Viral Variants: How mutations affect function
A new study reveals how naturally occurring mutations affect the biophysical properties of nucleocapsid proteins in SARS-CoV-2.
- Department of Physics and Astronomy, University of California, Riverside, United States
The COVID-19 pandemic has inflicted enormous societal, economic, and personal costs to populations around the world. It has resulted in hundreds of millions of infections, millions of deaths as well as lingering effects such as persistent symptoms and long COVID. Combatting the virus that causes COVID-19, known as SARS-CoV-2, as well as other viruses that could cause future pandemics, requires an understanding of the basic mechanisms through which viruses evolve and exert their effects.
Every person infected with COVID-19 carries their own pool of viral variants, which each contain a unique set of mutations in their genome. State-of-the-art sequencing technology has allowed an organization known as the Global Initiative on Sharing All Influenza Data to create a SARS-CoV-2 repository, which currently contains around 17 million different viral genome sequences (Elbe and Buckland-Merrett, 2017). This provides unparalleled insight into how mutations correlate with the evolution and emergence of SARS-CoV-2 variants of concern, such as delta and omicron.
However, despite this wealth of available sequencing information, our understanding of how these genetic changes actually impact how variants behave, such as their ability to infect cells and the severity of symptoms they cause, is lacking. Now, in eLife, Peter Schuck and colleagues at the National Institutes of Health – including Ai Nguyen as first author – report how mutations affect a structural protein in SARS-CoV-2 that is responsible for packaging and protecting the viral genome (Nguyen et al., 2024).
Nguyen et al. focused their study on nucleocapsid protein, the most abundant SARS-CoV-2 protein in infected cells, which is typically the molecule probed by at-home tests for the virus (Finkel et al., 2021; Frank et al., 2022). The primary role of the nucleocapsid protein is to package the genomic material of SARS-CoV-2 into the shell of the viral particle. But nucleocapsid protein also binds to many proteins in the host cell, which can modify stress and immune responses that may kill the virus or affect the severity of the infection (Chen et al., 2020; Gordon et al., 2020; Li et al., 2020). These interactions are determined by the structure and biophysical properties of nucleocapsid protein, such as its electrical charge and hydrophobicity.
By studying available genetic sequences of SARS-CoV-2 nucleocapsid proteins, Nguyen et al. found that most mutations lie in intrinsically disordered regions of the protein (Figure 1). These parts of the protein are highly flexible and do not fold into any rigidly defined structure. As such, they are more tolerant to a wider diversity of changes and mutations than other, more highly ordered segments of the protein.
Figure 1
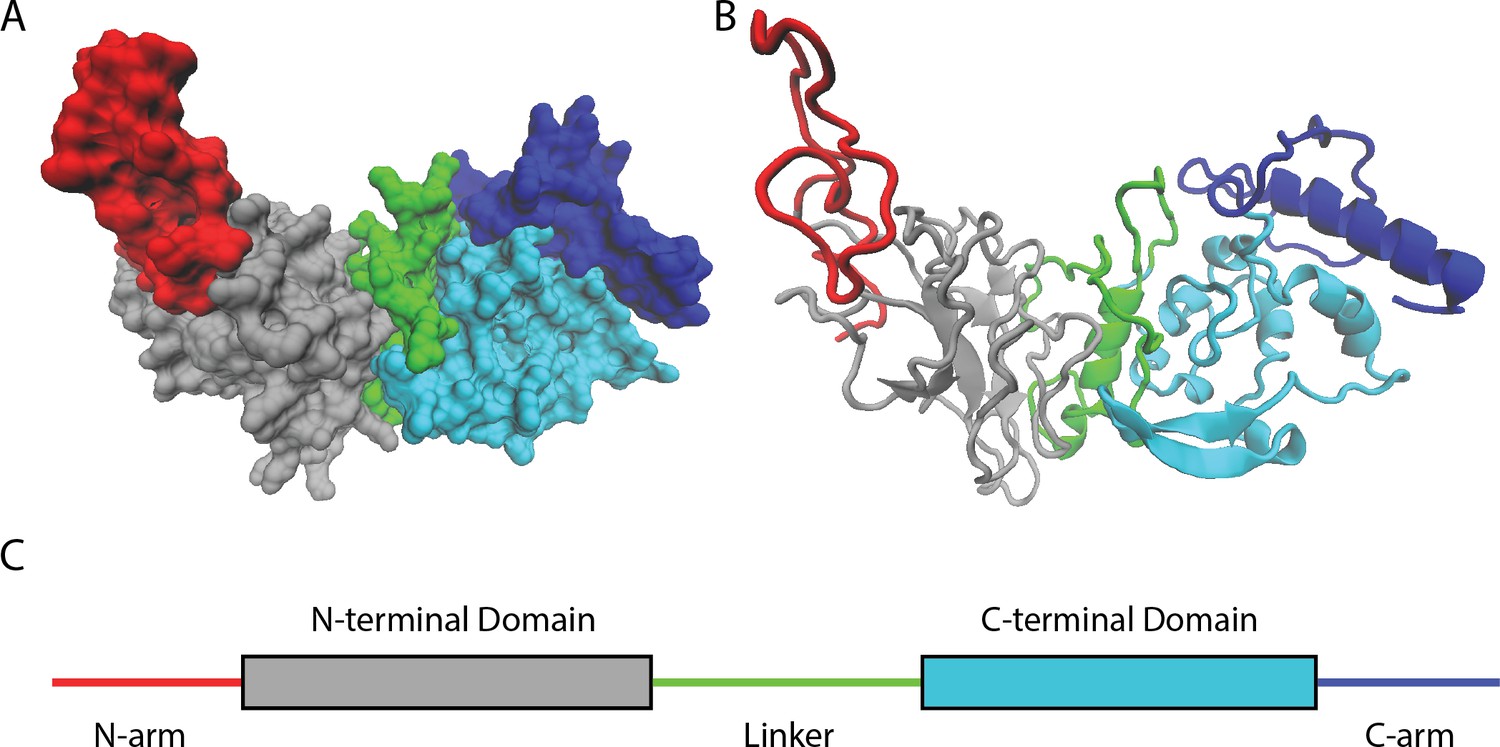
Naturally occurring mutations in the nucleocapsid protein of SARS-CoV-2.
(A) Space filling, (B) ribbon, and (C) domain representation of a single SARS-CoV-2 nucleocapsid protein. Nguyen et al. identified that most mutations occur in intrinsically disordered regions of the protein – the N-arm (red), linker (green), and C-arm (blue) – as opposed to more structured regions, such as the N-terminal (grey) and C-terminal (cyan) domains.
