Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins
- The Scripps Research Institute, United States
- San Diego State University, United States
Abstract
Pharmacologic arm-selective unfolded protein response (UPR) signaling pathway activation is emerging as a promising strategy to ameliorate imbalances in endoplasmic reticulum (ER) proteostasis implicated in diverse diseases. The small molecule N-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (147) was previously identified (Plate et al., 2016) to preferentially activate the ATF6 arm of the UPR, promoting protective remodeling of the ER proteostasis network. Here we show that 147-dependent ATF6 activation requires metabolic oxidation to form an electrophile that preferentially reacts with ER proteins. Proteins covalently modified by 147 include protein disulfide isomerases (PDIs), known to regulate ATF6 activation. Genetic depletion of PDIs perturbs 147-dependent induction of the ATF6-target gene, BiP, implicating covalent modifications of PDIs in the preferential activation of ATF6 afforded by treatment with 147. Thus, 147 is a pro-drug that preferentially activates ATF6 signaling through a mechanism involving localized metabolic activation and selective covalent modification of ER resident proteins that regulate ATF6 activity.
https://doi.org/10.7554/eLife.37168.001Introduction
When protein misfolding or aggregation within the endoplasmic reticulum (ER) becomes excessive, cells respond to this ER stress or imbalance in protein homeostasis (or ‘proteostasis’) by activating the Unfolded Protein Response (UPR), a three-armed stress-responsive signaling pathway (Hetz et al., 2015; Plate and Wiseman, 2017; Walter and Ron, 2011). Signaling within these arms is initiated by the ER stress-sensing transmembrane proteins, ATF6, IRE1 and PERK, for which the arms are named. A key role of the UPR is to restore ER proteostasis in response to pathologic insults that induce ER stress, through a combination of transcriptional and translational mechanisms. IRE1 and ATF6 activate the stress-responsive transcription factors, spliced XBP1 (XBP1s, a spliced variant of XBP1) and ATF6 (a cleaved product of full-length ATF6), respectively, that induce the expression of overlapping, and distinct subsets of ER chaperones, folding enzymes, and degradation factors involved in maintaining ER proteostasis (Shoulders et al., 2013; Lee et al., 2003; Yamamoto et al., 2004). PERK activation induces a transient attenuation of new protein synthesis and activation of transcription factors, such as ATF4 that induce stress-responsive genes involved in both ER proteostasis maintenance and other aspects of cellular physiology, including redox regulation and amino acid synthesis (Harding et al., 1999; Harding et al., 2003). Through these adaptive signaling mechanisms, the UPR functions to promote ER proteostasis and alleviate ER stress.
The capacity of the UPR signaling arms to distinctly influence ER proteostasis and function suggests that selective activation of these pathways has significant potential to alleviate pathologic imbalances in ER proteostasis associated with etiologically diverse human diseases (Hetz et al., 2015; Plate and Wiseman, 2017). In particular, activation of the ATF6 signaling arm has been shown to be useful for ameliorating disease-associated imbalances in ER proteostasis and function. We previously showed that stress-independent activation of the ATF6 transcription factor using a chemical genetic approach induces protective remodeling of ER proteostasis pathways to selectively reduce secretion and extracellular aggregation of destabilized, amyloid disease-associated proteins, such as transthyretin and immunoglobulin light chain, without significantly impacting the secretion of the endogenous proteome (Shoulders et al., 2013; Chen et al., 2014; Cooley et al., 2014; Plate et al., 2016). Overexpression of the active ATF6 transcription factor in the heart also has been shown to improve cardiac performance in mouse models of ischemic heart disease, through a mechanism involving ATF6-dependent regulation of the antioxidant gene, catalase (Jin et al., 2017). Similarly, overexpression of the active ATF6 transcription factor in the liver improves insulin sensitivity in obese mice (Ozcan et al., 2016). These results indicate that ATF6 activation offers a unique therapeutic opportunity to ameliorate ER proteostasis defects implicated in diverse diseases.
The potential for ATF6 activation as a therapeutic approach is further demonstrated by human genetic studies. These show that patients harboring ATF6α mutations that render this pathway constitutively active present with the retinal development disorder, achromatopsia, but notably do not show defects in other organs or systemic tissues resulting from constitutive ATF6 activity (Chiang et al., 2017; University of Washington Center for Mendelian Genomics et al., 2015; Kohl et al., 2015). This indicates that pharmacologic activation of ATF6 in adults is also likely to be well tolerated and not lead to systemic defects. The therapeutic potential for ATF6 activation demonstrated by these genetic results combined with the potential to correct imbalances in ER proteostasis associated with diverse diseases through ATF6 activation has led to a significant effort to identify small molecule pharmacologic activators of ATF6 that similarly correct pathologic imbalances in ER proteostasis implicated in disease.
