RETRACTED: Protein kinase C is a calcium sensor for presynaptic short-term plasticity
- Harvard Medical School, United States
- University of California, Davis, United States
- University of Oslo, Norway
Abstract
In presynaptic boutons, calcium (Ca2+) triggers both neurotransmitter release and short-term synaptic plasticity. Whereas synaptotagmins are known to mediate vesicle fusion through binding of high local Ca2+ to their C2 domains, the proteins that sense smaller global Ca2+ increases to produce short-term plasticity have remained elusive. Here, we identify a Ca2+ sensor for post-tetanic potentiation (PTP), a form of plasticity thought to underlie short-term memory. We find that at the functionally mature calyx of Held synapse the Ca2+-dependent protein kinase C isoforms α and β are necessary for PTP, and the expression of PKCβ in PKCαβ double knockout mice rescues PTP. Disruption of Ca2+ binding to the PKCβ C2 domain specifically prevents PTP without impairing other PKCβ-dependent forms of synaptic enhancement. We conclude that different C2-domain-containing presynaptic proteins are engaged by different Ca2+ signals, and that Ca2+ increases evoked by tetanic stimulation are sensed by PKCβ to produce PTP.
https://doi.org/10.7554/eLife.03011.001eLife digest
Brain function is dependent upon the rapid transfer of information from one brain cell to the next at junctions known as synapses. When an electrical signal called an action potential is generated by the cell before the synapse, the presynaptic cell, it triggers an influx of calcium ions into that cell. These ions activate specific calcium sensors, triggering release of molecules called neurotransmitters from the presynaptic cell through exocytosis of synaptic vesicles. These neurotransmitters bind to receptors on the membrane of the postsynaptic cell, and produce an electrical signal whose size is a measure of synaptic strength.
The strength of a synapse can change over time—a property that is called plasticity. Synapses can undergo both long-term and short-term increases in strength. Post-tetanic potentiation is a short-term increase in strength that lasts for tens of seconds: it is triggered by a calcium increase in the presynaptic cell and involves an increase in the amount of neurotransmitter released in response to each presynaptic action potential. Post-tetanic potentiation is thought to underlie short-term memory. However, the identity of the sensor that detects the build-up of calcium in post-tetanic potentiation was not known.
Now, Fioravante, Chu et al. have provided the first direct evidence that an enzyme called protein kinase C is responsible. Electrophysiological recordings in brain slices from genetically modified mice revealed that animals that lack protein kinase C do not show post-tetanic potentiation. However, potentiation can be restored by re-introducing the enzyme into presynaptic cells. Importantly, a mutated version of protein kinase C that lacks the ability to bind calcium is unable to trigger post-tetanic potentiation.
Protein kinase C represents a new class of presynaptic calcium sensors that supports short-term plasticity. It is likely that future studies will identify additional members of this class of sensors that allow different synapses to have different forms of short-term plasticity. Further research is also needed to clarify the mechanisms underlying short-term plasticity and to understand how different forms of short-term plasticity are associated with different functions and behaviors.
https://doi.org/10.7554/eLife.03011.002Introduction
The complex manner in which patterns of action potentials (AP) are transformed into neurotransmitter release suggests the existence of multiple presynaptic calcium (Ca2+) sensors (Kaeser and Regehr, 2013). Synaptotagmin-1, synaptotagmin-2, and synaptotagmin-9 have been identified as Ca2+ sensors for synchronous release (Sudhof, 2013), but the Ca2+ sensors that regulate short-term use-dependent plasticity remain elusive. For a widespread form of short-term plasticity termed post-tetanic potentiation (PTP), a high-frequency burst of presynaptic APs enhances subsequent AP-evoked release for tens of seconds. PTP requires sustained elevation of presynaptic Ca2+, and in most cases synaptic enhancement outlives Ca2+ increases (Regehr et al., 1994; Brager et al., 2003; Korogod et al., 2005; Habets and Borst, 2007; Fioravante et al., 2011, 2012). Although PTP is thought to contribute to short-term memory (Silva et al., 1996; Abbott and Regehr, 2004), the Ca2+ sensor that mediates this plasticity has not been identified.
Three Ca2+-dependent isoforms of protein kinase C (PKCCa; PKCα, PKCβ, and PKCγ) play crucial roles in PTP (Fioravante et al., 2011, 2012; Chu et al., 2014). Because these isoforms contain Ca2+-binding C2 domains (Shao et al., 1996; Sutton and Sprang, 1998), we hypothesize that they function as Ca2+ sensors for PTP. However, it is unclear whether the Ca2+-binding properties of the C2 domain of PKCCa (Kohout et al., 2002) are well-suited to mediate PTP, which is thought to rely, at least in part, on the waning residual Ca2+ after the AP burst (Fioravante and Regehr, 2011). Moreover, diacylglycerol (DAG) binding to the C1 domain of PKCCa (Figure 1A) can regulate the activity of PKCCa (Newton, 2010), and it has been proposed that PKC could play a permissive role in PTP rather than function as the Ca2+ sensor (Saitoh et al., 2001). Indeed, presynaptic C2-domain proteins do not necessarily function as Ca2+ sensors; rather, they can regulate release independent of their Ca2+-binding properties (for an example, see Groffen et al., 2010; Pang et al., 2011). Furthermore, additional Ca2+-binding proteins have been implicated in short-term plasticity (Sakaba and Neher, 2001; Junge et al., 2004; Mochida et al., 2008; He et al., 2009; Shin et al., 2010), but it has not been established that Ca2+ binding to these proteins is required for short-term plasticity. Thus, in order to determine whether PKCCa isoforms are Ca2+ sensors that mediate PTP, it must be determined if PTP relies on Ca2+ binding to the PKCCa C2 domain.
