Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells
- University of Michigan School of Medicine, United States
- Howard Hughes Medical Institute, Johns Hopkins University School of Medicine, United States
- Johns Hopkins University School of Medicine, United States
Abstract
Human SEMAPHORIN 5A (SEMA5A) is an autism susceptibility gene; however, its function in brain development is unknown. In this study, we show that mouse Sema5A negatively regulates synaptogenesis in early, developmentally born, hippocampal dentate granule cells (GCs). Sema5A is strongly expressed by GCs and regulates dendritic spine density in a cell-autonomous manner. In the adult mouse brain, newly born Sema5A−/− GCs show an increase in dendritic spine density and increased AMPA-type synaptic responses. Sema5A signals through PlexinA2 co-expressed by GCs, and the PlexinA2-RasGAP activity is necessary to suppress spinogenesis. Like Sema5A−/− mutants, PlexinA2−/− mice show an increase in GC glutamatergic synapses, and we show that Sema5A and PlexinA2 genetically interact with respect to GC spine phenotypes. Sema5A−/− mice display deficits in social interaction, a hallmark of autism-spectrum-disorders. These experiments identify novel intra-dendritic Sema5A/PlexinA2 interactions that inhibit excitatory synapse formation in developmentally born and adult-born GCs, and they provide support for SEMA5A contributions to autism-spectrum-disorders.
https://doi.org/10.7554/eLife.04390.001eLife digest
Neurons communicate with one another at specialized junctions called synapses. There are two types of synapses, called excitatory synapses and inhibitory synapses, and the density and strength of both are tightly regulated because small deviations from the normal density and/or strength may lead to illness. For example, an excess of excitatory synapses has been observed in patients who have autism spectrum disorders and exhibit difficulties in social interaction.
The gene that codes for a protein called SEMA5A has been identified as an autism susceptibility gene in humans. SEMA5A is a transmembrane protein that regulates the development of connections between neurons, but it is not known how mutations in the gene for SEMA5A might lead to brain illnesses such as autism spectrum disorders.
Now, Duan et al. report that Sema5A selectively inhibits the formation of excitatory synapses in neurons called dentate granule cells in mice. Moreover, Sema5A reduces the frequency and amplitude of the signals that pass through excitatory synapses in certain granule cells. Sema5A directly binds to the receptor PlexA2, a protein that is involved in controlling the density of synapses. In behavioral studies, Sema5A mutant mice displayed altered patterns of social interaction compared to control animals, being less willing to interact with unfamiliar mice.
The presence of increased numbers of excitatory synapses in the brains of Sema5A mutant mice implies that expression of the Sema5A gene normally prevents the formation of too many excitatory synapses. The fact that these animals also show altered social behavior suggests that an excess of synapses—whether as a result of increased synapse formation and/or reduced synapse elimination—can lead to changes in brain circuitry that give rise to patterns of behavior that are characteristic of autism spectrum disorders.
https://doi.org/10.7554/eLife.04390.002Introduction
In the mammalian CNS, most excitatory neurotransmission takes place at spiny synapses, and our understanding of the mechanisms that control the density and strength of excitatory glutamatergic synapses remains incomplete. Many synaptogenic molecules have been identified that can induce pre- or postsynaptic differentiation (Allen and Barres, 2005; Shen and Cowan, 2010; Siddiqui and Craig, 2011; de Wit et al., 2011). Much less is known, however, about molecular players that prevent formation of supernumerary spine synapses (Chung and Barres, 2012; Mironova and Giger, 2013). Precise regulation of CNS synapse density is critical for proper brain function and mental health, and imbalances in excitatory and inhibitory synaptic transmission are associated with neurodevelopmental disorders such as autism-spectrum disorders (ASD) and schizophrenia (Penzes et al., 2011).
The dentate gyrus (DG) is one of two neurogenic areas in the adult mammalian brain (Altman and Bayer, 1990; Ming and Song, 2011). Proper insertion of adult-born granule cells (GCs) into a pre-existing synaptic network offers a unique opportunity to study mechanisms governing axon guidance, dendrite elaboration and formation of synaptic contacts that contribute to neuronal plasticity in mature nervous tissue. Despite recent progress (Tran et al., 2009; Siddiqui et al., 2013; de Wit et al., 2013), our understanding of the molecular programs that regulate GC synaptogenesis is incomplete, including understanding the extent to which the same molecular cues can direct developmentally born and adult-born GCs to establish proper synaptic connectivity (Toni and Sultan, 2011; Kim et al., 2012; Schnell et al., 2012).
One family of extracellular cues known to regulate the morphology of developmentally born GCs is the semaphorins. Semaphorin 6A (Sema6A) and Sema6B inhibit growth of GC axons in vitro and are necessary for laminar targeting of mossy fiber (MF) projections in the CA3 subregion early during postnatal development (Suto et al., 2007; Tawarayama et al., 2010). Further, dendritic elaboration, spine density, and synaptic transmission in GCs are regulated, at least in part, by secreted class 3 semaphorins (Sahay et al., 2005; Tran et al., 2009; Ng et al., 2013).
Sema5A and Sema5B are two closely related transmembrane proteins with an extracellular sema-domain followed by a cluster of seven type-1 thrombospondin repeats (TSRs). The cytoplasmic domains of Sema5A and Sema5B are ∼85 amino acid residues in length and harbor no obvious signaling motifs (Adams et al., 1996; Tran et al., 2009). In vitro, Sema5A functions as an axon guidance molecule for various subclasses of primary neurons (Goldberg et al., 2004; Kantor et al., 2004), and Sema5B regulates the development of synaptic contacts in hippocampal neurons in vitro (O'Connor et al., 2009). In addition, Sema5A and Sema5B together insure proper stratification of murine retinal neuron projections, utilizing both Plxna1 and Plxna3 as receptors (Matsuoka et al., 2011).
Genome wide-association studies identify SEMA5A as an ASD susceptibility gene (Weiss et al., 2009). However, the role of Sema5A in mammalian brain development and physiology has not been addressed in vivo. Here, we find that Sema5A, but not Sema5B, negatively regulates synaptic density in both developmentally born and adult-born dentate GCs. PlexinA2 is a novel receptor for Sema5A, and we show that loss of Sema5A leads to increased excitatory synaptic transmission and ASD-like behavioral phenotypes.
Results
Class 5 semaphorins are expressed in dentate GCs and enriched in the post-synaptic density
Sema5A and Sema5B are expressed in the rodent hippocampus (Simmons et al., 1998; O'Connor et al., 2009). To augment these data, we conducted a detailed analysis of Sema5A expression in the postnatal hippocampus and entorhinal cortex (EC) using a Sema5A reporter mouse expressing nuclear lacZ (Gunn et al., 2011). In the P18 and P30 hippocampus, strong β-gal activity is observed in the granule cell layer (GCL), the hilus and in CA3 pyramidal neurons. Moderate-to-strong β-gal activity is also observed in deep EC layers (Figure 1A), however more superficial EC layers, including layer II/III neurons that give rise to the perforant path, do not show Sema5A promoter activity. In situ hybridization for Sema5A revealed a similar expression pattern (Figure 1B). Sema5B expression in the postnatal hippocampus is largely confined to the DG subgranular zone (SGZ). Subcellular fractionation of hippocampal tissue revealed that Sema5A protein is present in synaptic and extra-synaptic membrane fractions and is enriched in detergent-resistant post-synaptic density (PSD) fractions. Sema5B is enriched postsynaptically and only found in the PSDIII fraction (Figure 1C).
Figure 1 with 2 supplements see all
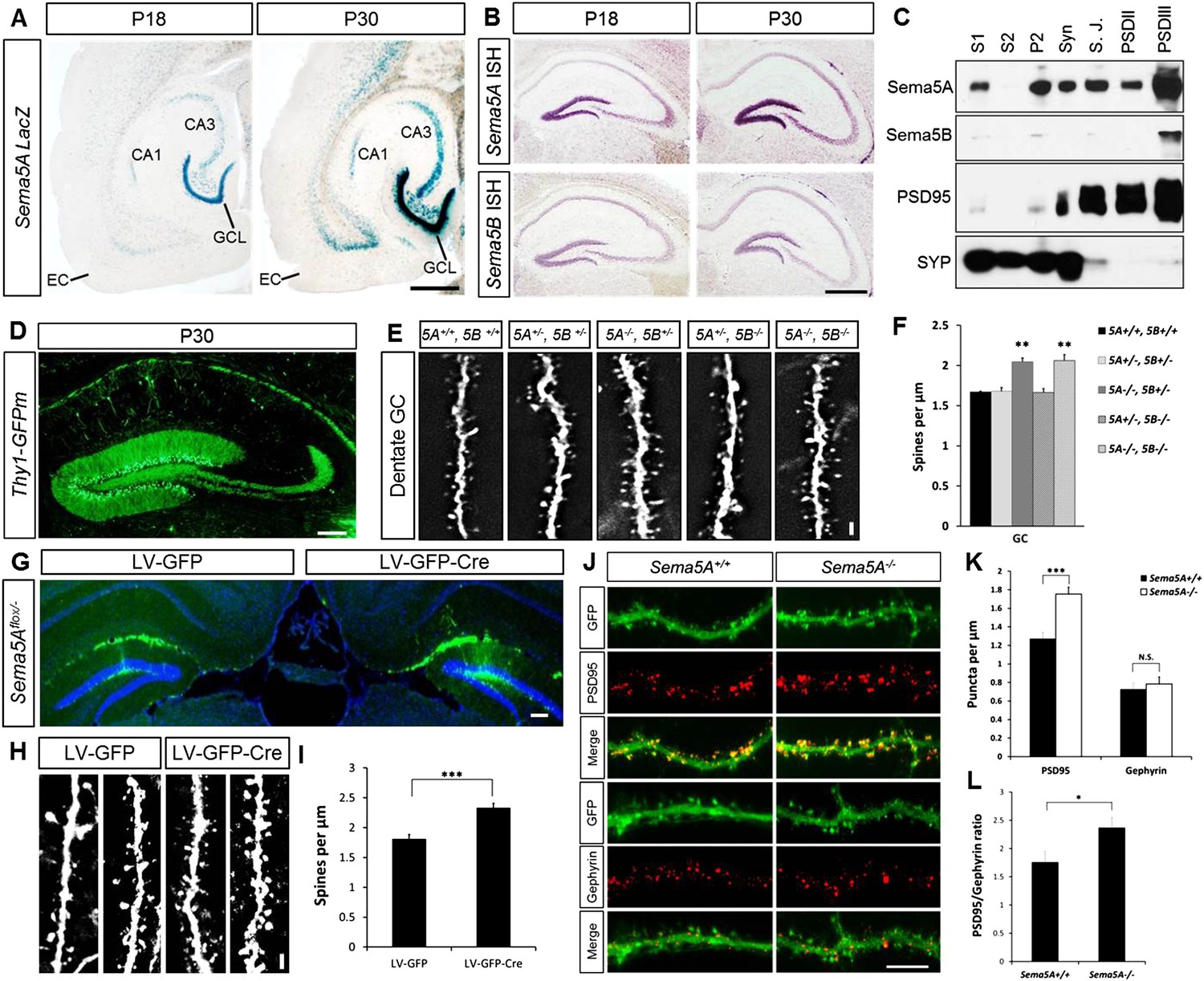
Sema5A, but not Sema5B, negatively regulates dendritic spine density of hippocampal GCs in vivo.
(A) Horizontal sections through the hippocampus of Sema5ALacZ/+ brains show robust β-gal activity in the GCL at P18 and P30. Weaker labeling is observed in the pyramidal cell layer of CA3 and a small segment of CA1. In the EC, labeling is confined to deep cortical layers. (B) In situ hybridization of P18 and P30 coronal sections with probes specific for Sema5A and Sema5B. (C) Western blot analysis of synaptic density fractions prepared from P18 mouse hippocampus. S1, homogenate; S2, cytosolic fraction; P2, membrane fraction; Syn, synaptosomal fraction; S.J., synaptic junction, and PSD purified postsynaptic fractions. Anti-PSD95 and anti-Synaptophysin (SYP) are shown as post- and pre-synaptic markers. (D) Coronal section of the P33 Thy1-GFPm hippocampus showing labeling of GCs. (E) Representative images of GFP-positive dendrites of GCs of WT (5A+/+, 5B+/+), Sema5A+/−, Sema5B+/− (5A+/−, 5B+/−); Sema5A−/−, Sema5B+/− (5A−/−, 5B+/−); Sema5A+/−, Sema5B−/− (5A+/−, 5B−/−) and double mutant (5A−/−, 5B−/−) mice. (F) Quantification of dendritic spine density of GCs shown in E. Values are represented as mean ± SEM from 3 to 4 mice per genotype (for details on spine quantification see Table 1). **indicates p < 0.01, two-tailed unpaired Student's t test. (G) Representative image of the DG following stereotaxic injection of LV-GFP (left side) or LV-syn-GFP-IRES-Cre (right side) viral vector into Sema5Aflox/− mice. (H) High magnification images of LV transduced GC dendrites. (I) Quantification of spine density of GFP+ GC dendrites. Values are represented as mean ± SEM from three independent mice per condition. (J) Cultured mouse hippocampal neurons at DIV21 obtained from Sema5A+/+ and Sema5A−/− pups. Cultures were transfected at DIV4 with a GFP expression construct and fixed at DIV21. GCs were identified by anti-Prox1 labeling (data not shown). Cultures were stained with anti-PSD95 or anti-gephyrin to identify excitatory synapses confined to dendritic spines, and inhibitory synapses confined to the dendritic shaft of GCs. (K) Quantification of PSD95 positive puncta reveals a significant increase in Sema5A−/− GCs. No significant (N.S.) difference in gephyrin positive puncta was observed between Sema5A+/+ and Sema5A−/− GCs. (L) The ratio of excitatory/inhibitory synapses is significantly increased in Sema5A−/− GCs. Number of neurons quantified: n = 32–34 neurons per condition from three mice per genotype. Values are represented as mean ± SEM. ***p < 0.001; *p < 0.05 two-tailed unpaired Student's t test. Scale bars, A and B = 500 µm, D and G = 200 µm, J = 10 µm, E and H = 1 µm.
