The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes
- University of California, Los Angeles, United States
- UCL Institute of Child Health, United Kingdom
- Karlsruhe Institute of Technology, Germany
- David Geffen School of Medicine, University of California, Los Angeles, United States
Abstract
Altered Ca2+ handling is often present in diseased hearts undergoing structural remodeling and functional deterioration. However, whether Ca2+ directly regulates sarcomere structure has remained elusive. Using a zebrafish ncx1 mutant, we explored the impacts of impaired Ca2+ homeostasis on myofibril integrity. We found that the E3 ubiquitin ligase murf1 is upregulated in ncx1-deficient hearts. Intriguingly, knocking down murf1 activity or inhibiting proteasome activity preserved myofibril integrity, revealing a MuRF1-mediated proteasome degradation mechanism that is activated in response to abnormal Ca2+ homeostasis. Furthermore, we detected an accumulation of the murf1 regulator FoxO in the nuclei of ncx1-deficient cardiomyocytes. Overexpression of FoxO in wild type cardiomyocytes induced murf1 expression and caused myofibril disarray, whereas inhibiting Calcineurin activity attenuated FoxO-mediated murf1 expression and protected sarcomeres from degradation in ncx1-deficient hearts. Together, our findings reveal a novel mechanism by which Ca2+ overload disrupts myofibril integrity by activating a Calcineurin-FoxO-MuRF1-proteosome signaling pathway.
https://doi.org/10.7554/eLife.27955.001Introduction
The establishment and maintenance of rhythmic cardiac contractions require tightly regulated Ca2+ signaling and intact contractile machinery. In the heart, a small amount of Ca2+ enters cardiomyocytes upon stimulation by an action potential. This Ca2+ influx induces the release of a larger amount of Ca2+ from the sarcoplasmic reticulum (SR) resulting in an abrupt increase in cytosolic Ca2+ levels and muscle contraction. The re-sequestration of Ca2+ to the SR by SERCA2 and extrusion of Ca2+ from the cell by NCX1 allows the muscle to relax (Bers, 2002). Abnormal Ca2+ handling has been associated with cardiac diseases including heart failure and arrhythmia in humans and animal models (Luo and Anderson, 2013) and structurally defective myofibrils are also often observed in diseased hearts (Lopes and Elliott, 2014). However, whether or not there is a causal relationship between abnormal Ca2+ handling and myofibril disarray in diseased myocytes has not yet been established.
The RING finger protein MuRF1 (also known as TRIM63) is a muscle-specific E3 ubiquitin protein ligase involved in the regulation of muscle turnover in normal physiology and under pathological conditions. MuRF1 acts on several sarcomeric target proteins, tagging them with polyubiquitin chains for proteasome-dependent degradation (Kedar et al., 2004; Clarke et al., 2007; Cohen et al., 2009; Mearini et al., 2010). Through this mechanism, MuRF1 regulates normal sarcomere protein turnover and removes misfolded and/or damaged proteins in skeletal and cardiac muscles (Lyon et al., 2013; Pagan et al., 2013; Willis et al., 2014). Murf1 expression is elevated under muscle catabolic conditions and overexpression of murf1 in the heart results in a thin ventricular wall and a rapid transition to heart failure upon transaortic constriction, suggesting that MuRF1 is a major player in muscle catabolic processes (Bodine et al., 2001; Labeit et al., 2010; Baehr et al., 2011; Files et al., 2012; Gomes et al., 2012; Bodine and Baehr, 2014). Conversely, knockout of murf1 promotes resistance to muscle atrophy and an exaggerated hypertrophic response to pressure overload (Willis et al., 2007; Willis et al., 2009a; Willis et al., 2009b). In humans, patients with specific murf1 gene variants develop hypertrophic cardiomyopathy at a younger age (Chen et al., 2012; Su et al., 2014), revealing a pathological role for MuRF1 in the progression of cardiac diseases.
In skeletal muscles, the Forkhead box O (FoxO) transcription factor family serves as a nodal point controlling muscle degradation via the regulation of MuRF1 expression. Under catabolic conditions, the PI3K-Akt pathway is suppressed and hypophosphorylated FoxO translocates into the nucleus causing murf1 induction and muscle atrophy (Lecker et al., 2004; Waddell et al., 2008). Conversely, upon IGF stimulation, the phosphorylation of FoxO by activated AKT sequesters FoxO in the cytoplasm, resulting in reduced murf1 expression and an increase in myocyte mass (Sacheck et al., 2004; Stitt et al., 2004). Similarly, an AKT-FoxO-mediated suppression of murf1 expression in response to insulin has been noted in cardiac muscles (Skurk et al., 2005; Paula-Gomes et al., 2013).
In this study, we used the zebrafish tremblor/ncx1h mutant to explore the regulatory relationship between Ca2+ homeostasis and the maintenance of cardiac muscle integrity. We have previously shown that ncx1h (also known as slc8a1a) encodes a cardiac specific sodium-calcium exchanger 1 (NCX1) in zebrafish and that the tremblor (tre) mutant lacks functional NCX1h (Langenbacher et al., 2005). NCX1 is a primary Ca2+ efflux mechanism in cardiomyocytes (Ottolia et al., 2013), and consistent with this important role in Ca2+ homeostasis, cytosolic Ca2+ levels are increased and cyclic Ca2+ transients are abolished in tre/ncx1h cardiomyocytes resulting in fibrillation-like chaotic cardiac contractions (Ebert et al., 2005; Langenbacher et al., 2005; Shimizu et al., 2015). Like NCX1-/- mice, tre/ncx1h zebrafish hearts also develop severe myofibril disarray (Koushik et al., 2001; Wakimoto et al., 2003; Ebert et al., 2005), suggesting that a conserved molecular link exists between aberrant Ca2+ handling and myofibril disarray. From a microarray analysis, we found that the expression of murf1 is significantly upregulated in ncx1h-deficient hearts. This MuRF1 upregulation was responsible for the myofibril disarray in ncx1h-deficient hearts, and normal cardiac myofibrils could be restored by genetic and pharmacological manipulation of MuRF1 or proteasome activity. We also found that elevated intracellular Ca2+ levels enhanced murf1 expression via activation of Calcineurin signaling, which dephosphorylates the murf1 transcriptional regulator FoxO, leading to its nuclear translocation. Our findings reveal a novel signaling pathway in which Ca2+ homeostasis modulates the integrity of cardiac muscle structure via murf1 regulation.
Results and discussion
NCX1 is required for the maintenance of myofibril integrity in cardiomyocytes
Zebrafish ncx1h mutant embryos lack functional NCX1 in myocardial cells resulting in aberrant Ca2+ homeostasis and a fibrillating heart (Ebert et al., 2005; Langenbacher et al., 2005; Shimizu et al., 2015). Similar to the myofibril phenotype observed in NCX1-/- mice, sarcomeres in zebrafish ncx1h mutant cardiomyocytes are damaged (Koushik et al., 1999; Wakimoto et al., 2003; Ebert et al., 2005). To investigate whether NCX1 activity affects the assembly or the maintenance of sarcomeres in myocardial cells, we examined the distribution of α-actinin protein. In striated muscles, α-actinin is localized to the Z-line and is a good marker for assessing sarcomere structure. We found that α-actinin is organized into a periodic banding pattern in both wild type and ncx1h mutant cardiomyocytes at 30 hpf (Figure 1A), suggesting that sarcomere assembly is initiated properly in the absence of NCX1 activity. Interestingly, the sarcomeres degenerate in ncx1h mutant cardiomyocytes a day later resulting in a sporadic distribution of α-actinin (Figure 1A). Zebrafish myocardial cells of the outer curvature normally assume an elongated, flat shape by two days of development (Auman et al., 2007; Cavanaugh et al., 2015). However, ncx1h mutant cardiomyocytes fail to elongate (Figure 1B) and both atrial and ventricular chambers become dysmorphic (Figure 1C), indicating a requirement for NCX1 activity in the maintenance of myofibril integrity and cardiac chamber morphology.
Figure 1
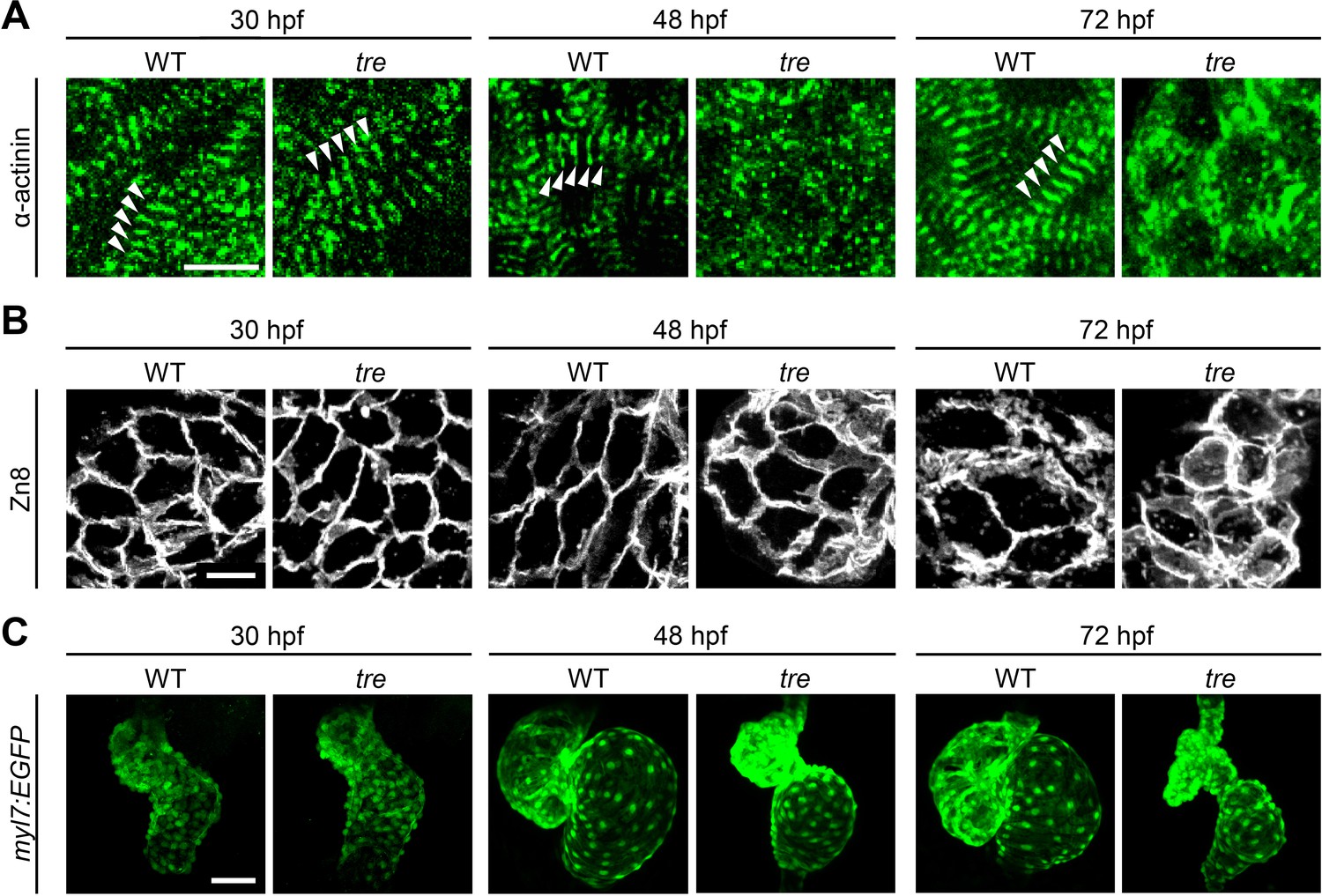
Disorganized myofibril structure in tre/ncx1h cardiomyocytes.
