Connexin26 hemichannels with a mutation that causes KID syndrome in humans lack sensitivity to CO2
- University of Warwick, United Kingdom
- University of Connecticut Health Center, United States
- University of Connecticut, United States
Abstract
Mutations in connexin26 (Cx26) underlie a range of serious human pathologies. Previously we have shown that Cx26 hemichannels are directly opened by CO2 (Meigh et al., 2013). However the effects of human disease-causing mutations on the CO2 sensitivity of Cx26 are entirely unknown. Here, we report the first connection between the CO2 sensitivity of Cx26 and human pathology, by demonstrating that Cx26 hemichannels with the mutation A88V, linked to Keratitis-Ichthyosis-Deafness syndrome, are both CO2 insensitive and associated with disordered breathing in humans.
https://doi.org/10.7554/eLife.04249.001Main text
Connexin26 (Cx26) is one of 21 connexin genes found in humans (Cruciani and Mikalsen, 2006). The canonical function of connexins is to form gap junctions in which two hexameric connexons, or hemichannels, in closely apposed membranes dock together to form an intercellular channel. However connexins can also function as hemichannels, thereby providing large conductance channels, which allow passage of small molecules such as ATP into the extracellular space (Stout et al., 2004; Wang et al., 2013). We have recently shown that Cx26 hemichannels are directly sensitive to CO2 (Huckstepp et al., 2010a; Meigh et al., 2013). When CO2 binds to Cx26, it carbamylates K125, forms a salt bridge to R104 and opens the hemichannel (Meigh et al., 2013). Cx26 hemichannels are thus a source of CO2-gated ATP release (Huckstepp et al., 2010a).
Mutations of Cx26 are the commonest cause of non-syndromic hearing loss (Cohn and Kelley, 1999; Kelley et al., 2000; Xu and Nicholson, 2013). Some of these mutations cause loss of functional protein, while other mutations result in gap junctions and hemichannels with altered properties. However the effect of these mutations on the CO2 sensitivity of Cx26 has never been examined. Some missense mutations of Cx26 cause serious pathologies in humans, such as the very rare ectodermal disorder, Keratitis-Ichthyosis-Deafness (KID) syndrome. KID syndrome involves a combination of deafness, visual impairment, and dermatological abnormalities (Caceres-Rios et al., 1996). About 100 cases have been reported in the literature, and of these around 70% are caused by de novo mutations in Cx26, with the remainder being inherited in an autosomal dominant manner or via germ line mosaicism (Sbidian et al., 2010). To date there are nine missense mutations that can cause KID syndrome (Xu and Nicholson, 2013). The severity of the symptoms of KID syndrome depends on the particular mutation in Cx26 (Janecke et al., 2005; Jonard et al., 2008).
The mutation, Cx26A88V, is linked to a very severe form of KID syndrome, which is fatal in infancy (Haruna et al., 2010; Koppelhus et al., 2010). In one of the original reports linking Cx26A88V to KID syndrome, the patient required mechanical ventilation (Koppelhus et al., 2010), suggesting a possible effect of the mutation on the neural control of breathing. In KID syndrome caused by a different missense mutation (G45E), which is fatal within the first year of life, there are also reports of breathing problems. One patient required mechanical ventilation immediately after birth (Janecke et al., 2005) and a second died from breathing failure (Sbidian et al., 2010). Nevertheless, without detailed recordings of cardiorespiratory activity, it is not possible to know whether these patients experienced inadequate central respiratory drive. For other mutations linked to KID syndrome there are no reports of abnormal breathing in the literature.
The reason why the A88V and G45E mutations should cause such pervasive and severe pathology remains unclear as subunits of Cx26A88V and Cx26G45E form both functional gap junctions and hemichannels (Gerido et al., 2007; Mhaske et al., 2013). Expression of Cx26A88V in HeLa cells gives rise to enhanced hemichannel-mediated currents (compared to wild type Cx26, Cx26WT) at positive transmembrane potentials and in the absence of extracellular Ca2+, leading to the suggestion that this mutation represents a gain of function (Mhaske et al., 2013). The G45E mutation, also causes enhanced hemichannel activity in the absence of extracellular Ca2+, and increased permeability to Ca2+ (Gerido et al., 2007; Sanchez et al., 2010). A gain of function has therefore been suggested as underlying the actions of this mutation too. Although the absence of extracellular Ca2+ opens connexin hemichannels, this condition is unlikely to occur in physiological systems. Thus the consequences of the A88V and G45E mutations on physiologically relevant gating of Cx26 remain unclear.