Analysis of Sema5A and Sema5B null mutants (Matsuoka et al., 2011) at the macroscopic level revealed no obvious defects in the hippocampal formation (Figure 1—figure supplement 1). GCs in compound heterozygotes and null mutants were analyzed using anti-doublecortin, anti-calretinin, and anti-calbindin labeling. No defects were identified using these markers, suggesting that Sema5A and Sema5B are dispensable for GC maturation and for general patterning of GC axonal and dendritic processes in vivo.
Loss of Sema5A, but not Sema5B, increases GC dendritic spine density
We next examined dendritic spine distribution in P30–33 hippocampal neurons in Sema5;Thy1-GFPm reporter mice, analyzing dendritic spines in the middle and outer thirds of supra-pyramidal GC dendrites within the rostral hippocampus (Figure 1D). We also quantified spine density along apical dendrites of hippocampal CA1 pyramidal neurons. Spine density in WT GCs (1.67 ± 0.01 spines/µm), Sema5A+/−;Sema5B+/− (1.68 ± 0.05 spines/µm), and Sema5A+/−;Sema5B−/− mice (1.66 ± 0.05 spines/µm) is similar. However, GC spine density in Sema5A−/−;Sema5B+/− mice (2.05 ± 0.05 spines/µm) is increased by 23 ± 3% compared to WT, and no further increase is observed in Sema5A−/−;Sema5B−/− double-mutants (2.06 ± 0.08 spines/µm) (Figure 1E,F, Table 1). Spine density along CA1 pyramidal neuron primary and secondary apical dendrites is not altered by the loss of Sema5s (Figure 1—figure supplement 2 and Table 1). To conditionally ablate Sema5A in P15 GCs, neuron-specific recombination in Sema5Aflox/− mice was achieved by stereotaxic injection of a Cre recombinase-expressing lentiviral vector. Similar to Sema5A germline null mice, conditional Sema5A ablation in GCs leads to increased spine density (Figures 1G-I). The vast majority of excitatory inputs onto GCs and pyramidal neurons form on dendritic spines (Harris and Kater, 1994; Trommald and Hulleberg, 1997). Our results suggest that Sema5A inhibits the formation of excitatory synapses on GCs. To assess the density of excitatory and inhibitory GC synapses, Sema5A WT and null neurons were cultured and labeled with anti-PSD95 and anti-gephyrin (Figure 1J–L). We found that Sema5A−/− GCs exhibit a selective increase in excitatory, but not inhibitory, synapses.
Table 1
Quantification of dendritic spine density in the hippocampus of Thy1-eGFP WT, Sema5 and Plxna mice
| Genotype | Age | Dentate GC dendrite | CA1 primary dendrite | CA1 secondary dendrite | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N | p Value (vs wt) | Spine density (mean ± SEM) | Distance from soma | n | p Value (vs wt) | Spine density (mean ± SEM) | Distance from soma | n | p Value (vs wt) | Spine density (mean ± SEM) | Distance from soma | ||
| WT | P30–33 | 4 animals (74 neurons) | 1.67 ± 0.01 | 50–100 μm | 3 animals (26 neurons) | 2.76 ± 0.07 | 50–100 μm | 3 animals (15 neurons) | 2.93 ± 0.02 | 50–100 μm | |||
| Sema5A+/−; Sema5B+/− | P30–33 | 3 animals (41 neurons) | 0.8795 | 1.68 ± 0.05 | 50–100 μm | 3 animals (24 neurons) | 0.6549 | 2.79 ± 0.019 | 50–100 μm | 3 animals (24 neurons) | 0.6333 | 2.99 ± 0.09 | 50–100 μm |
| Sema5A−/−; Sema5B+/− | P30–33 | 4 animals (65 neurons) | 0.0012 | 2.05 ± 0.05 | 50–100 μm | 3 animals (27 neurons) | 0.5122 | 2.81 ± 0.03 | 50–100 μm | 3 animals (21 neurons) | 0.618 | 2.99 ± 0.09 | 50–100 μm |
| Sema5A+/−; Sema5B−/− | P30–33 | 3 animals (46 neurons) | 0.8844 | 1.66 ± 0.05 | 50–100 μm | 3 animals (26 neurons) | 0.9797 | 2.76 ± 0.15 | 50–100 μm | 3 animals (21 neurons) | 0.9661 | 2.94 ± 0.02 | 50–100 μm |
| Sema5A−/−; Sema5B−/− | P30–33 | 3 animals (30 neurons) | 0.0066 | 2.06 ± 0.08 | 50–100 μm | 3 animals (27 neurons) | 0.5389 | 2.81 ± 0.02 | 50–100 μm | 3 animals (23 neurons) | 0.4006 | 2.98 ± 0.03 | 50–100 μm |
| Plxna1−/− | P30–33 | 3 animals (56 neurons) | 0.154 | 1.79 ± 0.05 | 50–100 μm | ||||||||
| Plxna2−/− | P30–33 | 3 animals (54 neurons) | <0.0001 | 2.24 ± 0.07 | 50–100 μm | ||||||||
| Plxna2+/− | P30–33 | 4 animals (74 neurons) | 0.0359 | 1.86 ± 0.05 | 50–100 μm | ||||||||
| Sema5A+/−; Plxna2+/− | P30–33 | 4 animals (72 neurons) | <0.0001 | 2.39 ± 0.05 | 50–100 μm | ||||||||
Loss of Sema5A negatively regulates dendritic spine density and alters synaptic transmission in adult-born GCs
We next used retrovirus-mediated birth-dating to permanently label newly born DG neurons in the adult mouse (Ge et al., 2006). We injected an onco-retroviral vector harboring a GFP transgene into the hilus of 6- to 8-week-old mice, labeling proliferating neural progenitors (Figure 2A–F). GC dendritic spine density was assessed 19–21 days after injection, the peak time for the formation of glutamatergic synaptic inputs onto adult-born GCs (Toni et al., 2007). At this developmental time point, adult-born GCs have spiny dendrites that reach the outer ML and begin acquiring excitatory glutamatergic input from the EC (Esposito et al., 2005; Zhao et al., 2006). In WT mice, the dendritic spine density of adult-born WT GCs (0.96 ± 0.07 spines/µm) was lower than that of fully mature WT GCs (1.67 ± 0.01 spines/µm). However, in Sema5A−/− mice, spine density of adult-born GCs was significantly increased (1.65 ± 0.08 spines/µm) and comparable to mature WT GCs (1.67 ± 0.01 spines/µm) (Figure 2G). This shows that Sema5A negatively regulates dendritic spine development in adult-born GCs in vivo.
Figure 2
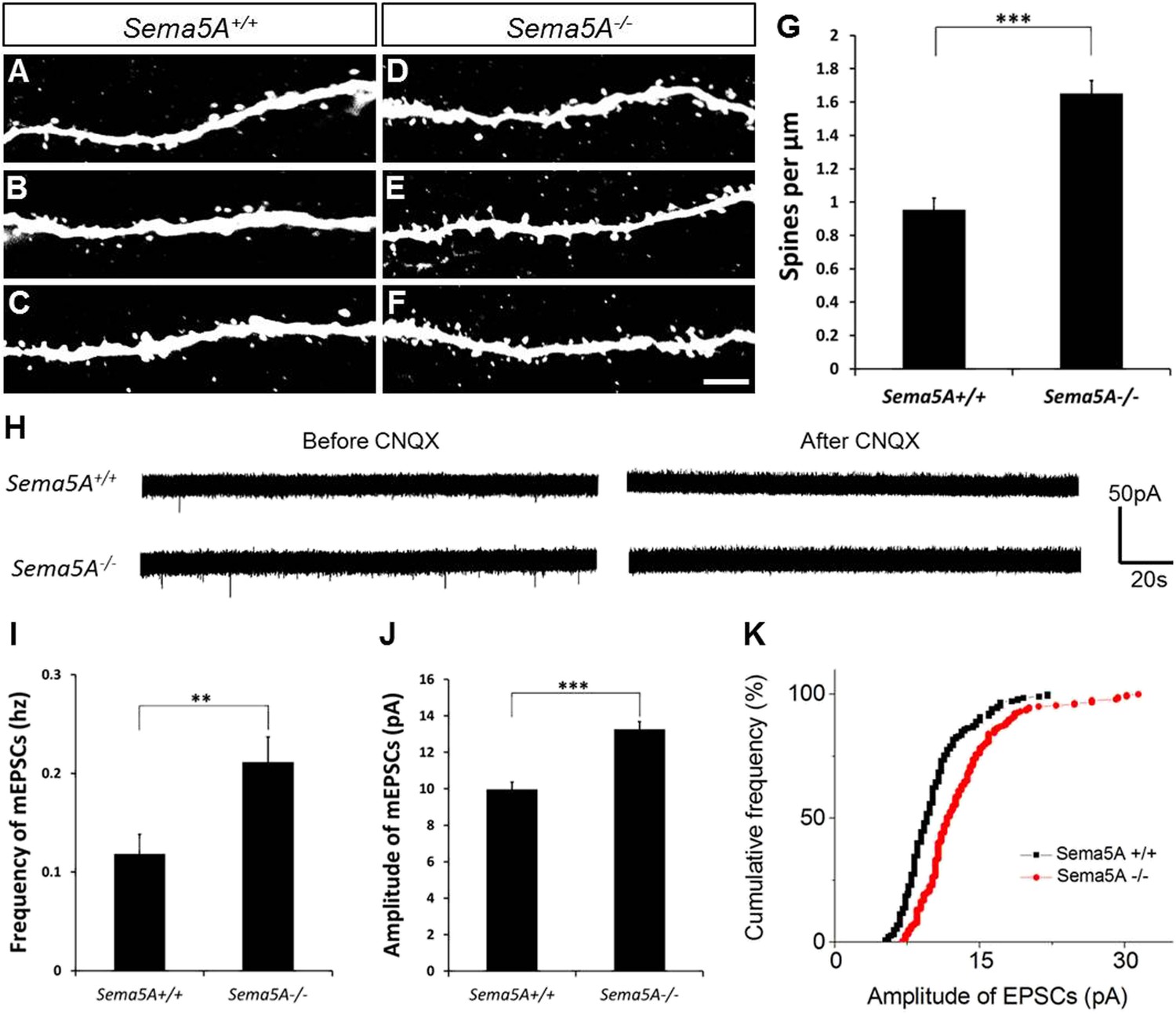
In adult-born GCs, Sema5A negatively regulates dendritic spine density and affects their electrophysiological properties.
(A–F) Representative images of dendritic segments of adult-born, 19- to 21-day-old retrovirally-labeled GCs in 3-month-old Sema5A+/+ and Sema5A−/− mice. (G) Quantification of dendritic spine density in adult-born GCs of Sema5A−/− (n = 34 neurons) and Sema5A+/+ (n = 35 neurons) mice. Error bars represent ± SEM, n = 3 independent animals per genotype. ***indicates p < 0.0001, two-tailed unpaired Student's t test. Scale bar = 5 µm. (H–K) Recordings of spontaneous mEPSCs from retrovirally-labeled adult-born GCs at 19–21 days postmitosis. (H) Shown are sample voltage-clamp whole-cell recording traces (Vm = −65 mV) from Sema5A+/+ (top) and Sema5A−/− (bottom) animals before and after bath application of CNQX (20 μM). (I) Quantification of AMPA-type mEPSC mean frequency and (J) mean amplitude. (K) Relative cumulative frequency distribution of mEPSC amplitudes in Sema5A+/+ (black) and Sema5A−/− (red) GCs. The distribution of mEPSC amplitudes in mutants is significantly shifted to the right, indicating an increase in the proportion of larger amplitude events. Recordings were performed in the presence of TTX (1 μM) and bicuculline (10 μM). Values are represented as mean ± SEM. Sema5A+/+, n = 13 cells/6 mice and Sema5A−/−, n = 13 cells/6 mice. **p < 0.01, ***p < 0.001, Student's t test.
