Hypoxia-inducible factor cell non-autonomously regulates C. elegans stress responses and behavior via a nuclear receptor
- Massachusetts Institute of Technology, United States
Abstract
The HIF (hypoxia-inducible factor) transcription factor is the master regulator of the metazoan response to chronic hypoxia. In addition to promoting adaptations to low oxygen, HIF drives cytoprotective mechanisms in response to stresses and modulates neural circuit function. How most HIF targets act in the control of the diverse aspects of HIF-regulated biology remains unknown. We discovered that a HIF target, the C. elegans gene cyp-36A1, is required for numerous HIF-dependent processes, including modulation of gene expression, stress resistance, and behavior. cyp-36A1 encodes a cytochrome P450 enzyme that we show controls expression of more than a third of HIF-induced genes. CYP-36A1 acts cell non-autonomously by regulating the activity of the nuclear hormone receptor NHR-46, suggesting that CYP-36A1 functions as a biosynthetic enzyme for a hormone ligand of this receptor. We propose that regulation of HIF effectors through activation of cytochrome P450 enzyme/nuclear receptor signaling pathways could similarly occur in humans.
https://doi.org/10.7554/eLife.36828.001Introduction
The capacity to sense and respond to oxygen deprivation, or hypoxia, is crucial to normal physiological function and survival of aerobic organisms, which require oxygen to perform respiration and generate energy in the form of ATP. The fundamental importance of a mechanism to detect and react to low oxygen is reflected in the presence of a conserved hypoxia-response pathway in most animal cells. This pathway consists of the transcription factor HIF, or hypoxia-inducible factor, and its negative regulator, the prolyl hydroxylase EGLN, which together mediate a diversity of metabolic and physiological adaptations to hypoxia. The three human EGLNs, which were identified as homologs of the C. elegans protein EGL-9, function as oxygen sensors. In the presence of oxygen, EGLN hydroxylates the HIF α-subunit (HIFα), allowing the von Hippel-Lindau (VHL) E3 ubiquitin ligase to promote HIFα degradation (Maxwell et al., 1999; Jaakkola et al., 2001; Epstein et al., 2001; Ivan et al., 2001). In conditions of low oxygen, HIFα is stabilized and acts with its partner HIFβ to drive adaptations to hypoxia through activation of its transcriptional targets (Kaelin and Ratcliffe, 2008; Semenza, 2011; Wang et al., 1995).
The canonical function of the EGLN/HIF pathway is to regulate genes that either increase oxygen availability, for example by promoting erythropoiesis and angiogenesis, or reduce the cellular requirement for oxygen, for example by driving a shift from oxidative phosphorylation to glycolytic metabolism. However, a growing body of work has found roles for the EGLN/HIF pathway in controlling other aspects of animal physiology and behavior. HIF promotes the response to numerous stressors, including infection, proteotoxicity, and oxidative stress (Palazon et al., 2014; Schito and Rey, 2018; Nakazawa et al., 2016; Powell-Coffman, 2010). HIF activation is associated with increased lifespan in C. elegans (Mehta et al., 2009; Zhang et al., 2009; Chen et al., 2009; Lee et al., 2010); this longevity phenotype likely stems from the improved stress resistance associated with HIF activity, as is often the case for pathways regulating longevity (Leiser et al., 2015; Shore and Ruvkun, 2013). The EGLN/HIF pathway also modulates several behaviors of C. elegans following prolonged hypoxia exposure, suggesting a role for this pathway in tuning neural circuit function (Chang and Bargmann, 2008; Pocock and Hobert, 2010; Ma et al., 2012). The mechanisms by which HIF mediates these non-canonical physiological and behavioral changes remain poorly defined.
Here we report the discovery of an endocrine signaling pathway that regulates multiple aspects of physiology and behavior downstream of HIF in C. elegans. From a genetic screen for suppressors of an egl-9(lf) mutant behavioral defect, we identified a cytochrome P450 gene, cyp-36A1, that is required for modulation of egg-laying behavior by the egl-9/hif-1 pathway. cyp-36A1 is transcriptionally upregulated in hypoxia or egl-9(lf) mutants, in which HIF-1 is constitutively active, and appears to be a direct HIF-1 target. cyp-36A1 controls expression of more than a third of HIF-1-upregulated genes, demonstrating that cyp-36A1 acts broadly downstream of hif-1. Regulation of gene expression and behavior by cyp-36A1 occurs cell non-autonomously, and the downstream effector of cyp-36A1 is the nuclear hormone receptor nhr-46, indicating that the likely function of CYP-36A1 is to generate a diffusible signal that controls NHR-46 activity. In addition to modulating behavior and gene expression, cyp-36A1 and nhr-46 mediate multiple forms of stress resistance associated with HIF activation. We conclude that CYP-36A1 and NHR-46 are important downstream effectors of the EGL-9/HIF pathway and function together to regulate a wide range of HIF-mediated physiology.
Results
A screen for suppressors of the egl-9(lf) egg-laying defect identifies the cytochrome P450 gene cyp-36A1
To identify novel, functionally important HIF effectors, we analyzed the modulation of C. elegans egg laying, the behavior that led our laboratory to discover the first EGLN gene, egl-9, and the first known functional role for any member of the EGLN/HIF pathway (Trent et al., 1983). egl-9(lf) mutants, in which HIF-1 is constitutively active, are defective in egg laying and become bloated with eggs as adults. Although the egg-laying defect of egl-9(lf) mutants is well-established, the downstream effectors of EGL-9 and HIF-1 in regulating egg-laying behavior remain unknown.
We performed a mutagenesis screen to identify genes that act in response to egl-9 to control egg laying. Specifically, we screened for second-site mutations that suppressed the egg-laying defect of egl-9(lf) animals (Figure 1A). Such suppressors could define genes that function downstream of egl-9. Two isolates from this screen were allelic to hif-1 (Figure 1—figure supplement 1A and B), consistent with a previous observation that hif-1(lf) suppresses the egl-9(lf) egg-laying defect (Bishop et al., 2004) and validating the screen as a means of identifying components of the HIF-1 pathway. A third isolate, n5666, was not allelic to hif-1 and had a G106R missense mutation in the gene cyp-36A1, which encodes a cytochrome P450 enzyme (Figure 1—figure supplement 1A and C). A transgene carrying a wild-type copy of cyp-36A1 fully rescued the suppression by n5666 of the egl-9(lf) egg-laying defect, demonstrating that the mutation in cyp-36A1 is the causative mutation and suggesting that the suppression phenotype is caused by reduction of cyp-36A1 function (Figure 1B–1E). Confirming these conclusions, a nonsense allele of cyp-36A1 also suppressed the egl-9(lf) egg-laying defect (Figure 3—figure supplement 1B and C). cyp-36A1(lf) single mutants did not exhibit hyperactive egg-laying behavior (Figure 1F and Figure 3—figure supplement 1A and D), indicating that suppression of the egl-9(lf) egg-laying defect by cyp-36A1(lf) is not a consequence of a nonspecific increase in egg-laying rate. We further showed that cyp-36A1(lf) suppressed the previously reported egg-laying defect of hypoxia-exposed worms (Miller and Roth, 2009), demonstrating a role for CYP-36A1 under physiological conditions of HIF-1 activation (Figure 1—figure supplement 2). We then analyzed the role of cyp-36A1 in regulating other behaviors. We observed that egl-9(lf) mutants have reduced locomotion and defecation rates, both of which were suppressed by hif-1(lf) (Figure 1G and H). cyp-36A1(lf) partially suppressed the slow locomotion and defecation rates of egl-9(lf) mutants, showing that CYP-36A1 modulates not only egg laying but also multiple other HIF-1-regulated behaviors.
Figure 1 with 2 supplements see all
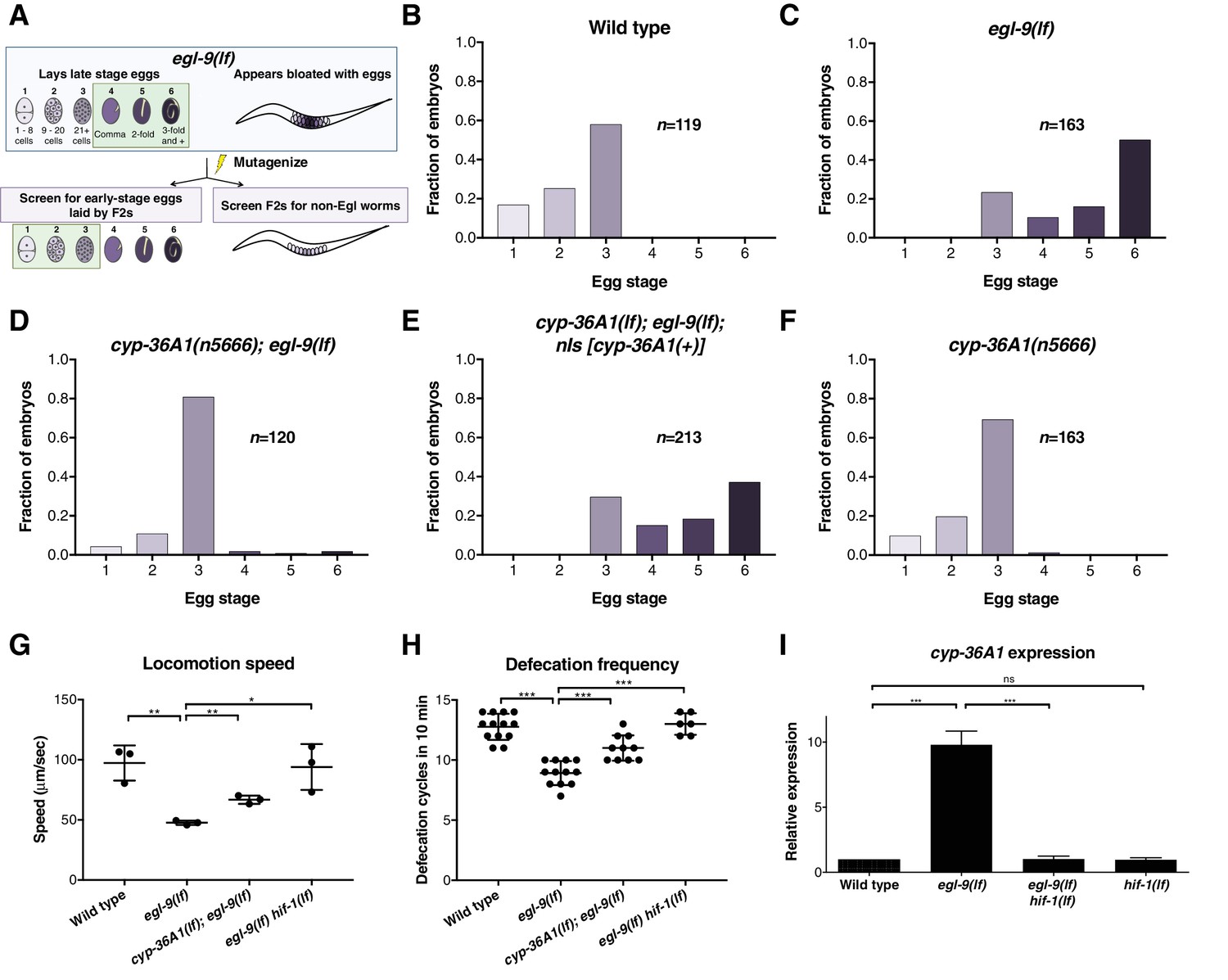
The cytochrome P450 gene cyp-36A1 is an effector of the hypoxia-response pathway that regulates behavior.