Wild type (WT) and tre/ncx1h (tre) mutant hearts at 30, 48 and 72 hpf. (A) Zebrafish hearts stained for α-actinin to visualize Z-lines. At 30 hpf, periodic α-actitin staining was observed in wild type and tre hearts (arrowheads). By 48 hpf, sarcomeres are disassembled in tre hearts. Scale bar, 10 μm. (B) The cell shape of cardiomyocytes was visualized by Zn8 staining. Scale bar, 10 μm. (C) Embryonic fish hearts were visualized by GFP expression in the myl7:EGFP transgenic background. Note that tre hearts become dysmorphic after two days of development. Scale bar, 50 μm.
Elevated MuRF1 expression in ncx1h-deficient hearts
To explore molecular pathways by which NCX1 influences myofibril integrity, we isolated hearts from wild type and ncx1h mutant embryos and compared their gene expression profiles. We found that the expression of Muscle Ring-finger protein-1 (MuRF1, also known as TRIM63) is significantly elevated in ncx1h mutant hearts. There are two highly homologous murf1 genes in zebrafish (murf1a/trim63a and murf1b/trim63b) (Macqueen et al., 2014). Phylogenetic analysis showed that zebrafish murf1a and murf1b cluster with other vertebrate murf1 genes (Figure 2A). Both genes span a single exon encoding peptides highly homologous to each other and to their mammalian orthologs (Figure 2B) (Postlethwait, 2007) and are expressed in striated muscles (Figure 2C) (Willis and Patterson, 2006). In situ hybridization and quantitative RT-PCR analyses further confirmed that both murf1a and 1b are upregulated in ncx1 mutant hearts (Figure 3).
Figure 2
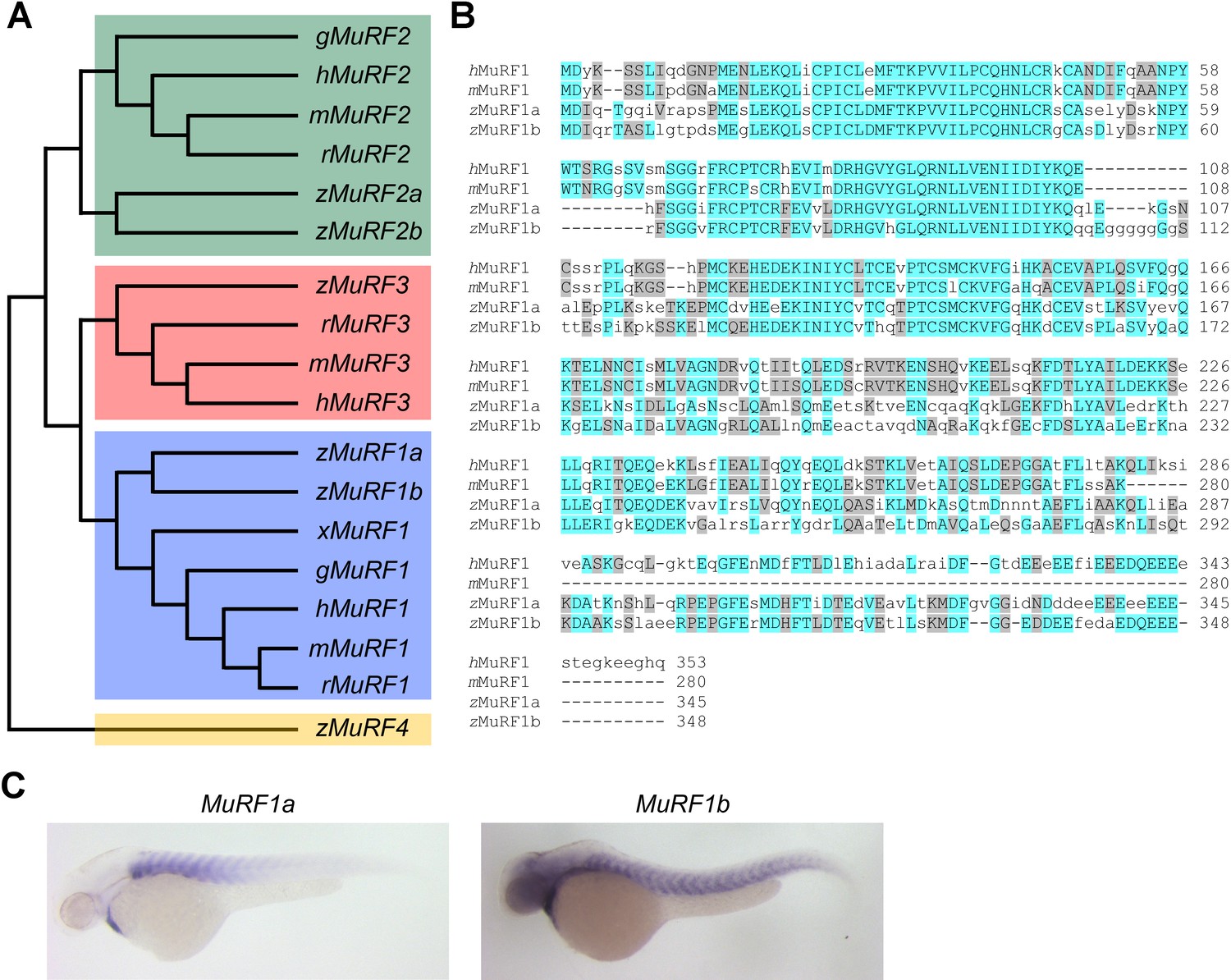
Zebrafish murf1 genes.
(A) Phylogenetic tree of vertebrate murf1, 2, 3 and 4 (also known as trim63, 55, 54 and 101, respectively). The tree was constructed using ClustalX with the neighbor-joining method. Zebrafish (z), Human (h), mouse (m), rat (r), chick (g), frog (x). (B) Alignment of murf1 genes from human, mouse and zebrafish. Blue boxes highlight identical amino acids and grey boxes indicate similar residues. (C) Whole-mount in situ hybridization demonstrating the expression patterns of murf1a and murf1b in the zebrafish embryo.
Figure 3
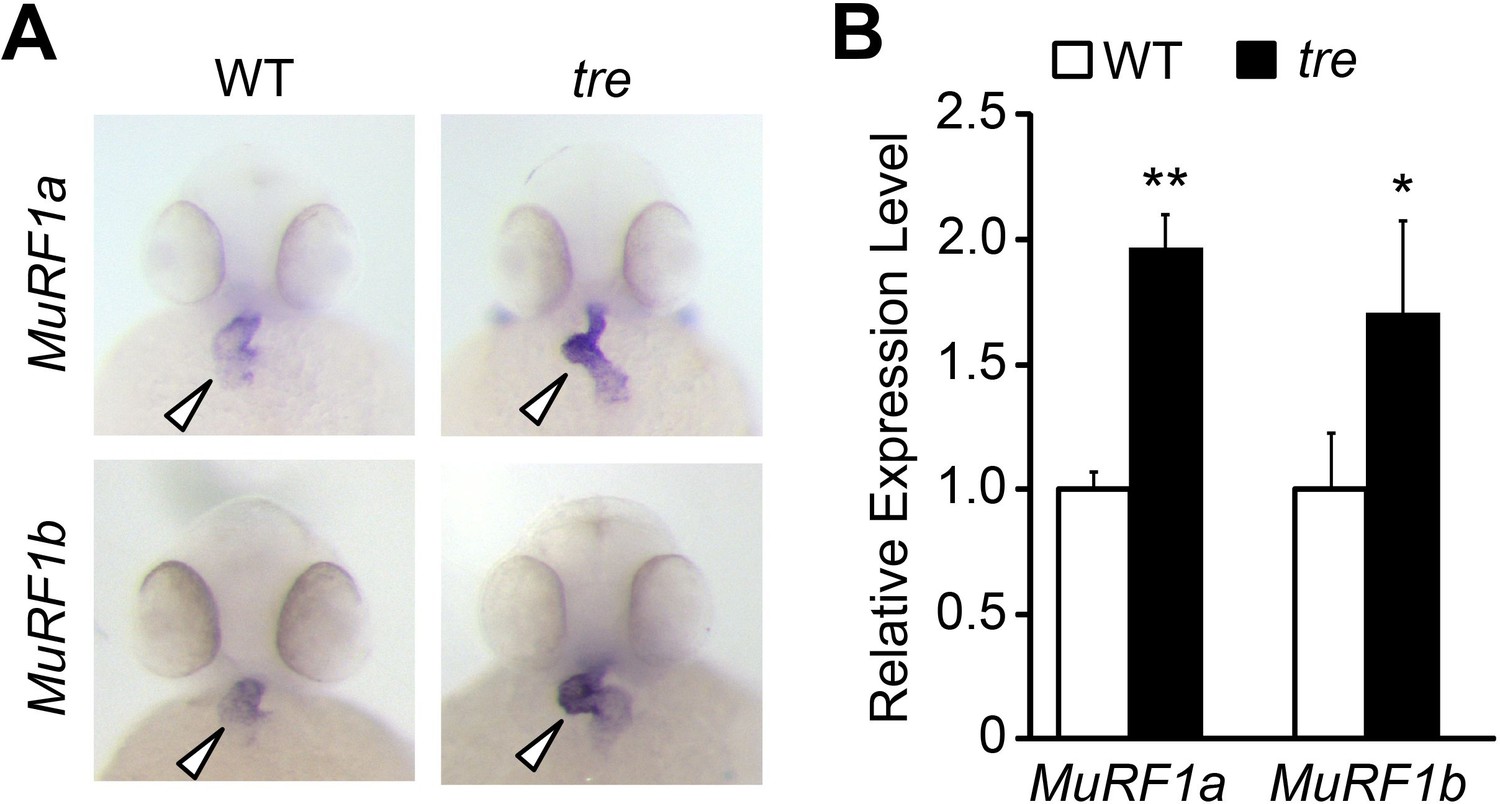
Upregulation of MuRF1 in tre/ncx1h deficient hearts.