We recently reported the discovery of several small molecules that preferentially activate the ATF6 arm of the UPR, which were identified using a cell-based-reporter high-throughput screening (HTS) strategy (Plate et al., 2016). One of the more promising of these compounds, N-(2-hydroxy-5-methylphenyl)-3-phenylpropanamide (147), was further characterized by whole cell RNAseq and proteomic profiling to confirm preferential activation of the ATF6 transcriptional program. Importantly, 147 phenocopied protective benefits associated with genetic ATF6 activation. For example, treatment with 147 reduced the secretion and extracellular aggregation of destabilized, amyloidogenic variants of transthyretin and immunoglobulin light chain in amyloid disease relevant cell models (Plate et al., 2016). And notably, 147 did not influence secretion of the endogenous secretory proteome. Furthermore, 147-dependent ATF6 activation was recently shown to promote stem cell differentiation of embryonic stem cells to mesodermal lineages, revealing a previously unknown role for ATF6 in dictating stem cell specification (Kroeger et al., 2018). Thus, 147 has emerged as a promising compound for further development as a pharmacologic ATF6 activator that can be employed to enhance ER proteostasis in the context of human disease.
Despite this promise, the mechanism of how 147 preferentially activates the ATF6 arm of the UPR is unclear. In response to ER stress, ATF6 is activated through a process involving reduction of inter- and intra-molecular disulfides that results in formation of a reduced ATF6 monomer that is trafficking competent (Nadanaka et al., 2007; Nadanaka et al., 2006). This reduced monomer then traffics to the Golgi where it is processed by the Site 1 and Site 2 proteases (S1P and S2P, respectively), releasing the N-terminal, active ATF6 transcription factor responsible for upregulation of the ATF6 transcriptional program (Ye et al., 2000). Previously, we showed that treatment with 147 activates endogenous ATF6 through a similar mechanism involving increased processing by S1P and S2P to release the active ATF6 transcription factor (Plate et al., 2016). However, the protein target(s) and molecular mechanism(s) responsible for the 147-dependent increases in ATF6 activation are currently unknown. The ATF6 protein has no known small molecule binding sites that could serve to allosterically activate this protein, so it seems likely that 147 initiates signaling by interacting with proteins critical for regulating ATF6 activity. However, this hypothesis remains untested.
Here, we report our efforts focused on defining the molecular mechanism of 147-dependent ATF6 activation. We find that the acylated 2-amino-p-cresol substructure of 147, which is shared by several other pharmacologic ATF6 activators identified in our HTS screen (Plate et al., 2016), is required for pharmacologic ATF6 activation by this compound. Given that chemicals comprising p-alkyl phenol substructures are oxidized in cells by cytochrome P450s to yield highly reactive p-quinone methides (Bolton, 2014), the dependence of 147-dependent ATF6 activation on the 2-amino-p-cresol moiety suggested that metabolic oxidation could be involved in the activity of 147. We find that 147 is metabolically activated in a process sensitive to the P450 inhibitor, resveratrol. This activation results in the formation of an electrophile that reacts with multiple proteins to form covalent conjugates. A key observation is that the proteins modified by 147 are primarily localized to the ER, which is not a general property of reactive electrophiles (Trott et al., 2008), and is consistent with the metabolic activation of this compound by an ER-localized cytochrome P450(s). Interestingly, ER protein disulfide isomerases (PDIs), a class of proteins involved in regulating and catalyzing disulfide formation and breakage within the ER, are enriched among the proteins modified by 147. PDIs have been previously implicated in regulating the formation and dissolution of ATF6 disulfides critical for initiating ATF6 trafficking and subsequent ATF6 transcription factor activation during ER stress (Higa et al., 2014). This suggests that 147-dependent ATF6 activation is mediated by targeting PDI activity. Consistent with this notion, shRNA-mediated knockdown of select PDIs impairs 147-dependent induction of the ATF6-target gene, BiP. Our results indicate that 147-dependent ATF6 activation proceeds through a mechanism requiring metabolic activation close to or within the ER to produce a reactive electrophile that selectively modifies a subset of ER proteins, including multiple PDIs, that enhances ATF6 activity by increasing the ER population of the reduced, trafficking-competent ATF6 monomer.
Results
The 2-amino-p-cresol substructure of 147 is necessary for compound-dependent ATF6 activation
Compound 147 consists of a 2-amino-p-cresol substructure (i.e. the A-ring) connected to a second aromatic ring (the B-ring) via a carbon-based chain (the linker; see Figure 1A). We prepared analogs of 147 where the A-ring was varied by converting 3-phenylpropanoic acid to 3-phenylpropionyl chloride using oxalyl chloride and then coupling it to a variety of aromatic amines (Figure 1—figure supplement 1A). The activities of these compounds were determined by measuring their ability to activate a previously described ATF6 transcriptional reporter that consists of a fragment of the ATF6-regulated BiP promoter driving expression of firefly luciferase (referred to as ERSE.FLuc) in HEK293T cells (Plate et al., 2016; Yoshida et al., 1998). Interestingly, removal of the phenolic hydroxyl group in the A-ring (147-1) or replacement of it with a fluoro group (147-2) yields compounds that do not activate this ATF6 reporter (Figure 1B). Compounds where the methyl group of 147 is removed (147-3) or where the methyl is replaced with a trifluoromethyl group (147-4) also do not activate this reporter. Compound 147–4 also does not induce ATF6 target genes such as BiP in human embryonic stem cells (Kroeger et al., 2018). Furthermore, moving the hydroxyl group to the ortho or meta positions of the cresol moiety (147–5 and 147–6) also results in compounds that do not activate the ATF6 reporter. These results indicate that the 2-amino-p-cresol substructure of 147, in its entirety, is required for compound-dependent ATF6 activation.
