Drug Discovery: How to exploit the recycling system of a cell
Nature has inspired the design of improved inhibitors for cancer-causing proteins.
- Department of Chemistry, Faculty of Natural, Mathematical and Engineering Sciences, King’s College London, United Kingdom
Each of our trillions of cells bustles with activity in a carefully choreographed quest to keep us healthy. At their core, quality control mechanisms ensure that the right proteins are in the right place at the right time and that they are rapidly removed and recycled when necessary (Thompson and De-Souza, 2023). This relies on waste disposal systems such as the ubiquitin-proteasome pathway. In a modern therapeutic twist, these systems can sometimes be hijacked to specifically remove disease-causing proteins (Koszła and Sołek, 2024).
Ubiquitin is a small molecule aptly named for its ubiquitous presence throughout the body. It serves as a molecular ‘rubbish tag’, with long chains of ubiquitin being added onto a protein to earmark it for destruction. This labelling process requires three types of enzymes working in sequence. E1s and E2s first work generically across the cell to activate and prepare ubiquitin for attachment. E3 ligases then ensure that the tag is added to the right protein; as such, hundreds of specific human E3 enzymes exist, each with precise targets that ensure specificity (Müller and Hoppe, 2024).
E3 ligases are considered important candidates for modern therapeutic development, as they are mutated or present in abnormal levels in a range of diseases. APC/C (short for anaphase promoting complex/cyclosome), for example, operates during cell division and is implicated in certain types of cancer (Greil et al., 2022). E3 inhibitors, created to bind the enzymes and stop them from acting on their targets, therefore represent a promising approach. However, designing compounds that can act on E3 ligases is often a challenge. The activity of these large, multi-unit enzymes heavily relies on flexible regions that lack the neat crevices into which small molecules can be designed to fit. Various strategies are afoot to try and get past these problems (Rodríguez-Gimeno and Galdeano, 2025). Now, in eLife, Laura Itzhaki and colleagues – including Rohan Eapen as first author – report important advances in designing compounds that can block E3 activity (Eapen et al., 2025b).
The researchers, who are based at the University of Cambridge, AstraZenecaCambridge, the MRC Laboratory of Molecular Biology and the p53 Laboratory in Singapore, focused their efforts on the enzyme APC/C and its substrates (Okoye et al., 2022; Höfler et al., 2024). They zoned in on Cdc20, a key region in APC/C that recognises a motif, aptly known as the Destruction box (D-Box), on proteins targeted for disposal (Okoye et al., 2022). Binding takes place by coordinating multiple such weak interactions, a common biological strategy that confers strength in numbers while allowing for versatility (Isaacson and Díaz-Moreno, 2018). The goal here was to create molecules that would bind tightly to Cdc20, displacing its natural substrate.
The team started by designing ‘D-Box peptides’ that could bind to this APC/C region, based on analyses of the protein regions it naturally recognises. Different methods were applied to measure how strongly each of these peptides could associate with APC/C. This, in turn, revealed which amino acids were most important for tight binding. Peptide design was refined by introducing unnatural amino acids thought to provide a better fit and stronger binding. These are distinct from the 20 amino acids naturally encoded by our genes and include additional chemical groups that can nestle more closely into the protein’s surface. The atomic structure of the best four candidates was examined via X-ray crystallography, revealing where and how the interactions occurred (Figure 1). Further experiments in live cells confirmed that the D-Box peptides could stabilise APC/C and inhibit its function.