(A) In situ hybridization analysis shows a significant increase of murf1a and murf1b expression in tre hearts. Arrowheads point to the heart. (B) Quantitative RT-PCR analysis shows an upregulation of murf1a and murf1b in tre hearts. * p<0.05; ** p<0.01.
MuRF1 regulates myofibril integrity in cardiomyocytes
Elevated murf1 expression is associated with muscle atrophy and can induce the breakdown of myofibrils in cultured cardiomyocytes (Kedar et al., 2004). We thus asked whether MuRF1 overexpression in the heart is sufficient to induce cardiomyopathy. To this end, we generated a transgenic fish, myl7:MuRF1a-IRES-GFP, in which MuRF1 and a GFP reporter are expressed under the control of the cardiac-specific myl7 promoter (Figure 4A). As shown in Figure 4B, murf1a expression is upregulated in myl7:MuRF1a-IRES-GFP transgenic hearts. Interestingly, α-actinin failed to maintain a striated pattern in cardiomyocytes of myl7:MuRF1a-IRES-GFP embryos (Figure 4C), demonstrating that overexpression of MuRF1 leads to sarcomere disassembly in the heart. Consequently, MuRF1-overexpressing hearts become dilated (Figure 4D) and their cardiac function is compromised (Videos 1 and 2). The atrial fractional shortening of myl7:MuRF1a-IRES-GFP hearts was reduced by approximately 10% and ventricular fractional shortening by ~15% compared to non-transgenic siblings and the heart rate was also reduced by 10% (Figure 4E). Together, these findings demonstrate that overexpression of MuRF1 is sufficient to disrupt myofibril structure and impair cardiac function in vivo.
Figure 4
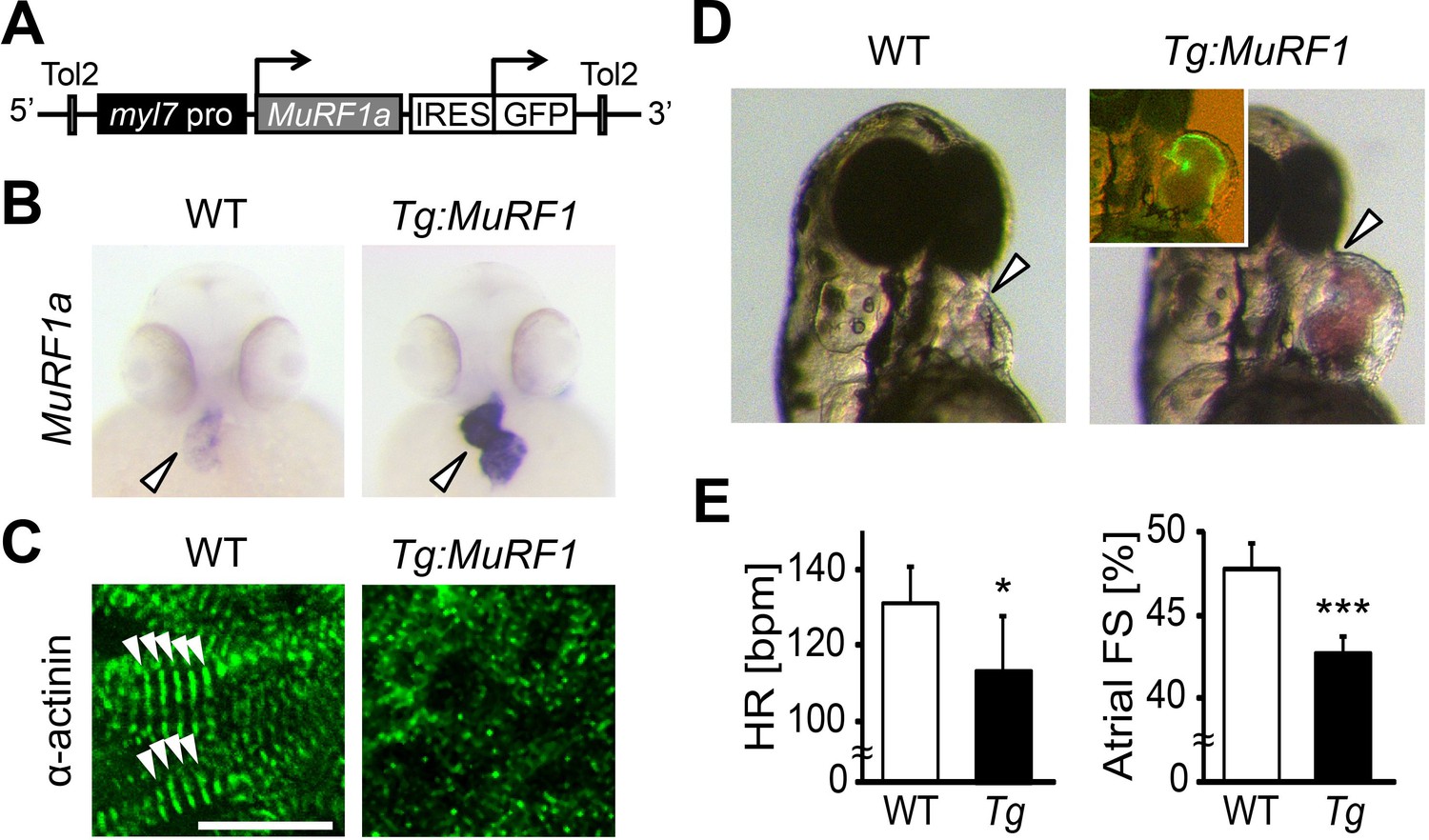
Upregulation of MuRF1a leads to myofibril disarray.
(A) Schematic representation of the construct that drives cardiac-specific MuRF1a expression. (B) Murf1a expression is upregulated in the 2-day-old myl7:MuRF1a-IRES-GFP heart (right panel) compared to the wild type heart (left panel). (C) α-actinin staining in 3-day-old wild type (left panel) and transgenic (right panel) cardiomyocytes. Note that sarcomeres are disassembled in myl7:MuRF1a-IRES-GFP cardiomyocytes. (D) Live images of wild type and Tg(myl7:MuRF1a-IRES-GFP) fish at 72 hpf (left panels). Transgenic hearts are GFP positive and become dilated (inset). (E) Heart rate (HR) and atrial fractional shortening (FS) in wild type and myl7:MuRF1a-IRES-GFP embryos at 72 hpf. *p<0.05; ***p<0.001.
Video 1
Three-day-old myl7:EGFP transgenic heart.
https://doi.org/10.7554/eLife.27955.006
Video 2
Three-day-old myl7:MuRF1a-IRES-GFP transgenic heart.
https://doi.org/10.7554/eLife.27955.007Blocking MuRF1-induced protein degradation preserves myofibril integrity in ncx1h mutant hearts
MuRF1’s upregulation upon loss of ncx1h activity, along with its established function as a muscle-specific E3 ubiquitin protein ligase that targets sarcomeric proteins for proteasome degradation, make it a good candidate for the cause of the myofibril disarray present in ncx1h deficient hearts. If MuRF1 upregulation indeed causes sarcomere disassembly, one would predict that blocking MuRF1 activity or its downstream protein degradation pathway might ameliorate the myofibril defects in ncx1 mutant hearts. Since both murf1a and murf1b are upregulated in ncx1h deficient hearts, we knocked down these genes simultaneously. Western blot analysis showed that murf1a/murf1b morpholino knockdown reduced overall MuRF1 protein levels by 27% (Figure 5C). Interestingly, even this modest level of reduction in MuRF1 expression improved sarcomere integrity in ncx1h deficient hearts; ~80% of ncx1h/murf1a/murf1b triple-deficient embryos had intact sarcomeres (n = 24), a significant increase compared to ncx1h mutant hearts (~35%, n = 21; p<0.001) (Figure 5A,B). Similarly, treatment with the proteasome inhibitor MG132 suppressed the myofibril disarray caused by NCX1 deficiency. Approximately 72% of MG132-treated ncx1h mutants had a striated α-actinin pattern, indicating the presence of intact sarcomeres (n = 18; p<0.001) (Figure 5A,B), suggesting that upregulation of MuRF1 induces myofibril degradation via a proteasome-dependent mechanism in ncx1h-deficient cardiomyocytes. Prior studies have implied a connection between cardiac contraction and myofibril integrity (Berdougo et al., 2003; Auman et al., 2007; Nishii et al., 2008; Yang et al., 2014). Interestingly, ncx1h/murf1a/murf1b triple-deficient hearts never establish coordinated contractions (Video 3), demonstrating that sarcomere integrity can be uncoupled from the loss of cardiac contractions in the context of aberrant Ca2+ handling-induced heart failure.