Figure 1 with 6 supplements see all
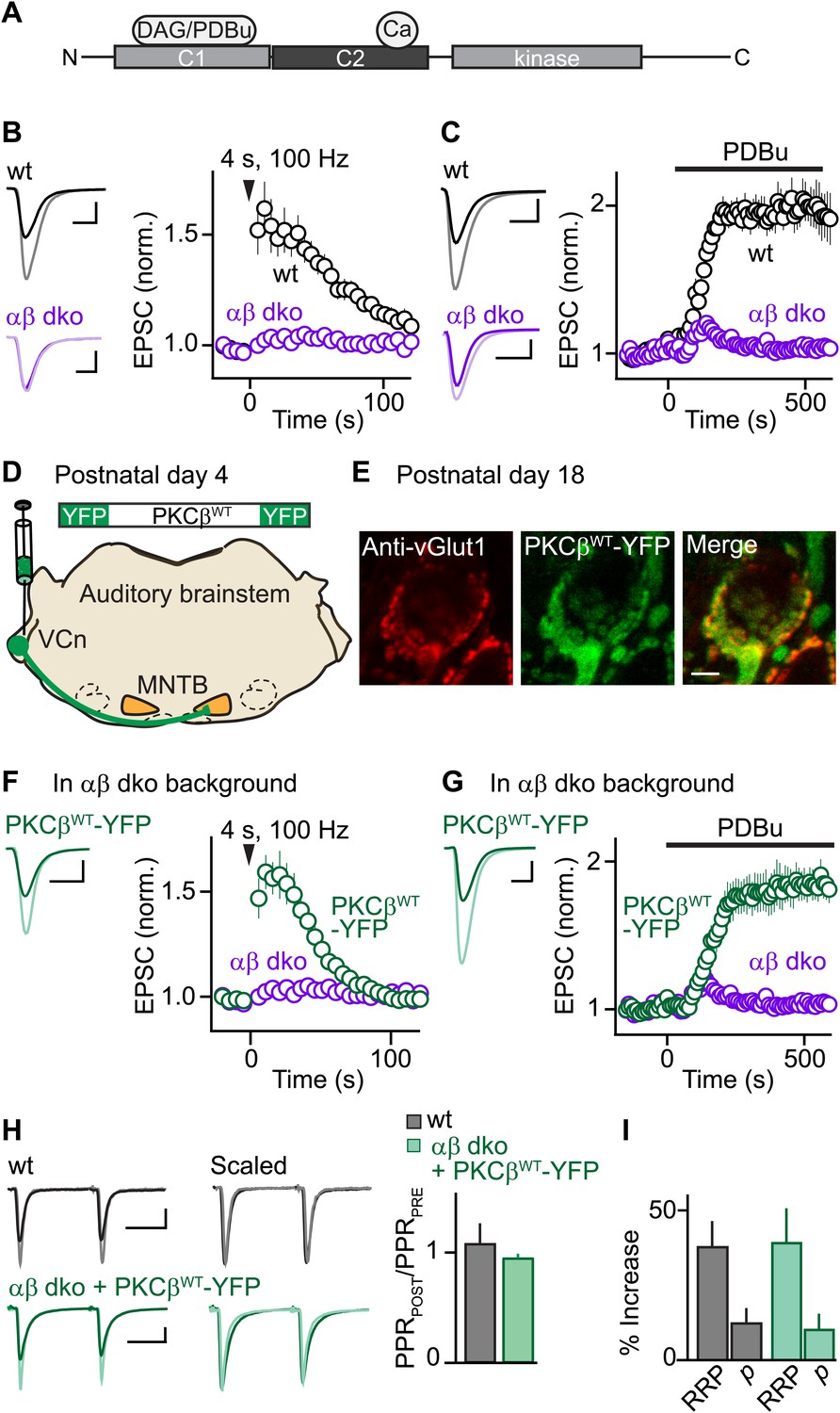
Expression of PKCβ rescues synaptic potentiation in animals lacking calcium-dependent PKCs.
Synaptic plasticity was examined at the calyx of Held following tetanic stimulation (B and F) or bath application of the phorbol ester PDBu (C and G) for wild-type (wt, black), PKCαβ dko animals (purple), and PKCαβ dko animals expressing PKCβWT-YFP (green). (A) Domain arrangement of PKCCa. DAG and PDBu bind to the C1 domain and Ca2+ binds to the C2 domain. (B, C, F, G) Left, example EPSCs recorded prior to (bold traces) and after (light traces) synaptic enhancement for each experimental condition. Right, EPSCs are plotted as a function of time (mean ± SEM). For (B), wild-type: 62 ± 12%; αβ dko: 2.4 ± 1.8%. Also see Figure 1—figure supplement 1 and accompanying legend for PTP induced under elevated-temperature conditions. Similar to PTP induced at room temperature, PTP at near-physiological temperature requires PKCCa (Figure 1—figure supplement 2). For (C), at steady state: wild-type: 97 ± 12%; αβ dko: 3.2 ± 3.4%; for (F), PKCβWT-YFP: 61 ± 7%; for (G), 84 ± 11%. In F and G, the αβ dko group data from B and C respectively are re-plotted for comparison. Also see Figure 1—figure supplement 3 and Figure 1—figure supplement 4. (D) In this schematic of the auditory brainstem, the ventral cochlear nucleus (VCn) and medial nuclei of the trapezoid body (MNTB) are labeled. An AAV expressing PKCβWT-YFP was injected in the VCn at postnatal day 4. (E) Confocal images of a brain section labeled with an antibody against vGlut1 (red) are shown for a calyx of Held expressing PKCβWT-YFP (green) in a PKCαβ dko animal at postnatal day 18. Scale bar: 10 µm. (H and I) The synaptic mechanism through which PKCβ rescues PTP was examined under conditions that relieve AMPA receptor desensitization and saturation. (H) Left, overlay of EPSCs (10 ms inter-stimulus interval) delivered prior to (bold traces) and 10 s after (light traces) PTP-inducing tetanus. Middle, traces are normalized to the first EPSC to allow comparison of PPR. Right, PPRPOST (after tetanus) over PPRPRE (before tetanus) (mean ± SEM, see Figure 1—source data 1 and 2). Wild-type: p=0.49; αβ dko expressing PKCβWT-YFP: p=0.68. (I) Summary of the readily releasable pool (RRP) and release probability (p) contributions to PTP (mean ± SEM, also see Figure 1—figure supplements 5 and 6 and Figure 1—source data 1 and 2). RRPWT: 37 ± 9%; RRPPKCβWT-YFP: 39 ± 12%; p=0.88. Scale bars in B, C, F, and G: 2 nA, 1 ms. Scale bars in H: 2 nA, 5 ms.
-
Figure 1—source data 1
Summary and statistical analyses of synaptic properties during PTP and PDBu-induced potentiation.
- https://doi.org/10.7554/eLife.03011.004
-
Figure 1—source data 2
Summary and statistical analyses of basal synaptic properties.
- https://doi.org/10.7554/eLife.03011.005
Results
To investigate the function of PKCCa isoforms in PTP, we first examined their role at the functionally mature calyx of Held synapse (postnatal day 17–22) (Fedchyshyn and Wang, 2005; Yang et al., 2010) using double knockout mice for PKCα and β (αβ dko). We recorded excitatory postsynaptic currents (EPSCs) from principal neurons in the medial nucleus of the trapezoid body (MNTB) in response to extracellular stimulation. Tetanic stimulation induced PTP in wild-type animals (Figure 1B, black) but not in PKCαβ dko animals (Figure 1B, purple). Thus, in contrast to the immature calyx of Held where a substantial component of PTP (∼20%) is independent of PKCCa (Fioravante et al., 2011), PTP at the functionally mature calyx of Held relies entirely on PKCCa isoforms.