(A) Schematic of the screen design. Stages of embryonic development adapted from Ringstad and Horvitz (2008) and Paquin et al. (2016). (B–F) Distribution of stages of eggs newly laid by adult hermaphrodites, used as a proxy for egg retention time in utero, of animals of the indicated genotypes. (B) Stages of eggs laid by wild-type animals. (C) egl-9 loss-of-function (lf) mutants laid later stage eggs than the wild type (p<0.001, Chi-square test with Holm-Bonferroni correction). (D) cyp-36A1(n5666) suppressed the egg-laying defect of egl-9(lf) mutants (p<0.001). (E) The cyp-36A1(+) transgene, which contains wild-type cyp-36A1, rescued the suppression of the egg-laying defect observed in cyp-36A1(n5666); egl-9(lf) mutants (p<0.001). (F) cyp-36A1(n5666) mutants displayed wild-type egg laying (p>0.05). (G) egl-9(lf) mutants were defective in locomotion rate, and this defect was suppressed by hif-1(lf) and cyp-36A1(lf) mutations. Mean ± SD of n = 3 biological replicates, *p<0.05, **p<0.01 considered significant (Student’s t-test with Holm-Bonferroni correction). (H) egl-9(lf) mutants were defective in defecation rate, and this defect was suppressed by hif-1(lf) and cyp-36A1(lf). Mean ± SD of n ≥ 6 animals, ***p<0.001 considered significant (Student’s t-test with Holm-Bonferroni correction). (I) Relative expression of cyp-36A1 mRNA in the wild type, egl-9(lf), egl-9(lf) hif-1(lf), and hif-1(lf) mutants, measured by qRT-PCR and normalized to the expression of the large ribosomal subunit rpl-32. Mean ± SD of n = 3 biological replicates, ***p<0.001 considered significant. ns, not significant (Student’s t-test with Holm-Bonferroni correction). Alleles used for (B–F) were cyp-36A1(n5666), egl-9(n586), and nIs674 (nIs [cyp-36A1(+)]), and all strains used in (B–F) contained the nIs470 (Pcysl-2::gfp) transgene. Alleles used for (G–I) were egl-9(sa307), hif-1(ia4), and cyp-36A1(gk824636).
Next we observed that cyp-36A1 expression is increased in egl-9(lf) mutants in a hif-1-dependent manner (Figure 1I), consistent with results from an earlier genome-wide microarray study that identified cyp-36A1 as one of 63 genes regulated by hif-1 in hypoxia-exposed worms (Shen et al., 2005). ChIP-seq of HIF-1 by the modERN project showed HIF-1 binding at two sites near the cyp-36A1 coding region, one 5’ to the start of the gene and one in the first intron (Kudron et al., 2018); both of these sites contain the HIF binding motif 5’RCGTG (Kaelin and Ratcliffe, 2008). We conclude that cyp-36A1 is a downstream effector of the hypoxia-response pathway that regulates multiple behaviors and that cyp-36A1 likely is a direct transcriptional target of HIF-1.
CYP-36A1 regulates gene expression changes and stress resistance downstream of HIF-1
We sought to determine if CYP-36A1 regulates other HIF-1-dependent processes. Based on sequence identity, CYP-36A1 is most closely related to the CYP2 family of cytochrome P450 enzymes, which function in both detoxification of xenobiotics and metabolism of endogenous molecules (Nebert et al., 2013). CYP2 family members and other CYPs can act on endogenous substrates to generate diffusible signaling molecules that regulate gene expression, such as eicosanoids and steroid hormones (Rendic and Guengerich, 2015; Dennis and Norris, 2015; Evans and Mangelsdorf, 2014). We hypothesized that CYP-36A1 might function in a transcriptional cascade to mediate aspects of HIF-1-dependent gene regulation. We performed an RNA-seq experiment comparing the wild type, egl-9(lf) mutants, egl-9(lf) hif-1(lf) double mutants, and cyp-36A1(lf); egl-9(lf) double mutants. We found that hif-1(lf) suppressed egl-9-dependent gene expression for 93% of egl-9(lf)-downregulated genes and 87% of egl-9(lf)-upregulated genes, indicating that most but not all regulation of transcription by egl-9 occurs through hif-1, consistent with previous work (Angeles-Albores et al., 2018). We further found that 36% of HIF-1-upregulated genes (i.e. genes that are upregulated in egl-9(lf) mutants and suppressed by hif-1(lf)) and 10% of HIF-1-downregulated genes were also regulated by cyp-36A1 (Figure 2A and B and Supplementary files 1 and 2). We focused on the HIF-1-upregulated genes, for which CYP-36A1 function appeared to be more broadly required. Gene ontology (GO) enrichment analysis of these HIF-1/CYP-36A1-upregulated genes suggested a role for cyp-36A1 in regulating stress resistance downstream of egl-9 and hif-1 (Figure 2—source data 1). The EGL-9/HIF-1 pathway has previously been implicated in responses to numerous stressors in both nematodes and mammals, with crosstalk occurring between HIF and regulators of the immune response, unfolded protein response, and other stress-response pathways (Palazon et al., 2014; Schito and Rey, 2018; Wouters and Koritzinsky, 2008; Nakazawa et al., 2016; Powell-Coffman, 2010). We tested whether CYP-36A1 is involved in the response to three stressors for which HIF-1 is known to mediate resistance in C. elegans: infection by the pathogenic bacteria Pseudomonas aeruginosa (Darby et al., 1999; Bellier et al., 2009; Shao et al., 2010; Budde and Roth, 2011; Kirienko et al., 2013), tunicamycin-induced ER stress (Leiser et al., 2015), and oxidative stress from tert-butyl hydroperoxide (Bellier et al., 2009). Animals in which HIF-1 is constitutively active because of mutation in egl-9 or the C. elegans VHL homolog vhl-1 are resistant to these stressors relative to wild-type animals: such mutants survive longer when grown on Pseudomonas aeruginosa strain PA14 (Bellier et al., 2009), display reduced tunicamycin-induced growth inhibition (Leiser et al., 2015), and survive exposure to tert-butyl hydroperoxide at a higher rate than the wild type (Bellier et al., 2009). We found that cyp-36A1(lf); egl-9(lf) double mutants are more sensitive than egl-9(lf) mutants to all three of these stressors (Figure 2C–2E and Figure 2—figure supplement 1), suggesting that CYP-36A1 mediates responses to these stressors downstream of HIF-1. Together the CYP-36A1-dependent changes in gene expression and stress resistance indicate that CYP-36A1 plays a major role in regulating HIF-1-mediated physiology.
Figure 2 with 1 supplement see all

CYP-36A1 acts downstream of HIF-1 to regulate gene expression changes and stress responses.
(A) Blue circle: Genes that were at least twofold downregulated in egl-9(lf) mutants. Purple and orange circles: Subset of egl-9(lf)-downregulated genes that were significantly upregulated in egl-9(lf) hif-1(lf) double mutants (purple) or cyp-36A1(lf); egl-9(lf) double mutants (orange) vs. egl-9(lf) single mutants. (B) Blue circle: Genes that were at least twofold upregulated in egl-9(lf) mutants. Purple and orange circles: Subset of egl-9(lf)-upregulated genes that were significantly downregulated in egl-9(lf) hif-1(lf) double mutants (purple) or cyp-36A1(lf); egl-9(lf) double mutants (orange) vs. egl-9(lf) single mutants. Significance for all comparisons in (A) and (B) was based on two biological replicates and determined by the Benjamini-Hochberg procedure with a false-discovery rate of 0.05. (C) Survival of animals grown from the L4 larval stage on the pathogen Pseudomonas aeruginosa. Wild type (n = 86 animals) vs. egl-9(lf) (n = 154) p<0.001; egl-9(lf) vs. cyp-36A1(lf); egl-9(lf) (n = 83), p<0.001; cyp-36A1(lf); egl-9(lf) vs. cyp-36A1(lf); egl-9(lf); nIs [cyp-36A1(+)] (n = 97), p<0.001; wild type vs. cyp-36A1(lf) (n = 61) p<0.05, as determined by the log-rank (Mantel-Cox) test, correcting for multiple comparisons with the Holm-Bonferroni method. See figure supplement for replicate data. (D) Survival of animals to the L4 larval stage or later after growth for three days from the L1 larval stage on plates containing 5 μg/ml tunicamycin. Mean ± SD of n = 3 biological replicates. **p<0.01, ***p<0.001 considered significant. ns (p>0.05), not significant (Student’s t-test with Holm-Bonferroni correction). (E) Survival of animals exposed to 7.5 mM tert-butyl hydroperoxide for 10 hr as young adults. Mean ± SD of n = 5 biological replicates. *p<0.05 considered significant. ns (p>0.05), not significant (Student’s t-test with Holm-Bonferroni correction). Alleles used for (A) and (B) were egl-9(sa307), hif-1(ia4), and cyp-36A1(gk824636). Alleles used for (C–E) were egl-9(n586), cyp-36A1(n5666), and nIs674 (nIs [cyp-36A1(+)]), and all strains used in (C–E) contained the nIs470 (Pcysl-2::gfp) transgene.