Figure 1 with 1 supplement see all
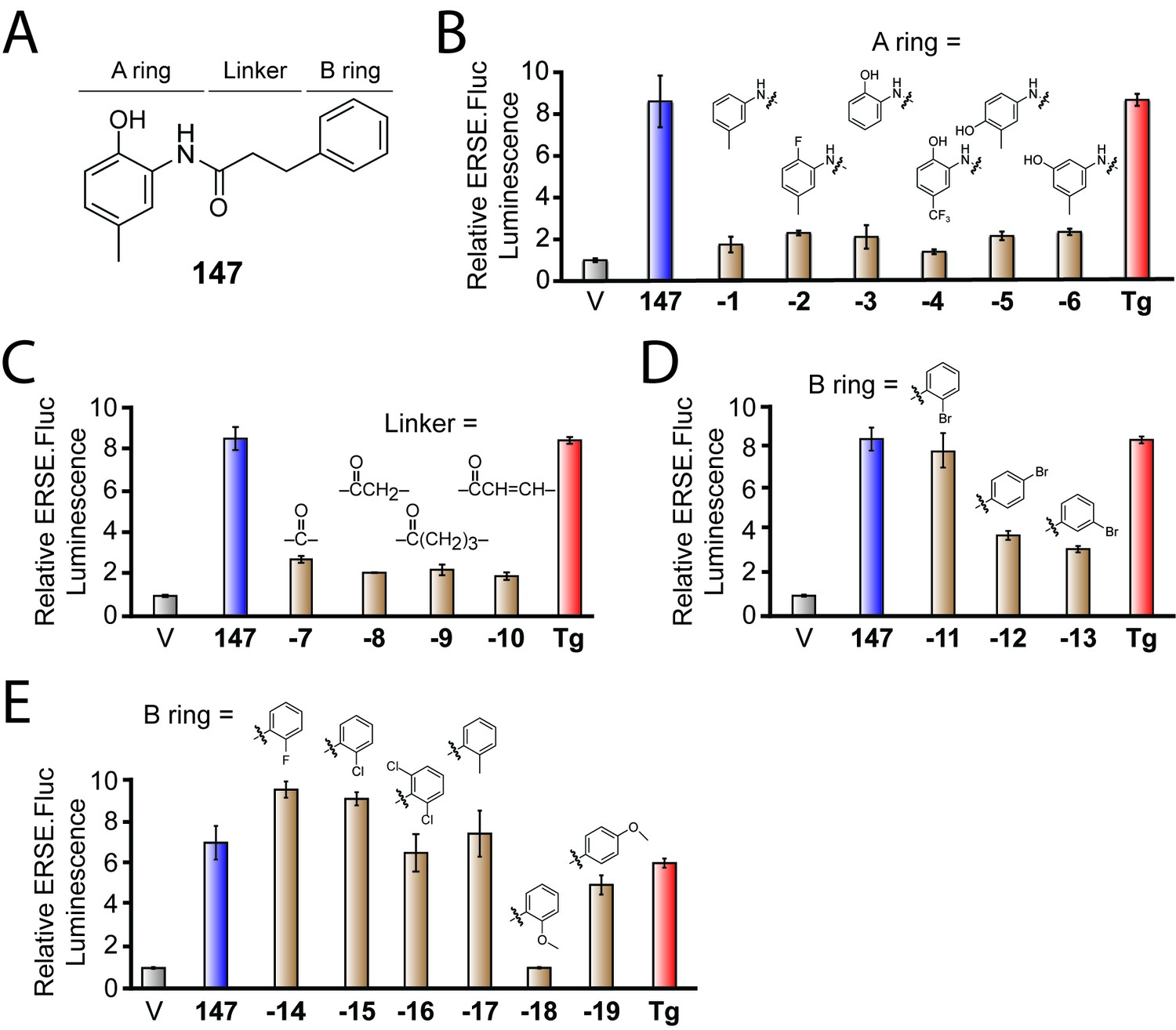
Activities of 147 analogs toward ATF6 activation.
(A) Structure of 147 with its three components – the A-ring, B-ring, and linker – indicated. (B) Bar graph showing the relative activation of the ERSE.FLuc ATF6 transcriptional reporter in HEK293T cells treated with 147 (10 µM; 18 hr), the ER stressor thapsigargin (Tg; 0.5 µM; 18 hr), or the indicated analogs of 147 where the A-ring was varied (10 µM; 18 hr). Error bars show SEM for three technical replicates. (C) Bar graph as in (B) where HEK293T cells were treated with the indicated 147 analogs where the linker was varied (10 µM; 18 hr). Error bars show SEM for three technical replicates. (D) Bar graph as in (B) where HEK293T cells were treated with the indicated 147 analogs in which a bromine atom was placed in the ortho, para, and meta positions of the B-ring (10 µM; 18 hr). Error bars show SEM for three technical replicates. (E) Bar graph as in (B) where HEK293T cells were treated with the indicated 147 analogs with a variety of substituents incorporated onto the B-ring. Error bars show SEM for three technical replicates.