We identified a patient with KID syndrome caused by a heterozygous Cx26 A88V mutation. This patient failed to breathe spontaneously at birth and initially required mechanical ventilation. Later when he started to breathe spontaneously, he continued to demonstrate periods of apnea and bradycardia. A pneumogram performed at a post-menstrual age of 40 weeks showed abnormal persistence of central apnea lasting ≥20 s and accompanied by periods of bradycardia and prolonged oxygen desaturation (Figure 1). This respiratory pattern is abnormal for the age of the infant and is suggestive of blunted chemosensory control of breathing. Given the previously described role of Cx26 in mediating the CO2-dependent drive to breathe (Huckstepp et al., 2010b; Wenker et al., 2012), we considered whether the mutation A88V might alter the CO2-sensitivity of Cx26.
Figure 1
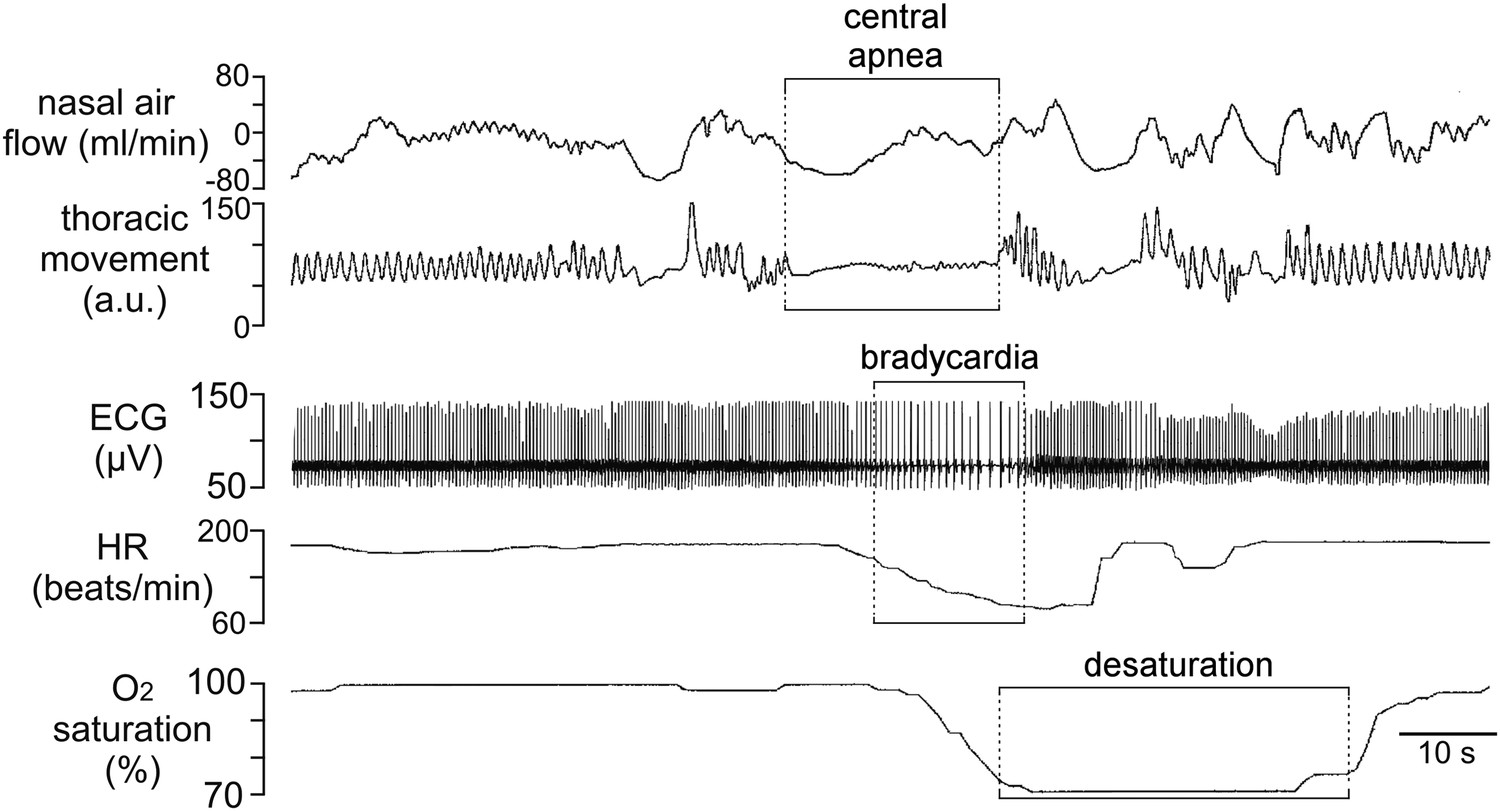
Incidence of central sleep apnea in a patient with Cx26A88V.