-
Figure 2—source data 1
RNA-seq GO enrichment analysis.
Gene ontology enrichment analysis terms for genes more than twofold upregulated in egl-9 mutants and significantly suppressed by hif-1(lf) and cyp-36A1(lf).
- https://doi.org/10.7554/eLife.36828.007
CYP-36A1 functions cell non-autonomously to regulate gene expression
We next sought to identify the site of action of CYP-36A1. We hypothesized that CYP-36A1 might function cell non-autonomously, as is the case for other cytochrome P450 enzymes that generate signaling molecules (Nebert et al., 2013; Evans and Mangelsdorf, 2014; Gerisch and Antebi, 2004). We observed cyp-36A1 expression in many tissues, including neurons, intestine, hypoderm, and muscle (Figure 3—figure supplement 2). To test the hypothesis of cell non-autonomous CYP-36A1 function, we focused on a cyp-36A1-mediated abnormality of egl-9(lf) mutants for which the site of dysfunction is well defined. Specifically, we examined expression of a GFP transcriptional reporter for the gene T24B8.5 (Shivers et al., 2009), which is expressed in only the intestine and based on our RNA-seq data is upregulated in egl-9(lf) mutants in a cyp-36A1-dependent manner. Interestingly, T24B8.5 expression is also upregulated in response to infection, ER stress, and oxidative stress (Shivers et al., 2009; Lim et al., 2014; Park et al., 2009). Expression of the reporter recapitulated the T24B8.5 expression changes observed by RNA-seq: increased expression of GFP was observed in egl-9(lf) mutants, which was suppressed by a second mutation in either hif-1 or cyp-36A1 (Figure 3A and C and Figure 3—figure supplement 3). To determine the site of action of cyp-36A1 for regulation of intestinal T24B8.5 expression, we expressed wild-type cyp-36A1 cDNA using tissue-specific promoters. We found that the low PT24B8.5::gfp expression of cyp-36A1(lf); egl-9(lf) double mutants was rescued by expressing cyp-36A1(+) either cell autonomously in the intestine or cell non-autonomously in neurons, hypoderm or body-wall muscle (Figure 3B and C and Figure 3—figure supplement 3). cyp-36A1(+) expression in all four tissues also rescued the suppression of the egg-laying defect of cyp-36A1(lf); egl-9(lf) mutants (Figure 3—figure supplement 1). Next we found that expressing a nondegradable constitutively active HIF-1 mutant protein (P621A) (Pocock and Hobert, 2008) in any of the same four tissues also promoted intestinal expression of the GFP reporter and that this HIF-1-mediated increase in expression required cyp-36A1 (Figure 3D and E and Figure 3—figure supplement 4). Thus, CYP-36A1 can function cell non-autonomously to regulate gene expression downstream of HIF-1, consistent with the hypothesis that CYP-36A1 acts by generating a diffusible signal.
Figure 3 with 4 supplements see all
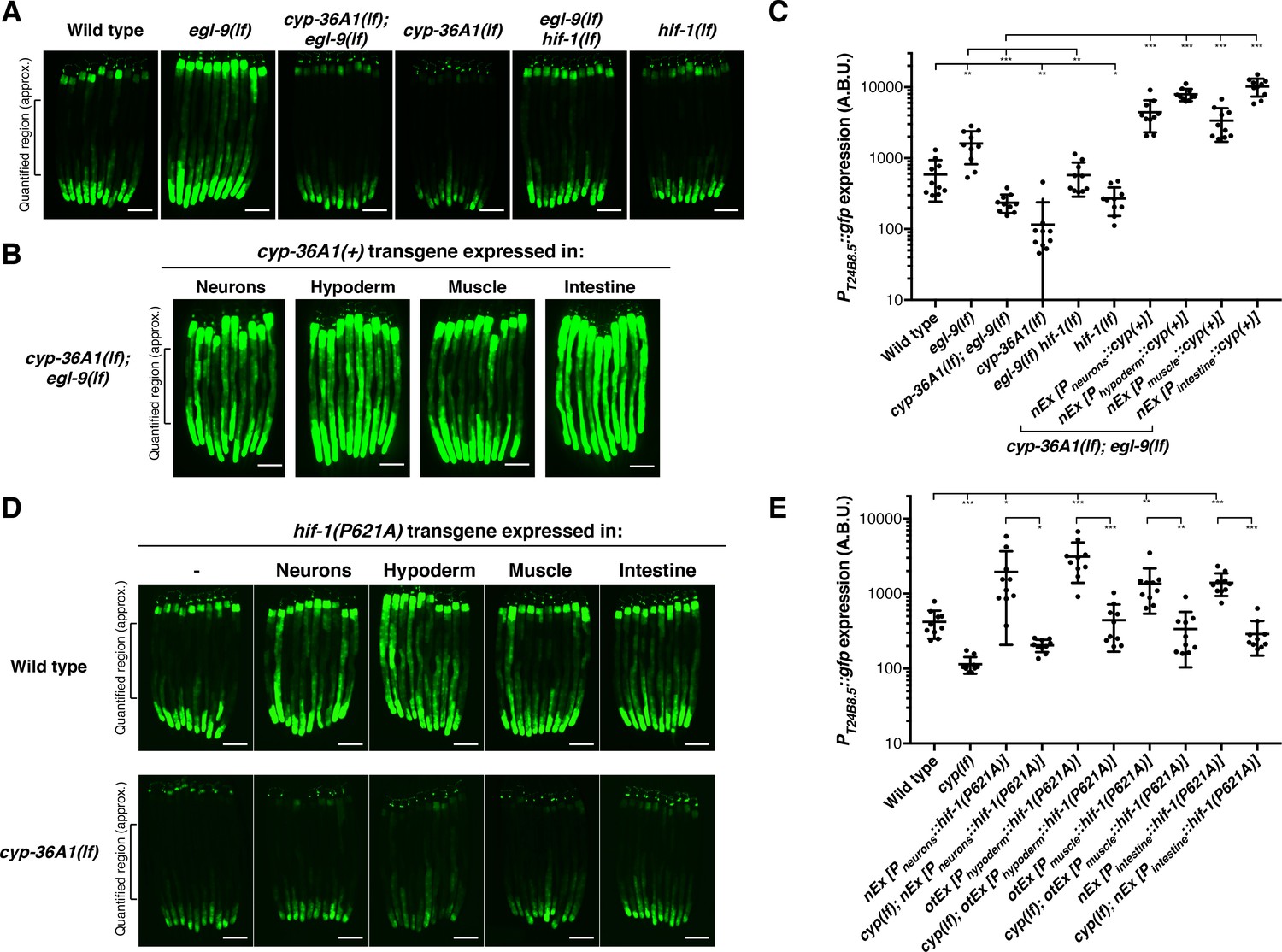
HIF-1 and CYP-36A1 cell non-autonomously regulate expression of a stress-responsive gene.
(A) PT24B8.5::gfp expression of the indicated genotypes (n = 10 animals per image). Scale bars, 100 μm. (B) Expression of cyp-36A1(+) specifically in neurons, hypoderm, muscle, or intestine of cyp-36A1(lf); egl-9(lf) animals increased expression of PT24B8.5::gfp in intestine (n = 10 animals per image). Scale bars, 100 μm. (C) Quantification of fluorescence intensity for (A) and (B), measured as average intensity for a 300 μm section of the intestine in the midbody of each animal, as indicated. *p<0.05, **p<0.01, ***p<0.001 considered significant (Student’s t-test with Holm-Bonferroni correction). Mean ± SD of n = 10 animals. See figure supplement for replicate data. (D) Expression of hif-1(P621A), which encodes a stable variant of HIF-1 (Pocock and Hobert, 2008), specifically in neurons, hypoderm, muscle, or intestine increased expression of PT24B8.5::gfp in intestine; increased expression was suppressed by cyp-36A1(lf) (n = 10 animals per image). Scale bars, 100 μm. (E) Quantification of fluorescence intensity for (D), measured as average intensity for a 300 μm section of the intestine in the midbody of each animal, as indicated. *p<0.05, ***p<0.01, ***p<0.001 considered significant (Student’s t-test with Holm-Bonferroni correction). Mean ± SD of n = 10 animals. See figure supplement for replicate data. Alleles used were egl-9(sa307), hif-1(ia4), cyp-36A1(gk824636), nEx2699[Pneurons::hif-1(P621A)], otEx3156 [Phypoderm::hif-1(P621A)], otEx3165 [Pmuscle::hif-1(P621A)], nEx2860 [Pintestine:: hif-1(P621A)], nEx2853 [Pneurons::cyp(+)], nEx2856 [Phypoderm::cyp(+)], nEx2859 [Pmuscle::cyp(+)], and nEx2849 [Pintestine::cyp(+)]. All strains contained the agIs219 (PT24B8.5::gfp) transgene.