We next explored the dependence of 147 activity on the linker connecting the A- and B-rings (Figure 1C). Analogs of 147 in which the linker length or structure was varied were prepared by coupling 2-amino-p-cresol to benzoic acid (147-7), phenylacetic acid (147-8), 4-phenylbutanoic acid (147-9) and cinnamic acid (147-10) via their respective acyl chlorides (Figure 1—figure supplement 1B). Interestingly, all of these analogs showed diminished capacity to activate the ATF6 reporter relative to 147, indicating that the length and flexibility of the 3-carbon linker are critical for the activity of 147 (Figure 1C).
Finally, we explored how substitutions on the B-ring influence 147-dependent ATF6 activation. Analogs of 147 with B-ring substituents were prepared by coupling 2-amino-p-cresol to appropriately substituted 3-phenylpropanoic acids (Figure 1—figure supplement 1C). Incorporating a single bromine atom at different positions around the B-ring demonstrated that the ortho position is amenable to substitution, as the o-Br analog (147-11) showed activation of the ATF6 reporter to levels comparable to the parent compound (Figure 1D). In contrast, the p-Br (147-12) or m-Br (147-13) analogs showed lower levels of reporter activation. Fluorine, chlorine, and methyl substituents at a single ortho position or Cl atoms at both ortho positions were also tolerated (o-F: 147–14; o-Cl: 147–15; o-Cl2: 147–16; o-CH3: 147–17; Figure 1E). Methoxy groups were tolerated at the para position of the B ring, but not the ortho, position (o-OCH3: 147–18; p-OCH3: 147–19).
These medicinal chemistry efforts demonstrate that the 2-amino-p-cresol A ring is required for 147-dependent ATF6 activation, whereas the B-ring is more tolerant of structural perturbation. Interestingly, we also found that the length and flexibility of the linker is important for the activity of 147, likely reflecting the need for a specific conformation and relative positioning of the two rings for 147-dependent ATF6 activation.
147-dependent ATF6 activation is inhibited by exogenous thiols and a cytochrome P450 inhibitor
The requirement for the 2-amino-p-cresol substructure for ATF6 activation indicates that this component is central to the activity of 147. Interestingly, some compounds with similar substructures are oxidized to highly protein reactive structures in cells and in vivo (Bolton, 2014). Eugenol (4-allyl-2-methoxyphenol; Figure 2—figure supplement 1A) is a particularly well-studied example of a p-alkyl phenol that is oxidized to a p-quinone methide by cytochrome P450s in cells (Bolton et al., 1995; Thompson et al., 1991; Thompson et al., 1995a; Thompson et al., 1995b; Thompson et al., 1993). This p-quinone methide (Figure 2—figure supplement 1A) can react with nucleophiles resulting in depletion of glutathione and modification of cellular proteins, presumably through reactions with thiol side chains of reactive cysteines (Thompson et al., 1991). In addition, p-cresol itself can be oxidized to a p-quinone methide in rat and human microsomes in a process that is catalyzed by cytochrome P450s (Thompson et al., 1995b; Yan et al., 2005). These observations, combined with the requirement of the 2-amino-p-cresol substructure for 147 activity, suggest that 147-dependent ATF6 activation could similarly involve oxidation to the p-quinone methide (147-QM) shown in Figure 2A.
