Coordinated control of terminal differentiation and restriction of cellular plasticity
- Howard Hughes Medical Institute, Columbia University, United States
Abstract
The acquisition of a specific cellular identity is usually paralleled by a restriction of cellular plasticity. Whether and how these two processes are coordinated is poorly understood. Transcription factors called terminal selectors activate identity-specific effector genes during neuronal differentiation to define the structural and functional properties of a neuron. To study restriction of plasticity, we ectopically expressed C. elegans CHE-1, a terminal selector of ASE sensory neuron identity. In undifferentiated cells, ectopic expression of CHE-1 results in activation of ASE neuron type-specific effector genes. Once cells differentiate, their plasticity is restricted and ectopic expression of CHE-1 no longer results in activation of ASE effector genes. In striking contrast, removal of the respective terminal selectors of other sensory, inter-, or motor neuron types now enables ectopically expressed CHE-1 to activate its ASE-specific effector genes, indicating that terminal selectors not only activate effector gene batteries but also control the restriction of cellular plasticity. Terminal selectors mediate this restriction at least partially by organizing chromatin. The chromatin structure of a CHE-1 target locus is less compact in neurons that lack their resident terminal selector and genetic epistasis studies with H3K9 methyltransferases suggest that this chromatin modification acts downstream of a terminal selector to restrict plasticity. Taken together, terminal selectors activate identity-specific genes and make non-identity-defining genes less accessible, thereby serving as a checkpoint to coordinate identity specification with restriction of cellular plasticity.
https://doi.org/10.7554/eLife.24100.001Introduction
The acquisition of differentiated cell identities in a multicellular organism is accompanied by the loss of developmental plasticity. Undifferentiated cells are pluripotent and have the potential to activate diverse categories of genes. As differentiation progresses, cells activate cellular identity-specific genes as well as repress non-identity-specific genes, thereby losing pluripotency and attaining a differentiated state that is restricted. In this work, we aimed to uncover molecules and mechanisms that coordinate identity specification with the restriction of cellular plasticity.
The progressive loss of developmental plasticity in differentiating cells was first observed in pioneering somatic nuclear transplant experiments (Briggs and King, 1952; Gurdon, 1960, 1962). When nuclei from progressively more differentiated frog intestinal cells were transplanted into enucleated eggs, it was observed that egg cells containing less differentiated nuclei developed into tadpoles more efficiently than eggs containing more differentiated nuclei (Gurdon, 1960, 1962). Similar trends have been noted in somatic nuclear transplant experiments performed since (Gurdon and Wilmut, 2011).
Experiments in which transcription factors (TFs) are ectopically expressed in progressively more differentiated cells also demonstrate restriction of plasticity. For instance, in C. elegans, the overexpression of myogenic helix-loop-helix TF hlh-1/MyoD early in embryonic development leads to the activation of muscle genes in all somatic cells (Fukushige and Krause, 2005). However, the ability of hlh-1 to activate its target genes decreases and is completely lost by the time the embryonic cells are post-mitotic. Similar observations are made when endodermal GATA TFs end-1 and elt-7 (Zhu et al., 1998), ectodermal GATA TFs elt-1 and elt-3 (Gilleard and McGhee, 2001; Gilleard et al., 1999; Page et al., 1997) and pharyngeal FoxA TF pha-4 (Horner et al., 1998) are ectopically expressed in the worm embryo at different developmental time points. Likewise, the overexpression of pluripotency TFs c-Myc, Klf4, Sox2, and Oct4 in non-terminally differentiated Pro-B cells leads to the formation of iPSCs, but expression in differentiated B lymphocytes does not (Hanna et al., 2008). In all of these cases, restriction of plasticity is apparent in the inability of more differentiated cells to activate non-identity-specific genes in response to ectopically expressed TFs.
There is accumulating evidence that gene repression mediated by chromatin structure and organization plays an important role in restricting cellular plasticity (Meister et al., 2011). In C. elegans and mice, electron microscopy shows mostly open euchromatin in cells of early embryos and an accumulation of closed heterochromatic regions in differentiated cells (Efroni et al., 2008; Leung et al., 1999; Park et al., 2004). Differentiation is also coupled with changes in covalent histone modifications. These histone modifications, in conjunction with one another and their effector proteins can influence chromatin structure, nucleosome-DNA interactions, as well as accessibility to TFs (Greer et al., 2014; Tessarz and Kouzarides, 2014). The presence of activating and repressive chromatin marks can divide the genome of a differentiated cell into genes that can be expressed and not expressed, thereby contributing to restriction of plasticity (Mohn and Schübeler, 2009).
The repressive histone modifications H3K27me3 and H3K9me3 have been specifically implicated in the loss of plasticity. These marks are generally found in distinct chromatin regions in various species including humans, Drosophila, mice, and C. elegans (Ernst and Kellis, 2010; Filion et al., 2010; Ho et al., 2014). H3K27me3 is enriched on developmentally regulated, cell-type-specific genes (Boyer et al., 2006; Filion et al., 2010; Ho et al., 2014; Oktaba et al., 2008). A correlation between loss of H3K27me3, chromatin compaction, and plasticity has been shown in C. elegans embryos. The depletion of H3K27me3 in embryos leads to an extension of the time during which chromatin stays more ‘open’ and a correlated increase in the window of time during which an ectopically expressed TF, pha-4, can activate its target genes in embryonic cells (Yuzyuk et al., 2009). In previous studies, we have also shown that loss of H3K27me3 depositing polycomb repressive complex 2 (PRC2) in the germ cells of C. elegans leads to a gain of plasticity, as measured by the ability of three different identity-specifying TFs to activate their target genes (Patel et al., 2012). On the other hand, H3K9me3 is enriched at repeat-rich regions which remain repressed in all cell types, but there is accumulating evidence that it is also involved in the repression of cell-type-specific genes (Allan et al., 2012; Feldman et al., 2006; Garrigues et al., 2015; Ho et al., 2014; Loh et al., 2007; Martens et al., 2005; Towbin et al., 2012). As for restricting plasticity, it has been observed that removing methyl transferases that deposit H3K9me3 increases the efficiency of fibroblast to iPSC reprogramming (Soufi et al., 2012). Furthermore, H3K9me3 is enriched at targets of iPSC-inducing ‘Yamanaka’ factors that fail to become activated in fibroblasts that do not convert into iPSCs (Soufi et al., 2012; Soufi and Zaret, 2013).
During differentiation, cells activate identity-specific genes and form active and repressive chromatin landscapes (Meister et al., 2010; Mohn and Schübeler, 2009). How is the establishment of this chromatin-level organization coordinated with the transcription of identity-specific effector genes? Certain identity-specifying TFs facilitate the formation of active chromatin landscapes at cis-regulatory regions that they bind. The binding of these TFs leads to formation of more ‘open’ euchromatin by facilitating changes in histone modifications and chromatin accessibility (Fakhouri et al., 2010; Iwafuchi-Doi and Zaret, 2014; Zaret and Carroll, 2011, 2016). These findings have led to the hypothesis that TFs that activate identity-specific genes might also function as organizers of identity-specific active chromatin (Natoli, 2010). TFs that regulate the formation of open chromatin landscapes are straight-forward to identify since their loss generally leads to the loss of cellular identity. Much less is known about the organization of heterochromatin that mediates restriction of plasticity. Genetic removal of heterochromatin organizers is not necessarily expected to lead to the derepression (i.e. activation) of numerous non-identity-specific genes under wildtype conditions. However, the loss of heterochromatin organizers might make genes more accessible, so that they can be activated if trans-activators are also present in the cell. For this reason, the ectopic expression of TFs represents an experimental strategy for uncovering mechanisms that regulate heterochromatin organization and plasticity restriction.
In this work, we use a TF overexpression assay to show that cellular identity-specifying TFs also regulate the restriction of cellular plasticity at least partially by directing the formation of repressive chromatin in C. elegans neurons. In various animal species, it has been shown that the terminal differentiation of individual neuron types is carried out by one or more TFs called terminal selectors, which activate expression of a variety of identity-specific effector genes whose protein products shape the terminally differentiated state of a cell (Hobert, 2011, 2016). In the absence of these TFs, neurons still migrate properly, extend processes, and acquire a ‘generic’ neuronal state characterized by the expression of pan-neuronal markers; but these neurons do not express neuron-type-specific genes such as ion channels, neurotransmitters, neurotransmitter receptors, etc. (Hobert, 2011; Stefanakis et al., 2015). We demonstrate here that terminal selector TFs also function to restrict cellular plasticity. To assay restriction, we have used the overexpression of terminal selector CHE-1, a Zn-finger TF, which is normally only expressed in two glutamatergic ASE sensory neurons and which specifies the identity of these neurons by activating expression of ASE-specific genes (Etchberger et al., 2007; Patel et al., 2012; Serrano-Saiz et al., 2013; Uchida et al., 2003). We show here that if CHE-1 is ectopically expressed during embryonic and early larval development, when cells are dividing and differentiating, it can activate expression of ASE-specific effector genes in many cell types. If CHE-1 is expressed at later stages when all cells have differentiated, it is no longer able to activate ASE effector genes in most cells, showing that they are restricted. We then expressed CHE-1 in wildtype and mutant animals in which terminal selectors of specific neuron types are genetically eliminated to assay whether the cells affected in the terminal selector mutants would remain responsive to CHE-1. We found this to be the case in five of seven terminal selector mutants tested, showing that several terminal selector TFs are required for the loss of plasticity.
To understand the mechanistic basis of the plasticity-restricting function of terminal selectors, we focused on unc-3, a COE (Collier/Olf1/Ebf) TF which specifies the identity of cholinergic motor neurons (MNs). We identified an allele of unc-3 in which cholinergic MNs express identity-specific markers and function normally, yet are still plastic. This finding strongly suggests that even in an otherwise appropriately specified neuron, terminal selector function is required to restrict plasticity. Next, using a LacI/LacO spot assay (Meister et al., 2010; Towbin et al., 2012; Yuzyuk et al., 2009) to tag transgenic loci, we found that the locus of a reporter of a CHE-1 target gene is less condensed in unc-3 mutant neurons compared to wildtype neurons, suggesting that terminal selector TFs are required for the formation of identity-specific heterochromatin. We also observe that removal of H3K27me and H3K9me depositing enzymes leads to loss of restriction in otherwise normally differentiated neurons, further emphasizing that chromatin does play a role in restricting plasticity of neurons. While the loss of H3K27 methyltransferase showed an additive effect with unc-3, the loss of H3K9 methyltransferases did not modify the unc-3 phenotype suggesting that correct deposition of H3K9me3 may be downstream of unc-3 function. Overall, this work demonstrates a novel role for terminal selector-like TFs in coordinating the processes of identity-specific gene activation with restriction of cellular plasticity, the latter being mediated at least partially through the correct organization of chromatin in differentiated cells.