We further tested whether the contribution of PKCCa to a related form of potentiation that occludes PTP also increases with development. Phorbol 12,13-dibutyrate (PDBu), a DAG analog, can enhance transmission by activating not only PKCCa (Figure 1A) but also Ca2+-insensitive PKC isoforms and other presynaptic proteins (Brose and Rosenmund, 2002; Newton, 2010). At immature calyces, ∼35% of PDBu-mediated enhancement is independent of PKCCa (Fioravante et al., 2011). We found that PDBu enhances release at functionally mature wild-type calyces (Figure 1C, black) but not at age-matched αβ dko calyces (Figure 1C, purple). Thus, at the functionally mature calyx of Held, both PTP and PDBu-mediated enhancement rely entirely on PKCCa, suggesting that the contributions of parallel mechanisms to these forms of plasticity (e.g., Wierda et al., 2007; Shin et al., 2010) diminish with development.
Although most of the studies presented here were performed at room temperature, we also examined PTP at near-physiological temperatures (34°C). Higher stimulus frequencies were required to induce PTP (Figure 1—figure supplement 1), but PTP was still dependent on PKCCa (Figure 1—figure supplement 2).
We next assessed whether presynaptic expression of PKCβ in αβ dko animals rescues PTP. PKCβ was chosen because genetic deletion of PKCα had little effect on PTP (Figure 1—figure supplement 3), suggesting that PTP is mediated primarily by PKCβ at the functionally mature calyx. We generated an adeno-associated virus (AAV), which we used to express wild-type PKCβ fused to yellow fluorescent protein (PKCβWT-YFP) in αβ dko animals (Figure 1D). Two weeks after injection, virally expressed PKCβWT-YFP localized to glutamatergic terminals positive for the marker vGlut1 (Figure 1E). In contrast to non-injected αβ dko animals, we observed reliable PTP at synapses expressing PKCβWT-YFP (Figure 1F, green; compare to non-injected age-matched αβ dko animals, purple). Expression of PKCβWT-YFP did not alter basal synaptic properties (Figure 1—figure supplement 4). Moreover, PKCβWT-YFP expression supported PDBu-induced potentiation in αβ dko mice (Figure 1G, green), which was very similar in amplitude to that observed in wild-type animals (Figure 1C, black; Figure 1—source data 1). Thus, expression of PKCβ is sufficient to rescue PTP and PDBu-mediated enhancement in PKC αβ dko animals.
To determine whether rescued PTP and PTP in wild-type animals are mediated by the same synaptic mechanism, we calculated the paired-pulse ratios (PPR = EPSC2/EPSC1) before and at the peak of PTP. If PTP reflects an increase in vesicular release probability (p), which is inversely related to PPR, then PPRPOST/PPRPRE should decrease. However, PPRPOST/PPRPRE was unchanged in both wild-type (Figure 1H, black) and αβ dko animals expressing PKCβWT-YFP (Figure 1H, green). This suggests that in both groups PTP is not mediated by an increase in p. We next examined the contribution of the readily releasable pool (RRP) of vesicles to PTP by evoking EPSCs with AP trains before and at the peak of PTP (Figure 1—figure supplement 5) and found that PTP was mediated by equivalent increases in the RRP in wild-type and rescued groups (Figure 1I, Figure 1—figure supplement 6). Thus, at the functionally mature calyx, the same mechanism mediates PTP in wild-type animals and at synapses in PKCαβ dko animals that express PKCβWT-YFP.
To determine if PKCβ is the Ca2+ sensor for PTP, it is necessary to abolish Ca2+ binding to PKCβ. To this end, we mutated five C2-domain aspartates (D) to alanines (A) and examined the effect on Ca2+ binding using purified recombinant wild-type C2 (C2WT) and mutant C2D/A domains (Figure 2A,B, Figure 2—figure supplement 1). These aspartates are predicted to mediate Ca2+ binding based on structural similarity (Nalefski and Falke, 1996; Ubach et al., 1998). We assessed Ca2+ binding through changes in intrinsic fluorescence of tryptophan residues adjacent to the predicted Ca2+-binding sites (Figure 2—figure supplement 1; Nalefski and Newton, 2001). In the absence of Ca2+, C2WT displayed a characteristic intrinsic fluorescence emission spectrum that peaked around 340 nm (Figure 2C, dark green). A similar basal emission spectrum was observed for C2D/A (Figure 2C, dark blue), suggesting that D-to-A mutations did not affect domain folding (Nalefski and Newton, 2001). Addition of 1 mM Ca2+ increased the fluorescence intensity of C2WT (Figure 2C, light green) but not C2D/A (light blue, Figure 2C), indicating that D-to-A mutations prevent Ca2+-dependent rearrangements.