Recording of cardiorespiratory activity during sleep from an infant at a post-menstrual age of 40 weeks diagnosed with KID syndrome. Traces of nasal air flow, thoracic movement, electrocardiogram (ECG), heart rate (HR) and arterial O2 saturation show that this patient exhibited a prolonged period during which no effort was made to breathe and this was followed by pronounced bradycardia and arterial O2 desaturation, all of which are characteristic of central sleep apnea. Unfortunately, at 2 months of age this patient died from overwhelming sepsis.
We introduced the A88V mutation into Cx26 and then tested the CO2 sensitivity of Cx26A88V hemichannels expressed in HeLa cells via an established and sensitive dye-loading protocol (Huckstepp et al., 2010a; Meigh et al., 2013). Under conditions of normal extracellular Ca2+, HeLa cells expressing wild type Cx26 hemichannels readily load with carboxyfluorescein when exposed to a moderately hypercapnic saline (PCO2 55 mmHg) (Huckstepp et al., 2010a; Meigh et al., 2013). However HeLa cells expressing Cx26A88V showed no such CO2-dependent dye loading even when exposed to higher levels of PCO2 (70 mmHg, Figure 2). The failure to exhibit CO2-dependent dye loading was not due to a lack of functional hemichannels as the positive control of removing extracellular Ca2+, which opens all connexin hemichannels, caused robust dye loading (Figure 2). HeLa cells transfected with an empty vector do not show any dye loading in response to a CO2 stimulus or removal of extracellular Ca2+ (Figure 2—figure supplement 1). Surprisingly therefore, the conservative mutation A88V caused Cx26 hemichannels to lose their sensitivity to CO2. As this mutation is far from the residues involved in CO2 binding (K125 and R104), the mechanism for the loss of CO2 sensitivity is unclear.