Results
Differential ability of CHE-1 to activate target genes
In order to gain insights into the mechanisms that restrict plasticity, we probed cellular plasticity in many cell types of C.elegans throughout different developmental stages. To this end, we ubiquitously expressed Zn-finger TF CHE-1 at different time points in developing worms. CHE-1 is normally expressed only in the ASEs, two sensory neurons located in the head, and acts as a terminal selector by activating expression of numerous ASE-specific effector genes (Chang et al., 2003; Etchberger et al., 2007; Rinn et al., 2007; Tursun et al., 2009; Uchida et al., 2003). We used a previously described transgenic worm strain which contains a heatshock inducible promoter driving che-1 expression (CHE-1hs), and a reporter of a CHE-1 target gene, gcy-5, that is normally expressed in ASER (Patel et al., 2012; Tursun et al., 2011). In this assay, non-ASE cells that activate expression of gcy-5 in response to CHE-1hs expression are considered plastic, whereas cells that do not activate gcy-5 expression are considered restricted. In this study, we used several tools to detect the expression of gcy-5, including two kinds of transgenic reporters (gcy-5prom::gfp and gcy-5fosmid::gfp), smFISH probes against gcy-5 mRNA, and an allele of gcy-5 in which the endogenous locus is tagged with mNeonGreen using CRISPR/Cas9-mediated genome engineering (Figure 1—figure supplement 1A,B,C). gcy-5prom::gfp uses 3.2 kb of regulatory sequences directly upstream of the start codon of the gcy-5 gene, while the gcy-5fosmid::gfp contains ~33 kb of DNA surrounding the gcy-5 locus, which should include all potential cis-regulatory regions (Figure 1—figure supplement 1A). smFISH and the mNeonGreen tagged allele validate the endogenous transcription of gcy-5, but because the mNeonGreen tagged allele was difficult to detect by standard microscopy, we utilized smFISH for the detection of the endogenous transcript in our CHE-1 misexpression experiments. gcy-5 is normally expressed in the ASER and RIGL/R neurons. Expression of gcy-5 in the ASER is che-1 dependent (Chang et al., 2003; Uchida et al., 2003), while expression in the RIG neurons is dependent on lim-6 (Figure 1—figure supplement 1D), and should not be relevant in CHE-1 overexpression assays.
When CHE-1hs is induced during early embryonic stages (<300 min), gcy-5prom::gfp is broadly activated in numerous cells (Figure 1Aiii). However, these embryos become deformed and arrest, making it difficult to score exactly which and to what extent non-ASE cells are CHE-1-responsive. Induction of CHE-1hs during larval development results in an increasingly restricted activation of gcy-5, and the animals retain a normal overall morphology (Figure 1Aiv–viii). With the exception of a few neurons in the head and tail, cells that express gcy-5 in response to larval CHE-1hs are those that have just been generated and hence are still differentiating (Figure 1B). For instance, CHE-1hs induction in the L2/L3 stage activates gcy-5 in seam cells and ventral nerve cord (VNC) neurons, which are dividing and differentiating at this time. As some VNC neurons are born during embryonic stages, we examined whether VNC cells that activate gcy-5 at L2 are those that are newly born. To this end, we used a pan-neuronal marker, rab-3prom::rfp, which is brightly expressed in VNC neurons that were born during embryonic stages, but is dim in cells that are still undergoing differentiation. In these worms, activation of gcy-5fosmid::gfp after L2 CHE-1hs expression is enriched in rab-3low cells (Figure 1—figure supplement 2A), showing that cells that are dividing and differentiating are more receptive to CHE-1 than cells that have already differentiated. We also observe that vulval muscle cells, which are dividing and differentiating at the L3 stage are receptive to CHE-1hs at this stage (Figure 1B). At the L4 and adult stages, when all tissues have differentiated, the only cells that activate gcy-5prom::gfp in response to CHE-1hs are a handful of neurons and pharyngeal muscle cells, suggesting that these differentiated cells provide some molecular context which makes them amenable to che-1 activity (Figure 1Aviii and B). Such context-dependent activity has been previously observed for TF elt-7, which when overexpressed, leads to a context-dependent activation of endodermal identity in specific adult tissues (Riddle et al., 2016, 2013). The CHE-1 responsive neurons include the RIS, CEP, ASK, and ASI neurons. These neurons do not bear any obvious relationship to ASE, and it is unclear why these cells remain responsive to CHE-1.
Figure 1 with 3 supplements see all
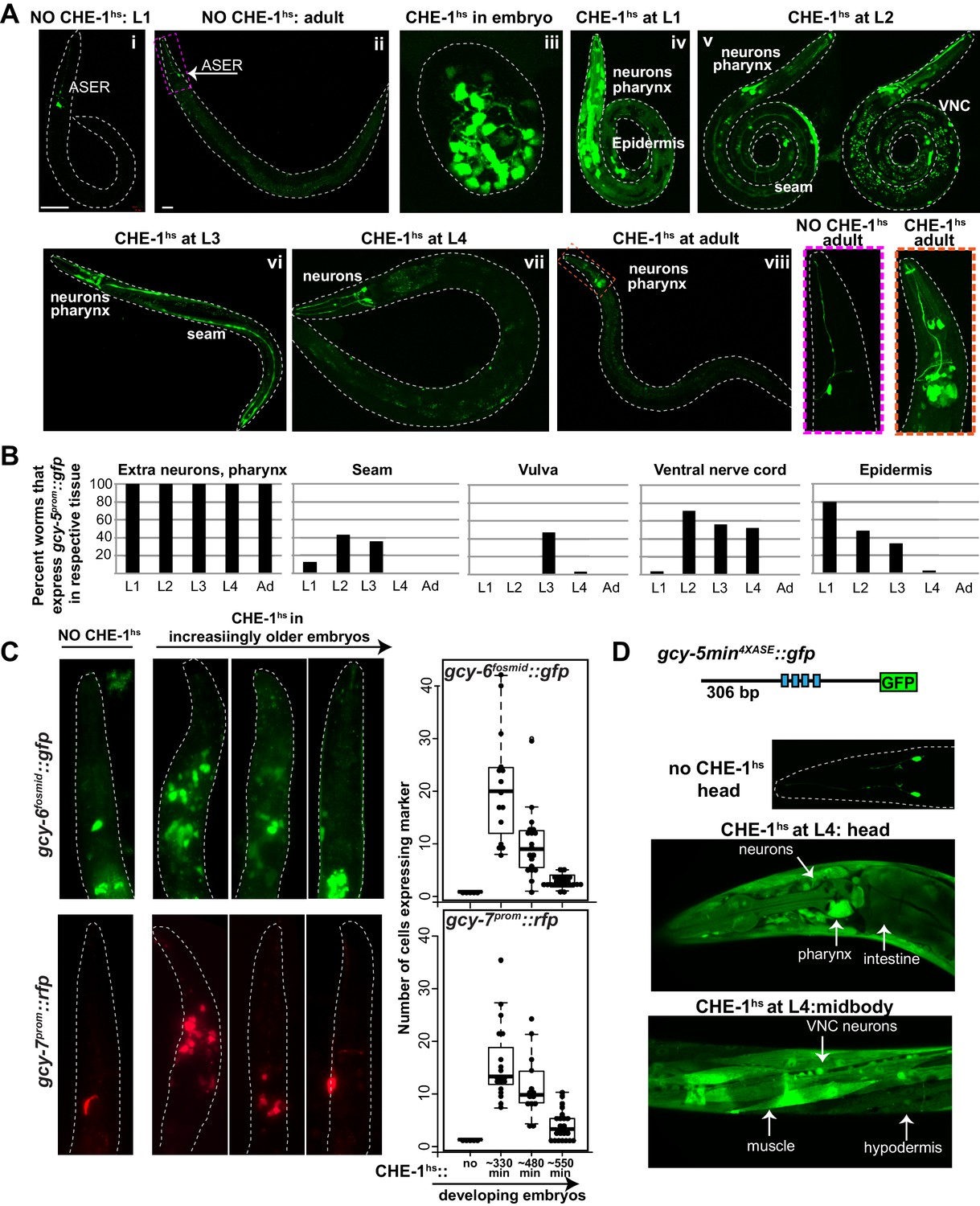
Expression of ASE markers in response to CHE-1hs at different developmental stages.
(A) Expression of gcy-5prom::gfp becomes less broad as CHE-1hs is induced at later stages. (i) L1 worm without CHE-1hs, bright expression in ASER; (ii) adult without CHE-1hs, expression in ASER (arrow); (iii) embryonic CHE-1hs, broad gcy-5prom::gfp induction in deformed embryos; (iv) L1 CHE-1hs,expression of gcy-5prom::gfp in various tissues including hypodermis, various neurons, muscles, pharynx. (v) L2 CHE-1hs at two different focal points, gcy-5prom::gfp in seam, muscles, ventral nerve cord, other neurons and pharynx; (vi) L3 CHE-1hs, gcy-5prom::gfp in seam, vulva cells, neurons and pharynx; (vii) L4 and (viii) adult CHE-1hs, gcy-5prom::gfp is seen only in neurons and pharynx. Higher magnification of the heads in (i) and (viii). (B) Quantification of the expression of gcy-5prom::gfp in various tissues after CHE-1hs induction at various ages, as listed on the X-axis. n => 20 for each stage. (C) Expression of gcy-6fosmid and gcy-7prom in response to CHE-1hs at different stages. Quantification of the ectopic expression: every dot in this graph represents a worm and the number on the y-axis represents the total number of marker-expressing neurons counted. Ectopic expression decreases as the age of the embryos being heatshocked increases. (D) A synthetic ~300 bp region of the gcy-5 promoter in which the ASE motif has been multimerized four times can be broadly induced by CHE-1hs at all stages, including L4s, as shown here. Some of the tissues that express gcy-54XASE::gfp are labeled. gcy-54XASE::gfp is normally expressed only in the two ASEs.
The same pattern of gcy-5prom::gfp induction is also seen when CHEhs is induced with a single copy hsprom::che-1 transgene generated by MiniMos-mediated genome engineering (Frøkjær-Jensen et al., 2014). Additionally, gcy-5 smFISH of these worms shows that expression of gcy-5prom::gfp overlaps with the expression of endogenous gcy-5 mRNA, indicating that the endogenous gcy-5 gene is induced by ectopic CHE-1hs and that the transgene gcy-5prom::gfp reliably replicates the activity of the endogenous gcy-5 locus (Figure 1—figure supplement 2B).
We confirmed that the restricted activation of gcy-5 in adults is not due to an inability to efficiently induce CHE-1hs. Ater heatshock induction in adults, both che-1 mRNA and protein can be detected in all cell types (Figure 1—figure supplement 2C,D). Furthermore, to ensure that the expressed CHE-1 protein is functional, we generated a synthetic strong reporter that contains a 4X multimerized CHE-1 binding site (‘ASE motif’), taken from the gcy-5 locus, driving gfp. We find that this gcy-54XASE::gfp reporter is expressed broadly, in all somatic cell types at every stage of development, in response to CHE-1hs expression from both the multi-copy and single-copy heatshock-inducible transgenes (Figure 1D). Therefore, the restricted activation of gcy-5 reflects the inability of CHE-1hs to activate its target genes in differentiated cells.
To determine the breadth of this phenomenon, we examined expression of two additional che-1 target genes, gcy-6 and gcy-7, in the heatshock assay(Chang et al., 2003; Uchida et al., 2003). These genes are receptor guanylate cyclases expressed exclusively in the ASEL neuron during normal development (Yu et al., 1997). If CHE-1hs is induced during early embryonic stages, the resulting arrested embryos and larvae activate broad expression of gcy-6fosmid::gfp and gcy-7prom::rfp. Expression of these markers also becomes more restricted as CHE-1hs is induced at later stages. However, ectopic induction of these genes is not as broad as gcy-5 and no ectopic expression is detected when CHE-1hs is induced in larval stages. (Figure 1C). Previous analysis has shown that while CHE-1 is sufficient to activate ASER-specific genes, it may require other co-activators to activate ASEL-specific genes (Etchberger et al., 2009; Hobert, 2014). This may explain the difference in the breadth of ectopic CHE-1 activity.