Figure 5
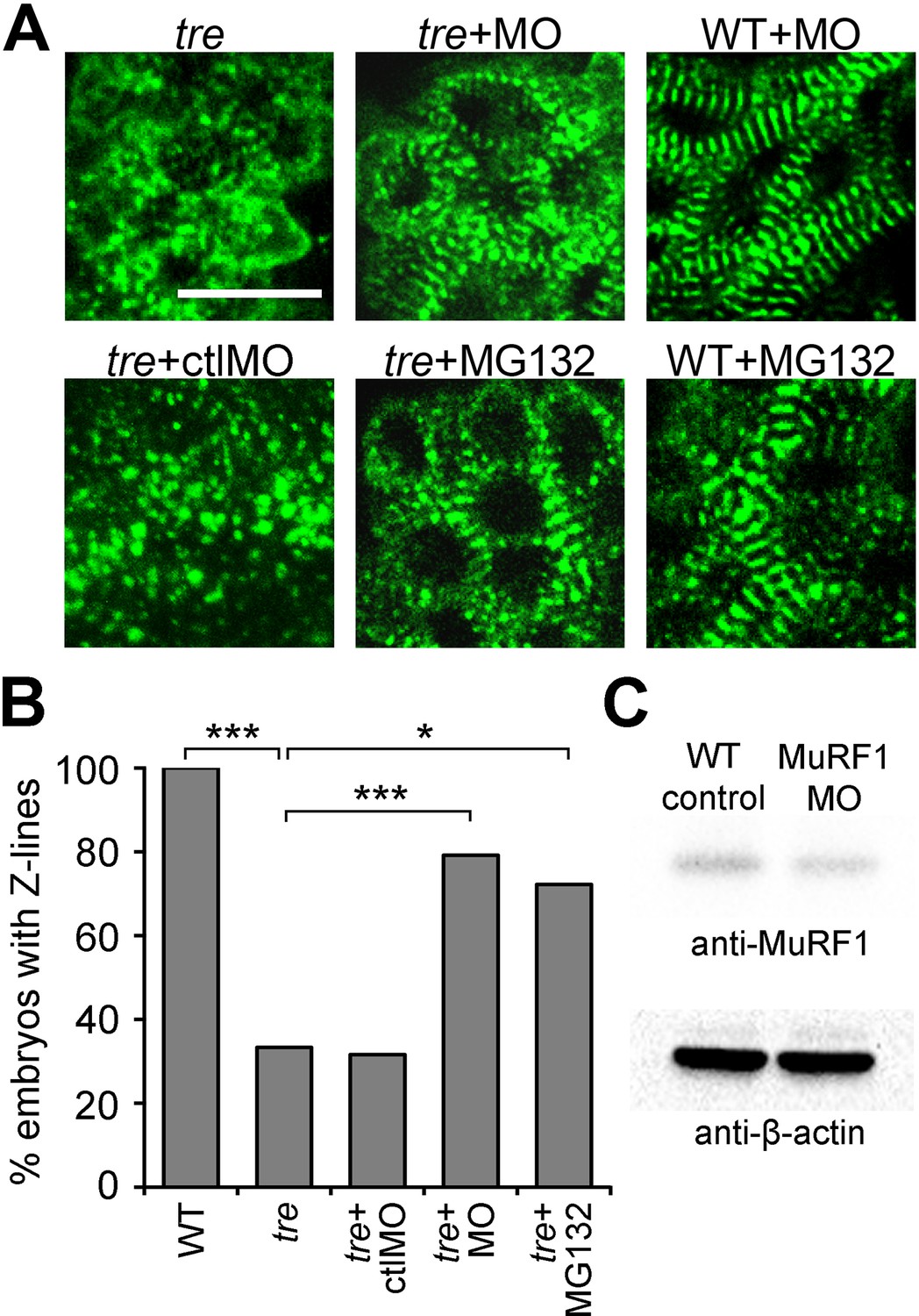
Blocking MuRF1-mediated proteasome degradation preserves myofibril integrity in tre/ncx1 deficient hearts.
(A) Z-lines were visualized by α-actinin staining. By 72 hpf, sarcomeres are disassembled in hearts of uninjected (tre) and control morpholino-injected (tre +ctlMO) tremblor embryos. Murf1a/murf1 b knockdown does not affect sarcomere integrity in wild type embryos (WT +MO), but prevents sarcomere degradation in tre (tre +MO). Similarly, treatment with a proteasome inhibitor, MG132, preserves myofibril integrity in tre cardiomyocytes (tre +MG132). Scale bar, 10 μm. (B) Graph shows % of embryos with periodic α-actinin staining at 72 hpf. (C) Western blot detecting MuRF1 and β-actin proteins in uninjected control (WT control) and murf1a/murf1 b knockdown (MuRF1 MO) embryos. Chi-squared test, *p<0.05; **p<0.01; ***p<0.001.
Video 3
Two-day-old ncx1h/murf1a/murf1b triple deficient heart.
https://doi.org/10.7554/eLife.27955.009Ca2+ induces MuRF1a expression
Our study showed that while ncx1h mutant hearts suffer from Ca2+ handling defects and never establish normal Ca2+ cycles or heartbeats, the initial assembly of sarcomeres proceeds properly. Since the Ca2+ handling defects precede the breakdown of sarcomeres, we hypothesized that Ca2+ overload induces murf1 expression in cardiomyocytes and thereby leads to sarcomere disassembly and cardiomyopathy. To examine this hypothesis, we isolated a 6.9 kb genomic fragment upstream of murf1a, MuRF1a (−6906). Transgenic analysis showed that this genomic fragment was sufficient to drive GFP expression in cardiac and skeletal muscles (Figure 6A and B), a pattern resembling the endogenous murf1a expression pattern (Figure 2C), indicating that critical regulatory elements are present in this genomic fragment. We then created a MuRF1a (−6906) Luciferase reporter construct to test whether this murf1 upstream regulatory element is responsive to Ca2+ signaling. We transfected the MuRF1a (−6906) Luciferase reporter into HEK293T cells and induced Ca2+ flux by treatment with the Ca2+ ionophore A23187. Interestingly, the luciferase activity was significantly enhanced by A23187 induction (Figure 6C), demonstrating that murf1 transcription is sensitive to intracellular Ca2+ levels.
Figure 6
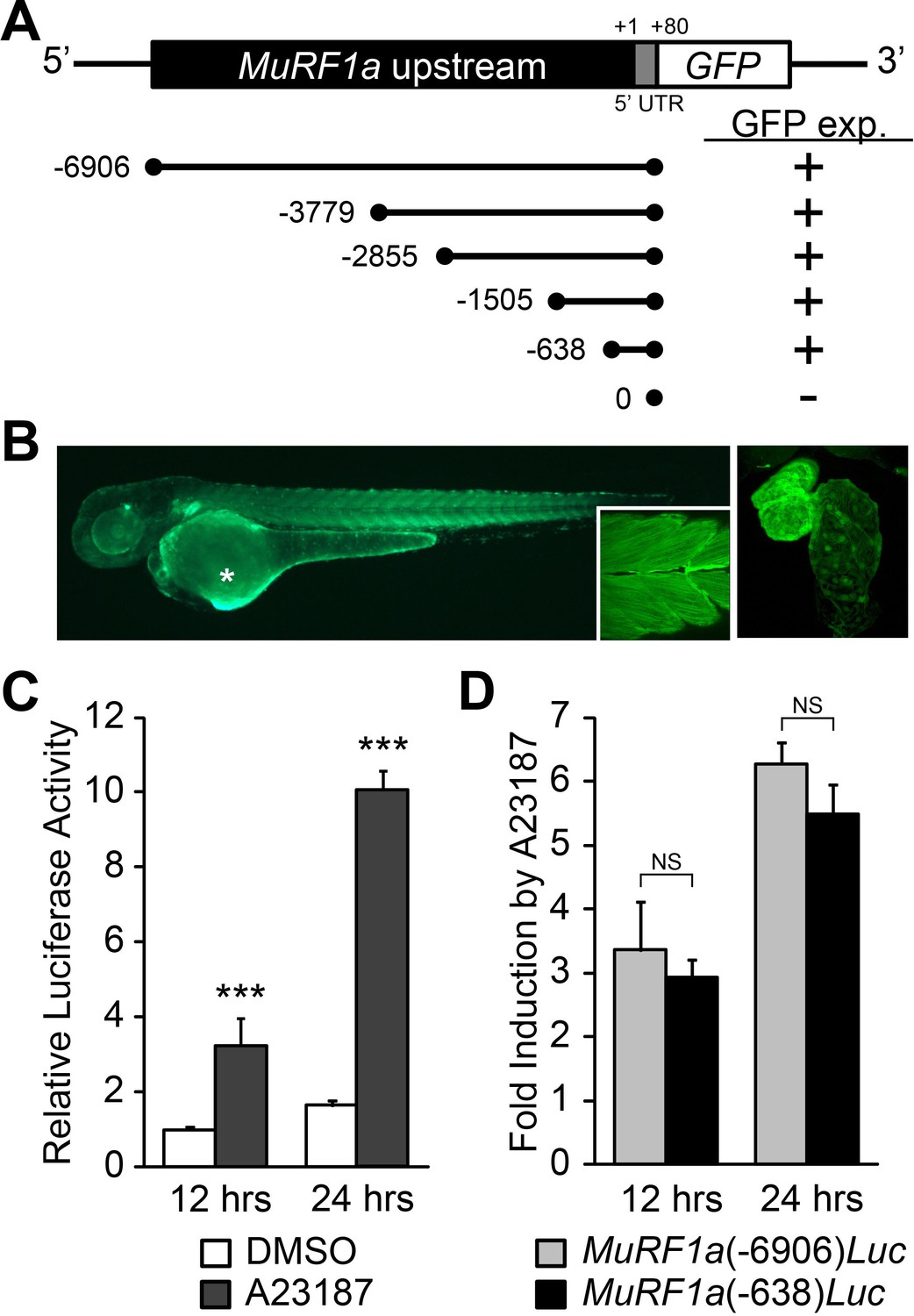
Identification of a MuRF1a regulatory element.