A screen for suppressors of cyp-36A1(lf) identifies the nuclear receptor gene nhr-46
We performed a mutagenesis screen to identify CYP-36A1 effectors that regulate egg-laying behavior, stress responses, and gene expression. We screened for mutations that suppressed both the low PT24B8.5::gfp expression and normal egg laying of cyp-36A1(lf); egl-9(lf) double mutants, looking for triple mutants that, like egl-9(lf) single mutants, had high GFP expression and were egg-laying defective. By screening for suppressors of the two abnormalities simultaneously, we were able to focus on effectors of CYP-36A1 rather than finding genes that affect only egg laying or only expression of T24B8.5 independently of the EGL-9/HIF-1/CYP-36A1 pathway. From this screen we identified one nonsense and one missense allele of the nuclear receptor gene nhr-46 (Figure 4A–4D and Figure 4—figure supplement 1), both of which caused an egg-laying defect and high expression of the PT24B8.5::gfp reporter. We tested whether nhr-46 also functions in regulating stress responses downstream of cyp-36A1 and found that cyp-36A1(lf); nhr-46(lf); egl-9(lf) triple mutants were more resistant to Pseudomonas infection, ER stress, and oxidative stress than cyp-36A1(lf); egl-9(lf) double mutants (Figure 4E–4G and Figure 4—figure supplement 2). Thus, NHR-46 is a downstream effector of CYP-36A1 in regulation of stress resistance as well as of behavior and gene expression. Interestingly, nhr-46(lf) single mutants displayed wild-type egg laying (Figure 4D), tunicamycin resistance (Figure 4F), and oxidative stress resistance (Figure 4G), and nearly wild-type survival on Pseudomonas (Figure 4E and Figure 4—figure supplement 2), indicating that in addition to nhr-46 at least one other pathway is required to transduce egl-9-mediated modulation of egg laying and stress resistance. For example, in egl-9(lf) mutants or hypoxia, HIF-1 might drive expression of two (or more) targets that act together to inhibit egg laying and promote stress resistance. Thus, cyp-36A1 activation and consequent nhr-46 inhibition would promote egg-laying inhibition and stress resistance in egl-9(lf) mutant animals – in which this second pathway was also activated – but not in nhr-46(lf) single mutants – in which this second pathway was not activated. nhr-46(lf) single mutants had increased expression of the PT24B8.5::gfp reporter (Figure 4B and C and Figure 4—figure supplement 1), suggesting that at least some gene expression changes can be mediated by nhr-46 alone.
Figure 4 with 2 supplements see all
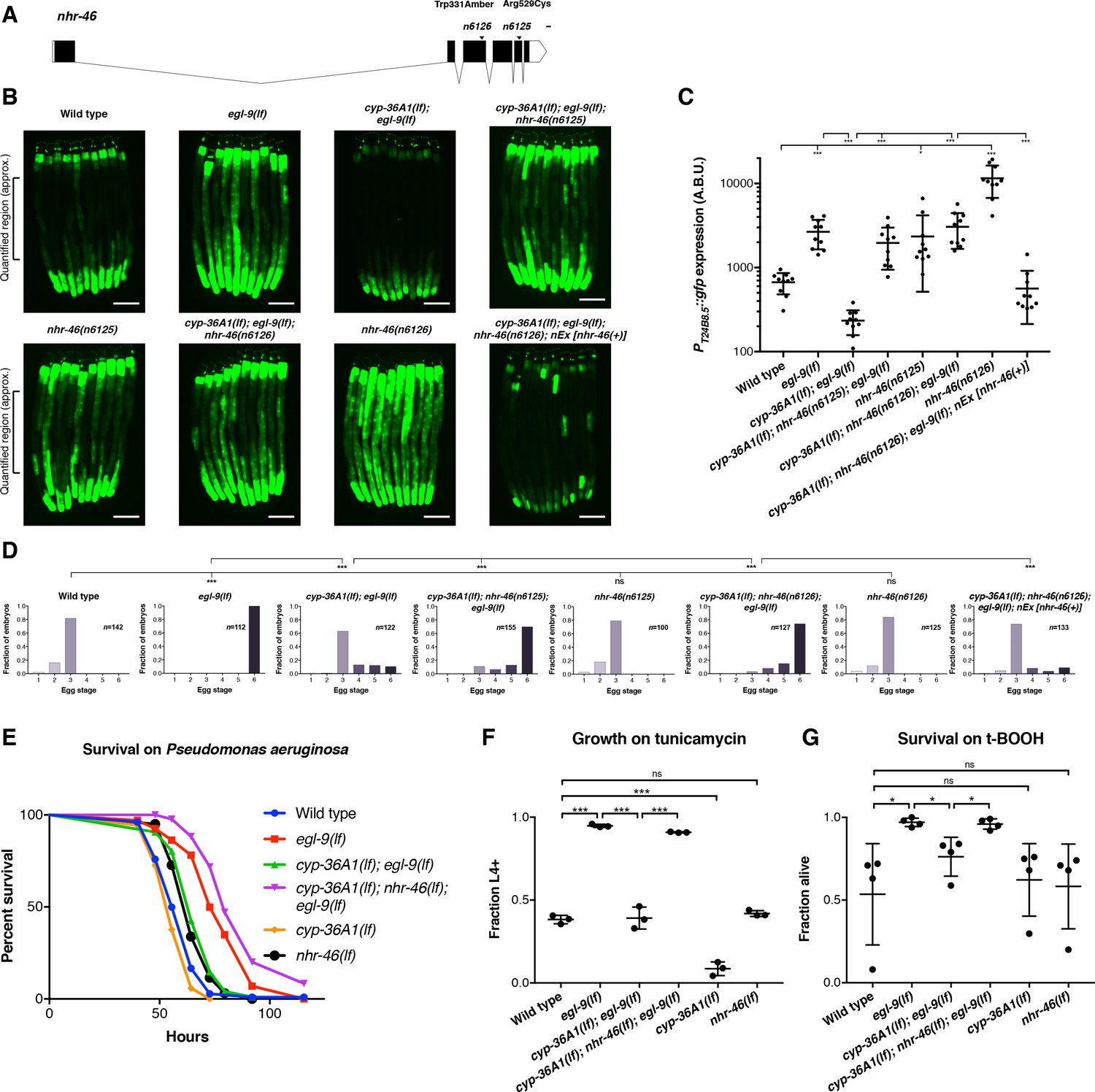
The nuclear hormone receptor NHR-46 acts downstream of CYP-36A1.
(A) nhr-46 gene diagram; isoform C45E5.6b is shown. n6125 and n6126 are in the NHR-46 ligand-binding domain. Scale bar, 100 bases. (B) PT24B8.5::gfp fluorescence of the indicated genotypes (n = 10 animals per image). Scale bars, 100 μm. (C) Quantification of fluorescence intensity for (B), measured as average intensity for a 300 μm section of the intestine in the midbody of each animal, as indicated. *p<0.05, ***p<0.001 considered significant (Student’s t-test with Holm-Bonferroni correction). Mean ±SD of n = 10 animals. See figure supplement for replicate data. (D) Distribution of stages of eggs newly laid by adult hermaphrodites of the indicated genotypes. ***p<0.001 considered significant. ns (p>0.05), not significant (Chi-square test with Holm-Bonferroni correction). (E) Survival of animals grown from the L4 larval stage on the pathogen Pseudomonas aeruginosa. Wild type (n = 129) vs. egl-9(lf) (n = 67) p<0.001; egl-9(lf) vs. cyp-36A1(lf); egl-9(lf) (n = 129), p<0.001; cyp-36A1(lf); egl-9(lf) vs. cyp-36A1(lf); nhr-46(lf); egl-9(lf) (n = 94), p<0.001; wild type vs. cyp-36A1(lf) (n = 106), p<0.05; wild type vs. nhr-46(lf) (n = 121), p<0.001, as determined by the log-rank (Mantel-Cox) test, correcting for multiple comparisons with the Holm-Bonferroni method. egl-9(lf) allele was egl-9(n586). See figure supplement for replicate data. (F) Survival of animals to the L4 larval stage or later after growth for three days from the L1 larval stage on plates containing 5 μg/ml tunicamycin. Mean ± SD of n = 3 biological replicates. ***p<0.001 considered significant. ns (p>0.05), not significant (Student’s t-test with Holm-Bonferroni correction). (G) Survival of animals exposed to 7.5 mM tert-butyl hydroperoxide for 10 hr as young adults. Mean ± SD of n = 4 biological replicates. *p<0.05 considered significant. ns, not significant (Student’s t-test with Holm-Bonferroni correction). Alleles used for (B–G) were egl-9(sa307), cyp-36A1(gk824636), nhr-46(n6126), and nEx2586 (nEx [nhr-46(+)]) except where otherwise noted. All strains in (B), (C), (D), (F), and (G) contained the agIs219 (PT24B8.5::gfp) transgene.
nhr-46 functions tissue-specifically to regulate gene expression and behavior
nhr-46 is expressed in many tissues, including neurons, hypoderm, muscle, intestine, and the spermatheca (Feng et al., 2012). Tissue-specific expression of nhr-46 in the intestine, but not in neurons or muscle, rescued the high GFP expression caused by nhr-46(lf), indicating that nhr-46 acts cell autonomously in the intestine to control intestinal expression of T24B8.5 (Figure 5A and B and Figure 5—figure supplement 1). nhr-46 expression in neurons fully rescued the egg-laying defect of cyp-36A1(lf); nhr-46(lf); egl-9(lf) triple mutants (Figure 5C), demonstrating that nhr-46 function in neurons is sufficient to regulate egg-laying behavior. nhr-46 expression in intestine, but not muscle, also partially rescued the egg-laying defect of cyp-36A1(lf); nhr-46(lf); egl-9(lf) triple mutants (Figure 5C). In combination with the tissue-specific CYP-36A1 and HIF-1 experiments described above, these results suggest that a CYP-36A1-regulated cell non-autonomous signal from any tissue can act on NHR-46 in the intestine to drive intestinal T24B8.5 expression and in either the nervous system or the intestine to regulate egg-laying behavior. That nhr-46 can act in either of two different tissues to regulate egg laying suggests that a second intercellular signal, for example a peptide or other hormone, might regulate egg laying downstream of NHR-46.