Since embryonic expression of CHEhs leads to deformation and developmental arrest, we expressed CHE-1 using tissue-specific promoters to understand more thoroughly its activity in various differentiating cell types. We utilized a muscle (unc-27prom), hypodermal (dpy-7prom), pan-neuronal (ric-19prom), and two neuron-type-specific drivers (unc-47prom expressed in GABAergic MNs, ift-20prom expressed in sensory neurons). With the exception of the hypodermal promoter, these promoters all become activated only in post-mitotic cells at the time that cells are initiating expression of various identity-specific genes. CHE-1 driven by all neuronal promoters resulted in broad ectopic expression of gcy-5prom::gfp, gcy-5fosmid::gfp, and ceh-36fosmid::yfp (another che-1 target, a homeodomain TF expressed in ASEL/R and AWC neurons) . However, gcy-6fosmid::gfp and gcy-7prom::rfp show restricted activation in only 1–2 extra cells (Figure 1—figure supplement 3A). Transgenic animals expressing CHE-1 in the hypodermis or muscle also showed gcy-5prom::gfp expression in the respective tissues (Figure 1—figure supplement 3b,c). In combination with the heatshock experiments, these experiments show that a wide range of cell types are receptive to CHE-1 activity after post-mitotic division while they are terminally differentiating. Once terminal differentiation is accomplished, most cells lose their multipotency and become refractory to CHE-1 activity.
Terminal selector unc-3 restricts plasticity of cholinergic motor neurons
Our finding that cells lose their receptivity to ectopically expressed CHE-1 after terminal differentiation led us to hypothesize that the loss of cellular plasticity might be coordinated with the acquisition of a terminal identity. In C. elegans, the expression of identity-specific genes in many neurons is established by single or combinations of terminal selector TFs. These TFs are generally expressed after the terminal cell division and are required for both the activation and maintenance of identity-specific effector gene batteries (Hobert, 2011). In the absence of such TFs, the mutant neurons are born and express numerous pan-neuronal genes that normally get activated at the same time as identity-specific genes, suggesting that multiple aspects of terminal differentiation proceed normally. However, the lack of identity- and function-specific genes which include neurotransmitter synthesis genes, ion channels, etc., indicate that these neurons fail to acquire their neuron-type-specific identity. We asked whether, in addition to specifying identity, these terminal selector TFs might also facilitate the process of restriction, thereby coordinating it with terminal differentiation. If this were the case, in the absence of a terminal selector gene, a cell would remain in a more plastic state, and in our experimental paradigm, retain its receptivity to CHE-1hs. Alternatively, if an independent mechanism triggers the restriction of cellular plasticity, cells would lose their receptivity to CHE-1hs regardless of the presence of the terminal selector.
To test these alternative hypotheses, we induced CHE-1hs in unc-3 mutants. unc-3 is a COE-type TF that acts as the terminal selector for A/B-type and AS cholinergic MN identity (Kratsios et al., 2011; Prasad et al., 1998). unc-3(-) cholinergic MNs are born and found in approximately their wildtype positions in the ventral nerve cord (VNC), retro-vesicular ganglion (RVG), and pre-anal ganglion (PAG) of the worm. They express pan-neuronal identity markers, and form axonal projections. However, these neurons lack expression of all the acetylcholine pathway genes, numerous other terminal identity genes, show defects in synaptogenesis and are non-functional (Kratsios et al., 2011; Prasad et al., 1998). To test if such unc-3(-) neurons retain plasticity, we induced CHE-1hs in wildtype and unc-3 mutant worms at various larval stages and scored the expression of gcy-5prom::gfp in the RVG, VNC, and PAG. In addition to fifty unc-3 dependent MNs, these ganglia contain 19 GABAergic MNs and 13 other unc-3-independent neurons (Figure 2A). To decrease the ambiguity in scoring, we used a red fluorescent GABAergic marker in the background and counted the number of gcy-5-positive cells that were non-GABAergic. In wildtype worms, we find that an average of 11 neurons activate gcy-5prom::gfp at the L2 stage upon ectopic CHE-1hs expression, and this number is reduced to an average of 2 neurons by the L4 and adult stages. In unc-3 mutants, a significantly higher number of neurons express gcy-5prom::gfp at every developmental stage upon ectopic CHE-1hs expression (Figure 2B). gcy-5prom::gfp expression in the VNC was not seen when unc-3 mutants were heatshocked in the absence of the hsprom::che-1 transgene, showing that the loss of unc-3 is not sufficient for an identity switch. We repeated these experiments using the single copy heatshockprom::che-1 transgene, and again observed that gcy-5prom::gfp expression is induced in significantly more unc-3(-) than wildtype cells. Additionally, smFISH of these worms showed that expression of endogenous gcy-5 mRNA was also induced in unc-3(-) cells to a similar extent (Figure 2C). On average, about 30% of affected neurons in unc-3 mutants express gcy-5 in response to CHE-1hs induction.
Figure 2
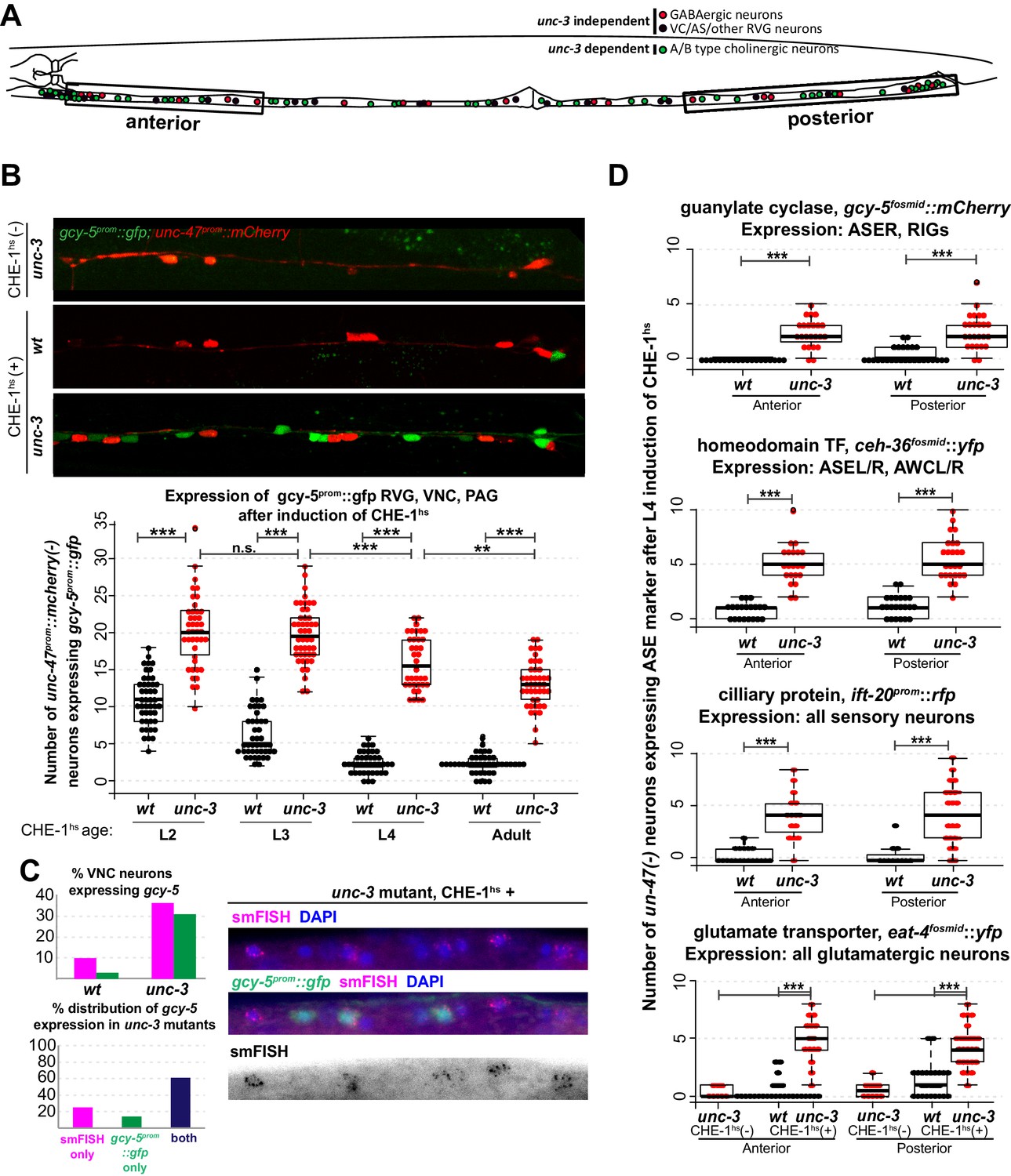
Several CHE-1 targets retain their CHE-1 responsiveness in unc-3 mutant cholinergic MNs.
For all heatshock experiments, three biological replicates were performed. The student t-test is used for statistical comparisons, *p<0.05, **p<0.01, ***p<0.001, n.s.= p>0.05. (A) A schematic representation of the neurons in the RVG, VNC, and PAG of the worm. In heatshock experiments represented in B, C, and D, the number of ASE marker expressing cells in the RVG, VNC, and PAG were scored. In B and D, unc-47, a marker of GABAergic identity (is used in the background and only unc-47(-) neurons are scored. This strategy ensures proper scoring of the ~50 unc-3 dependent cholinergic MNs . (B) Images of wildtype and unc-3 worms with and without CHE-1hs and a quantification of gcy-5prom induction at various stages of development. Every dot in this plot represents an individual worm. The entire RVG, VNC, and PAG of the worm is scored. A significantly higher number of gcy-5prom::gfp neurons are seen in unc-3 mutants as compared to wildtype in response to CHE-1hs induction. No gcy-5prom::gfp neurons are seen in unc-3 mutant worms that get heatshocked but do not contain the heatshockprom::che-1 array (0 cells in >20 worms). (C) gcy-5prom transgene (green bar) and gcy-5 mRNA (smFISH; pink bar) induction in heatshocked worms carrying a single copy insertion of heatshockprom::che-1. Induction of gcy-5 endogenous mRNA (smFISH) is similar to the induction of the gcy-5prom reporter. (D) Expression of various other ASE markers is seen in a significantly higher number of unc-47(-) RVG, VNC, and PAG neurons in unc-3 mutants as compared to wildtype after CHE-1hs induction. The scored markers and their wildtype (without CHE-1hs) expression patterns are listed above each plot. Every dot in these plots represents an individual worm. Markers are scored only in the anterior and posterior regions of the worm, as represented in A. These worms were heatshocked at the L4 stage.
To assess the breadth of CHE-1hs activity, we also examined the activation of other CHE-1 target genes in the VNCs of unc-3 mutants. Genes that showed increased induction after L4 CHE-1hs in unc-3 included the glutamate transporter eat-4fosmid::yfp, the homeodomain TF ceh-36fosmid::yfp, and the ciliary protein ift-20prom::rfp (Figure 2D). No induction of gcy-6fosmid::gfp or gcy-7prom::rfp was observed in wildtype or unc-3(-) neurons, which is consistent with the idea that che-1 is not sufficient for the induction of these genes, as discussed earlier (0 neurons induced in >20 worms checked). As the transgenic markers that show increased induction in unc-3(-) neurons are randomly integrated into the different chromosomes of the worm, this phenotype suggests that several regions of the genome remain receptive to CHE-1hs activity in the mutant condition.
unc-3 is required broadly for activation of both generic and sub-type-specific cholinergic MN identity features. The further diversification of cholinergic MNs into sub-types is controlled by the activity of various class-specific repressors, such as paired homeodomain TF unc-4, the nuclear receptor TF unc-55, and the T-box TF mab-9 (Kerk et al., 2017; Miller and Niemeyer, 1995; Pocock et al., 2008). The loss of these TFs results in MNs with mixed class identities and subsequent functional defects. We asked if these repressor TFs also contribute to the restriction of plasticity. Neither unc-4 mutants, nor unc-55; mab-9 double mutants, however, show increased induction of gcy-5prom::gfp in cholinergic MNs in response to CHE-1hs (Figure 3—figure supplement 1A). These data demonstrate that a disruption of function and identity-specific transcriptional programs does not automatically make cells more plastic.
