An IS-mediated, RecA-dependent, bet-hedging strategy in Burkholderia thailandensis
- Department of Microbiology and Immunology, University of North Carolina at Chapel Hill, United States
Abstract
Adaptation to fluctuating environmental conditions is difficult to achieve. Phase variation mechanisms can overcome this difficulty by altering genomic architecture in a subset of individuals, creating a phenotypically heterogeneous population with subpopulations optimized to persist when conditions change, or are encountered, suddenly. We have identified a phase variation system in Burkholderia thailandensis that generates a genotypically and phenotypically heterogeneous population. Genetic analyses revealed that RecA-mediated homologous recombination between a pair of insertion sequence (IS) 2-like elements duplicates a 208.6 kb region of DNA that contains 157 coding sequences. RecA-mediated homologous recombination also resolves merodiploids, and hence copy number of the region is varied and dynamic within populations. We showed that the presence of two or more copies of the region is advantageous for growth in a biofilm, and a single copy is advantageous during planktonic growth. While IS elements are well known to contribute to evolution through gene inactivation, polar effects on downstream genes, and altering genomic architecture, we believe that this system represents a rare example of IS element-mediated evolution in which the IS elements provide homologous sequences for amplification of a chromosomal region that provides a selective advantage under specific growth conditions, thereby expanding the lifestyle repertoire of the species.
Editor's evaluation
This paper reports a bet hedging strategy in bacteria based on chromosomal duplications and rearrangements that confer advantages in certain growth conditions. The work is of fundamental importance for understanding the role of genetic and biological variation in bacteria. The experimental work is exceptionally strong and convincing. The paper will be of interest to a broad audience including bacteriologists, geneticists and evolutionary biologists.
https://doi.org/10.7554/eLife.84327.sa0eLife digest
Bacterial populations are often diverse, even when originating from a single cell. This diversity helps microbes survive in fluctuating environmental conditions by increasing the odds of population survival. For example, if environmental conditions change such that only a subpopulation with unique abilities survives, the entire population will be saved.
Genomes are naturally dynamic. For example, mobile sections of DNA, called transposable elements, can change their position within a genome. If a transposable element jumps into a gene, it can harm the cell. But if it moves into a different site, it may provide an organism with new features that can help it survive.
Most organisms contain multiple copies of transposable elements in their DNA. For example, a subtype of the soil bacterium Burkholderia thailandensis, strain E264, has two identical transposable elements that book-end a region of DNA that contains 157 genes. Lowrey et al. studied this bacterial strain in different environmental conditions to find out more. The experiments revealed that in growing populations of E264, some bacteria had one copy of the region, while others had two or three. In a rich environment, most bacteria had just one copy of the region. However, when grown in challenging conditions, most bacteria contained two or three copies of the region.
Moreover, bacteria required at least two copies to form dense communities known as biofilms, which are advantageous for bacterial survival in challenging conditions. Bacteria with only one copy, however, were better adapted to a free-swimming lifestyle.
Lowrey et al. further showed that the DNA repair system was required for duplicating the region. Usually, this system finds and recombines identical DNA sequences to repair broken DNA. However, if two identical DNA sequences (a pair of transposable elements) are present, the repair system can recombine them during DNA replication, resulting in the duplication of the DNA between the identical sequences. The same system also reduces the copy number of the region from three or two to just one.
Since the repair system is constantly working and DNA recombination is always occurring at a low level, B. thailandensis E264 maintains a genetically diverse population with bacteria containing different copy numbers of the region. This diversity ensures that the strain survives in fluctuating environmental conditions.
Transposable elements are hotspots of evolution. They are known to interrupt genes and shrink genomes. Lowrey et al. showed that transposable elements also influence evolution by providing DNA sequences that the DNA repair system can use to duplicate DNA. This process of duplicating genes is more frequent than random genetic mutations, expediting adaptation.
Introduction
Microbes often exist in complex communities wherein productivity and resilience rely on population diversity. This diversity commonly appears as mixed-species populations. However, diversity can also exist within otherwise clonal populations through genotypic and phenotypic heterogeneity. Heterogeneity can increase the fitness of a population by contributing to the division of labor, or it can serve as a bet-hedging strategy to increase the odds of population survival when alteration of environmental conditions, such as nutrient availability, host response, or antimicrobial concentrations, occur quickly (Magdanova and Golyasnaya, 2013).
A common facilitator of phenotypic heterogeneity is phase variation, in which reversible genetic alterations occur that function as ‘ON/OFF’ switches that control specific phenotypes (van der Woude, 2011; van der Woude and Bäumler, 2004). These genetic alterations typically occur at frequencies that are orders of magnitude greater than spontaneous mutations. One common generator of phenotypic heterogeneity is recombinase-mediated, site-specific recombination, such as invertible switches and transposable elements, which can influence the expression of coding sequences by altering the orientation of promoters or introducing new regulatory sequences, respectively (Trzilova and Tamayo, 2021; Schneider and Lenski, 2004; Darmon and Leach, 2014).
Phenotypic heterogeneity within a population can also be achieved by recombination between homologous sequences at different genomic loci, a phenomenon referred to as unequal or non-allelic recombination. Unlike site-specific recombination, which relies on a specific recombinase catalyzing strand exchange between specific target sequences, any homologous sequences greater than 50 bp can act as substrates for unequal homologous recombination. These homologous sequence substrates are often ribosomal RNA operons or transposable elements, which are commonly found in multiple copies per genome (Andersson and Hughes, 2009; Raeside et al., 2014). The DNA repair and maintenance recombinase, RecA, identifies homologous sequences and catalyzes strand exchange which, when unequal homologous sequences are recombined, leads to modifications in genomic architecture through sequence inversions, deletions, or duplications (Darmon and Leach, 2014; Bourque et al., 2018). These adjustments to genome architecture can similarly influence the orientation of promoters through sequence inversion, but also change copy number of genes by deleting and duplicating intervening sequences (Vandecraen et al., 2017). Changes in gene copy number can impact protein production, and thus cell phenotypes – a phenomenon referred to as gene dosage effects (Andersson and Hughes, 2009; Sandegren and Andersson, 2009).
Phenotypic heterogeneity has been observed in a wide range of Burkholderia species. The bioterrorism agent B. pseudomallei displays at least seven distinct morphologies when propagated on Ashdown’s agar, and virulence-associated phenotypes, such as intracellular survival, colonization ability, and antigen presentation, differ between the various morphotypes (Chantratita et al., 2007; Tandhavanant et al., 2010; Austin et al., 2015; Wikraiphat et al., 2015). The cystic fibrosis-associated opportunistic pathogen B. cenocepacia similarly exhibits phenotypic and genotypic heterogeneity during growth in the lab as well as during growth in the cystic fibrosis lung (Häussler et al., 2003; Lee et al., 2017; Springman et al., 2009).
Burkholderia thailandensis strain E264 (BtE264) displays distinct phenotypes, including aggregation, biofilm formation, Congo Red binding, and the production of a gold-brown ‘pigment’, that were attributed initially to the activity of the bcpAIOB-encoded contact-dependent inhibition system and hence we named them contact-dependent signaling phenotypes (Anderson et al., 2012; Garcia et al., 2016). Subsequent analyses, however, showed that these phenotypes occur when a 208.6 kb region on chromosome I that contains 157 predicted coding sequences plus one of the flanking IS2-like elements (shown in Figure 1) is present in two or more copies (Ocasio and Cotter, 2019), so we will now refer to those phenotypes as duplication-dependent phenotypes. Our previous data suggested that amplification of the 208.6 kb region required BcpAIOB activity and the IS2-like element at the 5′ end (ISβ), and that the region amplified as extrachromosomal, circular DNA molecules (Ocasio and Cotter, 2019). Our investigations into the mechanism underlying the amplification, however, caused us to completely re-evaluate our previous conclusions. Here, we present data indicating that BtE264 possesses a homologous recombination-mediated phase variation mechanism that facilitates rapid adaptation to disparate growth conditions by altering the copy number of the 208.6 kb region on chromosome I in a manner that is independent of both ISβ and BcpAIOB activity.
Figure 1

Burkholderia thailandensis E264 from ATCC is a genotypically heterogeneous population.
(A) Graphical representation of the 208.6 kb duplicating region bounded by homologous IS2-like elements referred to as ISβ (LO74_RS09425) and ISα (LO74_RS28540). The Ctrl1 and Ctrl2 primers amplify a 1 kb DNA product. Junc1 and Junc2 primers amplify a 2.2 kb product if the region has duplicated. (B–E) Twelve colonies from stocks of E264 from four different laboratories (Cotter, Mougous, Chandler, and Manoil) were used as templates for PCR using either Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (F, G) Twelve colonies from B. thailandensis strain 2002721643 and 2002721723 were used as templates for PCR using either Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers.
-
Figure 1—source data 1
Uncropped DNA gel displaying junction PCR that was used to make Figure 1B.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data1-v2.zip
-
Figure 1—source data 2
Uncropped DNA gel displaying control PCR that was used to make Figure 1B, C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data2-v2.zip
-
Figure 1—source data 3
Uncropped DNA gel displaying junction PCR that was used to make Figure 1C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data3-v2.zip
-
Figure 1—source data 4
Uncropped DNA gel displaying junction PCR that was used to make Figure 1D.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data4-v2.zip
-
Figure 1—source data 5
Uncropped DNA gel displaying control PCR that was used to make Figure 1D, E.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data5-v2.zip
-
Figure 1—source data 6
Uncropped DNA gel displaying junction PCR that was used to make Figure 1E.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data6-v2.zip
-
Figure 1—source data 7
Uncropped DNA gel displaying junction PCR that was used to make Figure 1F, G.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data7-v2.zip
-
Figure 1—source data 8
Uncropped DNA gel displaying control PCR that was used to make Figure 1F, G.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig1-data8-v2.zip
Results
Burkholderia thailandensis E264 from ATCC is a genotypically heterogeneous population
Our investigations led us to suspect that not all of the bacteria in our stock of wild-type BtE264 from ATCC contained multiple copies of the 208.6 kb DNA region. To assess the potential heterogeneity of our stock, we conducted PCR analyses on individual colonies after plating an aliquot of the stock on LSLB agar. To detect the presence of multiple copies of the 208.6 kb region, we use outward-facing primers that anneal near the ends of the 208.6 kb region, but not within the IS elements (Figure 1A, Junc1 and Junc2). PCR with these primers yields a 2.2 kb DNA product containing one IS element flanked by the 5′ and 3′ ends of the region if two or more copies of the region are present (Ocasio and Cotter, 2019). While a product of the expected 1 kb size was amplified from all colonies when control primers (Figure 1A, Ctrl 1 and Ctrl 2) were used (Figure 1B), the 2.2-kb ‘junction PCR’ product was obtained from only approximately 50% of the colonies (Figure 1B). These results indicate that our BtE264 stock from ATCC is a genotypically heterogeneous population with approximately half of the bacteria containing a single copy of the 208.6 kb region and half containing two or more copies of the 208.6 kb region.
To determine if this genotypic heterogeneity is unique to our stock of BtE264, we conducted PCR analyses on colonies of BtE264 that we obtained from three separate laboratories. Junction PCR products of the expected 2.2 kb size were obtained for 0/12, 6/12, and 6/12 colonies from BtE264 obtained from the Mougous, Chandler, and Manoil laboratories, respectively, while all colonies yielded 1 kb PCR products when the control primers were used (Figure 1C–E). We suspect that the difference in stocks between laboratories can be explained by how the laboratories prepared their wild-type frozen stocks. Each laboratory had obtained its sample of BtE264 from ATCC, suggesting that the ATCC stock of BtE264 is a genotypically heterogeneous population composed of bacteria with either a single (‘junction PCR negative’) or multiple (‘junction PCR positive’) copies of the 208.6 kb region.
A small number of other B. thailandensis strains for which genome sequence is available also possess an IS-bounded region similar to that in BtE264. Two of these strains, 2002721643 and 2002721723, have genomic synteny of 99.0% and 98.9% and region synteny of 96.2% and 96.3% compared to BtE264, respectively. We conducted PCR analyses on colonies from 2002721643 and 2002721723. Junction PCR products of the expected 2.2 kb size were obtained for 5/12 colonies from 2002721643 and 0/12 colonies from 2002721723, while all colonies yielded 1 kb PCR products when the control primers were used (Figure 1F, G). These results suggest that B. thailandensis strains closely related to BtE264 also exhibit variation in copy number of a similar region.
Genotypic heterogeneity correlates with phenotypic heterogeneity
We noticed that although colonies from our BtE264 stock appeared identical after 24 hr growth on LSLB agar, three distinct morphologies were apparent after 48 hr. Nearly half of the colonies were flat with slightly raised centers (i.e., umbonate) (Figure 2A, white arrow), the other half were raised and rough (Figure 2A, black arrow), and about 1/75 were raised and smooth (Figure 2A, striped arrow). Restreaking flat colonies yielded only daughter colonies with the same flat morphology (Figure 2B), while restreaking raised rough and raised smooth colonies yielded daughter colonies wherein 90% exhibited the parental colony morphology and the remainder exhibited other morphologies (Figure 2B). All raised colonies (rough and smooth), and no flat colonies, yielded 2.2 kb junction PCR products (Figure 2C), demonstrating a correlation between colony morphology and the presence of two or more copies of the 208.6 kb region.