(A) Schematic representation of murf1a reporter constructs. + denotes the presence of GFP expression in the heart and somites. (B) A MuRF1a (−6906)-GFP transgenic embryo exhibits GFP expression in the heart and the somites (inset shows higher magnification image of the somites). The right panel shows a higher magnification image of the heart. The asterisk denotes auto-fluorescence from the yolk. (C) A23187 treatment induces luciferase activity driven by the MuRF1a (−6906) promoter. Values on the y-axis represent the luciferase activity relative to cells treated with DMSO for 12 hr. (D) Comparison of Ca2+ responsiveness between the MuRF1a (−638) and MuRF1a (−6906) promoters. Values on the y-axis represent the fold increase in luciferase activity in response to A23187 treatment compared to DMSO-treated cells at each time point. ***p<0.001; NS, not significant.
We next tested the expression patterns of a series of deletion constructs of MuRF1a (−6906) and found that the 638 bp region immediately upstream of the transcription initiation site, MuRF1a (−638), was sufficient to drive reporter gene expression in cardiac and skeletal muscles (Figure 6A). MuRF1a (−638)-Luc also displayed enhanced luciferase activity upon A23187 induction at levels comparable to MuRF1a (−6906)-Luc (Figure 6D), indicating that the 638 bp murf1a proximal region is sufficient to direct Ca2+-mediated murf1 transcription.
Ca2+ regulates MuRF1 expression via the Calcineurin-FoxO signaling pathway
Calmodulin-dependent protein kinase II (CaMKII) and the calmodulin-dependent protein phosphatase calcineurin (Cn) are two major transducers of Ca2+ signals in cardiomyocytes (Heineke and Molkentin, 2006; Maillet et al., 2013). We asked whether either of these pathways is involved in the regulation of murf1 gene expression. We treated MuRF1a (−638)-Luc-transfected HEK293T cells with either KN62, a chemical inhibitor of CaMKII, or FK506, an inhibitor of Cn. KN62 treatment did not have a significant impact on MuRF1a (−638)-driven expression, but FK506 treatment potently attenuated the A23187-induced increase of MuRF1a (−638)-Luc reporter activity (Figure 7A), suggesting that MuRF1 expression is regulated by a Cn-mediated mechanism. This interpretation is further supported by the observations that Cn overexpression enhances A23187-induced murf1 reporter activity and that the A23187-induced MuRF1 expression is blunted by overexpression of a dominant negative form of Cn (DN-Cn) (Figure 7B) (Kahl and Means, 2004).
Figure 7
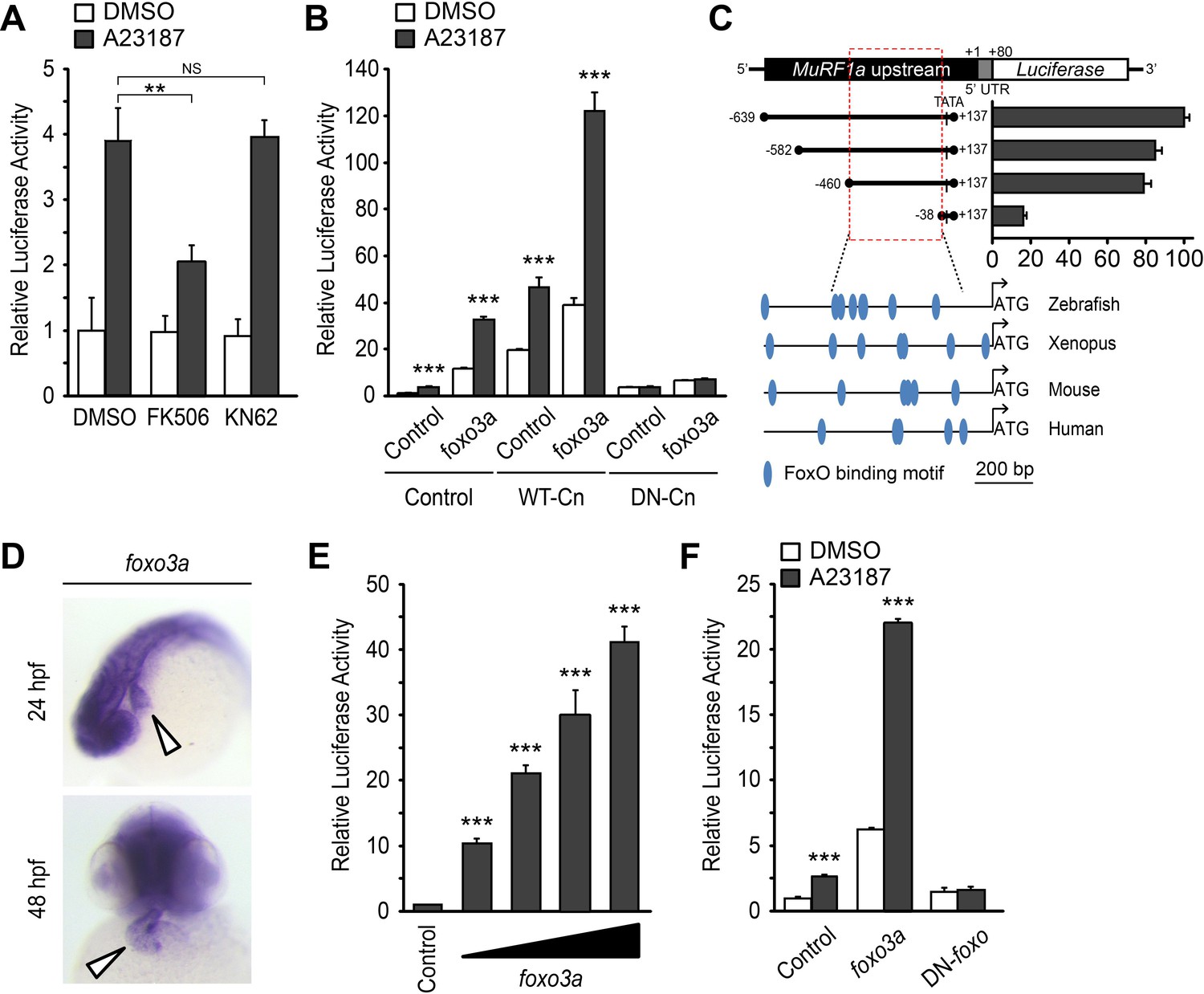
A Ca2+-Cn-FoxO signaling pathway regulates MuRF1 expression.
(A) HEK293T cells were transiently transfected with the MuRF1a (−638) luciferase reporter construct. Cells were incubated with FK506 or KN62 before DMSO or A23187 treatment. (B) Luciferase assay of the MuRF1a (−638) reporter cotransfected with foxo3a, wild type calcineurin (WT-Cn) or dominant-negative calcineurin (DN-Cn). (C) Diagram of the MuRF1a (−638) reporter serial deletion constructs generated for this study. The bar graph (right) shows the luciferase activity of each reporter construct relative to that of the empty expression plasmid. The red dotted box indicates the minimal cis-regulatory element of MuRF1a. The lower diagrams represent an alignment of the zebrafish, Xenopus, mouse and human MuRF1 promoters. Blue circles indicate putative FoxO binding sites (D) Whole-mount in situ hybridization detects foxo3a expression in the zebrafish heart. White arrowheads point to the heart. (E) HEK293T cells were transfected with the MuRF1a (−638) luciferase reporter and foxo3a expression plasmid. (F) HEK293T cells were transfected with the MuRF1a (−638) reporter plasmid and either a wild type or dominant negative foxo3a expression plasmid. Values on the y-axis are expressed relative to the luciferase activity of DMSO treated cells. **p<0.01; ***p<0.001; NS, not significant.
We next explored the potential molecular mechanisms by which Cn influences MuRF1 expression. We found multiple putative FoxO binding sites located the minimal regulatory regions of zebrafish murf1a/b and within the 1 kb region immediately upstream of the transcription initiation sites of the frog, mouse and human murf1 genes (Figure 7C). Since FoxO is a downstream mediator of Cn signaling (Hudson and Price, 2013) and is involved in the regulation of MuRF1 in skeletal muscles (Stitt et al., 2004; Waddell et al., 2008), we examined the possibility that FoxO mediates Cn’s regulation of MuRF1 expression in cardiomyocytes. There are seven foxo genes in zebrafish (Wang et al., 2009), all of which are expressed in the developing heart (Figure 7D and Figure 8). When co-transfected with the MuRF1a(−638)-Luc reporter, all zebrafish foxo genes tested were capable of enhancing the A23187-induced increase in MuRF1a(−638) luciferase activity (Figure 9). FoxO3a promoted strong murf1a promoter activity (Figure 9) and was used for the remainder of our analyses. We found that FoxO3a enhanced MuRF1a(−638)-Luc reporter activity in a dose-dependent manner (Figure 7E) whereas overexpression of a dominant-negative form of FoxO (DN-foxO), which lacks the transactivation domain but harbors an intact DNA binding domain (Medema et al., 2000; van den Heuvel et al., 2005), abrogated the A23187-induced MuRF1a(−638)-Luc activity (Figure 7F). We next asked whether FoxO mediates Cn signaling to control MuRF1 expression. We found that cotransfection of Cn and FoxO3a enhances murf1a promoter activity (Figure 7B,E) and that FoxO could no longer induce MuRF1a expression in the presence of a dominant negative form of Cn (Figure 7B,E), demonstrating that Ca2+ influences MuRF1 expression via the Cn-FoxO signaling axis.