Figure 2 with 1 supplement see all
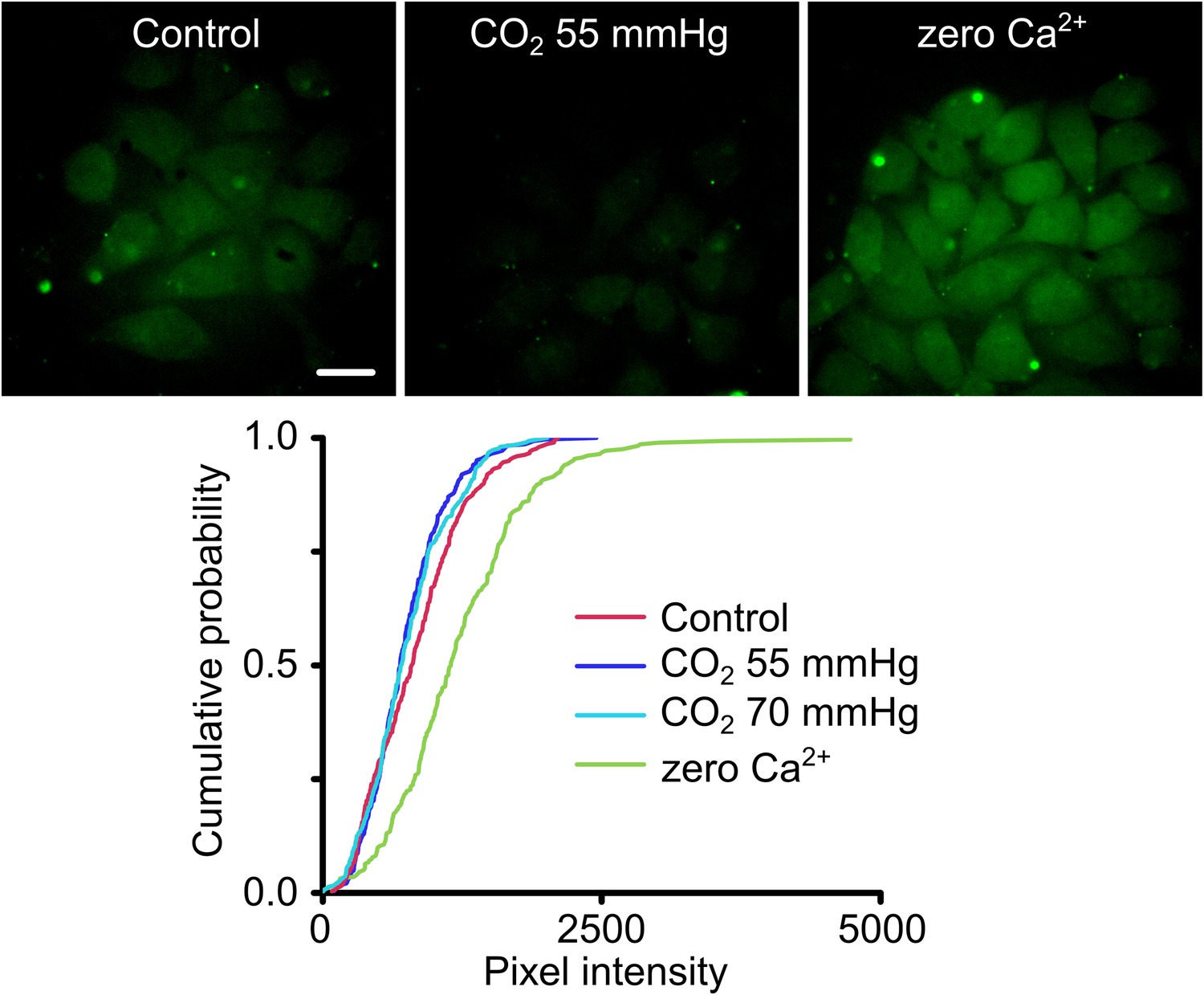
Cx26A88V hemichannels are no longer sensitive to CO2.
(Top) Images of HeLa cells expressing Cx26A88V under control, hypercapnic and zero Ca2+ conditions. The cells were exposed to 200 µM carboxyfluorescein (CBF) for 5 min under each condition before being washed. Some low background loading of CBF is seen under control conditions. In presence of CO2 no loading is seen. The positive control of removal of extracellular Ca2+ causes robust dye loading demonstrating the presence of functional hemichannels. (Bottom) Cumulative probability distributions of pixel intensity of HeLa cells expressing Cx26A88V under control, hypercapnia (two levels of PCO2) and zero Ca2+. Only the removal of extracellular Ca2+ causes dye loading as shown by the rightward shift of the curve to higher pixel intensities (p = 0.004, Mann Whitney U test compared to control). These distributions show all of the measurements made (minimum 40 cells each from five independent repetitions).