Glutamatergic inputs onto adult-born GCs appear between 14 and 28 days post mitosis (Esposito et al., 2005; Ge et al., 2006), and we next performed whole-cell patch clamp recordings in acute hippocampal slices prepared from Sema5A−/− mice and control littermates (Figure 2H–K). To identify adult-born GCs, and also to ensure that recordings were from neurons at the same postmitotic stage, we combined electrophysiological recordings with retrovirus-mediated birth-dating. Analysis of the frequency and amplitude of miniature excitatory postsynaptic currents (mEPSCs) in 19- to 21-day postmitotic GCs of 9- to 11-week-old mice revealed a significant increase in mEPSC frequency and amplitude in Sema5A−/− hippocampal slices compared to WT slices (Figure 2I–K). Thus, in Sema5A−/− mice, increased GC spine density (Figure 1E) is correlated with an increase in mEPSC frequency, suggesting that supernumerary spines receive presynaptic input and are electrically active. Moreover, since these mEPSCs are fully blocked in the presence of CNQX, this suggests that loss of Sema5A leads to increased AMPA receptor responses.
Sema5A cell-autonomously inhibits GC dendritic spine density
The expression of Sema5A in dentate GCs, but not EC superficial layers, coupled with our observation that conditional ablation of Sema5A in the DGL increases dendritic spine density, strongly suggest that Sema5A functions in a GC-autonomous manner. To address whether Sema5A influences spine density through interactions in trans between neighboring GCs or in cis within the same cell, we employed primary hippocampal neurons stained for the GC marker prospero homeobox protein 1 (prox1) (Elliott et al., 2001) (Figure 3). At DIV21, Sema5A−/− GCs show a significant (33.5%) increase in spine density (2.23 ± 0.08 spines/µm) as compared to WT controls (1.67 ± 0.04 spines/µm). To ask whether the supernumerary GC spine phenotype is rescued by recombinant Sema5A, neuronal cultures were transfected at DIV4 and analyzed at DIV21 (Figure 3A). Because only a small fraction (∼1%) of the neurons in culture is transfected, and since spine density was assessed in isolated, prox1+, neurons whose processes did not overlap with any other Sema5A-transfected neurons, trans intercellular Sema5A interactions do not figure in this analysis. Recombinant Sema5A was present on dendrites and cell bodies (Figure 3—figure supplement 1A,B) and decreased GC spine density by 34% in Sema5A−/− (1.10 ± 0.04 spines/µm) and by 29% in Sema5A+/+ (1.18 ± 0.04 spines/µm) hippocampal cultures (Figure 3A,B). This suggests that a Sema5A cis interaction cell-autonomously constrains the formation of supernumerary GC spines. In these same cultures, expression of recombinant Sema5A in pyramidal neurons leads to a reduction in spine density (Figure 3—figure supplement 2). To examine whether or not Sema5A serves as a receptor (Pasterkamp, 2012), we expressed a Sema5A deletion mutant lacking its cytoplasmic domain (S5AΔcyto) and asked if this altered Sema5A protein was sufficient to rescue the Sema5A−/− GC spine phenotype. Similar to full-length Sema5A (1.10 ± 0.04 spines/µm), S5AΔcyto rescues the Sema5A−/− spine phenotype (1.09 ± 0.03 spines/µm) (Figure 3A,B). We next generated Sema5A deletion constructs lacking either the sema-domain (S5AΔSema) or the 7 TSRs (S5AΔTSR), confirmed that they were appropriately expressed (data not shown), and found that S5AΔSema (1.94 ± 0.05 spines/µm), but not S5AΔTSR (1.11 ± 0.05 spines/µm), fails to rescue the Sema5A−/− GC spine phenotype (Figure 3A,B). These results show that the sema-domain of Sema5A is necessary and sufficient to regulate GC dendritic spine density. Further, they suggest that Sema5A functions as a ligand or co-receptor that interacts in cis with a transmembrane-spanning signal transducing receptor to constrain GC spine formation. Independent evidence that Sema5A functions as a ligand stems from experiments in which we bath applied soluble Sema5A-Fc fusion protein to DIV21 GCs. Sema5A-Fc, but not control IgG protein, results in a rapid loss of dendritic spines (Figure 3C,D).
Figure 3 with 2 supplements see all
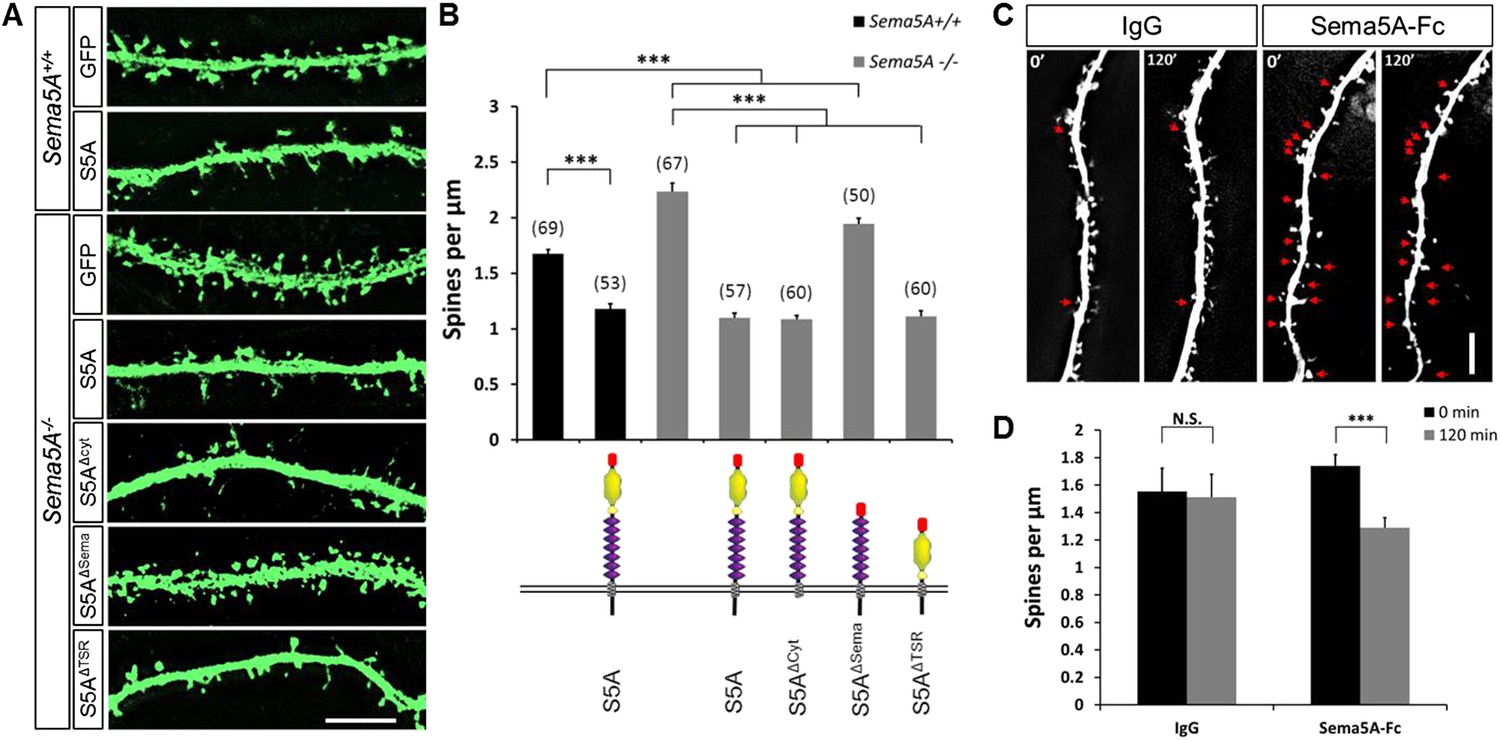
Sema5A inhibits dendritic spine density in primary dentate GCs.
(A) High power deconvolution images of dendritic segments of GCs from WT (Sema5A+/+) and mutant (Sema5A−/−) hippocampal cultures transfected either with eGFP only, or eGFP plus full-length Sema5A (S5A), the Sema5A deletion constructs S5AΔcyt, S5AΔSema, or S5AΔTSR plasmid DNA. (B) Quantification of dendritic spine density of DIV21 prox1+ GCs. Numbers in brackets indicate the number of cells analyzed per condition. (C) Time-lapse study of Sema5A-Fc induced GC dendritic spine loss. DIV21 dentate cultures were treated for 120 min with Sema5A-Fc or control IgG. (D) Quantification of dendritic spine density in prox1+ GCs. Values are represented as mean ± SEM from 3 to 4 cultures, established from 3 to 4 different animals for each condition. ***p < 0.001, two-tailed unpaired Student's t test. Scale bars in A and C = 10 µm.
Sema5A binds directly to PlexA1 and PlexA2 with high affinity
We next assessed Sema5A interactions with PlexA family members since class 5 semas are known to utilize class A plexins as receptors in vivo (Matsuoka et al., 2011). Sema5A binds directly and with low-nanomolar affinity to PlexinA1 and PlexinA2 ectopically expressed in COS7 cells, and the sema-domain of Sema5A binds to the sema-domains of PlexA1 and PlexA2 (Figure 4A–C, Figure 4—figure supplement 1). We next assessed hippocampal neurons from Plxna1, Plxna2, and Plxna3 homozygous mutant pups for their ability to support Sema5A-Fc binding. Sema5A-Fc bound strongly to WT and Plxna3−/− hippocampal cultures, however, binding to Plxna1−/− and Plxna2−/− neurons was greatly reduced, and virtually no binding was observed to Plxna1−/−;Plxna2−/− double mutant neurons. In the same assay, binding of an unrelated Fc-fusion protein, NgROMNI-Fc (Robak et al., 2009), is not influenced by the loss of Plxna family members (Figure 4D–F). Therefore, PlexA1 and PlexA2 are the main Sema5A-Fc binding partners in cultured hippocampal neurons. Conversely, PlexA2sema-Fc binds to neurites of WT hippocampal neurons, but not to Sema5A−/− neurons, showing that endogenous Sema5A is localized at the neuronal cell surface and that it is also present on neurites (Figure 4—figure supplement 2).
Figure 4 with 2 supplements see all
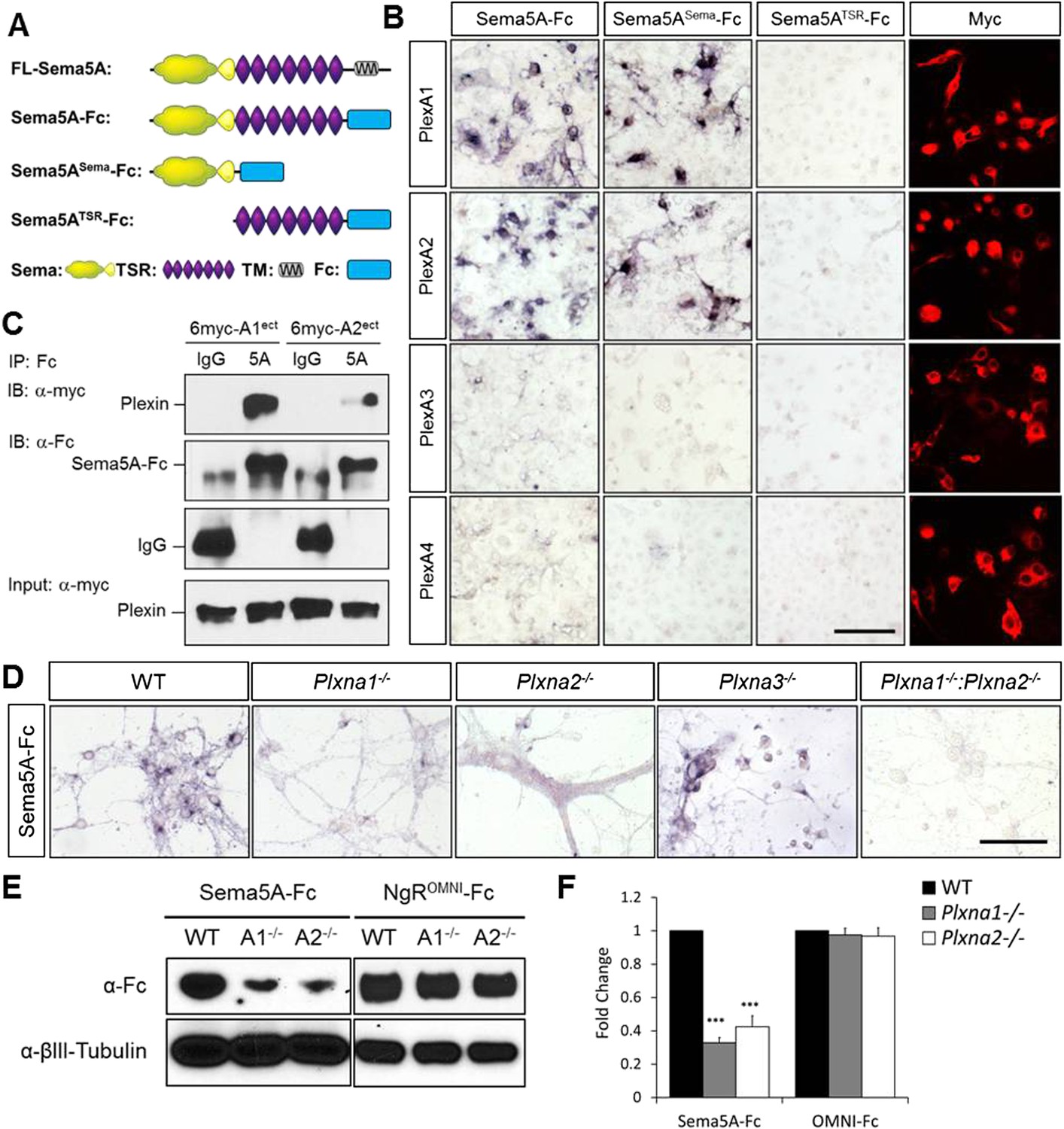
Direct binding of Sema5A to PlexA1 and PlexA2.