Figure 2 with 1 supplement see all
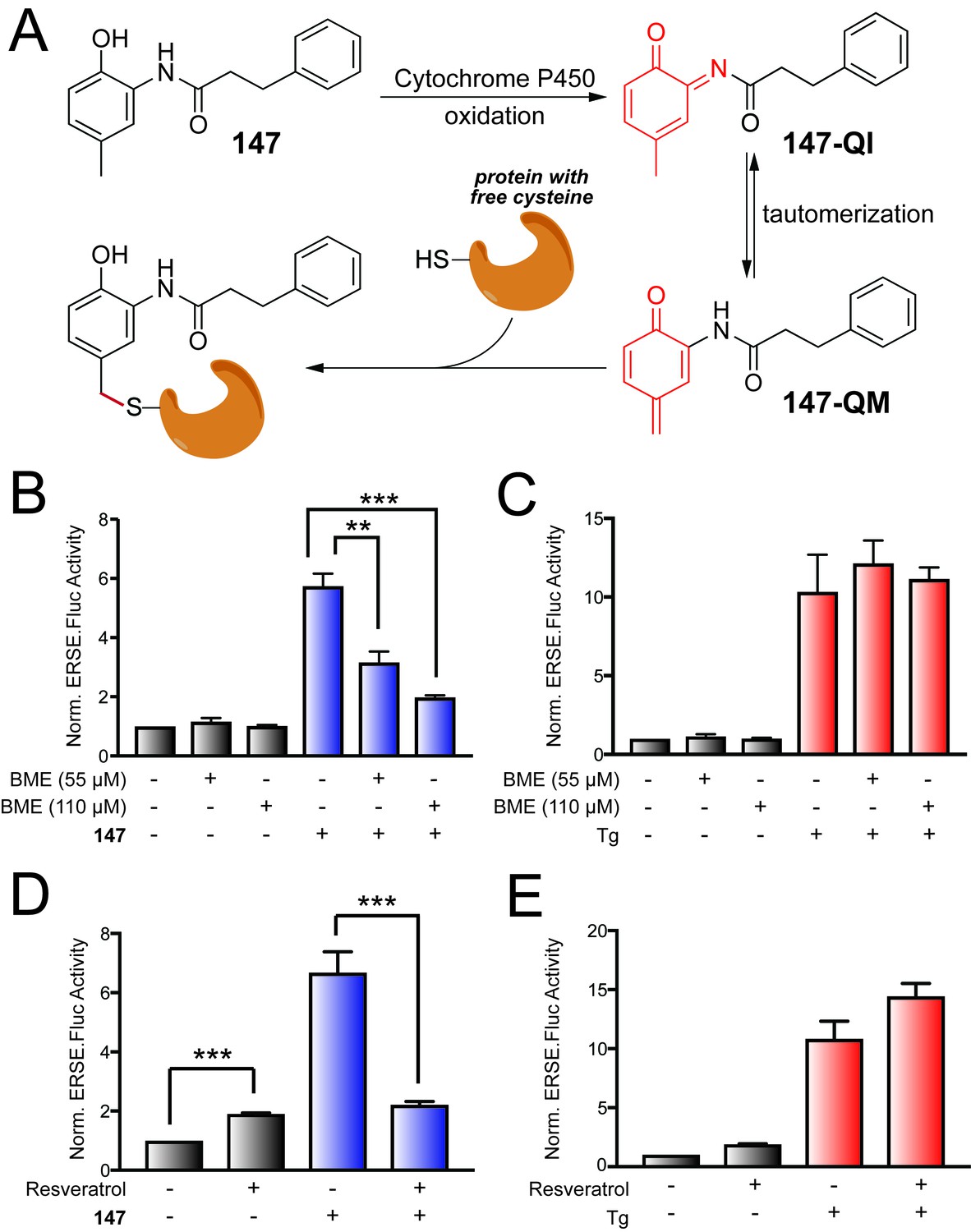
147-dependent ATF6 activation is sensitive to increases in cellular free thiols and resveratrol.
(A) Illustration showing a mechanism whereby 147 is converted to an o-quinone imine (147-QI) followed by tautomerization to a quinone methide (147-QM), which can then react with cellular proteins through nucleophiles such as free Cys side chains. (B) Bar graph showing the activation of the ERSE.FLuc ATF6 reporter in HEK293T cells treated with 147 (10 µM) and/or β-mercaptoethanol (BME; 55 μM or 110 μM) for 18 hr. Error bars show SEM for 3 independent experiments. *p<0.05. (C) Bar graph showing the activation of the ERSE.FLuc ATF6 reporter in HEK293T cells treated with thapsigargin (Tg; 0.5 µM) and/or β-mercaptoethanol (BME; 55 μM or 110 μM) for 18 hr. Error bars show SEM for 3 independent experiments. (D) Bar graph showing the activation of the ERSE.FLuc ATF6 reporter in HEK293T cells treated with 147 (10 µM) and/or resveratrol (10 µM) for 18 hr. Error bars show SEM for 4 independent experiments. ***p<0.005. (E) Bar graph showing the activation of the ERSE.FLuc ATF6 reporter in HEK293T cells treated with Tg (500 nM) and/or resveratrol (10 µM) for 18 hr. Error bars show SEM for 4 independent experiments.
Alternatively, metabolic activation of 147 could occur by oxidation of the 2-acylated amino p-cresol to an o-quinone imine (147-QI; Figure 2A). This process is directly analogous to the well-known oxidation of the analgesic antipyretic acetaminophen (Figure 2—figure supplement 1B) to N-acetyl-p-benzoquinone imine (Dahlin et al., 1984). The 147-QI intermediate can spontaneously tautomerize to the reactive p-quinone methide 147-QM, which could initiate ATF6 activation through the covalent modification of proteins (Figure 2A) (Bolton, 2014). The cellular components most likely responsible for initiating this oxidation mechanism are cytochrome P450s, which are generally responsible for metabolizing xenobiotic compounds, although other phase 1 enzymes are also capable of catalyzing the necessary oxidation steps (Bolton, 2014; Cao and Peng, 2014).
We initially scrutinized the metabolic activation mechanism for 147 shown in Figure 2A by monitoring activation of the ERSE.FLuc ATF6 reporter in HEK293T cells pre-treated with the free thiol-containing compound β-mercaptoethanol (BME). If 147 activity requires oxidation to an electrophile that covalently modifies Cys residues of proteins involved in ATF6 regulation, the exogenous addition of BME should disrupt this mechanism either by directly reacting with the quinone methide or by increasing the reducing potential in the cell so free thiols on a competing molecule (e.g. glutathione) react with 147-QM before it can react with its protein targets. Consistent with this notion, increasing doses of BME significantly reduced 147-dependent activation of the ATF6 reporter (Figure 2B), but did not influence activation of the ATF6 reporter afforded by treatment with the global ER stressor, thapsigargin (Tg; Figure 2C). Similar results were observed upon co-treatment with the alternative free thiol containing compound, N-acetyl cysteine (NAC; Figure 2—figure supplement 1C). Collectively, these results show that increases in intracellular free thiols reduce 147-dependent ATF6 activation.
