulation was observed when 6R (→←→→←←), 5R (→→→←←), or 4R (→→←←) was added along ParBpSM (Figure 1A). No significant difference was observed among 5R, 6R, or 7R; both half-saturation concentrations and the apparent kcat were comparable (Figure 1A). ParBpSM-4R-parSpSM also induced similar rates of ParApSM ATP hydrolysis, but a significantly higher concentration was required for full stimulation (Figure 1A). Different heptad orientation arrangements of 4R-parSpSM (→→←→ and →→→→) stimulated ParApSM ATPase to a similar extent (Figure 1B). In contrast, 3R- (→→←), 2R- (→←), and 1R- (→) parSpSM were poor cofactors for ParBpSM-dependent ATPase stimulation (apparent kcat = 3.2–3.6 ± 1.3–2.1 h–1, N = 4–7*, Figure 1A), not significantly different from the scrambled sequence DNA. 3R-parSpSM fragments with different repeat arrangements behaved similarly to each other (Figure 1C). The ATPase stimulation by parSpSM DNA with 1–3 parSpSM repeats also appeared to saturate at around 2 μM ParBpSM, indicating that the affinity of the nsDNA-bound ParApSM to ParBpSM in the presence of truncated parSpSM was not limiting above ~2 μM ParBpSM. Previously it has been shown that ParBpSM bound 3R, 4R DNA fragments of different sequence orientation combinations, or a 10R DNA fragment with roughly similar affinity that was >50-fold higher than nsDNA or 1R DNA (de la Hoz et al., 2004). We confirmed that ParBpSM binding was similarly strong for 4R and 3R duplex DNA (KD ~ 17 nM) and weak for nsDNA (KD ~ 1 μM, Figure 1—figure supplement 3). Therefore, the affinity of ParBpSM for the 3R-parSpSM DNA is not limiting the ParApSM ATPase stimulation. In the above experiment, the length of parSpSM DNA fragments decreased as the number of parSpSM repeats decreased (Figure 1E, Figure 1—figure supplement 2). We tested longer 3R-parSpSM DNA fragments containing an additional non-parSpSM heptamer sequence (non-consensus, nc) (3R-1nc) and confirmed that parSpSM-DNA fragment size was not a significant factor for the ATPase stimulation efficiency (Figure 1C).
These results suggested that ParApSM ATP turnover is fine-tuned by the structural arrangement of the ParBpSM and parSpSM within the PC. ParBpSM assembles as a left-handed spiral to wrap parSpSM DNA without significantly distorting the DNA backbone geometry (Weihofen et al., 2006), implying that after approximately four repeats, ParBpSM dimers would make a full turn around parSpSM, positioning themselves on the same face of the nucleoprotein filament. Thus, the position of the fourth repeats relative to that of the first repeat might be functionally important. To test this possibility, we examined the ParApSM ATPase stimulation efficiency of parSpSM DNA fragments containing two copies of 3R sequences separated by 7 bp or 14 bp of non-consensus sequence (3R-1nc-3R and 3R-2nc-3R) (Figure 1E). The 3R-1nc-3R-parSpSM fragments showed only slightly higher stimulation (kcat = 8.3 ± 3.5 h–1, N = 8*) compared to the single 3R-parSpSM fragment, and the 3R-2nc-3R was functionally indistinguishable from the single 3R-parSpSM fragment (Figure 1C). We conclude that disrupted 6R-parSpSM fragments are unable to recover the full stimulation of ParApSM ATPase activity. These findings indicate that the ParBpSM-ParApSM interactions differ when ParBpSM dimers are bound to ≥4 contiguous heptad repeats compared to ParBpSM dimers unbound to the repeats or bound to fewer or a disrupted array of heptad repeats.
Substoichiometric concentration of parSpSM relative to ParBpSM is sufficient to fully activate ParApSM ATPase
We originally assumed that for efficient ParApSM ATPase activation by the ParBpSM-parSpSM complex all ParBpSM dimers need to be bound to parSpSM and accordingly maintained stoichiometrically excess parSpSM-sequence concentration relative to ParBpSM-dimers in the reaction. To test this assumption, we next changed the concentration of the 7R-parSpSM DNA fragment while keeping the concentrations of ParApSM and ParBpSM both at 2 μM. Contrary to our expectation, significantly lower heptad consensus sequence concentrations of the 7R-parSpSM DNA fragment compared to ParBpSM dimers (B2) were sufficient for ATPase activation (Figure 1D). If the original assumption was correct, 2 μM ParBpSM should have required 1 μM consensus sequence repeats (143 nM 7R-parSpSM) for full activation. According to the results of Figure 1A, at high 7R-parSpSM concentration, half-activation by ParBpSM required ~650 nM ParBpSM, which would have required ~325 nM parSpSM consensus sequence if full binding was needed. Observed half-saturation parSpSM repeat concentration was ~130 nM (~19 nM 7R-parSpSM) or less. Thus, assuming most of the ParBpSM molecules in our preparations are active, it appears that at any given time, less than half of the ParBpSM dimers need to be in complex with 7R-parSpSM to exert full ATPase activation.
Stimulation of ParApSM release from nsDNA-carpet by ParBpSM requires parSpSM DNA with four or more heptad repeats
Next, we examined how the dissociation of ParApSM-ATP dimers from nsDNA is influenced by ParBpSM in the presence of parSpSM DNA. For these experiments, we used an nsDNA-carpeted two-inlet flow cell observed under TIRF microscopy as described in ‘Materials and methods’ (Vecchiarelli et al., 2013; Figure 2A). We used a ParApSM-GFP fusion protein and a ParBpSM-cys conjugated to Alexa647 to facilitate visualization of protein association-dissociation to/from the nsDNA-carpet. Stimulation of ParApSM-GFP ATPase activity by ParBpSM and 7R-parS was comparable to wild-type ParApSM (Figure 2—figure supplement 1). First, we tested the association/dissociation of individual proteins (bound at 1 μM) to/from the nsDNA-carpet. The affinity of ParBpSM-Alexa647 (1% labeled) to nsDNA was weak. ParBpSM-Alexa647 in the absence of ParApSM accumulated on the DNA-carpet reaching a density of ~3000 ± 500 dimers/μm2 after 15 min of constant flow (5 μl/min) (Figure 2—figure supplement 2A, bottom). When the sample solution was switched to a buffer without protein, most ParBpSM rapidly dissociated with kinetics that can be fitted to a double-exponential function (koff-fast = 5.1 ± 0.7 min–1 [73%], koff-slow = 0.11 ± 0.04 min–1, N = 3; all koffs reported in this study represent apparent pseudo-first order rate constants) (Figure 2—figure supplement 2B, bottom). ParApSM-GFP preincubated with ATP and Mg2+ reached a saturation density on the nsDNA-carpet of 4.25 (±0.06) × 104 dimers/μm2 (N = 4*), whereas in the absence of ATP, less than 1000 dimers/μm2 of ParApSM bound to the nsDNA-carpet (Figure 2—figure supplement 2A, top). Next, 1 μM ParApSM-GFP preincubated with ATP was flowed onto the nsDNA-carpet until 5–10% of saturation density (~4000 dimers/μm2), at which point the flow was switched to buffer containing ATP without ParApSM-GFP. A small fraction of ParApSM-GFP (less than ~5%) dissociated within ~1 min and the remainder dissociated slowly with koff of <0.013 min–1 (N = 3, Figure 2B).
Figure 2 with 3 supplements see all
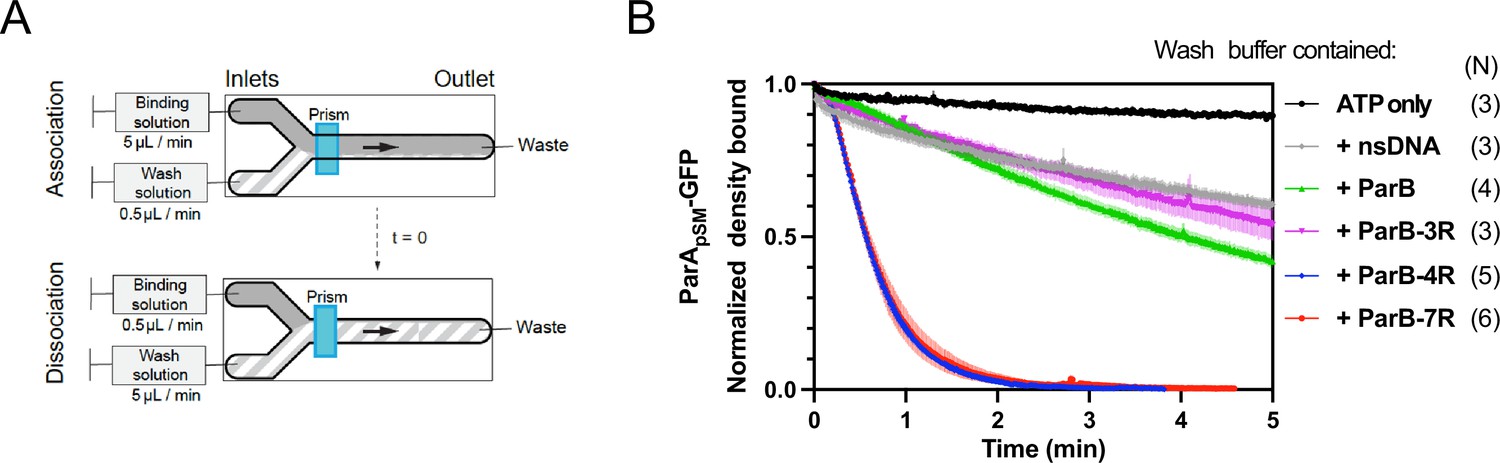
Kinetics of ParApSM disassembly from the nsDNA-carpet.
(A) Schematic of two-inlet flow cell used for visualizing the association and dissociation of fluorescent proteins on nsDNA-carpet. Binding and washing solutions each containing proteins, double-stranded DNA fragments or buffer alone as specified were infused at different flow rates (5 μl/min or 0.5 μl/min) from two inlets on the left into a Y-shaped flow cell. At the middle of the flow channel just downstream of the flow convergence point where the observations are made, content of the faster infusion syringe flows over the observation point. By switching the flow rates of the two syringes, protein binding to the nsDNA-carpet and dissociation during the washing cycle can be recorded. (B) ParBpSM in the presence of parSpSM with at least four contiguous ParBpSM-binding sequence repeats stimulates the ParApSM-GFP dissociation from the nsDNA-carpet. ParApSM-GFP (1 μM) preincubated with 1 mM ATP was infused into the nsDNA-carpeted flow cell at 5 μl/min, while the washing solution containing the specified components was infused at 0.5 μl/min. When the ParApSM-GFP density on the nsDNA-carpet reached 5–10% of the saturation density (t = 0) (~4000 ParApSM dimers/μm2), the flow rates were switched to start the wash with solution containing: buffer alone, 55 bp scramDNA fragment (65 nM in bp), or with ParBpSM (1 μM) without or with different parSpSM fragment (0.5 μM parSpSM heptad repeat sequence). The Y-axis shows the ParApSM-GFP intensity normalized to that at t = 0. Each time point represents the mean with error bar corresponding to the standard errors of mean (SEM) of N repeated experiments.