(A) Schematic diagrams of Sema5A domain deletion constructs used for binding studies. (B) Binding of different Sema5A-Fc fusion proteins to COS7 cells transiently expressing myc-tagged PlexA1, PlexA2, PlexA3, or PlexA4. Sema5A-Fc and Sema5ASema-Fc bind selectively to PlexA1 and PlexA2. PlexA expression was confirmed by anti-myc immunostaining. (C) Co-precipitation of purified ectodomain of Sema5A (Sema5A-Fc) with the ectodomains of PlexA1 (6x-myc-PlexA1ect) or PlexA2 (6x-myc-PlexA2ect). Immunoprecipitation (IP) with anti-Fc shows that Sema5A-Fc (5A), but not control IgG, forms a complex with the ectodomains of PlexA1 and PlexA2. The precipitated plexin fragments can be detected by anti-myc immunoblotting (top blot). Immunoblotting with an antibody specific for the Fc fragment detects Sema5A-Fc and control IgG in the precipitate (second blot from top). Note that the higher molecular weight band is Sema5A-Fc and the lower molecular weight band is intact (not fully reduced ∼150-kDa) IgG. Anti-IgG immunoblotting shows that control IgG at 50-kDa was successfully pulled down with protein A/G agarose-beads (third blot from top). To demonstrate that equal amounts of plexinA ectodomain were used for the IP experiment, an aliquot of the input was probed with anti-myc (blot at bottom). (D) Binding of Sema5A-Fc to primary hippocampal neurons established from WT, Plxna1, Plxna2 and Plxna3 single null mice as well as Plxna1; Plxna2 double null mice. (E) Hippocampal neurons from WT, Plxna1−/−, and Plxna2−/− mice were incubated with either Sema5A-Fc (50 nM) or NgROMNI-Fc (50 nM) for 1 hr. Cells were rinsed extensively, lysed, and subjected to WB analysis. Sema5A-Fc binding to Plxna1−/− and Plxna2−/− neurons is reduced compared to WT neurons. NgROMNI-Fc binding was strong to hippocampal neurons and independent of the Plxna genotype. Anti-βIII tubulin in lysates is shown as a loading control. (F) Quantification of Western blot signals from three independent experiments. Scale bars in B and D = 50 µm.
PlexA2, but not PlexA1, is strongly expressed in the postnatal dentate GCL
We next explored which PlexA family members serve as functional receptors for Sema5A in dentate GCs. We analyzed Plxna1, Plxna2, and Plxna3 gene expression in the hippocampus by in situ hybridization at P7 and P30 (Figure 5A) since the peak period of synaptogenesis between EC and GC neurons occurs between postnatal week two and four (Esposito et al., 2005). Plxna2, but not Plxna1, transcripts are abundantly found in the DG before the onset (P7) and toward the end (P30) of early postnatal GC synaptogenesis (Figure 5A). Plxna3 expression in the DG is quite robust at P7, and at P30 it is largely confined to the SGZ, suggesting higher expression in immature GCs (Figure 5A). In hippocampal lysates, PlexA2 protein is most abundant during early postnatal synaptogenesis and continues to be present, but at a lower level, in adulthood (Figure 5B).
Figure 5 with 3 supplements see all
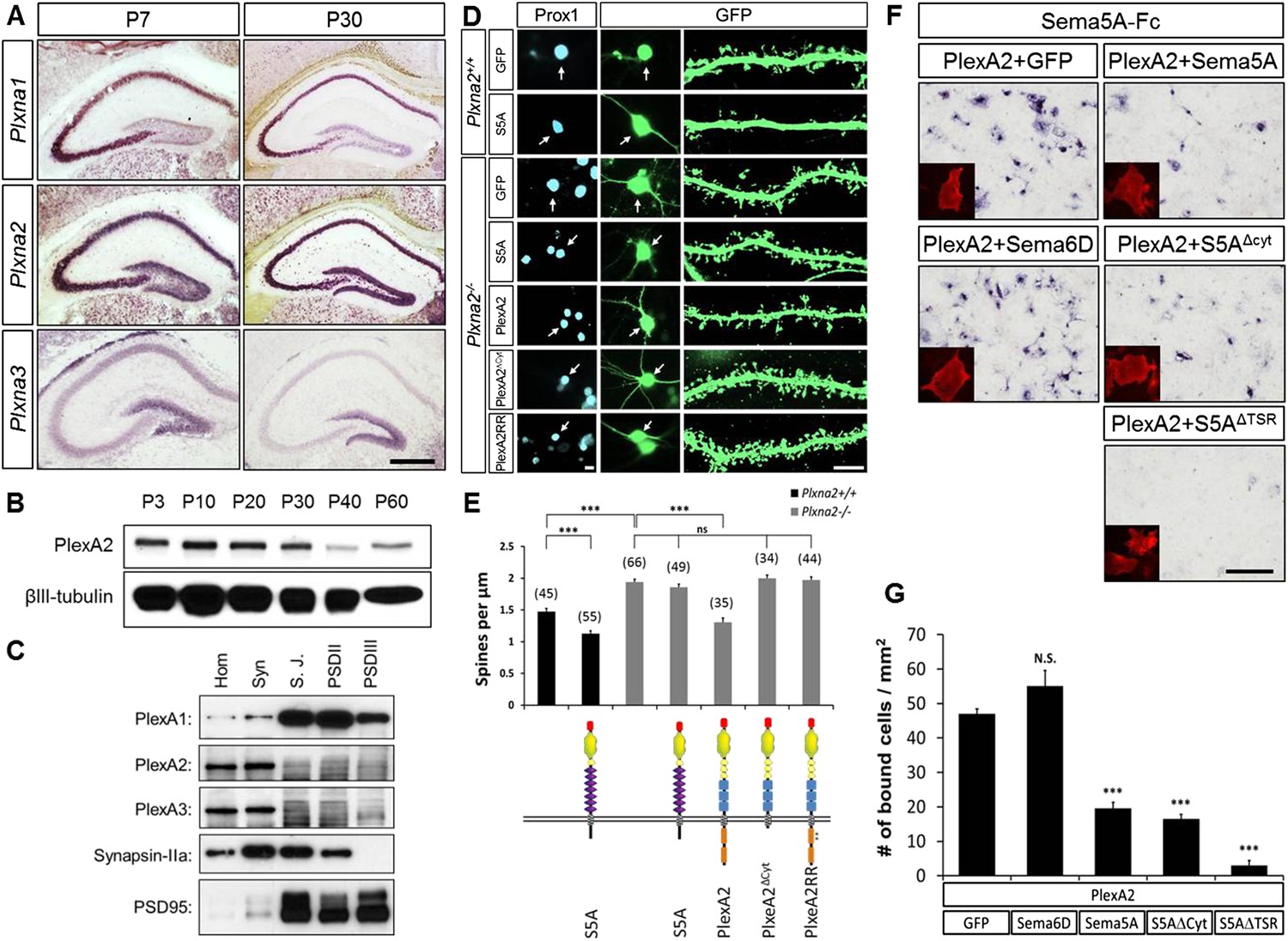
In dentate GCs, PlexA2 is necessary for Sema5A-mediated regulation of dendritic spine density.
(A) Coronal sections of P7 and P30 mouse hippocampus labeled with riboprobes specific for Plxna1, Plxna2, and Plxna3 transcripts. In the DG, strong expression of Plxna2 is observed, while Plxna1 is virtually absent. Plxna3 mRNA expression is largely confined to more immature GCs located near the inner surface of the GCL. (B) Developmental time course of PlexA2 protein expression in the mouse hippocampus between P3 and P60, normalized to βIII-tubulin. (C) Distribution of PlexA1, PlexA2, and PlexA3 in synaptic density fractions prepared from P23 mouse hippocampus. The three PlexA family members are present in the synaptosomal (syn) fraction. PlexA1, but not PlexA2 or PlexA3, is enriched in the synaptic junction (S.J.) and postsynaptic density (PSD) fractions. As controls, markers enriched in postsynaptic fractions (anti-PSD95) and presynaptic fractions (anti-Synapsin-IIa) are shown. n = 4 independent experiments. (D) Representative images of DIV21 GCs from Plxna2+/+ and Plxna2−/− mice transfected with GFP alone or GFP plus myc-tagged FL-Sema5A (S5A), FL-PlexA2 (PlexA2), PlexA2Δcyt, or PlexA2RR expression constructs. Cultures were fixed and stained with anti-Prox1 (blue) and anti-GFP (green). The cell bodies of GFP-expressing GCs are labeled with an arrow and a dendritic segment of the same neuron is shown. (E) Quantification of spine density of GCs shown in panel D. WT neurons (black bars) and Plxna2−/− mutant neurons (grey bars). Numbers in brackets indicate GCs analyzed per condition. Values are represented as mean ± SEM from three animals for each condition. (F) Cis interaction between Sema5A and PlexinA2. Membrane-bound Sema5A presented in cis, and soluble Sema5A-Fc presented in trans, compete for binding to PlexinA2. Sema5A-Fc binding to COS7 cells transiently transfected to co-express PlexA2 and GFP, PlexA2 and Sema6D, PlexA2 and Sema5A, PlexA2 and Sema5A lacking the cytoplasmic domain (S5AΔcyt), or PlexA2 and Sema5A lacking the seven thrombospondin type-1 domains (S5AΔTSR). Prior to binding, Sema5A-Fc was super-clustered with anti-Fc conjugated to human placental alkaline phosphatase (Robak et al., 2009). Surface expression of recombinant PlexA2 was confirmed in a parallel experiment by immunofluorescence labeling of its N-terminal myc epitope (small inserts). (G) Quantification of Sema5A-Fc bound to COS7 cells co-expressing PlexA2 together with GFP, Sema6D, Sema5A, S5AΔcyt, or S5AΔTSR. Cis expression of Sema5A constructs that harbor the sema domain significantly attenuates the binding of bath applied Sema5A-Fc to PlexA2. Error bars represent SEM from three independent experiments. ***indicates p < 0.0001. Two-tailed unpaired Student's t test. Scale bar in A = 50 µm.
PlexA1, PlexA2, and PlexA3 are all found in hippocampal homogenates; however, the subcellular distribution patterns of these proteins in P23–P26 synaptic density fractions are quite distinct (Figure 5C). PlexA1, but not PlexA2 or PlexA3, is enriched in the synaptic junction (SJ) and detergent-resistant postsynaptic density (PSD) fractions. PlexA2 and PlexA3 are present in synaptosomes (syn) but are not enriched in the SJ or PSD fractions. This suggests PlexA2 and PlexA3 are preferentially localized at extra-synaptic sites, including the dendritic shaft and spine neck, while PlexA1 is enriched in the PSD near the active zone. Therefore, Sema5A and PlexinA2 are both expressed by GCs and show overlapping distributions in synaptosomal density fractions. Plxna2 in situ hybridization on adult Sema5AlacZ reporter brains followed by anti-β-galactosidase staining revealed double-labeled cells (Figure 5—figure supplement 1). This suggests that Sema5A and PlexA2 are co-expressed by GCs.
PlexA2 is necessary for Sema5A-mediated regulation of dentate GC dendritic spine density
To investigate whether PlexA2 is a functional Sema5A receptor, we analyzed spine density in cultured Plxna2−/− GCs at DIV21 and found a significant increase (1.94 ± 0.04 spines/µm) compared to WT controls (1.47 ± 0.05 spines/µm). This increase in Plxna2−/− GC spine density is similar to that observed in DIV21 Sema5A−/− GCs (2.23 ± 0.08 spines/µm) (Figure 5D,E, Table 1). Further, transfection of WT GCs with full-length Sema5A leads to a significant decrease in spine density (1.13 ± 0.05 spines/µm) (Figures 3B and 5E). However, Plxna2−/− GCs are resistant to the reduction in spine density caused by Sema5A overexpression (1.88 ± 0.05 spines/µm), showing that Plxna2 is indeed required for Sema5A-mediated regulation of GC spine density (Figure 5D,E). Since we quantified spine density only in isolated prox1+ neurons that overexpress Sema5A and do not contact any nearby Sema5A-overexpressing neurons, these observations strongly suggest that Sema5A functions in cis to regulate dendritic spine density. To directly show that the PlexA2 cytoplasmic domain is required to inhibit GC spine density, Plxna2−/− cultures were transfected with full-length PlexA2 or PlexA2Δcyt, which lacks the cytoplasmic domain. Though expression of full-length PlexA2 is sufficient to reduce spine density (1.38 ± 0.10 spines/µm) in Plxna2−/− GCs, no significant reduction was observed following transfection with PlexA2Δcyt (2.03 ± 0.09 spines/µm) (Figure 5D,E).