Figure 2
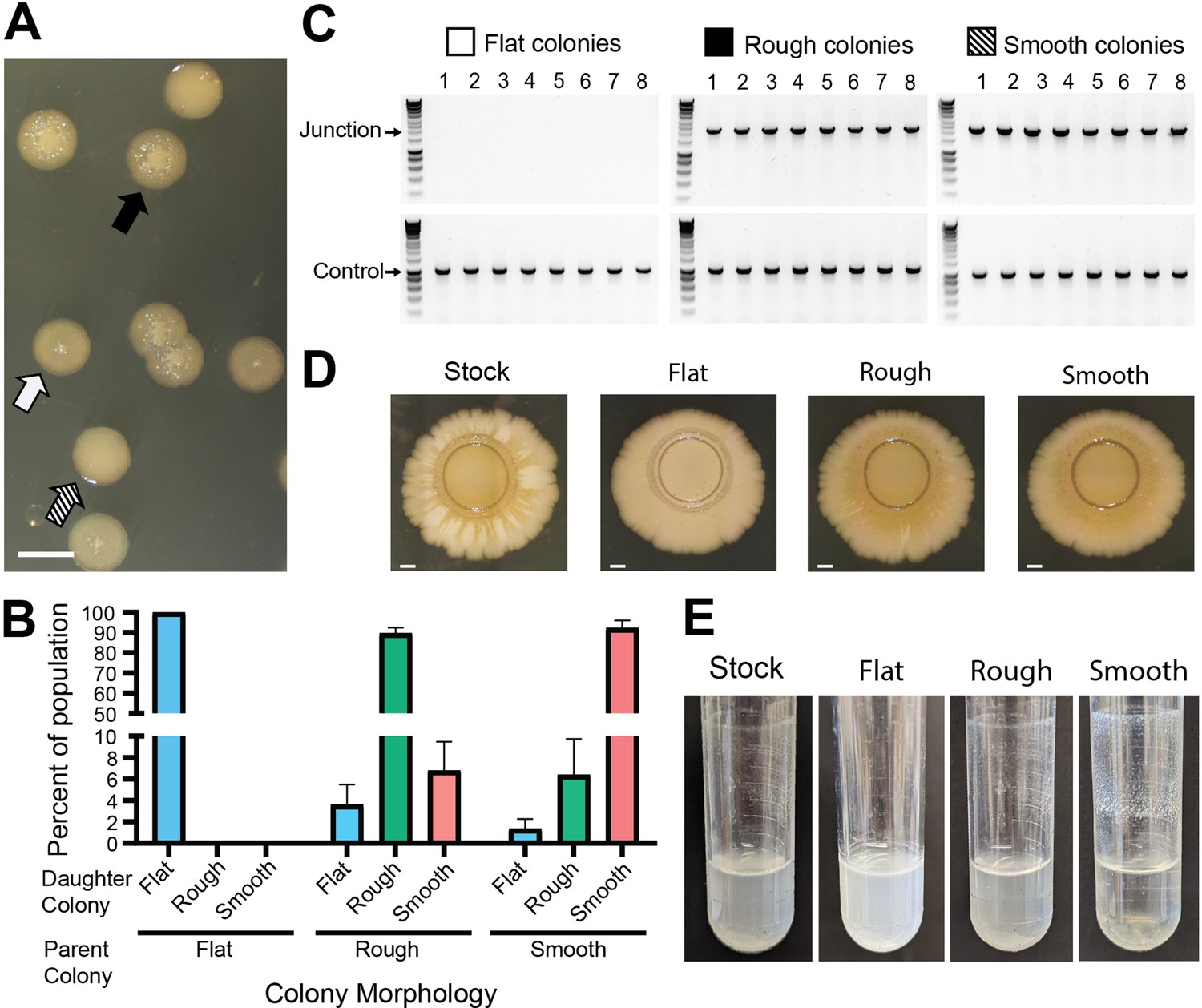
Genotypic heterogeneity correlates with phenotypic heterogeneity.
(A) Image of the three BtE264 colony phenotypes – flat (white arrow), raised rough (black arrow), and raised smooth (striped arrow) – observed on LSLB agar. Scale bar: 2 mm. (B) Colonies of each morphology were restreaked and the percentage of each morphology within the population of daughter colonies was calculated. (C) Eight colonies of each morphology were used as templates for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (D) Images of colony biofilms from the BtE264 stock, flat colonies, rough raised colonies, and raised smooth colonies. Scale bar: 2 mm. Data are representative of three independent biological replicates. (E) Images of test tube aggregation seeded from the BtE264 stock, flat colonies, raised rough colonies, and raised smooth colonies. Data are representative of three independent biological replicates.
-
Figure 2—source data 1
Uncropped DNA gel displaying junction and control PCRs of flat colonies that was used to make Figure 2C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig2-data1-v2.zip
-
Figure 2—source data 2
Uncropped DNA gel displaying junction and control PCRs of rough colonies that was used to make Figure 2C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig2-data2-v2.zip
-
Figure 2—source data 3
Uncropped DNA gel displaying junction and control PCRs of smooth colonies that was used to make Figure 2C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig2-data3-v2.zip
We investigated duplication-dependent phenotypes and found that bacteria taken directly from our stock of BtE264 displayed intermediate phenotypes compared with junction PCR positive and junction PCR negative bacteria. For example, the brownish-gold coloration developed after 28 days in colony biofilms established by bacteria from raised (junction PCR positive) colonies, but not in colony biofilms established by bacteria from flat (junction PCR negative) colonies, while colony biofilms established by bacteria taken directly from our frozen stock developed the brownish-gold coloration in the center and in alternating areas radiating outward from the original spot (Figure 2D). When grown in M63 medium on a roller, bacteria from raised colonies, but not flat colonies, aggregated and adhered to the walls of the test tube at the air–liquid interface (Figure 2E). Aggregation was apparent in the culture initiated with bacteria from our stock of BtE264, but it was substantially less than that of the culture from raised colonies. These data further demonstrate the heterogeneity of our stock of BtE264 and confirm previous observations that specific phenotypes correlate with the presence of two or more copies of the 208.6 kb region.
The 208.6 kb region is present as directly oriented, tandem repeats in the chromosome of junction PCR positive bacteria
We previously concluded that the 208.6 kb region amplified as extrachromosomal, circular, DNA molecules (Ocasio and Cotter, 2019). However, subsequent genetic experiments investigating the requirements for the formation of the extrachromosomal circle led us to question that conclusion. To distinguish between extrachromosomal circles and tandem duplications in the chromosome (both of which would generate the same junction and hence yield the same junction PCR product), we engineered strains with unique I-SceI restriction endonuclease sites located approximately 50 kb outside the boundaries of the 208.6 kb region (Figure 3A). Agarose plugs of cultures from flat, raised rough, and raised smooth colonies containing the I-SceI sites were digested with I-SceI, then the DNA was separated by pulsed-field gel electrophoresis (PFGE). A single band of ~310 kb was present in I-SceI-digested DNA from flat colonies, indicating the presence of one copy of the 208.6 kb region in the chromosome (Figure 3B). I-SceI digestion of DNA from raised rough colonies resulted in a prominent band of ~520 kb, and faint bands of ~310 and ~730 kb. This result indicates that a majority of the bacteria in the raised rough colony contain two tandem copies of the 208.6 kb region, and minor populations in the colony contain either a single copy (~310 kb band) of the region or three tandem copies (~730 kb band) of the region. DNA from the raised smooth colonies yielded a prominent ~730 kb band, with faint bands corresponding to ~520, ~310, and ~970 kb, indicating that a majority of the bacteria in raised smooth colonies contain three copies of the 208.6 kb region. No bands of ~210 kb, the size expected for an extrachromosomal circle, were detected in any sample. While junction PCR suggests that tandem repeats are oriented in a direct orientation, we wanted to further verify the directionality of the duplications. Plugs from flat, raised rough, and raised smooth colonies were digested with SpeI which is native to a locus ~143 kb within the duplicating region (Figure 3—figure supplement 1A, B). Directionality of the duplication will lead to differently sized fragments (~208 kb for a direct orientation and ~132 kb for an inverted orientation). We observed ~208 kb fragments indicating that the region amplifies as directly oriented copies. Our data demonstrate that the 208.6 kb region is present in directly oriented tandem copies in the chromosome and not extrachromosomal circular DNA molecules. These results suggest that amplification and resolution of the 208.6 kb region within a growing population of bacteria is dynamic. For simplicity, we will henceforth refer to junction PCR positive bacteria (which contain two or more copies of the 208.6 kb region) as Dup+ and junction PCR negative bacteria (which contain a single copy of the region) as Dup−.
Figure 3 with 1 supplement see all
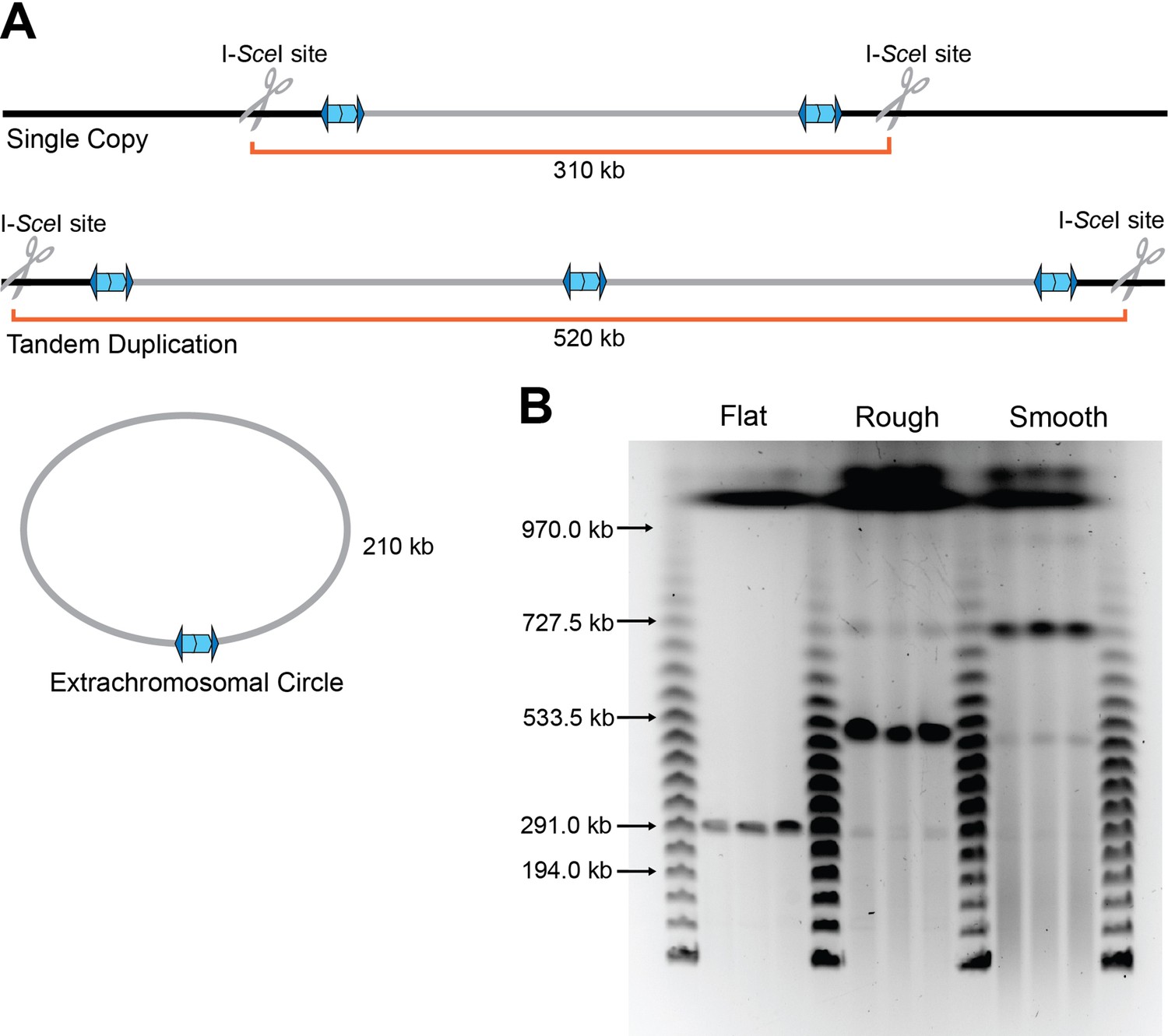
The duplication is present as a directly oriented tandem repeat of the 208.6 kb region in the chromosome.
(A) Graphical representation of BtE264 with engineered unique I-SceI cut sites ~50 kb outside of the 208.6 kb duplicating region creating differently sized fragments depending on the form of the duplication. (B) Pulsed-field gel electrophoresis separated duplicating region fragments, post I-SceI digestion, from three flat, raised rough, and raised smooth colonies each alongside lambda ladder. Data are representative of two independent biological replicates.
-
Figure 3—source data 1
Uncropped pulsed-field gel electrophoresis (PFGE) DNA gel that was used to make Figure 3B.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig3-data1-v2.zip
Resolution of tandem duplications occurs through RecA-dependent homologous recombination
When we restreaked Dup− colonies on LSLB agar, we found that of hundreds of daughter colonies tested, none yielded a junction PCR product, indicating the presence of only a single copy of the 208.6 kb region (Figure 4A). When we restreaked Dup+ colonies on LSLB agar, ~95% of the daughter colonies yielded a positive junction PCR product, while ~5% of the daughter colonies failed to yield a junction PCR product, indicating that the tandem duplication had resolved to a single copy in ~5% of the bacteria in each Dup+ colony. These results indicate that resolution of tandem copies of the region to a single copy occurs during growth as a single colony on LSLB agar.
