The common Sting1 HAQ, AQ alleles rescue CD4 T cellpenia, restore T-regs, and prevent SAVI (N153S) inflammatory disease in mice
- Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of Florida, United States
- Division of Vascular Surgery & Endovascular Therapy, Department of Surgery, University of Florida, United States
eLife assessment
This study describes useful mouse models of knock-ins of human STING1 variants and an assessment of these variants' action in mouse immune cells. While the implications of the variants in the inflammatory response are of significant interest, limitations are still found in the authors' interpretation and conclusions made, and the evidence for the conclusion remains incomplete.
https://doi.org/10.7554/eLife.96790.3.sa0Significance of the findings:
Useful: Findings that have focused importance and scope
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Incomplete: Main claims are only partially supported
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
The significance of STING1 gene in tissue inflammation and cancer immunotherapy has been increasingly recognized. Intriguingly, common human STING1 alleles R71H-G230A-R293Q (HAQ) and G230A-R293Q (AQ) are carried by ~60% of East Asians and ~40% of Africans, respectively. Here, we examine the modulatory effects of HAQ, AQ alleles on STING-associated vasculopathy with onset in infancy (SAVI), an autosomal dominant, fatal inflammatory disease caused by gain-of-function human STING1 mutations. CD4 T cellpenia is evident in SAVI patients and mouse models. Using Sting1 knock-in mice expressing common human STING1 alleles HAQ, AQ, and Q293, we found that HAQ, AQ, and Q293 splenocytes resist STING1-mediated cell death ex vivo, establishing a critical role of STING1 residue 293 in cell death. The HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice did not have CD4 T cellpenia. The HAQ/SAVI(N153S), AQ/SAVI(N153S) mice have more (~10-fold, ~20-fold, respectively) T-regs than WT/SAVI(N153S) mice. Remarkably, while they have comparable TBK1, IRF3, and NFκB activation as the WT/SAVI, the AQ/SAVI mice have no tissue inflammation, regular body weight, and normal lifespan. We propose that STING1 activation promotes tissue inflammation by depleting T-regs cells in vivo. Billions of modern humans have the dominant HAQ, AQ alleles. STING1 research and STING1-targeting immunotherapy should consider STING1 heterogeneity in humans.
Introduction
STING1 drives cytosolic DNA-induced type I IFNs production (Barber, 2015). Recent research revealed that STING1 promotes inflammation in a variety of inflammatory diseases, including nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, kidney injury, neurodegenerative diseases, cardiovascular diseases, obesity, diabetes, and aging (Skopelja-Gardner et al., 2022; Bai and Liu, 2021; Li et al., 2023; Gulen et al., 2023; Szego et al., 2022; Gao et al., 2023b; Yan et al., 2022; Gao et al., 2023a). The type I IFNs-independent function of STING1 has also emerged (Gao et al., 2023a; Yamashiro et al., 2020; Wu et al., 2020; Pham et al., 2024). For example, initially described as a type I interferonopathy (Liu et al., 2014), recent studies in STING-associated vasculopathy with onset in infancy (SAVI) mouse models showed that SAVI is largely independent of type I IFNs (Luksch et al., 2019; Stinson et al., 2022; Warner et al., 2017; Frémond et al., 2021). In a Sting1 N153S mouse model of SAVI, crossing N153S mice to IRF3/IRF7, and IFNAR1 knockout mice, N153S mice still developed spontaneous lung diseases (Luksch et al., 2019). JAK inhibitors were used to block type I IFNs signaling for SAVI patients with mixed success (Frémond et al., 2021; Kim et al., 2022; Volpi et al., 2019; Tang et al., 2020). For example, in a review of JAK inhibition in 18 SAVI patients, incomplete response to treatment happened in 7/18 (38%) of patients (Dai et al., 2020). Furthermore, two patients died of respiratory failure despite this treatment (Dai et al., 2020). Both radioresistant parenchymal and/or stromal cells and hematopoietic cells influence SAVI pathology in mice (Gao et al., 2024; Gao et al., 2022). The observation is important because it predicts that allogeneic stem cell transplantation may not work in human SAVI patients. Indeed, lung transplantations did not show improvement in SAVI patients (Kim et al., 2022; Picard et al., 2016). Patients died at 3- and 9 months post-lung transplant (Kim et al., 2022; Picard et al., 2016). How STING1 drives inflammation in vivo, independent of type I IFNs, remains unknown. Consequently, SAVI has no curative care.
Characterized as an innate immune sensor, STING1 expression is, paradoxically, high in CD4 T cells (Liu et al., 2014; Jin et al., 2013). Furthermore, STING1 activation kills mouse and human CD4 T cells ex vivo (Larkin et al., 2017; Cerboni et al., 2017; Kuhl et al., 2023). SAVI patients and mouse models had CD4 T cellpenia (Liu et al., 2014; Kuhl et al., 2023). STING1 was first discovered as MPYS for its cell growth inhibition and cell death function in mouse B lymphoma cells (Jin et al., 2008). STING1-mediated cell death is cell type dependent. For example, while STING1 activation kills human endothelial cells, primary and cancerous T cells, it does not kill mouse MEFs, BMDCs, or BMDMs (Gulen et al., 2017; Kabelitz et al., 2022; Murthy et al., 2020). Second, STING1-mediated cell death is type I IFNs-independent (Kuhl et al., 2023; Gulen et al., 2017; Murthy et al., 2020). Multiple cell death pathways, that is apoptosis, necroptosis, pyroptosis, ferroptosis, and PANoptosis, are proposed (Murthy et al., 2020; Li et al., 2021; Song et al., 2022). Last, the in vivo biological significance of STING1-mediated CD4 T cell death is not clear (Kuhl et al., 2023; Murthy et al., 2020). In humans, SAVI patients with constitutively activated STING1 have low CD4 T cell numbers (Liu et al., 2014), and type I IFNs are dispensable for STING1-mediated human CD4 T cell death (Kuhl et al., 2023). Different from SAVI mice, SAVI patients (N154S or V155M) had normal counts of CD8 T and B cells (Liu et al., 2014).
The human STING1 gene is highly heterogeneous (Jin et al., 2011; Patel et al., 2017a). Approximately 50% of people in the U.S. carry at least one copy of non-WT STING1 allele (Jin et al., 2011). Among them, the R71H-G230A-R293Q (HAQ) is the second most common STING1 allele carried by ~23% of people in the U.S. (Jin et al., 2011). However, in East Asians, WT/HAQ (34.3%), not WT/WT (22.0%), is the most common STING1 genotype (Patel et al., 2017a). Critically, the HAQ allele was positively selected in modern humans outside Africa Mansouri et al., 2022. Anatomically modern humans outside Africa are descendants of a single Out-of-Africa Migration 50,000~70,000 years ago. ~1.4% of Africans have the HAQ allele, while ~63.9% of East Asians are HAQ carriers (Mansouri et al., 2022). Haplotype analysis revealed that HAQ was derived from G230A-R293Q (AQ) allele (Mansouri et al., 2022). Importantly, the AQ allele was negatively selected outside Africa (Mansouri et al., 2022). Approximately 40.1% of Africans are AQ carriers, while ~0.4% of East Asians have the AQ allele (Mansouri et al., 2022). STING1 alleles often have a dominant negative effect likely because the protein STING1 exists as a homodimer (Jin et al., 2008; Patel and Jin, 2019). SAVI is an autosomal dominant inflammatory disease (Liu et al., 2014). WT/HAQ individuals had reduced Pneumovax23-induced antibody responses compared to WT/WT individuals (NCT02471014) (Sebastian et al., 2020). Notably, AQ responds to CDNs and produces type I IFNs in vivo and in vitro (Mansouri et al., 2022; Yi et al., 2013; Patel et al., 2017b), but the AQ allele was negatively selected in non-Africans (Mansouri et al., 2022). In contrast, the HAQ allele, defective in CDNs-type I IFNs responses (Jin et al., 2011; Patel et al., 2017a; Sebastian et al., 2020; Nissen et al., 2018; Ruiz-Moreno et al., 2018), was positively selected in non-Africans (Mansouri et al., 2022), indicating that the CDNs-type I IFNs independent function of STING1 was essential for the survival of early modern humans outside of Africa.
In this study, we discovered, surprisingly, that the HAQ, AQ splenocytes are resistant to STING1-mediated cell death. We generated HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice and found that the HAQ, AQ alleles prevent CD4 T cellpenia, increasing/restoring T-regs and alleviating/stopping tissue inflammation in SAVI mice, thus providing evidence for the in vivo significance of type I IFNs-independent, STING1-mediated cell death and potential AQ-based curative therapy for SAVI patients.