Figure 2 with 1 supplement see all
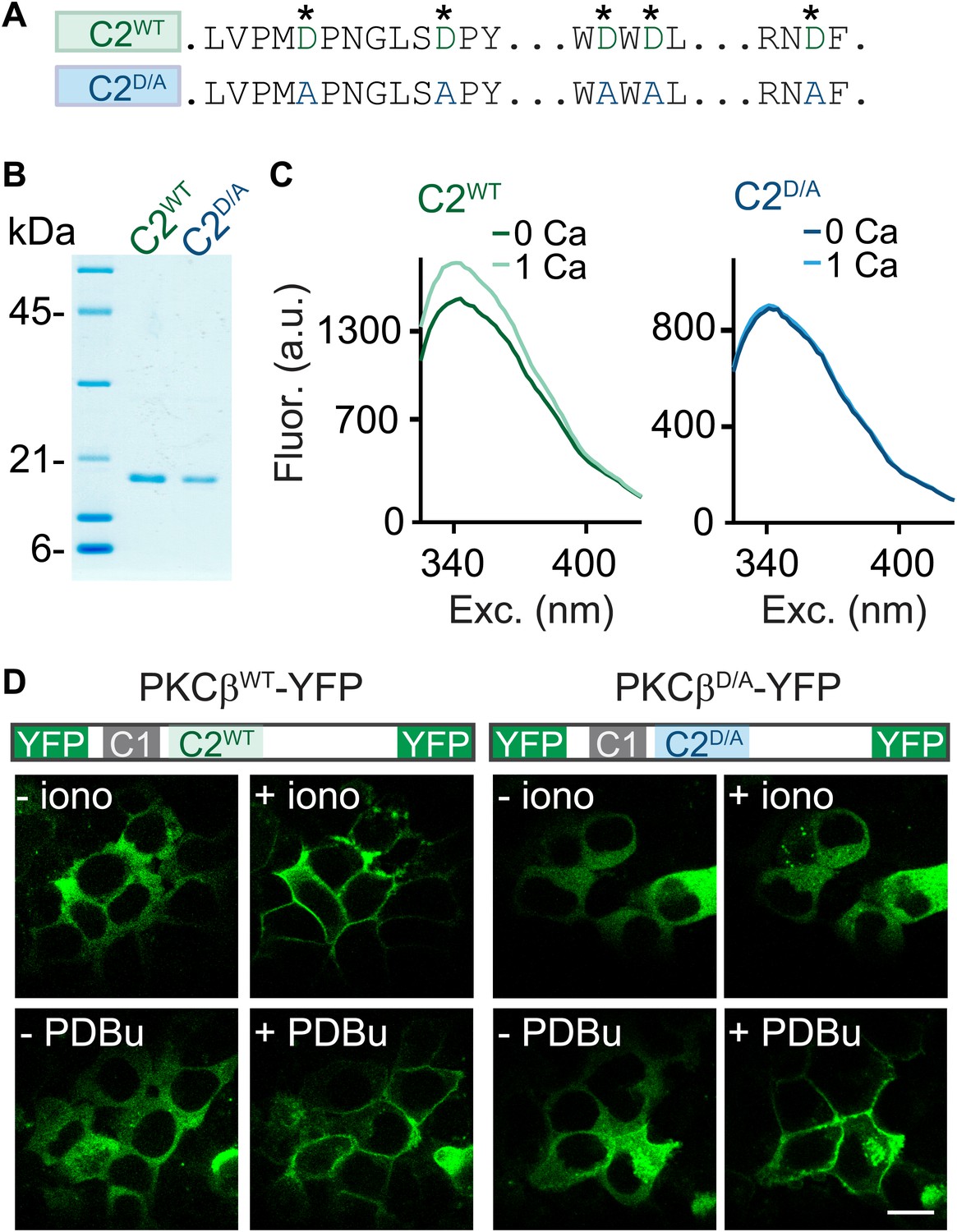
C2-domain mutations of PKCβ abolish Ca2+ binding and Ca2+-induced translocation without impairing phorbol ester-induced translocation.
(A) A partial sequence of the PKCβ C2 domain is shown with Ca2+-coordinating aspartates in green. These aspartates were mutated to alanines (blue) in the C2D/A construct. Also see Figure 2—figure supplement 1. (B) Coomassie blue-stained gel of recombinant wild-type (C2WT) and mutant (C2D/A) PKCβ C2 domains. (C) Averaged intrinsic tryptophan fluorescence is shown for C2WT and C2D/A. Fluorescence emission spectra were recorded in 0 mM Ca2+ (bold traces) and 1 mM Ca2+ (light traces). Peak fluorescence intensity change: C2WT:17 ± 1.3%; C2D/A: −1.3 ± 2.0%. (D) Translocation of PKCβWT-YFP (left) and PKCβD/A-YFP (right) in HEK293T cells was monitored in response to the Ca2+ ionophore ionomycin and in response to PDBu. Ca2+ increases caused PKCβWT-YFP to translocate, but not PKCβD/A-YFP. Both PKCβWT-YFP and PKCβD/A-YFP translocated in response to PDBu. Scale bar: 10 µm.
When activated by either phorbol esters or Ca2+, PKC translocates from the cytoplasm to the plasma membrane (Newton, 2010). We utilized this property to test the effects of the D-to-A mutations on the response of PKCβ to Ca2+ increases. We expressed PKCβWT-YFP or PKCβD/A-YFP in HEK293T cells and monitored the subcellular distribution of the kinase. The Ca2+ ionophore ionomycin induced translocation of PKCβWT-YFP (Figure 2D, top left), but did not alter the intracellular distribution of PKCβD/A-YFP (Figure 2D, top right). In contrast, PDBu, which binds to the C1 domain, caused both PKCβWT-YFP and PKCβD/A-YFP to translocate. This result indicates that Ca2+ binding to the PKC C2 domain is necessary for Ca2+-induced, but not PDBu-induced, translocation of PKCβ. Moreover, it suggests that D-to-A mutations in the C2 domain prevent Ca2+ activation of PKCβ without interfering with C1-domain-mediated membrane recruitment of PKCβ.
We next tested whether Ca2+ binding to the PKCβ C2 domain is required for PTP. Using AAV to express PKCβD/A-YFP, we found that PKCβD/A-YFP localized to vGlut1-positive areas and distributed similarly to PKCβWT-YFP (Figure 3A, compare to Figure 1E). Expression of PKCβD/A-YFP in αβ dko calyces, similar to wild-type PKCβ, did not affect basal synaptic properties (Figure 3—figure supplement 1, Figure 3—source data 2). However, in stark contrast to wild-type PKCβ, PKCβD/A failed to rescue PTP (Figure 3B, blue; also see Figure 3D, left). The inability of PKCβD/A-YFP to support PTP could be due to a loss of Ca2+ binding to PKCβ; alternatively, the D-to-A mutations may have induced more profound impairments of PKCβ, rendering it unable to enhance neurotransmitter release. To distinguish between these possibilities, we tested PDBu-induced potentiation in PKCβD/A-YFP-expressing calyces. Compellingly, PDBu-induced potentiation in PKCβD/A-YFP-expressing calyces was rescued to wild-type levels (Figure 3C, blue; also see Figure 3D, right). This indicates that PKCβD/A-YFP retained its ability to enhance synaptic transmission. We conclude that PKCβD/A-YFP is unable to mediate PTP because it is unable to bind Ca2+, and that Ca2+ binding to the C2 domain of PKCβ is required for PTP. Therefore, PKCβ is a Ca2+ sensor for PTP.