The addition of competitor nsDNA (71 nM scrambled 55 bp duplex DNA) in the wash buffer slightly increased the koff of majority (~93%) fraction of ParApSM-GFP (koff = 0.075 ± 0.001 min–1, N = 3), perhaps in part by competing with rebinding of ParApSM-GFP-ATP to the nsDNA-carpet (Figure 2B). When 1 μM ParBpSM (unlabeled or 1% Alexa647 labeled) was added to the wash buffer without competitor nsDNA, koff of ParApSM-GFP from nsDNA-carpet reached 0.17 ± 0.01 min–1 (N = 4, Figure 2B). ParApSM-GFP dissociation from the nsDNA-carpet was accelerated when wash solution contained 71 nM 7R-parSpSM DNA (0.5 μM of the consensus sequence repeat) and 1 μM ParBpSM (koff = 1.97 ± 0.09) after a brief period of initial slower dissociation rate (see below for details) (Figure 2B). This was ~12 or ~26-fold higher koff compared to ParBpSM or nsDNA wash, respectively. Similarly, 4R-parSpSM DNA supported accelerated ParApSM-GFP release by ParBpSM, but 3R-parSpSM DNA did not (Figure 2B). The results paralleled those shown in Figure 1A solidifying the notion that a functional parSpSM must carry four copies of ParBpSM dimer-binding sequence repeats.
The dependence of ParApSM dissociation behaviors on the heptad repeat number of the parSpSM fragment combined with 1 μM ParBpSM in the wash solution when the wash was started with ParApSM bound to the nsDNA-carpet at a saturating density was qualitatively similar. However, the dissociation kinetics were significantly slower (Figure 2—figure supplement 3A), likely reflecting the approximately tenfold higher starting ParApSM-GFP density on the nsDNA-carpet, for which the concentration of ParBpSM used likely was limiting.
Accelerated release of ParApSM from DNA-carpet starts only after accumulation of ParBpSM on the carpet in the presence of 7R- or 4R-parSpSM
The accelerated ParApSM-GFP release from the nsDNA-carpet when washed with ParBpSM-4R or –7R parSpSM complexes started after an initial slower rate of ParApSM release (Figure 2B). This time lag prompted us to examine the binding kinetics of ParBpSM to the ParApSM-bound nsDNA-carpet at different concentrations of ParBpSM. Upon switching the flow to the washing solution without parSpSM, ParBpSM bound to the ParApSM-bound nsDNA-carpet to a peak density of ~3000 dimers/μm2 and then quickly decreased to a plateau density of ~2000 dimers/μm2 (Figure 3Ad). The initial overshoot of ParBpSM binding appeared to roughly coincide with the fast dissociation phase of ParApSM. During the subsequent slow dissociation of ParApSM, ParBpSM density on the nsDNA-carpet remained relatively constant with the protein ratio reaching ~1 after several min of washing with 1 μM ParBpSM (Figure 3Ad).
Figure 3
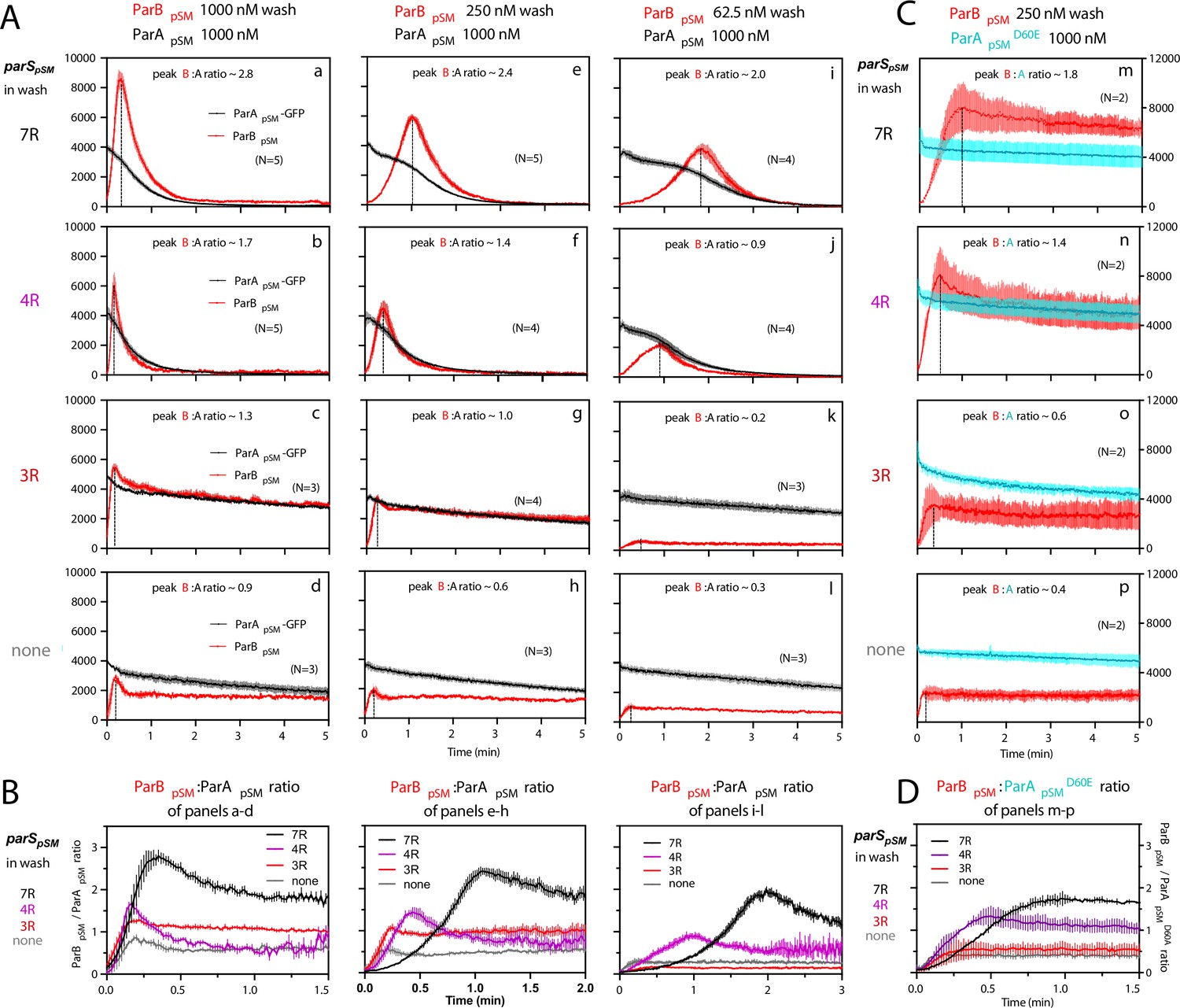
ParBpSM-parSpSM concentration affects kinetics of ATP hydrolysis-dependent ParApSM disassembly from the nsDNA-carpet.
(A) ParApSM-GFP (1 μM) preincubated with 1 mM ATP was infused into the nsDNA-carpeted flow cell at 5 μl/min and when the ParApSM-GFP density on the nsDNA-carpet reached ~10% of the saturation density, the flow over the observation area was switched (t = 0) to wash solution containing ParBpSM ([a–d] 1000 nM, [e–h] 250 nM, or [i–l] 62.5 nM) and the stoichiometric concentration of parSpSM fragments indicated in the ‘parSpSM in wash’ column on the left side of each row of panels. Fluorescence signal was converted to protein density and plotted (dimers/μm2, ParApSM-GFP: black; and ParBpSM-Alexa647: red). Each time point represents mean and standard errors of mean (SEM) (vertical spread) of N repeated experiments. Dashed vertical lines indicate the peak ParBpSM density on the nsDNA-carpet. (B) The time course of the ParBpSM:ParApSM molar ratio (B/A) for the four panels in the columns in (A) with matching ParBpSM concentration with different parSpSM (7R: black; 4R: purple, 3R: red; none: gray) in the wash solution. (C) ATP hydrolysis is required for accelerated ParApSM release from the nsDNA-carpet. The experiments shown in (A) (middle column) were repeated using ParApSMD60E-GFP (1 μM) bound to the nsDNA-carpet and ParBpSM (250 nM) plus parSpSM fragment, (m) 7R, (n) 4R, (o) 3R, or (p) without parSpSM (125 nM heptad concentration) in the wash solution. Fluorescence signal was converted to protein density (dimers/μm2, ParApSMD60E-GFP: cyan; ParBpSM-Alexa647: red) and plotted. Each time point represents mean and SEM (vertical spread) of N experiments. (D) The time course of the ParBpSM:ParApSMD60E molar ratio (B/AD60E) for the four panels of (C) (7R: black; 4R: purple; 3R: red; none: gray). Each time point represents the mean with error bar corresponding to the SEM of N repeated experiments.
In the presence of 1 μM ParBpSM and 7R-parSpSM in the wash solution, ParApSM-GFP release from the nsDNA-carpet took place in two phases (Figure 3Aa). First, ParBpSM accumulated on the ParApSM-bound nsDNA-carpet to a density twofold or more in excess of the carpet-bound ParApSM. During this phase, ParApSM dissociated from the nsDNA-carpet relatively slowly. Next, as the binding of ParBpSM stopped and began to quickly dissociate (koff = 2.4 ± 0.1 min–1, N = 5), ParApSM dissociation also accelerated to a higher rate (koff = 1.8 ± 0.05 min–1, N = 5) (Figure 3Aa). Near-complete dissociation of both proteins from the nsDNA-carpet occurred within a few minutes. Biphasic dissociation kinetics of ParApSM was clearer when the wash solution contained lower concentrations of the 7R-parSpSM-ParBpSM complex, as ParBpSM accumulated on the ParApSM-bound nsDNA-carpet slower, and it took longer to transition to the accelerated dissociation phase (Figure 3Ae,i). This indicates that the initial nsDNA-carpet-bound ParApSM-ParBpSM complex is not activated for ParApSM dissociation from nsDNA, and slow transition of the nsDNA-bound complex is necessary to start the ParApSM disassembly.