Figure 4
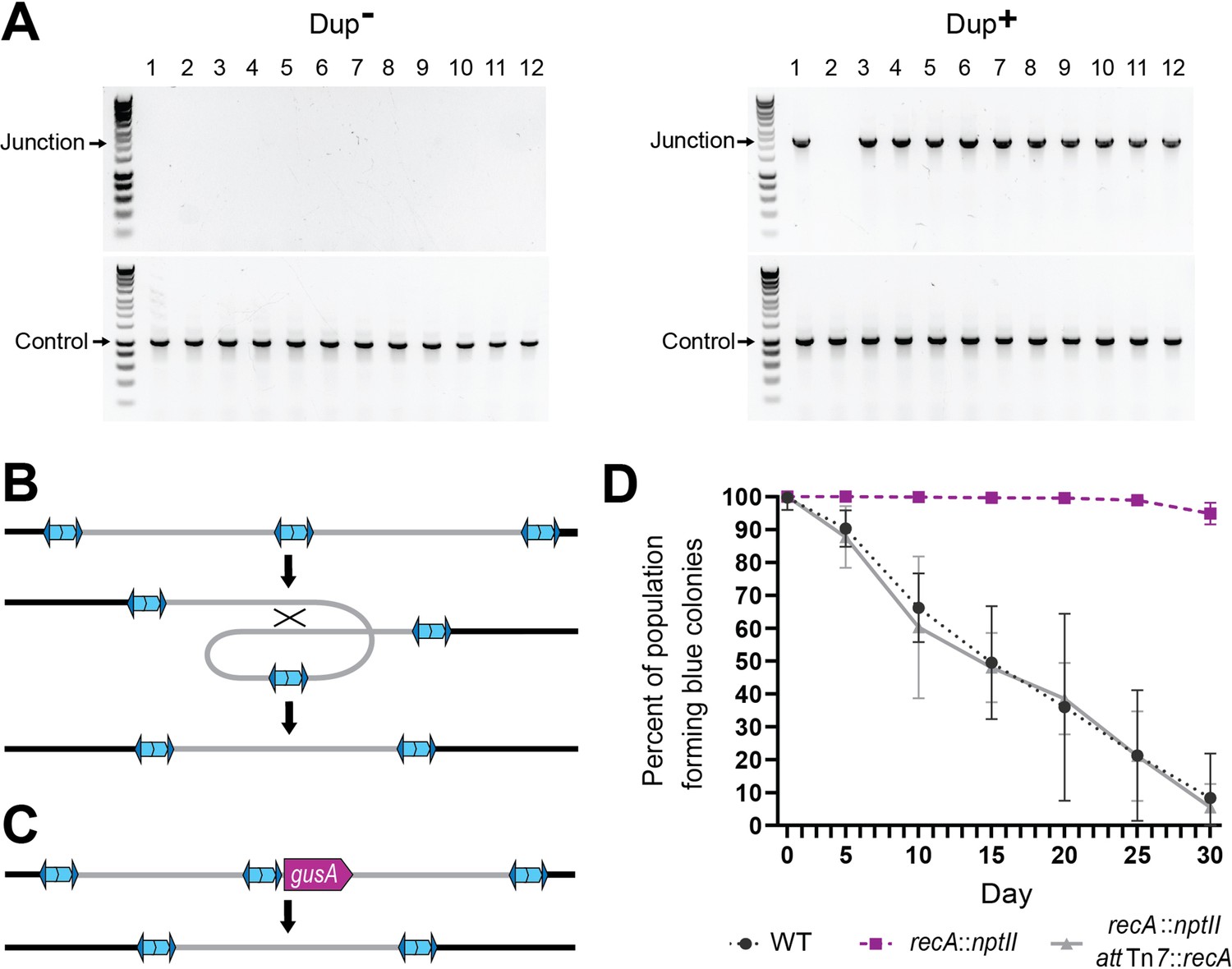
Resolution of tandem duplications occurs through RecA-dependent homologous recombination.
(A) Twelve daughter colonies from a Dup+ and Dup− colony were used as templates for PCR using either Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (B) Model for tandem resolution by recombination between homologous sequences in the duplicated region. (C) Schematic of the gusA reporter strain used to visualize colonies that have resolved the tandem duplication. (D) Percentage of WT, recA::nptII, and recA::nptII attTn7::recA colonies containing gusA throughout daily liquid subculturing. Mean and standard deviation plotted. From day 5 onward, the percent of blue colonies are significantly different between strains with and without functional recA, p values <0.01 at days 5–30 as analyzed by Kruskal–Wallis test.
-
Figure 4—source data 1
Uncropped DNA gel displaying junction PCR of colonies from Dup+ parent colonies that was used to make Figure 4A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig4-data1-v2.zip
-
Figure 4—source data 2
Uncropped DNA gel displaying junction PCR of colonies from Dup− parent colonies that was used to make Figure 4A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig4-data2-v2.zip
-
Figure 4—source data 3
Uncropped DNA gel displaying control PCR of colonies from Dup+ and Dup− parent colonies that was used to make Figure 4A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig4-data3-v2.zip
We hypothesized that resolution of tandem copies of the 208.6 kb region to a single copy occurs through RecA-dependent homologous recombination, which could occur anywhere within the 208.6 kb of homologous sequences, resulting in a deletion of an entire copy of the region (Figure 4B). To test this hypothesis, we constructed a reporter strain in which we inserted gusA adjacent to the unique junction in a Dup+ strain (Figure 4C). The product of gusA hydrolyzes X-Gluc, releasing chloro-bromindigo and resulting in the formation of blue colonies. With gusA located near the center of the duplicated region, resolution of a tandem duplication to a single copy should, in the vast majority of cases, result in loss of gusA and hence the formation of white colonies on plates containing X-Gluc. We also constructed a recA-deficient derivative of this strain (∆recA::nptII), and a derivative of the ∆recA::nptII strain in which recA, under the control of its native promoter, was inserted into both of the attTn7 sites in the BtE264 genome (∆recA::nptII attTn7::recA). We inoculated LSLB with the gusA-containing strains, and every 24 hr we collected aliquots for subculturing into fresh LSLB and for plating on X-Gluc-containing agar. The proportion of blue colonies obtained from our otherwise wild-type gusA insertion strain decreased from 100% on day 1 to ~10% by day 30 (Figure 4D). The proportion of blue colonies was maintained at nearly 100% over the 30-day course of the experiment in the recA-deficient strain, but the complemented strain mirrored wild-type and dropped to ~10% by day 30. These data indicate that resolution of tandem copies of the 208.6 kb region to a single copy during growth in LSLB occurs by recA-dependent homologous recombination.
Duplication of the 208.6 kb region is enriched in the center of colony biofilms
We observed that when colony biofilms of Dup− cells were grown at room temperature for an extended amount of time (>4 weeks), brownish-gold spots appeared in the center (Figure 5A). The coloration was reminiscent of that which appears throughout the middle of colony biofilms initiated with Dup+ bacteria. We hypothesized that these spots were clusters of cells in which the 208.6 kb region had amplified. To attempt to speed up the process, we seeded colony biofilms with a 5× denser culture of cells. We established colony biofilms with 3.6 × 107 cfu instead of 7.2 × 106 cfu in a 20 µl droplet. After 7 days of growth, we picked bacteria from the center and edge of the colony biofilm for PCR analysis (Figure 5B). In colony biofilms established with Dup+ bacteria, junction PCR products were obtained from bacteria picked from both the center and the edge (Figure 5C). In Dup− colony biofilms, junction PCR products were obtained from bacteria picked from the center, but not the edge. We also tested colony biofilms established with Dup− 2002721643, 2002721723, and BtE264 from the Mougous laboratory and similarly observed that Junction PCR products were obtained from bacteria picked from the center, but not the edge, of the colony biofilms (Figure 5D). These results demonstrate that Dup− BtE264 bacteria from our laboratory and the Mougous laboratory, and strains 2002721643 and 2002721723, are capable of duplicating the 208.6 kb region and suggest that either duplication occurs more frequently during growth in the center of a colony biofilm, or that duplication of the region confers a growth or survival advantage to bacteria growing in the center of a dense colony biofilm.
Figure 5
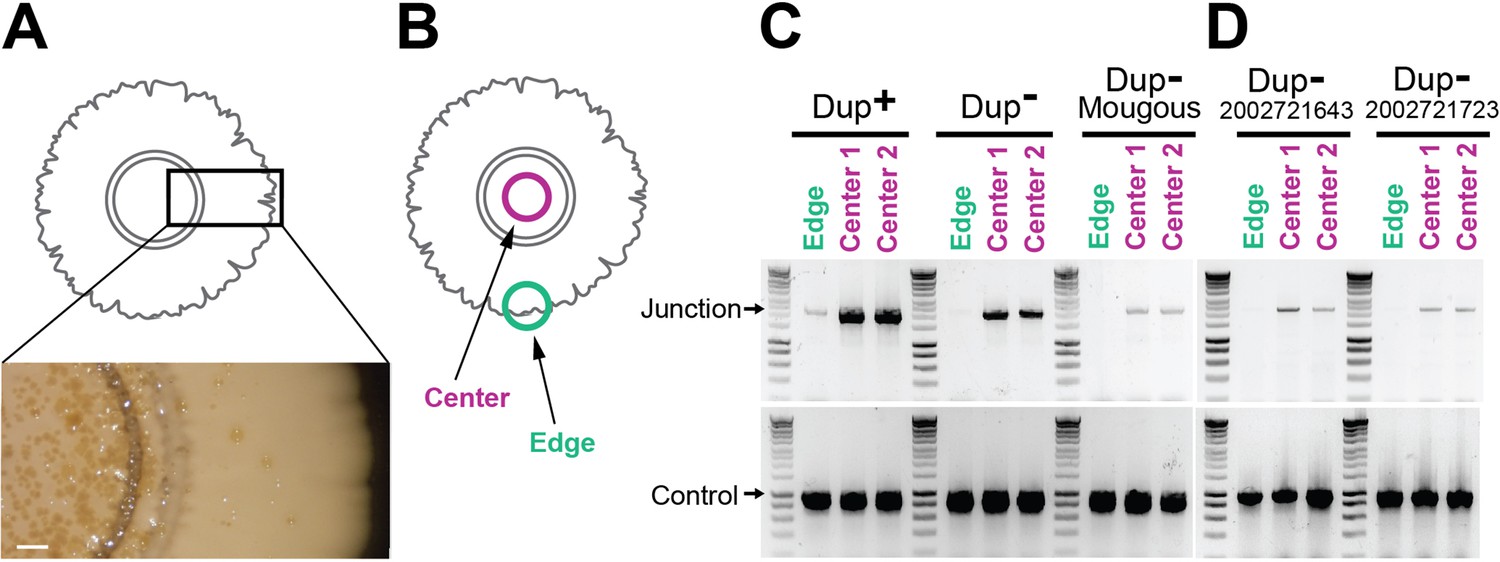
Duplications are enriched for in the center of colony biofilms.
(A) Colony biofilm schematic and image depicting brownish-gold spots form primarily in the center of Dup− colony biofilms. Scale bar: 2 mm. (B) Schematic demarcating the center and the edge portions of colony biofilms. The center and edge of colony biofilms from (C) wild-type Dup+, wild-type Dup−, and Mougous laboratory BtE264 and (D) B. thailandensis 2002721643 and 2002721723 were used as templates for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers.
-
Figure 5—source data 1
Uncropped DNA gel displaying junction PCR from wild-type Dup+, wild-type Dup−, and Mougous Laboratory’s wild-type strains that was used to make Figure 5C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig5-data1-v2.zip
-
Figure 5—source data 2
Uncropped DNA gel displaying control PCR from wild-type Dup+, wild-type Dup−, and Mougous Laboratory’s wild-type strains that was used to make Figure 5C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig5-data2-v2.zip
-
Figure 5—source data 3
Uncropped DNA gel displaying junction PCR from strains 2002721643 and 2002721723 that was used to make Figure 5C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig5-data3-v2.zip
-
Figure 5—source data 4
Uncropped DNA gel displaying control PCR from strains 2002721643 and 2002721723 that was used to make Figure 5C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig5-data4-v2.zip
BcpAIOB activity is not involved in amplification of the 208.6 kb region
We previously concluded that the BcpAIOB contact-dependent inhibition system was required for amplification of the 208.6 kb region, and that the amplification occurred by the production of extrachromosomal, circular, DNA molecules (Ocasio and Cotter, 2019). Having demonstrated that the 208.6 kb region in junction PCR positive wild-type E264 bacteria are present as tandem copies in the chromosome, we re-evaluated the form of the DNA amplification in bcpAIOB mutants. We introduced I-SceI sites into the previously constructed ∆bcpAIOB and PS12-bcpAIOB strains, which are Dup− and Dup+, respectively, digested chromosomal DNA with I-SceI, and examined the digested DNA by PFGE (Anderson et al., 2012). The digested DNA from the ∆bcpAIOB strain contained a fragment of ~310 kb, corresponding to a single copy of the region, while digested DNA from the PS12-bcpAIOB strain contained a prominent ~520 kb band, corresponding to the size expected for a tandem duplication of the 208.6 kb region, and faint bands at ~310 and ~730 kb, corresponding to fragments containing one and three copies of the region, respectively (Figure 6—figure supplement 1). These results indicate that the presence of the unique junction sequence in our bcpAIOB mutant strains correlates with multiple or single chromosomal copies of the 208.6 kb region in the chromosome, as it does for the flat and raised colonies from our frozen stock of BtE264.
We were not aware of the heterogeneity in our wild-type BtE264 stock when constructing the original ∆bcpAIOB and PS12-bcpAIOB strains, causing us to generate strains with inconsistent copy number of the 208.6 kb region. We therefore re-evaluated whether a correlation between BcpAIOB activity and copy number of the 208.6 kb region (and duplication-dependent phenotypes) actually exists by constructing new ∆bcpAIOB and PS12-bcpAIOB strains. Introduction of the S12 promoter upstream of the bcpAIOB genes in a Dup− strain resulted in a Dup− strain that lacked duplication-dependent phenotypes (Figure S2B), and deletion of both copies of bcpAIOB in a Dup+ strain (∆bcpAIOB::ble ∆bcpAIOB::nptII) resulted in a Dup+ strain that displayed duplication-dependent phenotypes (Figure S2C). We also grew Dup− strains with the bcpAIOB mutations as colony biofilms and conducted PCR analyses. Junction PCR products were obtained from bacteria picked from the center of all colony biofilms, including the ∆bcpAIOB strain (Figure 6A). Together, these results indicate that duplication-dependent phenotypes correlate with the presence of two or more copies of the 208.6 kb region and not with BcpAIOB activity (even indirectly), and they show definitively that amplification of the 208.6 kb region does not require BcpAIOB activity.