Figure 1
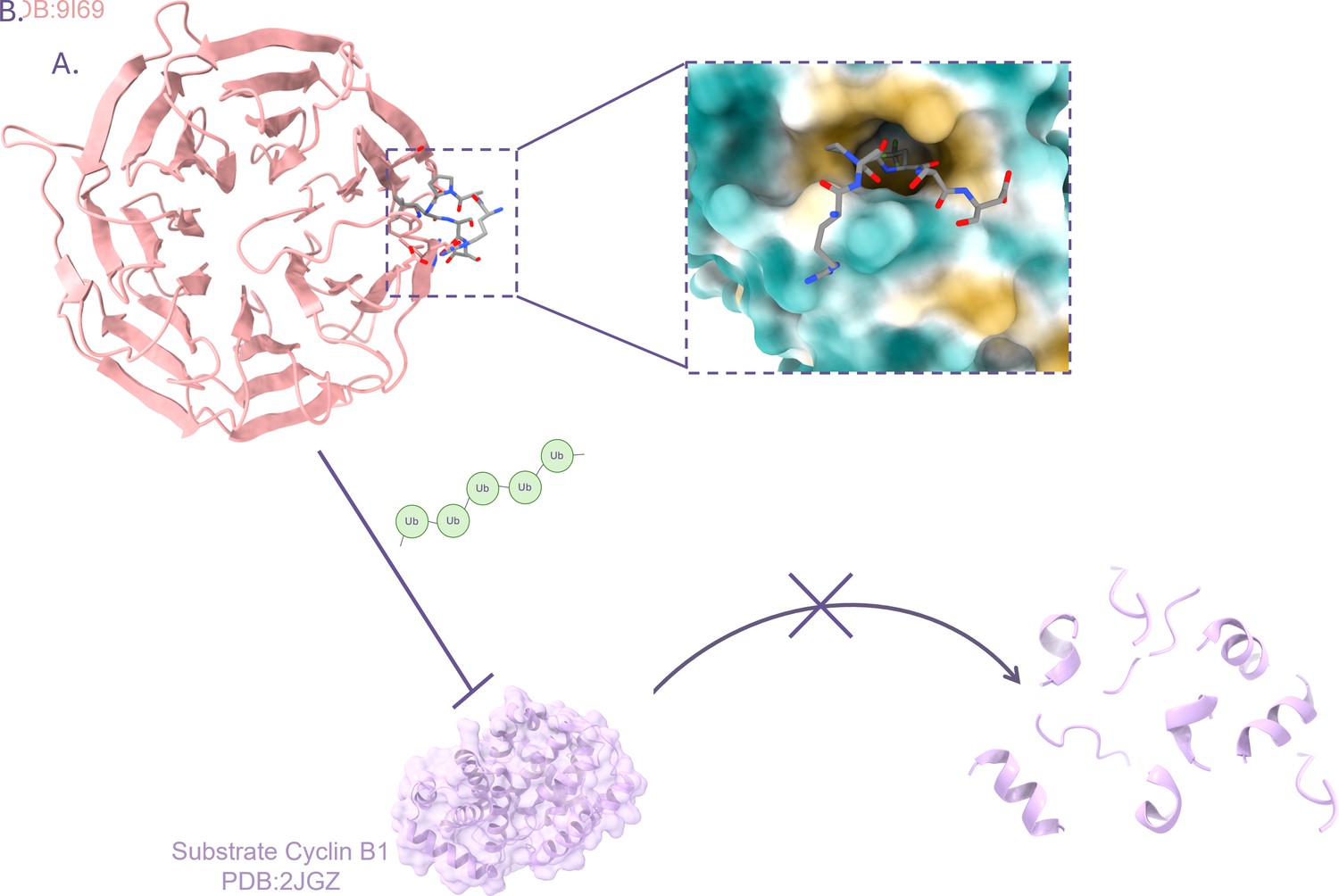
Designing artificial peptides that block the activity of the E3 ligase APC/C.
(A) The E3 ligase APC/C is often implicated in cancers. It targets certain proteins important for cell division, such as Cyclin B1. Eapen et al. focused on a region in APC/C, known as the Cdc20 WD40 domain (pink banded structure), which is part of an element that recognises ‘D-box’ regions in target substrates. Based on information from natural APC/C, the team designed artificial peptides (inset), such as the D20 peptide (grey), that could bind to Cdc20 by recognizing a hydrophobic pocket (yellow) which is part of the Cdc20 D-box (whose hydrophilic areas are in turquoise). (B) The binding of the D20 peptide to the Cdc20 D-box prevents the addition of chains of ubiquitin (green) onto the APC/C substrate Cyclin B1 (lilac). Loss of ubiquitination prevents Cyclin B1 from being targeted for degradation.
Image credit: Crystal structure of human Cdc20 WD40 domain bound to synthetic D-box peptide D20 based on data deposited by Eapen et al., 2025a (CC BY 4.0); crystal structure of Cyclin B1 created using atomic coordinates deposited by Brown et al., 2007 (CC0). Figure created using Chimera X (Meng et al., 2023).
These findings represent an exciting launch pad for a range of scientific tools and potential therapeutics. Beyond the design of E3 inhibitors, these results could also inform the design of PROTACs, a new class of drugs launched in 2001 that feature a module designed to bind a specific target, such as a cancer-causing protein, attached to a module that recruits the ubiquitin-proteasome pathway to destroy it (Li and Crews, 2022). As for E3 inhibitors, the hope is that PROTACs have minimal side effects as they target only the disease culprit and nothing else. However, the challenge to find effective and selective versions of both modules persists. The work by Eapen et al. potentially offers solutions to the refinement of these promising therapeutics.
References
-
DataCrystal structure of phospho-CDK2 in complex with Cyclin BWorldwide Protein Data Bank.https://doi.org/10.2210/pdb2jgz/pdb
-
DataCrystal structure of human Cdc20 bound to synthetic D-box peptide D20Worldwide Protein Data Bank.https://doi.org/10.2210/pdb9i69/pdb
-
Editorial: Weak interactions in molecular machineryFrontiers in Molecular Biosciences 5:117.https://doi.org/10.3389/fmolb.2018.00117
-
PROTACs: past, present and futureChemical Society Reviews 51:5214–5236.https://doi.org/10.1039/d2cs00193d
-
UPS-dependent strategies of protein quality control degradationTrends in Biochemical Sciences 49:859–874.https://doi.org/10.1016/j.tibs.2024.06.006
-
Counting degrons: lessons from multivalent substrates for targeted protein degradationFrontiers in Physiology 13:913063.https://doi.org/10.3389/fphys.2022.913063
-
Drug discovery approaches to target E3 ligasesChembiochem 26:e202400656.https://doi.org/10.1002/cbic.202400656
-
A year at the forefront of proteostasis and agingBiology Open 12:bio059750.https://doi.org/10.1242/bio.059750
Article and author information
Author details
Publication history
Copyright
© 2025, Evans et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 521
- views
-
- 48
- downloads
-
- 0
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Drug Discovery: How to exploit the recycling system of a cell
eLife 14:e105995.
https://doi.org/10.7554/eLife.105995




{kind=link}