We next examined the ability of 4R- and 3R-parSpSM DNA to accelerate ParApSM-GFP dissociation in the presence of ParBpSM. Switching 7R-parSpSM in the wash solution to 4R-parSpSM (1 μM ParBpSM, 125 nM 4R DNA) yielded qualitatively similar two-step dissociation curves (Figure 3Ab). The koff of ParApSM for the accelerated dissociation phase (1.9 ± 0.06 min–1, N = 5) was roughly the same as in the presence of 7R-parSpSM. After the peak of binding, ParBpSM dissociated from the nsDNA-carpet with koff of 3.9 ± 0.2 min–1, N = 5. The dissociation burst of ParApSM-GFP was triggered in the presence of 4R-parSpSM at ParBpSM/ParApSM molar ratio of ~1.7 compared to ~2.8 in the presence of 7R-parSpSM (at 1 μM ParBpSM). This difference likely in part reflects the fact that an individual ParBpSM-parSpSM complex with 4R-parSpSM contains fewer ParBpSM dimers than the 7R-parSpSM complex contains. Also, at a given ParBpSM concentration in the wash solution, washing with 7R-parSpSM took roughly twice as long to reach the peak ParBpSM/ParApSM ratio to start the rapid ParApSM dissociation phase compared to the 4R-parSpSM wash (Figure 3B). We believe this likely reflects, in part, a lower concentration of the larger ParBpSM-7R-parSpSM complex than the complex with 4R-parSpSM. The accelerated phase of ParApSM koff appeared higher in the presence of higher concentrations of 7R- or 4R-parSpSM-ParBpSM complex in the wash solution (Table 1).
Table 1
The accelerated ParApSM dissociation phase of the curves in Figure 3Aa,c,i and b,f,j after the peak ParBpSM/ParApSM ratio points were fitted to single exponential curves to estimate the koff values.
The means with error bars corresponding to the standard errors of mean (SEM) of N repeated experiments in Figure 3 are shown.
| [ParBpSM] (nM) | 7R-parSpSM | 4R-parSpSM |
|---|---|---|
| ParApSM koff (min–1) | ParApSM koff (min–1) | |
| 1000 | 1.808 ± 0.045 | 1.909 ± 0.057 |
| 250 | 1.495 ± 0.024 | 1.462 ± 0.040 |
| 62.5 | 1.184 ± 0.041 | 1.101 ± 0.033 |
Unlike the results above using 7R- or 4R-parSpSM in the wash solution, washing experiments with 3R-parSpSM (1 μM ParBpSM, 166 nM 3R-parSpSM) did not exhibit an accelerated ParApSM-GFP dissociation phase, as in the absence of parSpSM (Figure 3Ac,d). ParBpSM quickly accumulated onto the ParApSM-bound nsDNA-carpet up to a ParBpSM/ParApSM ratio of ~1.3 (1 μM ParBpSM). Thus, ParBpSM appears to be able to associate with nsDNA-bound ParApSM to a stoichiometry, which is unlikely to be the factor preventing stimulation of ParApSM dissociation. Both ParApSM-GFP and ParBpSM dissociated from their peak density on the carpet with double-exponential kinetics roughly in parallel (ParApSM-GFP; koff-fast = 3.8 min–1 ± 0.4 min–1 [~19%], koff-slow = 0.07 ± 0.003 min–1: ParBpSM; koff-fast = 1.4 ± 0.3 min–1 [~24%], koff-slow = 0.05 ± 0.004 min–1, N = 3) (Figure 3Ac).
ATP hydrolysis triggers fast ParApSM dissociation from the nsDNA-carpet
We next asked whether the accelerated dissociation of ParApSM from the nsDNA-carpet in the presence of parSpSM fragments depends on ATP hydrolysis by ParApSM. Among ParApSM homologues, a conserved Asp in the Walker A’ motif is critical for ATP hydrolysis (Pratto et al., 2008; Park et al., 2012). We prepared the ParApSMD60E-GFP fusion protein; it exhibited no significant residual ATPase activity and faster ATP-dependent nsDNA binding compared to the wild-type protein (Figure 2—figure supplement 1, Figure 2—figure supplement 2A, top). The apparent koff of the majority fraction (>80%) of nsDNA-bound ParApSMD60E-GFP washed with ATP-containing buffer (0.015 ± 0.007 min–1, N = 2) was not significantly different from ParApSM-GFP (0.014 ± 0.002 min–1, N = 2) (Figure 2—figure supplement 2B, top). However, when the wash solution contained 250 nM ParBpSM and 36 nM 7R-parSpSM, ParApSMD60E-GFP dissociation from the nsDNA-carpet was only a factor of ~2 faster (Figure 3Ca; koff = 0.029 ± 0.005 min–1, N = 2) than washing with ATP-buffer. As washing started, the ParBpSM associated with the carpet-bound ParApSMD60E-GFP to a density well beyond that of ParApSMD60E-GFP with similar association kinetics as with the carpet-bound ParApSM-GFP (Figure 3Ae). The ParBpSM/ParApSM ratio bound to the nsDNA-carpet at the initial ParBpSM overshoot peak was ~1.75 in the presence of 7R-parSpSM, ~1.3 with 4R-parSpSM, and ~0.6 with 3R-parSpSM (Figure 3D). After the peak density, ParBpSM dissociated from the nsDNA-carpet slowly (koff = 0.036 ± 0.003 min–1, N = 2) (Figure 3Ca). Thus, the accelerated DNA dissociation of ParApSM by the active ParBpSM-parSpSM complex is coupled to ATP hydrolysis, and in the absence of hydrolysis, ParApSMD60E-ATP dimers stayed stably on nsDNA while associated with active parSpSM-ParBpSM complex.
ParBpSM associates with nsDNA-bound ParApSM more stably in the presence of 7R- or 4R-parSpSM prior to ATP hydrolysis
In the experiments of Figures 2 and 3, ParApSM-ATP dimers were bound to the nsDNA-carpet first without ParBpSM, and during the wash phase of the experiments, the carpet-bound ParApSM was constantly exposed to a solution containing ParBpSM and parSpSM. Because ParBpSM and parSpSM dissociating from carpet-bound ParApSM could be exchanged by those in the wash solution, the data did not report the stability of ParApSM-ParBpSM interaction on the nsDNA-carpet. In the next set of experiments, we preincubated an equimolar ratio of ParApSM-GFP (1 μM), ParBpSM (1 μM), and 7R-, 4R-, or 3R-parSpSM (0.5 μM total concentration of the consensus sequence repeats) in the presence of ATP for 30 min at room temperature. The samples were then infused into the nsDNA-carpeted flow cell at 5 μl/min for 15 min. At this point, the incoming components interacting with the nsDNA-carpet would be approaching a steady state. The carpet-bound protein complexes were then washed with a buffer containing ATP without ParApSM, ParBpSM, or parSpSM. ParApSM-GFP-ATP in the absence of ParBpSM reached saturation on the nsDNA-carpet in 5–10 min and then was released from the carpet slowly during the wash phase with dissociation kinetics that fit a double exponential function, majority fraction dissociating slowly (koff-fast=1.7 ± 1.1 min–1 [~7%], koff-slow=0.014 ± 0.002 min–1, N = 2) (Figure 2—figure supplement 2B, top) as observed starting with lower ParApSM-GFP density (Figure 2B).
When ParApSM-GFP was mixed with ParBpSM and ATP in the absence of parSpSM and infused into the flow cell, ParApSM-GFP-ATP reached near saturation density on the nsDNA-carpet in ~10 min. During the wash phase, ParApSM-GFP dissociated from the nsDNA-carpet with double exponential kinetics (koff-fast = 2.3 ± 0.5 min–1, koff-slow = 0.057 ± 0.01 min–1, N = 4) as in the absence of ParBpSM except for the several fold faster slow phase koff and increased fraction of faster-dissociating population (~30%) (Figure 4Ad). When the reaction mixture also contained 3R-parSpSM, ParApSM-GFP dissociation kinetics were similar to the above, except reduced fraction of the faster dissociating ParApSM-GFP population (Figure 4Ac). In these experiments, ParBpSM first accumulated on the nsDNA-carpet to roughly two-thirds of the density of ParApSM. When washing started, the majority (~75%) of ParBpSM dissociated quickly (~4 or ~7 min–1, with or without 3R-parSpSM). Thus, even in the presence of 3R-parSpSM fragment, most ParBpSM dissociates from ParApSM bound to nsDNA-carpet within half of a minute (Figure 4Ac and B).
Figure 4
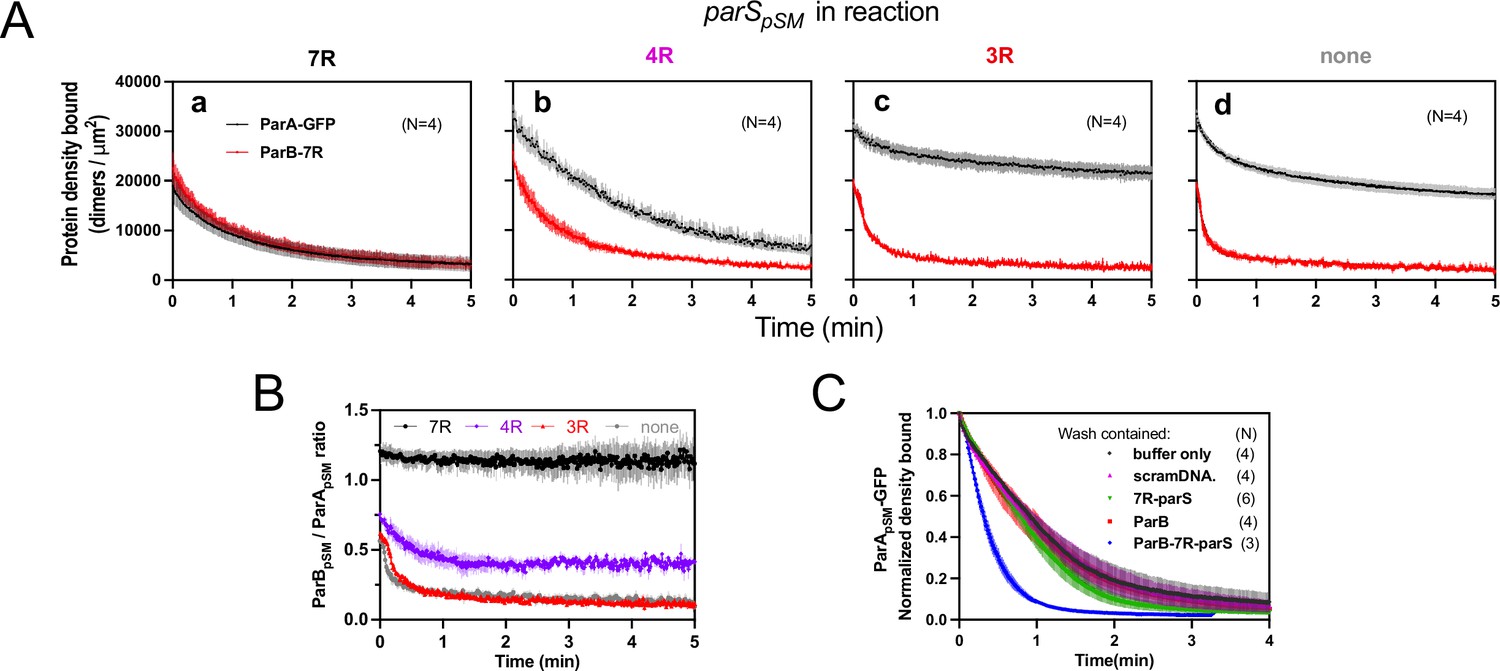
Stable ParApSM-ParBpSM complex is formed prior to ATP hydrolysis in the presence of functionally active parSpSM.
