The RAM signaling pathway links morphology, thermotolerance, and CO2 tolerance in the global fungal pathogen Cryptococcus neoformans
- Department of Plant Biology, University of Georgia, United States
- Department of Microbiology, University of Georgia, United States
- Department of Pediatrics, Carver College of Medicine, University of Iowa, United States
- Department of Microbiology and Immunology, Carver College of Medicine, University of Iowa, United States
Abstract
The environmental pathogen Cryptococcus neoformans claims over 180,000 lives each year. Survival of this basidiomycete at host CO2 concentrations has only recently been considered an important virulence trait. Through screening gene knockout libraries constructed in a CO2-tolerant clinical strain, we found mutations leading to CO2 sensitivity are enriched in pathways activated by heat stress, including calcineurin, Ras1-Cdc24, cell wall integrity, and Regulator of Ace2 and Morphogenesis (RAM). Overexpression of Cbk1, the conserved terminal kinase of the RAM pathway, partially restored defects of these mutants at host CO2 or temperature levels. In ascomycetes such as Saccharomyces cerevisiae and Candida albicans, transcription factor Ace2 is an important target of Cbk1, activating genes responsible for cell separation. However, no Ace2 homolog or any downstream component of the RAM pathway has been identified in basidiomycetes. Through in vitro evolution and comparative genomics, we characterized mutations in suppressors of cbk1Δ in C. neoformans that partially rescued defects in CO2 tolerance, thermotolerance, and morphology. One suppressor is the RNA translation repressor Ssd1, which is highly conserved in ascomycetes and basidiomycetes. The other is a novel ribonuclease domain-containing protein, here named PSC1, which is present in basidiomycetes and humans but surprisingly absent in most ascomycetes. Loss of Ssd1 in cbk1Δ partially restored cryptococcal ability to survive and amplify in the inhalation and intravenous murine models of cryptococcosis. Our discoveries highlight the overlapping regulation of CO2 tolerance and thermotolerance, the essential role of the RAM pathway in cryptococcal adaptation to the host condition, and the potential importance of post-transcriptional control of virulence traits in this global pathogen.
Editor's evaluation
This paper reports the identification of molecular determinants of CO2 tolerance in the human fungal pathogen Cryptococcus neoformans. The results are important for our understanding of how the fungus adapts from the ambient atmosphere to the CO2-enriched environment in the human host, and the findings are convincing and rely on biochemical, molecular, and genetic techniques. The results should be of interest to a broad community in the life sciences including microbiologists and infectious diseases investigators.
https://doi.org/10.7554/eLife.82563.sa0Introduction
There are over 278,000 cases of cryptococcal meningitis every year, causing over 180,000 deaths (Rajasingham et al., 2017). Cryptococcal meningitis is primarily caused by the ubiquitous environmental fungus Cryptococcus neoformans. Airborne spores or desiccated yeast cells of C. neoformans are inhaled into the lungs, where they are cleared or remain dormant until reactivation upon host immunosuppression (Casadevall and Perfect, 1998; Zhao et al., 2019).
Litvintseva et al. found that most environmental Cryptococcus isolates cannot cause fatal disease in mouse models of cryptococcosis, despite having similar genotypes and in vitro phenotypes to known virulent isolates, including thermotolerance, melanization, and capsule production (Litvintseva and Mitchell, 2009). Mukaremera et al., 2019 also observed that in vitro phenotype assays for thermotolerance, capsule production, titan cell formation, or fluconazole heteroresistance could not differentiate high-virulence strains from low-virulence strains. These observations raise the possibility that other, unidentified virulence traits are important for Cryptococcus pathogenesis. Tolerance to host levels of CO2 (~5% CO2 in the host vs. ~0.04% in ambient air) is likely a significant factor separating the potentially virulent natural isolates from the non-pathogenic environmental isolates that Litvintseva et al. tested (Krysan et al., 2019; Litvintseva and Mitchell, 2009).
The ability to adapt to host conditions is a prerequisite for cryptococcal pathogenesis. For instance, the ability of C. neoformans to replicate at human body temperature (≥37°C) has been extensively investigated. Many genes have been shown to be essential for thermotolerance (Perfect, 2006; Stempinski et al., 2021; Yang et al., 2017), including calcineurin which is currently being explored for antifungal drug development (Gobeil et al., 2021). By contrast, the underlying mechanisms or genes that play a role in CO2 tolerance have yet to be identified. Here, we set out to identify CO2-sensitive mutants and to gain the first insight into the genetic components involved in CO2 tolerance in C. neoformans.
Results
CO2 sensitivity is independent of pH
Our previous work indicates that many C. neoformans environmental strains are sensitive to 5% CO2 when grown on buffered RPMI media, commonly used for mammalian cell cultures and testing antifungal susceptibility (Krysan et al., 2019). CO2 at host concentrations also acts synergistically with the commonly used antifungal drug fluconazole in inhibiting cryptococcal growth on buffered RPMI media. Because CO2 lowers the pH of aqueous environments, it is possible that the CO2 growth inhibitory effect or its synergy with fluconazole is simply due to lower medium pH. To address this question, we tested sensitivity to fluconazole of wild-type (WT) strain H99 using E-test on buffered RPMI media of either pH 6 or pH 7, with or without 5% CO2. In this E-test, the size of halo (clearance zone) reflects fungal susceptibility to fluconazole. As shown in Figure 1A, clearance zones were much larger in 5% CO2 relative to those in ambient air at both pH 6 and pH 7, indicating that CO2 sensitizes cryptococcal susceptibility to fluconazole. Furthermore, CO2 inhibits the growth of H99 at both pH 6 and pH 7 (smaller colony size in 5% CO2 relative to that in ambient air). Additionally, growth of CO2-sensitive environmental strain A7-35-23 (Krysan et al., 2019) was severely inhibited by 5% CO2 at both pH 6 and pH 7 (Figure 1B). In general, C. neoformans grows better at acidic pH (can grow well in pH 3), and both A7-35-23 and H99 grew better at pH 6 than at pH 7 in ambient air (Figure 1B). Taken together, these results suggest that cryptococcal growth inhibition by CO2 is not simply due to lowered pH.
Figure 1
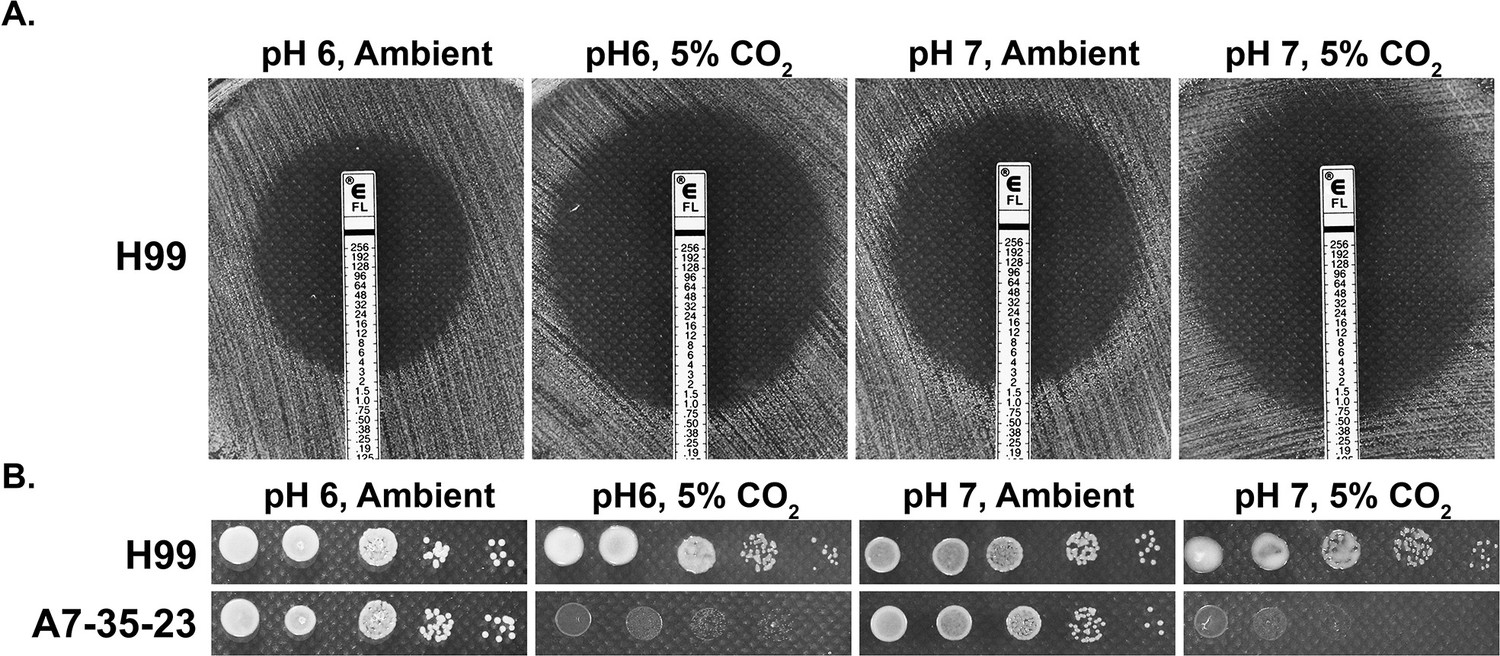
CO2 sensitivity is not simply due to lowered medium pH.
(A) H99 cells were plated onto RPMI solid medium buffered to either pH 6 or pH 7. Fluconazole containing E-test strips were placed onto the lawn of H99 cells, and the plates were incubated at 37°C in ambient air or in 5% CO2. The larger the halo surrounding the E-strip, the more sensitive the cells are to fluconazole. The intercept value of the halo with the E-strip is the minimal inhibitory concentration. (B) Cells of the previously identified CO2-tolerant H99 and CO2-sensitive A7-35-23 were serial diluted, spotted onto RPMI media buffered to pH 6 or pH 7, and incubated at 37°C in ambient air or in 5% CO2.