Next, we probed the dependence of ATF6 activation by 147 on cytochrome P450s by assessing activation in the presence of a P450 inhibitor. We co-treated cells with 147 and resveratrol – a compound that is both an anti-oxidant and an inhibitor of CYP1A1, CYP1A2, and CYP1B1 (Chang et al., 2001). Resveratrol alone modestly increased basal activation of the ERSE.Fluc reporter, indicating that this compound either mildly activates ATF6 or stabilizes the firefly luciferase reporter (Figure 2D). In contrast, co-treatment with resveratrol and 147 significantly reduced the 147-dependent increase in ATF6 reporter activation (Figure 2D). Notably, co-treatment with resveratrol and Tg did not reduce Tg-dependent activation of the ATF6 reporter (Figure 2E). The reduction in 147-dependent ATF6 activation afforded by resveratrol co-treatment is consistent with 147 activity involving P450-dependent oxidation to a reactive electrophile(s), as indicated in Figure 2A.
The oxidation of 147 results in the covalent modification of cellular proteins
The above results support the model shown in Figure 2A whereby 147 is metabolically activated by cytochrome P450s to produce a reactive electrophile such as a quinone methide (147-QM), either directly, or through the intermediacy of 147-QI. The 147-QM could covalently modify proteins important for regulating ATF6 activation. We further tested this prediction in cell culture models using affinity purification mass spectrometry experiments.
We installed an alkyne substructure onto 147 by converting the Br of the methoxymethyl-protected variant of 147–11 to an alkyne (147-20a) using a Songashira coupling with trimethylsilyl acetylene followed by deprotection and desilylation (Figure 3—figure supplement 1A) (Hundertmark et al., 2000). The resulting alkyne-substituted 147 analog (147-20) had activity comparable to 147 in our ATF6 reporter assay, demonstrating that the inclusion of the alkyne did not diminish the compound’s ability to activate ATF6 (Figure 3A). The alkyne in 147–20 allows for a copper-catalyzed azide-alkyne cyclization (CuAAC) ‘click’ reaction (Besanceney-Webler et al., 2011) with an azide-functionalized biotin (Yang et al., 2010), which can be used as a handle to purify proteins covalently modified by 147–20 through the same mechanism shown in Figure 2A (Figure 3B). A cleavable diazo linker allows 147–20-protein conjugates to be cleaved from the affinity column.
Figure 3 with 1 supplement see all

Compound 147 covalently modifies cellular proteins.
(A) Bar graph showing the relative activation of the ERSE.FLuc ATF6 reporter in HEK293T cells treated with 147 (10 µM), 147–20 (10 µM), or thapsigargin (Tg; 0.5 µM) for 18 hr. Error bars show SEM for three technical replicates. (B) Schematic showing the protocol for affinity purification of proteins covalently modified by 147–20. (C) Coomassie-stained SDS-PAGE of affinity purified proteins from ALMC-2 cells treated with 147 (10 µM), or 147–20 (10 µM), or the combination of the 147–20 (10 µM) and 147 (50 µM), or 147–20 (10 µM) and 147–4 (50 µM) in combination for 18 hr. Specific proteins identified by mass spectrometry of excised bands are indicated.
In order to identify proteins covalently modified by 147–20, we prepared lysates from ALMC-2 cells treated with 147–20 for 18 hr. ALMC-2 cells are a plasma cell line derived from a light chain amyloidosis patient, in which it has been shown that 147 activates the ATF6 arm of the UPR (Plate et al., 2016; Arendt et al., 2008). These lysates were treated with an excess of biotin–diazo-linker–azide (Figure 3B) under ‘click’ chemistry conditions, enabling proteins covalently modified by 147–20 to be affinity isolated using streptavidin beads. Numerous 147–20-based protein conjugates isolated by affinity chromatography were identified by SDS-PAGE (Figure 3C). Co-treatment of cells with 147–20 together with a 5-fold excess of the parent compound 147 effectively eliminated these bands, indicating that 147 and 147–20 are reacting with the same subset of proteins. Notably, co-treatment with a 5-fold excess of the inactive analog 147–4 (Figure 1B), having a trifluoromethyl group on the A-ring instead of a methyl group, did not block 147–20 labeling, suggesting that conjugate formation with a subset of proteins is what is activating the ATF6 arm of the UPR. As expected, co-treatment with BME or resveratrol reduced 147–20 conjugate formation, further confirming the sensitivity of the oxidative ATF6 activation mechanism to these compounds (Figure 3—figure supplement 1B). In gel digestion of the 147–20 conjugates shown in Figure 3C followed by mass spectrometry characterization identified four ER protein disulfide isomerases (PDIA1, PDIA4, PDIA6, and TXNDC5), a proteasome subunit (PSME2), and a chloride channel (CLIC1) as the most prevalent bands appearing in Figure 3C that were efficiently eliminated by adding a 5-fold excess of 147. Interestingly, we did not observe covalent modification of a FLAG-tagged ATF6 in HEK293T cells treated with 147 or 147–20 or the combination of 147 and 147–20, indicating that 147 does not appear to directly modify ATF6 (Figure 3—figure supplement 1C). Instead, these results suggest that 147-dependent ATF6 activation involves 147-mediated conjugate formation with a few select proteins that regulate ATF6 activity.