Figure 3 with 1 supplement see all
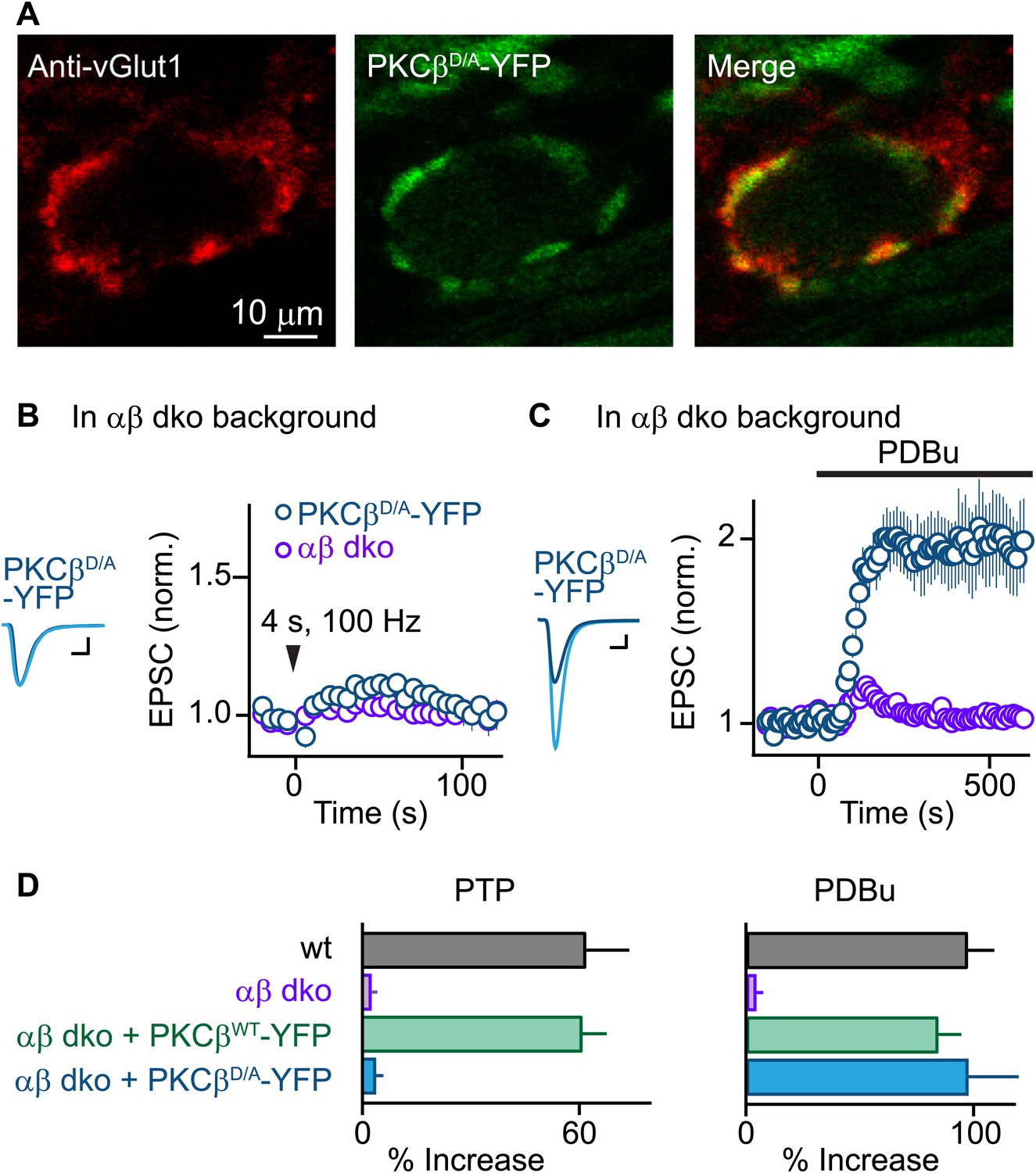
PTP requires Ca2+ binding to PKCβ but phorbol ester-induced potentiation does not.
(A) Confocal images of a brain section labeled with an antibody against vGlut1 (red) are shown for a calyx of Held expressing PKCβD/A-YFP (green) in a PKCαβ dko animal. (B and C) Synaptic plasticity was examined in PKCαβ dko animals at calyces of Held expressing Ca2+-insensitive PKCβ (PKCβD/A-YFP, blue traces). Representative traces and time-courses (mean ± SEM) are shown following tetanic stimulation (B) and during bath application of PDBu (C). For (B), PKCβD/A: 3.6 ± 2.2%; for (C), PKCβD/A: 98 ± 23%. Scale bars: 1 nA, 1 ms. In (B and C), the αβ dko group data from Figure 1B,C respectively are re-plotted for comparison. For basal synaptic properties of the PKCβD/A-YFP-expressing group, see Figure 3—figure supplement 1 and Figure 3—source data 2. (D) Summary plots (mean ± SEM) of the magnitude of synaptic enhancement produced by tetanic stimulation (left) and by PDBu (right). Source data are provided in Figure 3—source data 1 and 2. See also Figure 1—source data 1 and 2.
-
Figure 3—source data 1
Summary and statistical analyses of synaptic properties during PTP and PDBu-induced potentiation.
- https://doi.org/10.7554/eLife.03011.015
-
Figure 3—source data 2
Summary and statistical analyses of basal synaptic properties.
- https://doi.org/10.7554/eLife.03011.016
Discussion
To the best of our knowledge, PKCβ is the first Ca2+ sensor to be identified specifically for short-term synaptic plasticity. Similar to synaptotagmin-1, synaptotagmin-2, and synaptotagmin-9, PKCβ requires binding of Ca2+ to its C2 domain for its Ca2+-sensing function (Figure 3). However, PKCβ acts upstream of vesicle fusion (de Jong and Verhage, 2009), does not regulate basal transmission or paired-pulse plasticity (Figure 1—figure supplement 4), and is not expected to be activated by single stimuli. How do PKCβ and synaptotagmins respond to such different activity patterns and, consequently, such different Ca2+ signals? It is likely a combination of differences in Ca2+-binding properties and subcellular localization that underlie these contrasting responses (Nalefski et al., 2001). Synaptotagmin-1 binds Ca2+ cooperatively with low affinity and fast kinetics, and localizes close to release sites on synaptic vesicles; therefore, it is poised to detect large, transient Ca2+ signals near open voltage-gated Ca2+ channels (Sudhof, 2013). In contrast, PKCβ is activated by lower Ca2+ levels with lower cooperativity and is cytosolic (Nalefski and Newton, 2001; Kohout et al., 2002). Prolonged stimulation is necessary to produce a sufficient buildup of Ca2+ to activate PKCβ, which is consistent with the prolonged activity requirement for PTP (Habets and Borst, 2005; Korogod et al., 2005).
It is unlikely that PKCβ is the sole Ca2+ sensor that triggers PTP. At the granule cell to Purkinje cell synapse, PTP can be dependent on either PKCα or PKCβ (Fioravante et al., 2012), and at the calyx of Held synapse prior to the onset of hearing PTP depends on PKCγ (Chu et al., 2014). These findings suggest that Ca2+-sensitive PKC isoforms, including PKCα and PKCγ, may constitute a class of proteins that acts as Ca2+ sensors for PTP, much as multiple isoforms of synaptotagmin act as Ca2+ sensors for fast synaptic transmission (Sudhof, 2012). Further studies are required to determine whether PKCα, PKCγ, and other Ca2+-sensitive proteins implicated in PTP, such as Munc13 (Wierda et al., 2007), calmodulin (Junge et al., 2004), and synaptotagmin-2 (He et al., 2009; Xue and Wu, 2010), can also act as Ca2+ sensors to produce PTP.