As the missense mutations which underlie KID syndrome act in a dominant manner (Jonard et al., 2008; Xu and Nicholson, 2013), we tested whether the expression of Cx26A88V subunits might have a dominant negative action on the CO2 sensitivity of Cx26WT. We transfected HeLa cells that stably expressed Cx26WT with the Cx26A88V subunit and documented their sensitivity to CO2 following transfection. 4 days after transfection with Cx26A88V, the HeLa cells still exhibited sensitivity to CO2 (Figure 3A), but this was reduced compared to the Cx26WT HeLa cells that had not been transfected with Cx26A88V (Figure 3B). 5 and 6 days after transfection, the HeLa cells showed no sensitivity to CO2 (Figure 3A). Nevertheless functional hemichannels were still present as the removal of extracellular Ca2+ caused dye loading (Figure 3A). The loss of CO2 sensitivity was not simply a consequence of days in culture, as Cx26WT HeLa cells that had not been transfected with Cx26A88V retained their sensitivity to CO2 over the whole period examined (Figure 3B). We therefore conclude that Cx26A88V subunits are able to act in a dominant negative manner to cause loss of CO2 sensitivity from wild type Cx26 hemichannels.
Figure 3
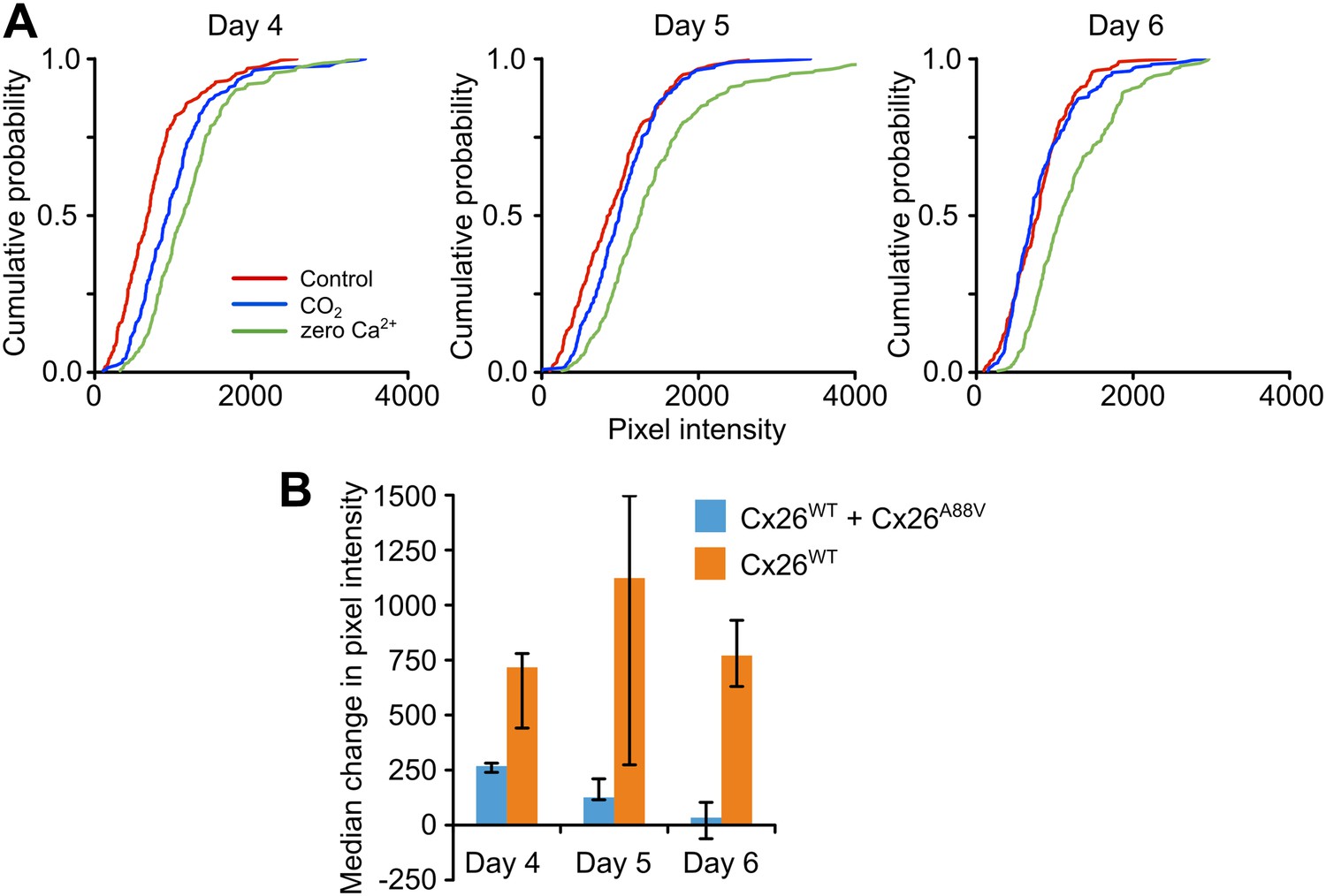
Cx26A88V hemichannels act in a dominant negative manner to remove CO2 sensitivity from Cx26WT.