Restriction of cellular plasticity is a function shared by several terminal selectors
We examined the expression of gcy-5 in response to adult CHE-1hs after removal of six other terminal selectors that control the identity of a wide range of different neuron types in the head and tail ganglia of the worm. Four of the six mutants showed a phenotype similar to unc-3 in that their removal allowed ectopic, CHE-1hs-dependent gcy-5 expression. These terminal selectors are ETS-domain TF ets-5, and homeodomain TFs ttx-1, ceh-14, and lin-11. Specifically, ets-5 is a terminal selector for the CO2 sensing BAG neurons (Guillermin et al., 2011). In our assay, 30% of ets-5(-) BAG neurons express gcy-5fosmid::gfp in response to adult CHE-1hs as opposed to 4% of wildtype BAG neurons (Figure 3A). ttx-1 is a terminal selector for thermosensory AFD neurons (Satterlee et al., 2001; Serrano-Saiz et al., 2013) and 23% of ttx-1(-) AFD neurons express gcy-5fosmid::gfp after adult CHE-1hs whereas 2% of wildtype AFD neurons do (Figure 3B). Removing both ttx-1 and ceh-14, the co-factor of ttx-1 in AFD specification, results in a 34.3% phenotype compared to 4.8% in matched wildtypes. ceh-14 also acts as a terminal selector in several distinct tail neuron classes (Serrano-Saiz et al., 2013). We found an average increase of five gcy-5fosmid-positive neurons in the tails of ceh-14 mutants upon CHE-1 induction (Figure 3C). Lastly, lin-11 is required for terminal identity specification of ASG neurons (Serrano-Saiz et al., 2013), and 30% of lin-11(-) ASG neurons express gcy-5fosmid::gfp as opposed to 2% of wildtype ASG neurons (Figure 3d).
Figure 3 with 1 supplement see all
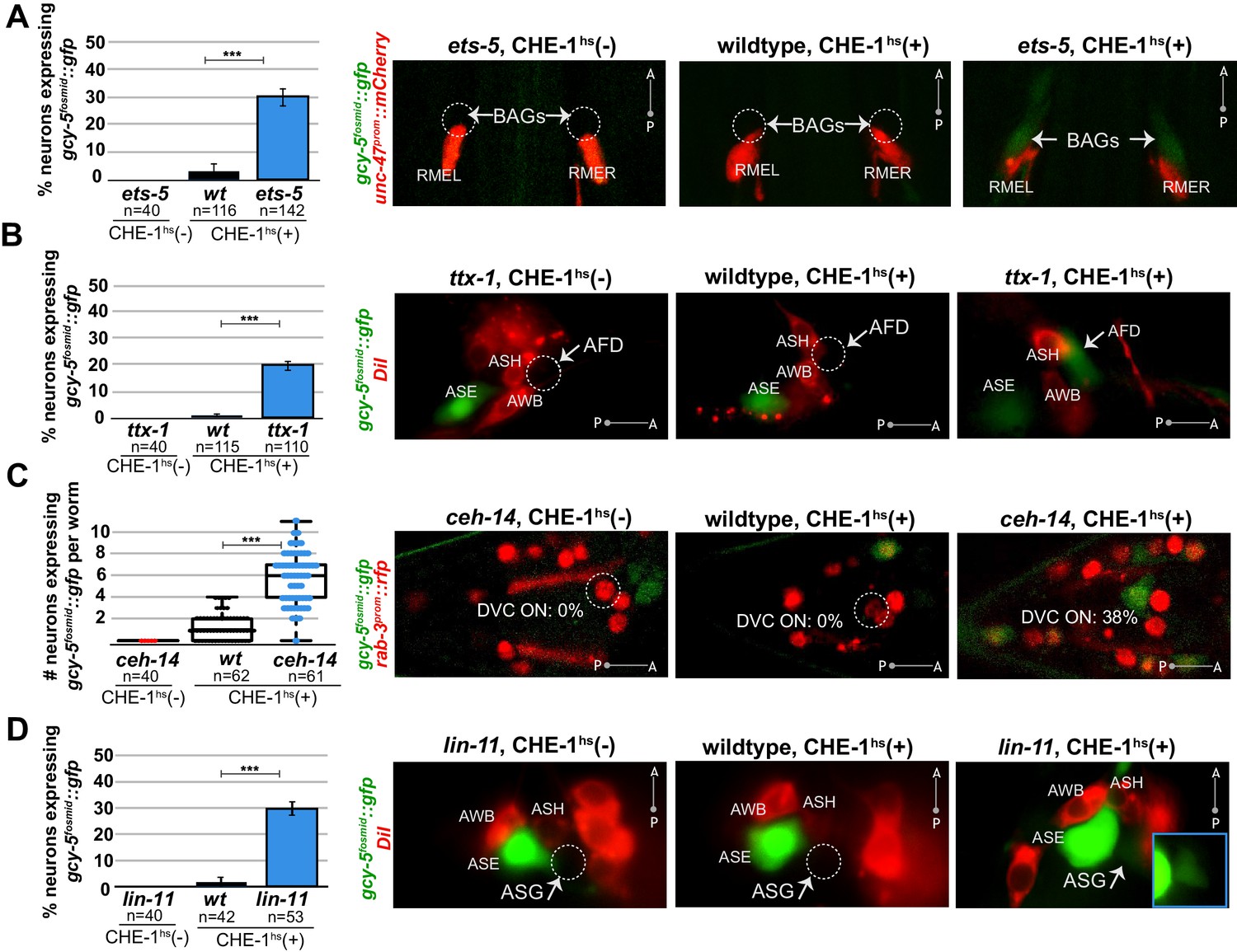
Several distinct terminal selectors control restriction of cellular plasticity in distinct neuron types.
CHE-1hs was induced in adults for all experiments except lin-11, for which L4s were heatshocked (lin-11 mutant adults are very short-lived due to internal hatching of progeny). The markers in red are used to help identify the neuron of interest. gcy-5fosmid::gfp is more frequenctly activated by CHE-1hs in BAG neurons of ets-5 mutants (panel A), in AFD neurons of ttx-1 mutants (panel B), in DVC and a few other unidentifiable neurons in the tail of ceh-14 mutants (panel C), and in ASG neurons of lin-11 mutants (panel D). All error bars represent SEM. Data are accumulated from three independent heatshock experiments. The student t-test is used, ***p<0.0001, n.s.= p>0.01.
Two terminal selector mutants, the homeodomain TFs unc-30 and unc-42, did not share this phenotype. unc-30 is the terminal selector for GABAergic MNs in the VNC and unc-42 is the terminal selector for ASH sensory neurons (Baran et al., 1999; Cinar et al., 2005; Jin et al., 1994; Serrano-Saiz et al., 2013). No increases in induction of gcy-5prom::gfp or ceh-36fosmid::yfp were detected in response to CHE-1hs in the GABAergic MNs of unc-30 (Figure 3—figure supplement 1C, data only shown for ceh-36fosmid::yfp). In the case of unc-42 mutants, neither gcy-5fosmid::rfp, nor ceh-36fosmid::yfp was detected in the ASH neurons after CHE-1 misexpression (Figure 3—figure supplement 1D, data only shown for gcy-5fosmid::rfp). Expression of che-1 early in the development of GABAergic MNs and ASH neurons does lead to activation of target genes, based on tissue-specific promoter driven expression of che-1 (Figure 3—figure supplement 1A). Additionally, activation of CHE-1hs in L4s/adults results in the expression of gcy-54XASE::gfp in both sets of neurons (Figure 3—figure supplement 1E,F). This means that CHE-1 is competent to activate target genes in these neurons while they are differentiating, and that its activity as a TF is uncompromised in these neurons during the adult stage. Therefore, we conclude that while unc-30 and unc-42 are required for terminal differentiation, they are dispensable for restricting cellular plasticity.
Overall, these results suggest that numerous terminal selector genes play a role in restricting the activity of ectopically expressed CHE-1. The affected neurons in all these TF mutants lack expression of certain identity-specific effector genes. The negative results with unc-30 and unc-42 demonstrate that the loss of a terminal identity in itself it not sufficient to keep a neuron in a plastic state. This suggests that the restriction of plasticity is a distinct and parallel function carried out by certain terminal selector TFs. In the next sections, we provide more evidence that supports this functional distinction.
The identity-specifying and plasticity-restricting functions of UNC-3 can be genetically separated
Two alternative hypotheses can explain the function of terminal selectors in the restriction of plasticity. Terminal selectors could simply be required for terminal differentiation, and it would be the completion of differentiation that then indirectly triggers restriction of plasticity. Alternatively, these transcription factors could be required for restricting plasticity in a mechanism that is separate from inducing effector genes of terminal cellular identity. A hypomorphic allele of unc-3 that we inadvertently generated by CRISPR-Cas9 mediated transgenesis allowed us to separate the identity-specifying function from the plasticity-restricting function of unc-3. This allele contains an insertion of a plant-specific AID degron into the unc-3 locus that allows for protein degradation in the presence of exogenous plant hormone auxin and the Arabidopsis F-box protein TIR1 (Zhang et al., 2015) (Figure 4A). In the absence of TIR1 and auxin, the unc-3::mNG::AID worms display wildtype locomotion and express cholinergic MN markers at near wildtype levels (Figure 4A,B). However, cholinergic MNs of these worms were receptive to CHE-1hs, even in the absence of TIR1 and auxin (Figure 4C,D). When CHE-1hs is overexpressed in adult worms containing unc-3::mNG::AID, expression of gcy-5prom::gfp is observed at a penetrance indistinguishable from that observed in the unc-3 null allele. Hence, in this allele the cholinergic MNs differentiate largely normally, and yet retain plasticity in response to CHE-1hs activity. This allele therefore supports the hypothesis that terminal selector-like TFs control restriction independently of specification. While it is unclear how the AID allele affects the unc-3 protein, we propose that this allele results in lower levels of active protein, perhaps by degradation through an auxin/TIR1 independent pathway.