147 preferentially modifies ER-localized proteins
To more comprehensively identify the cellular proteins forming covalent conjugates with 147–20, we performed an affinity purification–mass-spectrometry-based proteomics analysis in three distinct cell types treated with 147–20 using the work flow outlined in Figure 3B. We individually treated ALMC-2, HEK293T, and liver-derived HepG2 cells with 147 (10 μM), or 147–20 (10 μM), or 147–20 (10 μM) in combination with 147 (50 μM) for 18 hr (147 was previously shown to activate ATF6 in all three cell lines employed [Plate et al., 2016]). The lysates were collected, treated with the diazo biotin-azide linker under ‘click’ chemistry conditions, and the 147–20-protein conjugates were affinity purified as described above, cleaved with 50 mM sodium dithionite from the affinity resin, and eluted in 1% SDS. The eluted protein conjugates were subjected to Tandem Mass Tag (TMT)-Multidimensional Protein Identification Technology (MuDPIT) analysis, as we have described previously (Mortenson et al., 2018). Several hundred proteins were identified in each cell line. However, only 90 proteins were identified in all eight replicates (two biological sets of technical duplicates in ALMC-2 cells and one set of technical duplicates each in HEK293T and HepG2 cells). Of these 90 proteins, 19 exhibited significant depletion in cells incubated with 5-fold excess of 147, indicating specific labeling. Hereafter, we define the competition TMT reporter ion ratio as the ratio between the samples treated with 147–20 to those co-treated with 147–20 and a 5-fold excess of 147. Significant depletion is defined as a competition TMT reporter ion ratio that is significantly greater than 1.0 with a one-tailed Wilcoxon signed-rank test p value < 0.05. These 19 proteins are listed in Table 1. Their median competition TMT reporter ion ratios vary from 1.4 to 2.8. The full list of 90 proteins is available in Table 1—source data 1 and the list of all identified proteins is available in Table 1—source data 2. Notably, ATF6 itself was not identified in any of the cell types, consistent with our results showing 147–20 does not appear to covalently modify ATF6 (Figure 3—figure supplement 1B).
Table 1
Protein targets that are covalently modified by 147.
https://doi.org/10.7554/eLife.37168.008| Protein name | Symbol | Uniprot accession | Competition Ratio* | Redox?† | ER-localization?‡ |
|---|---|---|---|---|---|
| Cytoskeleton-associated protein 4 | CKAP4 | Q07065 | 2.8 (1.7, 3.5) | N | Y |
| Protein disulfide isomerase A4 | PDIA4 | P13667 | 2.4 (1.8, 3.3) | Y | Y |
| Protein disulfide isomerase A6 | PDIA6 | Q15084 | 2.4 (2.0, 3.0) | Y | Y |
| Protein disulfide isomerase A1 (a.k.a. Prolyl 4-hydroxylase subunit beta) | PDIA1 (a.k.a. P4HB) | P07237 | 2.3 (2.1, 3.1) | Y | Y |
| Protein disulfide isomerase A3 | PDIA3 | P30101 | 2.2 (1.8, 3.1) | Y | Y |
| Thioredoxin | TXN | P10599 | 2.2 (1.2, 3.5) | Y | N |
| Thioredoxin domain-containing protein 5 | TXNDC5 | Q8NBS9 | 2.2 (2.0, 2.8) | Y | Y |
| Heme oxygenase 2 | HMOX2 | P30519 | 2.2 (1.4, 4.3) | N | Y |
| Chloride intracellular channel 1 | CLIC1 | O00299 | 1.9 (1.5, 2.3) | N | N |
| Thioredoxin-related transmembrane protein 1 | TMX1 | Q9H3N1 | 1.9 (1.5, 4.3) | Y | Y |
| Voltage dependent anion channel 2 | VDAC2 | P45880 | 1.6 (1.2, 1.9) | N | N§ |
| Poly(rC)-binding protein 1 | PCBP1 | Q15365 | 1.6 (1.4, 1.8) | N | N |
| Ribosome-binding protein 1 | RRBP1 | Q9P2E9 | 1.5 (1.3, 1.8) | N | Y |
| Proteasome subunit alpha type 1 | PSMA1 | P25786 | 1.5 (1.1, 1.7) | N | N |
| d-3-phosphoglycerate kinase | PHGDH | O43175 | 1.5 (1.4, 1.9) | N | N |
| Histocompatibility minor 13 | HM13 | Q8TCT9 | 1.5 (1.2, 2.0) | N | Y |
| Polyadenylate-binding protein 4 | PABPC4 | Q13310 | 1.5 (1.1, 1.7) | N | N |
| Ribophorin 1 | RPN1 | P04843 | 1.4 (1.3, 1.8) | N | Y |
| ER resident protein 29 | ERP29 | P30040 | 1.4 (1.0, 1.8) | N | Y |
-
*Competition ratio = Ratio of TMT reporter ions for the protein in question between samples treated with 147–20 and those co-treated with 147–20 and 147. The median value across all cell lines and replicates are shown, with the values at the 1st and 3rd quartiles shown in parenthesis.
†Indicates whether the protein in question has the ‘cell redox homeostasis’ GO annotation (GO:0045454).
-
‡Indicates whether the protein in question has the ‘endoplasmic reticulum part’ GO annotation (GO:0044432).