(A) Cumulative probability distributions for CO2-dependent dye loading in HeLa cells that stably express Cx26WT, which have been transfected with Cx26A88V. 4 days after transfection with Cx26A88V the cells still exhibit significant sensitivity to 55 mmHg PCO2 stimulus (p = 0.048 CO2 compared to control, Mann Whitney U test). 5 and 6 days after transfection the CO2 sensitivity of the HeLa cells was abolished. On all 3 days, the positive control of zero Ca2+ caused dye loading, demonstrating the presence of functional hemichannels. The graphs show all of the measurements made from 5 independent repetitions of the experiment. (B) Comparison of the sensitivity to CO2 of HeLa cells stably expressing Cx26WT which have been transfected with Cx26A88V (Cx26WT + Cx26A88V, n = 5) with those that have not (Cx26WT, n = 7). In the absence of transfection, the Cx26WT-expresssing HeLa cells retain sensitivity to CO2 on all 3 days. By contrast Cx26A88V causes significantly depressed CO2 sensitivity 4 days after transfection (p = 0.001), and loss of sensitivity on days 5 (p = 0.024) and 6 (p = 0.001). Comparisons of Cx26WT with Cx26WT + Cx26A88V via Mann Whitney U test, and False Discovery Rate procedure for multiple comparisons (Curran-Everett, 2000). Error bars upper and lower quartiles.
This is the first instance in which a mutation linked to serious human pathologies has been demonstrated to abolish the CO2 sensitivity of Cx26. This in turn suggests that Cx26-mediated CO2 sensing may be important for human physiology in the range of contexts that are associated with the diverse pathologies linked to this mutation. In the closely related β connexin, connexin30 (Cx30), the mutation A88V connected to Clouston's Syndrome (Bosen et al., 2014), may result in constitutively open Cx30 hemichannels (Essenfelder et al., 2004). However Cx30 is also opened by CO2 (Huckstepp et al., 2010a) and the effect of this mutation on the CO2 sensitivity of Cx30 has not yet been investigated. There are no reports in the literature of disordered breathing in patients with Clouston's syndrome.
Previous studies suggesting that the A88V mutation gave a gain of function in Cx26, examined hemichannel function in the absence of extracellular Ca2+ (Mhaske et al., 2013). As the CO2 sensitivity of the mutated hemichannel was not specifically examined in this previous study, it is likely that both sets of findings are correct—an enhancement of macroscopic hemichannel currents (Mhaske et al., 2013), and a loss of CO2 sensitivity. However under physiological conditions of normal extracellular Ca2+ and in the presence of physiological CO2/HCO3− buffering, we suggest that A88V should be considered as a loss-of-function mutation that effectively removes the capacity for CO2-evoked ATP release via Cx26 hemichannels.
Our report is the first to document altered central respiratory drive in a KID syndrome patient. In rodents, CO2-sensitivity of Cx26 contributes to the chemosensory control of breathing (Huckstepp et al., 2010b; Wenker et al., 2012). Although we do not know if the loss of CO2 sensitivity in Cx26 contributed to the aberrant respiratory drive exhibited by this patient, these results are consistent with this possibility, and represent the first evidence to suggest that Cx26 hemichannels are a requisite component of the drive to breathe in humans. Overall the ability of physiological levels of PCO2 to permit ATP release via Cx26 hemichannels may be important in the epidermis, cochlea and brain. Investigation of whether the absence of this mechanism of ATP release in patients with Cx26A88V contributes to the serious pathological abnormalities that they suffer would seem to be warranted.