(A) ParApSM-GFP (1 μM) and ParBpSM-Alexa647 (1% labeled, 1 μM) were preincubated with ATP (1 mM) plus (a) 7R-, (b) 4R-, (c) 3R-parSpSM, or (d) without parSpSM. The preincubated sample was infused into the nsDNA-carpeted flow cell at 5 μl/min for 15 min and then the solution flowing over the observation area was switched to a buffer containing ATP (t = 0). Fluorescence signals of ParApSM (black) and ParBpSM (red) were converted to protein density on the nsDNA-carpet (dimers per μm2) and plotted. Each time point represents mean and SEM (vertical spread) of N experiments. (B) Time course of the carpet-bound ParBpSM:ParApSM molar ratio for the four panels of (A) (7R: black; 4R: purple; 3R: red; none: gray). (C) Continued supply of ParBpSM and parSpSM in the washing solution is required to sustain high rate of ParApSM disassembly. The experiment in the presence of 7R-parSpSM in panel (Aa) was repeated with addition of ParBpSM and/or 7R-parSpSM in the wash solution. Fluorescence signals of ParApSM were normalized to the value at t = 0. Each time point represents mean and standard errors of mean (SEM) (vertical spread) of N experiments.
When ParApSM-GFP and ParBpSM were preincubated with 7R-parSpSM, the steady-state ParApSM accumulation on the nsDNA-carpet was suppressed by nearly 50%, and ParBpSM and ParApSM accumulated on the nsDNA-carpet at ~1A:1.2B ratio. When the wash was started, the two proteins dissociated from the carpet maintaining roughly constant protein stoichiometry with kinetics that can be fit to a double-exponential curve (koff-fast-A [~73%] 0.92 ± 0.18 min–1, koff-slow-A 0.075 ± 0.04 min–1, and koff-fast-B [~70%] 1.1 ± 0.14 min–1, koff-slow-B 0.13 ± 0.03 min–1, N = 4; Figure 4Aa). Thus, before ATP hydrolysis disassembles ParApSM dimer from the nsDNA-carpet, ParApSM-ParBpSM protein interaction appears to be significantly more stable in the presence of 7R-parSpSM compared to the complex involving 3R-parSpSM (Figure 4B).
In the presence of 4R-parSpSM, carpet binding and dissociation dynamics of the two proteins were qualitatively similar to those in the presence of 7R-parSpSM, except for the following differences: 4R-parSpSM did not suppress ParApSM binding to the nsDNA-carpet as effectively as 7R-parSpSM, the ParBpSM/ParApSM ratio at the end of 15 min sample infusion was ~0.75, which dropped to ~0.5 within 1 min of washing after which the ratio remained constant. Observed koff were ParApSM (koff-fast-A [60%] 0.67 ± 0.14 min–1, koff-slow-A 0.17 ± 0.05 min–1; and koff-fast-B [66%] 1.9 ± 0.2 min–1, koff-slow-B 0.23 ± 0.02 min–1, N = 4; Figure 4Ab). It appears that ParApSM bound to the nsDNA-carpet interacts slightly less stably with ParBpSM in the complex assembled with 4R-parSpSM compared to those assembled with 7R-parSpSM, but it was still significantly more stable compared to the majority of the ParApSM-ParBpSM complexes that accumulate in the presence of 3R-parSpSM or without parSpSM DNA.
In this experiment, the absence of ParBpSM and 7R- or 4R-parSpSM in the wash solution flowing over the nsDNA-carpet appeared to have caused a slowdown of dissociation of the ParApSM-ParBpSM complexes from nsDNA compared to the experiments of Figures 2B and 3A. This prompted us to examine whether partial loss of the parSpSM DNA and/or ParBpSM from the complex during the washing caused this kinetic change. We repeated the experiments of Figure 4Aa with the addition of ParBpSM (1 μM) and/or 7R-parSpSM DNA fragment (500 nM consensus repeats) in the washing solution. The addition of ParBpSM and 7R-parSpSM in the washing solution significantly accelerated ParApSM koff to 2.5 ± 0.25 min–1 (N = 3), a slightly higher level than seen in Figure 3Aa (1.8 ± 0.05 min–1), which might have been an underestimate due to the preceding slow complex assembly steps. The addition of ParBpSM or 7R-parSpSM separately to the washing solution did not have significant effects on the ParApSM dissociation kinetics (Figure 4C). Thus, partial loss of ParBpSM-parSpSM complex from the nsDNA-bound ParApSM at the onset of the wash (7R- and 4R-parSpSM) and also during the wash (4R-parSpSM) appears to have compromised the efficiency of ParApSM dissociation from nsDNA in the experiments of Figure 4Aa,b. This indicates that not all the ParBpSM-parSpSM complexes that can contribute to dissociation of ParApSM from nsDNA are stably retained via ParApSM to the nsDNA-carpet through the course of reaction.
ParApSM-ATP in the wash solution stabilizes functional ParBpSM-parSpSM complexes associated with carpet-bound ParApSM
When preformed ParApSM-ParBpSM complexes bound to the nsDNA-carpet in the presence of 7R- or 4R-parSpSM DNA were washed with buffer containing ATP-activated ParApSM-GFP (0.5 μM), freshly arriving ParApSM-ATP dimers on the nsDNA-carpet replacing the dissociating ParApSM partially stabilized ParBpSM (Figure 5a, b). This observation indicates that when ParApSM dissociates from nsDNA following ATP hydrolysis, some fraction of ParBpSM in the presence of 7R- or 4R-parSpSM that induced ATP hydrolysis can capture the newly arriving ParApSM-ATP dimers on the nsDNA-carpet. The complex containing 7R-parSpSM was more efficient for recapture than the 4R-parSpSM complex. We propose larger numbers of ParBpSM dimers complexed with parSpSM with more sequence repeats can interact simultaneously with multiple ParApSM-nsDNA mini-filaments, more readily capturing newly arrived ParApSM-ATP dimers at nearby nsDNA sites. Evidence supporting such multivalent interactions between nsDNA-bound ParApSM and parSpSM-bound ParBpSM has been reported previously (Pratto et al., 2009). ParBpSM stabilization by replenishment of ParApSM-ATP was not observed in the presence of 3R-parSpSM or in the absence of parSpSM, consistent with the notion that ParApSM-ParBpSM interactions under these conditions are unstable (Figure 5c,d).
Figure 5
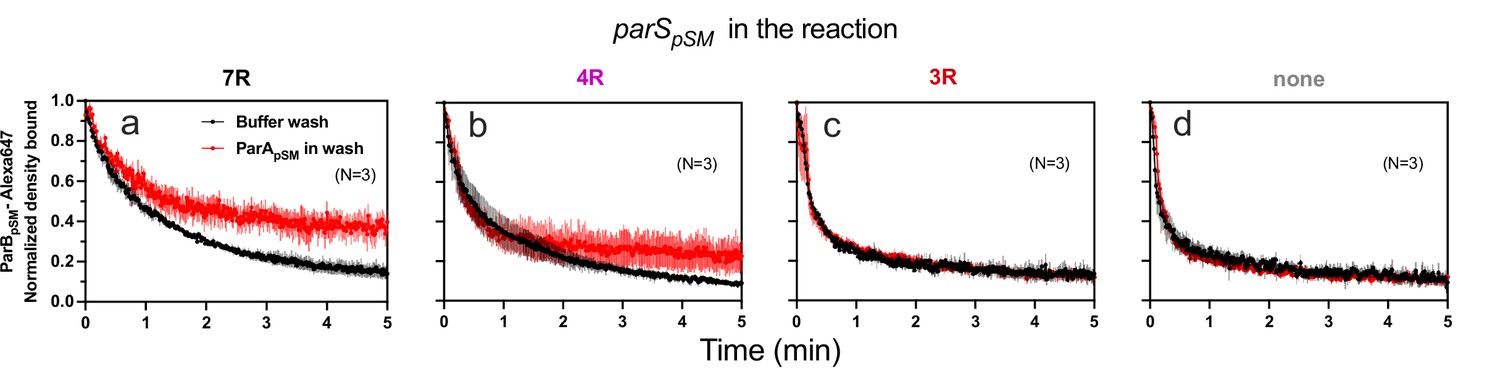
ParApSM-ATP stabilizes functionally active parSpSM-bound ParBpSM on the nsDNA-carpet.
Complexes containing ParApSM-GFP, and ParBpSM-Alexa647 bound to the DNA-carpet together with different parSpSM DNA fragments, (a) 7R, (b) 4R, (c) 3R, or (d) without parSpSM, were washed with either a buffer containing 1 mM ATP (black) or the same buffer containing ParApSM-GFP (0.5 μM, red), in addition to ATP. The ParBpSM-Alexa647 dissociation curves normalized to the protein density at t = 0 were plotted. Each time point represents mean and standard errors of mean (SEM) (vertical spread) of N experiments.
Discussion
Using a combination of steady-state ATPase assays and nsDNA dissociation kinetic measurements of ParApSM-ParBpSM complexes in the presence of a variety of modified parSpSM-DNA fragments, we studied the molecular requirements for activation of ParApSM. This work revealed distinct parSpSM structural requirements for efficient activation of ParApSM ATPase by ParBpSM and a multistep assembly process for the active parSpSM-ParBpSM-ParApSM complex. The parSpSM DNA must contain at minimum four contiguous consensus sequence repeats to enable ParBpSM dimer binding in a state that induces ATP hydrolysis and accelerated dissociation of ParApSM from nsDNA. The structural requirements for parSpSM function assure high specificity of the centromere site for PC function. Considering the clear biochemical functionality difference depending on the parSpSM sequence repeat number and the repeat contiguity, we believe in vivo plasmid stability would also require parSpSM site(s) at minimum four or possibly larger number of repeats in contiguous arrangements. However, this point remains to be experimentally confirmed. Considering that there are six copies of parSpSM sites in pSM19035, it also would be useful to learn the number of parSpSM sites required for the plasmid stability and whether this requirement depends on the plasmid copy number.