Figure 8
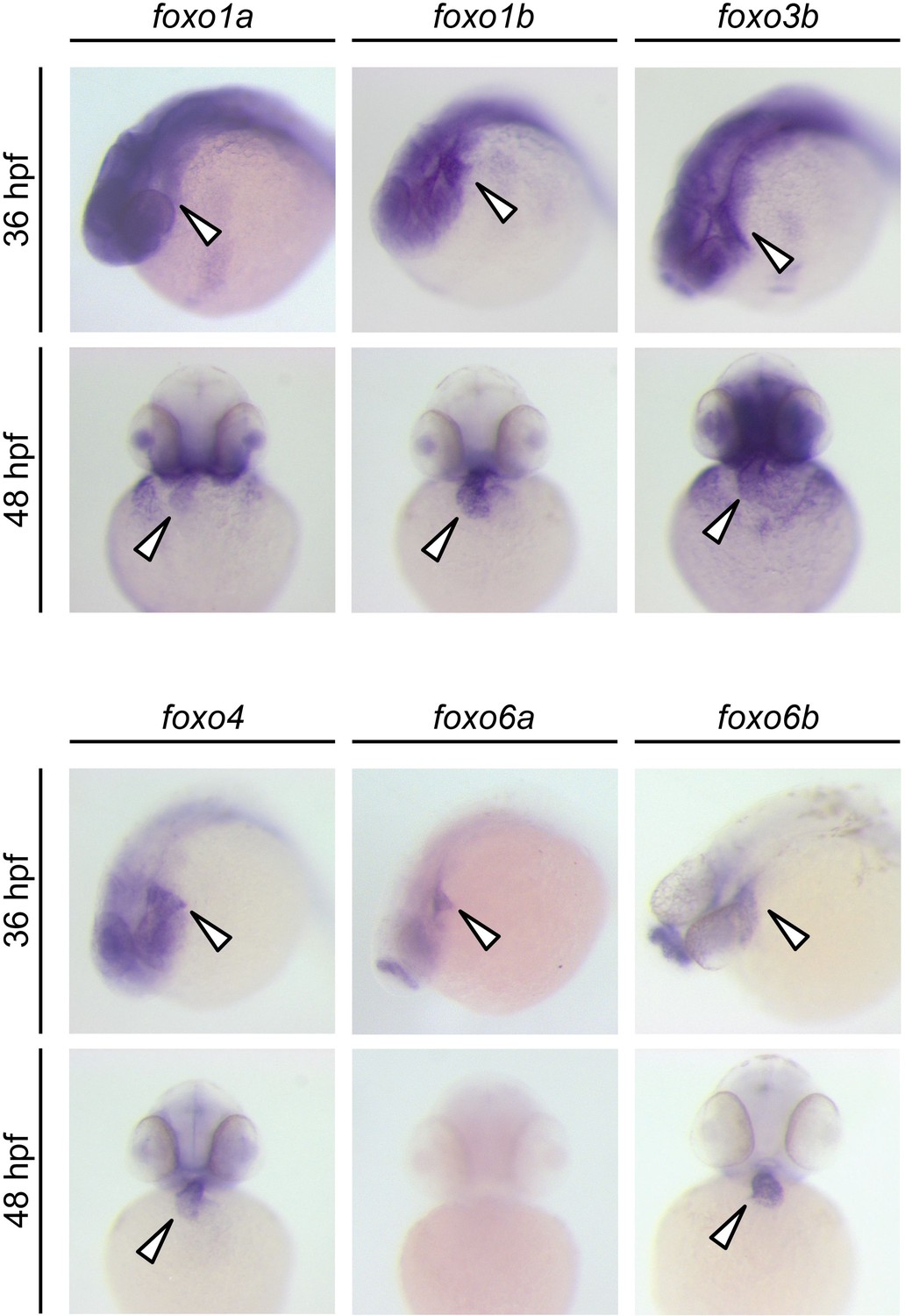
Expression patterns of zebrafish foxo genes.
Whole-mount in situ hybridization analysis showing foxo expression in zebrafish embryos. While foxo6a expression is diminished by 48 hpf, all the other foxo genes examined (foxo1a, 1b, 3b, 4, 6a and 6b) are persistently expressed in the heart. Arrowheads point to the heart.
Figure 9

FoxO induces MuRF1 expression.
Luciferase activity of the MuRF1a (−638) reporter cotransfected with different foxo genes. Values on the y-axis are expressed relative to the luciferase activity of DMSO treated cells. ***p<0.001.
Cn and FoxO regulate MuRF1 expression in the heart
Based on our finding that a Cn-FoxO-MuRF1 regulatory pathway is activated in response to elevated intracellular Ca2+ levels in cultured cells, we explored the significance of the Cn-FoxO-MuRF1 pathway in the regulation of myofibril integrity in myocardial cells in vivo. The subcellular localization of FoxO is controlled by its phosphorylation status (Huang and Tindall, 2007). We reasoned that the Ca2+ extrusion defect in ncx1h mutant hearts could activate Cn resulting in the dephosphorylation and nuclear translocation of FoxO. To assess the subcellular localization of FoxO in cardiomyocytes, we injected the myl7:FLAG-foxo3a-IRES-EGFP plasmid into zebrafish embryos and used the FLAG-epitope as a proxy to assess the localization of FoxO. Indeed, while FoxO was primarily sequestered in the cytoplasm of cardiomyocytes in wild type zebrafish hearts, FoxO protein was enriched in the nuclei of ncx1h mutant cardiomyocytes (Figure 10A). This nuclear accumulation of FoxO correlated with the increased MuRF1 expression in ncx1h mutant hearts (Figure 3). In addition, we found that MuRF1 expression could also be induced in the heart by overexpression of FoxO3a or a constitutively active form of FoxO3a in which three phosphorylation sites were replaced by with alanines (CA-FoxO3a: T29A, S236A, S299A) (Figure 10B) (Brunet et al., 1999). Conversely, pharmacological inhibition of Cn activity by treatment with FK506 or overexpression of DN-FoxO blunted MuRF1 expression in ncx1h mutant embryos (Figure 10B). Finally, we used α-actinin as a proxy to examine whether the correlation between FoxO and MuRF1 expression translates to the preservation of sarcomere structure. We found that overexpression of CA-FoxO3a in wild type embryos resulted in a sporadic α-actinin distribution in cardiomyocytes that resembled the phenotype observed in ncx1h mutant hearts whereas overexpression of DN-FoxO restored a striated α-actinin pattern in ncx1h mutant hearts (Figure 10C).
Figure 10
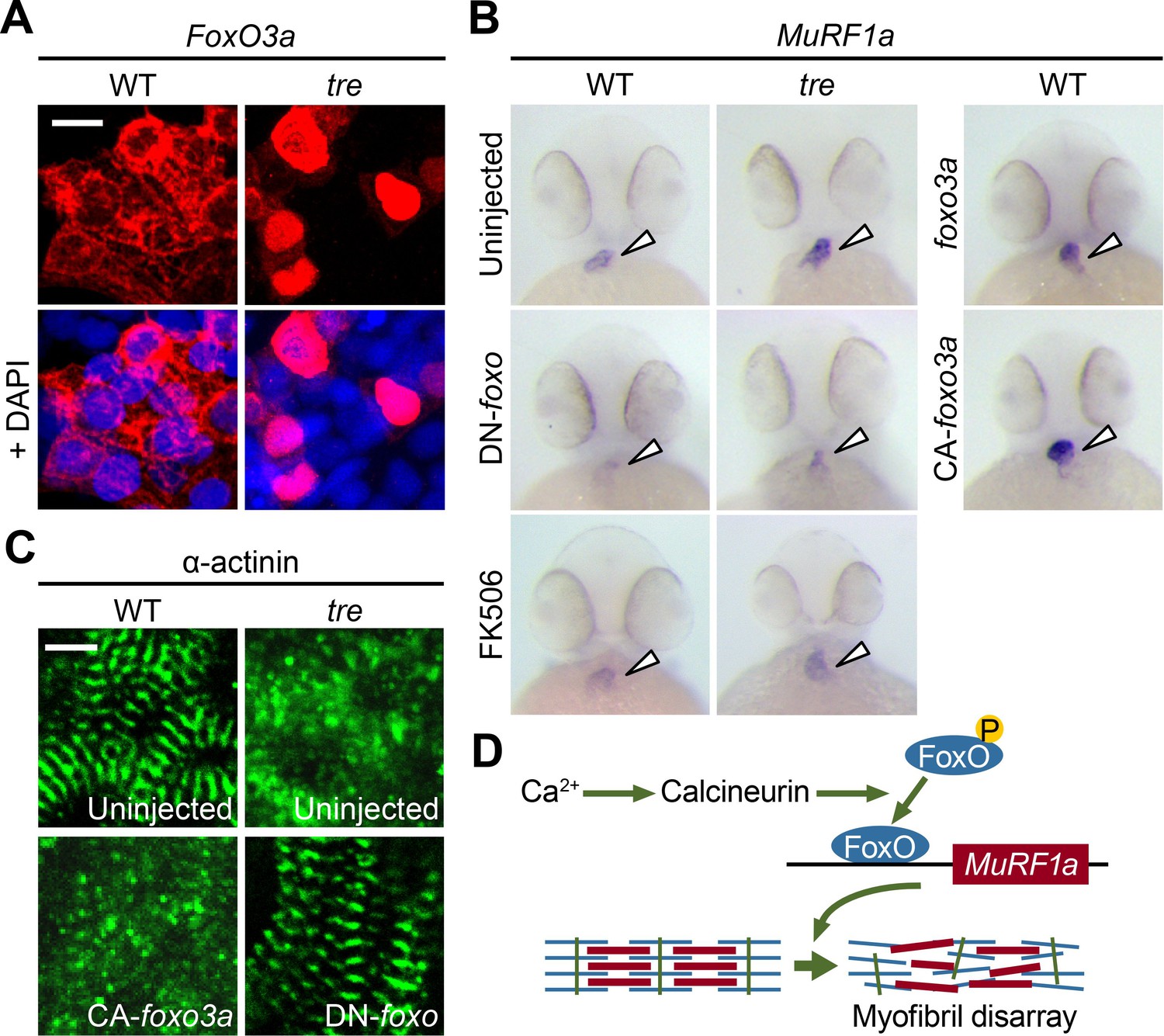
FoxO3a regulates MuRF1 expression in the heart.