Materials and methods
Case study
Request a detailed protocolThe Institutional Review Board of the Connecticut Children's Medical Center considered this under the category of a case report and thus exempt from formal review.
Mutant connexin production
Request a detailed protocolPuc19 Cx26A88V was produced from wild type Cx26 via the Quikchange protocol using the following primers: forward 5′ TGT CCA CGC CGG TCC TCC TGG TAG C 3′ reverse 5′ GCT ACC AGG AGG ACC GGC GTG GAC A 3′. Cx26A88V was subcloned into a pCAG-GS mCherry vector for mammalian cell transfection. Successful mutation of Cx26 was confirmed by sequencing which also verified that apart from the desired mutation the sequence was identical to the wild type.
HeLa cell culture
Request a detailed protocolHeLa cells were cultured by standard methods in DMEM, 10% FCS with addition of 3 mM CaCl2. For experimentation, cells were plated onto coverslips at a density of 5 × 104 cells per well. Transient transfections were performed using the genejuice protocol.
Solutions used
Control aCSF
Request a detailed protocol124 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 10 mM D-glucose, 1 mM MgSO4, 1 mM CaCl2.
Zero Ca2+ aCSF
Request a detailed protocol124 mM NaCl, 26 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 10 mM D-glucose, 1 mM MgSO4, 1 mM MgCl2, 1 mM EGTA.
Hypercapnic (55 mmHg CO2) aCSF
Request a detailed protocol100 mM NaCl, 50 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 10 mM D-glucose, 1 mM MgSO4, 1 mM CaCl2.
Hypercapnic (70 mmHg CO2) aCSF
Request a detailed protocol70 mM NaCl, 80 mM NaHCO3, 1.25 mM NaH2PO4, 3 mM KCl, 10 mM D-glucose, 1 mM MgSO4, 1 mM CaCl2.
Hypercapnic aCSF was saturated with sufficient CO2 (the remaining balance being O2) to adjust the final pH (pH 7.5) to that of the control aCSF removing any potential effects of changes in extracellular pH.
All other solutions were saturated with 95% O2/5% CO2.
Dye loading protocols
Request a detailed protocolCoverslips plated with HeLa cells transiently transfected with Cx26A88V were exposed to Hypercapnic aCSF (55 mmHg or 70 mmHg) containing 200 µM CBF for 10 min. This was followed by control aCSF with 200 µM CBF for 5 min and a 30 min wash with control aCSF to ensure that all dye is removed from the outside of the cells.
A control comparison was used to establish any baseline loading occurring in the absence of a stimulus. HeLa cells expressing Cx26A88V were exposed to 200 µM CBF in control aCSF for 15 min, followed by 30 min of washing.
A zero Ca2+ positive control was also performed to ensure functional connexin hemichannels were being expressed. Cx26A88V expressing HeLa cells were exposed to 200 µM CBF in zero Ca2+ aCSF for 10 min. This was followed by control aCSF with 200 µM CBF for 5 min and 30 min of washing with aCSF.
Imaging and analysis
Request a detailed protocolFor each condition cells were imaged by epifluorescence (Scientifica Slice Scope, Cairn Research OptoLED illumination, 60× water Olympus immersion objective, NA 1.0, Hamamatsu ImageEM EMCCD camera, Metafluor software). Using ImageJ, the extent of dye loading was measured by drawing a region of interest (ROI) around individual cells and calculating the mean pixel intensity for the ROI. The mean pixel intensity of the background fluorescence was also measured in a representative ROI, and this value was subtracted from the measures obtained from the cells. All of the images displayed in the figures reflect this procedure in that the mean intensity of the pixels in a representative background ROI has been subtracted from every pixel of the image. The analysis of the CO2 sensitivity of Cx26A88V was performed as five independent repetitions in which at least 40 cells were measured in each condition, and the mean pixel intensities plotted as cumulative probability distributions.