Results
STING1 activation kills mouse spleen CD4, CD8 T, and CD19 B cells ex vivo
We first used the synthetic non-CDNs STING1 agonist diABZI (Ramanjulu et al., 2018) to induce lymphocyte death because diABZI induces cell death without the need for lipid transfection or detergent for cell permeabilization (Kabelitz et al., 2022; Messaoud-Nacer et al., 2022) and diABZI is in clinical trials (NCT05514717). Splenocytes from C57BL/6 N mice were treated with diABZI in culture, and cell death was determined by Annexin V and Propidium Iodide stain. Splenocyte cell death could be detected at 5 hr post diABZI treatment (Figure 1—figure supplement 1A). Dosage responses showed that ~25 ng/ml diABZI could kill 70% of splenocytes (Figure 1—figure supplement 1B). Similarly, STING1 agonists DMXAA and synthetic CDNs RpRpss-Cyclic di-AMP killed mouse spleen CD4, CD8 T cells, and CD19 B cells (Figure 1A). Thus, STING1 activation readily induces mouse lymphocyte death ex vivo.
Figure 1 with 1 supplement see all
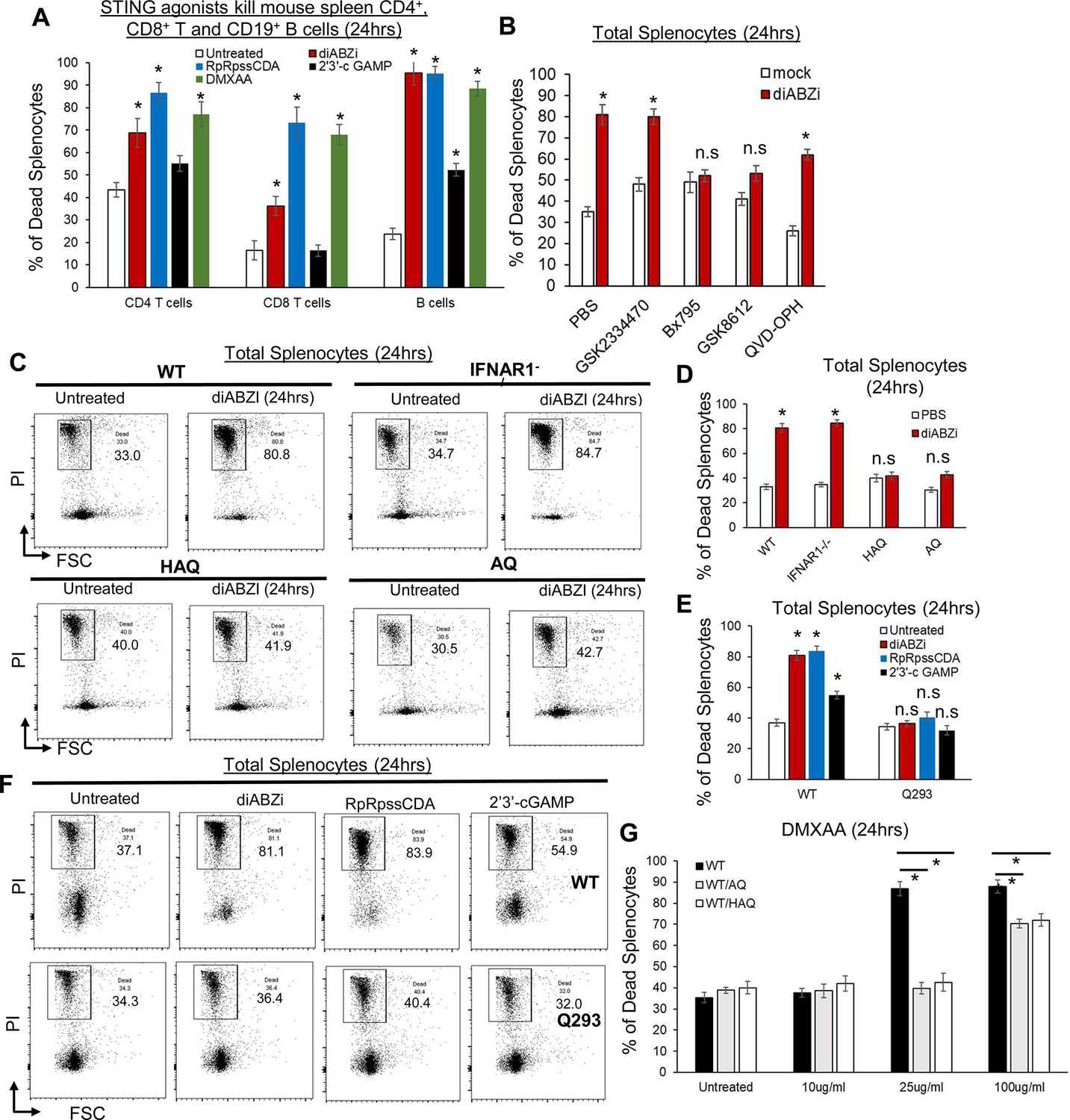
Splenocytes from HAQ, AQ, and Q293 mice are resistant to STING1-mediated cell death ex vivo.
(A) C57BL/6 N splenocytes were treated directly (no transfection) with diABZI (100 ng/ml), RpRpss-Cyclic di-AMP (5 μg/ml) or 2′3′-cGAMP (10 μg/ml), DMXAA (25 μg/ml) for 24 hr in culture. CD4, CD8 T cells and CD19 B cells death were determined by PI staining. (B). Splenocytes from C57BL/6 N mice were pre-treated with indicated small molecules, GSK2334470 (1.25 µM), GSK8612 (2.5 µM), Bx-795 (0.5 µM), QVD-OPH (25 µM) for 2 hrs. diABZI (100 ng/ml) was added in culture for another 24 hr. Dead cells were determined by PI staining. (C–D). Flowcytometry of HAQ, AQ, IFNAR1-/- or C57BL/6 N splenocytes treated with diABZI (100 ng/ml) for 24 hrs. Cell death was determined by PI staining. (E–F). Q293 or the WT littermates splenocytes were treated with diABZI (100 ng/ml), RpRpss-Cyclic di-AMP (5 μg/ml) or 2′3′-cGAMP 10 μg/ml for 24 hr. Cell death was determined by PI staining. (G). WT/HAQ, WT/AQ, or WT/WT littermates splenocytes were treated with DMXAA (10, 25 or 100 µg/ml) for 24 hr. Cell death was determined by PI staining. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values are determined by one-way ANOVA Tukey’s multiple comparison test (A, E, G) or unpaired student T-test (B, D) * p<0.05. n.s: not significant.
TBK1 activation is required for STING1-mediated mouse spleen cell death ex vivo
STING1 activation can lead to apoptosis, pyroptosis, necroptosis, or ferroptosis (Kuhl et al., 2023; Jin et al., 2008; Gulen et al., 2017; Kabelitz et al., 2022; Murthy et al., 2020; Li et al., 2021; Song et al., 2022; Messaoud-Nacer et al., 2022; Tang et al., 2016). We then treated mouse splenocytes with apoptosis, pyroptosis, necroptosis inhibitors, STING1 inhibitors H-151, C-176, and palmitoylation inhibitor 2-bromopalmitate (2 BP), followed by diABZI stimulation. Inhibitors for NLRP3 (MCC950), RIPK1 (Necrostatin-1), RIPK3 (GSK872), Caspase-1 (VX-795), Caspase-3 (Z-DEVD-FMK), Caspase 1,3,8,9 (Q-VD-Oph), ferroptosis (liproxstatin-1) did not affect diABZI-induced splenocyte cell death ex vivo (Figure 1—figure supplement 1B, C). The STING1 inhibitors H-151, C-176, and 2 BP also could not prevent diABZI-induced cell death (Figure 1—figure supplement 1C), although they inhibited diABZI-induced IFNβ production (Figure 1—figure supplement 1D). Instead, the TBK1 inhibitor BX-795 abolished diABZI-induced splenocyte death (Figure 1—figure supplement 1C).
BX-795 is a multi-kinase inhibitor, including 3-phosphoinositide-dependent protein kinase 1 (PDK1) and TBK1 (IC50s = 6 and 11 nM, respectively). However, the treatment of PDK1 inhibitor GSK2334470 (IC50=10 nM) did not prevent diABZI-induced splenocyte death (Figure 1B). In contrast, GSK8612, a highly potent and selective inhibitor for TBK1, prevented diABZI-induced splenocyte death (Figure 1B). Thus, TBK1 activation is likely critical for STING1-mediated splenocyte cell death ex vivo.