(A) FoxO3a is predominantly localized in the cytoplasm of 2-day-old wild type cardiomyocytes, but is concentrated in the nuclei of tre cardiomyocytes. FoxO3a is pseudo colored in red and nuclei are labeled by DAPI in blue. Scale bar: 10 μm. (B) In situ hybridization showing stronger MuRF1 signals in tre mutant and CA-foxo3a injected two dpf hearts compared to wild type siblings. Murf1a expression in tre hearts is suppressed by DN-foxo overexpression or FK506 treatment. (C) Immunostaining of α-actinin in three dpf hearts. Intact sarcomeres were detected in control (uninjected) and DN-foxo3a injected tre hearts whereas disassembled sarcomeres were observed in tre and CA-foxo3a injected hearts. Scale bar: 5 μm. (D) Model for Ca2+ overload-induced myofibril disarray. Calcineurin dephosphorylates FoxO leading to FoxO nuclear translocation, MuRF1 expression and sarcomere disassembly.
Conclusion
Compromised Ca2+ homeostasis and damaged cardiac muscle fibers are often observed in deteriorating diseased hearts, but a causative relationship between these outcomes has not previously been demonstrated. In this study, we used the zebrafish ncx1h mutant as an animal model to explore the molecular link between Ca2+ signaling and myofibril integrity in the heart. We showed that NCX1 activity is dispensable for the initial assembly of sarcomeres, but the maintenance of myofibril structure in myocardial cells requires tightly controlled Ca2+ homeostasis and MuRF1 expression.
Our molecular analyses using cultured cells and in vivo studies in zebrafish reveal a FoxO-MuRF1 signaling axis that is critically involved in the Ca2+-dependent regulation of myofibril integrity in the heart. We propose that under normal physiological conditions where the cytosolic diastolic Ca2+ level is low, FoxO is sequestered in the cytoplasm and MuRF1 expression is maintained at a basal level to support the normal turnover of sarcomeric proteins. Under pathological conditions, when diastolic Ca2+ is elevated, the activation of Cn dephosphorylates FoxO and allows its nuclear translocation, leading to upregulation of MuRF1 and the degradation of myofibrils (Figure 10D). Interfering with the Cn-FoxO-MuRF1-proteosome pathway by pharmacological or genetic means can protect the sarcomeric integrity of cardiomyocytes suffering from Ca2+ dysregulation, indicating that the FoxO-MuRF1 signaling axis is a central regulator of the Ca2+-dependent growth and degradation of striated muscles. The activity of the Cn-FoxO-MuRF1 signaling pathway identified in this study is consistent with the roles of the FoxO-MuRF1 pathway in hypertrophy and atrophy responses in skeletal muscles (Sacheck et al., 2004; Stitt et al., 2004; Waddell et al., 2008) and suggests that FoxO-MuRF1 signaling is critical to the maintenance of tissue homeostasis and the response of myocytes to pathological stimuli. Furthermore, cardiac-specific overexpression of MuRF1 results in phenotypes resembling those observed in cardiomyopathy, including the breakdown of sarcomeres and a dilated heart with reduced heart rate and decreased contractility, raising the possibility that misregulation of MuRF1 contributes to the pathological progression of cardiovascular diseases. Interestingly, cardiac patients carrying specific murf1 gene variants have a poor prognosis (Chen et al., 2012; Su et al., 2014), suggesting that MuRF1 has a conserved role in the regulation of cardiac structure and function from lower vertebrates to humans and raising an intriguing possibility that the Cn-FoxO-MuRF1-proteosome pathway may be an attractive point of therapeutic intervention for cardiomyopathies. The complete loss of ncx1h activity in tremblor mutant cardiomyocytes eliminates Ca2+ cycling and causes embryonic lethality, an extreme condition that is more severe than what is observed in patients with chronic heart disease (Ebert et al., 2005; Langenbacher et al., 2005). Future studies using clinically relevant mammalian models will further illuminate the therapeutic potential of targeting the Cn-FoxO-MuRF1-proteosome pathway in the context of cardiovascular disease.
Materials and methods
Zebrafish husbandry, chemical treatment and morpholino knockdown
Request a detailed protocolZebrafish tremblor tc318 heterozygotes were bred in the Tg(myl7:EGFP) background and raised as previously described (Westerfield, 2000). Embryos were raised at 28.5°C and staged as previously described (Kimmel et al., 1995). For Cn or proteasome inhibition, embryos were treated with 10 μM FK506 (Sigma-Aldrich, St. Louis, MO) or 50 μM MG132 (Sigma-Aldrich) at 24 hpf. The morpholino-modified antisense oligonucleotides targeting the translation initiation sites of murf1a and murf1b (Table 1, Gene Tools) were microinjected at the 1- to 2 cell stage (8 ng each). This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols of the University of California, Los Angeles. The protocol was approved by the Chancellor's Animal Research Committee of the University of California, Los Angeles.
Table 1
Primers and morpholinos used in this manuscript
https://doi.org/10.7554/eLife.27955.015| Experiment | Target gene | Sequence |
|---|---|---|
| Quantitative RT-PCR | murf1a | F: 5'- GGAAGAAAACTGCCAGGCACAG −3' R: 5'- CTGGGTGATCTGCTCCAGAAGATG −3' |
| murf1b | F: 5'- CAGGACAATGCTCAACGTGCC −3' R: 5'- CTTGCTCTTTGCCAATACGCTCTAAGAG −3' | |
| Molecular cloning* | murf1a | F: 5'- CTGAGGTACCAAGCAGTGAAGGTTA −3' R: 5'- GCTAGGTACCAGTCTCTCATTGCTT −3' |
| murf1b | F: 5'- CTATGGATCCCTGCAGGGAATCATTTAC −3' R: 5'- GTTACTCGAGCATTTGTCAATGACCTTG −3' | |
| foxo1a | F: 5'- GTCTGAATTCCAGTATTGCTGGTACCATG −3' R: 5'- CATTGCTAGCACTACCCAGACACCCAG −3' | |
| foxo1b | F: 5'- GTATGGATCCTTGGTGATGGCAGAACC −3' R: 5'- GTATCTCGAGCAGCAGATGACATGTCTATC −3' | |
| foxo3a | F: 5'- GTATGGATCCGGAGTCGAGGAAATATGG −3' R: 5'- GTATCTCGAGCAGTTGCTTTACAGTGGAC −3' | |
| foxo3b | F: 5'- GTATGGATCCCGACCAAGACAGTAAAGAG −3' R: 5'- GTATCTCGAGCTGAGCAATTCCCATCAG −3' | |
| foxo4 | F: 5'- GTCTGAATTCCATCGCACAATGGAGG −3' R: 5'- CATTGCTAGCCAACAGTGGAGTTAGCT −3' | |
| foxo6a | F: 5'- ATGAGGATCCAACTCCATTAGACACAACC −3' R: 5'- GCTAGAATTCGTGTGATTGTTGAGGTCC −3' | |
| foxo6b | F: 5’- ATGAGGATCCCGGTTTCTTAAGCACAGAAG- 3’ R: 5’- ATGAGAATTCGACATTTATCCAGGCACC- 3’ | |
| MuRF1a(−6906) MuRF1a(−639) MuRF1a(−582) MuRF1a(−460) MuRF1a(−38) Common Reverse primer for MuRF1 | F: 5'- GTTAGCTAGCCGACTTACTCACTCC −3' F: 5'- GTACTTGGAGCGGCCGCAATAA −3' F: 5'- GTCAGCTAGCCCAACCCAGACAATATATTACT −3' F: 5'- GTCGGCTAGCGGGAAATAATAATATTGTGATTG −3' F: 5'- GACTGCTAGCCGGCTGGTATATAAGAC −3' R: 5'- GAATCTCGAGTGCTGAGGTAGAGTC −3' | |
| Morpholino | Control murf1a murf1b | 5’-CCTCTTACCTCAGTTACAATTTATA-3’ 5’-TTTGACCCGTTTGGATGTCCATTGC-3 5’-AAGAGGCAGTTCGCTGAATGTCCAT-3’ |
-
*Restriction enzyme sites are underlined.
Zebrafish transgenesis
Request a detailed protocolTransgenic constructs, myl7:MuRF1a-IRES-EGFP and myl7:FLAG-foxo3a-IRES-EGFP, were generated using the Tol2kit (Kwan et al., 2007). Wild type embryos were injected at the 1 cell stage with 10–20 pg of the transgene plasmid and 20 pg of mRNA encoding Tol2 transposase. Embryos with cardiac-specific EGFP expression were raised as founders.
Microarray and quantitative PCR
Request a detailed protocolWild type and tre mutant hearts were isolated at 48 hpf as previously described (Geoffrey Burns and MacRae, 2006). Total RNA was purified using the RNeasy micro kit (Qiagen, Valencia, CA). Microarray hybridization was performed in triplicate using the Affymetrix Zebrafish GeneChip containing 15,617 genes. Data were analyzed using scripts written in the statistical programming language R (R Development Core Team, 2014). Differentially expressed genes were identified using linear models and multiple testing correction implemented in the Limma package (Smyth, 2004). The relative expression levels of murf1a and murf1b in the wild type and tre hearts were determined by quantitative PCR using the LightCycler 480 System (Roche Applied Science). GAPDH served as the internal control for normalization. Primer sequences used in this study are listed in Table 1.
In vivo GFP reporter assay
Request a detailed protocolAn approximately 7.0 kb genomic fragment upstream of the zebrafish murf1a gene (ranging from –6906 to +80 bp) was amplified from genomic DNA (Table 1). A deletion series of MuRF1a-EGFP construct was generated using the ERASE-A-BASE system (Promega, Madison, WI). For transient expression analysis, each deletion construct was digested with NheI and SalI to release the MuRF1a-EGFP reporter and microinjected into 1 cell stage embryos. A minimum of 20 EGFP-positive embryos of each group were examined at 1 and 2 days post fertilization using a Zeiss SV-11 epifluorescence microscope.