ParBpSM-parSpSM recognition by ParApSM
The parSpSM sequence features required for the function of ParBpSM, an RHH-ParB protein contrast with those for the HTH-ParB proteins. Whereas natural parSF sequence for the F-plasmid is composed of 12 repeats of the ParBF dimer binding sequence, a single copy of this consensus sequence is able to support faithful plasmid partition (Biek and Shi, 1994). Further, HTH-ParB proteins can spread around parS sites, forming large PCs containing many ParB molecules, most of which have moved away from parS sequence. In contrast, RHH-ParB proteins lack the CTPase domain that is critical for HTH-ParB spreading (Soh et al., 2019; Jalal et al., 2020; Osorio-Valeriano et al., 2019) and they cannot spread from parS sites to flanking DNA (Pratto et al., 2009). Systems involving RHH-ParB proteins perhaps evolved a fundamentally different system architecture for PC dynamics to accomplish robust partitioning of replicated plasmid copies. It is currently unclear what constitutes the defining feature of the ParBpSM molecules bound to four or more contiguous parSpSM heptad-sequence-repeats: we offer a possible scenario below. It is interesting to note that parSpSM-dependence of ParApSM-ATPase activation is high compared to F-plasmid Par system involving HTH-ParB, which exhibits either only less than twofold higher ATPase activation in the presence of parSF (in the absence of CTP) or no difference of the maximum ATP turnover rate in the presence of CTP (Ah-Seng et al., 2009; Taylor et al., 2021). This is reasonable for HTH-ParB systems; most ParB molecules in the PC are not parS-associated at the time it interacts and activates the ParAF-ATPase and only limited number of ParBF molecules are outside of PCs. In contrast, fewer PC-associated RHH-ParB molecules and perhaps comparatively more non-PC-associated RHH-ParB molecules exist inside a cell, necessitating tighter parS-dependent ATPase activation for the nonspreading ParB systems.
Assembly/disassembly dynamics of ParABpSM complex on the nsDNA
ParApSM-ATP is slow to dissociate from the nsDNA-carpet in the presence of ParBpSM alone, or ParBpSM-3R-parSpSM complex in the wash solution. We showed that ParBpSM can quickly interact with nsDNA-bound ParApSM in the absence of functional parSpSM DNA (Figure 3Ac). However, this ParApSM-ParBpSM interaction does not fully activate the ATPase (Figure 1). All wash curves of ParApSM-ATP dimers bound to nsDNA-carpet under conditions that do not trigger efficient ATPase activation exhibited double-exponential dissociation kinetics, although the small amplitude of the fast dissociation phase, typically ~5%, made quantitative comparison difficult (Figures 2B and 3A). The fast dissociation phase had a time scale of less than 1 min, while the majority fraction dissociated much slower under conditions that do not activate the ATPase. This indicated that the carpet-bound ParApSM dimers were composed of two distinct state populations, transitions between which are slow. ParBpSM with or without 3R-parSpSM, after the initial rapid binding to nsDNA-bound ParApSM, also dissociated with double-exponential kinetics; a fraction of the carpet-associated ParBpSM quickly dissociated within 1 min before settling to a quasi-steady state with the constant supply of ParBpSM or ParBpSM-3R-parSpSM complex in the wash solution and slowly dissociating ParApSM on the nsDNA-carpet (Figure 3Ac,d). We speculate that this small initial binding overshoot reflects ParBpSM association to the less populated fast-dissociating fraction of the carpet-bound ParApSM dimers, which presumably are in a state with faster ParBpSM-association rate, lower nsDNA affinity, and perhaps closer to the ATP hydrolysis-competent state.
When ParApSM-ATP-ParBpSM complexes bound to the nsDNA-carpet in a steady state with or without 3R-parSpSM was washed with a simple buffer (Figure 4Ac,d), a double-exponential dissociation of ParApSM was again observed with increased fraction (up to ~30%) of the fast-dissociating population. This supports the notion that the fast-dissociating ParApSM population is in a state closer to, but not committed to, ATP hydrolysis. We hypothesize that this ParApSM transitory state becomes more populated during preincubation with ParBpSM on the nsDNA-carpet accounting for the moderate stimulation of the DNA-bound ParApSM ATPase by ParBpSM or ParBpSM-3R-parSpSM complex at steady state (Figure 1A). However, this ‘fast’ dissociation does not depend on ATP hydrolysis, considering the ATPase-defective mutant ParApSMD60E also exhibits clear double-exponential dissociation (Figure 2—figure supplement 2B, top, Figure 2—figure supplement 3B).
When ParApSM-ATP prebound to nsDNA-carpet in the flow cell was washed with a solution containing fully functional 7R-parSpSM-ParBpSM complexes, accelerated release of ParApSM from nsDNA was observed (Figure 2B, Figure 3Aa,e,i). However, the initial ParApSM dissociation kinetics resembled the ParBpSM wash without fully active parSpSM and accelerated disassembly started with a delay. This clear transition of the dissociation mode indicates the initial complex between the nsDNA-bound ParApSM and parSpSM-bound ParBpSM does not immediately trigger ATP hydrolysis. Rather a series of steps must take place subsequent to the initial association of the two protein-DNA complexes to trigger ATP hydrolysis and complex disassembly. This process appears to involve participation of additional ParBpSM dimers and/or parSpSM beyond the initial complex, considering that the delay time before the accelerated ParApSM dissociation phase depends on their concentration in the wash solution while clear biphasic kinetic feature was observed even at limiting ParBpSM concentrations.
The ParApSM-ParBpSM interaction is not stable in the absence of 7R- or 4R- parSpSM (Figure 4B). In contrast, interaction between nsDNA-bound ParApSM and ParBpSM in the presence of 7R-parSpSM is significantly more stable prior to ATP hydrolysis-dependent dissociation of ParApSM from nsDNA (Figure 4Aa). Thus, ParApSM-ATP interacts with ParBpSM in the presence of fully active parSpSM in a distinct manner than in its absence. For simplicity, we propose this transition to stably interacting ParApSM-ParBpSM complex bound to nsDNA is the limiting step necessary before ATP hydrolysis-dependent acceleration of ParApSM dissociation. Slower koff of ParApSM from nsDNA in the experiments of Figure 4Aa,b compared to those in Figure 2B and Figure 3Aa,b, and recovery of faster koff by the replenishment of ParBpSM and parSpSM in the wash solution (Figure 4C) indicated that ParApSM-ParBpSM complex must turn over to maintain high ParApSM koff. This suggests that not all the nsDNA-bound ParApSM dimers are interacting with ParBpSM in the state committed for ATPase activation in the initial set of active complexes that form on the nsDNA-carpet.
A model for ParBpSM activation of ParApSM-ATPase
Without further experimental constraints, our consideration of the mechanism of ParApSM-ATPase activation by ParBpSM remains speculative. Unlike other members of ParA-family of ATPases studied before, such as ParAF (Taylor et al., 2021) or MinD (Vecchiarelli et al., 2016), simple interaction of ParBpSM-N-terminal domain, ParBpSM1-27, with ParApSM dimers did not efficiently activate the ATPase (Figure 1—figure supplement 1B). Because of the helical nature of the ParBpSM-parSpSM complex (Weihofen et al., 2006) and ParApSM-nsDNA mini-filament formation (Pratto et al., 2009), two ParApSM dimers within a mini-filament might be approximately in position to interact with ParBpSM dimers separated by two intervening parSpSM sequence repeats on one face of the helical filament. Let us assume, however, that the pitch and/or the angular arrangements of these two pairs of protein dimers are not in perfect match allowing only partial interaction between ParApSM dimers and ParBpSM dimers in the basal state structures of the two mini-filaments (Figure 6A). Then, the establishment of divalent interactions and full engagement between the two protein-DNA complexes perhaps would force distortions of the structures of both of the protein-DNA mini-filaments (Figure 6B). The force imposed upon ParApSM-mini-filament might act as a steppingstone for the conformational change of ParApSM necessary for ATPase activation. Such a scenario explains the requirement for the fourth parSpSM consensus sequence repeat for the ATPase activation. We propose the specific mechanical properties of the contiguous ParBpSM-parSpSM mini-filament, likely different from gapped mini-filaments, is required for efficient ATPase activation, explaining the requirement for the parSpSM sequence repeat contiguity. The divalent interactions between one ParBpSM-parSpSM mini-filament with a ParApSM-nsDNA mini-filament, with a non-interacting middle segment separating the two interacting pairs, might allow the ParBpSM-parSpSM complex to semi-processively activate multiple ParApSM-nsDNA complexes in succession by an inchworm-like transfer to a new ParApSM-nsDNA mini-filament. The ability of the otherwise dissociating 7R-parSpSM-ParBpSM complex after ATPase activation and dissociation of one partner ParApSM mini-filament to recapture a freshly arriving ParApSM-ATP dimers on the nsDNA-carpet (Figure 5a) is consistent with such a possibility.
Figure 6
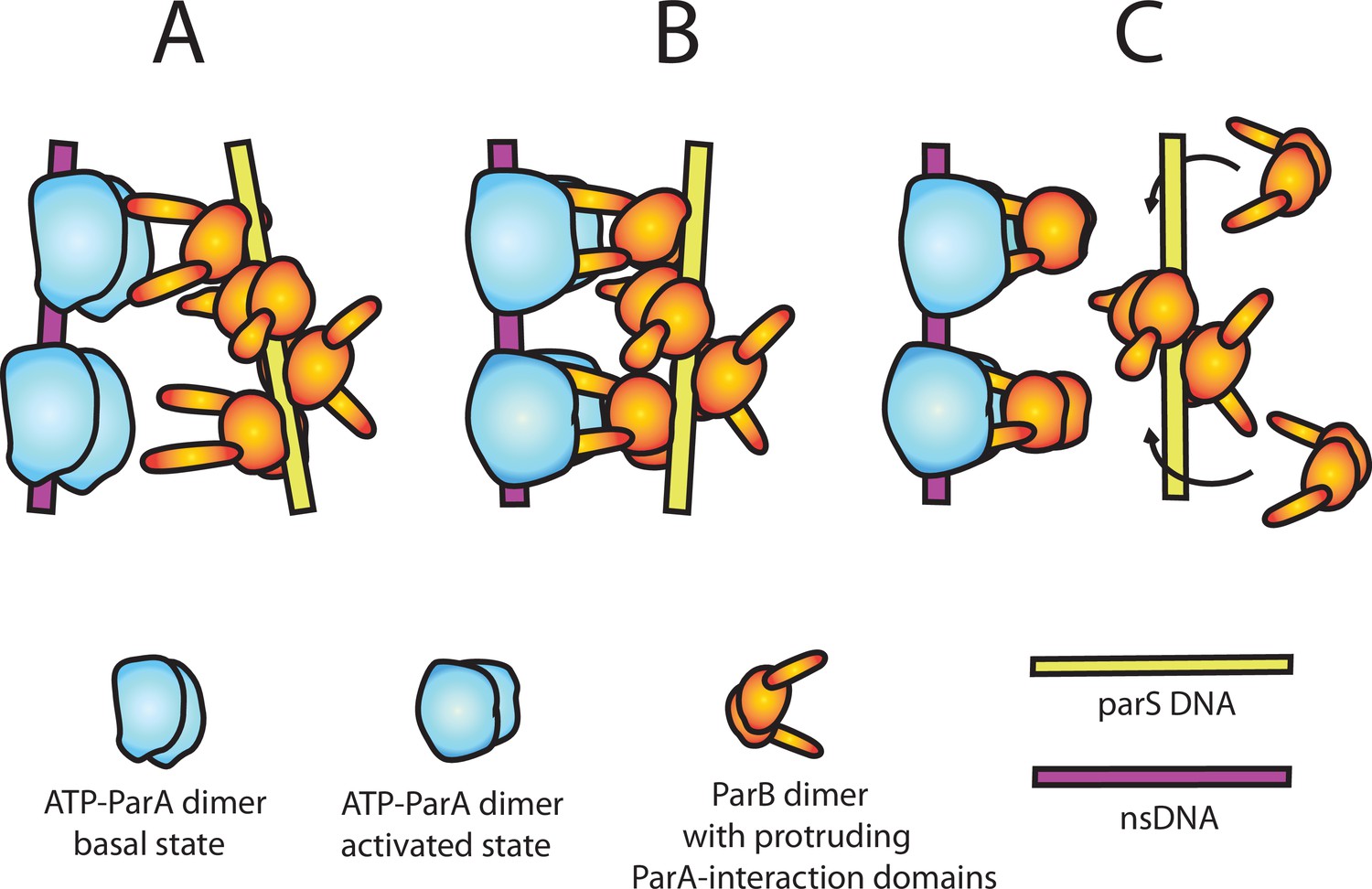
A model of nsDNA-bound ParApSM-ATPase activation by parSpSM-bound ParBpSM.