We next addressed the role played by the PlexA2 RasGAP domain in the regulation of dendritic spine density since this plexin cytoplasmic domain has been shown to regulate the cytoskeleton during semaphorin signaling (Rohm et al., 2000; He et al., 2009; Pasterkamp, 2012). We generated a point-mutation in the PlexA2 cytoplasmic domain that abolishes its RasGAP activity (PlexA2RR) but not Sema5A-Fc binding when expressed in COS7 cells. The PlexA2 rasGAP activity is required for the ‘collapse’ of COS7 cells (Figure 5—figure supplement 2). In primary GCs, PlexA2RR fails to rescue the spine phenotype observed in Plxna2−/−; Prox1+ GCs (1.97 ± 0.05 spines/µm) (Figure 5D,E), suggesting that the RasGAP activity directly participates in PlexA2-mediated regulation of dendritic spine density.
Transmembrane semaphorin–plexin receptor interactions in cis regulate neuronal repulsion in mouse sympathetic neurons, synaptic tiling in Caenorhabditis elegans, and starburst amacrine cell morphology and function (Haklai-Topper et al., 2010; Mizumoto and Shen, 2013; Sun et al., 2013). To better define Sema5A-PlexA2 signaling, we co-expressed PlexA2 with full-length Sema5A or membrane-bound Sema5A deletion mutants lacking either the cytoplasmic domain (S5AΔcyt) or the 7TSRs (S5AΔTSR) in COS7 cells. Co-expression of PlexA2 with Sema5A constructs that contain the Sema5A sema domain significantly attenuates binding of bath applied Sema5A-Fc (Figure 5F,G). In low-density HEK293T cell cultures, co-expression of individual PlexA family members with membrane-bound full-length Sema5A shows that PlexA1 and PlexA2, but not PlexA3, are part of a Sema5A complex that can be immunoprecipitated from cell lysates (Figures 5—figure supplement 3A). A membrane-bound Sema5A deletion construct lacking the 7 TSRs (S5AΔTSR) interacts with full-length PlexA1 and PlexA2 when co-expressed with these receptors, but not PlexA3. The cytoplasmic domain of PlexA1 and PlexA2 are dispensable for this interaction with Sema5A (Figures 5—figure supplement 3B). Together, these observations strongly suggest that the sema-domain of Sema5A forms a complex with PlexA2 when presented in cis or in trans.
Plxna2−/− mice show increased dentate GC synapse density
To examine the in vivo roles of PlexA1 and PlexA2 in GCs, we generated Plxna1−/−;Thy1-eGFP and Plxna2−/−;Thy1-eGFP mice and quantified spine density in these mutants at P33. Compared to WT mice (1.70 ± 0.04 spines/µm), we observed a significant increase in spine density in Plxna2−/− (2.24 ± 0.07 spines/µm), but not in Plxna1−/− (1.79 ± 0.05 spines/µm), mutants (Figure 6A and B, Table 1). This increase in GC spine density in Plxna2−/− mutants is comparable to that observed in Sema5A−/− mutants (2.05 ± 0.05 spines/µm). To analyze synaptic profiles at the ultra-structural level, and also to obtain a direct comparison between the Sema5A−/− and Plxna2−/− GC phenotypes, we performed scanning electron microscopy (sEM) followed by three-dimensional (3D) reconstructions of GC dendritic segments in 1-month-old WT, Plxna2−/− and Sema5A−/− mice. We analyzed dendrites in the outer one-third of the supragranular ML (Figure 6—figure supplement 1A–I), as we did with Plxna2−/−;Thy1eGFP and Sema5A−/−;Thy1eGFP mice, using immunofluorescence microscopy (Figures 1E and 6A). sEM revealed a robust supernumerary GC spine phenotype in Plxna2−/− mice (2.44 ± 0.19 spines/µm) and Sema5A−/− mice (2.65 ± 0.20 spines/µm) compared to WT (1.75 ± 0.11 spines/µm) (Figure 6C,D). Most dendritic spines reconstructed from WT (89.8%), Plxna2−/− (92.3%) and Sema5A−/− (93.9%) mice had a clearly identifiable PSD, a synaptic cleft and an opposed presynaptic bouton with vesicles (Figure 6—figure supplement 1G,H,I). The density of spines with no identifiable PSD was comparable among WT, Sema5A−/−, or Plxna2−/− GCs (Figure 6—figure supplement 1J), and the mostly thin and filopodia-like structure of spines in these mutants suggests that they had not yet developed into mature synapses. No significant differences in spine head volume or PSD area were detected among WT, Plxna2−/−, or Sema5A−/− mutants (Figures 6—figure supplement 1K,L). We found no evidence of dendritic spines that are contacted by more than one presynaptic bouton (Figure 6—figure supplement 1M). The increase in synapse density in Plxna2 mice detected using sEM was independently confirmed with 2D EM (Figures 6—figure supplement 1N–P). These experiments show that synapse density in the GC outer ML is significantly increased in Plxna2−/− and Sema5A−/− mice, and they also show that Sema5A and PlexA2 negatively regulate excitatory synapse density in vivo.
Figure 6 with 2 supplements see all

Sema5A and Plxna2 genetically interact, and Plxna2−/− and Sema5A−/− mice show increased synapse density in dentate GCs.
(A) Representative images of outer dendritic segments of GCs from P30 Thy1-eGFPm WT, Plxna1−/− and Plxna2−/− hippocampal sections. (B) Quantification of GC dendritic spine density reveals a significant increase in Plxna2−/− mice compared to Plxna1−/− and WT controls. Numbers in brackets indicate the dendritic segments analyzed per condition. Values are represented as mean ± SEM from three to four animals of each genotype. ***p < 0.001, two-tailed unpaired Student's t test. (C) Three-dimensional reconstruction of GC dendritic segments from P32 WT, Plxna2−/− and Sema5A−/− mice. The dendritic shaft is colored in gray, dendritic spines in blue and the PSDs in orange. A total of 110, 208, and 165 spines were reconstructed and analyzed for WT, Plxna2−/− and Sema5A−/− mice, respectively (n = 1 animal per genotype). (D) Quantification of spine density. Values are represented as mean ± SEM from three to four dendritic segments (each 20 to 22 µm in length) per genotype, *p < 0.05, two-tailed unpaired Student's t test. (E) Representative images of GC dendritic segments of P30 Thy1-eGFPm WT, Sema5A+/−, Plxna2+/−, and compound heterozygous Sema5A+/−; Plxna2+/− mice. (F) Quantification of GC spine densities of genotypes in E reveals significantly more spines in compound heterozygotes than WT and single heterozygotes. Numbers in brackets indicate the number of dendrites analyzed per condition. Values are represented as mean ± SEM from four animals per genotype. ***p < 0.0001, two-tailed unpaired Student's t test. Scale bars in A, C and D = 5 µm.
Sema5A and Plxna2 genetically interact
We next assessed GC dendritic spine density in P33 heterozygous Sema5A+/− (1.65 ± 0.06 spines/µm) and Plxna2+/− (1.86 ± 0.05 spines/µm) mice, comparing these observations to compound heterozygous Sema5A+/−;Plxna2+/− mice (2.39 ± 0.05 spines/µm) (Figure 6E,F). Only compound heterozygous mice show a significant increase in spine density, and this increase is comparable to that observed in Sema5A−/− and PlexA2−/− single mutants (Table 1). Therefore, Sema5A and Plxna2 genetically interact, suggesting they function in the same signaling pathway in vivo. Together with our observations of direct Sema5A and PlexA2 binding, co-expression in GCs, localization to synaptosomal fractions, and function in primary GCs to regulate dendritic spine density, these data demonstrate that PlexA2 is a functional receptor for Sema5A, mediating Sema5A constraint of GC excitatory synapse density in vivo.
Sema5A−/− mice exhibit impaired social behavior
A human genome-wide association study identified a significant association between a SNP near the SEMA5A locus and autism (Weiss et al., 2009). However, behavioral studies with a Sema5A gene-trap insertion mouse mutant (Sema5AlacZ/lacZ) on a mixed C57Bl/6/129P2 background showed no behavioral differences compared to C57Bl/6 control mice (Gunn et al., 2011). This gene-trap insertion is in the last coding exon of the Sema5A gene, generating a deletion mutant lacking the C-terminal portion of the cytoplasmic domain, a region of Sema5A that we find is not necessary for the regulation of dendritic spine density (Figure 3B). Indeed, when we analyzed GC spine density in these mice we did not observe a significant difference compared to WT controls, suggesting this Sema5A allele is not a null, or strong loss of function, mutation (Figure 6—figure supplement 2). Therefore, we extensively backcrossed our Sema5A−/− mice to C57Bl/6 mice. These congenic Sema5A−/− mutants were confirmed to be >99% C57Bl/6 and were subjected to a panel of behavioral assays. Sema5A−/− congenic mutants show normal locomotor behavior in the open field test (Figure 7—figure supplement 1A), and mutant and WT mice exhibit similar anxiety levels (Figures 7—figure supplement 1B,C). They also spend a similar percentage of time in the open and closed arms of the elevated plus maze (Figure 7—figure supplement 1D). Contextual and cued fear conditioning revealed no difference between Sema5A WT and null mice (Figure 7A,B). In the three-chambered apparatus (Yang et al., 2011), a simple social approach task, both C57Bl/6 WT and Sema5A−/− mice spent significantly more time engaged in nose-to-nose interactions with a mouse than sniffing an inanimate object (Figure 7C), a behavior representing normal sociability (Roullet and Crawley, 2011). We next assessed the time of direct nose-to-nose interaction of WT C57Bl/6 and Sema5A−/− mice with a stranger vs a familiar mouse. WT mice show a strong preference for nose-to-nose contact with the stranger mouse, a behavior representing normal social interaction (Yang et al., 2011). This preference was not observed in Sema5A−/− mice (Figure 7D), indicating that Sema5A−/− mice exhibit deficits in sociability. These behavioral studies establish an endophenotype in Sema5A−/− mice commonly associated with ASD.
Figure 7 with 1 supplement see all
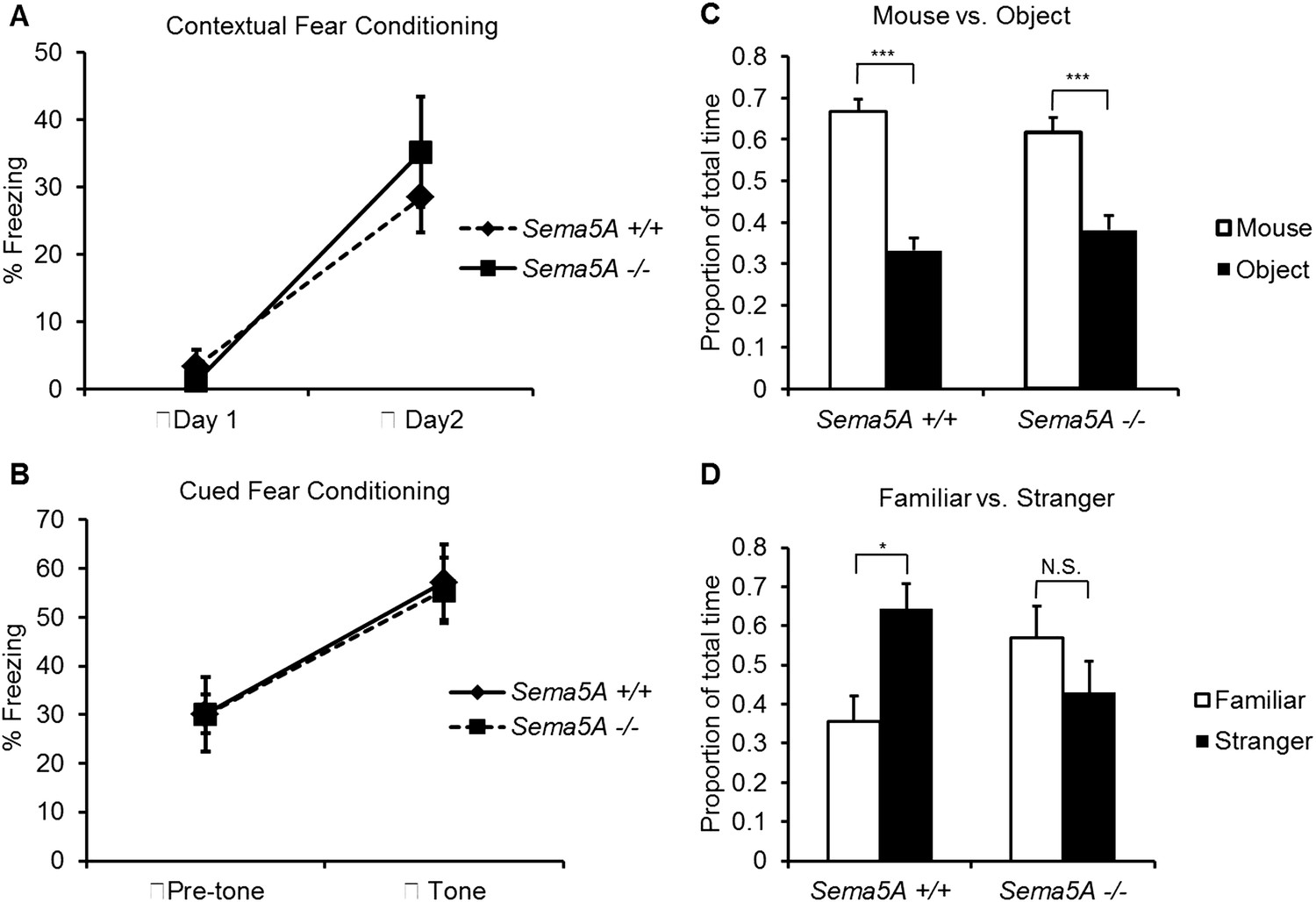
Sema5A−/− mice show abnormal social interaction.