HAQ, AQ, Q293 STING1 knock-in mouse splenocytes are resistant to STING1-mediated cell death ex vivo
HAQ and AQ are common human STING1 alleles (Jin et al., 2011; Patel et al., 2017a; Mansouri et al., 2022). Previously, we reported that HAQ knock-in mice are defective in CDNs-induced immune responses, while CDNs responses in AQ knock-in mice are similar to WT mice (Mansouri et al., 2022). We treated splenocytes from HAQ and AQ mice with diABZI ex vivo and found, surprisingly, that both HAQ and AQ splenocytes were resistant to diABZI-induced cell death (Figure 1C and D). In comparison, IFNAR1-/- splenocytes were killed by diABZI, confirming that STING1-mediated lymphocytes death are type I IFNs-independent (Figure 1C and D; Kuhl et al., 2023; Gulen et al., 2017; Murthy et al., 2020).
HAQ and AQ share the common A230 and Q293 residues changes. We thus generated a Q293 Sting1 knock-in mouse. Notably, the Q293 splenocytes were resistant to STING1 agonists 2’3’-cGAMP, RpRpss-Cyclic di-AMP, and diABZI-induced cell death (Figure 1E and F). Thus, the residue 293 of STING1 is critical for its cell death function.
WT/HAQ, WT/AQ mouse splenocytes are partially resistant to STING1-mediated cell death ex vivo
WT/HAQ (34.3%) is the most common human STING1 genotype in East Asians, while WT/AQ (28.2%) is the 2nd most common STING1 genotype in Africans (Patel et al., 2017a). We generated WT/HAQ, WT/AQ mice and treated their splenocytes with mouse STING1 agonist DMXAA. WT/HAQ and WT/AQ splenocytes were protected from 25 µg/ml DMXAA-induced cell death (Figure 1G). A total of 100 µg/ml DMXAA could kill WT/HAQ and WT/AQ splenocytes, albeit less than WT/WT cells (Figure 1G). Thus, the HAQ and AQ alleles are dominant and likely impact STING1 activation even in heterozygosity.
STING1 activation kills primary human CD4 T cells but not CD8 T or CD19 B cells
STING1 agonists-based clinical trials in humans have been disappointing (NCT02675439, NCT03010176, NCT05514717; Meric-Bernstam et al., 2023; Meric-Bernstam et al., 2022). We showed that the human STING1 gene might undergo natural selection during the out-of-Africa migration (Mansouri et al., 2022) sensitive to evolutionary pressure. Thus, we investigated STING1-mediated death in primary human lymphocytes.
Human explant lung cells from the WT(R232)/WT(R232) donors were treated with STING1 agonists 2’3 c GAMP, RpRpss-Cyclic di-AMP, diABZI for 24 hr in culture. Lymphocyte cell death was determined by Propidium Iodide staining. Different from mouse lymphocytes, diABZI and RpRpss-Cyclic di-AMP killed human CD4 T but not CD8 T or CD19 B cells (Figure 2A and B). Human CD8 T and CD19 B cells are resistant to 500 ng/ml diABZI-induced cell death (Figure 2—figure supplement 1A).
Figure 2 with 1 supplement see all

HAQ, AQ, and Q293 human cells are resistant to STING1 agonists-induced death.
(A–B) Total human Lung cells from WT/WT individuals were treated with diABZi (100 ng/ml) for 24 hr. Cell death in CD4, CD8 T cells and CD19 B cells were determined by PI staining. (C). Total lung cells from a WT/HAQ (2 individuals) and a WT/WT (3 individuals) were treated with diABZi (25, 100 ng/ml) for 24 hr. Cell death in CD4 T cells was determined by PI staining. (D–E). STING1-KO THP-1 cells (Invivogen,, cat no. thpd-kostg) were stably reconstituted with human WT (R232), HAQ, AQ, Q293. Cells were treated with diABZI (200 ng/ml) in culture for 24 hr. Dead cells were determined by Annexin V staining. (F). STING1-KO THP-1 cells stably reconstituted with human WT (R232), Q293 were treated with indicated dose of diABZI for 24 hr in culture. Dead cells were determined by Annexin V staining. (G). STING1-KO THP-1 cells stably reconstituted with human WT (R232), Q293 were treated with indicated dose of diABZI for 2hs in culture. STING1 activation was detected by anti-STING1 antibody (Proteintech, #19851–1-AP). (H). STING1-KO THP-1 cells stably reconstituted with human WT (R232), HAQ, AQ, Q293 were treated with 50 ng/ml diABZI in culture for 24 hr. ISG-54 reporter luciferase activity was determined in cell supernatant and normalized to 10 ng/ml IFNβ-stimulated ISG-54 luciferase activity. (I). STING1-KO THP-1 cells stably reconstituted with human WT (R232), HAQ, AQ were treated with 50 ng/ml diABZI in culture for 2 hr. STING1 and IRF3 activation were determined by anti-STING1 antibody and anti-p IRF3 antibody (CST, Ser396, clone 4D4G). Densitometry was determined by ImageLab 5. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values determined by one-way ANOVA Tukey’s multiple comparison test (B, C, F, H, G) or unpaired student T-test (E, I). * p<0.05, n.s: not significant.
WT/HAQ human CD4 T cells are resistant to low doses of diABZI-induced cell death
WT/HAQ mouse splenocytes are resistant to low-dose diABZI-induced cell death (Figure 1G). To extend our observation into primary human T cells, we obtained lung explants from WT/WT and WT/HAQ individuals (Figure 2—figure supplement 1B) and treated them with diABZI in culture. 25 ng/ml diABZI killed WT/WT, but not WT/HAQ, human lung CD4 T cells (Figure 2C).
diABZI induces cell death in STING1-KO human THP-1 cells reconstituted with WT human STING1 (R232) but not HAQ, AQ or Q293 human STING1 allele
To further determine cell death influenced by human STING1 alleles HAQ, AQ, and Q293, we used the STING1-KO THP-1 cell line because STING1 agonist induces type I IFNs and cell death in STING1-KO THP-1 cells expressing WT human STING1 (Song et al., 2022; Figure 2—figure supplement 1C and D). We, thus, generated stable THP-1 STING1-KO lines expressing HAQ, AQ, WT, or Q293 STING1 allele. Cell death was determined by Annexin-V staining. diABZI killed THP-1 STING1-KO lines expressing WT but not HAQ, AQ, or Q293 STING1 allele (Figure 2D and E). No cell death was induced in the Q293 THP-1 cells stimulated by 20–200 ng/ml of diABZI (Figure 2F). diABZI also did not induce STING1 activation in Q293 THP-1 cells (Figure 2G). Notably, 50 ng/ml diABZI induced p-IRF3 activation and type I IFNs in AQ THP-1 cells but not HAQ THP-1 cells (Figure 2H and I), indicating that the STING1-cell death and STING1-IRF3-Type I IFNs pathways can be uncoupled.
HAQ and AQ alleles rescue the lymphopenia and suppress myeloid cell expansion in SAVI(N153S) mice
The in vivo significance of the STING1/MPYS-cell death is unclear. Furthermore, multiple cell death pathways, that is apoptosis, necroptosis, pyroptosis, ferroptosis, and PANoptosis, are proposed (Murthy et al., 2020; Li et al., 2021; Song et al., 2022). The uncertainty likely results from studies using different cell types (primary cells vs cancer cell lines); species (human vs mouse); STING1 agonists (cGAMP, which requires cell permeabilization by detergents or lipid transfection, vs diABZi, DMXAA that can directly cross the membrane; Larkin et al., 2017; Cerboni et al., 2017; Gulen et al., 2017; Kabelitz et al., 2022; Wu et al., 2019). Critically, which mechanism is relevant in vivo, causing T cellpenia is not known. To clarify the in vivo significance and mechanisms of STING1-mediated cell death, we turned to SAVI mice.
SAVI is an autosomal dominant, inflammatory disease caused by one copy of a gain-of-function STING1 mutant (WT/SAVI) Liu et al., 2014). CD4 T cellpenia was found in SAVI patients and SAVI mouse models (Liu et al., 2014; Luksch et al., 2019. STING1 activation in SAVI mice is independent of ligands and happens in vivo. We thus generated HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice aiming to establish the in vivo significance and mechanism of STING1-cell death.
First, HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice had reduced splenomegaly compared to WT/SAVI(N153S) mice though their spleens were still larger than the littermates WT/HAQ and WT/AQ (Figure 3A and B). Next, HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice had similar spleen B cells and CD4 T cell numbers as the WT/HAQ, WT/AQ littermates (Figure 2B and C). Their CD8+ T cells were lower than their WT littermates but much higher than the WT/SAVI(N153S) mice (Figure 3D). Third, spleen myeloid cell numbers, that is neutrophils, Ly6Chi monocytes and F4/80 macrophages, were all reduced by half compared to WT/SAVI(N153S) mice (Figure 3E, H). Last, splenocytes from HAQ/SAVI, AQ/SAVI mice were partially resistant to diABZI, DMXAA-induced cell death ex vivo (Figure 3—figure supplement 1). Notably, the HAQ/SAVI(N153S) and AQ/SAVI(N153S) mice also had restored bone marrow monocytes (Figure 4—figure supplement 1). Thus, HAQ and AQ alleles prevent lymphopenia and suppress myeloid cell expansion in SAVI(N153S) mice.