(A) ParApSM-ATP dimers bound to nsDNA in their basal state can interact with ParA-activation domains protruding from ParBpSM dimers bound to parSpSM sequence repeats. However, multiple ParApSM dimers in a mini-filament cannot simultaneously interact with ParBpSM dimers bound to a set of repeated sequence copies within a parSpSM site and the two proteins dissociate quickly. We propose the interacting pair of proteins at this stage are not fully engaged and these ParApSM dimers are not in the ATPase-activated state. Here, we assume two ParA-interacting domains belonging to one ParBpSM dimer binds one ParApSM dimer. (B) We propose torsional (or other conformational) thermal Brownian dynamics of the mini-filaments allow the ParBpSM dimer at the fourth position to establishes interaction with another ParApSM dimer, locking in the non-equilibrium conformation of the individual mini-filaments prior to the formation of this second bridge. The distortion promotes conformational transition of the ParApSM dimers to the ATPase active state with fully engaged ParBpSM. (C) The conformational transition also destabilizes ParBpSM-parS interaction, releasing the parSpSM DNA from the activated nsDNA-ParApSM-ParBpSM complex prior to ATP hydrolysis and disassembly of the complex. This allows reloading of fresh ParBpSM to the released parSpSM, which recycles to disassemble the remaining ParApSM on the nsDNA. Meanwhile, fully engaged ParBpSM dimers left on the ParApSM dimers cause ATP hydrolysis and disassemble ParApSM dimers from nsDNA.
The concentration mismatch between ParBpSM and parSpSM needed for full activation of ParApSM ATPase (Figure 1D) suggests additional details; the functional parSpSM DNA might be needed only to deliver ParBpSM dimers onto ParApSM-nsDNA mini-filament generating a complex committed to ATP hydrolysis. After ParBpSM delivery, parSpSM might release the delivered ParBpSM dimers, perhaps helped by the mini-filament distortion discussed above, without waiting for ATP hydrolysis (Figure 6C). The emptied consensus sequence on the parSpSM would then be quickly recharged with free ParBpSM dimers in solution to activate another ParApSM-nsDNA complex, explaining the higher concentration demand for ParBpSM over parSpSM for ATPase activation. The requirement for parSpSM and ParBpSM replenishment from the washing solution to sustain maximum koff of the nsDNA-bound ParApSM dimers (Figure 4C) supports the notion that the initial active ParApSM-ParBpSM-parSpSM complex assembled on the nsDNA perhaps does not induce ATP hydrolysis of all the ParApSM dimers within the complex. Full disassembly of the complex likely involves release of the parSpSM fragment, which gets reloaded with ParBpSM in solution and returns to the ParApSM dimers left on the nsDNA. This local recycling of ParBpSM and parSpSM would become inefficient when ParBpSM and parSpSM are removed from reaction by the buffer wash. We note that dependence of the accelerated phase of ParApSM koff on the concentration of the parSpSM-ParBpSM complex (Table 1) is consistent with the above observation. In vivo, local off-rate of ParApSM from nucleoid where a PC is located is likely to be significantly faster than observed here, considering the local density of the nucleoid-bound ParApSM near a PC would be lower and the ParBpSM-parSpSM concentration of a PC, with six copies of parS sites, much higher compared to the conditions in this study.
Earlier we described our model considering one ParBpSM-parSpSM mini-filament acting on one ParApSM cluster on nsDNA for simplicity. However, this does not immediately explain the clearly biphasic disassembly kinetics of the nsDNA-bound ParApSM during washing with ParBpSM-parSpSM complex (Figure 2B, Figure 3Aa,e,i). This kinetics is highly reminiscent of activation and membrane dissociation kinetics of membrane-bound MinD-ATPase, a member of ParA-ATPase family, when washed by MinE-containing buffer (Vecchiarelli et al., 2016), which has been proposed to reflect requirement for two MinE dimers for the activation of one MinD dimer. Thus, we suspect two ParBpSM-parSpSM mini-filaments may need to cooperate from two sides of one ParApSM cluster on nsDNA, and the second binding is kinetically limiting.
Lastly, irrespective of the details of the model, we need to pay attention to a few aspects of the pSM19035 Par system in the context of the diffusion-ratchet concept. As mentioned in the ‘Introduction,’ plasmid or chromosome partition by a diffusion-ratchet mechanism requires the balance between the stability of each ParA/ParB-mediated cargo-nucleoid bridge and the number of these bridges per cargo. In the pSM19035 system, prior to ATPase-activating nsDNA-ParApSM-ParBpSM-parSpSM complex assembly, the ParApSM-ParBpSM link in the bridge is expected to have a stability close to that in the presence of inactive 3R-parSpSM (koff = ~4 min–1, Figure 4Ac). This stability is comparable to that of F-plasmid ParAF- ParBF bridge between the nucleoid and cargo, which we believe to involve one ParAF dimer, and therefore DNA-carpet-bound ParAF FRAP rate in the presence of ParBF (~4 min–1, Vecchiarelli et al., 2013) would approximate the stability of the bridge. In contrast, we believe that the effective stability of a unit of the cargo-nucleoid bridge by the ATPase-activating nsDNA-ParApSM-ParBpSM-parSpSM complex containing many ParApSM dimers and ParBpSM dimers is likely to be significantly higher. If one assumes processive rounds of ATPase activation for subsets of ParApSM dimers within a complex as considered in our model, individual cargo-nucleoid bridge would have substantially longer lifetime than individual ParApSM dimer dissociation rate constant of up to ~2 min–1 observed by the experiments of Figure 3. At the same time, the number of individual units of bridges to the nucleoid per cargo would be very small compared to up to a few hundred in cases of HTH-ParB plasmid systems. Long-range transfer of the PC from one ParApSM cluster to another via simultaneous multiple ParApSM cluster interactions involving nucleoid DNA looping without bridge dissolution discussed above would further extend the lifetime of individual bridges, enhancing the effective PC diffusion suppression capability of very small number of bridges without blocking the PC motion. In addition, very slow dissociation of ParApSM dimers from nsDNA prior to formation of fully active complex with a PC (koff < 0.1 min–1, Figures 2 and 3) compared to ParAF or ParAP1 (koff = ~5 min–1, Hwang et al., 2013, Vecchiarelli et al., 2013) suggests their very slow diffusion on the nucleoid (practically no hopping). Together with expected slow overall ATP hydrolysis rate, it predicts slow ParApSM depletion zone development and refilling. While these parameter shifts compared to the F- or P1-Par systems are generally speaking in the mutually compensatory directions (Hu et al., 2015; Hu et al., 2017), whether they are balanced or not needs to be carefully evaluated as more details of the mechanism come to light in future.