Figure 4
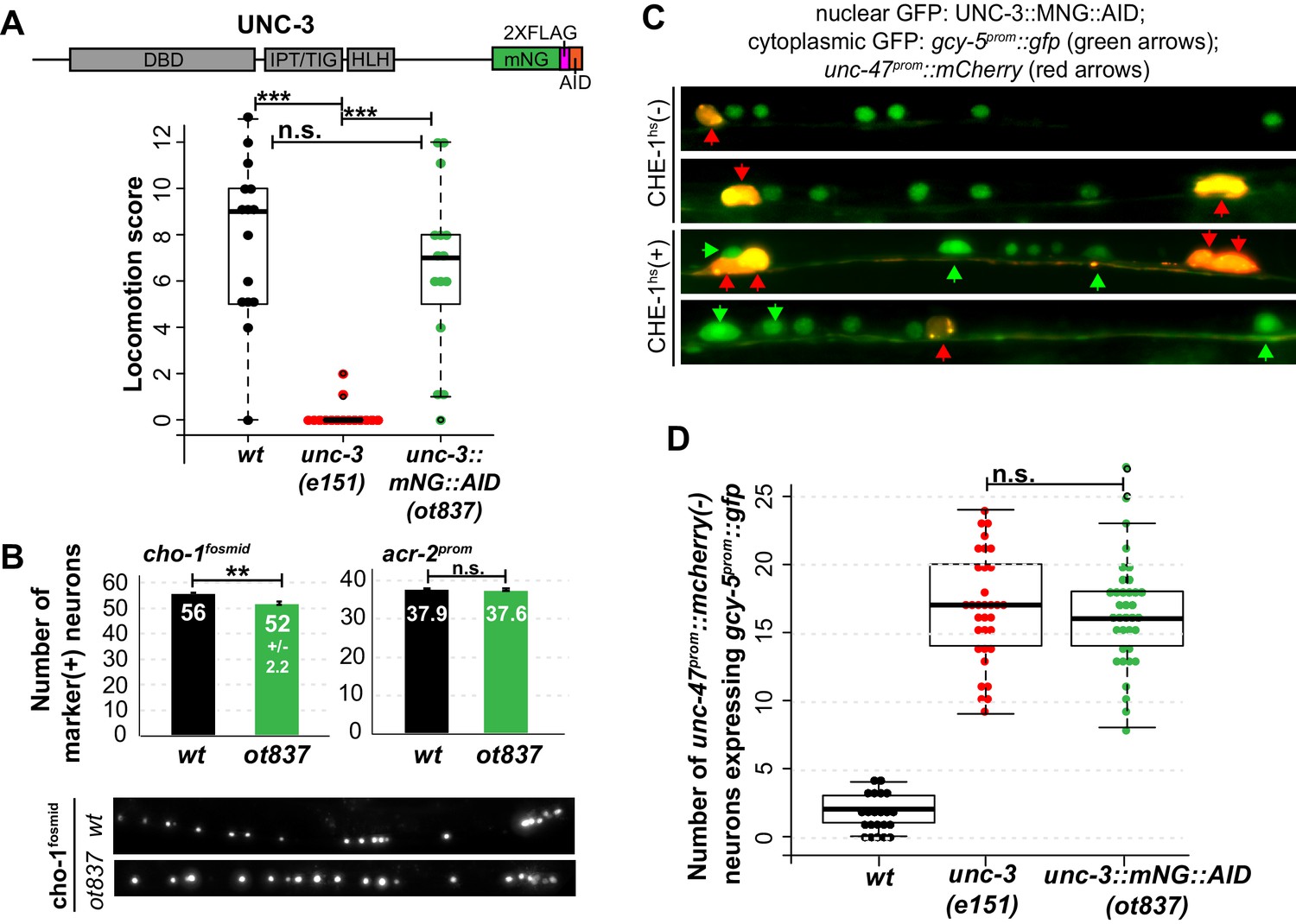
Genetic separation of identity-specification and plasticity restriction.
All error bars represent SEM. The student t-test is used, *p<0.05, **p<0.01, ***p<0.001, n.s.= p>0.05. (A) An mNeonGreen::AID tagged allele of unc-3, ot837, does not have the locomotion defects of an unc-3 null allele. (B) ot837 worms show near wildtype and wildtype induction of cho-1fosmid and acr-2prom respectively, whereas in unc-3 null mutants these genes are strongly repressed (Kratsios et al., 2011). cho-1fosmid expression is missing in ~4 neurons in ot837, but levels of expression in the neurons that retain expression look wildtype. (C) ot837 behaves like the unc-3 null with respect to restriction of plasticity. Induction of gcy-5prom is indistinguishable in ot837 and the unc-3 null allele after CHE-1hs induction. (D) Images of gcy-5prom expression in ot837 worms after CHE-1hs induction. In these images the nuclear green signal comes from the mNG tagged UNC-3 protein, the cytoplasmic GFP from gcy-5prom (green arrows), and the red cells are GABAergic (red arrows).
unc-3 regulates chromatin compaction
We next asked if terminal selectors restrict cellular plasticity by controlling chromatin accessibility at genes that are not expressed in differentiated cells. We focused again on unc-3 and hypothesized that in the wildtype cholinergic MNs CHE-1 targets would be present in a repressed, inaccessible chromatin state while in unc-3 mutants they would be present in a more open state, such that they are more likely to be activated by CHE-1. To visualize the chromatin state with single cell resolution, we used a chromosome tagging method that has previously been used to study the localization and state of compaction of transgenic loci in C. elegans (Cochella and Hobert, 2012; Fakhouri et al., 2010; Meister et al., 2010; Yuzyuk et al., 2009). We created a repetitive transgene array containing gcy-5prom::rfp and bacterial LacO sites and randomly integrated it into the genome. These transgenic worms also carry a separate transgene to ubiquitously express a LacI::GFP fusion protein. Thus, in every nucleus of this worm, LacI::GFP binds to the LacO sites surrounding the gcy-5 reporter, thereby labeling the gcy-5 transgenic loci as two green dots (Figure 5A,B). We also generated LacO arrays for an unc-3-dependent cholinergic gene, ace-2prom::rfp, as a control (Figure 5C). This technique has previously been used to show a correlation between the compaction state of the LacI::GFP spots and the repressed or active state of the locus in the array (Cochella and Hobert, 2012; Fakhouri et al., 2010; Meister et al., 2010; Yuzyuk et al., 2009).
Figure 5

Assessing chromatin state of a gcy-5 transgenic locus with the LacI/LacO dot assay.
The student t-test is used for statistical comparisons, *p<0.05, **p<0.01, ***p<0.001, n.s.= p>0.05. (A) Schematic representation of the spot assay. A transgene containing the gcy-5prom::rfp sequence and LacO binding sites is integrated into the genome of worms that ubiquitously express LacI::GFP. In the resulting transgenic worm, every nucleus contains two GFP spots representing the LacI::GFP bound to the gcy-5 LacO array. The structure and location of this locus can be visualized by GFP. (B) The gcy-5 arrays localize to the nuclear periphery in all cells in which expression was checked, including the ASEs. The array is transcriptionally active in the ASER, as evident by the presence of RFP, suggesting that this locus localizes to the nuclear periphery regardless of its transcriptional state. An identically built ace-2prom::rfp LacO array shows sub-nuclear localization that correlates to its transcriptional state. In cells that express ace-2prom::rfp, as seen by the presence of RFP, the GFP spots are diffused and in the nuclear lumen, suggesting that the locus is decompacted. In cells that do not express ace-2prom::rfp, the GFP spots are compact and localized to the nuclear periphery. Two independent array integrants examined for both ace-2 and gcy-5 showed similar results and one is shown here. (C) Worms were fixed and stained with DAPI for quantification of the GFP spots as an estimation of array structure. (D) ace-2 arrays were significantly larger when transcriptionally active as opposed to inactive in the VNC neurons of wildtype worms compared to unc-3 mutants. In (D, E, F) each dot represents a single nucleus and the number on the y-axis is the total pixel count occupied by the LacI/O spots or H3K27me3 per neuron. (E) gcy-5 arrays were significantly larger in unc-3 mutant neurons than in wildtype. (F) Control staining for H3K27me3 in gcy-5 LacO array containing worms showed no difference between wildtype and unc-3 and the size of the gcy-5 LacI/O spots do not change in the hypodermal nuclei of wildtype vs. unc-3 worms.
To facilitate quantification of the state of compaction, wildtype and unc-3 mutant animals containing the ace-2 and gcy-5 LacO arrays were fixed with paraformaldehyde and stained with GFP and DAPI. The total number of GFP pixels per VNC neuron was then calculated as an approximation of the space occupied by the ace-2 and gcy-5 LacO transgenes in wildtype and unc-3 mutant neurons. This quantification was performed blinded. The ace-2 LacO array is transcriptionally active in wildtype cholinergic neurons but not in unc-3 mutants (Figure 5B,C). Consistent with its expression, the ace-2 LacO array showed a significantly higher GFP pixel count in wildtype VNC neurons as compared to unc-3 (Figure 5C,D). By showing that the ace-2 transgenic locus is more open when it is transcriptionally active, this experiment indicated that the LacI/O dot assay is indeed indicative of the state of chromatin in cholinergic MNs. We then quantified GFP spots on the LacO array that contains gcy-5, the ASE-specific CHE-1 target gene that is normally not expressed in VNC MNs. In this case, the size of the GFP spot was increased in unc-3 mutant MNs compared to wildtype (Figure 5E). These data show that the gcy-5 locus is in a less compact state in unc-3 mutant cholinergic MNs, which may make it more amenable to activation by CHE-1. This change in compaction was not as dramatic as the one observed in the ace-2 array, but this may be because while the ace-2 array is actively transcribed in wildtype neurons, the gcy-5 array is not being actively transcribed in unc-3 mutants, rather it is simply in a more accessible state so that it is more likely to be transcribed when CHE-1 is ectopically induced. The quantified worms were also co-stained with H3K27me3 to control for staining efficiency and we did not observe a significant change in the number of H3K27me3 pixels between wildtype and unc-3 in the same neurons (Figure 5F).
If the change in compaction of the gcy-5 array were caused by unc-3 function, this change should be specific to neurons in which unc-3 acts as a terminal selector. Indeed, the size of the gcy-5 LacO array in hypodermal cells of wildtype and unc-3 mutants show no statistically significant difference (Figure 5F). It is also important to note that not all VNC neurons contain larger GFP spots, which is consistent with the variability we observe in the number of cells per worm that activate gcy-5 in response to CHE-1 (Figure 5E, Figure 2B).
Interplay between unc-3 and chromatin modifiers in regulating the restriction of plasticity
To confirm and further understand the role of chromatin compaction in our restriction assay, we probed the function of H3K27me and H3K9me. Both of these modifications are associated with silent and condensed chromatin (Towbin et al., 2012; Yuzyuk et al., 2009).
We have previously shown that the removal of the C. elegans PRC2 complex, which functions as an H3K27methyl transferase, makes germ cells more plastic and receptive to CHE-1 activity (Patel et al., 2012). Here, we tested the activity of CHE-1 in the ventral nerve cord of mes-2/E(z) mutants. mes-2 is the enzyme responsible for the histone methyl transferase activity of PRC2 and is known to be required for H3K27me3 in the C. elegans germline at all stages, and in somatic cells during embryonic and early larval stages, but not in the adult (Bender et al., 2004). We found that loss of mes-2 permits CHE-1hs to induce gcy-5prom::gfp to the same extent as removal of unc-3 does (Figure 6A), indicating that mes-2 is indeed required for restriction of plasticity in cholinergic MNs. To determine whether mes-2 acts in parallel or in the same pathway as unc-3, we tested mes-2; unc-3 double mutants and found that they have an additive phenotype. Removal of both unc-3 and mes-2 leads to the induction of gcy-5prom::gfp in twice as many cells as seen in unc-3 or mes-2 mutants alone. This suggests that unc-3 and mes-2 work in parallel genetic pathways to ensure plasticity restriction in differentiated cholinergic MNs.
Figure 6 with 1 supplement see all

Interactions of unc-3 with chromatin mutants.
CHE-1hs was induced at the L4 stage. For each graph, the data cumulatively represent three independent heatshock experiments in which all genotypes were scored in parallel. Every dot represents an individual worm. One way ANOVA with post-hoc Bonferroni and Scheffé tests for multiple comparisons were performed independently for each group of strains tested together. *p<0.05, **p<0.01, ***p<0.001, n.s.= p>0.05. (A) mes-2 mutants show increased induction of gcy-5prom::gfp in response to CHE-1hs and show an additive phenotype with unc-3, suggesting H3K27me3 regulates restriction in a parallel pathway with unc-3. The mes-2 and unc-4 mutations are genetically linked. unc-4 does not play a role in restriction, but it was used in all strains tested in this set of experiments to ensure similar genetic backgrounds. (B) Both met-2 and set-25 mutants have significantly more gcy-5prom::gfp expressing cholinergic MNs compared to wildtype. However, for set-25 the increase is only modest, and a met-2 set-25 double mutant is not significantly different from met-2, suggesting met-2, has a greater role in making the gcy-5prom::gfp locus inaccessible to CHE-1. An additive phenotype is not evident in met-2; unc-3, set-25; unc-3, or met-2 set-25; unc-3, suggesting that these factors might act in the same pathway to restrict the accessibility of gcy-5prom::gfp. No ectopic expression of gcy-5prom::gfp is seen in these mutants in the absence of CHE-1hs (not shown, n > 50 worms). (C) HP1 mutants also show increased induction of gcy-5prom::gfp, and slightly enhance the phenotype of unc-3 suggesting they have roles independent of met-2 set-25 dependent H3K9me. No gcy-5prom::gfp is induced in hpl-1;hpl-2 double mutants in the absence of CHE-1hs (not shown, n > 50 worms). (D) Representative images of scored worms. (E) Induction of CHE-1hs with the single copy transgene also results in increased induction of gcy-5prom::gfp in the VNC of met-2 set-25 mutants and the number of gcy-5prom::gfp and gcy-5 mRNA expression cells in the double mutant worms are similar showing that the phenotype in (B) is not simply a result of derepressed repetitive transgenes.