Identifying genes important for CO2 tolerance
To identify genes involved in CO2 tolerance in C. neoformans, we screened gene deletion mutants constructed in the CO2-tolerant clinical reference strain H99. For large-scale screening, we used the nutrient rich yeast peptone dextrose (YPD) medium on which C. neoformans grows well. Accordingly, we tested the growth of two CO2-sensitive environmental strains and the CO2-tolerant H99 strain in different levels of CO2 when cultured on YPD. As expected, relative to H99, the CO2-sensitive strains A7-35-23 and A1-38-2 grew poorly at 5% CO2 and worse at 20% CO2 (Figure 2A). Using this approach, the following deletion mutant libraries were screened at 20% CO2 on YPD media: a set of strains previously constructed in our lab, the collections constructed by the Madhani lab, and a set generated in the Lodge Lab (Chun and Madhani, 2010). As some mutants are known to be temperature sensitive, we carried out the screens at 30°C rather than 37°C. From over 5000 gene knockout mutants screened (~7000 protein coding genes in the H99 genome), 96 were found to be sensitive to CO2 by visual observation (Supplementary file 1). We noticed that knockout mutants for multiple pathways known to be activated by heat stress are CO2 sensitive, including the Ras1-Cdc24 pathway, calcineurin, cell wall integrity (CWI), and Regulator of Ace2 and Morphogenesis (RAM). This finding indicates an overlapping nature of these two traits.
Figure 2 with 3 supplements see all
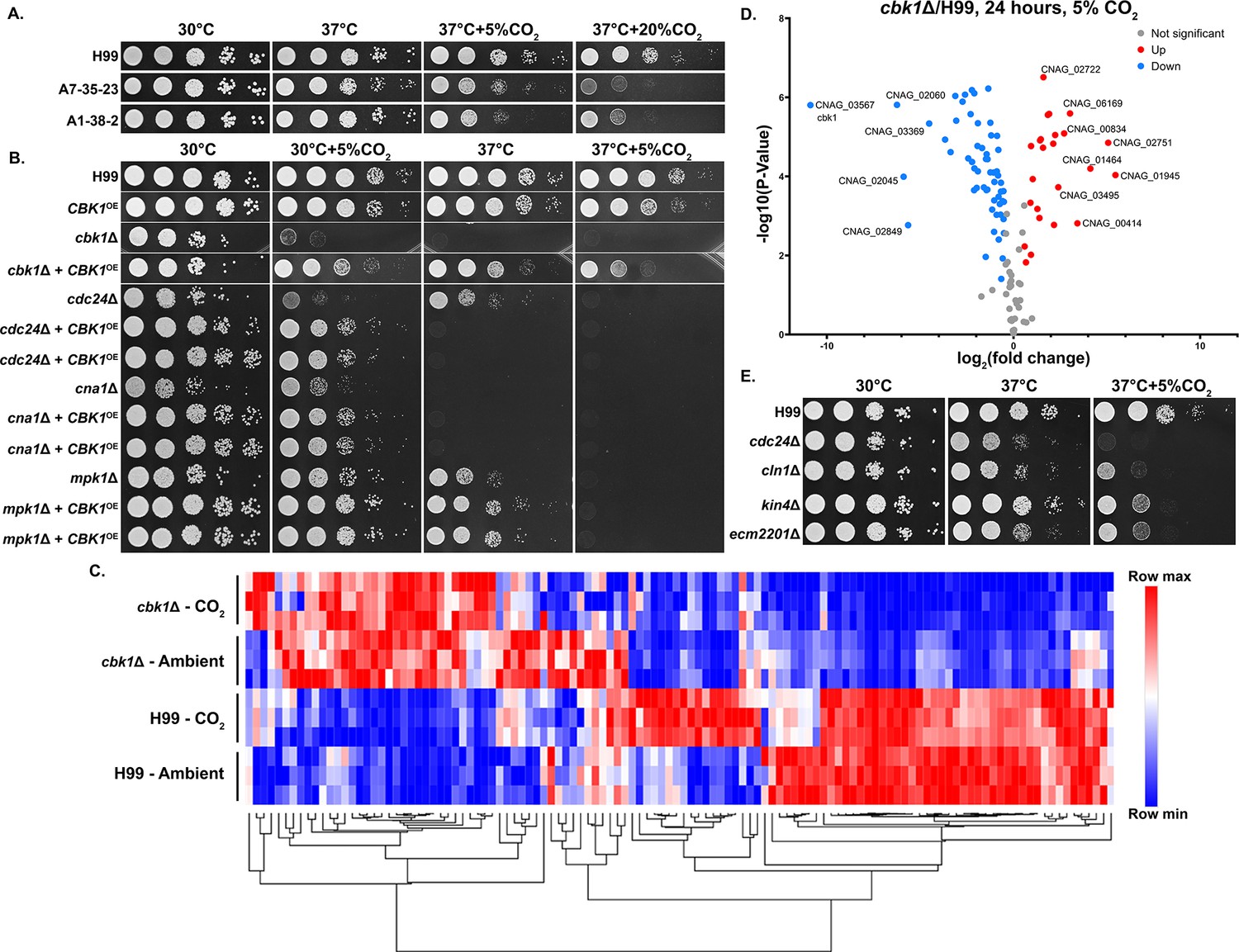
The Regulator of Ace2 and Morphogenesis (RAM) pathway effector kinase Cbk1 is critical for CO2 tolerance.
(A). The clinical reference strain H99 and environmental strains A7-35-23 and A1-38-2 were grown overnight in yeast peptone dextrose (YPD), serially diluted, and spotted onto solid YPD media plates. Photographs were taken 2 days after incubation in the indicated condition. (B) This serial dilution spotting assay was similarly performed for H99 and the mutants indicated. Two independent overexpression transformants for each mutant background were included as biological replicates. (C) Heatmap showing normalized total RNA counts of NanoString targets in H99 and cbk1Δ cultured at either ambient or 5% CO2, red indicates higher and blue indicates lower transcript abundance. (D) Volcano plot showing significantly differentially expressed transcripts (p-value of <0.05) in the cbk1Δ compared to H99 in the 5% CO2 condition. (E) Serial dilution spotting assay of H99 and four of the mutants found in the deletion set screening to be CO2 sensitive which also correspond to significantly downregulated genes shown in the volcano plot.
We were surprised by the absence of components of adenylyl cyclase-PKA(protein kinase A) pathway from the set of hits. In Candida albicans, the adenylyl cyclase pathway is crucial for the yeast-hypha transition in response to host levels of CO2 (Klengel et al., 2005). This pathway has also been proposed to play an important role for Cryptococcus to sense CO2, and the carbonic anhydrase Can1 is required for growth at low concentrations of CO2 (Bahn et al., 2005; Mogensen et al., 2006). However, we found that adenylyl cyclase pathway mutants showed no growth defects at host levels CO2, including the adenylyl cyclase mutant cac1Δ, the adenylyl cyclase associated protein mutant aca1Δ, the alpha G protein subunit mutant gpa1Δ, and the cAMP-dependent protein kinase mutant pkr1Δ (Figure 2—figure supplement 1). This indicates that growth defects in response to host levels of CO2 are likely independent of bicarbonate activation of adenylyl cyclase. This is not unexpected given that bicarbonate is not a limiting factor under the high level of CO2 used in our screen.
Because the calcineurin, Ras1-Cdc24, CWI, and RAM pathways are all activated at host temperature and were identified in our screen for CO2-sensitive mutants, we reasoned their downstream effectors may be related or genetically interact. As the RAM pathway effector kinase mutant cbk1Δ showed the most severe defect in thermotolerance and CO2 tolerance compared to the mutants of the other pathways, we first overexpressed the gene CBK1 in the following mutants, cdc24Δ (Ras1-Cdc24), mpk1Δ (CWI), cna1Δ (Calcineurin), and the cbk1Δ mutant itself and observed their growth at host temperature and host CO2 (Figure 2B). Overexpression was achieved by placing the CBK1 open reading frame after the inducible CTR4 promoter, which is highly activated in YPD media (Ory et al., 2004; Wang et al., 2014; Wang et al., 2012). The CBK1 overexpression construct was specifically integrated into the ‘safe haven’ locus SH2 (Lin et al., 2020; Upadhya et al., 2017) in each mutant strain background to avoid complications due to positional effects. We additionally confirmed overexpression of CBK1 by RT-PCR (Figure 2—figure supplement 2). As expected, the growth defects of the cbk1Δ mutant at 37°C with and without 5% CO2 were largely restored by CBK1 overexpression. At 30°C, overexpression of CBK1 restored the growth of the mpk1Δ mutant, the cna1Δ mutant, and the cdc24Δ mutant in the CO2 condition. In terms of thermotolerance, overexpression of CBK1 restored growth of mpk1Δ but not cna1Δ, while the growth defect of cdc24Δ at 37°C was exacerbated. CBK1 overexpression failed to rescue growth of any of these mutants when both stressors were present (37°C+5% CO2). We found that overexpression of CBK1 in the WT H99 background caused a modest growth defect at 37°C+5% CO2. Thus, the detrimental effects from CBK1 overexpression under this growth condition may partially explain its inability to fully rescue growth of these tested CO2-sensitive mutants. The reciprocal overexpression of CDC24, MPK1, or CNA1 in the cbk1Δ mutant background did not restore growth under 37°C and/or 5% CO2 (Figure 2—figure supplement 3). These results support a hypothesis that Cbk1 integrates multiple stress response pathways to regulate both CO2 tolerance and thermotolerance.
To determine the extent of Cbk1’s role in CO2 tolerance, we conducted NanoString gene expression profiling of the WT H99 and cbk1Δ mutant cultured in ambient air and in 5% CO2 at 30°C (Figure 2C). Transcript levels of 118 genes were measured, and those genes were chosen based on RNA sequencing results from a separate study (Ristow et al., in preparation). In that study, these genes were differentially expressed in CO2 vs. ambient air conditions in either two CO2-sensitive or two CO2-tolerant natural strains (Source data 1). Out of these 118 CO2-associated genes, 81 were found to be significantly differentially expressed in the cbk1Δ mutant in both ambient air and in 5% CO2, indicating they are intrinsically dysregulated in the cbk1Δ mutant. 57/81 of these genes are downregulated and 24/81 upregulated compared to the WT H99 strain (Figure 2D). Interestingly, 16/57 of the downregulated genes were also hits in our deletion set screening. We picked four of these deletion mutants which showed high sensitivity in our screen, to assay their sensitivity to host CO2 conditions by spotting assay (Figure 2E). Taken together, this transcriptomic profiling shows that loss of Cbk1 significantly affects the expression of CO2-related genes.