Figure 3 with 2 supplements see all
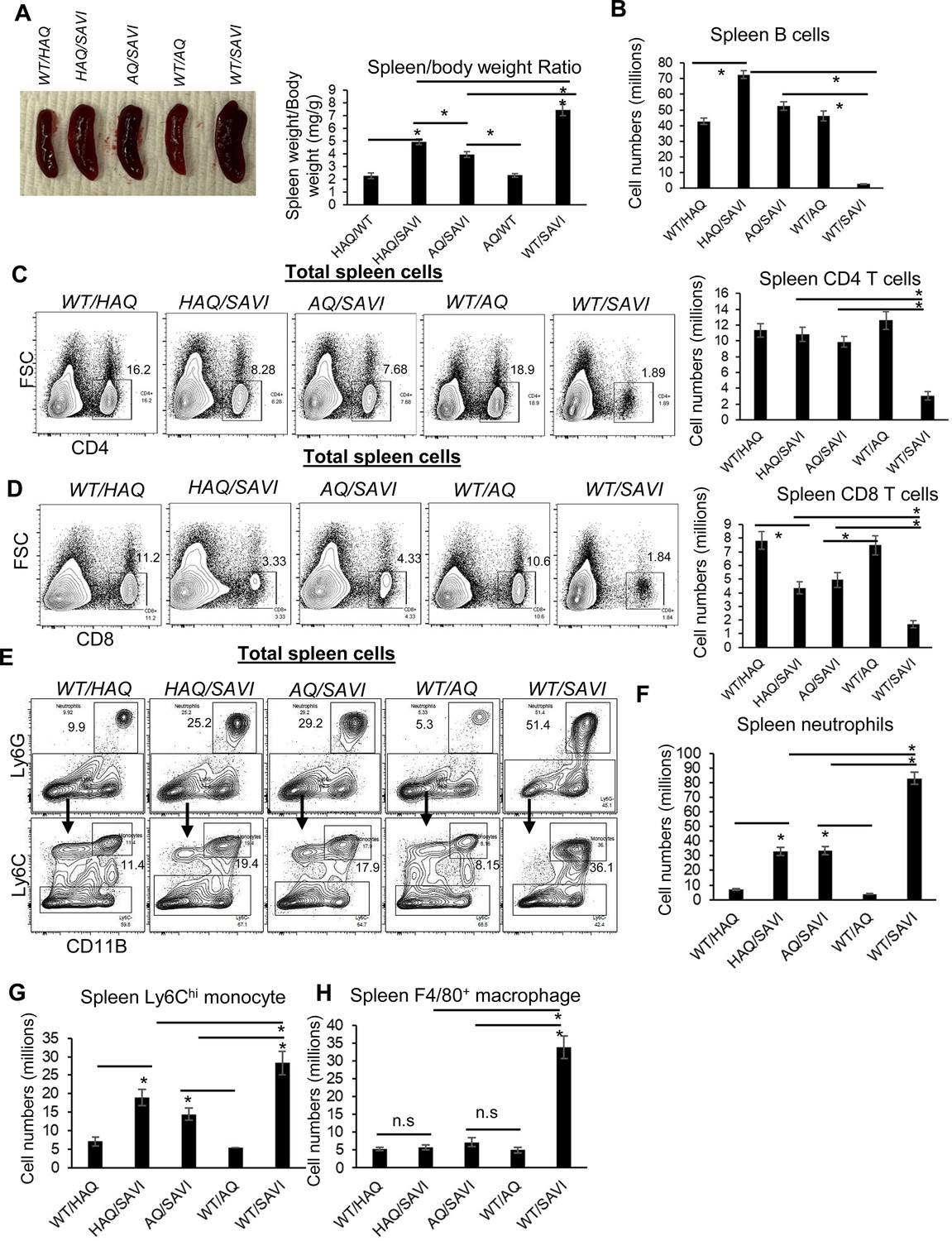
HAQ and AQ rescue the lymphopenia and suppress myeloid cell expansion in SAVI (N153S) mice.
(A) The size and weight of spleens from WT/HAQ, HAQ/SAVI, AQ/SAVI, WT/AQ, WT/SAVI. (B–D). Spleen CD19+ B cells, CD4, CD8 T cells were determined in the indicated mice by Flow. (E–H). Spleen Ly6G+ neutrophils, Ly6Chi monocytes and F4/80+ macrophage was determined in the indicated mice by Flow. Data are representative of three independent experiments. n=3–5 mice/group. Graphs represent the mean with error bars indication s.e.m. p values are determined by one-way ANOVA Tukey’s multiple comparison test. * p<0.05, n.s: not significant.
The HAQ allele alleviates and the AQ allele prevents SAVI(N153S) disease in mice
SAVI(N153S) disease is characterized by early onset, failure to thrive (low body weight), persistent lung inflammation, decreased lung function, and young death in humans and mouse models Liu et al., 2014; Frémond et al., 2021; Wu et al., 2019; Motwani et al., 2019. The HAQ/SAVI(N153S) mice weighed more and had an improved lifespan than the WT/SAVI(N153S) mice (Figure 4A and B). The lifespan, airway resistance, and tissue inflammation (lung, liver) were also improved in HAQ/SAVI(N153S) mice compared to the WT/SAVI(N153S) mice (Figure 4C, D, J and K). However, the pulmonary artery pressure was still elevated in HAQ/SAVI(N153S) mice (Figure 4E). Remarkably, the AQ/SAVI(N153S) mice had similar body weight and lifespan as the WT/AQ mice (Figure 4F and G). The airway resistance, pulmonary artery pressure, and tissue inflammation in AQ/SAVI(N153S) were similar to the WT/AQ littermates (Figure 4H, I, J and K). Thus, the HAQ allele alleviates and the AQ allele prevents inflammatory SAVI disease in mice. Interestingly, lungs from HAQ/SAVI, AQ/SAVI had similarly reduced infiltration of Ly6G+CD11B+ neutrophils and Ly6G-Ly6C+CD11B+ inflammatory monocytes compared to WT/SAVI mice (Figure 4—figure supplement 1).
Figure 4 with 1 supplement see all
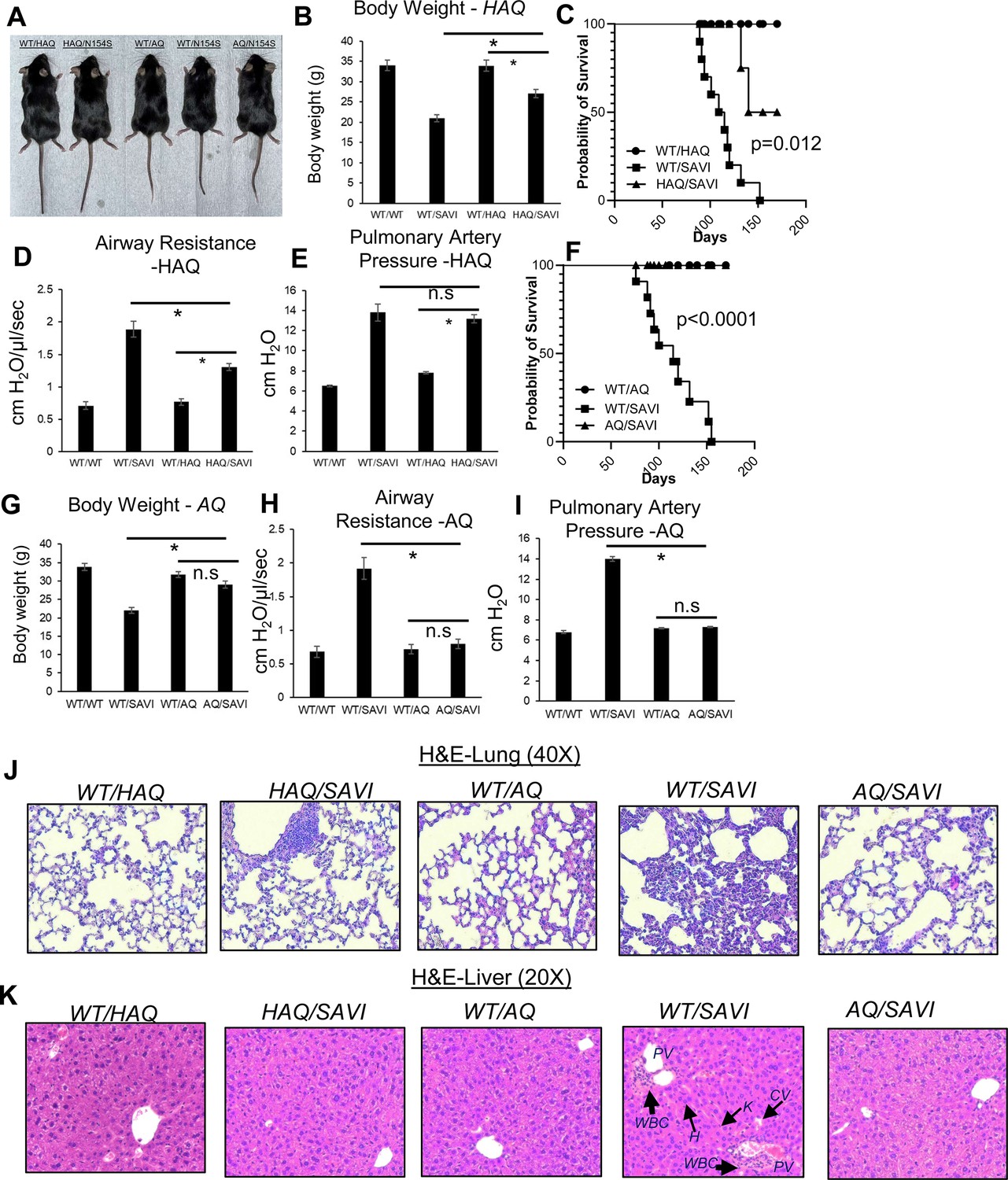
HAQ and AQ alleles prevent SAVI(N153S) disease in mice.