Figure 6 with 1 supplement see all
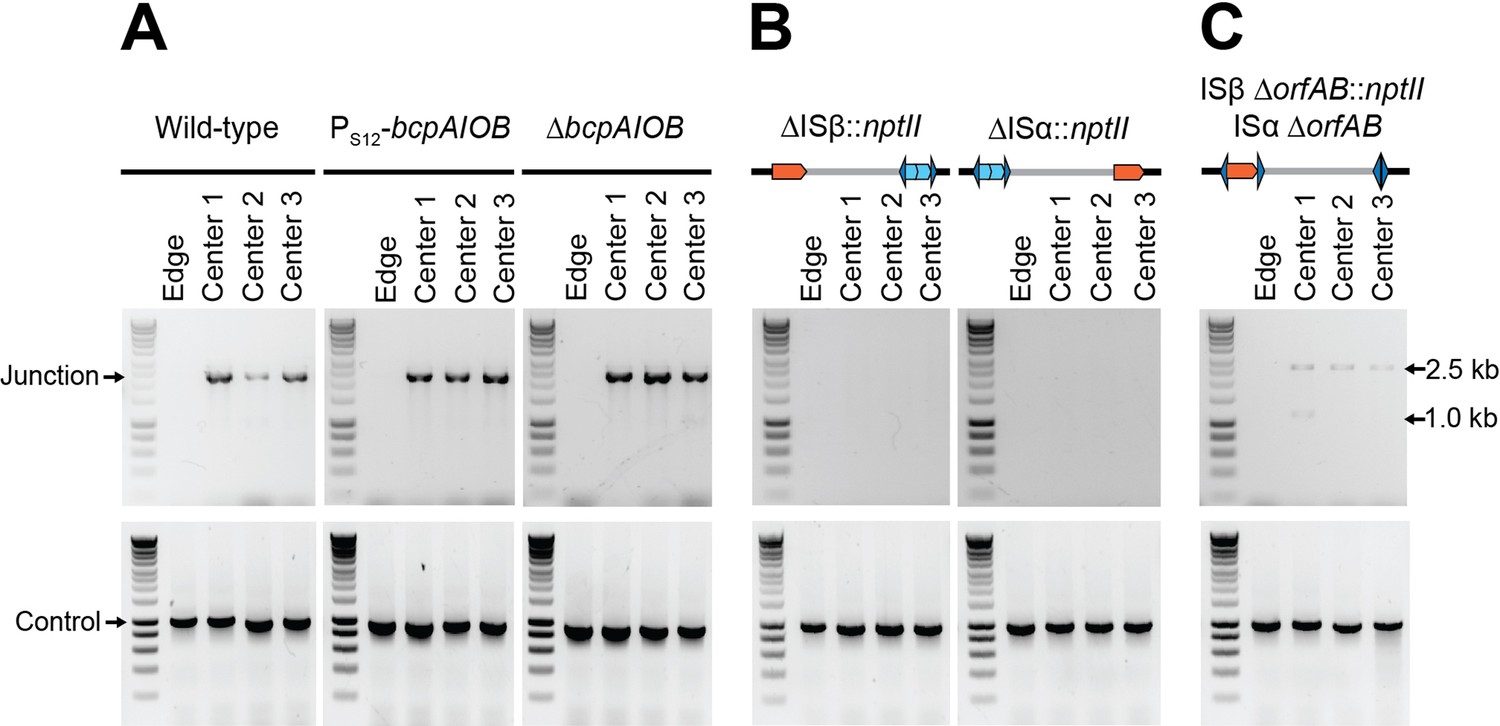
Contact-dependent growth inhibition (CDI) system-encoding bcpAIOB and transposase-encoding orfAB genes are not involved in duplication of the 208.6 kb region.
(A) The center and edge of colony biofilms from wild-type Dup+, wild-type Dup−, Dup+ PS12-bcpAIOB, and Dup− ΔbcpAIOB were used as templates for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (B) The center and edge of colony biofilms from ΔISβ::nptII and ΔISα::nptII were used as templates for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (C) The center and edge of colony biofilms from ISβ ΔorfAB::nptII ISα ΔorfAB were used as a template for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers.
-
Figure 6—source data 1
Uncropped DNA gel displaying junction PCR from wild-type, PS12-bcpAIOB, ΔbcpAIOB, and ISβ ΔorfAB::nptII ISα ΔorfAB strains that was used to make Figure 6A, C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig6-data1-v2.zip
-
Figure 6—source data 2
Uncropped DNA gel displaying junction PCR from ΔISβ::nptII and ΔISα::nptII strains that was used to make Figure 6B.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig6-data2-v2.zip
-
Figure 6—source data 3
Uncropped DNA gel displaying control PCR from wild-type, PS12-bcpAIOB, and ΔbcpAIOB strains that was used to make Figure 6A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig6-data3-v2.zip
-
Figure 6—source data 4
Uncropped DNA gel displaying control PCR from ΔISβ::nptII, ΔISα::nptII, and ISβ ΔorfAB::nptII ISα ΔorfAB strains that was used to make Figure 6B, C.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig6-data4-v2.zip
ISα and ISβ transposase activity is not required for amplification of the 208.6 kb region
We also previously concluded that ISβ was required for amplification of the 208.6 kb region, based on data from strains constructed before our awareness of copy number variability of the 208.6 kb region in our stock of BtE264. To re-evaluate the roles of ISα and ISβ in amplification of the 208.6 kb region, we separately replaced each IS element, including the inverted repeats that flank the transposase-encoding orfAB genes, with nptII (confers kanamycin resistance) in Dup− bacteria, grew the two strains as colony biofilms, and conducted PCR analyses. While PCR products were obtained from bacteria picked from the centers and edges of the colony biofilms when control primers were used, no junction PCR products were obtained from the ISα and ISβ mutants (Figure 6B). To determine specifically if the transposase encoded by either ISα or ISβ is required for amplification of the region, we constructed a strain (ISα ∆orfAB ISβ ∆orfAB::nptII) that lacked orfAB from both ISα and ISβ while leaving the flanking inverted repeats for both elements intact, and grew this strain as a colony biofilm. In all five colony biofilms tested, junction PCR yielded a~2.6 kb product that is of the predicted size for a junction that contains the nptII gene, and DNA sequence analysis confirmed that this fragment contains the nptII gene flanked by sequences representing the 5′ and 3′ ends of the duplicated region, matching the ISβ loci (Figure 6C). In one of the colony biofilms (center 1), an additional junction PCR product of ~1.1 kb was obtained, matching the ISα locus. DNA sequence analysis confirmed that this PCR product contained a junction containing the inverted repeats and not the nptII gene. These data show that the transposases encoded by ISα and ISβ are not required for amplification of the 208.6 kb region, but the inverted repeats are apparently necessary and sufficient for amplification of the 208.6 kb region.
Amplification of the 208.6 kb region occurs by RecA-mediated recombination between homologous sequences
Because transposases of the same family have been shown to only function in cis (Chandler et al., 2015), we thought it unlikely that inverted repeat-mediated duplication of the region was catalyzed by one (or more) of the four other IS2-like transposases encoded in the genome. Instead, we hypothesized that duplication, like resolution, occurs through RecA-mediated recombination between homologous sequences. To test this hypothesis, we grew colony biofilms from wild-type, recA-deficient (∆recA::nptII), and recA complemented (∆recA::nptII attTn7::recA) strains and conducted PCR analyses. No junction PCR products were produced from recA-deficient bacteria, while junction PCR products were obtained from the wild-type and recA complemented strains (Figure 7A). Amplification of the 208.6 kb region, therefore, requires recA.
Figure 7
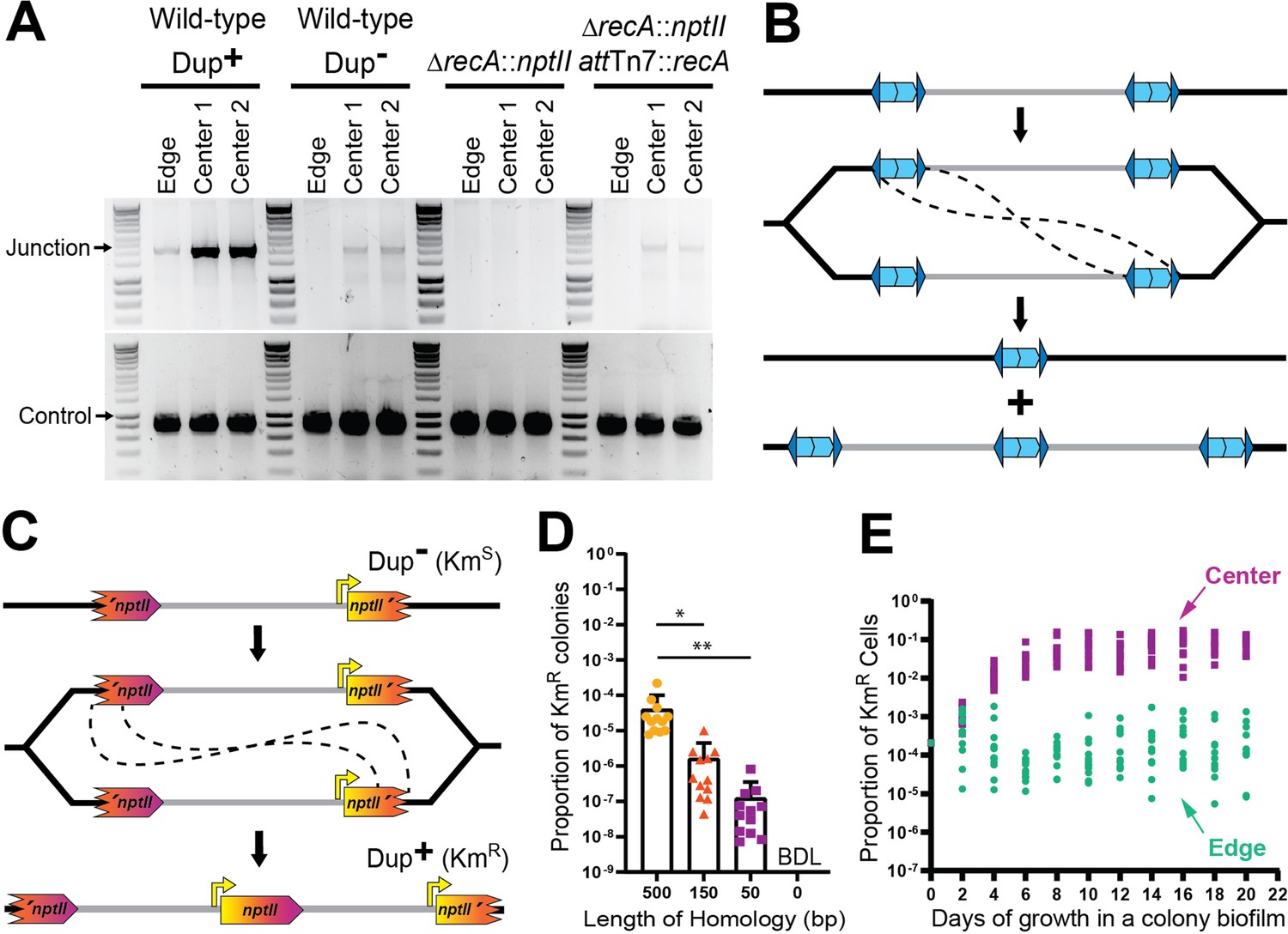
Duplications are formed through RecA-mediated recombination between homologous IS elements.
(A) The center and edge of colony biofilms from WT Dup+, WT Dup−, ΔrecA, and ΔrecA attTn7::recA were used as templates for PCR using Junction (Junc1 and Junc2) or Control (Ctrl1 and Ctrl2) primers. (B) Model for homologous recombination during replication resulting in two different genotypes within the daughter cells. One daughter cell contains a suspected fatal deletion of the 208.6 kb region while the other cell contains a tandem duplication of the 208.6 kb region. (C) Schematic of the fragmented nptII reporter system. Through recombination between homologous nptII sequences, a KmS cell can give rise to a KmR daughter cell with an intact nptII present at the junction. Orientation of the chromosome has been reversed for clarity and consistency. (D) KmR resistance frequency among fragmented nptII reporter strains with various amounts of homology (500, 150, 50, or 0 bp) following culture in LSLB broth. Mean and standard deviation indicated. ‘BDL’ indicates below detection limit. * and ** denote p values <0.05 and <0.01, respectively, as calculated by an analysis of variance (ANOVA). (E) Using the fragmented nptII reporter strain, calculated proportion of KmR cells from the center and edge of colony biofilms over time. From day 4 onwards, the proportion of KmR cells from the center and edge of the colony biofilm are significantly different; p values <0.0001 at days 4–20 as calculated by Mann–Whitney U.
-
Figure 7—source data 1
Uncropped DNA gel displaying junction PCR from wild-type Dup+, wild-type Dup−, ΔrecA::nptII, and ΔrecA::nptII attTn7::recA strains that was used to make Figure 7A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig7-data1-v2.zip
-
Figure 7—source data 2
Uncropped DNA gel displaying control PCR from wild-type Dup+, wild-type Dup−, ΔrecA::nptII, and ΔrecA::nptII attTn7::recA strains that was used to make Figure 7A.
The red box indicates the region of the gel used in the final figure. The lanes and identity of the band(s) are indicated.
- https://cdn.elifesciences.org/articles/84327/elife-84327-fig7-data2-v2.zip
These data support the hypothesis that amplification of the 208.6 kb region occurs by RecA-dependent recombination between the 1336 bp of homology shared by ISα and ISβ (Figure 7B). Homologous recombination can occur during DNA replication, through either unequal crossing over or during the repair of a collapsed replication fork to create DNA duplications (Hastings et al., 2009; Bzymek and Lovett, 2001). While replication fork repair leads to a deletion or duplication of the DNA region between the homologous sequences, unequal crossing over will generate one daughter chromosome with a duplication and one daughter chromosome with a deletion of those sequences (Hastings et al., 2009; Bzymek and Lovett, 2001). If at least one essential gene is present within the region, the daughter cell containing the chromosome with the deletion will be non-viable. To further evaluate if RecA-dependent recombination is duplicating the region, we developed a ‘fragmented reporter system’. We replaced ISβ with a 5′ portion of nptII and ISα with a 3′ portion of nptII (Figure 7C). The amount of overlap (in the center of nptII) in the strains varied between 500 and 0 bp. Recombination between the overlapping homologous sequences will result in the formation of an intact nptII gene, which will confer resistance to kanamycin (KmR). The KmR cfu and total cfu were obtained after overnight growth in LSLB and the proportion of KmR colonies was calculated (KmR cfu/total cfu = proportion KmR colonies) as 10−4, 10−6, and 10−7 for the strains with 500, 150, and 50 bp of homology, respectively (Figure 7D). The strain lacking homology produced no KmR colonies. These data indicate that duplication of the region occurs by RecA-dependent recombination between homologous sequences, independent of the specific sequence. Thus, the inverted repeats of ISα and ISβ are not required for duplication of the region, unless they are the only source of DNA sequence homology. And because homologous recombination regularly occurs at a low level, these data also suggest that wild-type BtE264 exists as a heterogeneous population.