Whole-mount in situ hybridization, immunostaining and Western analysis
Request a detailed protocolWhole mount in situ hybridization, immunostaining and Western analysis were performed as previously described (Langenbacher et al., 2011; Cavanaugh et al., 2015). The antisense RNA probes were synthesized from pCS2 +expression constructs containing a partial genomic fragment (foxo5a) or full-length cDNA fragments (murf1a, murf1b, foxo1a, foxo1b, foxo3a, foxo3b, foxo4, and foxo5b) (Table 1). Goat anti-MuRF1 (1:500 dilution, R and D Systems, AF5366) and rabbit anti-goat-HRP (1:15,000 dilution, Thermo Fisher Scientific, 81–1620) were used for Western analysis. Phalloidin (1:50, Sigma-Aldrich), anti-sarcomeric α-actinin (1:1000, clone EA53, Sigma-Aldrich), α-FLAG (1:100, clone M2, Sigma-Aldrich) and Zn8 (1:100, Developmental Studies Hybridoma Bank, Iowa City, IA) were used for immunostaining. Fluorescence images were acquired using an LSM 510 confocal microscope (Zeiss, Germany) with a 40x water objective. Embryos were classified as having intact sarcomeres if they exhibited at least five adjacent, clearly defined Z-lines marked by α-actinin in any area of the ventricle.
Cardiac imaging and analysis
Request a detailed protocolVideos of Tg(myl7:MuRF1-IRES-EGFP) and Tg(myl7:EGFP) hearts were taken at 30 frames per second. Cardiac parameters were assessed by line-scan analysis as previously described (Shimizu et al., 2015).
Cell-based luciferase assay
Request a detailed protocolHEK293T cells (ATCC, Manassas, VA) were plated into 96-well plates at a density of 32000 cells per well and transfected with 200 ng of the MuRF1a (−6906)- or MuRF1a (−638)-luciferase reporter construct, 50 ng of the SV40-Renilla luciferase reporter construct and expression vectors (Cn, DN-Cn, foxo3a, CA-foxo3a or DN-foxo3a). Cells were treated with 5 μM A23187 (Sigma-Aldrich), 0.5 μM FK506 (Sigma-Aldrich) or 0.5 μM KN62 (Sigma-Aldrich). Luciferase activities were determined with the Dual-Glo Luciferase Assay System (Promega) in triplicate at least three times, and the activity of firefly luciferase was normalized to that of Renilla luciferase for transfection efficiency and cell viability. The identity of the HEK293T cell line has been authenticated by STR profiling using the Promega PowerPlexX16 System recommended by the American Type Culture Collection (Laragen, Inc., Culver City, CA). No mycoplasma contamination was detected (Laragen, Inc.).
Statistics
Sample sizes with adequate statistical power were empirically determined based on previous experiments. Samples were randomly allocated into control and experimental groups. Experiments of each condition were performed at least three times on independent biological replicates. Results are presented as the mean ±S.E. p-values associated with all comparisons are based on unpaired two-sided Student’s t-tests (n ≥ 3) unless otherwise stated. All data values were included in the analysis.
References
-
Muscle sparing in muscle RING finger 1 null mice: response to synthetic glucocorticoidsThe Journal of Physiology 589:4759–4776.https://doi.org/10.1113/jphysiol.2011.212845
-
Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin-1AJP: Endocrinology and Metabolism 307:E469–E484.https://doi.org/10.1152/ajpendo.00204.2014
-
During muscle atrophy, thick, but not thin, filament components are degraded by MuRF1-dependent ubiquitylationThe Journal of Cell Biology 185:1083–1095.https://doi.org/10.1083/jcb.200901052
-
A critical role for muscle ring finger-1 in acute lung injury-associated skeletal muscle wastingAmerican Journal of Respiratory and Critical Care Medicine 185:825–834.https://doi.org/10.1164/rccm.201106-1150OC
-
Purification of hearts from zebrafish embryosBioTechniques 40:278–282.https://doi.org/10.2144/000112135
-
Regulation of cardiac hypertrophy by intracellular signalling pathwaysNature Reviews Molecular Cell Biology 7:589–600.https://doi.org/10.1038/nrm1983
-
Dynamic FoxO transcription factorsJournal of Cell Science 120:2479–2487.https://doi.org/10.1242/jcs.001222
-
Calcineurin: a poorly understood regulator of muscle massThe International Journal of Biochemistry & Cell Biology 45:2173–2178.https://doi.org/10.1016/j.biocel.2013.06.029
-
Calcineurin regulates cyclin D1 accumulation in growth-stimulated fibroblastsMolecular Biology of the Cell 15:1833–1842.https://doi.org/10.1091/mbc.E03-10-0730
-
Stages of embryonic development of the zebrafishDevelopmental Dynamics 203:253–310.https://doi.org/10.1002/aja.1002030302
-
The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructsDevelopmental Dynamics 236:3088–3099.https://doi.org/10.1002/dvdy.21343
-
Modulation of muscle atrophy, fatigue and MLC phosphorylation by MuRF1 as indicated by hindlimb suspension studies on MuRF1-KO miceJournal of Biomedicine and Biotechnology 2010:1–9.https://doi.org/10.1155/2010/693741
-
The PAF1 complex differentially regulates cardiomyocyte specificationDevelopmental Biology 353:19–28.https://doi.org/10.1016/j.ydbio.2011.02.011
-
Mechanisms of altered Ca²⁺ handling in heart failureCirculation research 113:690–708.https://doi.org/10.1161/CIRCRESAHA.113.301651
-
Breaking down protein degradation mechanisms in cardiac muscleTrends in Molecular Medicine 19:239–249.https://doi.org/10.1016/j.molmed.2013.01.005
-
Molecular basis of physiological heart growth: fundamental concepts and new playersNature Reviews Molecular Cell Biology 14:38–48.https://doi.org/10.1038/nrm3495
-
Atrogin-1 and MuRF1 regulate cardiac MyBP-C levels via different mechanismsCardiovascular Research 85:357–366.https://doi.org/10.1093/cvr/cvp348
-
Na/Ca exchange and contraction of the heartJournal of Molecular and Cellular Cardiology 61:28–33.https://doi.org/10.1016/j.yjmcc.2013.06.001
-
Role of the ubiquitin proteasome system in the heartCirculation Research 112:1046–1058.https://doi.org/10.1161/CIRCRESAHA.112.300521
-
Insulin suppresses atrophy- and autophagy-related genes in heart tissue and cardiomyocytes through AKT/FOXO signalingHormone and Metabolic Research 45:849–855.https://doi.org/10.1055/s-0033-1347209
-
The zebrafish genome in context: ohnologs gone missingJournal of Experimental Zoology Part B: Molecular and Developmental Evolution 308:563–577.https://doi.org/10.1002/jez.b.21137
-
IGF-I stimulates muscle growth by suppressing protein breakdown and expression of atrophy-related ubiquitin ligases, atrogin-1 and MuRF1AJP: Endocrinology and Metabolism 287:E591–E601.https://doi.org/10.1152/ajpendo.00073.2004
-
The FOXO3a transcription factor regulates cardiac myocyte size downstream of AKT signalingJournal of Biological Chemistry 280:20814–20823.https://doi.org/10.1074/jbc.M500528200
-
Linear models and empirical bayes methods for assessing differential expression in microarray experimentsStatistical Applications in Genetics and Molecular Biology 3:1–25.https://doi.org/10.2202/1544-6115.1027
-
Rare variants in genes encoding MuRF1 and MuRF2 are modifiers of hypertrophic cardiomyopathyInternational Journal of Molecular Sciences 15:9302–9313.https://doi.org/10.3390/ijms15069302
-
BookR: A Language and Environment for Statistical ComputingVienna, Austria: R Foundation for Statistical Computing.
-
Direct control of caveolin-1 expression by FOXO transcription factorsBiochemical Journal 385:795–802.https://doi.org/10.1042/BJ20041449
-
The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 geneAJP: Endocrinology and Metabolism 295:E785–E797.https://doi.org/10.1152/ajpendo.00646.2007
-
Na+/Ca2+ exchanger-deficient mice have disorganized myofibrils and swollen mitochondria in cardiomyocytesComparative Biochemistry and Physiology Part B: Biochemistry and Molecular Biology 135:9–15.https://doi.org/10.1016/S1096-4959(03)00057-5
-
FoxO gene family evolution in vertebratesBMC Evolutionary Biology 9:222.https://doi.org/10.1186/1471-2148-9-222
-
Into the heart: the emerging role of the ubiquitin-proteasome systemJournal of Molecular and Cellular Cardiology 41:567–579.https://doi.org/10.1016/j.yjmcc.2006.07.015
-
Muscle ring finger 1 mediates cardiac atrophy in vivoAJP: Heart and Circulatory Physiology 296:H997–H1006.https://doi.org/10.1152/ajpheart.00660.2008
-
Understanding Cardiac Sarcomere Assembly With Zebrafish GeneticsThe Anatomical Record 297:1681–1693.https://doi.org/10.1002/ar.22975
Article and author information
Author details
Adam D Langenbacher

Funding
National Institutes of Health (HL096980)
- Jau-Nian Chen
European Commission (ZF-MODELS)
- Robert Geisler
European Commission (ZF-HEALTH)
- Robert Geisler
National Institutes of Health (HL126051)
- Jau-Nian Chen
National Institutes of Health (HL108186)
- Yibin Wang
Nakajima Foundation
- Hirohito Shimizu
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank Dr. Chen Gao and members of the Chen Lab for stimulating discussions. This work was supported by grants from the Nakajima Foundation (to HS), the National Institute of Health (HL096980 and HL126051 to JNC and HL108186 to YW), European Commission’s Sixth Framework Programme (ZF-MODELS project to RG) and Seventh Framework Programme (ZF-HEALTH project to RG).
Ethics
Animal experimentation: This study was performed in strict accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols of the University of California, Los Angeles. The protocol was approved by the Chancellor's Animal Research Committee of the University of California, Los Angeles (ARC#2000-051-51A).
Copyright
© 2017, Shimizu et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,083
- views
-
- 339
- downloads
-
- 35
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 35
- citations for umbrella DOI https://doi.org/10.7554/eLife.27955
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The Calcineurin-FoxO-MuRF1 signaling pathway regulates myofibril integrity in cardiomyocytes
eLife 6:e27955.
https://doi.org/10.7554/eLife.27955




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}