To test the role of H3K9me3, we tested the ability of CHE-1hs to activate gcy-5 in the VNC of cholinergic MNs in met-2 and set-25 mutants. met-2 and set-25 are SET-domain proteins shown to be required for H3K9 methylation in C. elegans embryos, young larvae, and adult germ lines (Towbin et al., 2012; Zeller et al., 2016). met-2 mutant embryos lack H3K9me1/2/3 whereas set-25 embryos lack H3K9me3 (Towbin et al., 2012). Using antibody staining, we ensured that met-2; set-25 double mutants also lack H3K9me3 at the L4 and adult stages where we perform our experiments (Figure 6—figure supplement 1A). If H3K9me3 plays a role in decreasing the accessibility of a gcy-5prom::gfp, then this locus should remain more accessible in met-2 and set-25 mutants. We found that induction of CHE-1hs does lead to increased activation of gcy-5prom::gfp in the cholinergic MNs of met-2 single mutants and met-2 set-25 double mutants, but not in set-25 single mutants (Figure 6B). This suggests that H3K9me1/2 and H3K9me3 have to be perturbed for CHE-1hs to activate gcy-5prom::gfp. H3K9me3 is known to be involved in the repression of repetitive loci. To ensure that our phenotype is not an artifact of repetitive transgenes, we confirmed that the loss of met-2; set-25 in transgenic worms containing the single copy hs::che-1 transgene also results in the expression of gcy-5prom::gfp in the cholinergic MNs and that the expression of gfp in these worms overlaps with gcy-5 mRNA expression as determined by smFISH of endogenous gcy-5 transcripts (Figure 6E).
To test genetic interactions between unc-3, met-2 and set-25 we examined CHE-1hs dependent induction of gcy-5prom::gfp in met-2;unc-3, set-25;unc-3 and met-2 set-25;unc-3 double and triple mutants. None of these double and triple mutants show an additive effect that is significantly different from the phenotype of unc-3 single mutants (Figure 6B), indicating that met-2, set-25, and unc-3 act in the same pathway rather than in parallel pathways. Since the removal of met-2 and set-25 does not result in as strong a phenotype as an unc-3 single mutant, we furthermore infer that met-2;set-25 dependent H3K9 methylation is not the only pathway affected in the unc-3 mutants.
To investigate the mechanisms of H3K9 action further, we considered HP1, a conserved chromodomain containing protein that is known to bind methylated H3K9 (Bannister et al., 2001; Jacobs and Khorasanizadeh, 2002; Jacobs et al., 2001; Lachner et al., 2001; Nielsen et al., 2002). The C. elegans genome encodes two homologs of HP1, hpl-1 and hpl-2 (Couteau et al., 2002; Schott et al., 2006). hpl-1 has been shown to colocalize with H3K9me3 in embryos by immunofluorescence, and hpl-2 shows enriched binding at H3K9me1/2 regions by whole embryo ChIP analysis (Garrigues et al., 2015; Vandamme et al., 2015). Fluorescent-tagged HPL-1 and HPL-2 proteins localize to distinct foci on chromatin and these proteins seem to have both unique and redundant functions (Schott et al., 2006). We asked if mutants of these proteins phenocopy the loss of met-2 and unc-3 in MNs after CHE-1hs expression. Indeed, an increased induction of gcy-5prom::gfp was seen in MNs after removal of both hpl-1 and hpl-2, and this phenotype was enhanced in the double mutant, so that it was stronger than the phenotype of met-2 set-25 mutants (Figure 6c). An hpl-2; hpl-1 unc-3 triple mutant also shows a phenotype that is enhanced from the hpl-1; hpl-2 double or the unc-3 mutant alone, but it is not completely additive. This enhancement indicates that HP1 has functions independent of met-2 set-25 and H3K9me. Induction of gcy-5 in all of the mutants tested here required CHE-1hs induction. Taken together, both H3K9me and H3K27me contribute to the restriction of plasticity in differentiated cells. H3K9me might be acting along with terminal selectors to mediate this process, while H3K27me acts in parallel (Figure 7).
Figure 7
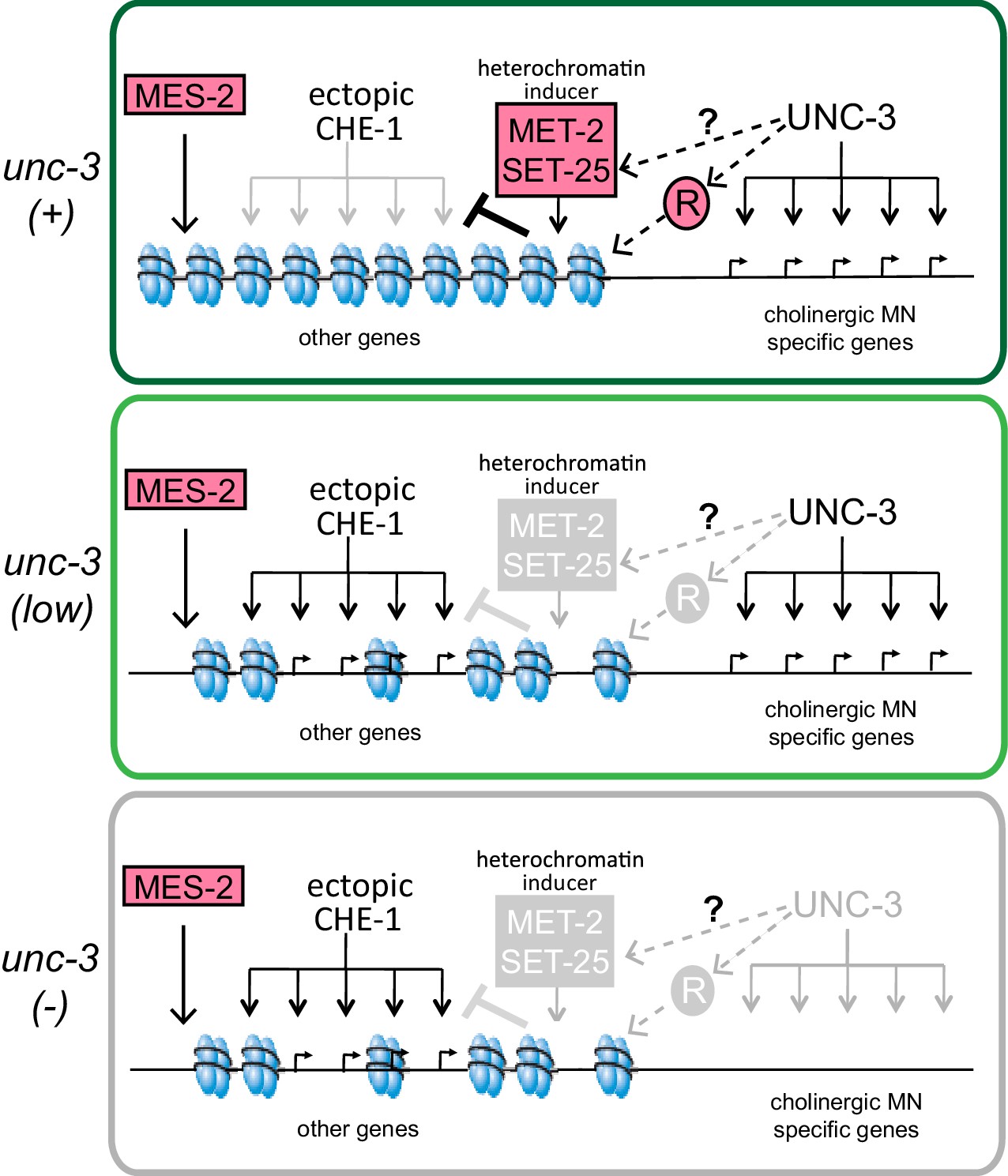
A model for the role of terminal selectors in restriction of plasticity.
Grey indicates lack of protein activity. In wildtype neurons, unc-3 activates expression of target genes to specify cholinergic MN identity but it also triggers the restriction of plasticity by activating pathways that repress genes not required for cholinergic MN identity. The repressed genes include ASE effector genes, which cannot be activated by ectopic CHE-1 in wildtype MNs. To restrict plasticity, unc-3 may regulate the correct distribution of H3K9 methylation through unknown pathways and may also activate additional repressors (R). These repressor mechanisms may then act to 'survey' the genome for genes that have not been activated during identity specification and transform them into a 'non-activatable' stage via H3K9-dependent heterochromatinization. unc-3 is required to trigger this surveillance mechanism by controlling the function or expression of the genes involved in this process. Independently, mes-2-dependent H3K27 methylation also plays a role in gene silencing. Upon lowering levels of unc-3 activity (ot837 allele) activation of cholinergic MN genes still takes place, however the unc-3 dependent pathways that repress other identity genes are not activated, as shown by the ability of CHE-1 to activate its target genes. When unc-3 is absent, neither specification nor restriction takes place so that cholinergic MN specific genes are not expressed and CHE-1 targets can be activated. A loss of both unc-3 and mes-2 results in even more open chromatin, and an increased activation of CHE-1 target genes.
We hypothesized that perhaps in unc-3 mutants, a reduction of histone modifications that are associated with active chromatin marks at unc-3-induced terminal differentiation genes might be causing a titration of negative histone marks over the genome, thereby increasing accessibility at genes that would otherwise have high levels of negative marks like H3K9me3. It has, for example, been observed that removal of positive mark H3K36 trimethylation leads to a titration of H3K27me3 from loci that are normally repressed (Gaydos et al., 2012). However, genetic removal of positive histone marks like H3K4me3 and H3K3me3 did not result in enhanced cellular plasticity (Figure 6—figure supplement 1B).
Further attempts to understand how unc-3 might regulate H3K9me have not yet been successful. First, hpl-1, hpl-2, and met-2 gene expression is unaffected in unc-3 mutants (Figure 6—figure supplement 1C). Second, H3K9me3 can be detected in both wildtype and unc-3 animals by immunostaining (Figure 6—figure supplement 1D). A third possibility is that the distribution of H3K9me3 over the genome may be affected in unc-3 mutant cholinergic MNs. We attempted to test this hypothesis by performing co-localization assays between the gcy-5 LacO transgene and H3K9me3, MET-2::mKATE2, or HPL-1/2::mKATE2 in wild-type and unc-3 deficient MNs. However, these tools were not sufficiently sensitive to answer this question. H3K9me3 localization at the gcy5 LacO array could not be quantified as the structure of the array was not well preserved after the H3K9me3 staining procedure (Materials and Methods). MET-2 and HPL-1/2 localization could not be quantified because of low expression levels in neurons. Therefore, it remains to be seen if terminal selectors like unc-3 regulate the correct deposition of H3K9me or if they regulate other functions mediated by met-2 and HP1 to make non-identity defining genes less accessible.
Discussion
Differentiated cells are defined by identity-specific transcriptional programs that are stably maintained throughout the life of a cell (Blau and Baltimore, 1991; Deneris and Hobert, 2014). Here, we provide evidence that in C. elegans neurons, identity-defining terminal selectors not only initiate and maintain the expression of effector genes that define a cellular phenotype but they also trigger the restriction of cellular plasticity. We propose that the coordination of identity specification and restriction of plasticity ensures the robustness of the differentiated state, protecting cells from accidental transcription of unwanted genes in response to fluctuations in the environment (Voss and Hager, 2014).