The RAM signaling pathway is critical for normal morphology, thermotolerance, and CO2 tolerance
The RAM pathway effector kinase Cbk1 is part of the NDR/LATS family of kinases, which is conserved from yeast to humans and affects a wide range of cellular functions including cell-cycle regulation. In C. neoformans, various virulence factors are impacted by deletion of CBK1, including urease activity and thermotolerance (Lee et al., 2016). Through our genetic screen for CO2-sensitive mutants, we found that all tested Cryptococcus RAM pathway mutants are extremely sensitive to 5% CO2 and high temperature, and they show no growth at 37°C+5% CO2 (Figure 3A). In ascomycetes such as Saccharomyces cerevisiae and C. albicans, RAM pathway mutants are defective in cytokinesis and exhibit loss of polarity, resulting in enlarged round cells that cluster together (Saputo et al., 2012; Figure 3—figure supplement 1A). In contrast, though defective in cytokinesis (Magditch et al., 2012; Walton et al., 2006), Cryptococcus RAM pathway mutants are hyper-polarized and constitutively form clusters of elongated pseudohyphal cells (Figure 3B). Moreover, we found that while the C. albicans homozygous cbk1ΔΔ mutant exhibits a general growth defect compared to the WT control, it shows no apparent specific growth defect at 37°C with or without 5% CO2 (Figure 3—figure supplement 1C). These results suggest that, although the RAM pathway is conserved in its role in cytokinesis, the effects of its downstream targets are divergent between ascomycetes and basidiomycetes.
Figure 3 with 1 supplement see all
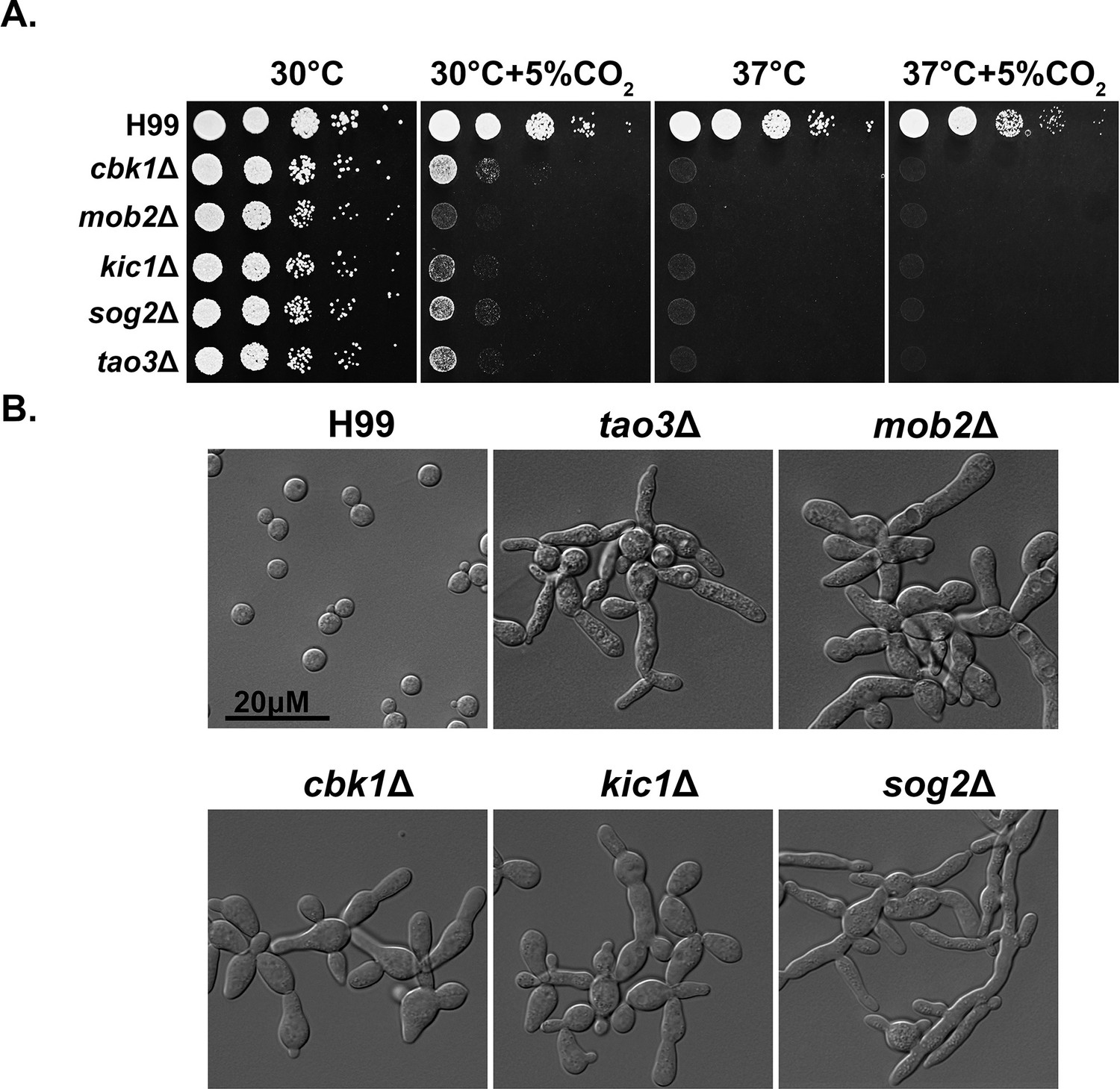
The Regulator of Ace2 and Morphogenesis (RAM) pathway is critical for normal morphology, thermotolerance, and CO2 tolerance.
(A) Cryptococcus neoformans WT H99 and RAM pathway mutants were serially diluted, spotted onto yeast peptone dextrose (YPD) medium, and incubated for 2 days at the indicated condition. (B) The cellular morphology of C. neoformans WT H99 and RAM pathway mutants cultured in YPD medium.
Suppressors of the cbk1Δ mutant show improved growth at host conditions
In ascomycetes, Ace2 is a key downstream transcription factor of the RAM pathway (hence in the name of RAM − Regulator of Ace2 and Morphogenesis), which is important for the activation of genes responsible for cell separation as well as a large number of genes with other functions (Mulhern et al., 2006; Wakade et al., 2020). However, no homolog to Ace2 has been identified in Cryptococcus or other basidiomycetes. Furthermore, no downstream targets of the RAM pathway have been identified in any basidiomycetes. To investigate potential downstream effectors of the RAM pathway in Cryptococcus, we screened for spontaneous suppressor mutants of cbk1Δ. To do so, cbk1Δ mutant cells from an overnight culture in liquid YPD at 30°C were plated onto solid YPD media and incubated for 2 days at 37°C+5% CO2. Out of >1×108 cells plated and cultured under this condition that is inhibitory for growth of the original cbk1Δ mutant, 11 suppressor colonies were isolated for further examination and sequencing. All the suppressor isolates showed dramatically improved growth over the original cbk1Δ mutant at 37°C and modestly improved growth at 37°C+5% CO2 (Figure 4C). Based on their distinctive phenotypes, the 11 suppressors were classified into two groups: sup1 (2/11) and sup2 (9/11). Shorter chains of cells in both groups indicate a partial restoration in cytokinesis (Figure 4D). The sup2 group has slightly improved growth at 37°C+5% CO2 and forms shorter chains of cells compared to the sup1 group (Figure 4C and D). Besides of these observations, sup1 and sup2 displayed similar phenotypes in growth assays including the cryptococcal virulence traits tested, including melanin production, capsule, urease activity, and cell wall stress tolerance. (Figure 4—figure supplement 1). Both sup1 and sup2 showed no improved growth compared to the cbk1Δ mutant at pH 7.4 37°C+5% CO2. This is likely due to the detrimental combination of high temperature, CO2, and high pH, as the WT also showed significantly reduced growth in this condition. (Figure 4—figure supplement 1A). Because RAM pathway suppressor mutants were previously identified after treatment with calcineurin inhibitor FK506 and showed improved growth in FK506 and restored mating (Magditch et al., 2012), we also tested our suppressors’ growth in FK506 and their ability to mate. We found that both sup1 and sup2 failed to restore growth of the cbk1Δ on media supplemented with FK506 or restore the ability to mate with the congenic strain H99a (Figure 4—figure supplement 1).