PKCβ mediates PTP by phosphorylating downstream targets (Genç et al., 2014), which could explain how PTP outlives elevations of presynaptic Ca2+ (Fioravante and Regehr, 2011). We suggest that PKCβ is a founding member of a new class of Ca2+ sensors that function upstream of vesicle fusion to regulate short-term plasticity.
Materials and methods
DNA constructs and viruses
Request a detailed protocolCloning was performed by Genscript. Viruses were generated by the University of Pennsylvania Vector Core. All constructs were verified by sequencing. Wild-type PKCβ-YFP (βWT-YFP) was obtained through PCR from Addgene plasmid #14866 (Violin et al., 2003), using the following primers: 5′-GACACAACAGTCTCGAACTTAATCGAACCCGCGGCACGAGCCTCGACG-3′; 3′-GGGAAAAAGATCGGATCCTCAGGCGTCGACGGGCCCTCTAGATTACTTG-5′. To generate an adeno-associated viral vector, PKCβWT-YFP was inserted into a pENN.AAV.CMV.TurboRFP.RBG cis-plasmid (courtesy of the University of Pennsylvania Vector Core) using SacII and SalI, after removal of the TurboRFP sequence with SpeI and XhoI. Mutant PKCβ-YFP (βD/A-YFP) was generated by replacing the 5 aspartates that coordinate calcium binding (Sutton and Sprang, 1998) with alanines through PCR.
To generate bacterial expression plasmids of the PKCβ C2 domain, the sequences for C2WT or C2D/A (a.a. 157–294) (Torrecillas et al., 2003) (see also Figure 2—figure supplement 1) were inserted into a pGEX-KG vector (Addgene database #2890) using XbaI and NcoI and the following primers: 5′-TCCGGTGGTGGTGGTGGAATTCTAGAAGAACGCCGTGGCCGCATC-3′ and 3’-AAGCTTGAGCTCGAGTCGACCCATGGTCATCCTTCCGGCGGCACCG-5′.
Animals
All animal experiments were conducted at Harvard Medical School and were completed in accordance with guidelines by the Harvard Medical Area Standing Committee on Animals. PKCαβ double knockout (dko) mice were obtained through breeding of PKCα and PKCβ single knockout (ko) animals generated by M Leitges (Leitges et al., 1996, 2002). The probability of obtaining an αβ double knockout animal from heterologous (het) crosses is very low (1:16), making viral injection experiments unfeasible. We therefore bred het-ko animals together to increase the probability of getting desired animals. Similarly, to increase the probability of obtaining wild-type mice, we crossed PKC het–het or het-wild-type mice to use as wild-type controls. Wild-type mice were derived from the same genetic line as αβ dko animals. To prevent genetic drift in the inbred ko lines, we backcrossed them every second generation to C57BL/6J or 129S2. For experiments, animals of both sexes were used and age-matched wild-type, PKCα ko and PKCαβ dko mice from our colony were interleaved.
Surgery
Request a detailed protocolP4 pups were stereotactically and unilaterally injected under isofluorane anesthesia with AAVs into the VCn (from lambda: 1.3 mm lateral, 0.9 mm caudal, 3 mm ventral), where globular bushy cells that give rise to calyx of Held synapses in the contralateral MNTB reside. Injections (600 nl at a rate of 1 nl/s) were performed with an UltraMicroPump (UMP3, WPI, Sarasota FL) and Wiretrol II capillary micropipettes (Drummond Scientific, Broomall PA) pulled to a fine tip (10–20 µm diameter). At this age, the skull is sufficiently soft so it can be penetrated with a 281/2–gage needle without the need for drilling. After the injection, the skin was closed with Gluture (Abbott Laboratories, Irving TX), and pups were allowed to recover on a heating pad prior to returning to the home cage. 14–18 days were allowed for expression prior to slice preparation.
Preparation of brain slices
Request a detailed protocolThe calyx of Held synapse in the auditory brainstem was chosen for this study because of its monosynaptic innervation and its amenability to viral manipulations. Indeed, our study would not be possible at more ‘conventional’ polysynaptic preparations such as the CA3-CA1 hippocampal synapse because it is not possible to infect 100% of synapses with viruses. Transverse 190-µm to 200-µm-thick brainstem slices containing the MNTB were made with a vibratome slicer (VT1000S, Leica, Buffalo Grove IL) from juvenile (postnatal day 17–22) mice deeply anesthetized with isoflurane. Brains were dissected and sliced at 4°C in cutting solution consisting of the following (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 0.1 CaCl2, 3 MgCl2, 25 glucose, 3 myo-inositol, 2 Na-pyruvate, 0.4 ascorbic acid, continuously bubbled with 95% O2/5% CO2 (pH 7.4). Slices were incubated at 32°C for 30 min in a bicarbonate-buffered solution composed of the following (in mM): 125 NaCl, 25 NaHCO3, 1.25 NaH2PO4, 2.5 KCl, 2 CaCl2, 1 MgCl2, 25 glucose, 3 myo-inositol, 2 Na-pyruvate, 0.4 ascorbic acid, continuously bubbled with 95% O2/5% CO2 (pH 7.4).
Electrophysiology
Request a detailed protocolSlices were transferred to a recording chamber at room temperature (21–24°C) under an upright microscope (Olympus, Center Valley PA) equipped with a 60× objective. During recordings, the standard perfusion solution consisted of the bicarbonate-buffered solution (see above) with 1 µM strychnine and 25 µM bicuculline (R&D Systems, Minneapolis MN) to block inhibition. Slices were superfused at 1–3 ml/min with this external solution. Whole-cell postsynaptic patch-clamp recordings were made from visually identified cells in the MNTB region using glass pipettes of 2–3 MΩ resistance, filled with an internal recording solution of the following (in mM): 20 CsCl, 140 Cs-gluconate, 20 TEA-Cl, 10 HEPES, 5 EGTA, 5 Na2-phosphocreatine, 4 ATP-Mg, 0.3 GTP-Na. Series resistance (Rs) was compensated by up to 70%, and the membrane potential was held at −70 mV.