Differentiation and the restriction of cellular plasticity
The loss of developmental plasticity in differentiating cells can be experimentally demonstrated as a cell’s inability to respond to ectopically expressed TFs. To assay plasticity, we have ubiquitously expressed a terminal selector TF, CHE-1, at various developmental time points in C. elegans and scored its ability to activate fluorescent reporters of target genes. CHE-1 is normally expressed only in 2 ASE sensory neurons where it activates an ASE-specific gene battery. We found that cells that are dividing and beginning to acquire lineage and identity-specific markers are receptive to the activity of ectopic CHE-1. Once cells are postmitotic, they remain receptive for a limited window of time while they are activating terminal identity genes, and then become refractory. A progressive loss of ectopic TF activity in postmitotic cells has also been reported in the mouse brain. Fezf-2, a master regulatory gene for corticospinal MNs, is capable of activating target genes in embryonic and early postnatal callosal projection neurons and retains its ability to act at postmitotic day 3 and 6. However, this ectopic activity progressively declines and is exhausted by day 21 (Rouaux and Arlotta, 2013). These data show that a change occurs in differentiated post-mitotic cells that makes non-identity-specific genes less likely to get activated. In this work, we find that this change is triggered by the resident terminal selector of the differentiated cells in conjunction with downstream and parallel chromatin changes.
We found that the loss or reduction of terminal selector TF activity keeps some neurons in a more plastic state. Five of seven terminal selectors tested in our CHE-1hs assay are required in distinct neuron types to make genes normally expressed in ASE neurons inaccessible to ectopically expressed CHE-1, suggesting that the coordination of differentiation and restriction of cellular plasticity is a commonly occurring phenomenon. The difference between TFs that do and do not function to restrict plasticity is not clear. The two terminal selector-depleted cell types that do not respond to CHE-1 are not lineally more distant from ASE neurons than the five terminal selector mutant cell types that do respond to CHE-1. A comprehensive expression profile of some of these neurons in the presence and absence of the respective TFs may resolve the mechanistic basis for this difference.
In mice, the terminal selector Pax5, which is required for the differentiation of B lymphocytes, also functions to restrict cellular plasticity. Non-terminally differentiated Pro-B cells are responsive to the pluripotency TFs c-Myc, Klf4, Sox2, and Oct4 and can be converted into iPSCs but differentiated B lymphocytes are refractory to the activity of these TFs. Pax5 mutant B lymphocytes, on the other hand, can be converted into iPSCs by the activity of the same TFs (Hanna et al., 2008). This suggests that the activity of TFs in restricting plasticity is conserved in other systems. There is, however, a significant difference that makes the function uncovered in this work novel. Pax5(-) B lymphocytes have been shown to retain characteristics of multipotent hematopoietic progenitor Pro-B cells, making it unclear whether it is the transformation into a less differentiated cell type or the lack of Pax5 itself that makes these cells plastic (Nutt et al., 1999). In contrast, we find that terminal selector-mutant cells can retain plasticity even if they are not in a progenitor-like state. While terminal selector mutant neurons do not express neuron-type-specific genes, they clearly differentiate into a neuron-like terminal end state. They are postmitotic, still adopt a neuronal morphology, and normally activate expression of pan-neuronal features such as presynaptic vesicle machinery (Cassata et al., 2000; Etchberger et al., 2007; Kratsios et al., 2011; Stefanakis et al., 2015). Additionally, cnd-1, a homolog of bHLH protein NeuroD, which is expressed transiently during the progenitor-like state in cholinergic MNs, is turned off normally in an unc-3 mutant (Hallam et al., 2000; Kratsios et al., 2011). Moreover, mutant neurons of most terminal selectors tested in this assay do not even show a complete loss of expression of terminal identity genes. In lin-11 mutants, for instance, expression of various terminal identity genes is reduced in ASG, but most tested markers are not completely eliminated (Sarafi-Reinach et al., 2001). Similarly, in ttx-1 mutant AFD neurons, the expression of the glutamate transporter is off in only ~20% of neurons, while in a ceh-14; ttx-1 double mutant it is off or dim in ~80% of neurons (Serrano-Saiz et al., 2013). However, the gcy-5fosmid::gfp locus is amenable to CHE-1 activation in both the ttx-1 single and ttx-1;ceh-14 double mutants to a similar extent. And finally, in the hypomorphic unc-3::mNG::AID allele generated in this work, the cholinergic MNs seem to express cholinergic markers and move like wildtype worms suggesting that the differentiation of these cells is minimally affected. Yet, the response to CHE-1 activity in this allele is indistinguishable from the unc-3 null allele in which MNs are entirely dysfunctional. Hence, the loss of a terminal selector does not simply eliminate the differentiated state, rendering cells more plastic by default. Rather, terminal selectors trigger plasticity restriction by a pathway that can be partially decoupled from identity acquisition.
Earlier acting selector genes of organ identity do not restrict cellular plasticity, as exemplified by the C. elegans pharyngeal organ selector pha-4 (Yuzyuk et al., 2009). This is in accordance with these earlier acting selector genes exerting their activity when cells still divide and have not yet acquired a stable and terminal identity.
Possible mechanism of action of terminal selectors in restricting plasticity
In all terminal selector mutants tested, CHE-1 activates ASE-identity genes in only about 30% of affected neurons. This suggests that restriction of cellular plasticity also requires input from terminal selector-independent factors. In the case of unc-3 dependent cholinergic neurons, the removal of both an H3K27 methyltransferase, mes-2, and an H3K9 methyltransferase, met-2, results in the gcy-5prom::gfp locus being more accessible to CHE-1. We find that unc-3 and mes-2 show an additive phenotype, suggesting that in these neurons, H3K27me3 is one of the terminal selector-independent mechanisms that contribute to restriction of cellular plasticity. On the other hand, a met-2; unc-3 double mutant phenocopies unc-3, suggesting that the incorrect distribution of H3K9 methylation could be a downstream effect of unc-3 function. This is congruent with the finding that unc-3 mutants display an expansion of the chromatin structure of a gcy-5 transgenic reporter.
The role of H3K27me3 in restricting plasticity has been well studied in the C. elegans embryo and the germline (Fakhouri et al., 2010; Patel et al., 2012). However, as overall levels of H3K27me3 do not seem to be affected in somatic cells of L4 stage mes-2 mutants (Bender et al., 2004), this would suggest that the lack or incorrect distribution of H3K27me3 in early stages of larval development is sufficient to interfere with restriction. H3K9me3 has been reported to function in preventing iPSC conversion of fibroblasts (Becker et al., 2016). However, in C. elegans, somatic developmental defects have not yet been observed in H3K9 methyltransferase mutants (Zeller et al., 2016). The current discovery that met-2 and the homologs of HP1 function to restrict plasticity highlights the utility of TF overexpression assays in uncovering mechanisms of development.
How terminal selectors might modulate chromatin compaction remains unresolved. In cholinergic MNs there is no obvious difference in the gross, overall levels of H3K9me3 between wildtype and unc-3 mutants. unc-3 does not seem to be playing a significant role in activating the expression of the met-2 or hpl-1/2 genes. Moreover, unc-3 does not appear to regulate the distribution of H3K9me3 marks indirectly by controlling the deposition of positive marks (H3K4me3 and H3K36me3) at genes that it transcriptionally activates, since we find that the loss of enzymes that generate these marks does not promote cellular plasticity.
Collectively, our data indicate that during identity specification, terminal selectors trigger a ‘surveillance mechanism’ which silences non-expressed, non-identity-specific genes. Genetic epistasis experiments suggest that this surveillance mechanism is (a) likely to be mediated by met-2 and hpl-1/2 and (b) carried out by the correct deposition of H3K9me on genes that are not activated in a given neuron type. This hypothesis can be tested in the future by performing cell-type-specific ChIP-seq experiments to determine the distribution of H3K9me3 in wild-type and terminal selector mutant neurons. How terminal selectors may modulate the deposition of chromatin marks remains to be discovered. One possibility is that terminal selectors directly activate target genes that define a specific neuronal identity while also directly silencing genes that are normally not expressed in the respective neuron type. Although some TFs can act as both activator and repressor depending upon the presence of other co-factors (Ip, 1995; Sakabe et al., 2012), we disfavor this model because it implies that to be kept in a ‘non-activatable’ state, all genes would need a unique terminal selector response element for every cell type that they are not expressed in. This mechanism implies an unreasonably complex gene regulatory architecture involving an exceptional number of cis-regulatory elements. We rather favor at this point a model in which (a) heterochromatin forms, perhaps by default mechanisms, on loci that are not activated during cellular differentiation and (b) unc-3 regulates the expression or function of some of the proteins that organize the heterochromatic state (Figure 7). These organizers may be broadly expressed heterochromatin regulators that are ‘plugged’ into cell-specific differentiation program via their activation by cell-specific terminal selectors. Such regulatory linkage would provide a checkpoint to coordinate the induction of cellular differentiation programs with the loss of cellular plasticity.
Materials and methods
Worm methods and strains
All strains were maintained using standard protocols. For heatshock experiments, worms were grown at 15°C, heatshocked at 32°C for 30 min, left at 20°C overnight and scored about 24 hr later.
Strains
OH9846: otIs305 [hsp16-41prom::che-1::: :3XHA::BLRP; rol-6(su1006)]; ntIs1 [gcy-5::gfp; lin-15b(+)]
OtTi6: hsp16-41prom::che-1::2xFLAG (miniMos single copy insertion)
OH13139: otIs587 [gcy-5fosmid::sl2::1xnlsgfp; ttx-3prom::mcherry]
OH13102: otIs586 [gcy-6fosmid::sl2::1xnlsgfp; ttx-3prom::mcherry]
OH13984: ot835 (gcy-5::sl2::1xnls::mNeonGreen) CRISPR engineered into the genome
OH14049: otIs629 [gcy-7prom::tagrfp; ttx-3prom::gfp]; ntIs1[gcy-5prom::GFP]
OH13099: otIs583 [gcy-5prom306bp4XASE::gfp; ttx-3prom::mcherry]
OH14218: otEx6597 [ric-19prom::che-1; myo-2prom::bfp]
OH14219: otEx6598 [unc-47prom::che-1; myo-2prom::bfp]
OH11979: otEx5430 [ift-20prom::che-1; rol-6(d)]
OH8882: otEx3909 [ceh-36fosmid::yfp]
OH10689: otis355 [rab-3prom::nlstagrfp]
OH10598: otis348 [unc-47prom::mchopti; pha-1]
OH11124: otis388 [eat-4fosmid::yfp::h2b]
OH13988: ieSi57 [Peft-3::TIR1::mRuby:;unc-54 3’UTR] II; ot837(unc-3::mNeonGreen::AID) protein tag CRISPR engineered into the genome
OH14021: ot841 (hpl-1::mKate2) protein tag CRISPR engineered into the genome
OH14220: ot860 (hpl-2:: mKate2) protein tag CRISPR engineered into the genome
OH14221: ot861 (met-2:: mKate2) protein tag CRISPR engineered into the genome
OH12738: otIs545 [gcy-5prom::rfp; LacO repeats]; otIs593 [let-858prom::gfp::LacI; ttx-3prom::mcherry]
OH11744: otis445 [ace-2prom::rfp; LacO repeats;] gwIs39 [baf-1prom::gfp::lacI; vit-5prom::gfp]
Mutant alleles
Request a detailed protocolunc-3(e151), ceh-14(ch3), lin-11(n389), unc-30(e191), unc-42(e419), ets-5(tm1734), ttx-1(p797), met-2(ok2307), set-25(n5021), hpl-1(tm1489), hpl-2(n4317), unc-55(e402), unc-4(e120), mab-9(gk396730), SS186: (mes-2(bn11) unc-4(e120)/mnC1 dpy-10(e128) unc-52(e444)II)
MiniMos
Request a detailed protocolMiniMos was performed exactly as presented in Frøkjær-Jensen et al. (2014). hsp16.41prom::che-1::2Xflag was cloned into the miniMOS vector pCFJ910 and integrated into the genome. Three independent insertions were obtained of which OTti6 was used for all experiments.