Figure 4 with 2 supplements see all
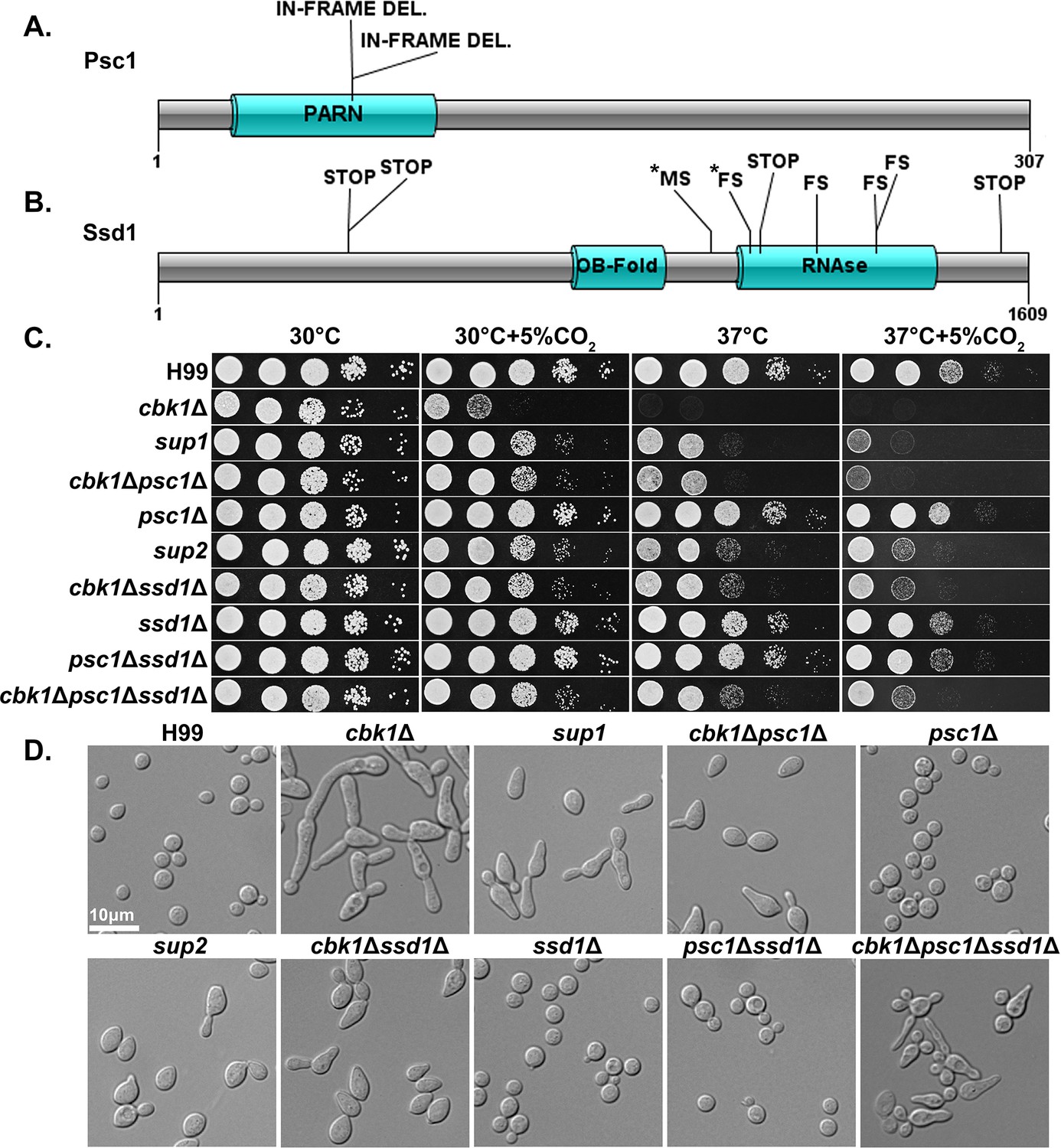
Natural suppressors of the Regulator of Ace2 and Morphogenesis (RAM) pathway cbk1Δ mutant restore multiple defects.
(A) Protein diagram of Psc1 showing the effects and positions of suppressor mutations in the two sup1 type natural suppressors. (B) Protein diagram of Ssd1 and the effects and positions of suppressor mutations in Ssd1 in the nine sup2 type natural suppressors. STOP indicates a non-sense mutation, MS a missense mutation, and FS a frameshift mutation. Asterisks (*) indicate mutations in the same suppressor strain. (C) Serial dilutions of H99 and the mutant strains were spotted onto yeast peptone dextrose (YPD) agar media and incubated for 2 days in the indicated condition to observe growth. (D) The cellular morphology of H99 and the mutant strains in liquid YPD cultures were examined under microscope.
Along with the original cbk1Δ mutant, we sequenced the genomes of the 11 cbk1Δ suppressors. By comparing their genome sequences with each other and with the original cbk1Δ mutant, we found that both sup1 type suppressor mutants contained a disruptive in-frame deletion at the same location in CNAG_01919, which encodes a putative Poly(A)-specific ribonuclease (PARN) domain-containing protein (Figure 4A). This domain was previously reported in S. pombe proteins (Marasovic et al., 2013). Interestingly, through a BLAST search of the PARN domain, we did not identify this domain in any protein in the genomes of S. cerevisiae, C. albicans, or other ascomycetes but found it in basidiomycetes and higher eukaryotes. The in-frame deletion results in a change of two amino acids within the predicted PARN domain, the only discernable domain present in this protein. We named this previously uncharacterized gene Partial Suppressor of cbk1Δ (PSC1). All nine sup2 isolates contained loss of function or missense mutations in the gene CNAG_03345 (Figure 4B), which encodes an RNA-binding protein homologous to S. cerevisiae Ssd1p, a known suppressor of cbk1Δ phenotypes in S. cerevisiae. ScSsd1p represses transcript translation and is negatively regulated by Cbk1p phosphorylation (Jansen et al., 2009; Wanless et al., 2014).
To confirm that the putative loss-of-function mutations in SSD1 and PSC1 are responsible for suppressing cbk1Δ phenotypes, we created cbk1Δssd1Δ and cbk1Δpsc1Δ double mutants together with the control single mutants ssd1Δ and psc1Δ. Indeed, relative to the cbk1Δ mutant, the double mutants showed reduced sensitivity to host temperature and CO2 levels (Figure 4C), similar to the natural suppressor mutants. Likewise, the morphology of the double mutants resembles that of the spontaneous suppressor mutants (Figure 4D). The deletion of SSD1 and PSC1 alone in the WT background did not yield any discernable phenotype. The results confirm that loss-of-function mutations in SSD1 and PSC1 are responsible for partial suppression of the cbk1Δ mutant’s growth defects observed in the isolated suppressor strains. Interestingly, sup2 and the cbk1Δssd1Δ mutants both grew noticeably better than sup1 and cbk1Δpsc1Δ at 37°C and 37°C+5% CO2. To test the genetic interaction between the two suppressor genes SSD1 and PSC1, we created a triple cbk1Δpsc1Δssd1Δ mutant and the control strain psc1Δssd1Δ. The psc1Δssd1Δ control strain did not exhibit any defect and grew similarly well to either single mutant or the WT (Figure 4C). The triple mutant cbk1Δpsc1Δssd1Δ grew similarly well as sup2 or cbk1Δssd1Δ at 37°C+5% CO2 (Figure 4C). However, the triple mutant displayed aberrant morphology and budding defects which are not observed in the natural suppressor mutants or the cbk1Δssd1Δ and cbk1Δpsc1Δ double mutants (Figure 4D). These results suggest that Psc1 and Ssd1 may function in the same pathway in regulating thermotolerance and CO2 tolerance, but their downstream effects on cell separation and/or polarized growth may be overlapping and distinct.
To determine if the suppressor mutations restore transcript abundance of the differentially expressed genes under CO2 in cbk1Δ, we compared the profiles of cbk1Δ to the two suppressor mutants: sup1 and sup2. Overall, we found that the spontaneous suppressors do not restore transcript abundances of most differentially expressed genes in cbk1Δ to WT levels (Figure 4—figure supplement 2), suggesting that suppressors affect post-transcriptional regulation of CO2 tolerance.
Spontaneous suppressors of cbk1Δ mutant show improved ability to survive and replicate in the host
RAM mutants have previously been found to be attenuated in virulence in the invertebrate wax moth larva infection model and mouse intranasal infection models (Lee et al., 2016; Magditch et al., 2012). Occasionally, cryptococcal strains with point mutations in RAM genes cause death of mice when revertant mutations occur, which restore the function of the RAM pathway (Magditch et al., 2012). As shown above and consistent with previous literature, the cbk1Δ mutant shows a severe growth defect at host temperature and CO2 concentrations (Figure 3C). Because sup1 and sup2 both largely restored growth to the cbk1∆ mutant at 37°C but only modestly restored growth at 37°C+5% CO2, we decided to test if, and by how much, these suppressor mutations would affect the virulence of the cbk1Δ mutant. We infected mice with 1×104 cells of WT, cbk1Δ, sup1, or sup2 intranasally. In this intranasal infection model, the WT H99 strain establishes lung infection first and typically disseminates to other organs including the brain by 7–10 days post-infection (DPI). Mice infected by H99 normally become morbidly ill by 3–4 weeks post-infection and have a high fungal burden in the lungs, brain, and kidney (Chadwick and Lin, 2020; Lin et al., 2022).
As expected, all mice infected with H99 were moribund by DPI 26 (Figure 5A), while those infected with the cbk1Δ mutant survived until the experiment was terminated at DPI 60. Surprisingly, sup1 and sup2 strains did not cause any mortality either. The organ fungal burden, however, revealed differences in virulence levels between these strains. At the time of euthanasia for H99-infected mice (prior to DPI 26), the median fungal burden in the lungs, brains, and kidneys was 2.1×108, 1.4×106, and 2.4×104 colony-forming units (CFUs) per organ, respectively (Figure 5B). As expected, mice completely cleared the cbk1Δ mutant at DPI 35. Surprisingly, despite largely restored growth at 37°C, sup1 was completely cleared from the mouse lungs by DPI 35, similar to the cbk1Δ mutant. In comparison, although sup2 did not cause any death during the study period, it was able to replicate in the mouse lungs. The median lung fungal burden at DPI 35 was 8.2×104, over eightfold higher than the original inoculum. The sup2 strain maintained the same high lung fungal burden at DPI 60 (Figure 5B), indicating that it can persist in the lung tissue. The only in vitro difference observed between sup1 and sup2 was better growth of sup2 at host CO2 levels which may explain the difference in their ability to propagate and persist in the mouse lung. However, it is worth nothing that due to the complex host environment, there could be other unrecognized factors contributing to the differences in vivo.
Figure 5

Suppressor mutants are partially restored for phagocytosis and can disseminate in the intravenous infection model of cryptococcosis.