(A, B, G) The size and body weight of HAQ/SAVI, AQ/SAVI, WT/SAVI and their littermates WT/HAQ, WT/AQ mice. (D, E, H, I). Airway resistance, and pulmonary artery pressure were determined as described in Materials and methods. (C, F). HAQ/SAVI, AQ/SAVI, WT/SAVI (10 mice/group), were monitored for survival by Kaplan-Meier. (J, K). Representative hematoxylin and eosin (H&E) staining of lung, liver sections from indicated mice. n=3–5 mice/group. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values are determined by one-way ANOVA Tukey’s multiple comparison test. * p<0.05, **p<0.01. n.s.: not significant. (WBC): white blood cells; H: hepatocytes; K: Kupper cells; PV: portal vein; CV: central vein.
diABZI induces similar STING1, TBK1, IRF3, NFκB activation in the AQ/SAVI(N153S) and WT/SAVI(N153S) bone-marrow-derived-macrophage (BMDM)
SAVI was characterized as type I interferonopathy (Frémond et al., 2021). However, several studies showed that type I IFN signaling and IRF3 activation were dispensable for SAVI disease (Luksch et al., 2019; Stinson et al., 2022; Gao et al., 2022; Motwani et al., 2019). AQ allele prevents SAVI disease (Figure 4). However, diABZI-treated AQ/SAVI(N153S) and WT/SAVI(N153S) BMDM had similar TBK1-IRF3 activation and IFNβ production (Figure 5A and B). diABZI treatment caused IκBα degradation, and similar TNF production in WT/SAVI and AQ/SAVI BMDM (Figure 5C and D). Furthermore, diABZI activation led to STING1 protein degradation in WT/SAVI and AQ/SAVI BMDM (Figure 5E). Last, using cleavable crosslinker dithiobis succinimidyl propionate (DSP), we showed that STING1 in WT/SAVI, AQ/SAVI BMDM forms a similar dimer in situ (Figure 5F). Thus, the AQ/SAVI BMDM had similar STING1 degradation, TBK1, IRF3, NFκB activation, and dimerization as the WT/SAVI BMDM.
Figure 5

AQ/SAVI(N153S) cells had similar TBK1-IRF3, NFκB activation and STING1 degradation as the WT/SAVI(N153S) cells.
(A, C). BMDM from WT/WT, WT/SAVI, HAQ/SAVI and AQ/SAVI were treated with 100 ng/ml diABZi in culture for 2 hr. Cells were lysed and run on a 4~20% Mini-PROTEAN TGX Stain-Free Precast Gel. The blot was probed for phospho-Thr172-TBK1 antibody (CST, clone D52C2), phosphor-Ser 396-IRF3 (CST, clone 4D4G), STING1 (Proteintech, #19851–1-AP) and IκBα (CST, clone 44D4) antibody. (E). BMDM from WT/WT, WT/SAVI, HAQ/SAVI and AQ/SAVI were treated with 100 ng/ml diABZi in culture for 24 hr. Cells were lysed and run on a 4~20% Mini-PROTEAN TGX Stain-Free Precast Gel. The blot was probed for STING1 antibody (Proteintech, 19851–1-AP). (B, D). IFNβ and TNF were determined by ELISA in the cell supernatant from E. (F). BMDM from WT/WT, WT/SAVI, HAQ/SAVI and AQ/SAVI were treated with 400 µM cleavable chemical crosslinker DSP (Pierce, cat no: PG82081) in PBS for 1 hr at 4 °C. Cells were washed with PBS and lysed in RIPA buffer. Whole cell lysate was mixed with 4 x Laemmli Sample Buffer (BioRad, cat no 1610747) containing 5% 2-mercaptoethanol, heated at 95 °C for 10 min and, run on a 4~20% Mini-PROTEAN TGX Stain-Free Precast Gel. The blot was probed for STING1 antibody (Proteintech, 19851–1-AP). Densitometry was determined by ImageLab 5. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values are determined by unpaired student T-test (A–E) or one-way ANOVA Tukey’s multiple comparison test (D, B). * p<0.05, **p<0.01, ***p<0.001. n.s.: not significant; n.d: not detected.
The HAQ allele increased, and the AQ allele restored T-regs in SAVI(N153S) mice
IFNγ was proposed to drive SAVI disease (Stinson et al., 2022; Gao et al., 2022; Patel and Jin, 2019). We confirmed that WT/SAVI CD4 T cells were enriched with IFNγ+ cells (Figure 6A). However, WT/SAVI mice have CD4 T cellpenia. Thus, the total numbers of spleen IFNγ+ CD4 T cells were comparable in WT/SAVI and AQ/SAVI mice (Figure 6A). In contrast, the HAQ/SAVI mice had decreased IFNγ+ CD4 T cells (Figure 6A).
Figure 6
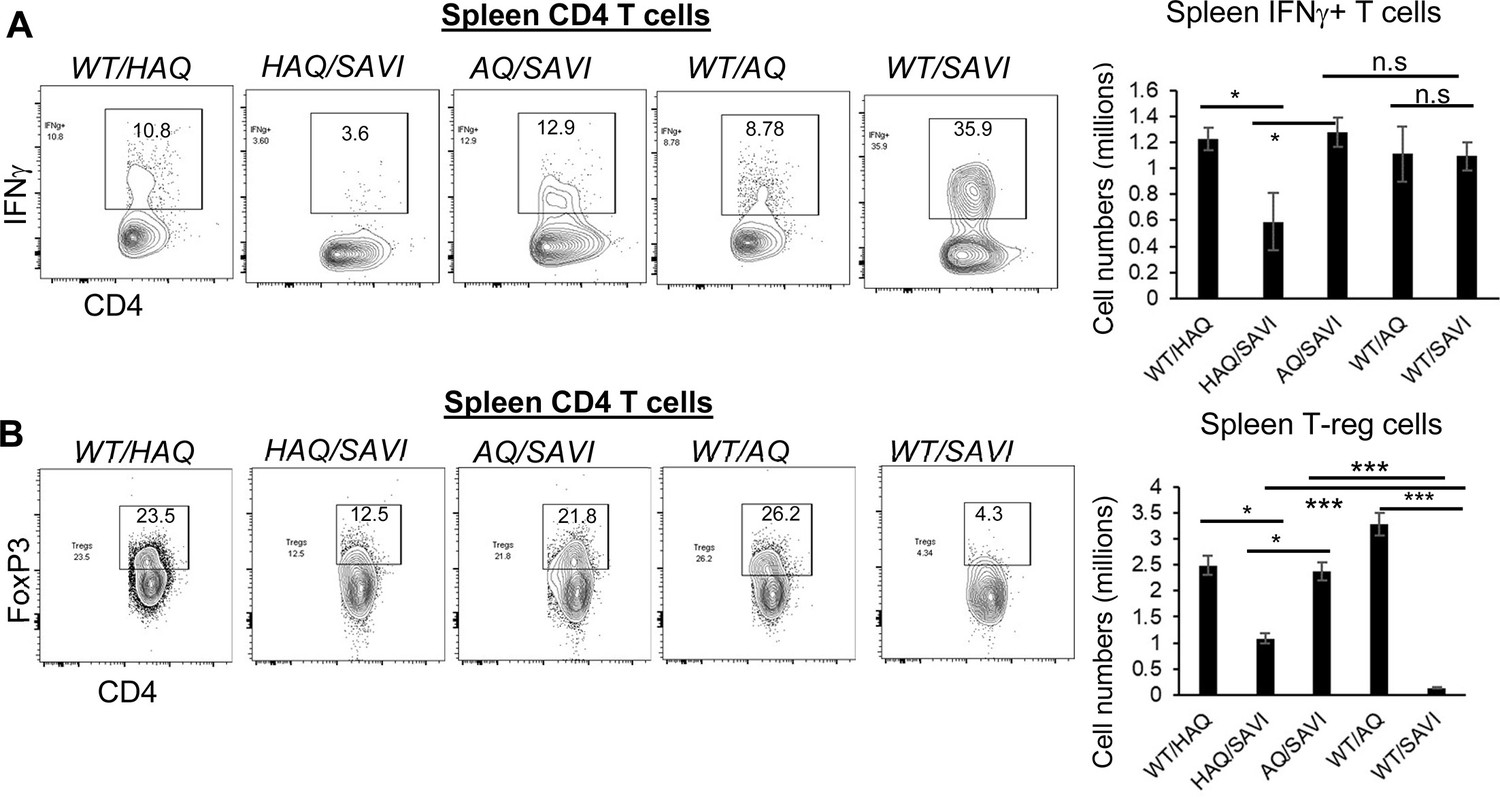
HAQ/SAVI(N153S) and AQ/SAVI(N153S) cells had 10-fold and 20-fold increased spleen T-regs compared to WT/SAVI mice.