Figure 5 with 1 supplement see all
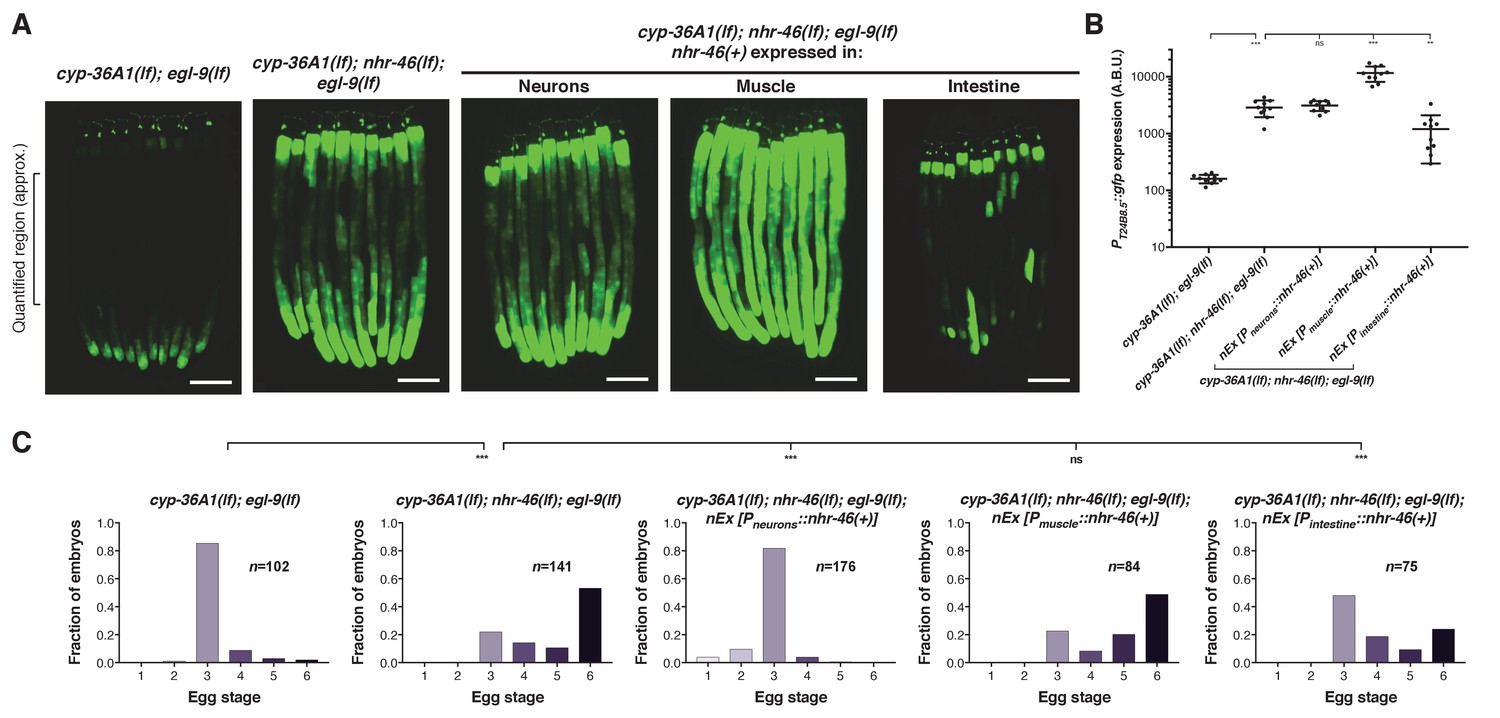
NHR-46 acts in different tissues to regulate T24B8.5 expression and egg laying.
(A) Expression of nhr-46(+) in the intestine but not in neurons or muscle rescued the high PT24B8.5::gfp expression in the intestine of cyp-36A1(lf); nhr-46(lf); egl-9(lf) mutants (n = 10 animals per image). Scale bars, 100 μm. (B) Quantification of fluorescence intensity for (A), measured as average intensity for a 300 μm section of the intestine in the midbody of each animal, as indicated. **p<0.01, ***p<0.001 considered significant. ns (p>0.05), not significant (Student’s t-test with Holm-Bonferroni correction). Mean ±SD of n = 10 animals. See figure supplement for replicate data. (C) Distribution of stages of eggs laid by adult hermaphrodites. Expression of nhr-46(+) in the intestine partially rescued the egg-laying defect of cyp-36A1(lf); nhr-46(lf); egl-9(lf) mutants. Neuronal nhr-46(+) expression fully rescued the egg-laying defect. Expression of nhr-46(+) in muscle did not rescue the egg-laying defect of cyp-36A1(lf); nhr-46(lf); egl-9(lf) mutants. ***p<0.001 considered significant. ns (p>0.05), not significant (Chi-square test with Holm-Bonferroni correction). Alleles used were egl-9(sa307), cyp-36A1(gk824636), nhr-46(n6126), nEx2713 [Pneurons::nhr-46(+)], nEx2715 [Pmuscle::nhr-46(+)], and nEx2864 [Pintestine::nhr-46(+)]. All strains contained the agIs219 (PT24B8.5::gfp) transgene.
Discussion
These studies define a novel molecular genetic pathway that mediates cell non-autonomous regulation of gene expression by the HIF-1 transcription factor. Our genetic analysis indicates that hif-1 activates the cytochrome P450 cyp-36A1, which in turn inhibits the nuclear receptor nhr-46 (Figure 6A). We speculate that the molecular function of CYP-36A1 is to generate an unidentified hormone that binds and regulates NHR-46, similar to other cytochrome P450 enzymes that function upstream of nuclear receptors (Evans and Mangelsdorf, 2014) and consistent with our observed cell non-autonomous function of CYP-36A1. We propose the following model (Figure 6B): In wild-type animals, EGL-9 inhibits HIF-1 activity, such that the HIF-1 target cyp-36A1 is not transcribed. The unliganded NHR-46 represses expression of genes that promote stress resistance and inhibit egg laying. In egl-9(lf) mutants, as in hypoxia-exposed worms, HIF-1 is stabilized and drives increased cyp-36A1 expression. A CYP-36A1-generated hormone then binds NHR-46 and antagonizes the repressive function of NHR-46, accounting for the observed negative regulatory relationship between cyp-36A1 and nhr-46. Ligand-bound NHR-46 is likely activated to promote the expression of target genes, by analogy to a well-established mechanism of nuclear receptor regulation in which ligand binding mediates a switch from repressive to activating nuclear receptor function (Evans and Mangelsdorf, 2014). Alternatively, CYP-36A1 might degrade a ligand that activates NHR-46.
Figure 6
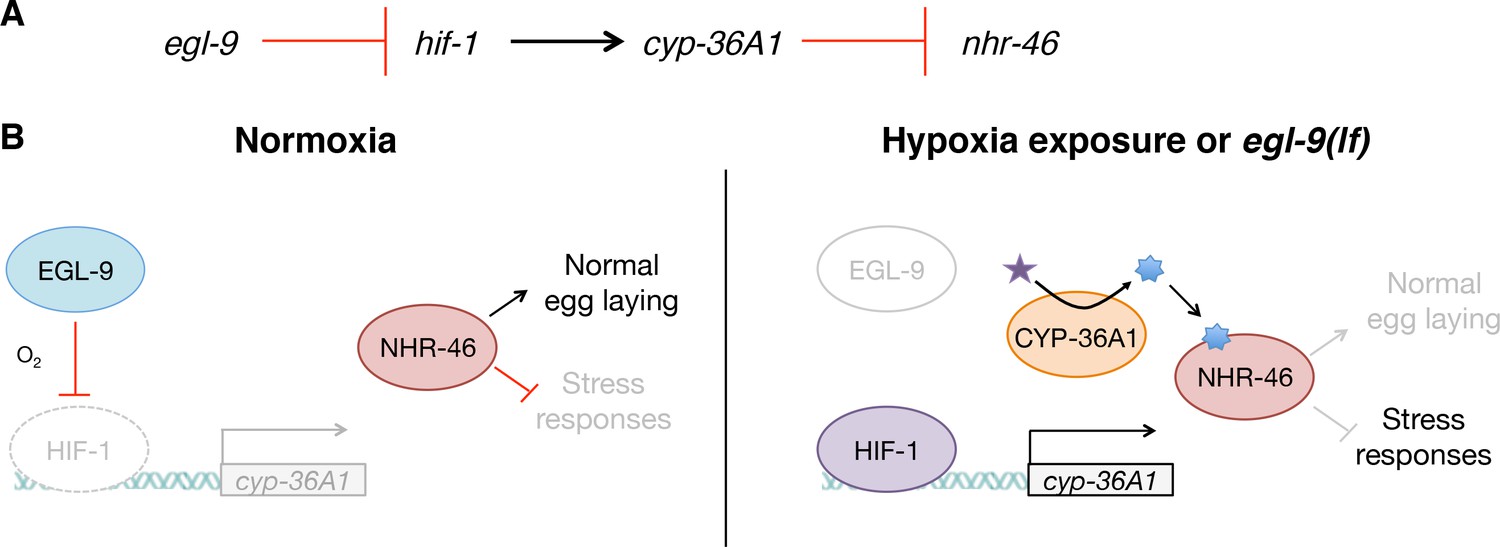
Model for the regulation of physiology and behavior by NHR-46 and CYP-36A1.
(A) The genetic pathway in which egl-9 inhibits hif-1, which activates cyp-36A1, which in turn inhibits nhr-46. (B) Model for how CYP-36A1 and NHR-46 function downstream of HIF-1. We suggest that CYP-36A1, which is transcriptionally upregulated by HIF-1, generates a hormone that binds NHR-46, thereby promoting transcriptional and physiological changes. See text for details.
Cell non-autonomous regulation of stress resistance by HIF involves multiple pathways
Numerous studies have reported that HIF-1 promotes longevity and stress resistance of C. elegans (Darby et al., 1999; Treinin et al., 2003; Bellier et al., 2009; Mehta et al., 2009; Zhang et al., 2009; Chen et al., 2009; Lee et al., 2010; Shao et al., 2010; Budde and Roth, 2011; Kirienko et al., 2013; Fawcett et al., 2015). Nonetheless, despite substantial interest in the role of this pathway in stress biology, few relevant HIF effectors have been identified. Interestingly, a recent study reported that HIF-dependent serotonin signaling from the nervous system cell non-autonomously drives expression of the xenobiotic detoxification enzyme flavin-containing monooxygenase-2 (FMO-2) in the intestine, resulting in increased stress resistance and consequent extension of lifespan (Leiser et al., 2015). Here we report that a different signal, likely a lipophilic hormone, acts cell non-autonomously downstream of HIF to regulate gene expression and stress resistance. Consistent with the findings of Leiser et al. (2015), our RNA-seq data showed that in egl-9(lf) mutants there is a strong induction of fmo-2 expression and that this induction is suppressed by hif-1 mutation (Supplementary file 2), that is HIF-1 upregulates fmo-2 expression. Notably, this HIF-dependent expression of fmo-2 does not require cyp-36A1: the high fmo-2 expression of egl-9(lf) mutants is not suppressed by cyp-36A1(lf). We therefore suggest that serotonin-mediated fmo-2 expression and cyp-36A1/nhr-46-mediated gene expression are parallel pathways downstream of HIF that regulate stress resistance.