Image credit: Atomic coordinates used from Casasanta et al., 2023 and graphics in (A) and (B) made using Visual Molecular Dynamics (Humphrey et al., 1996).
Nguyen et al. then used computational methods to calculate how the behaviour of nucleocapsid proteins would be expected to change in response to each of the mutations they had detected. This showed that while single mutations in the intrinsically disordered regions cause a broader distribution of effects than those in structured regions of the protein, the biophysical properties of the protein were, surprisingly, largely conserved. Moreover, Nguyen et al. demonstrated that the intrinsically disordered regions show little sequence similarity to related viruses, such as SARS-CoV-1, Middle Eastern Respiratory Syndrome and other coronaviruses. However, the biophysical properties of these regions, such as their polarity and hydrophobicity, were similar across the different viruses.
To investigate further, Nguyen et al. carried out experiments on nucleocapsid proteins which had been purified from SARS-CoV-2 and contained mutations found in several variants of interest. This revealed that when multiple mutations are present, this leads to significant – and sometimes compensatory – biophysical changes. This included changes in the thermal stability of the protein, its ability to self-associate into multi-protein complexes, and how it assembles into larger structures. These effects demonstrate how mutations in intrinsically disordered regions can impact the entire structure and function of a protein. Additionally, this emphasizes why it is difficult to assign variant properties and fitness effects to a single mutation.
The findings of Nguyen et al. suggest that the biophysical properties of intrinsically disordered regions – such as their charge, polarity and hydrophobicity – may contribute to determining the behavior and interactions of nucleocapsid proteins. This could also be the case for other SARS-CoV-2 proteins, as well as proteins in other infectious viruses. These properties may place constraints on the mutations that nucleocapsid proteins can tolerate while still remaining functional. Furthermore, the findings of Nguyen et al. could provide a foundation for understanding how genomic sequencing data can be used to predict the effects of mutations in viruses.
References
-
Structural insights of the SARS-CoV-2 nucleocapsid protein: Implications for the inner-workings of rapid antigen testsMicroscopy and Microanalysis 29:649–657.https://doi.org/10.1093/micmic/ozac036
-
VMD: visual molecular dynamicsJournal of Molecular Graphics 14:33–38.https://doi.org/10.1016/0263-7855(96)00018-5
Article and author information
Author details
Publication history
Copyright
© 2024, Kuhlman
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 679
- views
-
- 78
- downloads
-
- 1
- citation
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for umbrella DOI https://doi.org/10.7554/eLife.99991
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Viral Variants: How mutations affect function
eLife 13:e99991.
https://doi.org/10.7554/eLife.99991




{kind=link}