(A) Flow cytometry analysis of IFNγ producing spleen CD4+ T cells from WT/HAQ, WT/AQ, WT/SAVI, HAQ/SAVI and AQ/SAVI mice. (B) Flow cytometry analysis of CD4+ FoxP3+ spleen T-regs. n=3–5 mice/group. Data are representative of three independent experiments. Graphs represent the mean with error bars indication s.e.m. p values are determined by one-way ANOVA Tukey’s multiple comparison test. * p<0.05, **p<0.01, ***p<0.001. n.s.: not significant.
The induction of Foxp3 expression in T-reg cells during ongoing autoimmune inflammation resolved inflammation and pathology in mice (Hu et al., 2021). CD4 T cellpenia depletes CD4 T-regs. Indeed, WT/SAVI mice had ~20-fold reduction of spleen FoxP3+ T-regs compared to AQ/SAVI or WT/WT littermates (Figure 6B). The HAQ/SAVI mice also had ~10-fold more T-regs than the WT/SAVI littermate (Figure 6B).
Discussion
This study, using the HAQ, AQ, SAVI(N153S) Sting1 knock-in mice, reveals the in vivo significance and mechanism of STING1-mediated CD4 T cell death. HAQ, AQ alleles prevent CD4 T cellpenia, and increase/restore CD4 T-regs in SAVI mice. The results are consistent with previous finding that the impaired CD4 T cell proliferation by the SAVI(V155M) mutant could be rescued by the addition of the HAQ allele in vitro (Cerboni et al., 2017). STING1 has been increasingly implicated in inflammatory diseases such as nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, cardiomyopathy, obesity, diabetes, neurodegenerative diseases, aging, and kidney injury, many of which are independent of type I IFNs (Skopelja-Gardner et al., 2022; Bai and Liu, 2021; Gao et al., 2023a). It is tempting to suggest that STING1 activation in CD4 T cells leads to CD4 T-regs depletion that break tissue tolerance and exacerbates tissue inflammation.
Human immunodeficiency virus (HIV) primarily infects CD4 T cells and might activate the STING1 pathway in CD4 T cells (Monroe et al., 2014; Doitsh et al., 2014; Jakobsen et al., 2015; Silvin and Manel, 2015; Altfeld and Gale, 2015; Krapp et al., 2018). The loss of CD4 T cells is the hallmark of untreated HIV infection Gubser et al., 2019; Morou et al., 2019, and the measurement of CD4 T cell count is a central part of HIV care. We found that HAQ and AQ CD4 T cells are resistant to STING1-mediated cell death. Mogensen and colleagues reported that HAQ/HAQ was enriched in HIV-infected long-term nonprogressors (Nissen et al., 2018). These HAQ/HAQ individuals had reduced inhibition of CD4 T cell proliferation and a reduced immune response to DNA and HIV (Nissen et al., 2018). It is likely that HIV infection activates STING1-cell death pathway in CD4 T cells. In HAQ/SAVI and AQ/SAVI mice, one copy of HAQ, AQ allele suppressed CD4 T cell death. HAQ, AQ carriers might have fewer HIV-induced CD4 T cell death, thus being long-term nonprogressors in HIV infection-induced Acquired immunodeficiency syndrome (AIDS; Nissen et al., 2018). Targeting STING1 to prevent CD4 T cell death might be a valid therapy for AIDS.
Activating the STING1 pathway is a promising strategy for cancer immunotherapy (Hines et al., 2023; Samson and Ablasser, 2022; Liu et al., 2021; Zheng et al., 2020; Corrales et al., 2015; Fu et al., 2015; Barber, 2011). Multiple STING1 agonists are in clinical trials (Meric-Bernstam et al., 2023; Meric-Bernstam et al., 2022). Recently, the safety issue emerged in some STING1 agonist trials (Meric-Bernstam et al., 2023; Meric-Bernstam et al., 2022). For example, in the STING1 agonist, ADU-S100, clinical trial, Grade 3/4 treatment-related adverse events were reported in 12.2% of 41 pretreated patients (NCT02675439) (Meric-Bernstam et al., 2023; Meric-Bernstam et al., 2022). The National Institutes of Health defines grade 3 as ‘incapacitating; unable to perform usual activities; requires absenteeism or bed rest.’ In a clinical trial using STING1 antibody-drug-conjugate (ADC) that conjugates diABZI to anti-HER2 Ab, a Grade 5 (fatal) serious adverse event was recorded and deemed related to the STING1-ADC (NCT05514717). SAVI disease, driven by overreacting STING1, is often fatal Liu et al., 2014. AQ, to a less degree, HAQ, suppress mortality in SAVI mice. Future STING1 clinical trials should be based on human STING1 genotype to achieve safe and effective responses.
Mechanistically, apoptosis, pyroptosis, ferroptosis, necroptosis, and PANoptosis have all been reported in STING1-mediated cell death (Kuhl et al., 2023; Jin et al., 2008; Gulen et al., 2017; Kabelitz et al., 2022; Murthy et al., 2020; Li et al., 2021; Song et al., 2022; Messaoud-Nacer et al., 2022; Tang et al., 2016). Different cell types and STING1 agonists used likely contributed to the inconsistency and complexity. Here, we focused on lymphopenia in the SAVI mice that avoids ligand-dependent, non-physiological dosage in STING1-mediated cell death. HAQ and AQ alleles could prevent CD4 T cellpenia in the SAVI mice strongly indicating that residue A230 or Q293 prevent STING1-mediated CD4 T cell death in vivo. Splenocyte from Q293 mice were resistant to STING1 agonists-induced cell death ex vivo. Thus, it is likely that the Q293 residue is critical for STING1-mediate lymphopenia. Notably, Q293 is outside the C-terminal tail (CTT) (residues 341–379 of human STING1) critical for TBK1 recruitment and IRF3 phosphorylation (Liu et al., 2015) or miniCTT domain (aa343–354) (Cerboni et al., 2017), or the UPR motif (aa322–343) (Wu et al., 2019) important for T cells death in vitro. Further studies are needed to understand how the aa293 of STING1 mediates cell death in vivo. Noteworthy, AQ/SAVI cells had similar TBK1-IRF3, NFκB activation and STING1 degradation as the WT/SAVI cells. Yet, AQ/SAVI mice did not have CD4 T cellpenia as WT/SAVI mice suggeSting1 that the canonical STING1-TBK1-IRF3/NFκB pathway, likely STING1 oligomerization, is not sufficient for the induction of cell death at the physiological condition.
We used the WT/N153S knock-in SAVI mouse model that spontaneously develop lung inflammation, T cell cytopenia, and early mortality, mimicking pathological findings in human SAVI patients (Warner et al., 2017). Using the WT/N153S SAVI mouse model and human Jurkat T cell line, it was proposed that STING1 activation causes chronic ER stress and unfolded protein response, leading to T cell death by apoptosis (Wu et al., 2019). Furthermore, the study showed that crossing WT/N153S mice to the OT-I mice reduced ER stress and restored CD8+, but not CD4+, T cells (Wu et al., 2019). The restoration of CD8+T cells reduces inflammation and lung disease (Wu et al., 2019). However, human WT/N154S SAVI patients have normal CD8+ T cells numbers (Liu et al., 2014), and primary human CD8+ T cells are largely resistant to STING1-agonists-induced cell death ex vivo (Figure 2A; Kuhl et al., 2023). Thus, it is puzzling how restoring CD8+ T cells can rescue SAVI phenotypes since the SAVI patients already have normal CD8+ T cells numbers.