References
-
Clinical phenotype and mutations in connexin 26 (DFNB1/GJB2), the most common cause of childhood hearing lossAmerican Journal of Medical Genetics 89:130–136.https://doi.org/10.1002/(SICI)1096-8628(19990924)89:3<130::AID-AJMG3>3.0.CO;2-M
-
The vertebrate connexin familyCellular and Molecular Life Sciences 63:1125–1140.https://doi.org/10.1007/s00018-005-5571-8
-
Multiple comparisons: philosophies and illustrationsAmerican Journal of Physiology. Regulatory, Integrative and Comparative Physiology 279:R1–R8.
-
Connexin30 mutations responsible for hidrotic ectodermal dysplasia cause abnormal hemichannel activityHuman Molecular Genetics 13:1703–1714.https://doi.org/10.1093/hmg/ddh191
-
Aberrant hemichannel properties of Cx26 mutations causing skin disease and deafnessAmerican Journal of Physiology. Cell Physiology 293:C337–C345.https://doi.org/10.1152/ajpcell.00626.2006
-
Severe form of keratitis-ichthyosis-deafness (KID) syndrome associated with septic complicationsThe Journal of Dermatology 37:680–682.https://doi.org/10.1111/j.1346-8138.2010.00839.x
-
CO2-dependent opening of connexin 26 and related beta connexinsThe Journal of Physiology 588:3921–3931.https://doi.org/10.1113/jphysiol.2010.192096
-
GJB2 mutations in keratitis-ichthyosis-deafness syndrome including its fatal formAmerican Journal of Medical Genetics. Part A 133A:128–131.https://doi.org/10.1002/ajmg.a.30515
-
A familial case of Keratitis-Ichthyosis-Deafness (KID) syndrome with the GJB2 mutation G45EEuropean Journal of Medical Genetics 51:35–43.https://doi.org/10.1016/j.ejmg.2007.09.005
-
Connexin 26: required for normal auditory functionBrain Research. Brain Research Reviews 32:184–188.https://doi.org/10.1016/S0165-0173(99)00080-6
-
A novel mutation in the connexin 26 gene (GJB2) in a child with clinical and histological features of keratitis-ichthyosis-deafness (KID) syndromeClinical and Experimental Dermatology 36:142–148.https://doi.org/10.1111/j.1365-2230.2010.03936.x
-
The human Cx26-D50A and Cx26-A88V mutations causing keratitis-ichthyosis-deafness syndrome display increased hemichannel activityAmerican Journal of Physiology. Cell Physiology 304:C1150–C1158.https://doi.org/10.1152/ajpcell.00374.2012
-
Connexins: functions without junctionsCurrent Opinion in Cell Biology 16:507–512.https://doi.org/10.1016/j.ceb.2004.07.014
-
Paracrine signaling through plasma membrane hemichannelsBiochimica et Biophysica Acta 1828:35–50.https://doi.org/10.1016/j.bbamem.2012.07.002
-
Regulation of ventral surface CO2/H+-sensitive neurons by purinergic signallingThe Journal of Physiology 590:2137–2150.https://doi.org/10.1113/jphysiol.2012.229666
-
The role of connexins in ear and skin physiology - functional insights from disease-associated mutationsBiochimica et Biophysica Acta 1828:167–178.https://doi.org/10.1016/j.bbamem.2012.06.024
Article and author information
Author details
Funding
Medical Research Council (G1001259)
- Nicholas Dale
The funder had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Ethics
Human subjects: The Institutional Review Board of the Connecticut Children's Medical Center considered this under the category of a case report and thus exempt from formal review.
Copyright
© 2014, Meigh et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,339
- views
-
- 152
- downloads
-
- 48
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 48
- citations for umbrella DOI https://doi.org/10.7554/eLife.04249
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Connexin26 hemichannels with a mutation that causes KID syndrome in humans lack sensitivity to CO2
eLife 3:e04249.
https://doi.org/10.7554/eLife.04249




{kind=link}
{kind=link}
{kind=link}