EPSCs were evoked by stimulating presynaptic axons with a custom-made bipolar stimulating electrode midway between the medial border of the MNTB and the midline of the brainstem. For slice recordings from injected animals, principal neurons in the MNTB contralateral to the injection site were selected based on the presence of YFP-expressing presynaptic terminals. A Multiclamp 700B (Axon Instruments/Molecular Devices, Sunnyvale CA) amplifier was used. Recordings were digitized at 20 KHz with an ITC-18 A/D converter (Instrutech Corp./HEKA Elektronik, Bellmore NY) using custom macros (written by MA Xu-Friedman) in Igor Pro (Wavemetrics, Portland OR) and filtered at 8 kHz. Macros can be found on Dr Xu-Friedman's website http://biology.buffalo.edu/Faculty/Xu_Friedman/mafPC/sign_in.html.
The protocol for inducing PTP was as follows: an estimate of baseline synaptic strength was obtained through low-frequency stimulation at 0.2 Hz for 25 s. PTP was induced with a 4-s stimulus train at 100 Hz, followed by low-frequency stimulation to test for PTP. For phorbol ester experiments, PDBu (1 µM; Tocris, UK) was washed in for 10 min once a stable baseline of at least 3 min was established. Synaptic strength was evaluated by afferent fiber stimuli, repeated every 20 s. During the inter-trial intervals, 5 s stretches of postsynaptic current were recorded to assess the frequency and amplitude of mEPSCs. To assess PPR, pulses were delivered at an inter-stimulus interval of 10 ms. For all recordings, the access resistance and leak current were monitored, and experiments were rejected if either of these parameters changed significantly.
Data analysis
Request a detailed protocolData analysis was performed using routines written in IgorPro (WaveMetrics). PTP magnitude was calculated as the ratio of EPSC amplitude 10 s after the 4-s, 100 Hz train over the average baseline. The magnitude of PDBu-induced potentiation was estimated by averaging the steady-state responses, 430–600 s from wash-in onset. To analyze spontaneous events, mEPSCs were detected using a threshold (average peak to peak noise in the baseline) of the first derivative of the raw current trace and confirmed visually. Statistical analyses were done using one-way ANOVA tests for multiple group comparisons followed by Tukey post-hoc analysis, or Kruskal–Wallis non-parametric ANOVA for data sets that were not normally distributed. Pairwise comparisons were performed with Student's t tests. Level of significance was set at p<0.05.
To determine the contributions of RRP and p to wild-type and rescued PTP, stimulus trains were used in the presence of kynurenate (1 mM) and CTZ (0.1 mM) to prevent postsynaptic receptor saturation and desensitization. Briefly, the amplitude of the first 40 responses to the stimulus train used to induce PTP and to a stimulus train (400 ms, 100 Hz) 10 s later (at the peak of PTP) were measured, and a plot of the cumulative EPSC for each train vs the stimulus number was made. The key to this approach is that the EPSC amplitude eventually reaches a steady-state level, and under these conditions the RRP is depleted and the remaining release is due to replenishment from a recycling/reserve pool (Schneggenburger et al., 1999). The size of the RRP can then be determined by a linear fit to the steady-state responses (last 15 EPSCs), which is extrapolated back to the y-axis (Moulder and Mennerick, 2005; Thanawala and Regehr, 2013). p is then calculated from EPSC1/RRP.
Immunohistochemistry
Request a detailed protocol150-µm thick transverse brainstem slices were prepared as described above from P18–P22 animals injected with AAVs and fixed with 4% paraformaldehyde for 2 hr at 4°C. At the end of fixation, slices were transferred to phosphate buffered saline (Sigma-Aldrich, St. Louis MO) and stored at 4°C until further processing. Slices were then incubated in blocking solution (phosphate buffered saline +0.25% Triton X-100 [PBST] +10% normal goat serum) for 1 hr at room temperature. Slices were incubated with primary antibody (anti-vGlut1 guinea pig polyclonal [Synaptic Systems, Germany]) in PBST overnight at 4°C, followed by incubation with secondary antibody (goat anti-guinea pig Alexa 568-conjugated [Life Technologies, Carlsbad CA]) in PBST for 2 hr. Slices were mounted to Superfrost glass slides (VWR, Visalia CA) and air-dried for 30 min. Following application of Prolong anti-fade medium (Invitrogen), slices were covered with a top glass coverslip (VWR) and allowed to dry for 24 hr prior to imaging. Antibodies were used at 1:500 dilution.
Images were acquired with a Zeiss 510 Meta confocal microscope using a Plan-apochromat 1.4 NA 63x oil lens. Emission filters were BP570-670 nm for the red channel (vGlut1) and BP500-550 for YFP (PKCβ). Single optical sections at 1024 × 1024 (average of three scans) were obtained sequentially for the different channels. Color channels were split and merged in ImageJ to obtain the composite images in RGB.
Protein purification
Request a detailed protocolN-terminal GST fusion proteins of PKCβ C2WT and C2D/A were expressed in Escherichia coli BL21 cells. Pelleted bacteria were resuspended in ice-cold PBS supplemented with 500 µM EDTA, 0.5 mg/ml lysozyme (Amresco, Solon OH), and protease inhibitor cocktail (Easypack; Roche, South San Francisco CA), and the bacteria were lysed by sonication. After centrifugation at 11,200 RPM for 30 min, the soluble fraction was collected and incubated with glutathione sepharose 4B beads (GE healthcare, Pittsburgh PA) for 1 hr at 4°C. Samples were cleared from nucleic acid contaminants with benzonase (40 U/ml, Sigma) for 3 hr at RT, and subsequently eluted from the beads with solution containing 100 mM Tris, 10 mM CaCl2, 5 mM Glutathione (pH 7.4) for 1 hr at 4°C. GST was cleaved with thrombin-agarose (100 µl resin/mg protein, Sigma) for 24 hr at 4°C, and samples were dialyzed to solution containing 40 mM Tris–HCl pH 7.4, 100 mM NaCl, and 0.5 mM Na-EGTA. GST was removed from the samples using glutathione sepharose 4B beads. 10 µl of purified protein was run on a 12% SDS gel and Coomassie blue-stained to check for purity (Figure 2B).
Intrinsic tryptophan fluorescence assay
Request a detailed protocolIntrinsic tryptophan fluorescence of purified recombinant C2WT and C2D/A was monitored in dialysis buffer (see above). Emission spectra were recorded from 325 to 425 nm on a Spectramax M5 microplate reader (Molecular Devices). Excitation was set at 295 nm and peak intrinsic fluorescence change (ΔF) upon addition of 1 mM free Ca2+ was estimated at 341 nm. To correct for the effect of volume increase on fluorescence readings upon addition of Ca2+-containing buffer, ΔF in buffer-alone controls was subtracted from fluorescence values in buffer+Ca2+ groups. Experiments were repeated with two independently purified batches of protein, for a total of seven times. Similar results were obtained every time.