Finally, it is unexpected that both HAQ and AQ alleles are resistant to cell death. Our previous studies showed that the HAQ and AQ alleles have opposite functions (Mansouri et al., 2022). AQ-STING1, not HAQ-STING1, responds to CDNs (Jin et al., 2011; Patel et al., 2017a; Mansouri et al., 2022; Sebastian et al., 2020; Yi et al., 2013; Patel et al., 2017b; Nissen et al., 2018; Ruiz-Moreno et al., 2018; Movert et al., 2023). AQ mice are lean while HAQ mice are fat (Mansouri et al., 2022). Most importantly, HAQ was positively selected, while AQ was negatively selected, in modern humans outside Africans (Mansouri et al., 2022). Thus, the death pathway of STING1 is also distinct from the STING1 function that was naturally selected. It is worth noting that the AQ allele does better than the HAQ allele in suppressing SAVI disease. Thus, besides preventing cell death, additional mechanism by the AQ allele, for example fatty acid metabolism (Mansouri et al., 2022; Vila et al., 2022), is involved in curing SAVI.
The limitations of the study
The poor transferability of mouse to humans is a major issue in STING1 research (Meric-Bernstam et al., 2023; Meric-Bernstam et al., 2022). The present study used AQ/SAVI and HAQ/SAVI mice. Confirmation is needed in humans with the identification and evaluation of people who are AQ/SAVI, HAQ/SAVI.
Materials and methods
Experimental design
Request a detailed protocolThe study was designed to reveal (i) the in vivo significance of the type I IFNs-independent, STING1-dependent cell death function; (ii) the interplay between common STING1 alleles HAQ, AQ and the rare, gain-of-function SAVI STING1 mutation; (iii) the driver for the inflammatory SAVI disease. Mouse splenocytes, primary human lung cells, human THP-1 cells and HAQ, AQ, SAVI knock-in mice were used to establish the in vivo significance and human relevance. All the repeats were biological replications that involve the same experimental procedures on different mice. Where possible, treatments were assigned blindly to the experimenter by another individual in the lab. When comparing samples from different groups, samples from each group were analyzed in concert, thereby preventing any biases that might arise from analyzing individual treatments on different days. All experiments were repeated at least twice.
Mice
WT/SAVI(N153S) mice were purchased from The Jackson Laboratory. HAQ, AQ mice were previously generated in the lab (Patel et al., 2017a; Mansouri et al., 2022). The Q293 mice were generated by Cyagen Biosciences. Briefly, the linearized targeting vector was transfected into JM8A3.N1 C57BL/6 N embryonic stem cells. A positive embryonic stem clone was subjected to the generation of chimera mice by injection using C57BL/6 J blastocysts as the host. Successful germline transmission was confirmed by PCR sequencing. The heterozygous mice were bred to Actin-flpase mice [The Jackson Laboratory, B6.Cg-Tg (ACTFLPe)9205Dym/J] to remove the neo gene and make the Q293 knock-in mouse. Age- and gender-matched mice (2–6 month old, both male and female) were used for indicated experiments. WT/SAVI (male) x WT/HAQ (female), WT/SAVI (male) x WT/AQ (female) breeders were set up to generate HAQ/SAVI, AQ/SAVI mice. Mice were housed at 22 °C under a 12‐hr light‐dark cycle with ad libitum access to water and a chow diet (3.1 kcal/g, Teklad 2018, Envigo, Sommerset, NJ) and bred under pathogen-free conditions in the Animal Research Facility at the University of Florida. Littermates of the same sex were randomly assigned to experimental groups. All mouse experiments were performed by the regulations and approval of the Institutional Animal Care and Use Committee at the University of Florida, IACUC202200000058.
Reagent
Request a detailed protocolRecombinant human IFNβ (R&D, cat no. 8499-IF-010/CF), diABZI (Invivogen, cat no. 2138299-34-8), 2’3’-cGAMP (Invivogen, cat no. tlrl-nacga23-02), DMXAA (Invivogen, cat no. tlrl-dmx), H151 (Invivogen, cat no. inh-h151), RpRpSS-Cyclic di-AMP (Biolog, cat no. C118), THP1-Dual KO-STING1 Cells (Invivogen, cat no. thpd-kostg). All other chemical inhibitors are from Selleckchem. Mouse TNF alpha ELISA Ready Set Go. (eBioscience, cat no. 88–7324). Mouse IFN-Beta ELISA Kit (PBI, cat no. 42400).
Generation of THP-1 KO-STING1 cells stably expressing human STING1 alleles
Request a detailed protocolTHP1-Dual KO-STING1 cells were purchased from Invivogen (thpd-kostg). These cells are guaranteed mycoplasma-free and authenticated by the vendor. THP1-Dual KO-STING1 Cells in six-well plate were transfected with 1 µg STING1 plasmid (in pcDNA 3.1 vector) with Lipofectamine LTX and Plus Reagent (Invitrogen, cat no: A12621) according to the manufacturer’s instructions. Transfecting Plasmid DNA into THP-1 Cells Using Lipofectamine LTX Reagent | Thermo Fisher Scientific - US. Forty-eight hours after the transfection, the cell medium was changed. G418 (1 mg/ml) was added to the culture to select STING1 expressing THP-1 cells. The G418-resistant cells were established and expanded.
Histology
Request a detailed protocolLungs and livers were fixed in 10% formalin, paraffin-embedded, and cut into 4 µm sections. Lung, liver sections were then stained for hematoxylin-eosin. All staining procedures were performed by the histology core at the University of Florida. Briefly, tissue sectins were immersed Harris Hematoxylin for 10 s, then washed with tap water. Cleard sections were re-immersed in EOSIN stain for ~30 s. The sections were washed with tap water until clear, then dehydrate in ascending alcohol solutions (50%, 70%, 80%, 95% x 2, 100% x 2). Afterwards, the sections werer cleared with xylene (3–4 x). The sections were mounted on glass slide with permount organic mounting medium for visulization.
Lung function
Request a detailed protocolPulmonary function was evaluated using an isolated, buffer-perfused mouse lung apparatus (Hugo Sachs Elektronik, March-Huggstetten, Germany), as previously described (Cai et al., 2020). Briefly, mice were anesthetized with ketamine and xylazine and a tracheostomy was performed, and animals were ventilated with room air at 100 breaths/min at a tidal volume of 7 μl/g body weight with a positive end-expiratory pressure of 2 cm H2O using a pressure-controlled ventilator (Hugo Sachs Elektronik, March-Huggstetten, Germany).
Isolation of lung cells
Request a detailed protocolCells were isolated from the lung as previously described (Mansouri et al., 2020). The lungs were perfused with ice-cold PBS and removed. Lungs were digested in DMEM containing 200 μg/ml DNase I (Roche, 10104159001), 25 μg/ml Liberase TM (Roche, 05401119001) at 37 °C for 2 hr. Red blood cells were then lysed and a single-cell suspension was prepared by filtering through a 70 µm cell strainer.
BMDM activation
Request a detailed protocolBMDMs were induced from mouse bone marrow cells cultured in RPMI 1640 (cat#11965; Invitrogen) with 10% FBS, 2 mM L-glutamine, 1 mM sodium pyruvate, 10 mM HEPES buffer, 1% nonessential amino acids, 50 mM 2-ME, 1% Pen/Strep, with 20 ng/ml M-CSF (Kingfisher, RP0407M) for 10 days (Patel et al., 2017a). STING1 agonists were added into the culture (no transfection or membrane permeabilization).
Flow cytometry
Request a detailed protocolSingle-cell suspensions were stained with fluorescent-dye-conjugated antibodies in PBS containing 2% FBS and 1 mM EDTA. Surface stains were performed at 4 °C for 20 min. For intracellular cytokine or transcription factor staining of murine and human cells, cells were fixed and permeabilized with the Foxp3 staining buffer set (eBioscience, cat no 00-5523-00). Cells were washed and stained with surface markers. Cells were then fixed and permeabilized (eBioscience, cat no. 00-5523-00) for intracellular cytokine stain. Data were acquired on a BD LSRFortessa and analyzed using the FlowJo software package (FlowJo, LLC). Cell sorting was performed on the BD FACSAriaIII Flow Cytometer and Cell Sorter.