We can also use the fragmented reporter system to quantify population dynamics. When the fragmented nptII reporter strain with 500 bp of homology was grown as a colony biofilm, we observed that after 4 days of growth, the proportion of KmR bacteria from the center of the colony biofilm was consistently greater than the proportion of KmR bacteria from the edge (Figure 7E). These results further indicate that either duplication of the 208.6 kb region occurs more frequently at the center of the colony biofilm or, more likely, that bacteria with two or more copies of the region have a growth or survival advantage in the center of the colony biofilm.
Amplification of the 208.6 kb region greatly enhances static biofilm development
Our data suggest a correlation between amplification of the 208.6 kb region and biofilm formation. We therefore developed a fragmented gfp (encoding green fluorescent protein) reporter system (Figure 8A) and compared strains for their ability to form biofilms using a standard submerged, static biofilm assay on glass coverslips in M63 medium. Each strain contains rfp, encoding red fluorescent protein, at a location outside the 208.6 kb region. Biofilms were initiated with Dup+ or Dup− bacteria. In addition, to determine if the ability to amplify the 208.6 kb region is required for biofilm development, we used a Dup−-locked strain that has an intact ISβ at one boundary of the region and the 5′ fragment of gfp replacing ISα at the other, and hence cannot duplicate the region and form GFP+ bacteria. By 24 hr, the Dup+ bacteria had formed biofilms with characteristic pillars of bacteria, while the Dup− and Dup−-locked strains formed only a few, small aggregates of bacteria. By 72 hr, biofilms formed by Dup+ bacteria were significantly taller and covering significantly more surface area than biofilms formed by Dup− and Dup−-locked bacteria (Figure 8B–D). These data show that the presence of two or more copies of the 208.6 kb region greatly enhances biofilm formation by BtE264. Several clumps of RFP+GFP– cells were present in the biofilms formed by Dup+ bacteria, indicating that resolution to a single copy of the 208.6 kb region can occur during biofilm development, and that bacteria containing only a single copy of the region can be maintained in the biofilm, at least transiently. By contrast, RFP+GFP+ bacteria were rarely observed in the biofilms formed by Dup− bacteria. These results indicate that in this static biofilm experimental condition, Dup+ bacteria are not enriched within the population.
Figure 8

Duplication of the region greatly enhances biofilm development.
(A) Schematic of the fragmented gfp reporter system. Through recombination between homologous gfp sequences, a GFP− cell can give rise to a GFP+ daughter cell with an intact gfp present at the junction. (B) Z-stack confocal microscope images of biofilms from Dup+, Dup−, and locked fragmented gfp reporter strains at 24, 48, and 72 hr of growth. Scale bar: 30 µm. Average biofilm height (C) and surface area covered (D) from Dup+, Dup−, and locked fragmented gfp reporter strains at 72 hr. Columns indicate sample means and error bars correspond to standard deviation. *, **, and *** denote p values <0.05, <0.01, and <0.001, respectively, as produced by Kruskal–Wallis test. Data are representative of eight independent biological replicates.
Amplification of the 208.6 kb region in BtE264 provides a selective advantage during dynamic biofilm development
We next investigated the contribution of the 208.6 kb region to biofilm formation under more dynamic conditions using a fragmented gusA reporter system (Figure 9A). The fragmented gusA reporter strain was chosen for two reasons: (1) it allows us to screen, rather select than for cells with a duplication and (2) the gusA gene is long enough to create a fragmented reporter system with the same homologous sequence length provided by ISα and ISβ in wild-type cells (1336 bp). We inoculated M63 medium with Dup+ bacteria, incubated the cultures on a roller overnight, and maintained selection for biofilm growth by decanting the spent medium from the tube, sampling a portion of the bacteria at the air–liquid interface for plating on X-Gluc-containing plates, and then adding fresh medium to the same tube and repeating the process daily for 7 days. We also maintained selection for planktonic growth by sampling an aliquot from the liquid phase of the overnight culture for plating and for subculturing into fresh medium daily for 7 days. We quantified the proportion of blue colonies present on X-Gluc-containing LSLB agar to determine the frequency of cells with a duplication of the 208.6 kb region. The biofilm remained 100% Dup+ for the duration of the experiment, while the proportion of GusA+ bacteria in the liquid medium dropped to ~81% by day 1, ~16% by day 2, and ~0% by day 3. These data demonstrate that cells that resolve the tandem duplication do not remain in the biofilm. Under these dynamic conditions, growth in a biofilm selects for maintenance of a duplication of the 208.6 kb region, and planktonic growth selects for bacteria with a single copy of the region.
Figure 9
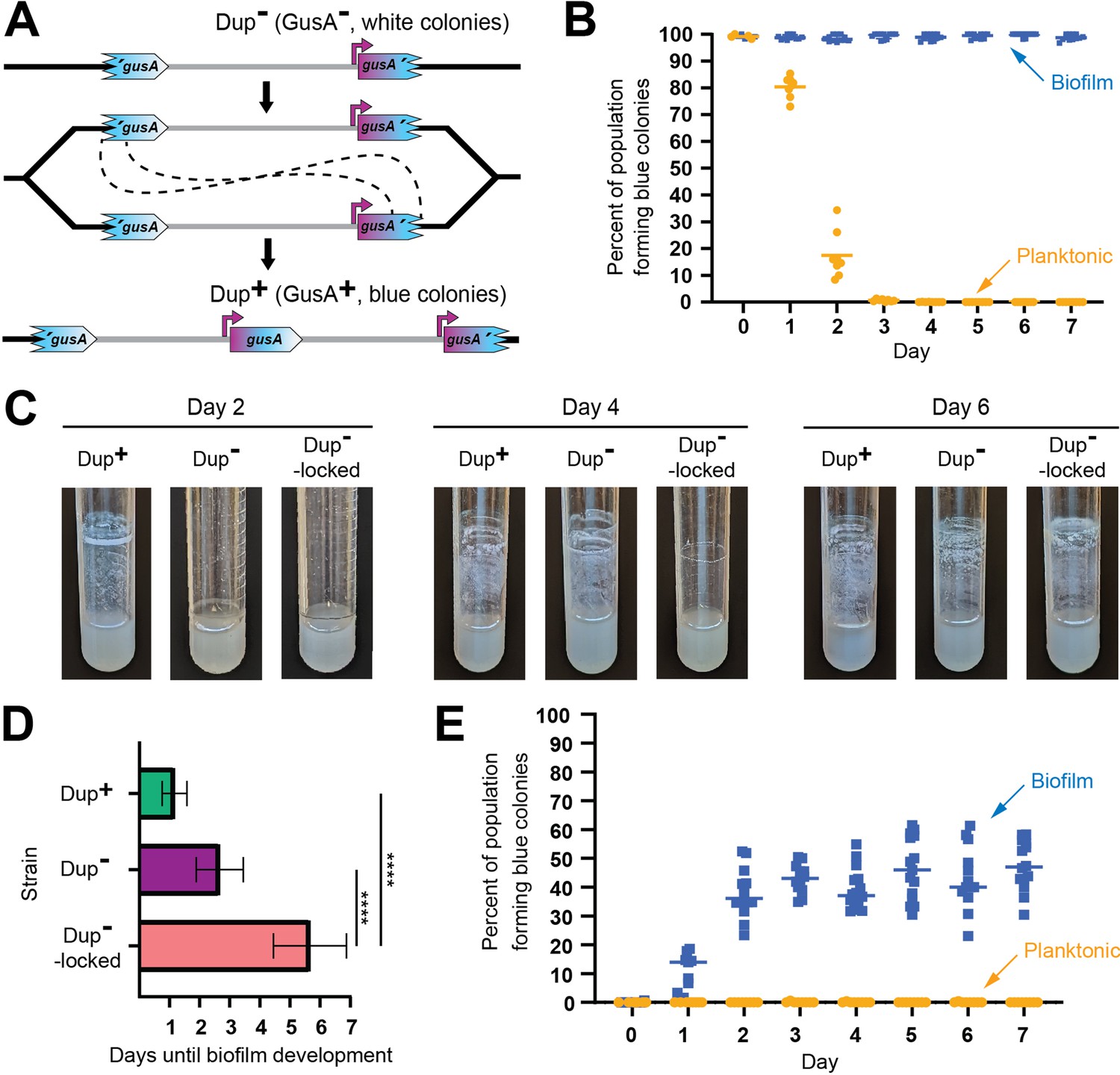
Planktonic growth enriches for a single copies of the 208.6 kb region while growth in a biofilm enriches for duplications.
(A) Schematic of the fragmented gusA reporter system. Through recombination between homologous gusA sequences, a GusA− cell can give rise to a GusA+ daughter cell with an intact gusA present at the junction. Orientation of the chromosome has been reversed for clarity and consistency. (B) Dup+ fragmented gusA reporter strain biofilms and planktonic cells were subcultured for 7 days and the proportion of blue colonies formed on X-Gluc media was calculated daily. Bars indicate sample mean. From day 1 onwards, the percent of the population forming blue colonies was significantly different, p values <0.01 at time points 1–7 as analyzed by Mann–Whitney U. (C) Images of representative air–liquid interface biofilms of Dup+, Dup−, and locked Dup− fragmented gusA strains at 2, 4, and 6 days of growth. (D) Days taken for Dup+, Dup−, and locked Dup− fragmented gusA strains to form visible, opaque biofilms at the air–liquid interface. Bars plot sample means, and error bars correspond to standard deviation. **** denotes p values <0.0001. Data are from six independent biological replicates. (E) Dup− fragmented gusA reporter strain biofilms and planktonic cells were subcultured for 7 days and the proportion of blue colonies formed on X-Gluc media was calculated daily. Bars indicate sample mean. Data are representative of three independent biological replicates. From day 1 onwards, the percent of the population forming blue colonies was significantly different, p values <0.01 at time points 1–7 as analyzed by Mann–Whitney U.
We next compared dynamic biofilm formation in M63 medium by Dup+, Dup−, and Dup−-locked bacteria. The Dup−-locked strain has an intact ISβ at one boundary of the region and the 3′ fragment of gusA at the other. As with the experiment described above, spent medium was replaced each day with fresh medium. At day 2, there were clearly aggregates of bacteria along the walls of the test tube that had been inoculated with Dup+ bacteria and only a barely visible film in the other cultures (Figure 9C). By day 4, however, cultures initiated with Dup+ and Dup− bacteria had both formed similar thick, opaque biofilms at the air–liquid interface, while the tube inoculated with Dup−-locked bacteria still had only a barely visible film (Figure 9C). By day 6, a biofilm was present even in the Dup−-locked culture (Figure 9C). The time to visible biofilm formation is graphed in Figure 9D. These data support the conclusion that Dup+ bacteria have a substantial advantage during biofilm formation. They also indicate, however, that even bacteria that cannot duplicate the 208.6 kb region can ultimately form biofilms, albeit much less efficiently than bacteria that can duplicate the region.
Because a strain capable of duplicating the 208.6 kb region constructed biofilms more rapidly than a strain that could not, we hypothesized that cells in the biofilm contained a duplication. To test this, we repeated the biofilm and planktonic growth selection experiment using a Dup− fragmented gusA reporter strain. Over the course of the experiment, the frequency of blue colonies from the broth culture remained around 0%, indicating that amplification of the 208.6 kb region in planktonically growing bacteria occurs at a very low frequency. Meanwhile, the proportion of blue colonies formed by bacteria recovered from the walls of the test tube increased to ~50% within 2 days, indicating a dramatic enrichment for Dup+ bacteria in the biofilm (Figure 9E).
Discussion
We have discovered a phase variation system that allows B. thailandensis to adapt quickly to environments that demand different (planktonic versus biofilm) lifestyles (Figure 10). We showed that a pair of IS2-like elements bounding a region containing 157 genes provide DNA sequence homology for RecA-mediated homologous recombination, resulting in the formation of tandem, directly repeated copies of the region in the chromosome. Resolution of tandem copies to a single copy also occurs by RecA-mediated homologous recombination. We showed that the presence of two or more copies of the 208.6 kb region promotes biofilm formation, while a single copy is advantageous for planktonic growth. Because amplification and resolution of the region occur at low and presumably constant (yet different) rates, all growing populations of BtE264 are heterogeneous with regard to region copy number, containing subpopulations that are optimized for either planktonic or sessile growth. This phenomenon, therefore, represents a bet-hedging strategy to improve population survival in fluctuating environments. Moreover, as it is likely that this phase variation system was formed by acquisition of the directly oriented IS2-like elements, we posit that it is an example of IS element-mediated evolution.