Human cytochrome P450 enzymes might act as mediators of HIF-dependent gene expression
We speculate that some human CYPs might serve as mediators of HIF-dependent gene expression changes through a mechanism analogous to that we describe above for CYP-36A1. In support of this hypothesis, previous studies have identified several human cytochrome P450 enzymes that are putative direct HIF targets based on whole-genome ChIP-chip and ChIP-seq analysis (Mole et al., 2009; Schödel et al., 2011). We note that the humoral nature of a CYP-generated molecule would make it a candidate mediator of non-autonomous regulation of hypoxia response by the EGLN/HIF pathway, such as is observed in remote ischemic preconditioning (Cai et al., 2013; Olenchock et al., 2016).
Cytochrome P450 enzymes might be major players in the hypoxia-response pathway
We previously identified another cytochrome P450 gene, cyp-13A12, as acting downstream of egl-9 in a locomotory behavior (Ma et al., 2013). In contrast to CYP-36A1, which is upregulated by HIF-1, CYP-13A12 is downregulated by HIF-1 upon hypoxia exposure or in egl-9(lf) mutants. The downstream effectors of CYP-36A1 and CYP-13A12 are also distinct, as we show here that CYP-36A1 regulates a nuclear receptor that controls transcription, whereas CYP-13A12 generates eicosanoids that act on a seconds-to-minutes timescale unlikely to require gene expression changes. Thus, different cytochrome P450 enzymes can act broadly, through multiple mechanisms, downstream of the EGL-9/HIF-1 hypoxia-response pathway in C. elegans. We propose that cytochrome P450 enzymes might similarly be important HIF effectors in mammals. Polymorphisms in numerous human cytochrome P450 genes have been associated with cardiovascular disease (Elbekai and El-Kadi, 2006; Rowland and Mangoni, 2014), for which HIF plays a protective role (Semenza, 2012), and with cancers (Agundez, 2004), for which HIF contributes to pathogenesis (Semenza, 2012). Furthermore, a study of genetic adaptations in humans to the environmentally hypoxic Tibetan plateau identified well-established members of the HIF pathway and, intriguingly, also noted positive selection at two cytochrome P450 loci (Simonson et al., 2010). Together these observations suggest that the cytochrome P450 family of enzymes is important in a wide range of hypoxia-associated contexts in humans. We suggest the presence of a mechanistic link between the canonical HIF pathway and the function of cytochrome P450 enzymes in humans and posit that an understanding of how these highly druggable enzymes (Schuster and Bernhardt, 2007) control processes downstream of HIF might reveal new therapeutic avenues for treating a broad array of disorders.
Cytochrome P450 enzymes require oxygen as a substrate and thus might be regulated by oxygen availability. A long-standing issue in the understanding of HIF function is ‘range finding,’ that is how activity of the EGLN/HIF pathway is modulated such that it responds to different oxygen set points depending on context to drive a diversity of biological outputs (Ratcliffe, 2013). We hypothesize that the oxygen sensitivity of HIF-regulated cytochrome P450 enzymes might enable them to function in such a range finding mechanism to narrow the range of oxygen concentrations at which a subset of HIF-regulated physiological changes are activated.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Caenorhabditis elegans) | AU78 | Dennis Kim | agIs9 III | |
| Strain, strain background (C. elegans) | CB6088 | Jonathan Hodgkin | egl-9(sa307) V hif-1(ia4) V | |
| Strain, strain background (C. elegans) | JT307 | Creg Darby | egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT20483 | Dengke Ma/Bob Horvitz | nIs470 IV | |
| Strain, strain background (C. elegans) | MT22836 | this paper | cyp-36A1(gk824636) I ; egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT23218 | this paper | nIs682 X | |
| Strain, strain background (C. elegans) | MT24164 | this paper | cyp-36A1(gk824636) I ; agIs9 III | |
| Strain, strain background (C. elegans) | MT24165 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT24166 | this paper | agIs9 III ; egl-9(sa307) V hif-1(ia4) V | |
| Strain, strain background (C. elegans) | MT24167 | this paper | agIs9 III ; egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT24177 | this paper | agIs9 III ; hif-1(ia4) V | |
| Strain, strain background (C. elegans) | MT24179 | this paper | cyp-36A1(n5666) I ; (nIs470) IV | |
| Strain, strain background (C. elegans) | MT24520 | this paper | cyp-36A1(n5666) I ; (nIs470) IV ; egl-9(n586ts) V ; nIs674 | |
| Strain, strain background (C. elegans) | MT24622 | this paper | cyp-36A1(n5666) I ; (nIs470) IV ; egl-9(n586ts) V | |
| Strain, strain background (C. elegans) | MT24684 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6126) IV ; egl-9(sa307) V ; nEx2586 | |
| Strain, strain background (C. elegans) | MT24690 | this paper | agIs9 III ; otEx3165 | |
| Strain, strain background (C. elegans) | MT24692 | this paper | agIs9 III ; otEx3156 | |
| Strain, strain background (C. elegans) | MT24911 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6126) IV ; egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT25047 | this paper | agIs9 III ; nEx2699 | |
| Strain, strain background (C. elegans) | MT25048 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nEx2699 | |
| Strain, strain background (C. elegans) | MT25050 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; otEx3156 | |
| Strain, strain background (C. elegans) | MT25051 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; otEx3165 | |
| Strain, strain background (C. elegans) | MT25104 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6126) IV ; egl-9(sa307) V ; nEx2713 | |
| Strain, strain background (C. elegans) | MT25107 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6126) IV ; egl-9(sa307) V ; nEx2715 | |
| Strain, strain background (C. elegans) | MT25196 | this paper | agIs9 III ; nhr-46(n6125) IV | |
| Strain, strain background (C. elegans) | MT25197 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6125) IV ; egl-9(sa307) V | |
| Strain, strain background (C. elegans) | MT25211 | this paper | cyp-36A1(gk824636) I | |
| Strain, strain background (C. elegans) | MT25212 | this paper | cyp-36A1(gk824636) I ; egl-9(n586ts) V | |
| Strain, strain background (C. elegans) | MT25213 | this paper | cyp-36A1(gk824636) I ; nhr-46(n6126) IV ; egl-9(n586ts) V | |
| Strain, strain background (C. elegans) | MT25214 | this paper | nhr-46(n6126) IV | |
| Strain, strain background (C. elegans) | MT25215 | this paper | egl-9(n586ts) V | |
| Strain, strain background (C. elegans) | MT25596 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; egl-9(sa307) V ; nEx2853 | |
| Strain, strain background (C. elegans) | MT25599 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; egl-9(sa307) V ; nEx2856 | |
| Strain, strain background (C. elegans) | MT25601 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; egl-9(sa307) V ; nEx2849 | |
| Strain, strain background (C. elegans) | MT25605 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; egl-9(sa307) V ; nEx2859 | |
| Strain, strain background (C. elegans) | MT25606 | this paper | agIs9 III ; nEx2860 | |
| Strain, strain background (C. elegans) | MT25610 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nhr-46(n6126) IV ; egl-9(sa307) V ; nEx2864 | |
| Strain, strain background (C. elegans) | MT25611 | this paper | cyp-36A1(gk824636) I ; agIs9 III ; nEx2860 | |
| Strain, strain background (C. elegans) | MT25627 | this paper | agIs9 III ; nhr-46(n6126) IV | |
| Strain, strain background (C. elegans) | ZG31 | Huaqi Jiang/Jo Anne Powell-Coffman | hif-1(ia4) V |
C. elegans strains and transgenes
Request a detailed protocolAll C. elegans strains were cultured as described previously (Brenner, 1974). We used the N2 Bristol strain as the reference wild-type strain, and the polymorphic Hawaiian strain CB4856 (Davis et al., 2005) for genetic mapping and SNP analysis. We used the following mutations and transgenes:
LGI: cyp-36A1(n5666, gk824636)
LGIII: agIs219[PT24B8.5::gfp::unc-54 3’UTR, Pttx-3::gfp::unc-54 3’UTR]
LGIV: nhr-46(n6125, n6126), nIs470[Pcysl-2::gfp, Pmyo-2::mCherry], him-8(e1489)
LGV: egl-9(n586, sa307), hif-1(ia4)
LGX: nIs682[Pcyp-36A1::gfp::unc-54 3’UTR, Pmyo-3::mCherry::unc-54 3’UTR]
Unknown linkage: nIs674[Pcyp-36A1::cyp-36A1(+) gDNA::cyp-36A1 3’UTR, Pmyo-3::mCherry::unc-54 3’UTR]
Extrachromosomal arrays
Request a detailed protocolotEx3156 [Pdpy-7::hif-1(P621A), Pttx-3::rfp], otEx3165 [Punc-120::hif-1(P621A), Pttx-3::rfp], nEx2699 [Prab-3::hif-1(P621A)::F2A::mCherry::tbb-2 3'UTR, Pttx-3::mCherry], nEx2860 [Pvha-6::hif-1(P621A)::F2A::mCherry::tbb-2 3'UTR, rol-6(su1006dm)], nEx2849 [Pvha-6::cyp-36A1 cDNA::F2A::mCherry::tbb-2 3'UTR, rol-6(su1006dm)], nEx2856 [Pdpy-7::cyp-36A1 cDNA::F2A::mCherry::tbb-2 3'UTR, rol-6(su1006dm)], nEx2853 [Prab-3::cyp-36A1 cDNA::F2A::mCherry::tbb-2 3'UTR, rol-6(su1006dm)], nEx2859 [Punc-54::cyp-36A1 cDNA::F2A::mCherry::tbb-2 3'UTR, Pttx-3::mCherry::tbb-2 3’UTR], nEx2586 [Pnhr-46::nhr-46(+) gDNA::nhr-46 3’UTR, Pmyo-3::mCherry::unc-54 3’UTR], nEx2715[Punc-54::nhr-46 cDNA::F2A::mCherry::tbb-2 3'UTR], nEx2713 [Prab-3::nhr-46 cDNA::F2A::mCherry::tbb-2 3'UTR], nEx2864 [Pvha-6::nhr-46 cDNA::F2A::mCherry::tbb-2 3'UTR, rol-6(su1006dm)]
Note on allele usage: For egl-9, the weaker n586 allele was used for the screen and in the initial phenotypic characterization of screen mutants (Figure 1A–1F and Figure 2C–2E). The stronger sa307 allele was used for all other experiments, except for in the slow killing assay, for which the sa307 allele is less protective than weaker alleles, as previously reported (Bellier et al., 2009). For cyp-36A1, the allele identified from the screen, n5666, was used in the initial phenotypic characterization (Figure 1D–1F and 2C–2E); the putative null allele gk824636 was used for all other experiments. Alleles for other genes were used as indicated in the figure legends.