Human lung explants
Request a detailed protocolHuman lung explants were procured at the Lung Transplant Center, Division of Pulmonary, Critical Care and Sleep Medicine, Department of Medicine, University of Florida. Donor and patients consent was obtained for a research protocol (UF IRB201902955-Treatment with IFNβ Induces Tolerogenic Lung Dendritic Cells in Human advanced lung disease). Healthy donor lungs were surgically removed postmortem, perfused, small pieces were cut from the right middle and lower lobes for research purpose, and stored in cold Perfadex at 4 °C for no more than 12 hr before processing. Ex planted lungs from emphysema lung transplant patients were stored in cold Perfadex at 4 °C for no more than 12 hr before the process. No lung explants were procured from prisoners.
Statistical analysis
Request a detailed protocolTo gain statistical power, we employ three ~four mice/groups to characterize lung immunity. Ten mice/group to monitor animal health. The statistical justification for group size was calculated using the SAS program to calculate the animal numbers. The analysis was carried out using a standard error of 0.5 for immunological assays, and a power of 0.9. All data are expressed as means ± SEM. Statistical significance was evaluated using Prism 9.0 software. Comparisons between two groups were analyzed by performing an unpaired Student’s t test. Comparisons between more than two groups were analyzed by performing a one-way analysis of variance (ANOVA) with Tukey’s multiple comparisons test.
Materials availability statement
Request a detailed protocolThe Q293 mouse is available upon request via a Material Transfer Agreement.
Data availability
Source data is deposited in Dryad https://doi.org/10.5061/dryad.m0cfxppcv.
-
Dryad Digital RepositoryData from: The common TMEM173 HAQ, AQ alleles rescue CD4 T cellpenia, restore T-regs, and prevent SAVI (N153S) inflammatory disease in mice.https://doi.org/10.5061/dryad.m0cfxppcv
References
-
Innate immunity against HIV-1 infectionNature Immunology 16:554–562.https://doi.org/10.1038/ni.3157
-
cGAS‒STING signaling and function in metabolism and kidney diseasesJournal of Molecular Cell Biology 13:728–738.https://doi.org/10.1093/jmcb/mjab066
-
STING: infection, inflammation and cancerNature Reviews. Immunology 15:760–770.https://doi.org/10.1038/nri3921
-
MicroRNA-206 antagomiR‒enriched extracellular vesicles attenuate lung ischemia‒reperfusion injury through CXCL1 regulation in alveolar epithelial cellsThe Journal of Heart and Lung Transplantation 39:1476–1490.https://doi.org/10.1016/j.healun.2020.09.012
-
Intrinsic antiproliferative activity of the innate sensor STING in T lymphocytesThe Journal of Experimental Medicine 214:1769–1785.https://doi.org/10.1084/jem.20161674
-
Overview of STING-associated vasculopathy with onset in infancy (SAVI) Among 21 PatientsThe Journal of Allergy and Clinical Immunology 9:803–818.https://doi.org/10.1016/j.jaip.2020.11.007
-
STING agonist formulated cancer vaccines can cure established tumors resistant to PD-1 blockadeScience Translational Medicine 7:283ra252.https://doi.org/10.1126/scitranslmed.aaa4306
-
CD4+ T cell signatures in HIV infectionNature Immunology 20:948–950.https://doi.org/10.1038/s41590-019-0447-5
-
Signalling strength determines proapoptotic functions of STINGNature Communications 8:427.https://doi.org/10.1038/s41467-017-00573-w
-
The Development of STING agonists and emerging results as a cancer immunotherapyCurrent Oncology Reports 25:189–199.https://doi.org/10.1007/s11912-023-01361-0
-
Regulatory T cells function in established systemic inflammation and reverse fatal autoimmunityNat Immunol 22:1163–1174.
-
Innate immune sensing of HIV-1 infectionCurrent Opinion in HIV and AIDS 10:96–102.https://doi.org/10.1097/COH.0000000000000129
-
Identification and characterization of a loss-of-function human MPYS variantGenes and Immunity 12:263–269.https://doi.org/10.1038/gene.2010.75
-
STING dependent sensing - Does HIV actually care?Cytokine & Growth Factor Reviews 40:68–76.https://doi.org/10.1016/j.cytogfr.2018.03.002
-
Cutting edge: activation of STING in T cells induces type I IFN responses and cell deathJournal of Immunology 199:397–402.https://doi.org/10.4049/jimmunol.1601999
-
STING1 promotes ferroptosis through MFN1/2-dependent mitochondrial fusionFrontiers in Cell and Developmental Biology 9:698679.https://doi.org/10.3389/fcell.2021.698679
-
Mini review: STING activation during non-alcoholic fatty liver diseaseFrontiers in Nutrition 10:1139339.https://doi.org/10.3389/fnut.2023.1139339
-
Activated STING in a vascular and pulmonary syndromeNew England Journal of Medicine 371:507–518.https://doi.org/10.1056/NEJMoa1312625
-
STING, A promising target for small molecular immune modulator: A reviewEuropean Journal of Medicinal Chemistry 211:113113.https://doi.org/10.1016/j.ejmech.2020.113113
-
STING-associated lung disease in mice relies on T cells but not type I interferonThe Journal of Allergy and Clinical Immunology 144:254–266.https://doi.org/10.1016/j.jaci.2019.01.044
-
MPYS modulates fatty acid metabolism and immune tolerance at homeostasis independent of type I IFNsJournal of Immunology 209:2114–2132.https://doi.org/10.4049/jimmunol.2200158
-
Crosstalk between cGAS-STING signaling and cell deathCell Death and Differentiation 27:2989–3003.https://doi.org/10.1038/s41418-020-00624-8
-
Multiple homozygous variants in the STING-encoding TMEM173 Gene in HIV long-term nonprogressorsJournal of Immunology 200:3372–3382.https://doi.org/10.4049/jimmunol.1701284
-
The Common R71H-G230A-R293Q human TMEM173 is a null alleleJournal of Immunology 198:776–787.https://doi.org/10.4049/jimmunol.1601585
-
Response to comment on “the common R71H-G230A-R293Q Human TMEM173 is a null allele.”Journal of Immunology 198:4185–4188.https://doi.org/10.4049/jimmunol.1700322
-
Non-interferon-dependent role of STING signaling in pulmonary hypertensionArteriosclerosis, Thrombosis, and Vascular Biology 44:124–142.https://doi.org/10.1161/ATVBAHA.123.320121
-
The cGAS-STING pathway and cancerNature Cancer 3:1452–1463.https://doi.org/10.1038/s43018-022-00468-w
-
Innate immune sensing of HIV infectionCurrent Opinion in Immunology 32:54–60.https://doi.org/10.1016/j.coi.2014.12.003
-
Role of the cGAS-STING pathway in systemic and organ-specific diseasesNature Reviews. Nephrology 18:558–572.https://doi.org/10.1038/s41581-022-00589-6
-
efficacy and adverse events during janus kinase inhibitor treatment of SAVI syndromeJournal of Clinical Immunology 39:476–485.https://doi.org/10.1007/s10875-019-00645-0
-
STING-associated vasculopathy develops independently of IRF3 in miceThe Journal of Experimental Medicine 214:3279–3292.https://doi.org/10.1084/jem.20171351
-
STING-mediated disruption of calcium homeostasis chronically activates ER stress and primes T cell deathThe Journal of Experimental Medicine 216:867–883.https://doi.org/10.1084/jem.20182192
Article and author information
Author details
Funding
National Heart, Lung, and Blood Institute (HL152163)
- Lei Jin
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
National Institutes of Health grant HL152163 (LJ).
Ethics
The donor lung and data are from deceased individuals. The specimens or data are de-identified for the purpose of this study. Under these conditions, the human study is exempt from Federal regulation, under category 4 (E4) Human lung explants were procured at the Lung Transplant Center, Division of Pulmonary,Critical Care and Sleep Medicine, Department of Medicine, University of Florida. Donor and patients consent was obtained for a research protocol (UF IRB201902955-Treatment with IFNs; Induces Tolerogenic Lung Dendritic Cells in Human advanced lung disease).
All mouse experiments were performed by the regulations and approval of the Institutional Animal Care and Use Committee at the University of Florida, IACUC202200000058.
Version history
- Preprint posted:
- Sent for peer review:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.96790. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2024, Aybar-Torres et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,254
- views
-
- 81
- downloads
-
- 8
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 3
- citations for umbrella DOI https://doi.org/10.7554/eLife.96790
-
- 1
- citation for Reviewed Preprint v1 https://doi.org/10.7554/eLife.96790.1
-
- 1
- citation for Reviewed Preprint v2 https://doi.org/10.7554/eLife.96790.2
-
- 3
- citations for Version of Record https://doi.org/10.7554/eLife.96790.3
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The common Sting1 HAQ, AQ alleles rescue CD4 T cellpenia, restore T-regs, and prevent SAVI (N153S) inflammatory disease in mice
eLife 13:RP96790.
https://doi.org/10.7554/eLife.96790.3




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}