(A and B) Fear conditioning. Sema5A+/+ and Sema5A−/− mice show no difference in freezing behavior under contextual fear conditioning (A) and cued fear conditioning (B) paradigms. Number of animals tested: n = 6 Sema5A+/+ mice and n = 8 Sema5A−/− mice. Error bars represent ± SEM. (C and D) Three-chambered social interaction test: (C) given the choice, both Sema5A+/+ and Sema5A−/− mice spent significantly more time engaged in nose-to-nose interaction with a mouse than sniffing an inanimate object. (D) Sema5A+/+ mice spent significantly more time engaged in nose-to-nose interaction with a stranger mouse compared to a familiar mouse, whereas Sema5A−/− mice showed no preference. Number of animals tested: n = 8 Sema5A+/+ mice and n = 8 Sema5A−/− mice for mouse vs inanimate object, and n = 7 Sema5A+/+ mice and n = 7 Sema5A−/− mice for familiar vs stranger mouse. Error bars represent ± SEM. n.s. not significant, ***p < 0.001 and *p < 0.05 by two-tailed Student's t test.
Discussion
We demonstrate here that Sema5A is a novel inhibitor of excitatory synapse formation and AMPA-type synaptic transmission. In the mouse hippocampus, Sema5A negatively regulates dendritic spine density in both developmentally born and adult-born dentate GCs. Loss of Sema5A-PlexA2 signaling results in the formation of supernumerary dendritic spines, exuberant excitatory synapses, and an increase in amplitude and frequency of excitatory synaptic transmission. Sema5A and PlexA2 are co-expressed in GCs and can both function cell-autonomously to prevent the formation of exuberant spine synapses. The rasGAP activity in the PlexA2 cytoplasmic domain is required for Sema5A-mediated inhibition of dendritic spine density. Sema5A−/− congenic mice show normal locomotion and no evidence for elevated fear or anxiety. However, the loss of Sema5A leads to deficiencies in sociability. Collectively, these results suggest that intradendritic semaphorin–plexin interactions inhibit synapse density and synaptic transmission, and when perturbed, the absence of this signaling pathway leads to impaired social behavior, a hallmark of ASD.
Sema5A negatively regulates GC synaptic strength
Developmentally born and adult-born GCs mature at different rates and in different environments, yet the sequence of neuronal maturation events is stereotyped and temporally segregated (Esposito et al., 2005). GC intrinsic programs may be advantageous in order to preserve the sequence of cellular maturation. Sema5A is strongly expressed in GCs during development and throughout adulthood. Similar to developmentally born GCs, we find that adult-born GCs employ Sema5A to negatively regulate dendritic spine density. In adult-born Sema5A−/− GCs 3-weeks postmitosis, dendritic spine density is increased and correlates with an increase in mEPSC frequency. In addition, the loss of Sema5A increases the amplitude of AMPA receptor currents in these adult-born GCs. Because of its postsynaptic localization, Sema5A may normally negatively regulate AMPA receptor surface expression, synaptic distribution, or downstream signaling events that limit mEPSC amplitude. In primary hippocampal neurons, we find that the loss of Sema5A does not lead to an increase in total GluA1 expression or localization to synapses (data not shown). Sema5A associates with HSPGs and CSPGs (Kantor et al., 2004), and proteoglycans are known to participate in the regulation of surface GluA1 distribution through inhibition of AMPA receptor lateral diffusion at synapses. In the absence of Sema5A, the barrier function of CSPGs may be increased, resulting in elevated synaptic strength (Allen and Barres, 2005; Frischknecht et al., 2009; Orlando et al., 2012). More indirect mechanisms may also apply here, since two recent studies show that presynaptic HSPGs, including members of the glypican and syndecan families, interact with the LRRTM4 protein to promote the formation of excitatory synapses on GCs (Siddiqui et al., 2013; de Wit et al., 2013). Sema5A can interact with neural syndecans (Kantor et al., 2004), and at GC synapses Sema5A may compete with the synaptogenic activity of LRRTM4. While the recombinant sema domain of Sema5A is sufficient to ‘rescue’ the increased dendritic spine density observed in primary GCs null for Sema5A−/− in vitro, TSR-dependent proteoglycan interactions with endogenous Sema5A may influence the strength of sema-domain dependent cis- and trans-complexes in vivo. It is interesting in this context that, similar to what we observe in Sema5A−/− mice, loss of LRRTM4 selectivity influences the density of excitatory synapses but has no effect on inhibitory synapse density. Additional studies will elucidate the precise mechanisms by which Sema5A negatively regulates synaptic strength, and whether it serves to directly counter synaptogenic pathways.
PlexA2 is a novel Sema5A receptor in dentate GCs
In non-neuronal cells, Sema5A signals in a PlexB3-dependent manner to elicit distinct cytoskeletal responses, including collapse of NIH3T3 cells, motility of glioma cells, and migration of primary endothelial cells (Artigiani et al., 2004; Li and Lee, 2010). Neurite outgrowth in primary retinal neurons is inhibited by Sema5A and Sema5B, and this response is abrogated in Plxna1, Plxna3 double null neurons (Oster et al., 2003; Goldberg et al., 2004; Matsuoka et al., 2011). Here, we identify PlexA2 as a high-affinity receptor for Sema5A in dentate GCs. Previous studies showed that Sema6A regulates GC axonal targeting in a Plxna2- and Plxna4-dependent manner (Suto et al., 2007), and also that the secreted molecule Sema3F signals through neuropilin-2/PlexA3 to control dendritic spine shape and density in developing GCs (Tran et al., 2009). Similar to Sema3F, Sema5A regulates GC spine density, however Sema3F, but not Sema5A, apparently regulates spine shape. Sema3F is a secreted molecule, and thus can exert long-range effects, while Sema5A is a transmembrane molecule and acts locally. Sema5A, but not Sema3F, continues to be expressed in the adult dentate, and we show that Sema5A regulates synaptic density and function in adult-born GCs. These studies highlight the intricacies of semaphorin–plexin interactions with respect to the regulation of hippocampal circuit assembly at the subcellular level.
Cell-autonomous Sema5A/PlexA2 signaling regulates GC synapse density
Many cis interactions between axon guidance molecules and their receptors, including ephrins/Ephs and Sema6s/PlexAs, attenuate receptor function (Haklai-Topper et al., 2010; Yaron and Sprinzak, 2012; Sun et al., 2013). Cis regulation of a membrane-bound semaphorin, Sema1, by a plexin receptor, Plxn-1, cell-autonomously controls the position of C. elegans synaptic boutons in developing DA9 motoneurons. Sema1 and Plxn-1 function to restrict synaptic bouton formation to appropriate subaxonal segments (Mizumoto and Shen, 2013). Similarly, the cis-interaction between PlexA2 and Sema5A on GC dendrites we describe here may suppress the formation of supernumerary protrusions that mature into excitatory synapses. In addition to this cis-interaction, Sema5A can also bind to PlexA2 when presented in trans. In cultured GCs this trans-interaction induces rapid dendritic spine elimination. In vivo, PlexA2-Sema5A cis-interactions may predominate since both molecules can be detected on the surface of cultured GC neurites. However, trans interactions may occur when filopodia or dendritic spines of two neighboring GCs compete for space on the same presynaptic bouton to form a multi-synapse bouton (Toni et al., 2007), or when different dendrites from the same GC neuron come into contact. Thus, our observations suggest that a combination of Sema5A-PlexA2 cis and trans interactions may ultimately determine GC spine density in vivo.
Sema5A and autism
Genome-wide ASD association analyses have identified an intergenic SNP near the SEMA5A locus (Weiss et al., 2009), and this same study showed that SEMA5A expression is reduced in Brodmann area 19 of autism brains. An independent study showed that expression of SEMA5A is down regulated in transformed lymphocytes of autistic subjects (Melin et al., 2006). We used Sema5A null mice for behavioral testing, including the three-chambered apparatus, which provides a simple social approach task. Sema5A−/− mice exhibit impaired sociability, a core trait of ASD that has been observed in several mouse models of autism (Roullet and Crawley, 2011). Although altered social behavior may be secondary to increased anxiety, this appears unlikely since Sema5A−/− mice show no signs of anxiety in the open field test or in the elevated plus maze compared to WT controls. Because Sema5A is expressed broadly in the CNS, including in neuronal and non-neuronal cell types (Zhang et al., 2014), additional studies are warranted to determine the cellular basis of the behavioral deficits we observed in Sema5A−/− mice. Studies utilizing Sema5A conditional mutants that lack Sema5A in specific neural cell-types or specific brain structures are required to identify where Sema5A function is required for proper neural circuit development and to pin point which of these regions participate in normal social interaction behaviors.
Linking the synaptic defects observed in Sema5A mutants to the observed behavioral phenotypes is difficult. However, a growing number of ASD-associated genes encode proteins that function at the synapse, suggesting that altered synapse assembly and density is causally linked to ASD (Peca and Feng, 2012; Voineagu and Eapen, 2013). Human SEMA5A has been identified as an ASD susceptibility gene (Weiss et al., 2009), and we show here that its mouse homologue, Sema5A, functions as a negative regulator of synaptic transmission and formation of spiny synapses. When coupled with the impaired social behavior we observe in Sema5A−/− mice, our studies begin to provide mechanistic insight into how altered SEMA5A function may lead to brain illness, including autism.
Materials and methods
Mice
All mice used in this study were housed and cared for in accordance with NIH guidelines, and all research conducted was done with the approval of the University of Michigan Medical School and The Johns Hopkins University Committees on Use and Care of Animals. Sema5A−/−, Sema5A(flox/flox), Sema5B−/−, Plxna1−/−, Plxna2−/−, Plxna3−/−, and Thy1-GFPm mice were previously described (Feng et al., 2000; Cheng et al., 2001; Yoshida et al., 2006; Matsuoka et al., 2011). Sema5Atm1Dgen/J mice express nuclear β-galactosidase (β-gal) under the control of the Sema5A promoter (Sema5AlacZ) (Gunn et al., 2011) and were purchased from Jackson Labs. For behavioral studies, Sema5A mice were backcrossed onto a C57Bl/6 background using the speed congenic services provided by Charles River. Male mice used for behavioral studies were >99% congenic, as assessed by single nucleotide polymorphism (SNP) analysis (Charles River).
DNA constructs
Request a detailed protocolRat Sema5A cDNA (Accession: NM_001107659) was used to generate expression constructs for Sema5A, including 6xMyc N-terminal-tagged full-length (FL)-Sema5A comprised of amino acid residues 23–1074 (23–1074 aa), membrane-bound Sema5A lacking the seven TSRs (6xMyc-Sema5AΔTSR; 23–539 and 951–1074 aa) and Sema5A lacking the cytoplasmic domain (6xMyc-Sema5AΔcyt; 23–999 aa). Constructs were obtained by PCR amplification of the corresponding Sema5A sequences and subsequently cloned into the pCAG expression vector in frame with the Ig kappa signal sequence and the N-terminal 6xMyc tag. The following Fc fusion constructs were assembled in pcDNA3.0 (Invitrogen, Grand Island, NY): Sema5A ectodomain (Sema5A-Fc; 1–961 aa), sema domain (Sema5ASema-Fc; 1–539 aa), and seven TSR repeats (Sema5ATSR-Fc; 536–960 aa). The expression and affinity purification of Fc fusion proteins, including NgROMNI-Fc, were carried out as described (Robak et al., 2009). Fusion proteins with C-terminal green fluorescent protein (GFP) in pcDNA3.0 included 6xMyc-FL-Sema5A-GFP and 6xMyc-Sema5AΔTSR-GFP. Mouse PlexA constructs with an N-terminal 6xMyc tag were generated by PCR cloning and included FL PlexA1, PlexA2, PlexA3, and PlexA4. The ectodomains of PlexA1 (PlexA1-Fc; 28–1237 aa), PlexA2 (PlexA2-Fc; 35–1232 aa), and the sema domain of PlexA1 (PlexA1SD-Fc; 28–580 aa) were fused to the Fc portion of human IgG1. The PlexA2SD-Fc and PlexA4SD-Fc constructs were generously provided by F Suto (Suto et al., 2007). To obtain deletion mutants that lack the cytoplasmic domain, the ectodomain plus transmembrane spanning domain of PlexA1 (28–1283 aa) and PlexA2 (35–1280) were cloned into the pCAG expression vector. The PlexA1 RasGAP-deficient mutant was generously provided by A Puschel (Rohm et al., 2000). The PlexA2 RasGAP-deficient mutant (PlexA2AA) was constructed by mutating two arginines, R1428A and R1429A, corresponding to key residues in the RasGAP catalytic domain of PlexA1 (Rohm et al., 2000).