Figure 10
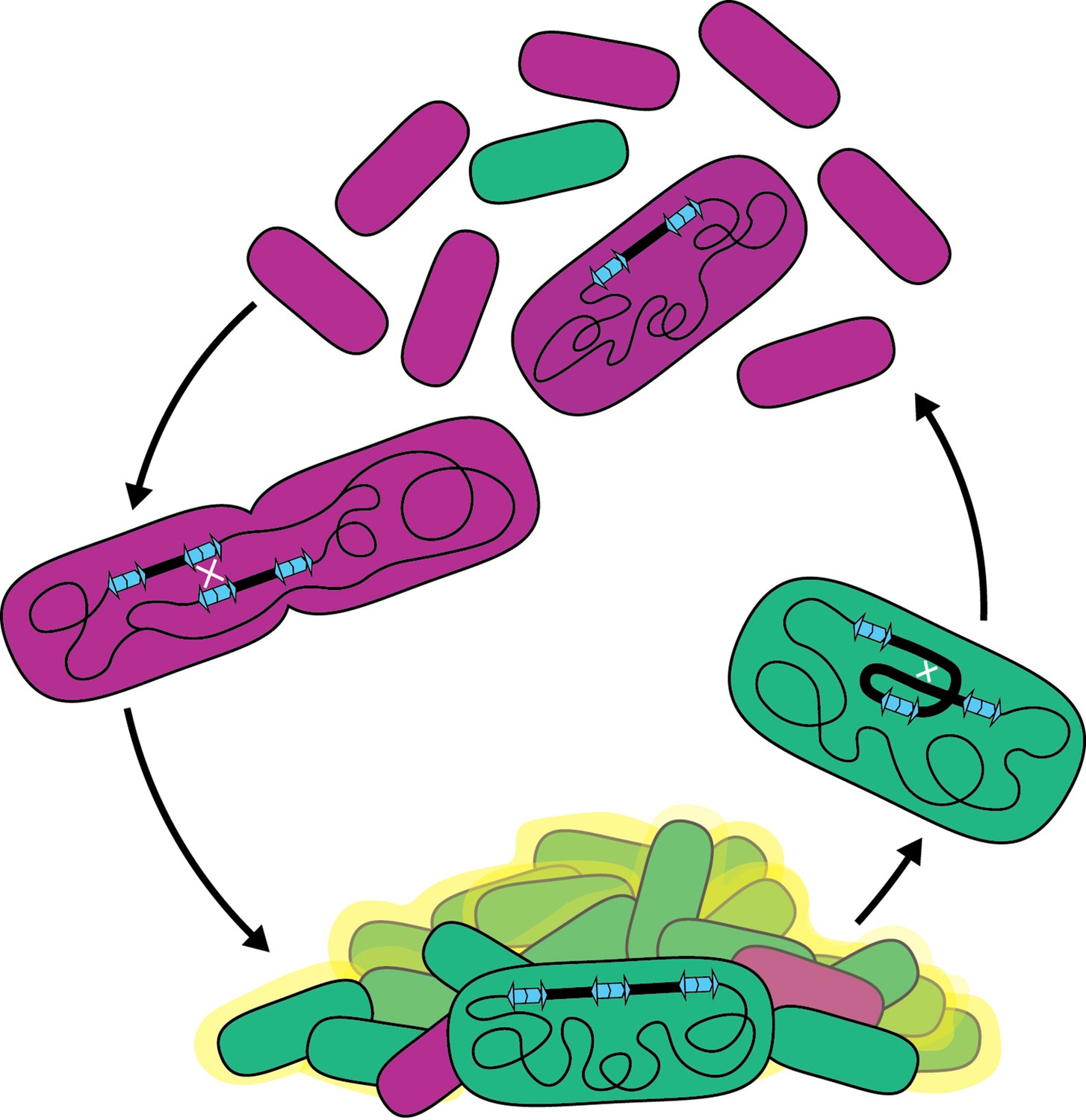
Model for the bet-hedging strategy in B. thailandensis strains containing ISα and ISβ.
Recombination between homologous IS sequences forms heterogeneous populations that allows B. thailandensis to adapt to disparate growth environments.Bacterial biofilms are ubiquitous in both natural and manufactured environments. Because biofilms can contribute to human disease and can cause environmental harm, it is important to understand mechanisms underlying their formation and maintenance. Our data indicate that the presence of two or more copies of the 208.6 kb region promotes biofilm formation, suggesting that the responsible gene(s) act as an insufficient haplotype when the region is present in single copy. The 157 genes within the region are functionally diverse: they include gene, or gene clusters, predicted to encode defense systems, antimicrobial resistance, amino acid, carbohydrate, and lipid biosynthesis, transport, and metabolism pathways, and transcription regulators. Determining which gene(s), when present in two or more copies, facilitates biofilm formation will be our next goal. Genes encoding regulatory factors, such as predicted transcription regulators lysR and luxR, are likely candidates as alteration in copy number can push signaling pathways over a threshold, leading to large changes in gene expression for all genes within the regulon. Other candidate genes include those predicted to encode fimbria or a polysaccharide transport system, which may be directly involved in biofilm formation, but whether two or three copies of these genes is sufficient for biofilm formation seems unlikely, although we cannot rule it out. While biofilm-associated growth is a single condition benefited by duplication of the 208.6 kb region, we anticipate that duplications may benefit cells in other conditions. For example, we do not know the nature or function of the molecule(s) causing the gold-brown coloration that correlates with the presence of multiple copies of the region, but we speculate that it may be advantageous for the bacteria under conditions other than, or in addition to, biofilm formation and maintenance. The diversity of predicted gene functions within the duplicating region suggests that amplification has potential to benefit BtE264 in multiple environments.
In the absence of a functional phase variation system (i.e., the Dup−-locked strain), BtE264 was still able to form a biofilm in the dynamic, test tube format, but it did so at a much slower rate compared to Dup− bacteria. The delay is consistent with these bacteria acquiring spontaneous mutations, which occur at much lower frequencies than homologous recombination. Also consistent with these bacteria possessing spontaneous, biofilm-promoting mutations, the colonies recovered from those biofilms appeared mucoid, and preliminary data indicate they form biofilms at least as fast as Dup+ bacteria. Identifying the genetic alterations in these biofilm-forming, Dup−-locked strains will shed light on the molecular mechanism underlying biofilm formation and will help us determine why duplication of the region positively influences biofilm formation.
Copy number dynamics differed within populations growing in static versus dynamic biofilms. In dynamic (culture tube-associated) biofilms, duplication of the region provided a strong selective advantage; bacteria that lost the duplication were not maintained in the biofilm and dispersed into the planktonic population. By contrast, in static (submerged coverslip) biofilms, the selective advantage was not as apparent, even though Dup+ bacteria clearly formed more robust biofilms and did so more quickly than Dup− bacteria. In static biofilms initiated with Dup+ bacteria, Dup− bacteria arose and were maintained for at least for 3 days. Moreover, static biofilms initiated with Dup− cells did not contain an appreciable number of Dup+ cells over the course of the experiment. We envisage four factors that may contribute to the difference in duplication dynamics between the two biofilm experiments.
Differences in culture conditions may impact the intensity of the selective pressure toward biofilm formation. While spent medium was replaced daily in both dynamic and static biofilms, the dynamic biofilms were constantly washed and agitated as the slanted culture tube rotated. We hypothesize that the constant washing of the dynamic biofilms removed less fit or less adherent Dup− cells from the biofilm, whereas the static biofilm scenario simply did not provide the physical force required to remove those cells.
Duplication rates are impacted by the amount of homology provided by the fragmented gfp (500 bp) and gusA (1336 bp) reporter strains used in the static and dynamic biofilms, respectively. Our data indicate that duplications form more frequently when more homologous sequences bounding the region are longer (Figure 7D), potentially contributing to why we see more Dup+ strains in the fragmented gusA dynamic biofilms.
Lack of agitation, and hence lack of aeration, during static biofilm growth may increase generation time. Because recombination to duplicate the region occurs during DNA replication, duplication frequency is directly related to growth rate.
Differences in growth conditions may impact recA expression or RecA activity levels, thereby affecting duplication frequency.
Data acquired in this study refute some of our previous conclusions. Although we were previously unable to definitively determine the form of the 206.8 kb duplication, we concluded that the extra copies likely existed as extrachromosomal circular DNA molecules based on the fact that IS2 elements transpose using a copy-out-paste-in mechanism and the observation that the region mobilized to a locus in chromosome II when we attempted to delete the 206.8 kb region from chromosome I (Ocasio and Cotter, 2019). Our PFGE data prove that the region is present as tandem, directly oriented copies in the chromosome in Dup+ bacteria, and we have also now shown that the transposases encoded by ISα and ISβ are not required for amplification of the region. Extrachromosomal, circular copies of the region will occur, however, when homologous recombination resolves merodiploids (tandem duplications) to a single copy, and these circular DNA molecules can integrate into the chromosome by homologous recombination between the IS element in the circular molecule and any of the four or two IS2-like elements in chromosome I or II, respectively. Although such an event would be rare, our attempt to delete essential genes within the 206.8 kb region apparently provided the necessary selective pressure, explaining how the region moved to chromosome II in our previous study.
Our previous conclusion that BcpA activity is required for amplification of the 208.6 kb region was based on the fact that strains with mutations that inactivate BcpA (e.g., deletion of the bcpAIOB locus or replacement of codons in bcpA required for BcpA catalytic activity) were junction PCR negative and did not display duplication-dependent phenotypes, and strains in which bcpAIOB was driven by the S12 promoter were strongly junction PCR positive and did display duplication-dependent phenotypes. It is now clear that when we constructed the BcpA-inactivating mutants (by either two-step allelic exchange or natural transformation and double homologous recombination) we chose Dup– strains when we confirmed that the mutants contained the intended mutations and not the wild-type sequences. When we introduced the S12 promoter 5′ to bcpAIOB through natural transformation, however, we confirmed that the S12 promoter was present at the intended location, but had no knowledge at the time that there could be another copy of the locus in the chromosome, so, by chance, had chosen a Dup+ strain. Our data now clearly show that duplication-dependent phenotypes are due to the duplication of the region and not BcpAIOB activity.
Bacterial populations are often more genotypically heterogeneous than presumed. It is estimated that 10% of cells in a growing culture carry a duplication of some region of the chromosome (Roth et al., 1996). Unfortunately, standard alignment techniques call out SNPs, deletions, and inversions, but not duplications. Identifying duplications requires close attention to sequencing read abundance or long-read sequencing, and hence they are often missed, even when their existence is considered a possibility. Because duplications have the potential to alter a wide variety of phenotypes, they can lead to flawed conclusions when cells with and without DNA duplications are unknowingly compared. Discovering the 208.6 kb DNA duplication in BtE264 allowed us to correct some of our previous conclusions, and also sheds light on other perplexing results. For example, a transposon screen of BtE264 yielded strains with transposon insertions in the gene (bcpI) encoding the immunity protein for the BcpAIOB contact-dependent inhibition system, which should have been fatal (Gallagher et al., 2013). Such a mutation could be tolerated, however, if a second copy of the bcpI gene is present, and hence the Tn insertion into bcpI may have selected for Dup+ cells. Our data suggest that duplications should always be considered when mutants are obtained that contain mutations in genes predicted or suspected to be essential.