(A) Mice were infected with 1×104 fungal cells intranasally, and their survival was monitored for 60 days post-infection. (B) At day 35 post-infection (DPI 35) and at the time of termination (DPI 60), 5 out of 10 mice per group for the cbk1Δ mutant, sup1 and sup2 groups were harvested for brains, kidneys, and lungs. For H99 infected mice, they were euthanized at their clinical end point (all before DPI 26). Tissue homogenate was serially diluted and plated onto YNB(yeast nitrogen base) medium to count the colony-forming units (CFUs) to measure the fungal burden per organ. (C) Murine macrophage J774A.1 cells were co-incubated with 2×106 cryptococcal cells opsonized with serum from naïve mouse for 2 hr. Non-adherent or phagocytosed cells were washed, and cryptococcal cells were released and then serially diluted before plating onto YNB medium for measurement of CFUs. (D) The same as above, except opsonization, was performed with serum of mice vaccinated against cryptococcosis. (E) Mice were challenged with 1×105 cryptococcal cells intravenously. At day 5 post-infection, five mice per group were sacrificed. Brains, kidneys, and lungs of euthanized mice were dissected and homogenized. Serial dilutions were plated to count CFUs for quantification of fungal burden per organ.
Although the spontaneous suppressor sup2 was able to replicate in the mouse lungs, no fungal burden was detected in the brain or the kidney at DPI 35 or 60 (no organisms were detected in any of the mice), indicating that the mutant was unable to disseminate. We considered two hypotheses: (1) inability of suppressor sup2 to disseminate from the lungs; (2) inability of suppressor sup2 to penetrate other organs from the blood. Because C. neoformans can disseminate from the lungs to other organs by a ‘Trojan Horse’ mechanism, where Cryptococcus travels within the mobile host phagocytes (Kechichian et al., 2007; Santiago-Tirado et al., 2017), we examined phagocytosis of the cbk1Δ mutant and its suppressors to test the first hypothesis. We expected that cryptococcal mutants defective in being phagocytosed by host cells might be defective in dissemination, and the cbk1Δ mutant was previously found to have a poor phagocytosis index (Lin et al., 2015). Here, we co-cultured murine macrophage JA774 cells with H99, cbk1Δ, sup1, sup2, the double mutant cbk1Δssd1Δ, or the control single mutant ssd1Δ. Because different types of opsonization can impact phagocytosis of C. neoformans, opsonization was performed using either naïve mouse serum (complement mediated phagocytosis) or serum from mice vaccinated against cryptococcosis (complement+antibody mediated phagocytosis). The serum (containing antibodies) from the vaccinated mice recognizes antigens present in the capsule of cryptococcal cells (Lin et al., 2022; Zhai et al., 2015). Consistent with our previous finding, phagocytosis of the cbk1Δ mutant was extremely low (~1% of the WT H99 level under complement mediated phagocytosis, Figure 5C). Opsonization with serum from vaccinated mice increased phagocytosis of cbk1Δ and the suppressor mutants, but the phagocytosis indexes of these mutants were still only 20% or less than that of the WT (Figure 5D). In both phagocytosis experiments, the suppressor mutants or the double mutants cbk1∆ssd1∆ and cbk1∆psc1∆ mutants showed increased phagocytosis relative to the cbk1∆ mutant. The poor phagocytosis of the cbk1Δ mutant and its suppressors may contribute to their lack of dissemination from the lungs to the other organs in the inhalation infection mouse model of cryptococcosis.
To test the second hypothesis, we infected mice intravenously with H99, cbk1Δ, sup1, sup2, the double mutant cbk1Δssd1Δ, or the control single mutant ssd1Δ. In this intravenous infection model, the barrier of the lungs is bypassed. H99 cells disseminate to the brain and other organs within hours (O’Connor et al., 2013). Because H99 rapidly disseminates in this model, infected mice typically reach moribundity after 1 week. Therefore, we euthanized mice at DPI 5 before H99-infected mice would have become moribund. As expected, H99-infected mice showed high fungal burdens in the lungs, brains, and kidneys, with the highest fungal burden in the brain (over 106 CFUs; Figure 5E). The cbk1Δ mutant failed to disseminate in this intravenous infection model as no viable cells were recovered in any organ. Similarly, we could not recover any sup1 cells from the lungs or the brain and only detected a few fungal cells in the kidney. In contrast, sup2 suppressor mutants were recovered in all three organs, albeit with reduced fungal burdens (~104 CFUs in the brain and a few hundred in lungs/kidney) compared to the WT H99 control group (Figure 5E). This finding indicates that the sup2 suppressor, once disseminated into the bloodstream, can invade other organs and replicate. Combined with the earlier observations that (1) both suppressors fully restore growth at host temperature and (2) sup2 is slightly more CO2 tolerant than sup1, the observation that only sup2 can survive, amplify, and persist in animals implicates an importance of CO2 tolerance in cryptococcal pathogenesis. Collectively, the results from phagocytosis, the inhalation infection model, and the intravenous infection model support the hypothesis that failure of the suppressor mutants to disseminate to other organs in the intranasal model is largely due to reduced phagocytosis and inability to escape the lungs. That said, other factors, such as increased systemic clearance by the immune system, could potentially contribute to the containment of the mutant in the lungs. Again, the cbk1Δssd1Δ mutant recapitulated the phenotype of the sup2 strain in intravenous infection model and other in vitro assays, demonstrating that our observed sup2 phenotypes are due to disruption of SSD1.
As mutants that are temperature sensitive have reduced virulence in the mouse model of cryptococcosis, we decided to test the virulence of these strains in the Galleria mellonella larvae infection model to remove temperature as a variable. We inoculated G. mellonella larvae with 5×104 cells of WT, cbk1Δ, sup1, or sup2 and maintained the larvae at 30°C as previously described (Magditch et al., 2012). PBS buffer inoculated larvae were included as a sham control. Infected larvae (n=20 per strain) were monitored for survival over a period of 15 days post-inoculation. All 20 of the larvae inoculated with the H99 strain died between DPI 3 and DPI 10 (Figure 6A). In comparison, only 2/20 of the cbk1Δ mutant-infected larvae died during this period. Interestingly, 5/20 sup1-infected larvae and 7/20 sup2-infected larvae died in this experiment (Figure 6A), indicating their partially restored virulence in this larva infection model. To further confirm the observed differences between these strains in this model conducted at 30°C, we infected five larvae with 5×104 cells per strain and measured their fungal burden at day 5 post-inoculation. At DPI 5, the mean fungal burden of WT-infected larvae was 1.5×107 CFUs (Figure 6B). In comparison, the mean fungal burden for the cbk1Δ-infected larvae was only 8.4×102 CFUs, which is almost 20,000-fold lower than the WT control group and about 60-fold lower than the original inoculum, indicating that most cbk1Δ cells have been cleared by this time point. The mean fungal burden of sup1-infected larvae was 9.6×103 CFUs, while the mean fungal burden sup2-infected larvae was 2.3×104 CFUs (Figure 6B). These results indicate that both sup1 and sup2 partially rescued virulence of the cbk1Δ mutant and that sup2 showed slightly better restoration of virulence compared to sup1 in this insect model, which is independent of tolerance to mammalian body temperature.
Figure 6
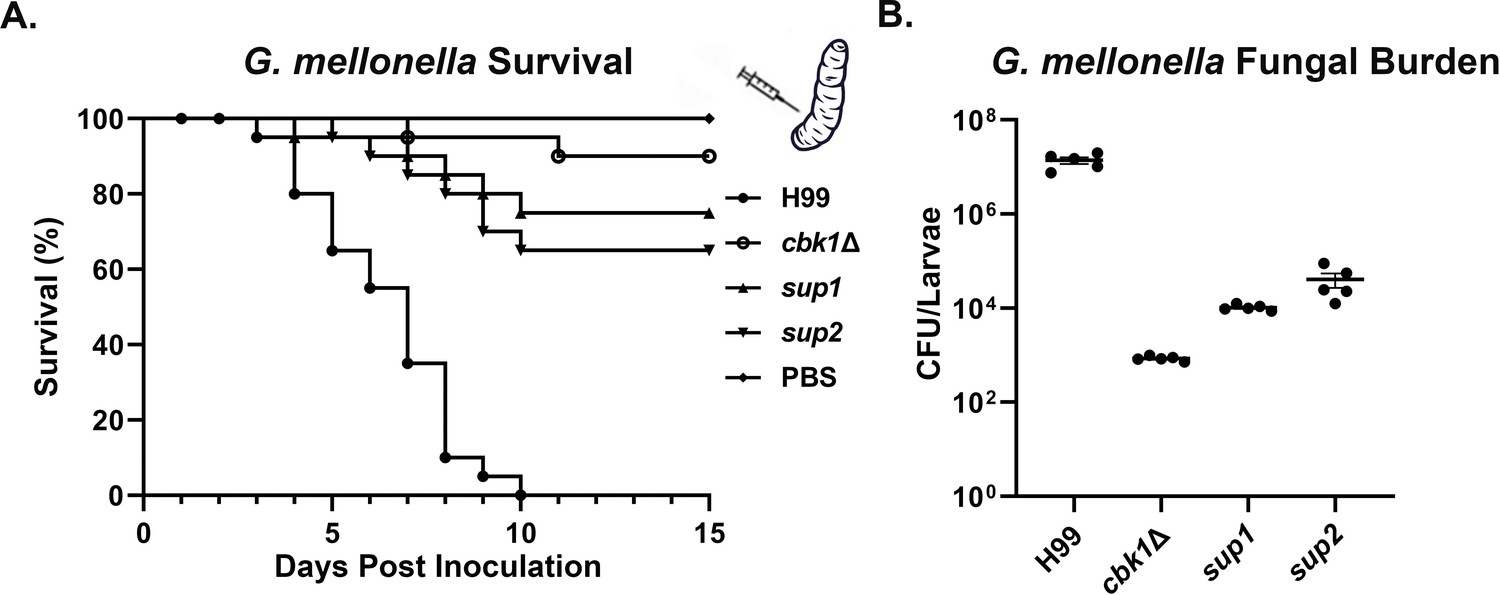
Suppressor mutants partially restore virulence in the G. mellonella model.
(A) G. mellonella larvae were infected with 5×104 fungal cells of the indicated strain, and their survival was monitored for 15 days post-inoculation. The cbk1Δ group survival curve was not significantly different from sup1 (p-value=0.2) and was significantly different from the sup2 curve (p-value=0.05). (B) At day 5 post-inoculation (DPI 5), 5 out of 25 larvae per group for the H99, the cbk1Δ mutant, sup1, and sup2 groups were homogenized, serially diluted, and plated to count the colony-forming units (CFUs) and measure the fungal burden of each larva. The fungal burden of the sup1 and sup2 groups was significantly higher than the cbk1Δ group (p-value<0.001).