CRISPR alleles
Request a detailed protocolCRISPR engineering was performed exactly as described in Dickinson et al. (2015). For gcy-5, SL2::1XNLS::mNeonGreen was inserted after the stop codon of gcy-5. For unc-3, mNeonGreen::AID tag was inserted into the C-terminus of the protein just before the stop codon. For hpl-1, mKate2 was inserted into the protein after the 12th amino acid. This tag was based on a published hpl-1 protein fusion (Couteau et al., 2002). For hpl-2, mKate2 was inserted into the protein after the 96th amino acid. This tag was based on a published hpl-1 protein fusion (Couteau et al., 2002). There are two isoforms of hpl-2, both are tagged in this allele. For met-2, mKate2 was added to the C-terminus of the protein followed by a small protein bridge present in the plasmids published in Dickinson et al. (2015). The start codon of met-2 was kept intact. The MET-2 protein seems to localize inside the nucleus in opposition to a previously published expression pattern (Towbin et al., 2012).
Fosmid recombineering
Request a detailed protocolFosmid recombineering to create otIs586 and otIs587 was performed as in Tursun et al. (2009). The recombineered fosmids were linearized and injected into hermaphrodites as complex arrays with 15 ng/µl of the linearized fosmid, 3 ng/µl of ttx-3prom::mcherry used as a co-injection marker and 100 ng/µl of digested bacterial genomic DNA. Extrachromosomal lines acquired after injection were integrated into the genome using gamma irradiation.
LacO arrays
Request a detailed protocolTo create the gcy-5 LacO arrays, 10 ng/µl of PCR product containing gcy-5prom::rfp were injected with 2 ng/µl of LacO repeats acquired by digesting out the10 kb Sph1/Kpn1 fragment from lacO multimeric plasmid pSV2-dhfr- 8.23, and 100 ng/µl of digested bacterial genomic DNA were injected into GW396 young adult hermaphrodites. Extrachromosomal arrays that formed nuclear LacI::GFP spots were chosen for integration with gamma irradiation. Integrants that showed nuclear spots were then chosen.
The ace-2 LacO arrays were similarly built. The injection mix contained: 10 ng/µl of ace-2prom::rfp PCR, 2 ng/µl of LacO repeats, 2 ng/µl of elt-2::nlsdsred, and 86 ng/µl of digested bacterial genomic DNA. Extrachromosomal and integrant lines were chosen as with gcy-5.
DiI filling
Request a detailed protocolThe amphid sensory neurons of worms can uptake fluorescent dyes (Shaham, 2006). DiI labeling was performed by washing worms into 1 mL of M9 butter containing 1:500 dilution of DiI. Worms were left to incubate with the dye for 1 hr at room temperature, washed three times with M9 and placed on NGM plates with E. coli for >1 hr to recover before imaging.
Antibody staining
Request a detailed protocolA modified version of the protocol published in Gendrel et al. (2016) was used for GFP and H3K27me3 staining. Worms were fixed in 4% paraformaldehyde/PBS for 15 min at 4°C, washed three times in 0.5% triton/PBS, rocked gently in 5% β-mercaptoethanol/1% Triton X-100/0.1 M Tris-HCl(pH 7.5) for 18 hr at 37°C, washed four times in 1% Triton X-100/0.1 M Tris-HCl(pH7.5) and once in 1 mM CaCl2/1% Triton X-100/0.1 M Tris-HCl(pH7.5). A worm pellet of 20–50 μL was then shaken in 1 mg/mL of collagenase type IV (C5138, Sigma)/1 mM CaCl2/1% Triton X-100/0.1 M Tris-HCl(pH7.5) at 37°C for 30 min. The worms were then washed three times in PBS/0.5% Triton X-100, blocked, and stained. Blocking was done in PBS/0.2% gelatin/0.25% Triton for 30 min at room temperature. Antibodies were diluted in PBS/0.1% gelatin/0.25% Triton. Primary antibody was left on overnight at 4°C or 1 hr at room temperature and secondary antibody was applied for 3 hr at room temperature. After washing off the secondary antibody, worms were incubated with DAPI for 15 min, washed again and mounted on glass slides. Primary antibodies used were: H3K27me3 (Millipore, 1:500) and GFP (Invitrogen, 1:1000 dilution). All secondary antibodies were Alexa Flour dyes used at 1:1000 dilution.
For H3K9me3 staining, we found that anti-H3K9me3 (Abcam,1:500 dilution) showed non-specific staining as observed by the presence of staining in met-2; set-25 embryos and L1s, which have been shown by Mass Spectroscopy to not have any H3K9me3 (Towbin et al., 2012). We found the Kimura H3K9me3 antibody, CMA318 purchased from MBL International, to be specific. For this antibody, the paraformaldehyde fixation described above did not work in somatic cells and a freeze crack antibody staining protocol on whole worms was used (Duerr, 2006). Worms were washed, suspended in 0.025% glutaraldehyde/ddH20 and spread out in between two frost-resistant glass slides. These slides were frozen on dry ice and cracked open to break the cuticle of the animals. Freeze cracked worms were fixed in ice-cold acetone for 5 min and then in ice-cold methanol for 5 min. The worms were then washed off the slides in PBS, blocked, stained, and mounted as above.
Microscopy
Request a detailed protocolµManager and Zen were used for image acquisition and processing (Edelstein et al., 2010).
Single molecule fluorescent in situ hybridization (smFISH)
Request a detailed protocolsmFISH was performed using Custom Stellaris FISH probes, purchased from Biosearch Technologies. Staining was performed according to the manufacturer's protocol.
References
-
The C. elegans homeodomain gene unc-42 regulates chemosensory and glutamate receptor expressionDevelopment 126:2241–2251.
-
H3K9me3-Dependent heterochromatin: barrier to cell fate changesTrends in Genetics 32:29–41.https://doi.org/10.1016/j.tig.2015.11.001
-
Differentiation requires continuous regulationThe Journal of Cell Biology 112:781–783.https://doi.org/10.1083/jcb.112.5.781
-
Maintenance of postmitotic neuronal cell identityNature Neuroscience 17:899–907.https://doi.org/10.1038/nn.3731
-
Computer control of microscopes using µmanagerCurrent Protocols in Molecular Biology Chapter 14:Unit14 20.https://doi.org/10.1002/0471142727.mb1420s92
-
The molecular signature and cis-regulatory architecture of a C. elegans gustatory neuronGenes & Development 21:1653–1674.https://doi.org/10.1101/gad.1560107
-
Activation of hypodermal differentiation in the Caenorhabditis Elegans embryo by GATA transcription factors ELT-1 and ELT-3Molecular and Cellular Biology 21:2533–2544.https://doi.org/10.1128/MCB.21.7.2533-2544.2001
-
The developmental capacity of nuclei taken from differentiating endoderm cells of xenopus laevisJournal of Embryology and Experimental Morphology 8:505–526.
-
The developmental capacity of nuclei taken from intestinal epithelium cells of feeding tadpolesJournal of Embryology and Experimental Morphology 10:622–640.
-
Nuclear transfer to eggs and oocytesCold Spring Harbor Perspectives in Biology 3:a002659.https://doi.org/10.1101/cshperspect.a002659
-
The C. elegans NeuroD homolog cnd-1 functions in multiple aspects of motor neuron fate specificationDevelopment 127:4239–4252.
-
Regulation of terminal differentiation programs in the nervous systemAnnual Review of Cell and Developmental Biology 27:681–696.https://doi.org/10.1146/annurev-cellbio-092910-154226
-
Terminal selectors of neuronal identityCurrent Topics in Developmental Biology 116:455–475.https://doi.org/10.1016/bs.ctdb.2015.12.007
-
pha-4, an HNF-3 homolog, specifies pharyngeal organ identity in Caenorhabditis ElegansGenes & Development 12:1947–1952.https://doi.org/10.1101/gad.12.13.1947
-
Pioneer transcription factors in cell reprogrammingGenes & Development 28:2679–2692.https://doi.org/10.1101/gad.253443.114
-
Organogenesis of the Caenorhabditis Elegans intestineDevelopmental Biology 216:114–134.https://doi.org/10.1006/dbio.1999.9471
-
Jmjd1a and Jmjd2c histone H3 lys 9 demethylases regulate self-renewal in embryonic stem cellsGenes & Development 21:2545–2557.https://doi.org/10.1101/gad.1588207
-
The spatial dynamics of tissue-specific promoters during C. elegans developmentGenes & Development 24:766–782.https://doi.org/10.1101/gad.559610
-
Locking the genome: nuclear organization and cell fateCurrent Opinion in Genetics & Development 21:167–174.https://doi.org/10.1016/j.gde.2011.01.023
-
Expression of the unc-4 homeoprotein in Caenorhabditis elegans motor neurons specifies presynaptic inputDevelopment 121:2877–2886.
-
Neuronal function of Tbx20 conserved from nematodes to vertebratesDevelopmental Biology 317:671–685.https://doi.org/10.1016/j.ydbio.2008.02.015
-
unc-3, a gene required for axonal guidance in Caenorhabditis Elegans, encodes a member of the O/E family of transcription factorsDevelopment 125:1561–1568.
-
Dual transcriptional activator and repressor roles of TBX20 regulate adult cardiac structure and functionHuman Molecular Genetics 21:2194–2204.https://doi.org/10.1093/hmg/dds034
-
The lin-11 LIM homeobox gene specifies olfactory and chemosensory neuron fates in C. elegansDevelopment 128:3269–3281.
-
Unique and redundant functions of C. elegans HP1 proteins in post-embryonic developmentDevelopmental Biology 298:176–187.https://doi.org/10.1016/j.ydbio.2006.06.039
-
Histone core modifications regulating nucleosome structure and dynamicsNature Reviews Molecular Cell Biology 15:703–708.https://doi.org/10.1038/nrm3890
-
H3K23me2 is a new heterochromatic mark in Caenorhabditis elegansNucleic Acids Research 43:9694–9710.https://doi.org/10.1093/nar/gkv1063
-
Dynamic regulation of transcriptional states by chromatin and transcription factorsNature Reviews Genetics 15:69–81.https://doi.org/10.1038/nrg3623
-
Pioneer transcription factors: establishing competence for gene expressionGenes & Development 25:2227–2241.https://doi.org/10.1101/gad.176826.111
-
Pioneer transcription factors, chromatin dynamics, and cell fate controlCurrent Opinion in Genetics & Development 37:76–81.https://doi.org/10.1016/j.gde.2015.12.003
Article and author information
Author details
Funding
Howard Hughes Medical Institute
- Oliver Hobert
National Institute of Neurological Disorders and Stroke
- Oliver Hobert
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Dylan Rahe for extensive discussions; Qi Chen for expert assistance in strain generation; Marie Gendrel for optimizing the immunostaining protocol; Baris Tursun for early guidance in the project; CGC for strains; members of the Hobert Lab for comments on the manuscript. TP was funded by a National Science Foundation GRFP fellowship. OH is an nvestigator of the Howard Hughes Medical Institute. This work was also supported by the National Institutes of Health (R21 NS076191-01).
Copyright
© 2017, Patel et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,179
- views
-
- 532
- downloads
-
- 81
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 81
- citations for umbrella DOI https://doi.org/10.7554/eLife.24100
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Coordinated control of terminal differentiation and restriction of cellular plasticity
eLife 6:e24100.
https://doi.org/10.7554/eLife.24100




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}