Histological procedures
Request a detailed protocolP18 and P30 Sema5Atm1Dgen/J mice were perfused transcardially with 4% paraformaldehyde (PFA). The brains were then dissected, post-fixed in 4% PFA for 30 min, cryoprotected in 30% sucrose in phosphate buffered saline pH7.4 (PBS) and flash frozen in dry-ice cooled isopentane. Horizontal sections were cut at 30-µm thickness, post-fixed in 4% PFA for 15 min, rinsed twice in 1× PBS, incubated with X-Gal solution (5 mM potassium ferricyanide, 5 mM potassium ferrocyanide, 2 mM MgCl2, and 1 mg/ml 5-bromo-4-chloro-3-indolyl-b-D-galactopyranoside [X-Gal] in PBS) and developed overnight at 37°C. Alternatively, β-galactosidase expression in brain sections was visualized by anti-β-galactosidase (1:500, Abcam, Cambridge, MA) immunohistochemistry. In situ hybrdization with digoxigenin-labeled riboprobes specific for Sema5A, Sema5B, Plxna1, Plxna2 and Plxna3 was carried out described previously (Matsuoka et al., 2011).
For immunohistochemistry, coronal brain sections, cryosectioned at 25 μm and lifted on Superfrost+ microscope slides, were fixed in 4% PFA for 15 min and rinsed twice with 1× PBS for 5 min each. The sections were blocked with 5% horse serum, 0.3% Triton X-100 in PBS for 1 hr at room temperature, and incubation overnight at 4°C with the following primary antibodies: rabbit anti-Calbindin (1:1000, Swant, Switzerland), goat anti-Calretinin (1:1000, Swant), goat anti-Doublecortin (1:200, Santa Cruz), rabbit anti-GFP antibody (1:1000, Life Science). The sections were rinsed times times with 1× PBS for 5 min each, and incubated with Alexa Fluoro-conjugated secondary antibodies for 1 hr at room temperature. The sections were imaged and analyzed with an Olympus IX71 inverted fluorescence microscope.
Dendritic spine quantification: to quantify spine density, Sema5A, Sema5B, Plxna1, and Plxna2 mice were crossed with Thy1-GFPm mice to genetically label a subset of postnatal hippocampal neurons. At P30–P33, mice were perfused transcardially with ice-cold 4% PFA in PBS and brains were postfixed for 2 hr in perfusion solution and cryoprotected in 30% sucrose in PBS. Coronal brain sections (8 µm thick) at the level of the rostral hippocampus were mounted on Superfrost+ microscope slides and fixed in 4% PFA for 15 min and processed for fluorescent microscopy. For imaging of GFP-labeled neurons, z-stacks at 0.3-µm intervals were taken using the Deltavision RT imaging system (Applied Precision), equipped with an Olympus IX-70 inverted microscope with a 100× Oil objective plus an extra 1.6× zoom. Captured images were processed with 3D deconvolution to reduce background signals. For GCs, dendritic spine density in the outer one-third of the dorsal ML was quantified. For CA1 pyramidal neurons, spine density on the first and second order branches was quantified separately. For retrovirus-mediated birth-dating of adult-born GC, virus was injected into the DG of 5- to 6-weeks-old mice (Ge et al., 2006). 3 weeks later, brains were dissected, cryosectioned coronally at 40-µm thickness, and stained with anti-GFP antibody. Images were acquired with a 40× oil objective plus 5× zoom on a Zeiss LSM 710 META multiphoton confocal system (Carl Zeiss, Thornwood, NY) using a multi-track configuration. Only GCs in the dorsal molecular layer of DG were imaged and dendritic spines quantified as described for Thy1-GFPm mice.
3D reconstruction of dendritic segments
Request a detailed protocolFor serial block-face scanning electron microscopy (SBF-SEM), P30 WT, Plxna2−/− and Sema5A−/− mice were perfused transcardially for 2 min with ice-cold PBS and then for 10 min with freshly prepared, ice-cold 4% PFA and 2.5% glutaraldehyde in 0.1 M Na+-cacodylate buffer (pH 7.4). Brains were dissected, postfixed in perfusion solution, and sectioned coronally in perfusion solution at 400 µm using a Leica vibrotome (TV1200). Brain slices that included the rostral hippocampus were fixed overnight in perfusion solution at 4°C and then processed for SBF-SEM according to instructions by Renovo Neural Inc. (Cleveland, OH). Briefly, tissue sections were rinsed in 0.1 M Na cacodylate buffer, stained with 0.1% tannic acid for 30 min that was followed by the staining regime developed by Renovo. Tissue was then dehydrated in ethanol and infiltrated and embedded in EPON 812. Approximately, 300 serial electron micrographs at 75-nm thickness were cut and imaged at a resolution of 7 nm/pix at Renovo Neural Inc., using a sigma VP scanning electron microscope (Carl Zeiss Microscopy, Jena, Germany) and a 3view door (Gatan Inc., Pleasanton, CA). For 3D reconstruction and quantification of dendritic segments, dendritic spine density, spine volume, spine head volume, and PSD surface, these structures were manually traced in individual sections using the PSD Reconstruct software (SynapseWeb).
Primary neuronal cell culture
Request a detailed protocolHippocampal cultures enriched for GCs were established from WT and mutant mouse pups (P1–P2) by microdissection of dentate gyri. Briefly, the hippocampus was dissected and the DG separated from CA3/CA1. Combined dentate gyri were trypsinized for 15 min at 37°C, rinsed twice in HBSS (Gibco, Grand Island, NY), and mechanically dissociated in neurobasal medium containing 2% B27 supplement, 2 mM Glutamax, 50 unit/ml Penicillin, and 50 µg/ml streptomycin (Invitrogen). Cells were rinsed once in HBSS and once in culture medium and plated onto glass coverslips (Carolina Biologicals) coated with 100 µg/ml Poly-D-Lysine (Sigma, St. Louis, MO) and 10 µg/ml laminin (Life Technologies), at a density of 300,000 cells per 18-mm coverslip in a 12-well plate. Using lipofectamine 2000 (LFA), DIV4 cultures were transfected for 4 hr with a mixture of 1 µg of eGFP-expressing construct and 2 µl LFA per well of a 12-well plate. At DIV21, cultures were fixed and immunostained for identification of GCs and quantification of dendritic spine density.
Immunocytochemistry
Request a detailed protocolBriefly, primary neurons were fixed with 4% PFA at DIV21 for 15 min at room temperature, and blocked in PBS with 1% horse serum and 0.1% Triton X-100 (PHT) for 1 hr. Cells were incubated with primary antibodies in PHT overnight at 4°C, followed by 1-hr incubation with fluorophore-conjugated secondary antibodies at room temperature. Primary antibodies used included: rabbit anti-Prox1 (1:5000, Millipore), mouse anti-myc (1:1000, Developmental Studies Hybridoma Bank at the University of Iowa), chicken anti-GFP (1:1000; AVES, Tigard, OR), goat anti-GFP (1:500; Rockland, Limerick, PA), guinea pig anti-vGlut1 (1:1000; Millipore), mouse anti-PSD95 (1:500; Millipore), rabbit anti-VGAT (1:1000; Synaptic Systems), and mouse anti-gephyrin (1:1000; Synaptic Systems). Species-specific and Alexa Fluor-conjugated secondary antibodies (Life Technologies) were used at 1:1000.
Isolation of synaptic density fractions and Western blot analysis
Request a detailed protocolFreshly dissected mouse brains were homogenized in ice-cold 4 mM HEPES pH 7.4, 0.32 M sucrose, and protease inhibitor cocktail (1:100, Roche). Brain homogenate was centrifuged at 2000×g for 10 min to obtain supernatant 1 (S1) and pellet 1 (P1). The S1 fraction was centrifuged at 37,000×g for 30 min resulting in S2 and P2. Then, P2 was resuspended in ice-cold 4 mM HEPES, pH 7.4 and fractionated by centrifugation (82,500×g for 2 hr) on a discontinuous sucrose gradient of 4 mM HEPES in 0.85 M, 1.0 M, and 1.2 M sucrose. The synaptosomal fraction (syn) was harvested at the 1.0/1.2 M sucrose interface, centrifuged at 150,000×g for 30 min, and resuspended in 80 mM TrisHCl pH 7.8. To isolate PSD fractions, synaptosomes were incubated with 40 mM TrisHCl pH 8.0, 0.5% Triton-X100 for 15 min on ice and then centrifuged at 32,000×g for 20 min. The pellet was resuspended in 40 mM TrisHCl pH 8.0 to obtain PSD I. The synaptic junction (SJ or PSD I) fraction was incubated with 40 mM TrisHCl pH 8.0, 0.5% Triton-X for 15 min on ice and centrifuged at 201,800×g for 1 hr. The pellet was resuspended in 40 mM TrisHCl pH 8.0 with 0.3% SDS to give PSD II fraction. To isolate the PSD III fraction, PSD I was incubated with 40 mM TrisHCl pH 8.0, 3% sarkosyl for 15 min and centrifuged at 201,800×g for 1 hr. The pellet was resuspended in 40 mM TrisHCl pH 8.0 to yield PSD III. Protein concentrations were measured with the BCA assay (Pierce) and samples mixed with 2× Laemmli buffer. 5 µg of each fraction was used for Western blot analysis. Primary antibodies used included mouse anti-PSD95 (1:2000; Millipore), rabbit anti-synaptophysin (1:1000; Santa Cruz), rabbit anti-Sema5A (1:1000, [Matsuoka et al., 2011]), rabbit anti-Sema5B (1:250, [Matsuoka et al., 2011]), rabbit anti-PlexA1 (1:2000; gift from Yutaka Yoshida), rabbit anti-PlexA2 (1:2000; gift from Hajime Fujisawa), rabbit anti-PlexA3 (1:500; Abcam), and mouse anti-neuropilin-2 (1:1000; Cell Signaling), mouse anti-Myc (1:1000, University of Iowa). Species-specific secondary antibodies were used at 1:5000 (Jackson, West Grove, PA).
Immunoprecipitation
Request a detailed protocolHEK293T cells were transfected with various combinations of 6xMyc-tagged FL-Sema5 and PlexA expression constructs, the deletion constructs PlexA-Δcyto, Sema5AΔTSR, or Sema5A-GFP. 24 hr after transfection, cells were incubated in ice-cold lysis buffer containing 20 mM TrisHCl (pH 7.5), 150 mM NaCl, 1% CHAPS (wt/vol), 1% Triton X-100 (vol/vol), and protease inhibitor cocktail. Cell lysates were precleared with Protein G Plus/Protein A–Agarose beads (Calbiochem), and then incubated with mouse anti-GFP antibody (Invitrogen) overnight at 4°C followed by incubation with Protein G Plus/Protein A–Agarose beads for an additional 4 hr. After the pull-down by centrifugation, beads were rinsed three times in lysis buffer, and bound proteins were analyzed by Western blotting. To assay for direct ligand–receptor interactions, Fc-tagged Sema5A ectodomain protein or Human IgG (50 nM) was incubated with 6xMyc-tagged PlexA1 or PlexA2 ectodomain produced in HEK293T cells, pulled down with Protein G Plus/Protein A–Agarose beads, and were analyzed for the presence of plexins by immunoblotting.
Fusion protein binding studies
Request a detailed protocolFor ligand–receptor interaction studies, binding of recombinant protein to transfected COS7 cells was carried out as previously described (Robak et al., 2009). Briefly, COS7 cells were plated onto poly-D-lysine-coated 24-well plates and cultured overnight before transfection with Lipofectamine 2000 (Life Technologies) and expression constructs for WT and mutant versions of PlexA or Sema5A. After 24–36 hr, cells were incubated for 75 min with Fc-tagged fusion proteins preclustered with anti-human IgG conjugated to human placental alkaline phosphatase (AP) in OptiMEM (Promega, Madison, WI). Unbound fusion protein was removed by extensive rinsing with OptiMEM. Cells were then fixed with 1% formaldehyde in 60% acetone, rinsed twice in HBHA (HBS supplemented with 0.05% horse serum, 0.5% NaN3), and once in HBS (Cellgro). Endogenous phosphatases were heat-inactivated by incubation at 65°C for 70 min. Binding of fusion protein was visualized by developing the AP reaction with NBT/BCIP substrate (Sigma). For binding of Sema5A-Fc and PlexinA2SD-Fc fusion proteins to non-transfected primary mouse hippocampal neurons (DIV5-7), the same protocol was followed; however, NeuroBasal medium was used instead of OptiMEM.
Dendritic spine collapse assay
Request a detailed protocolPrimary neuronal cultures enriched for GCs were established and transfected with an eGFP expression construct at DIV4 as described above. At DIV21, live imaging was performed and pictures taken with a Deltavision-RT Live Cell Imaging System at 5% CO2 and 37°C at the University of Michigan Imaging Laboratories (MIL). Briefly, neurons on glass coverslips were placed in the microscope incubation chamber with 0.5 ml prewarmed B27 growth medium. GFP transfected neurons were imaged with a 100× objective and pictures taken every 5 min for 10 min before ligand treatment and then for an additional 120 min. At 10 min, Sema5A-Fc or human IgG (Sigma) preclustered with anti-human IgG (Promega) was added to the culture medium at a final concentration of 10 µg/ml. 2 hr after ligand incubation, cultures were fixed and stained with anti-Prox1 to identify which of the imaged cells were GCs. For quantification of dendritic spine density, only prox1+ cells were included.