Discussion
Detection of and adaptation to changing CO2 levels are an important trait across biological kingdoms and may play a crucial role in the pathogenicity of fungi (Bahn and Mühlschlegel, 2006; Cummins et al., 2014; Hetherington and Raven, 2005; Krysan et al., 2019). Here, we report the identification of genes required for growth at high levels of CO2 in the fungal pathogen C. neoformans. Multiple pathways important for growth at high temperature, such as the Ras1-Cdc24, CWI, Calcineurin, and RAM pathways, were found to be required for growth in high CO2 concentrations, indicating that growth in response to host CO2 may be intricately coordinated and co-regulated with response to host temperature. It is therefore likely that both host CO2 and host temperature represent stressors that cryptococcal cells infecting mammalian hosts must overcome to cause disease.
Calcineurin and RAM pathways were both identified in our screen for mutants that affect cryptococcal CO2 sensitivity. A previous study found synthetic lethality between the RAM and calcineurin pathways in C. neoformans but not in S. cerevisiae (Walton et al., 2006). This corroborates our findings of the key differences between the basidiomycete C. neoformans and the ascomycete yeasts. In C. albicans, CO2 levels are sensed through bicarbonate or cAMP-dependent activation of adenylyl cyclase to increase hyphal growth (Du et al., 2012; Hall et al., 2010). While these pathways may also be functioning to sense CO2 in Cryptococcus (Bahn et al., 2005; Mogensen et al., 2006), our results indicate that these pathways do not play a significant role in host CO2 tolerance in C. neoformans. We also found that disruption of the RAM pathway effector kinase Cbk1 caused a severe growth defect at host CO2 in C. neoformans but not in C. albicans. The vast differences between these organisms in terms of growth response to CO2 may reflect the evolutionary distance between these species and/or the distinct niches they normally occupy. Indeed, C. albicans is a human commensal and has adapted to host CO2 concentrations. S. cerevisiae is a powerful fermenter that thrives in conditions with high levels of CO2. For the environmental fungus C. neoformans, however, the ability to grow in a CO2-enriched condition does not appear to be strongly selected for in the natural environment, and the host level of CO2 (~5% CO2) is over 100-fold higher than the ambient air (~0.04% CO2).
The RAM pathway mutants were among the most sensitive mutants to host levels of CO2. Remarkably, the growth defects of cbk1Δ could be partially restored by single mutations in the genes PSC1 or SSD1. While the PARN-encoding gene PSC1 represents an uncharacterized protein, SSD1 is a known suppressor of cbk1Δ phenotypes that has been extensively characterized in ascomycete yeasts to regulate the translation of numerous and diverse mRNA transcripts (Hu et al., 2018; Jansen et al., 2009; Lee et al., 2015; Li et al., 2009b; Wanless et al., 2014). Our genetic interaction analysis indicates that Psc1 likely functions in the same pathway as Ssd1. Interestingly, in S. cerevisiae, deletion of SSD1 can suppress the lethality of the cbk1Δ mutant but not the cell separation defect, which is regulated by the transcription factor Ace2 (Kurischko et al., 2005). However, an Ace2 homolog has not been identified in Cryptococcus or any other basidiomycete (Lin et al., 2015). In C. albicans, Ssd1 plays an important role in polarized growth and hyphal initiation by negatively regulating the transcription factor Nrg1 (Lee et al., 2015). The observation that cbk1Δpsc1Δ and cbk1Δssd1Δ suppressor mutants partially rescue cell separation defects or depolarized growth suggests that C. neoformans may primarily utilize Ssd1/Psc1 rather than a potential Ace2 homolog to regulate cell separation or polarization. Differential regulation of target mRNA transcripts by Ssd1 and Psc1 may explain the functional divergence of the RAM pathway we observed between the basidiomycete Cryptococcus and the ascomycete yeasts. Our observation that the natural suppressors do not restore transcript abundances of CO2-associated genes in cbk1Δ to WT levels supports a hypothesis that disruption of Ssd1 and Psc1 suppresses the cbk1Δ mutant’s defects at a post-transcriptional level. C. neoformans has been demonstrated to use post-transcriptional regulation to adapt to various host stresses (Bloom et al., 2019; Kalem et al., 2021; Stovall et al., 2021). A temperature-sensitive environmental species of Cryptococcus, Cryptococcus amylolentus, fails to initiate host stress-induced translational reprogramming and is non-pathogenic (Bloom et al., 2019). Whether or not translatome reprogramming is initiated in C. neoformans in response to host CO2, and whether such reprogramming, if occurs, relies on Ssd1 and/or Psc1, has yet to be determined.
Materials and methods
Strains, growth conditions, and microscopy examination
Request a detailed protocolStrains used in this study are listed in the key resources table. Unless stated otherwise, all C. neoformans cells were maintained at 30°C on YPD media or YPD + CuSO4 (25 μM) for strains transformed with PCTR4-CBK1. For morphological examination, all strains were examined under a Zeiss Imager M2 microscope, equipped with an AxioCam MRm camera. For spotting assays, the tested strains were grown overnight in liquid YPD medium at 30°C with shaking at 220 RPM. The cells were then adjusted to the same cell density of OD600=1 and serially diluted 10-fold. The cell suspensions were then spotted onto YPD agar medium and incubated at the indicated condition for 2 days. CO2 levels were controlled by a VWR CO2 incubator or by a Pro-CO2 controller (Biospherix, Lacona, NY, USA).
Genetic manipulation
Gene deletion constructs
Request a detailed protocolTo delete the gene SSD1, a deletion construct with a nourseothricin (NAT) resistance marker cassette with 5’ and 3’ homology arms to SSD1 was used. Primers Linlab7974 (gctgcctttgcgtcatctc) and Linlab7976 (ctggccgtcgttttactctcgccttccttctcctta) were used to amplify the 5’ arm from the H99 genome. The 3’ arm was amplified from H99 with primers Linlab7977 (gtcatagctgtttcctgcgattgacattgccgtcttag) and Linlab7979 (cgacctgatcaaactactcgc). The NAT marker was amplified with universal primers M13F and M13R from plasmid pPZP-NATcc. The three pieces were fused together by overlap PCR and amplified with nested primers Linlab7975 (acaatgagccactgccag) and Linlab7977 (tgcgtgttcactactgtagac). To disrupt the gene PSC1, a hygromycin (HYG) marker cassette was used to insert into the PARN domain. To generate the sgRNA for specific targeting to the SSD1 locus, the U6 promoter and sgRNA scaffold were amplified from JEC21 genomic DNA and the plasmid pDD162 using primers Linlab7980/Linlab4627 (ttgagtggggtgggtcaattaacagtataccctgccggtg and ggctcaaagagcagatcaatg) and Linlab7981/Linlab4628 (aattgacccaccccactcaagttttagagctagaaatagcaagtt and cctctgacacatgcagctcc). For sgRNA targeted mutation of PSC1, the primers Linlab8380/Linlab4627 (tagttgttttcgccgacgccaacagtataccctgccggtg and ggctcaaagagcagatcaatg) were used to amplify the U6 promoter and Linlab8381/Linlab4628 (ggcgtcggcgaaaacaactagttttagagctagaaatagcaagtt and cctctgacacatgcagctcc) to amplify the sgRNA scaffold. The U6 promoter and sgRNA scaffold were fused together by overlap PCR with primers Linlab4594/Linlab4595 (ccatcgatttgcattagaactaaaaacaaagca and ccgctcgagtaaaacaaaaaagcaccgac) to generate the final sgRNA construct as described previously (Fan and Lin, 2018; Lin et al., 2020).
Gene overexpression constructs
Request a detailed protocolThe CBK1 overexpression construct was generated by amplifying the CBK1 open reading frame with primers Linlab7005/BC (ataggccggccatgtcgtatcgcccaatccag) and Linlab7006/BC (cagcatctcgtatcgtcggaag) and cloning the fragment with FseI and PacI into the pXC plasmid backbone (Wang et al., 2012), which contains the promoter of CTR4 and neomycin resistance marker. The CTR4 promoter is highly induced on the copper limiting YPD media. The MPK1 overexpression construct was generated by amplifying the MPK1 open reading frame with primers Linlab8326/BC (ataggccggccatggacaatacccctagacac) and Linlab8327/BC (ccttaattaaggctatgataatttctgcctctcc) and cloning the fragment with FseI and AsiSI into a pUC19 plasmid backbone, containing the promoter of GPD1 and neomycin resistance marker. The CDC24 overexpression construct was generated by amplifying the CDC24 open reading frame with primers Linlab6674/BC (ataggccggccatgtctgtatccggtcccatctc) and Linlab6675/BC (ccttaattaaggataaatctctccttgtggggtacc) and cloning the fragment with FseI and PacI into a pUC19 plasmid backbone, containing the promoter of CTR4 and neomycin resistance marker. The overexpression constructs were integrated into the SH2 locus as described previously (Fan and Lin, 2018; Lin et al., 2020).
Transformation
Request a detailed protocolConstructs for overexpression and deletion were transformed into Cryptococcus strains by the TRACE method (Fan and Lin, 2018; Lin et al., 2020), and transformants were selected on YPD medium with 100 μg/mL of NAT,100 μg/mL of neomycin (NEO), or 200 μg/mL of HYG.
Quantitative real-time PCR
Request a detailed protocolWT H99 strain along with the cbk1Δ, CBK1OE strain were cultured by shaking at 220 RPM at 30°C overnight in liquid YPD medium containing 50 μM CuSO4 to suppress the CTR4 promoter of the CBK1OE construct. The cultures were then diluted to OD600=0.2 in fresh liquid YPD medium containing 50 μM BCS(bathocuproine disulfonate) to induce expression. After 5 hr of further incubation, cells were collected, flash frozen in liquid nitrogen, and lyophilized overnight. Three biological replicates per strain were used. Total RNA was isolated by using the PureLink RNA Mini Kit (Invitrogen), and first strand cDNA was synthesized using the GoScript Reverse Transcription System (Promega) following the manufacturer’s instructions. The Power SYBR Green system (Invitrogen) was used for RT-PCR. The following primers were used to target CBK1: Linlab9217/BC (gatgctctcactcctgattcc) and Linlab8641/BC (gtacgagtctgacttcaccga). The following primers were used to target the TEF1 housekeeping gene as an endogenous control for each sample: Linlab329/XL (cgtcaccactgaagtcaagt) and Linlab330/XL (agaagcagcctccatagg). Relative transcript level was determined using the ΔΔct method as described previously. Statistical significance was determined using a Student’s t-test.