Protein translocation assay
Request a detailed protocolHEK293T cells plated on glass coverslips were transfected with PKCβWT-YFP or βD/A-YFP expression vectors using Lipofectamine 2000 (Life Technologies). 24 hr after transfection, the coverslips were transferred to the imaging chamber of a custom-built 2-photon laser scanning microscope system and superfused with buffer (138 mM NaCl, 1.5 mM KCl, 10 mM HEPES, 1 mM MgCl2, 2 mM CaCl2, 10 mM glucose, pH 7.4) at 2 ml/min. YFP was excited at 840 nm with a Ti-Sapphire laser through a 60×, 1.1 NA water-immersion Olympus lens. A 500–550 BP emission filter was used. 512 × 512 frame scans were acquired at a rate of 1 line/4 ms, every 30 s. To stimulate translocation, the superfusion solution was switched to one containing 1 µM PDBu or 10 µM ionomycin (R&D Systems) for 15 min. The experiment was repeated three times for PKCβWT-YFP and twice for PKCβD/A-YFP, with similar results. Acquired images were exported to ImageJ and brightness/contrast was adjusted equally for all images within an experiment for display purposes.
References
-
Move over protein kinase C, you've got company: alternative cellular effectors of diacylglycerol and phorbol estersJournal of Cell Science 115:4399–4411.https://doi.org/10.1242/jcs.00122
-
Presynaptic signal transduction pathways that modulate synaptic transmissionCurrent Opinion in Neurobiology 19:245–253.https://doi.org/10.1016/j.conb.2009.06.005
-
Developmental transformation of the release modality at the calyx of Held synapseThe Journal of Neuroscience 25:4131–4140.https://doi.org/10.1523/JNEUROSCI.0350-05.2005
-
Short-term forms of presynaptic plasticityCurrent Opinion in Neurobiology 21:269–274.https://doi.org/10.1016/j.conb.2011.02.003
-
Post-tetanic potentiation in the rat calyx of Held synapseThe Journal of Physiology 564:173–187.https://doi.org/10.1113/jphysiol.2004.079160
-
Dynamics of the readily releasable pool during post-tetanic potentiation in the rat calyx of Held synapseThe Journal of Physiology 581:467–478.https://doi.org/10.1113/jphysiol.2006.127365
-
Molecular mechanisms for synchronous, asynchronous, and spontaneous neurotransmitter releaseAnnual Review of Physiology 76:333–363.https://doi.org/10.1146/annurev-physiol-021113-170338
-
Presynaptic Ca2+ requirements and developmental regulation of posttetanic potentiation at the calyx of HeldThe Journal of Neuroscience 25:5127–5137.https://doi.org/10.1523/JNEUROSCI.1295-05.2005
-
Knockout of PKC alpha enhances insulin signaling through PI3KMolecular Endocrinology 16:847–858.https://doi.org/10.1210/mend.16.4.0809
-
Reluctant vesicles contribute to the total readily releasable pool in glutamatergic hippocampal neuronsThe Journal of Neuroscience 25:3842–3850.https://doi.org/10.1523/JNEUROSCI.5231-04.2005
-
The C2 domain calcium-binding motif: structural and functional diversityProtein Science 5:2375–2390.https://doi.org/10.1002/pro.5560051201
-
Protein kinase C: poised to signalAmerican Journal of Physiology Endocrinology and Metabolism 298:E395–E402.https://doi.org/10.1152/ajpendo.00477.2009
-
The role of presynaptic calcium in short-term enhancement at the hippocampal mossy fiber synapseThe Journal of Neuroscience 14:523–537.
-
Activation of the epsilon isoform of protein kinase C in the mammalian nerve terminalProceedings of the National Academy of Sciences of the United States of America 98:14017–14021.https://doi.org/10.1073/pnas.241333598
-
Munc13 C2B domain is an activity-dependent Ca2+ regulator of synaptic exocytosisNature Structural & Molecular Biology 17:280–288.https://doi.org/10.1038/nsmb.1758
-
Impaired learning in mice with abnormal short-lived plasticityCurrent Biology 6:1509–1518.https://doi.org/10.1016/S0960-9822(96)00756-7
-
Calcium control of neurotransmitter releaseCold Spring Harbour Perspectives in Biology 4:a011353.https://doi.org/10.1101/cshperspect.a011353
-
Synthesis of anilino-monoindolylmaleimides as potent and selective PKCbeta inhibitorsBioorganic & Medicinal Chemistry Letters 14:5171–5174.https://doi.org/10.1016/j.bmcl.2004.07.061
-
A genetically encoded fluorescent reporter reveals oscillatory phosphorylation by protein kinase CThe Journal of Cell Biology 161:899–909.https://doi.org/10.1083/jcb.200302125
-
Post-tetanic potentiation is caused by two signalling mechanisms affecting quantal size and quantal contentThe Journal of Physiology 588:4987–4994.https://doi.org/10.1113/jphysiol.2010.196964
Article and author information
Author details
Funding
National Institute of Neurological Disorders and Stroke (R01NS032405)
- Wade G Regehr
National Institute of Neurological Disorders and Stroke (T32NS007484)
- Diasynou Fioravante
National Institute on Drug Abuse (K01DA029044)
- Pascal S Kaeser
National Institute on Deafness and Other Communication Disorders (F30-DC013716-01)
- YunXiang Chu
Nederlandse Organisatie voor Wetenschappelijk Onderzoek (NWO 825.12.028)
- Arthur PH de Jong
Howard Hughes Medical Institute (2011 Medical Fellows Program)
- YunXiang Chu
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank M Antal, R Held, S Jackman, S Rudolf, M Thanawala, C-C Wang, and L Witter for comments on a previous version of the manuscript. We particularly thank K McDaniels for help with genotyping, M Thanawala for help with two-photon imaging, and E Antzoulatos for help with Matlab.
Ethics
Animal experimentation: Animal experimentation: All procedures involving animals were performed in accordance with the guidelines of the National Institutes of Health. The protocol used (1493) was approved by the Harvard Medical Area (HMA) standing committee on animals.
Copyright
© 2014, Fioravante et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,539
- views
-
- 301
- downloads
-
- 17
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 17
- citations for umbrella DOI https://doi.org/10.7554/eLife.03011
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
RETRACTED: Protein kinase C is a calcium sensor for presynaptic short-term plasticity
eLife 3:e03011.
https://doi.org/10.7554/eLife.03011




{kind=link}
{kind=link}
{kind=link}