This study revealed a puzzlingly unique parSpSM DNA site structure required for the assembly of the fully active ParBpSM-parSpSM PC in the pSM19035 ParABS system. We propose one possible scenario to explain our findings. However, the model presented here is perhaps not the only possible explanation. Further studies are needed to support or refute the model to advance our mechanistic understanding of this family of partition systems. Direct examination of the effects of the PC torsional strain would be needed to test our current model. Further refinement of the cell-free partition reaction system is hoped to greatly assist our mechanistic understanding of the rich variations of the prokaryotic chromosome/plasmid partition systems.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Escherichia coli) | BL21 DE3 AI | Invitrogen | C607003 | Protein expression strain |
| Recombinant DNA reagent | pET11a | EMD Millipore | 9436 | Protein expression vector |
| Recombinant DNA reagent | pT712ω | Welfle et al., 2005 | ParBpSM overexpression plasmid | |
| Recombinant DNA reagent | pCB746 (pT712 vector) | Pratto et al., 2008 | ParApSM overexpression plasmid | |
| Recombinant DNA reagent | pCB755 (pT712 vector) | Pratto et al., 2008 | ParApSMD60A overexpression plasmid | |
| Recombinant DNA reagent | pET11a- ParApSMD60E | This work | ParApSMD60E overexpression plasmid | |
| Recombinant DNA reagent | pCB1033 (pT712 vector) | This work | ParBpSM-GCE overexpression plasmid | |
| Recombinant DNA reagent | pET11a- ParApSM-EGFP | This work | ParApSM-EGFP overexpression plasmid | |
| Recombinant DNA reagent | pET11a- ParApSMD60A-EGFP | This work | ParApSMD60A-EGFP overexpression plasmid | |
| Recombinant DNA reagent | pET11a- ParApSMD60E-EGFP | This work | ParApSMD60E-EGFP overexpression plasmid | |
| Sequence-based reagent | Scrambled 55mer DNA oligo + strand | This work | GGGATCAAACACTTGATAGACAAGTCTTTGACCTAATTGTGAAAATTATGAAGGG | |
| Sequence-based reagent | Scrambled 55mer DNA oligo - strand | This work | CCCTTCATAATTTTCACAATTAGGTCAAAGACTTGTCTATCAAGTGTTTGATCCC | |
| Sequence-based reagent | 7R parS DNA oligo + strand | This work | GGGAATCACAAATCACAAGTGATTAATCACAAATCACTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 7R parS DNA oligo - strand | This work | CCCAATCACAAATCACAAGTGATTTGTGATTAATCACTTGTGATTTGTGATTCCC | |
| Sequence-based reagent | 6R parS DNA oligo + strand | This work | GGGAATCACAAGTGATTAATCACAAATCACTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 6R parS DNA oligo - strand | This work | CCCAATCACAAATCACAAGTGATTTGTGATTAATCACTTGTGATTCCC | |
| Sequence-based reagent | 5R parS DNA oligo + strand | This work | GGGAATCACAAATCACAAATCACTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 5R parS DNA oligo - strand | This work | CCCAATCACAAATCACAAGTGATTTGTGATTTGTGATTCCC | |
| Sequence-based reagent | 4R(1) parS DNA oligo + strand | This work | GGGAATCACAAATCACTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 4R(1) parS DNA oligo - strand | This work | CCCAATCACAAATCACAAGTGATTTGTGATTCCC | |
| Sequence-based reagent | 4R(2) parS DNA oligo + strand | This work | GGGAATCACTTATCACAAGTGATTAATCACTGGG | |
| Sequence-based reagent | 4R(2) parS DNA oligo - strand | This work | CCCAGTGATTAATCACTTGTGATAAGTGATTCCC | |
| Sequence-based reagent | 4R(3) parS DNA oligo + strand | This work | GGGAATCACTTATCACAAATCACAAATCACTGGG | |
| Sequence-based reagent | 4R(3) parS DNA oligo - strand | This work | CCCAGTGATTTGTGATTTGTGATAAGTGATTCCC | |
| Sequence-based reagent | 3R-2nc-3R parS DNA oligo + strand | This work | GGGAATCACAAATCACAAATCACATCATAGTTCATAGTTGTGATTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 3R-2nc-3R parS DNA oligo - strand | This work | CCCAATCACAAATCACAAATCACAACTATGAACTATGATGTGATTTGTGATTTGTGATTCCC | |
| Sequence-based reagent | 3R-1nc-3R parS DNA oligo + strand | This work | GGGAATCACAAATCACAAATCACATCATAGTTGTGATTTGTGATTTGTGATTGGG | |
| Sequence-based reagent | 3R-1nc-3R parS DNA oligo - strand | This work | CCCAATCACAAATCACAAATCACAACTATGATGTGATTTGTGATTTGTGATTCCC | |
| Sequence-based reagent | 3R-1nc parS DNA oligo + strand | This work | GGGAATCACAAATCACTTGTGATTTCATAGTGGG | |
| Sequence-based reagent | 3R-1nc parS DNA oligo - strand | This work | CCCACTATGAAATCACAAGTGATTTGTGATTCCC | |
| Sequence-based reagent | 3R(1) parS DNA oligo + strand | This work | GGGAATCACAAATCACTTGTGATTGGG | |
| Sequence-based reagent | 3R(1) parS DNA oligo - strand | This work | CCCAATCACAAGTGATTTGTGATTCCC | |
| Sequence-based reagent | 3R(2) parS DNA oligo + strand | This work | GGGAATCACTTATCACAAATCACAGGG | |
| Sequence-based reagent | 3R(2) parS DNA oligo - strand | This work | CCCTGTGATTTGTGATAAGTGATTCCC | |
| Sequence-based reagent | 2R parS DNA oligo + strand | This work | GGGAATCACTTGTGATTGGG | |
| Sequence-based reagent | 2R parS DNA oligo - strand | This work | CCCAATCACAAGTGATTCCC | |
| Sequence-based reagent | 1R parS DNA oligo + strand | This work | GGGAATCACTGGG | |
| Sequence-based reagent | 1R parS DNA oligo - strand | This work | CCCAGTGATTCCC | |
| Peptide, recombinant protein | ParBpSM1-27 | This work | MIVGNLGAQKAKRNDTPISAKKDIMGD | |
| Peptide, recombinant protein | ParBpSM1-27 K10A | This work | MIVGNLGAQAAKRNDTPISAKKDIMGD | |
| Peptide, recombinant protein | ParBP11-30 | This work | MSKKNRPTIGRTLNPSILSGFDSSSASGDR | |
| Chemical compound, drug | [γ−32P]ATP | PerkinElmer | NEG002A250UC | |
| Chemical compound, drug | Biotin-17-dCTP | Invitrogen | 65601 | |
| Chemical compound, drug | Terminal transferase | New England Biolabs | M0315 | |
| Chemical compound, drug | Alexa Fluor 488 C5 maleimide | Thermo Fisher | A10254 | |
| Chemical compound, drug | Alexa Fluor 594 C5 maleimide | Thermo Fisher | A10256 | |
| Chemical compound, drug | Alexa Fluor 647 C2 maleimide | Thermo Fisher | A20347 | |
| Chemical compound, drug | Antifoam Y-40 emulsion | Sigma | A5758 | |
| Chemical compound, drug | EDTA-free Sigmafast protease inhibitor cocktail tablet | Sigma | S8830 | |
| Chemical compound, drug | DOPC | Avanti Polar Lipids | 850375P | |
| Chemical compound, drug | DOPE-Biotin | Avanti Polar Lipids | 870273C | |
| Chemical compound, drug | Biotin-14-dCTP | Thermo Fisher | 19518018 | |
| Software, algorithm | Prism 9 | GraphPad | Prism 9 | Used for curve fitting, and fitting parameters and their error estimation |
| Software, algorithm | MetaMorph 7 | Molecular Devices | MetaMorph 7 | Used for TIRF microscope data acquisition |
| Software, algorithm | ImageJ/Fiji | National Institutes of Health | ImageJ | Used for TIRF microscope image analysis |
| Other (instrument) | Prism type TIRF microscope | In-house; Vecchiarelli et al., 2013, Ivanov and Mizuuchi, 2010 | Used for ParAF-ParBF complex assembly–disassembly experiments |
Materials availability
Request a detailed protocolNewly generated materials from this study are available by request to the corresponding author, Kiyoshi Mizuuchi (kiyoshimi@niddk.nih.gov) until the lab stocks become exhausted or the lab group operation becomes terminated.
Proteins, peptides, and DNA
Request a detailed protocolNon-fluorescent ParApSM-His6 (wild-type and mutants) was purified as previously described (Pratto et al., 2008). ParApSM-GFP-His6 and ParApSMD60E-GFP-His6 were purified as described for ParAF-GFP-His6 (Vecchiarelli et al., 2010). ParBpSM wild-type and ParBpSM-cys, which had three residues (-GCE) added at the C-terminal, were purified essentially as previously described with the addition of reducing agent (2 mM DTT) in the buffers 50 mM Tris–HCl pH 7.5, 100 mM NaCl plus 5% glycerol. Protein concentrations were estimated based on OD280 and aromatic amino acid content.
Fluorescence labeling of ParBpSM was done as described for Alexa647-ParBF (Vecchiarelli et al., 2013). ParBpSM-GCE, in 50 mM Tris–HCl pH 7.4, 100 mM NaCl, 0.1 mM EDTA, 10% glycerol, was mixed with Alexa647 maleimide (Invitrogen) at a protein-to-dye ratio of 1:1 and then incubated for 1 hr at room temperature in the dark. Then, 20 mM DTT was added to stop the reaction. Free dye was removed by spin gel-filtration in a G-50 column. The dye labeling efficiency was determined by spectrophotometry to be ~15%, and the labeled protein was mixed with unlabeled protein to prepare 1%-labeled ParBpSM-Alexa647 used for the experiments. In vivo, ParBpSM-GCE in combination with ParApSM-GFP was fully competent for partition (Maria Moreno-del Álamo, personal communication), and in vitro, dye labeling did not affect its activities, as measured by its ability to stimulate ParApSM ATPase and by its parSpSM DNA binding activity (A. Volante, unpublished results).
The N-terminal peptides of ParBpSM (residues 1–27) and ParBP1 (residues 1–30) used in the ATPase assays were synthesized by GenScript. The sequence of ParBpSM1-27 and its variant ParBpSM1-27 K10A were NH2-MIVGNLGAQKAKRNDTPISAKKDIMGD-CO2H (≥97% purity) and NH2-MIVGNLGAQAAKRNDTPISAKKDIMGD-CO2H (≥96% purity), respectively. The sequence of ParBP11-30 was NH2-MSKKNRPTIGRTLNPSILSGFDSSSASGDR-CO2H (≥97% purity).
Two strands of each double-stranded DNA containing heptad repeat (5′-WATCACW-3′) or non-consensus scrambled sequences were synthetized, annealed, and purified by ITD (Integrated DNA Technologies). The forward sequences of a DNA duplex are listed in the following table:
| Name | Sequence (5′–3′) |
|---|---|
| Scram | 5′-GGG ATCAAAC ACTTGAT AGACAAG TCTTTGA CCTAATT GTGAAAA TTATGAA GGG-3′ |
| 7R | 5′-GGG AATCACA AATCACA AGTGATT AATCACA AATCACT TGTGATT TGTGATT GGG-3′ |
| 6R | 5′-GGG AATCACA AGTGATT AATCACA AATCACT TGTGATT TGTGATT GGG-3′ |
| 5R | 5′-GGG AATCACA AATCACA AATCACT TGTGATT TGTGATT GGG-3′ |
| 4R(1) | 5′-GGG AATCACA AATCACT TGTGATT TGTGATT GGG-3′ |
| 4R(2) | 5′-GGG AATCACT TATCACA AGTGATT AATCACT GGG-3′ |
| 4R(3) | 5′-GGG AATCACT TATCACA AATCACA AATCACT GGG-3′ |
| 3R – 2nc – 3R | 5′-GGG AATCACA AATCACA AATCACA TCATAGT TCATAGT TGTGATT TGTGATT TGTGATT GGG-3′ |
| 3R – 1nc – 3R | 5′-GGG AATCACA AATCACA AATCACA TCATAGT TGTGATT TGTGATT TGTGATT GGG-3′ |
| 3R – 1nc | 5′-GGG AATCACA AATCACT TGTGATT TCATAGT GGG-3′ |
| 3R(1) | 5′-GGG AATCACA AATCACT TGTGATT GGG-3′ |
| 3R(2) | 5′-GGG AATCACT TATCACA AATCACA GGG-3′ |
| 2R | 5′-GGG AATCACT TGTGATT GGG-3′ |
| 1R | 5′-GGG AATCACT GGG-3′ |
Plasmids
Plasmids encoding wild-type ParApSM (Delta, pCB746), mutant ParApSMD60A (pCB755), and ParBpSM (Omega, pT712ω) have been described previously (Welfe et al. 2005, Pratto et al., 2008; Volante and Alonso, 2015). The pET11a harboring the sequence of his-ParApSMD60E (pET11a-ParApSMD60E) his-ParApSM-eGFP (pET11a-ParApSM-eGFP) and its variants (D60A, pET11a-ParApSMD60A-eGFP and D60E, pET11a-ParApSMD60E-eGFP) were constructed by GenScript. The ParBpSM-GCE [72G, 73C, 74E] allele was created by site-directed mutagenesis and then cloned into the ParBpSM expression plasmid (pT712) to generate pCB1033.