Molecular biology and transgenic strain construction
Request a detailed protocolThe Pcyp-36A1::gfp::unc-54 3’UTR construct (transgene nIs682) was generated by using PCR fusion (Hobert, 2002) to fuse a PCR product containing the cyp-36A1 promoter fragment (4.4 kb of upstream sequence) to a PCR product containing gfp::unc-54 3’UTR. The cyp-36A1 rescuing construct (transgene nIs674) was generated by amplifying a PCR product from gDNA containing 4.4 kb upstream, the cyp-36A1 locus, and 1.6 kb downstream. The nhr-46 rescuing construct (transgene nEx2586) was generated by amplifying a PCR product from gDNA containing 1.9 kb upstream, the nhr-46 locus, and 0.9 kb downstream. All remaining constructs were generated using the Infusion cloning technique (Clontech). The promoter fragments used for dpy-7 (hypoderm) (Gilleard et al., 1997), vha-6 (intestine) (Allman et al., 2009) rab-3 (neurons) (Mahoney et al., 2006) and unc-54 (muscle) (D. Ma, personal communication) contain 1.3, 0.9, 1.4, and 1.9 kb, respectively, of sequence upstream of the start codons of each of these genes. Expression of tissue-specific rescuing transgenes was confirmed using mCherry tagging. We noticed expression in some unidentified cells outside the body-wall muscle near the junction of the pharynx and intestine in the strain cyp-36A1(gk824636); agIs9; egl-9(sa307); nEx2859 [Punc-54::cyp-36A1 cDNA::F2A::mCherry::tbb-2 3'UTR, Pttx-3::mCherry::tbb-2 3’UTR]; such expression might contribute to rescue of the mutant phenotype in that strain. C45E5.6b was used for nhr-46 cDNA. F38A6.3a with a P621A stabilizing mutation was used for hif-1 cDNA (Pocock and Hobert, 2008). Where present, the F2A sequence served as a ribosomal skip sequence to cause separation of the two peptides encoded before and after the F2A (Ahier and Jarriault, 2014). Transgenic strains were generated by germline transformation as described (Mello et al., 1991). All transgenic constructs were injected at 2.5–50 ng/μl.
Mutagenesis screen for suppressors of egl-9
Request a detailed protocolTo screen for suppressors of the egl-9 egg-laying defect, we mutagenized egl-9(n586) mutants with ethyl methanesulfonate (EMS) as described previously (Brenner, 1974). The starting strain contained the Pcysl-2::gfp (nIs470) transgene, which is highly expressed in egl-9(lf) mutants and served as a reporter for HIF-1 activity (Ma et al., 2012). We used a dissecting microscope to screen the F2 progeny for suppression of the egg-laying defect (i.e. the Egl phenotype), picking (1) adults that appeared less Egl than egl-9(n586) mutants, and (2) eggs laid by the F2 animals that were at an earlier developmental stage than those laid by egl-9(n586) mutants. Screen isolates were backcrossed to determine dominant vs. recessive and single-gene inheritance pattern and crossed to him-8(e1489); egl-9(sa307) hif-1(ia4) to test complementation with hif-1(lf). The screen allele n5666, which conferred a recessive phenotype and was not allelic to hif-1, mapped between SNPs pkP1052 and rs3139013 on LGI with SNP mapping (Davis et al., 2005) using a strain containing egl-9(n586) introgressed into the Hawaiian strain CB4856 (Ma et al., 2013). Whole-genome sequencing identified a mutation in cyp-36A1 in the n5666 interval, and transgenic rescue demonstrated that this cyp-36A1 mutation is the causative mutation, as described in the text.
Mutagenesis screen for suppressors of cyp-36A1
Request a detailed protocolTo screen for downstream effectors of cyp-36A1, we mutagenized cyp-36A1(gk824636); egl-9(sa307) with ethyl methanesulfonate (EMS). The starting strain contained the PT24B8.5::gfp (agIs219) transgene, which has low expression in cyp-36A1(gk824636); egl-9(sa307) mutants and served as a reporter for CYP-36A1 activity. We used a dissecting microscope equipped to examine GFP fluorescence to screen for F2 progeny with high GFP fluorescence and an Egl appearance. The only two isolates failed to complement and were found to be alleles of nhr-46 by whole-genome sequencing and transgenic rescue. The mutant phenotypes of cyp-36A1(lf); n6126; egl-9(lf) were rescued by an nhr-46(+) transgene, demonstrating that the mutation in nhr-46 is the causative mutation and suggesting that n6126 is a loss-of-function allele.
Behavioral assays
Request a detailed protocolTo quantify egg-laying behavior, we scored the developmental stages of eggs laid by young adult hermaphrodites as described previously (Ringstad and Horvitz, 2008). Egg-laying defective mutants retain eggs longer in the uterus, thus laying them at later developmental stages. To examine egg-laying behavior after exposure to hypoxia, young adult animals were placed in a hypoxia chamber (Coy Laboratory) at 1% O2 balanced by N2 for 24 hr, after which the egg-laying assay was performed in normoxia. Control animals were exposed to room air (21% O2) for 24 hr; animals were randomly allocated to 1% or 21% O2 treatment. Locomotion assays were performed on bacterial food and quantified using a custom worm tracker, as described previously (Paquin et al., 2016). Defecation assays were performed as described previously (Thomas, 1990), counting the number of defecation cycles in ten minutes.
Pseudomonas aeruginosa killing assay
Request a detailed protocolSensitivity to the Pseudomonas aeruginosa strain PA14 was assayed using the big lawn killing assay as described previously (Reddy et al., 2009). The big lawn killing assay was used to remove any influence of avoidance behavior on survival, as wild-type PA14 avoidance is dependent on normal aerotaxis behavior (Reddy et al., 2009), and egl-9(lf) mutants have previously been shown to display abnormal aerotaxis (Chang and Bargmann, 2008).
Tunicamycin survival assay
Request a detailed protocolSensitivity to tunicamycin was assayed by placing at least 100 starvation-synchronized L1 animals on NGM plates containing 5 μg/ml tunicamycin (Sigma), made using 10 mg/ml tunicamycin stock in DMSO and seeded with E. coli OP50 bacteria. Survival to the L4 larval stage or later was determined after three days.
t-BOOH survival assay
Request a detailed protocolSensitivity to tert-butyl hydroperoxide (t-BOOH) was assayed by placing ~60 young adult worms on NGM plates containing 7.5 mM t-BOOH, made using 70% t-BOOH solution (Sigma) and seeded with E. coli OP50 bacteria. Survival was evaluated after 10 hr.
Microscopy
Request a detailed protocolEpifluorescence images of PT4B8.5::gfp expression were obtained using an AxioImager Z2 upright microscope (Zeiss) and ZEN software (Zeiss). Confocal images of Pcyp-36A1::gfp expression were obtained using an LSM 800 instrument (Zeiss) and ZEN software. Fluorescence intensity for the PT24B8.5::gfp reporter was quantified by measuring average intensity in a 300 μm section of the intestine centered on the vulva using FIJI software. PT24B8.5::gfp reporter imaging conditions were optimized for observation of fluorescence in the midbody; fluorescence was not saturated in this region in quantified images.
RNA isolation for qRT-PCR and RNA-seq
Request a detailed protocolAll strains were maintained at 22.5°C for at least two generations without starvation prior to experiment. ~150 very young adults (0–1 eggs in uterus) were picked into M9 buffer and allowed to settle. M9 was aspirated, and worms were then rinsed twice with M9 and twice with RNase-free water. Excess liquid was aspirated and the pellet was frozen in liquid nitrogen. RLT buffer (QIAGEN) was added to the frozen pellet, and worms were lysed using a BeadBug microtube homogenizer (Sigma) and 0.5 mm zirconium beads (Sigma). RNA was extracted using the RNeasy Mini kit (QIAGEN) according to the manufacturer’s instructions.
cyp-36A1 mRNA expression analysis by qRT-PCR
Request a detailed protocolReverse transcription was performed using SuperScript III (Invitrogen). Quantitative PCR was performed using Applied Biosystems Real-Time PCR Instruments. Expression levels were normalized to the expression of the ribosomal subunit gene rpl-32.
Primers for qRT-PCR
Request a detailed protocolcyp-36A1 F: ACCAGCTTGTCCAACACCAA
cyp-36A1 R: CACGCTTTGGCTCCCATTTC
rpl-32 F: GGCTACACGACGGTATCTGT
rpl-32 R: CAAGGTCGTCAAGAAGAAGC
RNA-seq library preparation
Request a detailed protocolRNA integrity and concentration were checked on a Fragment Analyzer (Advanced Analytical). The mRNA was purified by polyA-tail enrichment, fragmented, and reverse transcribed into cDNA (Illumina TruSeq). cDNA samples were then end-repaired and adaptor-ligated using the SPRI-works Fragment Library System I (Beckman Coulter Genomics) and indexed during amplification. Libraries were quantified using the Fragment Analyzer (Advanced Analytical) and qPCR before being loaded for single-end sequencing using the Illumina HiSeq 2000.