NanoString RNA profiling
Request a detailed protocolOvernight YPD cultures of H99, cbk1Δ, cbk1Δssd1Δ, and cbk1Δpsc1Δ were washed 2× in PBS and resuspended in RPMI +165 mM MOPS, pH 7.4 before quantification on an Invitrogen Countess automated cell counter. Cells were diluted to 7.5×105 cells per mL in 3 mL per well in a 6-well plate. Two wells were used for each biological replicate (n=3) and condition (ambient or 5% CO2). Plates were sealed with BreatheEasy sealing membranes (Sigma #Z380059) and incubated in a static incubator at 30°C in ambient air or 5% CO2 for 24 hr. Cells were harvested, pelleted at 3200×g for 5 min, and the supernatant was removed. The pellets were then frozen at –80°C and lyophilized overnight. Lyophilized cells were disrupted for 45 s with 0.5 mm glass beads on an MP Biomedicals FastPrep-24 benchtop homogenizer. RNA was extracted following manufacturer instructions for the Invitrogen PureLink RNA mini-kit with on-column DNAse treatment. Purified RNA was quantified on a NanoDrop OneC spectrophotometer, and a total of 100 ng per sample was combined with a custom probeset (Source data 1) from NanoString Technologies according to manufacturer instructions. Probes were hybridized at 65°C for 18 hr, then run on a NanoString nCounter SPRINT profiler according to manufacturer instructions. Data from Reporter Code Count files were extracted with nSolver software (version 4.0), and raw counts were exported to Microsoft Excel. Internal negative controls were used to subtract background from raw counts (negative control average +2 SDs). Counts were normalized across samples by total RNA counts. Probes below background were set to a value of 1. Fold change and significance were calculated in Excel after averaging biological triplicates, using a Student t-test (p<0.05). Volcano plot was generated with transformed values (−log[p-value] and log2[fold change]) in GraphPad Prism 9. Normalized total counts were used in Morpheus (https://software.broadinstitute.org/morpheus/) to generate a heat map, with hierarchical clustering, one minus Pearson correlation, average linkage method, and clustered according to rows and columns.
Bioinformatics
Request a detailed protocolWhole genome sequencing was performed using the Illumina platform with NovaSeq 6000 at the University of California – Davis Sequencing Center, Novogene USA. A paired-end library with approximately 350 base inserts was constructed for each sample, and all libraries were multiplexed and run in one lane using a read length of 150 bases from either side.
The Illumina reads were first trimmed with Trim Galore v0.6.5 (Krueger, 2021) and then mapped to the C. neoformans H99 reference genome (FungiDB version 50) using the BWA-MEM algorithm of the BWA aligner v0.7.17 (Li, 2013). SAMtools v1.10 (Li et al., 2009a), Picard Tools v2.16.0 (Broad_Institute, 2022), and bcftools v1.13 (Danecek et al., 2021) were used for variant calling from each sample. Variants in the suppressor strains were called with the original cbk1Δ mutant as a reference.
The protein diagrams of Psc1 and Ssd1 were made with the illustrator of biological sequences software package (Liu et al., 2015).
Phagocytosis assays
Request a detailed protocolThe authenticated mouse macrophage cell line J774A.1 (ATCC TIB-67) was acquired from the American Type Culture Collection. Before being used, normal morphology, cell adhesion, and phagocytosis activity of the cell line was confirmed. Contamination by mycoplasma was not detected. Phagocytosis assays were performed using similar procedures as we described previously (Lin et al., 2015). Briefly, 1 mL of 2×105 J774A.1 macrophages (MΦ) in DMEM was seeded into a 24-well plate and incubated at 37°C with 5% CO2 for 24 hr. Cryptococcus strains with a starting OD600 of 0.2 in 3 mL of liquid YPD were cultured for 16 hr. Each strain had three technical replicates. The cells were washed three times in sterile H2O. 2×106 cryptococcal cells of each strain were opsonized in either 40 μL of 100% fetal bovine serum, naïve mouse serum, or mouse serum from LW10 vaccinated A/J mice (Lin et al., 2022; Zhai et al., 2015), for 30 min prior to co-incubation with MΦ. Old DMEM from MΦ was removed, and 1 mL of fresh DMEM with the opsonized Cryptococcus cells was added, followed by a 2 hr incubation at 37°C with 5% CO2. The co-culture was then washed six times with warm PBS to remove non-adherent Cryptococcus cells. To lyse the macrophages, the cell suspensions were washed with 1 mL of cold PBS +0.01% Triton X. Serial dilutions in PBS of the cell suspensions were then plated onto YNB agar medium and allowed to grow at 30°C for 2 days to count CFUs. Statistical analyses were performed using the program Graphpad Prism 8. A two-tailed t-test was applied to determine significance. A p-value of less than 0.05 was considered significant.
G. mellonella infection model
Request a detailed protocolG. mellonella larvae were purchased from Best Bait (Marblehead, OH, USA). The infection was performed as described previously described (Mylonakis et al., 2005). In brief, cryptococcal strains were inoculated in 3 mL of liquid YPD medium with the initial OD600=0.2 (approximately 106 cell/mL) and incubated for 15 hr at 30°C with shaking. Prior to infection, cells were washed with sterile PBS three times and adjusted to the final concentration of 1×107 cell/mL. 5 μL of the cell suspension (5×104 cells), or PBS for the control group, were injected into the last left proleg of the larvae. The proleg was cleaned with 70% ethanol prior to injection. Infected larvae were maintained at 30°C and monitored daily for survival.
Prior to fungal burden quantification, larvae were first cleaned with 70% ethanol. The larvae were cut open with sterile scissors and vortexed in a microcentrifuge tube containing 500 μL PBS and 100 μL of 0.5 mm diameter glass beads (RPI). Larval suspensions were then serially diluted in PBS and plated onto YNB agar medium containing 50 μg/mL kanamycin and 20 μg/mL chloramphenicol and incubated at 30°C for 2 days before counting the CFUs.
Statistical analyses were performed using the program Graphpad Prism 8. The log-rank Mantel-Cox test was used to assess statistical significance of survival curves for comparison between two groups. One-way ANOVA tests were used to compare groups of three or more and for fungal burden assays.
Murine models of cryptococcosis
Intranasal infection model
Request a detailed protocolFemale Balb/C mice of 8–10 weeks old were purchased from the Jackson Labs (Bar Harbor, Maine). Cryptococcal strains were inoculated in 3 mL of liquid YPD medium with the initial OD600=0.2 (approximately 106 cell/mL) and incubated for 15 hr at 30 °C with shaking. Prior to intranasal infection, cells were washed with sterile saline three times and adjusted to the final concentration of 2×105 cell/mL. Once the mice were sedated with ketamine and xylazine via intraperitoneal injection, 50 μL of the cell suspension (1×104 cells per mouse) were inoculated intranasally as previously described (Lin et al., 2022; Zhai et al., 2012; Zhai et al., 2013; Zhao et al., 2020; Zhu et al., 2013). Mice were monitored daily for disease progression. Surviving animals were euthanized at 35 or 60 DPI, and the brain, lungs, and kidneys, were dissected.
Intravenous infection model
Request a detailed protocolPrior to intravenous infections, cryptococcal cells were washed with sterile saline three times and adjusted to the final concentration of 1×106 cell/mL. Mice were sedated with Isoflurane. 100 μL of the cell suspension (1×105 cells per mouse) were injected intravenously as previously described (Zhai et al., 2012; Zhai et al., 2013; Zhao et al., 2020; Zhu et al., 2013). After DPI 5, animals were euthanized, and the brain, lungs, and kidneys were dissected.
For fungal burden quantifications, dissected organs were homogenized in 2 mL of cold sterile PBS using an IKA-T18 homogenizer as we described previously (Zhai et al., 2015; Zhai et al., 2012). Tissue suspensions were serially diluted in PBS and plated onto YNB agar medium and incubated at 30°C for 2 days before counting the CFUs.
Ethical statements
Request a detailed protocolThis study was performed according to the guidelines of NIH and the University of Georgia Institutional Animal Care and Use Committee (IACUC). The animal models and procedures used have been approved by the IACUC (AUP protocol numbers: A2017 08–023 and A2020 06–015).