ATPase assays
Request a detailed protocolUnless stated otherwise, all ATPase assays were performed as follows: the reaction contained 2 µM ParApSM, the indicated concentration of full-length ParBpSM (or ParBpSM1-27), nsDNA (plasmid pBR322 DNA, 60 µM in bp), and parSpSM DNA duplex in 50 mM Tris–HCl (pH 7.4), 100 mM KCl, 2 mM MgCl2, and 1.5 mM [γ-32P]ATP. Labeled ATP was purified before use as previously described (Vecchiarelli et al., 2010). Reactions were incubated at 37°C for 3 hr and analyzed by TLC as previously described (Pratto et al., 2008). The data points shown are the means with error bars (standard errors of mean) of repeat experiments (N) as indicated for each figure panel. N* indicates repeat number for majority of ParBpSM concentration points as detailed in the Source Data file. Datasets of repeated measurements were fit after subtraction of background measured without ParApSM to a modified Hill equation: v – v0 = (vmax [B]n)/(KAn + [B]n), and the fit parameters and symmetrized error ranges reflecting the larger error of the 95% confidence interval below and above the mean were estimated using Prism 9 (GraphPad). For [B], total concentration of ParBpSM was used instead of free ParBpSM concentration due to technical reasons.
nsDNA-carpeted flow cell preparation
Request a detailed protocolThe flow cells coated with lipid bilayer with attached biotin (DOPC plus DOPE-biotin [1%]) were prepared essentially as described in Han and Mizuuchi, 2010 and rinsed with a buffer containing 25 mM Tris–HCl pH 7.4, 150 mM NaCl, and 5 mM MgCl2 and 0.1 mM CaCl2. Sonicated and biotinylated DNA was prepared as follows: 250 μl of 10 mg/ml sonicated salmon sperm DNA (Sigma) was sonicated for an additional 5 min (Misonix sonicator 3000, output level 6, pulsed on/off for 10 s each at 16°C) to size-weighted average length of ~500 bp. In order to biotinylate the DNA ends, the sonicated DNA (1 mg/ml) was incubated with 40 μM biotin-17-dCTP (Invitrogen) and 0.6 units TdT (NEB) in the buffer specified by the enzyme manufacturer at 37°C for 30 min. The reaction was stopped by heating at 70°C for 10 min, and unincorporated biotin-17-dCTP was removed by using S-200 HR Microspin columns (GE Healthcare). The DNA was ethanol precipitated and resuspended in TE buffer. To coat the flow cell with sonicated DNA, the DNA prepared as above was dissolved to 1 mg/ml in 25 mM Tris–HCl pH 7.4, 150 mM NaCl, 5 mM MgCl2, and 0.1 mM CaCl2, infused into the assembled flow cell, and incubated overnight at 4°C. Unbound DNA was removed by rinsing with 50 mM Tris–HCl (pH 7.4), 100 mM KCl, 2 mM MgCl2, and 10% glycerol.
TIRFM setup and image processing
Request a detailed protocolTotal internal reflection fluorescence (TIRF) illumination and microscopy, as well as the camera settings, were essentially as described (Hwang et al., 2013). Combined beam of a 488 nm diode-pumped, solid-state laser (Coherent) and a 633 nm HeNe laser (Research Electro-Optics) was pointed through a fused silica prism onto the top side of the sample flow cell. Fluorescence emission was collected through a ×40 Plan Apo VC, NA 1.4 oil-immersion objectives (Nikon) and magnifier setting at ×1.5. The laser excitation lines were blocked below objective lens with notch filters (NF03-488E and NF03-633E, Semrock). The fluorescence images were captured by an EMCCD camera (Andor IXON +897) through a Dual-View module (630DCXR cube, Photometrics; short/long pass filters, SP01-633RS, LP02-633RE, Semrock). Microscopy experiments were carried out at room temperature (~23°C). Typical camera settings were digitization 16 bit at 1 MHz, preamplifier gain 5.2, vertical shift speed 2 MHz, vertical clock range: normal, EM gain 40, EMCCD temperature set at –98°C, baseline clamp ON. Images were acquired at exposure time 100 ms with frame rate 1, 0.4, or 0.2 Hz using MetaMorph 7 software (Molecular Devices) and analyzed using ImageJ software (NIH) as described (Hwang et al., 2013). Data were analyzed with Prism 9 (GraphPad).
Estimation of the nsDNA-carpet-bound protein densities from the observed fluorescence intensities
Request a detailed protocolParApSM and ParBpSM density on the nsDNA-carpet (dimers/μm2) were calculated from the fluorescence intensity of acquired images essentially according to the procedure described in Figure S4 legend in Vecchiarelli et al., 2016. The labeled protein samples used in the experiments were diluted to different concentrations (0–8 μM) in a buffer (50 mM Tris–HCl, pH 7.4, 100 mM KCl, 2 mM MgCl2, 10% glycerol, 1 mM DTT, 1 mg/ml α-casein, and 0.6 mg/ml ascorbic acid) and infused into flow cell coated with 1,2-dioleoyl-sn-glycero-3-phosphocholine. Fluorescence intensity data were collected before the protein arrival (background), with the protein sample in the flow cell, and after washing the flow cell with buffer (background) using the illumination and image acquisition parameter settings used for the series of experiments for which the conversion parameters were prepared. The fluorescence signal from the protein sample in the solution was obtained by subtracting the background signal with buffer only (mostly camera dark noise). From the wavelength, the refractive indices of fused silica slide glass and the reaction buffer, and the illumination angle, the evanescence penetration depths were calculated to be 131 and 170 nm for 488 and 633 nm excitation beams, respectively. The protein concentration and evanescence penetration volume yielded the number of protein molecules, and when bound to the flow cell surface, would produce the observed fluorescence signal, yielding the conversion factor used.
ParApSM and ParBpSM association and dissociation from the nsDNA-carpet
Request a detailed protocolTwo inlet flow cells were assembled and coated with sonicated salmon sperm DNA as described (Vecchiarelli et al., 2013). One inlet was connected to syringe A containing ‘binding solution’ and the other to syringe B containing ‘wash solution.’ For protein binding to the nsDNA-carpet, syringe A delivered at 5 μl/min, while syringe B at 0.5 μl/min. To start the wash, the flow rates of the two syringes were reversed keeping the total flow rate constant. Experiments were done at room temperature in pSM19035 buffer: 50 mM Tris–HCl (pH 7.4), 100 mM KCl, 2 mM MgCl2, 10% glycerol, 1 mM DTT, 0.1 mg/ml α-casein, 0.6 mg/ml ascorbic acid, and 1 mM ATP. Additional components of the binding and washing solutions were as specified in the figure legends. Syringe components were preincubated before infusion at 23°C for 30 min or longer. The association and dissociation data points represent the means with error bars (standard errors of mean) of repeat experiments (N) as indicated for each panel. The dissociation curves were fitted to a single- or a double-exponential equation after subtraction of background in the absence of protein with plateau level set to zero when necessary to avoid large plateau values, and the fraction of fast-dissociating population, the koff, and symmetrized error ranges reflecting the larger error of the 95% confidence interval below and above the mean were estimated using Prism 9 (GraphPad).
Data availability
All data generated or analysed during this study are included in the manuscript and supporting file; source data files have been provided for all figures presented.
References
-
Dual role of DNA in regulating ATP hydrolysis by the sopa partition proteinThe Journal of Biological Chemistry 284:30067–30075.https://doi.org/10.1074/jbc.M109.044800
-
A conserved mechanism drives partition complex assembly on bacterial chromosomes and plasmidsMolecular Systems Biology 14:e8516.https://doi.org/10.15252/msb.20188516
-
ParB spreading requires DNA bridgingGenes & Development 28:1228–1238.https://doi.org/10.1101/gad.242206.114
-
Partitioning of P1 plasmids by gradual distribution of the atpase paraMolecular Microbiology 78:1182–1198.https://doi.org/10.1111/j.1365-2958.2010.07398.x
-
Brownian ratchet mechanism for faithful segregation of low-copy-number plasmidsBiophysical Journal 112:1489–1502.https://doi.org/10.1016/j.bpj.2017.02.039
-
ParA-mediated plasmid partition driven by protein pattern self-organizationThe EMBO Journal 32:1238–1249.https://doi.org/10.1038/emboj.2013.34
-
Bacterial partition complexes segregate within the volume of the nucleoidNature Communications 7:12107.https://doi.org/10.1038/ncomms12107
-
A three-dimensional parf meshwork assembles through the nucleoid to mediate plasmid segregationNucleic Acids Research 45:3158–3171.https://doi.org/10.1093/nar/gkw1302
-
Mechanism of the asymmetric activation of the mind atpase by mineMolecular Microbiology 85:271–281.https://doi.org/10.1111/j.1365-2958.2012.08110.x
-
Single-molecule analysis of proteinxdna complexes formed during partition of newly replicated plasmid molecules in Streptococcus pyogenesThe Journal of Biological Chemistry 284:30298–30306.https://doi.org/10.1074/jbc.M109.035410
-
P1 plasmid segregation: accurate redistribution by dynamic plasmid pairing and separationJournal of Bacteriology 192:1175–1183.https://doi.org/10.1128/JB.01245-09
-
Molecular anatomy of the Streptococcus pyogenes psm19035 partition and segrosome complexesNucleic Acids Research 39:2624–2637.https://doi.org/10.1093/nar/gkq1245
-
Molecular anatomy of para-para and para-parb interactions during plasmid partitioningThe Journal of Biological Chemistry 290:18782–18795.https://doi.org/10.1074/jbc.M115.649632
Article and author information
Author details
Juan Carlos Alonso

Funding
National Institute of Diabetes and Digestive and Kidney Diseases
- Kiyoshi Mizuuchi
Ministerio de Ciencia e Innovación (2018-097054-B-I00)
- Juan Carlos Alonso
Ministerio de Ciencia e Innovación (2021AEP031)
- Juan Carlos Alonso
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to our colleagues Keir Neuman, Barbara Funnell, Anthony Vecchiarelli, James Taylor, Michiyo Mizuuchi, Min Li, Masaki Osawa, and William Carlquist for helpful suggestions and discussion, and to Maria Moreno-del Álamo for providing the information concerning the in vivo functionality of ParApSM-GFP/ParBpSM-GCE. This work was supported by the intramural research fund for National Institute of Diabetes and Digestive and Kidney Diseases (KM) and the Ministerio de Ciencia e Innovación, Agencia Estatal de Investigación (MCIN/AEI)/FEDER, 2018-097054-B-I00 and 2021AEP031 to JCA.
Copyright
This is an open-access article, free of all copyright, and may be freely reproduced, distributed, transmitted, modified, built upon, or otherwise used by anyone for any lawful purpose. The work is made available under the Creative Commons CC0 public domain dedication.
Metrics
-
- 550
- views
-
- 142
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Distinct architectural requirements for the parS centromeric sequence of the pSM19035 plasmid partition machinery
eLife 11:e79480.
https://doi.org/10.7554/eLife.79480




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}