Stereotaxic injection of viral vectors
Request a detailed protocolFor stereotaxic injection of Lentiviral vectors (LV)-eGFP or LV-synapsin-eGFP-IRES-Cre (Bateup et al., 2013) into the hippocampus of P15 Sema5A(flox/−) mice, an automated injector pump (Stoetling) connected to a 5-µl Hamilton syringe was used. Briefly, anesthesia was induced by inhalation of 4% isoflurane mixed with oxygen and maintained by 2% isoflurane during surgery. After the plane of anesthesia was reached, skin on top of the skull was cleaned three times with iodine and ethanol wipes alternately, and a 1-cm long skin incision was made near the midline. After exposure of skull bone, a small hole was drilled on each side of the midline (ML) with the following coordinates: ML = ±1.27 mm, anterior-posterior (AP) = −1.65 mm from bregma. A 30-gauge needle was slowly inserted into the brain at a dorsal–ventral depth (DV) of −2.25 mm from the skull surface. Then, 0.5 µl of high titer (500× concentrated) LV was injected at a speed of 0.5 µl/min. LV-eGFP was injected on the left side and LV-synapsin-eGFP-IRES-Cre on the right side. The injection needle was left in place for an additional 2 min after the injection was completed and then withdrawn slowly. The skin was closed using veterinary glue, and the animals were placed on a 37°C heating pad for recovery. After surgery, mice were monitored regularly for 14 days and killed at P29 to assess dendritic spine density of transduced dentate GCs.
Labeling of adult born GCs
Request a detailed protocolEngineered self-inactivating murine retroviruses with a GFP reporter under the control of ubiquitin promoter were used to label and birth-date the proliferating cells and their progeny as previously described (Ge et al., 2006). High titers of engineered retroviruses were produced by co-transfection of retroviral vectors and VSVG into HEK293gp cells followed by ultracentrifugation of viral supernatant. Young adult (5–6 weeks old) Sema5A−/− mice and their littermates housed under standard conditions were anaesthetized with ketamine/xylazine, and retroviruses were stereotaxically injected into the dentate gyrus at four sites (0.5 μl per site) with the following coordinates: AP = −2 mm from bregma; lateral from ML = ±1.5 mm; and DV = −2.5 mm.
Electrophysiology
Request a detailed protocolAdult mice housed under standard conditions were anaesthetized 19–21 days after retroviral injection and processed for slice preparation as previously described (Ge et al., 2006). The brains were quickly removed into the ice-cold solution (in mM: 110 choline chloride, 2.5 KCl, 1.3 KH2PO4, 25.0 NaHCO3, 0.5 CaCl2, 7 MgSO4, 20 dextrose, 1.3 sodium L-ascorbate, 0.6 sodium pyruvate, 5.0 kynurenic acid). Slices (275 μm thick) were cut using a vibrotome (Leica VT1200S) and transferred to a chamber containing the external solution (in mM: 125.0 NaCl, 2.5 KCl, 1.3 KH2PO4, 1.3 MgSO4, 25.0 NaHCO3, 2 CaCl2, 1.3 sodium L-ascorbate, 0.6 sodium pyruvate, 10 dextrose, pH 7.4, 320 mOsm), bubbled with 95% O2/5% CO2. Electrophysiological recordings were obtained at 32°C–34°C. GFP+ cells within the granule cell layer were visualized by DIC and fluorescence microscopy. The whole-cell patch-clamp configuration was employed in voltage-clamp mode (Vm = −65 mV). Microelectrodes (4–6 MΩ) were pulled from borosilicate glass capillaries and filled with the internal solution containing (in mM) 120 potassium gluconate, 15 KCl, 4 MgCl2, 0.1 EGTA, 10.0 HEPES, 4 ATP (magnesium salt), 0.3 GTP (sodium salt), 7 phosphocreatine (pH 7.4, 300 mOsm). The recordings were done in the presence of bicuculline (10 μM) to block GABAergic currents and TTX (1 μM) to block action potentials. Data were collected using an Axon 200B amplifier and acquired with a DigiData 1322A (Axon Instruments) at 10 kHz. Series and input resistances were monitored and only those with changes of less than 20% during experiments were analyzed. The series resistance ranged from 10 to 30 MΩ and was uncompensated.
Behavioral studies
All behavioral experiments were performed on 3- to 4-month-old male C57Bl/6 congenic Sema5A−/− mice and their WT littermates.
Open field
Request a detailed protocolThe test was performed using the Photobeam Activity System (San Diego Instruments). Mice were placed individually in the chambers and were allowed to move freely for 30 min. The number of beam interruptions caused by both horizontal and vertical movement was recorded. Total number of beam breaks was used to assess general locomotor activity. Percentage of time spent in the center was used to assess the anxiety level. Rearing represents the total number of vertical movements and was used to assess exploratory behavior.
Elevated plus maze
Request a detailed protocolElevated plus maze was used to assess anxiety levels. Mice were placed in the center of the elevated plus maze and were allowed to move freely in the maze for 5 min. The behavior of the mice was videotaped and analyzed by Any-maze video tracking system (Stoelting Co.).
Fear conditioning
Request a detailed protocolOn day 1, mice were placed in a fear-conditioning chamber for a total of 300 s. A 2000 Hz tone lasting 20 s was delivered at 210 s, followed by a 2-s foot shock. For contextual fear conditioning, mice were returned to the same chamber on day 2 and allowed to move freely for a total of 300 s. No tone or foot shocks were delivered. Freezing behavior was recorded and analyzed by FreezeScan software (Celver Sys Inc.) at 10-s intervals. For cued fear conditioning, mice were placed in a different chamber in a different room on day 2 and allowed to move freely for a total of 300 s. White noise was present for the duration of the test. A 2000 Hz tone lasting 20 s was delivered at 210 s. Freezing behavior was recorded and analyzed by FreezeScan software (Celver Sys Inc.) at 10-s intervals.
Social interaction studies
Request a detailed protocolThe three-chamber social interaction test was used as described previously (Yang et al., 2011). Briefly, the subject mouse was placed in the center chamber with nothing in the side chambers and allowed to move freely between chambers and acclimate for 5 min. Subsequently, a cage with a WT C57Bl/6 mouse and a cage with an inanimate object were placed in each of the side chambers. The subject mouse was allowed to enter each chamber freely and interact with the mouse or object for 5 min. Finally, the inanimate object was replaced with a new WT C57Bl/6 mouse, and the subject mouse was allowed to enter each chamber freely and interact with either mouse for 5 min. The behavior of the mice was videotaped and analyzed by Any-maze video tracking system (Stoelting Co.). Video of each subject mouse was reviewed and time engaged in nose-to-nose interaction with other mice and sniffing the inanimate object was quantified manually.
References
-
Signaling between glia and neurons: focus on synaptic plasticityCurrent Opinion in Neurobiology 15:542–548.https://doi.org/10.1016/j.conb.2005.08.006
-
Prolonged sojourn of developing pyramidal cells in the intermediate zone of the hippocampus and their settling in the stratum pyramidaleThe Journal of Comparative Neurology 301:343–364.https://doi.org/10.1002/cne.903010303
-
The role of glial cells in synapse eliminationCurrent Opinion in Neurobiology 22:438–445.https://doi.org/10.1016/j.conb.2011.10.003
-
Role of leucine-rich repeat proteins in the development and function of neural circuitsAnnual Review of Cell and Developmental Biology 27:697–729.https://doi.org/10.1146/annurev-cellbio-092910-154111
-
Neuronal differentiation in the adult hippocampus recapitulates embryonic developmentThe Journal of Neuroscience 25:10074–10086.https://doi.org/10.1523/JNEUROSCI.3114-05.2005
-
An oligodendrocyte lineage-specific semaphorin, Sema5A, inhibits axon growth by retinal ganglion cellsThe Journal of Neuroscience 24:4989–4999.https://doi.org/10.1523/JNEUROSCI.4390-03.2004
-
Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic functionAnnual Review of Neuroscience 17:341–371.https://doi.org/10.1146/annurev.ne.17.030194.002013
-
Crystal structure of the plexin A3 intracellular region reveals an autoinhibited conformation through active site sequestrationProceedings of the National Academy of Sciences of USA 106:15610–15615.https://doi.org/10.1073/pnas.0906923106
-
Semaphorin 5A and plexin-B3 inhibit human glioma cell motility through RhoGDIalpha-mediated inactivation of Rac1 GTPaseThe Journal of Biological Chemistry 285:32436–32445.https://doi.org/10.1074/jbc.M110.120451
-
Constitutional downregulation of SEMA5A expression in autismNeuropsychobiology 54:64–69.https://doi.org/10.1159/000096040
-
Where no synapses go: gatekeepers of circuit remodeling and synaptic strengthTrends in Neurosciences 36:363–373.https://doi.org/10.1016/j.tins.2013.04.003
-
Perisynaptic chondroitin sulfate proteoglycans restrict structural plasticity in an integrin-dependent manner18009–18017, The Journal of Neuroscience, 32, 18017a, 10.1523/JNEUROSCI.2406-12.2012.
-
Getting neural circuits into shape with semaphorinsNature Reviews Neuroscience 13:605–618.https://doi.org/10.1038/nrn3302
-
Cellular and synaptic network defects in autismCurrent Opinion in Neurobiology 22:866–872.https://doi.org/10.1016/j.conb.2012.02.015
-
Dendritic spine pathology in neuropsychiatric disordersNature Neuroscience 14:285–293.https://doi.org/10.1038/nn.2741
-
Mouse models of autism: testing hypotheses about molecular mechanismsCurrent Topics in Behavioral Neurosciences 7:187–212.https://doi.org/10.1007/7854_2010_113
-
Secreted semaphorins modulate synaptic transmission in the adult hippocampusThe Journal of Neuroscience 25:3613–3620.https://doi.org/10.1523/JNEUROSCI.5255-04.2005
-
Guidance molecules in synapse formation and plasticityCold Spring Harbor Perspectives in Biology 2:a001842.https://doi.org/10.1101/cshperspect.a001842
-
Synaptic organizing complexesCurrent Opinion in Neurobiology 21:132–143.https://doi.org/10.1016/j.conb.2010.08.016
-
Molecular cloning and mapping of human semaphorin F from the Cri-du-chat candidate intervalBiochemical and Biophysical Research Communications 242:685–691.https://doi.org/10.1006/bbrc.1997.8027
-
Roles of semaphorin-6B and plexin-A2 in lamina-restricted projection of hippocampal mossy fibersThe Journal of Neuroscience 30:7049–7060.https://doi.org/10.1523/JNEUROSCI.0073-10.2010
-
Synapse formation on adult-born hippocampal neuronsThe European Journal of Neuroscience 33:1062–1068.https://doi.org/10.1111/j.1460-9568.2011.07604.x
-
Synapse formation on neurons born in the adult hippocampusNature Neuroscience 10:727–734.https://doi.org/10.1038/nn1908
-
Converging pathways in autism spectrum disorders: interplay between synaptic dysfunction and immune responsesFrontiers in human Neuroscience 7:738.https://doi.org/10.3389/fnhum.2013.00738
-
Automated three-chambered social approach task for miceCurrent Protocols in Neuroscience, Chapter 8:Unit 8.26, 10.1002/0471142301.ns0826s56.
-
The cis side of juxtacrine signaling: a new role in the development of the nervous systemTrends in Neurosciences 35:230–239.https://doi.org/10.1016/j.tins.2011.12.003
-
An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortexThe Journal of Neuroscience 34:11929–11947.https://doi.org/10.1523/JNEUROSCI.1860-14.2014
Article and author information
Author details
Funding
National Institute of Neurological Disorders and Stroke
- Roman J Giger
National Institute of Mental Health
- Alex L Kolodkin
- Guo-li Ming
Howard Hughes Medical Institute
- Alex L Kolodkin
Dr. Miriam and Sheldon G. Adelson Medical Research Foundation
- Guo-li Ming
- Roman J Giger
Maryland Stem Cell Research Fund
- Juan Song
- Guo-li Ming
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Yutaka Yoshida for providing us with the Plxna1−/−, Plxna2−/−, and Plxna3−/− mice, Fumikazu Suto for PlexA2SD-Fc and PlexA4SD-Fc constructs and anti-PlexA2 antiserum, and Andreas Puschel for the PlexA1(RasGAP-RR) construct. We thank Onanong Chivatakarn and Ryota Matsuoka for their help with the initial analysis of the Sema5A hippocampal formation, Martín Riccomagno for assistance with viral vector injections, Xiao-Feng Zhao for help with histochemical procedures, and Dontais Johnson for technical assistance. We thank Alcino Silva, Jack Parent, and members of the Kolodkin and Giger laboratories for critical reading of the manuscript. This work is supported by the Maryland Stem Cell Research Fund (JS and G-lM); NS048271 and HD069184 (G-lM); MH59199 (ALK); NS081281 (RJG); and the Dr Miriam and Sheldon Adelson Medical Research Foundation (G-lM and RJG). ALK is an investigator of the Howard Hughes Medical Institute.
Ethics
Animal experimentation: All mice used in this study were housed and cared for in accordance with NIH guidelines, and all research conducted was done with the approval of the University of Michigan Medical School (UCUCA protocols PRO00002466 and PRO00001645) and The Johns Hopkins University (MO14M50 and MO12M381) Committees on Use and Care of Animals. All surgery was performed under sodium pentobarbital anesthesia, and every effort was made to minimize suffering.
Copyright
© 2014, Duan et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,012
- views
-
- 641
- downloads
-
- 122
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 122
- citations for umbrella DOI https://doi.org/10.7554/eLife.04390
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells
eLife 3:e04390.
https://doi.org/10.7554/eLife.04390




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}