RNA-seq data analysis
Request a detailed protocolReads were aligned against the C. elegans ce10 genome assembly using bwa 0.7.5a (Li and Durbin, 2009) and samtools/0.1.19 (1000 Genome Project Data Processing Subgroup et al., 2009) (bwa aln/bwa samse), and mapping rates, fraction of multiply-mapping reads, number of unique 20-mers at the 5’ end of the reads, insert size distributions and fraction of ribosomal RNAs were calculated using dedicated perl scripts and bedtools v. 2.17.0 (Quinlan and Hall, 2010). For expression analysis, reads were aligned against the C. elegans ce10 genome/ENSEMBL 65 annotation using RSEM 1.2.15 (Li and Dewey, 2011) and bowtie 1.0.1 (Langmead et al., 2009), with the following parameters: -p 6 --bowtie-chunkmbs 1024 --output-genome-bam. Raw expected read counts were retrieved and used for differential expression analysis with Bioconductor’s edgeR package in the R 3.2.3 statistical environment (Robinson et al., 2010). First, common, trended, and gene-specific read dispersion across sequencing libraries and genes was estimated using the estimateDisp function. Given the small number of replicates, a gene-wise negative binomial generalized linear model (GLM) with quasi-likelihood tests (as implemented in the glmQLFit function) was used to test for differential expression between conditions (Lun et al., 2016). Briefly, this statistical framework works by first fitting the observed and expected distributions of read counts for each gene across conditions using a GLM, which is based on the negative binomial distribution and the observed read dispersion. The significance of biases in read counts is then tested using the quasi-likelihood F-test (implemented in glmQLFTest). This test provides more robust and reliable error rate control at low number of replicates, because it reflects the uncertainty in read distribution better than the likelihood ratio test. Models were fitted across all conditions and relevant differential expression testing was performed using glmQLFTest between pairs of conditions of interest. P values were adjusted for multiple comparisons using the Benjamini-Hochberg procedure (Benjamini and Hochberg, 1995). Gene ontology enrichment analysis was performed using GOrilla (Eden et al., 2009), examining genes that were significantly downregulated in cyp-36A1(lf); egl-9(lf) double mutants vs. egl-9(lf) single mutants (i.e. orange circle in Figure 2B) as compared to genes that were at least twofold upregulated in egl-9(lf) mutants vs. wild type (adjusted p value<0.05) and significantly downregulated in egl-9(lf) hif-1(lf) vs. egl-9(lf) (adjusted p value<0.05) (i.e. purple circle in Figure 2B).
Statistical analysis
Request a detailed protocolChi-square tests were used to compare the distribution of stages of eggs laid by wild-type and mutant animals. Unpaired t-tests were used to compare cyp-36A1 mRNA expression between strains, survival on tunicamycin between strains, survival on t-BOOH between strains, and PT24B8.5::gfp reporter fluorescence intensity between strains. Log-rank (Mantel-Cox) tests were used to compare survival of different strains on Pseudomonas aeruginosa. In cases of multiple comparisons, a Holm-Bonferroni correction was applied. Statistical tests were performed using GraphPad Prism software (Graphpad Prism, RRID:SCR_002798) version 7.0a. Biological replicates were performed using separate populations of animals.
Accession numbers
Request a detailed protocolThe GEO accession number for the RNA-seq dataset in this paper is GSE108283.
Data availability
Sequencing data have been deposited in GEO under accession code GSE108283.
-
Hypoxia-inducible factor cell non-autonomously regulates C. elegans stress responses and behavior via a nuclear receptorPublicly available at the NCBI Gene Expression Omnibus (accession no: GSE108283).
References
-
Cytochrome P450 gene polymorphism and cancerCurrent Drug Metabolism 5:211–224.https://doi.org/10.2174/1389200043335621
-
Loss of the apical V-ATPase a-subunit VHA-6 prevents acidification of the intestinal lumen during a rhythmic behavior in C. elegansAmerican Journal of Physiology-Cell Physiology 297:C1071–C1081.https://doi.org/10.1152/ajpcell.00284.2009
-
Reconstructing a metazoan genetic pathway with transcriptome-wide epistasis measurementsProceedings of the National Academy of Sciences 115:E2930–E2939.https://doi.org/10.1073/pnas.1712387115
-
Controlling the false discovery rate: a practical and powerful approach to multiple testingJournal of the Royal Statistical Society. Series B 57:289–300.
-
Eicosanoid storm in infection and inflammationNature Reviews Immunology 15:511–523.https://doi.org/10.1038/nri3859
-
Cytochrome P450 enzymes: central players in cardiovascular health and diseasePharmacology & Therapeutics 112:564–587.https://doi.org/10.1016/j.pharmthera.2005.05.011
-
BookExpression pattern analysis of regulatory transcription factors in Caenorhabditis elegansIn: Deplancke B, Gheldof N, editors. Gene Regulatory Networks. Humana Press. pp. 21–50.
-
cis regulatory requirements for hypodermal cell-specific expression of the Caenorhabditis elegans cuticle collagen gene dpy-7Molecular and Cellular Biology 17:2301–2311.https://doi.org/10.1128/MCB.17.4.2301
-
PCR fusion-based approach to create reporter gene constructs for expression analysis in transgenic C. elegansBioTechniques 32:728–730.
-
The sequence alignment/Map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
Sumoylation regulates ER stress response by modulating calreticulin gene expression in XBP-1-dependent mode in Caenorhabditis elegansInternational Journal of Biochemistry & Cell Biology 53:399–408.https://doi.org/10.1016/j.biocel.2014.06.005
-
BookIt’s DE-Licious: A Recipe for Differential Expression Analyses of RNA-Seq Experiments Using Quasi-Likelihood Methods in edgeRStatistical GenomicsNew York, NY: Humana Press.
-
Regulation of synaptic transmission by RAB-3 and RAB-27 in Caenorhabditis elegansMolecular Biology of the Cell 17:2617–2625.https://doi.org/10.1091/mbc.e05-12-1170
-
Efficient gene transfer in C. elegans: extrachromosomal maintenance and integration of transforming sequencesThe EMBO Journal 10:3959.
-
C. elegans are protected from lethal hypoxia by an embryonic diapauseCurrent Biology 19:1233–1237.https://doi.org/10.1016/j.cub.2009.05.066
-
Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcriptsJournal of Biological Chemistry 284:16767–16775.https://doi.org/10.1074/jbc.M901790200
-
Oxygen availability and metabolic adaptationsNature Reviews Cancer 16:663–673.https://doi.org/10.1038/nrc.2016.84
-
Human cytochromes P450 in health and diseasePhilosophical Transactions of the Royal Society B: Biological Sciences 368:20120431.https://doi.org/10.1098/rstb.2012.0431
-
Oxygen levels affect axon guidance and neuronal migration in Caenorhabditis elegansNature Neuroscience 11:894–900.https://doi.org/10.1038/nn.2152
-
Hypoxia activates a latent circuit for processing gustatory information in C. elegansNature Neuroscience 13:610–614.https://doi.org/10.1038/nn.2537
-
Hypoxia signaling and resistance in C. elegansTrends in Endocrinology & Metabolism 21:435–440.https://doi.org/10.1016/j.tem.2010.02.006
-
Survey of human oxidoreductases and cytochrome P450 enzymes involved in the metabolism of xenobiotic and natural chemicalsChemical Research in Toxicology 28:38–42.https://doi.org/10.1021/tx500444e
-
FMRFamide neuropeptides and acetylcholine synergistically inhibit egg-laying by C. elegansNature Neuroscience 11:1168–1176.https://doi.org/10.1038/nn.2186
-
Cytochrome P450 and ischemic heart disease: current concepts and future directionsExpert Opinion on Drug Metabolism & Toxicology 10:191–213.https://doi.org/10.1517/17425255.2014.859675
-
Cell-Autonomous metabolic reprogramming in hypoxiaTrends in Cell Biology 28:128–142.https://doi.org/10.1016/j.tcb.2017.10.006
-
Inhibition of cytochromes P450: existing and new promising therapeutic targetsDrug Metabolism Reviews 39:481–499.https://doi.org/10.1080/03602530701498455
-
Oxygen sensing, homeostasis, and diseaseNew England Journal of Medicine 365:537–547.https://doi.org/10.1056/NEJMra1011165
-
Roles of the HIF-1 hypoxia-inducible factor during hypoxia response in Caenorhabditis elegansJournal of Biological Chemistry 280:20580–20588.https://doi.org/10.1074/jbc.M501894200
-
A cytoprotective perspective on longevity regulationTrends in Cell Biology 23:409–420.https://doi.org/10.1016/j.tcb.2013.04.007
-
Systemic stress signalling: understanding the cell non-autonomous control of proteostasisNature Reviews Molecular Cell Biology 15:211–217.https://doi.org/10.1038/nrm3752
-
Genetic analysis of defecation in Caenorhabditis elegansGenetics 124:855–872.
-
Egg-laying defective mutants of the nematode Caenorhabditis elegansGenetics 104:619–647.
-
Hypoxia signalling through mTOR and the unfolded protein response in cancerNature Reviews Cancer 8:851–864.https://doi.org/10.1038/nrc2501
Article and author information
Author details
Funding
National Institutes of Health (GM024663)
- Corinne L Pender
- H Robert Horvitz
Howard Hughes Medical Institute
- Corinne L Pender
- H Robert Horvitz
National Institutes of Health (T32GM007287)
- Corinne L Pender
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank K Burkhart, A Doi, S Sando, V Dwivedi, J Saul, E Lee, JN Kong, and D Ghosh for helpful discussion. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440).
Copyright
© 2018, Pender et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,145
- views
-
- 437
- downloads
-
- 38
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 38
- citations for umbrella DOI https://doi.org/10.7554/eLife.36828
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Hypoxia-inducible factor cell non-autonomously regulates C. elegans stress responses and behavior via a nuclear receptor
eLife 7:e36828.
https://doi.org/10.7554/eLife.36828




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}