Transposable elements are facilitators of evolution. In the short term, insertion into coding sequences can impact gene expression (Darmon and Leach, 2014). In the long term, homologous transposable elements can act as substrates for DNA recombination that can alter genome architecture (Vandecraen et al., 2017). Notably, recombination between transposable elements facilitates genome reduction in host-evolved bacteria, which has been observed, for example, in the evolution of the equine-infecting Burkholderia mallei from the broader host range pathogen Burkholderia pseudomallei (Losada et al., 2010). We hypothesize that acquisition of ISα and ISβ at their current locations in BtE264 represents an evolutionary step for BtE264 – creating a bet-hedging phase variation system that allows for population survival when conditions oscillate between favoring planktonic and biofilm lifestyles. Here, we show that acquisition of a pair of transposable elements has constructed a phase variation system that allows BtE264 to rapidly adapt to environments favoring planktonic or biofilm growth. Bioinformatic analysis of other B. thailandensis strains revealed that nearly all other related strains lack both ISα and ISβ. One exception that contains ISα but not ISβ and some adjacent sequences is a B. thailandensis strain that is unusually pathogenic called BPM (Chang et al., 2017). It is tempting to speculate that BPM represents an ancestral strain that has not acquired ISβ and the adjacent genes, although it may also represent a descendent that lost ISβ and the adjacent genes coincident with gaining virulence.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Burkholderia thailandensis) | Duplicating region | GCA_000012365.1 | LO74_RS28540–LO74_RS09425 | |
| Gene (Burkholderia thailandensis) | recA | GCA_000012365.1 | LO74_RS12520 | |
| Strain, strain background (Burkholderia thailandensis) | E264 | ATCC | 700388 | GCA_000012365.1 |
| Strain, strain background (Burkholderia thailandensis) | 2002721643 | USAMRIID | GCA_000959425.1 | |
| Strain, strain background (Burkholderia thailandensis) | 2002721723 | USAMRIID | GCA_000567925.1 | |
| Strain, strain background (Escherichia coli) | DH5α | GoldBio | CC-101-5x50 | Electrocompetent cells |
| Strain, strain background (Escherichia coli) | RHO3 | AddGene | 124700 | Electrocompetent cells |
| Genetic reagent (Burkholderia thailandensis) | WT with I-SceI cut sites 50 kb outside duplicating region | This study | Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | |
| Genetic reagent (Burkholderia thailandensis) | WT with gusA insertion | This study | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. | |
| Genetic reagent (Burkholderia thailandensis) | ΔrecA with gusA insertion | This study | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. Fused first 31 amino acids to last 38 amino acids of LO74_RS12520 (recA). | |
| Genetic reagent (Burkholderia thailandensis) | ΔrecA attTn7::recA with gusA insertion | This study | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. Fused first 31 amino acids to last 38 amino acids of LO74_RS12520. recA inserted at both attTn7 sites. | |
| Genetic reagent (Burkholderia thailandensis) | PS12-bcpAIOB Dup+ with I-SceI sites 50 kb outside duplicating region | This study | PS12 and nptII inserted between bcpA and its native promoter. nptII excised with Flp recombinase. Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | |
| Genetic reagent (Burkholderia thailandensis) | ΔbcpAIOB with I-SceI sites 50 kb outside duplicating region | This study | LO74_RS09295–LO74_RS09305 (bcpAIOB) replaced with nptII surrounded by FRT sites. nptII excised with Flp recombinase. Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | |
| Genetic reagent (Burkholderia thailandensis) | PS12-bcpAIOB Dup+ | Anderson et al., 2012 | PS12 promoter inserted between LO74_RS09305 (bcpA) and its native promoter. | |
| Genetic reagent (Burkholderia thailandensis) | ΔbcpAIOB::nptII Dup− | Anderson et al., 2012 | LO74_RS09295–LO74_RS09305 (bcpAIOB) replaced with nptII surrounded by FRT sites. | |
| Genetic reagent (Burkholderia thailandensis) | PS12-bcpAIOB Dup− | This study | PS12 promoter inserted between LO74_RS09305 (bcpA) and its native promoter. | |
| Genetic reagent (Burkholderia thailandensis) | ΔbcpAIOB::ble ΔbcpAIOB::nptII Dup+ | This study | LO74_RS09295–LO74_RS09305 (bcpAIOB) replaced with nptII surrounded by FRT sites. Additional copy of LO74_RS09295– LO74_RS09305 (bcpAIOB) replaced with ble. | |
| Genetic reagent (Burkholderia thailandensis) | ΔISβ::nptII | This study | Inverted repeats and LO74_RS09425 (ISβ) replaced with nptII surrounded by FRT sites. | |
| Genetic reagent (Burkholderia thailandensis) | ΔISα::nptII | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with nptII surrounded by FRT sites. | |
| Genetic reagent (Burkholderia thailandensis) | ISβ ΔorfAB::nptII ISα ΔorfAB | This study | LO74_RS28540 (ISα orfAB) replaced with nptII surrounded by FRT sites. nptII excised with Flp recombinase. LO74_RS09425 (ISβ orfAB) replaced with nptII surrounded by FRT sites. | |
| Genetic reagent (Burkholderia thailandensis) | ΔrecA | This study | Fused first 31 amino acids to last 38 amino acids of LO74_RS12520 (recA). | |
| Genetic reagent (Burkholderia thailandensis) | ΔrecA attTn7::recA | This study | Fused first 31 amino acids to last 38 amino acids of LO74_RS1252. recA inserted at both attTn7 sites. | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented nptII reporter 500 bp homology | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–545 of nptII (fwd). | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented nptII reporter 150 bp homology | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–195 of nptII (fwd). | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented nptII reporter 50 bp homology | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–95 of nptII (fwd). | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented nptII reporter 0 bp homology | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–45 of nptII (fwd). | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented gfp reporter attTn7::rfp | This study | PS12-rfp inserted an attTn7 site. Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 1–589 of gfp (rev). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 89–744 of gfp (rev). 500 bp of homology in gfp fragments. | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented gfp reporter attTn7::rfp locked | This study | PS12-rfp inserted an attTn7 site. Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 1–589 of gfp (rev). | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented gusA reporter | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 118–1691 of gusA (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–1454 of gusA (fwd) 1332 bp of homology in gusA fragments. | |
| Genetic reagent (Burkholderia thailandensis) | Fragmented gusA reporter locked | This study | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 118–1691 of gusA (fwd). | |
| Recombinant DNA reagent (plasmid) | pLL31 | This study | I-SceI recognition sequence and FRT-nptII-FRT flanked by 500 bp sequences homologous to portions of LO74_RS08325 and LO74_RS08330. | |
| Recombinant DNA reagent (plasmid) | pLL32 | This study | I-SceI recognition sequence and ble flanked by 500 bp sequences homologous to portions of LO74_RS09660 and LO74_RS09665. | |
| Recombinant DNA reagent (plasmid) | pLL44 | This study | gusA and ble flanked by 500 bp homologous to sequences within LO74_RS09420 and between LO74_RS09420 and LO74_RS31810. | |
| Recombinant DNA reagent (plasmid) | pABT84 | This study | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS12515 and LO74_RS12525. | |
| Recombinant DNA reagent (plasmid) | pLL46 | This study | recA under control of its native promoter | |
| Recombinant DNA reagent (plasmid) | pTNS3 | Choi et al., 2008 | Helper plasmid for mini-Tn7, oriT, R6K ori | |
| Recombinant DNA reagent (plasmid) | pABT86 | This study | PS12 promoter and FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS09305 and LO74_RS28675. | |
| Recombinant DNA reagent (plasmid) | pFlpTet | Garcia et al., 2013 | Rham-inducible flp, TS ori | |
| Recombinant DNA reagent (plasmid) | pLL37 | This study | ble flanked by 500 bp sequences homologous to LO74_RS09295- LO74_RS09305. | |
| Recombinant DNA reagent (plasmid) | pLL38 | This study | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS09295- LO74_RS09305. | |
| Recombinant DNA reagent (plasmid) | pLL29 | This study | FRT-nptII-FRT flanked by 500 bp sequences homologous to sequences between LO74_RS31810 and LO74_RS09435 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pABT104 | This study | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600. | |
| Recombinant DNA reagent (plasmid) | pABT66 | Ocasio and Cotter, 2019 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS31810 through 5′ ISβ inverted repeat and 3′ ISβ inverted repeat through LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pABT78 | Ocasio and Cotter, 2019 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS08585 through 5′ ISα inverted repeat and 3′ ISα inverted repeat through LO74_RS08600. | |
| Recombinant DNA reagent (plasmid) | pLL61 | This study | tet and nptII46–795 (fwd) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600. | |
| Recombinant DNA reagent (plasmid) | pLL62 | This study | ble and nptII1–545 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pLL63 | This study | ble and nptII1–195 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pLL64 | This study | ble and nptII1–95 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pLL74 | This study | ble and nptII1–45 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | pLL72 | This study | tet and gusA118–1691 (fwd) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600. | |
| Recombinant DNA reagent (plasmid) | pLL73 | This study | tet and gusA1–1454 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Recombinant DNA reagent (plasmid) | mini-Tn7-km-rfp | Norris et al., 2010 | PS12-Turborfp | |
| Recombinant DNA reagent (plasmid) | pLL65 | This study | tet and gfp1–589 (rev) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600. | |
| Recombinant DNA reagent (plasmid) | pLL66 | This study | tet and gusA89–744 (rev) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435. | |
| Sequence-based reagent | Junc1 | IDT | 5′ GCCGTGCTAGAGAGGCGCTA 3′ | |
| Sequence-based reagent | Junc2 | IDT | 5′ AGCAGAATCAGATGCACGCCATTCG 3′ | |
| Sequence-based reagent | Ctrl1 | IDT | 5′ TGATGCAGTTTCCGGCGCAGTAAC 3′ | |
| Sequence-based reagent | Ctrl2 | IDT | 5′ AATCGTGTCGGCGTGTGACGAA 3′ | |
| Chemical compound | X-Gluc (5-bromo-4-chloro-3-indolyl-beta-D-glucuronic acid, cyclohexyl ammonium salt) | Goldbio | B-735-250 | |
| Chemical compound, drug | Kanamycin Monosulfate | Chem-Impex International, Inc | 00195 | |
| Software | GraphPad Prism | GraphPad Prism (https://graphpad.com) | Version 9.5.0 | |
| Software | Imaris x64 | Imaris (https://imaris.oxinst.com/) | Version 9.9.1 |
Bacterial strains and culture conditions
Request a detailed protocolBtE264 is an environmental isolate (Brett et al., 1998). Strains 2002721643 and 2002721723 were obtained from the Department of Defense’s Unified Culture Collection (Johnson et al., 2015). Plasmids were maintained in E. coli DH5α. For insertion at the attTn7 sites or Flp-mediated FRT recombination, plasmids were introduced into BtE264 by conjugation with E. coli donor strain, RHO3 (López et al., 2009). BtE264 and E. coli strains were grown overnight with aeration at 37°C (unless indicated) in low-salt Luria-Bertani (LSLB, 0.5% NaCl). Antibiotics and supplements were added to cultures at the following concentrations: 50 μg/ml X-Gluc (5-bromo-4-chloro-3-indoxyl-beta-D-glucuronide), 200 μg/ml 2,6-diaminopimelic acid (DAP), 0.2% (wt/vol) rhamnose, 500 μg/ml (for BtE264) or 50 μg/ml (for E. coli) kanamycin (Km), 100 μg/ml ampicillin, or 200 μg/ml zeocin as appropriate. When indicated, BtE264 was cultured in M63 minimal medium (110 mM KH2PO4, 200 mM K2HPO4, 75 mM (NH4)2SO4, 16 nM FeSO4) supplemented with 1 mM MgSO4 and 0.2% glucose. For passaging experiments, M63 was further supplemented with 0.4% glycerol and 0.01% casamino acids.
Mutant construction techniques
Request a detailed protocolTo introduce a mutation by natural transformation, we linearized plasmids containing a gene-encoding antibiotic resistance and additional sequences, if necessary, flanked by ~500 bp sequences with homology to genomic regions of interest. Natural transformation was conducted following previously described protocols (Thongdee et al., 2008). Transformants were selected on LSLB containing the appropriate antibiotic and verified through PCR analysis.
Insertion of sequences to one or both attTn7 sites was conducted with triparental mating between BtE264, E. coli RHO3 containing the helper plasmid pTNS3, and E. coli RHO3 carrying a plasmid with sequences of interest according to previously described protocols (Choi et al., 2005). Mutants were confirmed with PCR analysis.
Removal of antibiotic resistance cassettes flanked by FRT sites was carried out through Flp-mediated FRT recombination according to previously described protocols (Choi et al., 2008).
Plasmids
| Name | Information | Resistance marker | Backbone | First appeared |
|---|---|---|---|---|
| pLL31 | I-SceI recognition sequence and FRT-nptII-FRT flanked by 500 bp sequences homologous to portions of LO74_RS08325 and LO74_RS08330 | Kanamycin, Ampicillin | pTWIST Amp High copy | This study |
| pLL32 | I-SceI recognition sequence and ble flanked by 500 bp sequences homologous to portions of LO74_RS09660 and LO74_RS09665 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL44 | gusA and ble flanked by 500 bp homologous to sequences within LO74_RS09420 and between LO74_RS09420 and LO74_RS31810 | Zeocin, Ampicillin | pJET2.1 | This study |
| pABT84 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS12515 and LO74_RS12525 | Kanamycin, Ampicillin | pJET2.1 | This study |
| pLL46 | recA under control of its native promoter | Tetracycline | pUC18tet | This study |
| pTNS3 | Helper plasmid for mini-Tn7, oriT, R6K ori | Ampicillin | Choi et al., 2008 | |
| pABT86 | PS12 promoter and FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS09305 and LO74_RS28675 | Kanamycin, Ampicillin | pJET2.1 | This study |
| pFlpTet | Rham-inducible flp, TS ori | Tetracycline | Garcia et al., 2013 | |
| pLL37 | ble flanked by 500 bp sequences homologous to LO74_RS09295- LO74_RS09305 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL38 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS09295–LO74_RS09305 | Kanamycin, Ampicillin | pJET2.1 | This study |
| pLL29 | FRT-nptII-FRT flanked by 500 bp sequences homologous to sequences between LO74_RS31810 and LO74_RS09435 and LO74_RS09435 | Kanamycin, Ampicillin | pJET2.1 | This study |
| pABT104 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600 | Kanamycin, Ampicillin | pJET2.1 | This study |
| pABT66 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS31810 through 5′ ISβ inverted repeat and 3′ ISβ inverted repeat through LO74_RS09435 | Kanamycin, Ampicillin | pJET2.1 | Ocasio and Cotter, 2019 |
| pABT78 | FRT-nptII-FRT flanked by 500 bp sequences homologous to LO74_RS08585 through 5′ ISα inverted repeat and 3′ ISα inverted repeat through LO74_RS08600 | Kanamycin, Ampicillin | pJET2.1 | Ocasio and Cotter, 2019 |
| pLL61 | tet and nptII46–795 (fwd) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600 | Tetracycline, Ampicillin | pTWIST Amp High copy | This study |
| pLL62 | ble and nptII1–545 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL63 | ble and nptII1–195 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL64 | ble and nptII1–95 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL74 | ble and nptII1–45 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
| pLL72 | tet and gusA118–1691 (fwd) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600 | Tetracycline, Ampicillin | pTWIST Amp High copy | This study |
| pLL73 | tet and gusA1–1454 (fwd) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