Appendix 1
Appendix 1—key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Genetic reagent (Cryptococcus neoformans KN99α, matα) | WT strain: H99 | Nielsen et al., 2003 | ||
| Genetic reagent (C. neoformans KN99α, matα) | WT strain: H99a | Nielsen et al., 2003 | ||
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δ | Walton et al., 2006 | FJW9 | H99alpha, CBK1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | mob2Δ | Walton et al., 2006 | FJW10 | H99alpha, MOB2::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | kic1Δ | Walton et al., 2006 | FJW8 | H99alpha, KIC1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | tao3Δ | Walton et al., 2006 | AI136 | MATalpha, TAO3:: NATr |
| Genetic reagent (C. neoformans KN99α, matα) | sog2Δ | Walton et al., 2006 | AI131 | MATalpha, SOG2:: NATr |
| Genetic Reagent (Candida albicans, SN250) | SN250 | Wakade et al., 2020 | SN250 | HIS-, LEU-, ARG- |
| Genetic Reagent (C. albicans, SN250) | cbk1ΔΔ | Wakade et al., 2020 | ΔΔcbk1 | SN250, ΔΔcbk1: HIS-, LEU-, ARG- |
| Genetic reagent (C. neoformans KN99α, matα) | cna1Δ | FGSC deletion set Plate 46 Well E12 | cna1Δ | H99alpha CNA1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | cdc24Δ | FGSC deletion set Plate 33 Well C4 | cdc24Δ | H99alpha, CDC24::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | mpk1Δ | FGSC deletion set Plate 11 Well A5 | mpk1Δ | H99alpha, MPK1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | CBK1OE | This study. | BC1449 | H99alpha, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cdc24Δ, CBK1OE | This study. | BC1281 | H99alpha, CDC24::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cdc24Δ, CBK1OE | This study. | BC1282 | H99alpha, CDC24::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cna1Δ, CBK1OE | This study. | BC1283 | H99alpha, CNA1::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cna1Δ, CBK1OE | This study. | BC1284 | H99alpha, CNA1::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | mpk1Δ, CBK1OE | This study. | BC1285 | H99alpha, MPK1::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | mpk1Δ, CBK1OE | This study. | BC1286 | H99alpha, MPK1::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cdc24Δ, CDC24OE | This study. | BC650 | H99alpha, CDC24::NATr, PCTR4-mNeonGreen-CDC24-NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δ, CBK1OE | This study. | BC669 | H99alpha, CBK1::NATr, PCTR4-CBK1-mCherry- NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | mpk1Δ, MPK1OE | This study. | BC1356 | H99alpha, MPK1::NATr, PGPD1-mNeonGreen-MPK1-NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cna1Δ, CNA1OE | Kozubowski et al., 2011 | LK214 | H99a, CNA1::NEOr, PH3-GFP-CNA1-NATr |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δ, CDC24OE | This study. | BC1357 | H99alpha, CBK1::NATr, PCTR4-mNeonGreen-CDC24-NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δ, MPK1OE | This study. | BC1358 | H99alpha, CBK1::NATr, PGPD1-mNeonGreen-MPK1-NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δ, CNA1OE | This study. | BC1359 | H99alpha, CBK1::NATr, PH3-GFP-CNA1-NATr |
| Genetic reagent (C. neoformans KN99α, matα) | sup1 | This study. | BC1068 | H99alpha, CBK1::NATr,SUP1 |
| Genetic reagent (C. neoformans KN99α, matα) | sup2 | This study. | BC1076 | H99alpha, CBK1::NATr,SUP2 |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δssd1Δ | This study. | BC1239 | H99alpha, CBK1::NATr, SSD1::NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | ssd1Δ | This study. | BC1241 | H99alpha, SSD1::NEOr |
| Genetic reagent (C. neoformans KN99α, matα) | cbk1Δpsc1Δ | This study. | BC1369 | H99alpha, CBK1::NATr, PSC1::HYGr |
| Genetic reagent (C. neoformans KN99α, matα) | psc1Δ | This study. | BC1393 | H99alpha, PSC1::HYGr |
| Genetic reagent (C. neoformans KN99α, matα) | cac1Δ | Bahn et al., 2006 | YSB42 | H99alpha, CAC1::NATr STM#159 |
| Genetic reagent (C. neoformans KN99α, matα) | aca1Δ | FGSC deletion set Plate 32 Well H6 | aca1Δ | H99alpha, ACA1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | pkr1Δ | FGSC deletion set Plate 33 Well H7 | pkr1Δ | H99alpha, PKR1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | pde1Δ | FGSC deletion set Plate 10 Well A12 | pde1Δ | H99alpha, PDR1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | pde2Δ | FGSC deletion set Plate 11 Well H7 | pde2Δ | H99alpha, PDR2::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | gpa1Δ | Bahn et al., 2005 | YSB83 | H99alpha, GPA1::NATr STM#5 |
| Genetic reagent (C. neoformans KN99α, matα) | cln1Δ | This study. | BZ36 | H99alpha, CLN1::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | ecm2201Δ | FGSC deletion set Plate 2 Well A10 | ecm2201Δ | H99alpha, ECM2201::NATr |
| Genetic reagent (C. neoformans KN99α, matα) | kin4Δ | Bahn Kinase Deletion Set Plate 4 Well G8 (Lee et al., 2016) | kin4Δ | H99alpha, KIN4::NATr |
| Recombinant DNA reagent | pPZP-NATcc | Dickinson et al., 2013 | pPZP-NATcc | |
| Recombinant DNA reagent | pDD162 | Walton et al., 2006 | pDD162 | |
| Recombinant DNA reagent | pFZ1-CDC24 | This study. | PCTR4-2-mNeonGreen-CDC24(H99)-NEOr | |
| Recombinant DNA reagent | pXC-CBK1-mCh | This study. | PCTR4-2-CDC24(H99)-mCherry-NEOr | |
| Recombinant DNA reagent | LKB61 | Kozubowski et al., 2011 | PGPD1-mCherry-CNA1-HYGr | |
| Recombinant DNA reagent | pUC19-MPK1-mNG | This study. | PGPD1-MPK1-mNeonGreen-NEOr | |
| Cell line (Mus. Musculus, macrophage cell line J774A.1) | J774A.1 | American Type Culture Collection | ATCC TIB-67 | |
| Chemical Compound and drug | Hygromycin | Research Products International | Cat. NO.: H75000 | |
| Chemical Compound and drug | G418 | Research Products International | Cat. NO.: G64000 | |
| Chemical Compound and drug | Nourseothricin | Jena Bioscience | Cat. NO.: AB-102–25 G | |
| Software and algorithm | Graphpad Prism 9 | Graphpad | ||
| Software and algorithm | nSolver software version 4.0 | NanoString Technologies | ||
| Software and algorithm | Trim Galore v0.6.5 | Krueger, 2021 | ||
| Software and algorithm | BWA aligner v0.7.17 | Li, 2013 | ||
| Software and algorithm | SAMtools v1.10 | Li et al., 2009a | ||
| Software and algorithm | Picard Tools v2.16.0 | Broad_Institute, 2022 | ||
| Software and algorithm | bcftools v1.13 | Danecek et al., 2021 | ||
| Software and algorithm | Illustrator of biological sequences (IBS) | Liu et al., 2015 |
Data availability
Sequences generated from this research has been deposited to the Sequence Read Archive (SRA) under project accession number: PRJNA791949.
-
NCBI BioProjectID PRJNA791949. Whole genome sequencing of C. neoformans H99 suppressor strains of the RAM pathway downstream kinase Cbk1 knockout mutant.
References
-
Co2 sensing in fungi and beyondCurrent Opinion in Microbiology 9:572–578.https://doi.org/10.1016/j.mib.2006.09.003
-
Carbon dioxide-sensing in organisms and its implications for human diseaseCellular and Molecular Life Sciences 71:831–845.https://doi.org/10.1007/s00018-013-1470-6
-
Twelve years of samtools and bcftoolsGigaScience 10:giab008.https://doi.org/10.1093/gigascience/giab008
-
The transcription factor FLO8 mediates CO2 sensing in the human fungal pathogen Candida albicansMolecular Biology of the Cell 23:2692–2701.https://doi.org/10.1091/mbc.E12-02-0094
-
Leveraging fungal and human calcineurin-inhibitor structuresBiophysical Data, and Dynamics To Design Selective and Nonimmunosuppressive FK506 Analogs. MBio 12:e0300021.https://doi.org/10.1128/mBio.03000-21
-
The biology of carbon dioxideCurrent Biology: CB 15:R406–R410.https://doi.org/10.1016/j.cub.2005.05.042
-
The sequence alignment/map format and samtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
Most environmental isolates of cryptococcus neoformans var. grubii (serotype A) are not lethal for miceInfection and Immunity 77:3188–3195.https://doi.org/10.1128/IAI.00296-09
-
Galleria mellonella as a model system to study Cryptococcus neoformans pathogenesisInfection and Immunity 73:3842–3850.https://doi.org/10.1128/IAI.73.7.3842-3850.2005
-
Cryptococcus neoformans: the yeast that likes it hotFEMS Yeast Research 6:463–468.https://doi.org/10.1111/j.1567-1364.2006.00051.x
-
Global burden of disease of HIV-associated cryptococcal meningitis: an updated analysisThe Lancet. Infectious Diseases 17:873–881.https://doi.org/10.1016/S1473-3099(17)30243-8
-
The antidepressant sertraline provides a promising therapeutic option for neurotropic cryptococcal infectionsAntimicrobial Agents and Chemotherapy 56:3758–3766.https://doi.org/10.1128/AAC.00212-12
-
Life cycle of cryptococcus neoformansAnnual Review of Microbiology 73:120210.https://doi.org/10.1146/annurev-micro-020518-120210
-
Activation of meiotic genes mediates ploidy reduction during cryptococcal infectionCurrent Biology: CB 30:1387–1396.https://doi.org/10.1016/j.cub.2020.01.081
-
Congenic strains for genetic analysis of virulence traits in cryptococcus gattiiInfection and Immunity 81:2616–2625.https://doi.org/10.1128/IAI.00018-13
Article and author information
Author details
Benjamin J Chadwick

Funding
National Institutes of Health (R01AI147541)
- Damian J Krysan
- Xiaorong Lin
National Institutes of Health (R01AI140719)
- Xiaorong Lin
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by National Institutes of Health (http://www.niaid.nih.gov) (R01AI147541 to DJK and XL, and R01AI140719 to XL). The funder had no role in study design, data collection, and interpretation, or the decision to submit the work for publication. We thank all Lin lab members for their helpful suggestions. We thank Dr. Fanglin Zheng for the plasmid pFZ1, and Dr. Lukasz Kozubowski for the plasmid LKB61.
Ethics
This study was performed according to the guidelines of NIH and the University of Georgia Institutional Animal Care and Use Committee (IACUC). The animal models and procedures used have been approved by the IACUC (AUP protocol numbers: A2017 08-023 and A2020 06-015).
Copyright
© 2022, Chadwick et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,693
- views
-
- 306
- downloads
-
- 32
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 32
- citations for umbrella DOI https://doi.org/10.7554/eLife.82563
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The RAM signaling pathway links morphology, thermotolerance, and CO2 tolerance in the global fungal pathogen Cryptococcus neoformans
eLife 11:e82563.
https://doi.org/10.7554/eLife.82563




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}