| mini-Tn7-km-rfp | PS12-Turborfp | Kanamycin, Ampicillin | pUC18km | Norris et al., 2010 |
| pLL65 | tet and gfp1–589 (rev) flanked by 500 bp sequences homologous to LO74_RS08585 and LO74_RS08600 | Tetracycline, Ampicillin | pTWIST Amp High copy | This study |
| pLL66 | tet and gusA89–744 (rev) flanked by 500 bp sequences homologous to LO74_RS09420 and LO74_RS31810 and LO74_RS09435 | Zeocin, Ampicillin | pJET2.1 | This study |
Mutants and mutant construction
Request a detailed protocol| Name | Information | Plasmid(s) used | Construction method | Resistance(s) | Figure referenced |
|---|---|---|---|---|---|
| WT with I-SceI cut sites 50 kb outside duplicating region | Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | pLL31, pLL32 | Two rounds of natural transformation | Kanamycin, Zeocin | 3A, 3B, 6A |
| WT with gusA insertion | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. | pLL44 | Natural transformation | Zeocin | 4C, 4D |
| ΔrecA with gusA insertion | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. Fused first 31 amino acids to last 38 amino acids of LO74_RS12520 (recA). | pLL44, pABT84 | Two rounds of natural transformation | Zeocin, Kanamycin | 4D |
| ΔrecA attTn7::recA with gusA insertion | gusA inserted between LO74_RS09420 and LO74_RS31810 near junction sequence. Fused first 31 amino acids to last 38 amino acids of LO74_RS12520. recA inserted at both attTn7 sites. | pLL44, pABT84, pTNS3, pLL46 | Two rounds of natural transformation and triparental mating | Zeocin, Kanamycin, Tetracycline | 4D |
| PS12-bcpAIOB Dup+ with I-SceI sites 50 kb outside duplicating region | PS12 and nptII inserted between bcpA and its native promoter. nptII excised with Flp recombinase. Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | pABT86, pFlpTet, pLL31, pLL32 | Natural transformation, Flp-FRT recombination, two rounds of natural transformation | Kanamycin, Zeocin | 6A |
| ΔbcpAIOB with I-SceI sites 50 kb outside duplicating region | LO74_RS09295- LO74_RS09305 (bcpAIOB) replaced with nptII surrounded by FRT sites. nptII excised with Flp recombinase. Inserted I-SceI recognition sites between LO74_RS08325 and LO74_RS08330 and LO74_RS09660 and LO74_RS09665. | pLL38, pFlpTet, pLL31, pLL32 | Natural transformation, Flp-FRT recombination, two rounds of natural transformation | Kanamycin, Zeocin | 6A |
| PS12-bcpAIOB Dup− | PS12 promoter inserted between LO74_RS09305 (bcpA) and its native promoter. | pABT86 | Natural transformation | Kanamycin | 6C |
| ΔbcpAIOB::ble ΔbcpAIOB::nptII Dup+ | LO74_RS09295– LO74_RS09305 (bcpAIOB) replaced with nptII surrounded by FRT sites. Additional copy of LO74_RS09295– LO74_RS09305 (bcpAIOB) replaced with ble. | pLL37, pLL38 | Two rounds of natural transformation | Zeocin, Kanamycin | 6D |
| ΔISβ::nptII | Inverted repeats and LO74_RS09425 (ISβ) replaced with nptII surrounded by FRT sites. | pLL29 | Natural transformation | Kanamycin | 6E |
| ΔISα::nptII | Inverted repeats and LO74_RS28540 (ISα) replaced with nptII surrounded by FRT sites. | pABT104 | Natural transformation | Kanamycin | 6E |
| ISβ ΔorfAB::nptII ISα ΔorfAB | LO74_RS28540 (ISα orfAB) replaced with nptII surrounded by FRT sites. nptII excised with Flp recombinase. LO74_RS09425 (ISβ orfAB) replaced with nptII surrounded by FRT sites. | pABT66, pFlptet, pABT78 | Natural transformation, Flp-FRT recombination, natural transformation | Kanamycin | 6F |
| ΔrecA | Fused first 31 amino acids to last 38 amino acids of LO74_RS12520 (recA). | pABT84 | Natural transformation | Kanamycin | 7A |
| ΔrecA attTn7::recA | Fused first 31 amino acids to last 38 amino acids of LO74_RS1252. recA inserted at both attTn7 sites. | pABT84, pTNS3, pLL46 | Natural transformation and triparental mating | Kanamycin, Tetracycline | 7A |
| Fragmented nptII reporter 500 bp homology | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–545 of nptII (fwd). | pLL61, pLL62 | Two rounds of natural transformation | Tetracycline, Zeocin | 7B, 7D, 7E |
| Fragmented nptII reporter 150 bp homology | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–195 of nptII (fwd). | pLL61, pLL63 | Two rounds of natural transformation | Tetracycline, Zeocin | 7B, 7D |
| Fragmented nptII reporter 50 bp homology | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–95 of nptII (fwd). | pLL61, pLL64 | Two rounds of natural transformation | Tetracycline, Zeocin | 7B, 7D |
| Fragmented nptII reporter 0 bp homology | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 46–795 of nptII (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–45 of nptII (fwd). | pLL61, pLL74 | Two rounds of natural transformation | Tetracycline, Zeocin | 7D |
| Fragmented gfp reporter attTn7::rfp | PS12-rfp inserted an attTn7 site. Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 1–589 of gfp (rev). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 89–744 of gfp (rev). 500 bp of homology in gfp fragments. | pLL65, pLL66, pTNS3, mini-Tn7-km-rfp | Triparental mating and two rounds of natural transformation | Tetracycline, Zeocin | 8A, 8B, 8C, 8D |
| Fragmented gfp reporter attTn7::rfp locked | PS12-rfp inserted an attTn7 site. Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 1–589 of gfp (rev). | pLL65, pTNS3, mini-Tn7-km-rfp | Triparental mating and natural transformation | Tetracycline | 8B, 8C, 8D |
| Fragmented gusA reporter | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 118–1691 of gusA (fwd). Inverted repeats and LO74_RS09425 (ISβ) replaced with ble and bases 1–1454 of gusA (fwd) 1332 bp of homology in gusA fragments. | pLL72, pLL73 | Two rounds of natural transformation | Tetracycline, Zeocin | 9A, 9B, 9C, 9D, 9E |
| Fragmented gusA reporter locked | Inverted repeats and LO74_RS28540 (ISα) replaced with tet and bases 118–1691 of gusA (fwd). | pLL72 | Two rounds of natural transformation | Tetracycline | 9C, 9D |
Polymerase chain reaction (PCR) analysis
Request a detailed protocolJunction PCR analyses were conducted as described previously (Ocasio and Cotter, 2019) using Junc1 (5′ GCCGTGCTAGAGAGGCGCTA 3′) and Junc2 (5′ AGCAGAATCAGATGCACGCCATTCG 3′) to amplify the junction sequence and Ctrl1 (5′ TGATGCAGTTTCCGGCGCAGTAAC 3′) and Ctrl2 (5′ AATCGTGTCGGCGTGTGACGAA 3′) to amplify a fragment inside the region (BTH_I2728) as a positive control. When necessary, PCR reactions were visualized by gel electrophoresis within 0.8% gels alongside 1 kb HyperLadder as a size standard.
Duplication-dependent phenotypic analysis
Request a detailed protocolColony morphologies were analyzed after 48 hr growth on LSLB agar and imaged at ×0.63 magnification on an Olympus MVX10 macroscope.
Test tube aggregation was assayed as previously described (Ocasio and Cotter, 2019).
Colony biofilms shown in Figures 2D and 6C, D were created by spotting 20 μl of an overnight culture diluted to an OD600 of 0.2 on LSLB agar. Biofilms were incubated at room temperature for 28 days and imaged at ×0.63 magnification on an Olympus MVX10 macroscope.
Colony biofilms shown in Figure 5A were created by spotting 10 μl of an overnight culture diluted to an OD600 of 0.2 on LSLB agar. Biofilms were incubated at room temperature for 30 days and imaged at ×0.63 magnification on an Olympus MVX10 macroscope.
For the results shown in Figures 5B, C, 6B, E, F, colony biofilms were created by spotting 20 μl of an overnight culture diluted to an OD600 of 1.0 on LSLB agar. Biofilms were incubated at room temperature for 7 days. Duplication formation in the center and edge of the colony biofilm (Figure 4B) was analyzed by PCR.
For the experiments, results in Figure 7E, colony biofilms were created by spotting 20 μl of an overnight culture diluted to an OD600 of 1.0 on LSLB agar. Biofilms were incubated at room temperature for up to 22 days. Every other day, bacteria were picked from the center and edge of the colony biofilm (Figure 4B), serially diluted to 109 and plated on LSLB agar and LSLB agar containing Km. After 16–18 hr of incubation at 37°C, colonies were counted and cfu was calculated on both media. The proportion on KmR colonies was calculated by dividing the cfu of KmR colonies by the total cfu.
Pulsed-field gel electrophoresis
Request a detailed protocolCell pellets from 2 ml overnight cultures were used to create gel plugs using the CHEF Bacterial Genomic DNA Plug Kit (Bio-Rad). DNA was digested with I-SceI. DNA fragments were resolved with 1% certified megabase agarose (Bio-Rad) containing GelRed Nucleic Acid Gel Stain (Biotium) in 0.5× Tris-borate_EDTA buffer (TBE). For Figure 3B, gels were run for 24 hr at 14°C, 6 V/cm, included angel of 120° with switch times gradually ascending from 75 to 155 s. For Figure 6A, gels were run for 24 hr at 14°C, 6 V/cm, included angel of 120° with switch times gradually ascending from 70 to 150 s. For Figure 3—figure supplement 1B, gels were run for 48 hr at 14°C, 3.6 V/cm, included angel of 120° with switch times gradually ascending from 50 to 60 s. Lambda ladder was used as a size standard.
Passage experiments
Request a detailed protocolFor the experiment, results in Figure 4D, overnight cultures of the gusA insertional strain in LSLB were washed in phosphate-buffered saline (PBS) and diluted down to an OD600 of 0.02 in 3 ml of LSLB. Following 24 hr of growth with aeration, an aliquot was subcultured at a OD600 of 0.02 in LSLB. Passaging was repeated every 24 hr until the end of the experiment. Every 5 days, an aliquot was diluted and plated for individual colonies on LSLB containing X-Gluc. The number of blue and white colonies was counted after the plate was incubated at 37°C for 24 hr and room temp for 48 hr.
For the experiment, results in Figure 7D, overnight cultures in LSLB of the fragmented nptII reporter strain were serially diluted to 109. Dilutions were plated on both LSLB agar and LSLB agar containing Km. After 16–18 hr of incubation at 37°C, colonies were counted and cfu was calculated for both media. The proportion on KmR colonies was calculated by dividing the cfu of KmR colonies by the total cfu.
For the experiment, results in Figure 9B–E, overnight cultures of the Dup+, Dup−, and Dup−-locked fragmented gusA reporter strains in LSLB were washed in PBS and diluted down to an OD600 of 0.2 in 3 ml of M63. Following 24 hr of growth with aeration, an aliquot was subculture at a OD600 of 0.2 in 3 ml of M63 for the liquid passage. For the biofilm passage, spent media was removed and 3 ml of fresh M63 was returned to the same tube. Passaging was repeated every 24 hr until the end of the experiment. Every 5 days, an aliquot was diluted and plated for individual colonies on LSLB containing X-Gluc. The number of blue and white colonies was counted after the plate was incubated at 37°C for 24 hr and room temperature for 48 hr.
Biofilm microscopy
Request a detailed protocolOvernight cultures of fragmented gfp reporter strains were diluted to an OD600 of 0.02 in 400 μl of M63 in 4-well chambered coverglass dishes. Biofilms were incubated at 45°C for 24–72 hr in humified chambers. Before imaging, biofilms were washed four times in PBS and overlaid with 400 μl PBS. Biofilms were imaged using confocal laser microscopy using a Zeiss LSM 780 with a ×40 objective. Z-stacks were processed and analyzed using Imaris x64 9.9.1 and the Biofilm Analysis Xtension (Bitplane Scientific Software). Red fluorescence was pseudo colored as magenta.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting file; Source Data files have been provided for all gels.
References
-
Gene amplification and adaptive evolution in bacteriaAnnual Review of Genetics 43:167–195.https://doi.org/10.1146/annurev-genet-102108-134805
-
Burkholderia thailandensis sp. nov., a Burkholderia pseudomallei-like speciesInternational Journal of Systematic Bacteriology 48 Pt 1:317–320.https://doi.org/10.1099/00207713-48-1-317
-
Human infection with Burkholderia thailandensis, China, 2013Emerging Infectious Diseases 23:1416–1418.https://doi.org/10.3201/eid2308.170048
-
Biological relevance of colony morphology and phenotypic switching by Burkholderia pseudomalleiJournal of Bacteriology 189:807–817.https://doi.org/10.1128/JB.01258-06
-
A tn7-based broad-range bacterial cloning and expression systemNature Methods 2:443–448.https://doi.org/10.1038/nmeth765
-
Genetic tools for select-agent-compliant manipulation of Burkholderia pseudomalleiApplied and Environmental Microbiology 74:1064–1075.https://doi.org/10.1128/AEM.02430-07
-
Bacterial genome instabilityMicrobiology and Molecular Biology Reviews 78:1–39.https://doi.org/10.1128/MMBR.00035-13
-
Mechanisms of change in gene copy numberNature Reviews. Genetics 10:551–564.https://doi.org/10.1038/nrg2593
-
Fatal outcome of lung transplantation in cystic fibrosis patients due to small-colony variants of the Burkholderia cepacia complexEuropean Journal of Clinical Microbiology & Infectious Diseases 22:249–253.https://doi.org/10.1007/s10096-003-0901-y
-
Versatile dual-technology system for markerless allele replacement in Burkholderia pseudomalleiApplied and Environmental Microbiology 75:6496–6503.https://doi.org/10.1128/AEM.01669-09
-
Continuing evolution of Burkholderia mallei through genome reduction and large-scale rearrangementsGenome Biology and Evolution 2:102–116.https://doi.org/10.1093/gbe/evq003
-
Stable, site-specific fluorescent tagging constructs optimized for Burkholderia speciesApplied and Environmental Microbiology 76:7635–7640.https://doi.org/10.1128/AEM.01188-10
-
BookEscherichia CColi and Salmonella: Cellular and Molecular Biology (2nd ed)Washington, D.C: American Society for Microbiology.
-
Bacterial gene amplification: implications for the evolution of antibiotic resistanceNature Reviews. Microbiology 7:578–588.https://doi.org/10.1038/nrmicro2174
-
Dynamics of insertion sequence elements during experimental evolution of bacteriaResearch in Microbiology 155:319–327.https://doi.org/10.1016/j.resmic.2003.12.008
-
Genetic diversity and multihost pathogenicity of clinical and environmental strains of Burkholderia cenocepaciaApplied and Environmental Microbiology 75:5250–5260.https://doi.org/10.1128/AEM.00877-09
-
Targeted mutagenesis of Burkholderia thailandensis and Burkholderia pseudomallei through natural transformation of PCR fragmentsApplied and Environmental Microbiology 74:2985–2989.https://doi.org/10.1128/AEM.00030-08
-
Phase and antigenic variation in bacteriaClinical Microbiology Reviews 17:581–611.https://doi.org/10.1128/CMR.17.3.581-611.2004
-
Phase variation: how to create and coordinate population diversityCurrent Opinion in Microbiology 14:205–211.https://doi.org/10.1016/j.mib.2011.01.002
-
The impact of insertion sequences on bacterial genome plasticity and adaptabilityCritical Reviews in Microbiology 43:709–730.https://doi.org/10.1080/1040841X.2017.1303661
Article and author information
Author details
Funding
National Institutes of Health (R35 GM136533)
- Peggy A Cotter
National Institutes of Health (R01 GM121110)
- Peggy A Cotter
The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Acknowledgements
Thank you to Colin Manoil, Josephine Chandler, Joseph Mougous, and the U.S. Army Medical Research Institute of Infectious Diseases (USAMRIID) for sending us B. thailandensis strains. We are also grateful to the members of the Cotter laboratory for their continued support and insightful discussions.
Microscopy was performed at the UNC Neuroscience Microscopy Core (RRID:SCR_019060), supported, in part, by funding from the NIH-NINDS Neuroscience Center Support Grant P30 NS045892 and the NIH-NICHD Intellectual and Developmental Disabilities Research Center Support Grant P50 HD103573. This research was supported by grants from the NIH: R35 GM136533 and R01 GM121110 to P.A.C.
Copyright
© 2023, Lowrey et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,265
- views
-
- 218
- downloads
-
- 17
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 17
- citations for umbrella DOI https://doi.org/10.7554/eLife.84327
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
An IS-mediated, RecA-dependent, bet-hedging strategy in Burkholderia thailandensis
eLife 12:e84327.
https://doi.org/10.7554/eLife.84327




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}