§VDAC2 is a mitochondrial membrane protein but has been shown to localize to sites of mitochondria that are associated with the ER.
-
Table 1—source data 1
Excel spreadsheet showing the competition ratio data for all 90 proteins that were identified as targets of 147 in every replicate and every cell type.
- https://doi.org/10.7554/eLife.37168.009
-
Table 1—source data 2
Excel spreadsheet showing all the proteins identified among the affinity-purified targets of 147.
- https://doi.org/10.7554/eLife.37168.010
It is immediately apparent that more than a third of the 19 identified proteins are involved in redox homeostasis, including six protein disulfide isomerases (PDIA1, PDIA3, PDIA4, PDIA6, TMX1 and TXNDC5) and thioredoxin (TXN). This observation can be quantified using the PANTHER Overrepresentation Test to identify significantly overrepresented Gene Ontology (GO) terms from the ‘biological process’ domain (Mi et al., 2017). We find that the seven redox proteins noted above are all annotated with the GO term ‘cell redox homeostasis’ (GO:0045454), corresponding to a > 100 fold enrichment (False Discovery Rate (FDR) = 6.6 × 10−9) relative to the expected number of occurrences of this annotation in a random sample of 19 genes. The cysteine residues in proteins of this class can react with electrophiles like the p-quinone methide predicted to be formed upon metabolic oxidation of 147 and 147–20 (Figure 2A). Other enriched GO terms include ‘response to ER stress’ (GO:0034976; 31-fold enriched, FDR = 7.9 × 10−6) and ‘protein folding’ (GO:0006457; 33-fold enriched, FDR = 8.2 × 10−6). In both cases, the enrichment is largely due to the presence of the protein disulfide isomerases (PDIs) in the list of 19 proteins identified in our TMT-MuDPIT proteomic analysis.
It is worth noting that many of the proteins in the list not involved in cellular redox homeostasis are nevertheless known to be regulated by the oxidation states of reactive cysteine residues. HMOX2 (heme oxygenase 2), unlike its paralog HMOX1 (which we did not find to be labeled by oxidized 147), has two Cys-Pro sequences near its C-terminus. When these cysteines are oxidized, the C-terminus is unstructured and HMOX2 binds a single heme in its catalytic site (Bagai et al., 2015; Fleischhacker et al., 2015). However, this region becomes helical when the cysteines are reduced to allow binding of a second heme (Bagai et al., 2015; Fleischhacker et al., 2015). Similarly, CLIC1 adopts two substantially different folded structures depending on the oxidation state of two of its cysteine residues (Littler et al., 2004). When the cysteine residues are reduced, it is a soluble monomer with a structure homologous to glutathione S-transferase. When the cysteines are oxidized to form a disulfide bond, CLIC1 undergoes a drastic structural transition, becoming a helical dimer that can insert into membranes and form chloride ion channels (Littler et al., 2004; Goodchild et al., 2009).
Twelve of the nineteen proteins (63%) in the target list are annotated as ER proteins (GO:0005783, 7.4-fold enriched, FDR = 2.0 × 10−6), demonstrating that 147–20 preferentially reacts with ER-localized proteins. These include the six PDIs noted above, along with HMOX2, HM13 (histocompatibility minor 13, also known as signal peptide peptidase or SPP; a peptidase that degrades signal peptides after they have been released from pre-proteins [Voss et al., 2013]), CKAP4 (cytoskeleton associated protein 4, also known as CLIMP-63; a transmembrane protein involved in the regulation of ER morphology [Sandoz and van der Goot, 2015]), RRBP1 (ribosome binding protein 1; a ribosome-binding protein on the rough ER [Savitz and Meyer, 1990]), RPN1 (ribophorin 1; a component of oligosaccharyl transferase, an enzyme complex that catalyzes protein N-glycosylation [Wilson et al., 2008]), and ERP29 (ER protein 29; a structural homolog of PDIs that lacks an active thioredoxin domain but retains chaperone activity [Mkrtchian and Sandalova, 2006; Suaud et al., 2011]). In addition, while VDAC2 is a mitochondrial protein, it has been shown to localize to sites of ER-mitochondrial contacts called mitochondrial-associated ER membranes or MAMs (Naghdi and Hajnóczky, 2016).
It is very important to note that the remarkable preference of 147–20 to react with ER-localized proteins (63% of the conjugates formed are with ER proteins) is not a general feature of other small molecule electrophiles (Figure 4A). Backus et al. examined the reactivity of a variety of fragment electrophiles containing cysteine-reactive chloroacetamide or acrylamide groups in live cells (Backus et al., 2016). Among the fragment electrophiles that modified 10 or more proteins with some degree of selectivity (as defined by Backus et al. [Backus et al., 2016]), proteins annotated with the GO term for ER localization (GO:0005783) generally constituted ≈ 10% of the targets that they modified (Figure 4A). This is comparable to the proportion of ER-localized proteins for the human proteome as a whole (8.6%). At most, ER-localized proteins made up 30% (4 out of 13) of the targets for compound B-51 (Figure 4A), but this still does not represent a significant enrichment after multiple-testing correction using the PANTHER Overrepresentation Test.
Figure 4 with 2 supplements see all
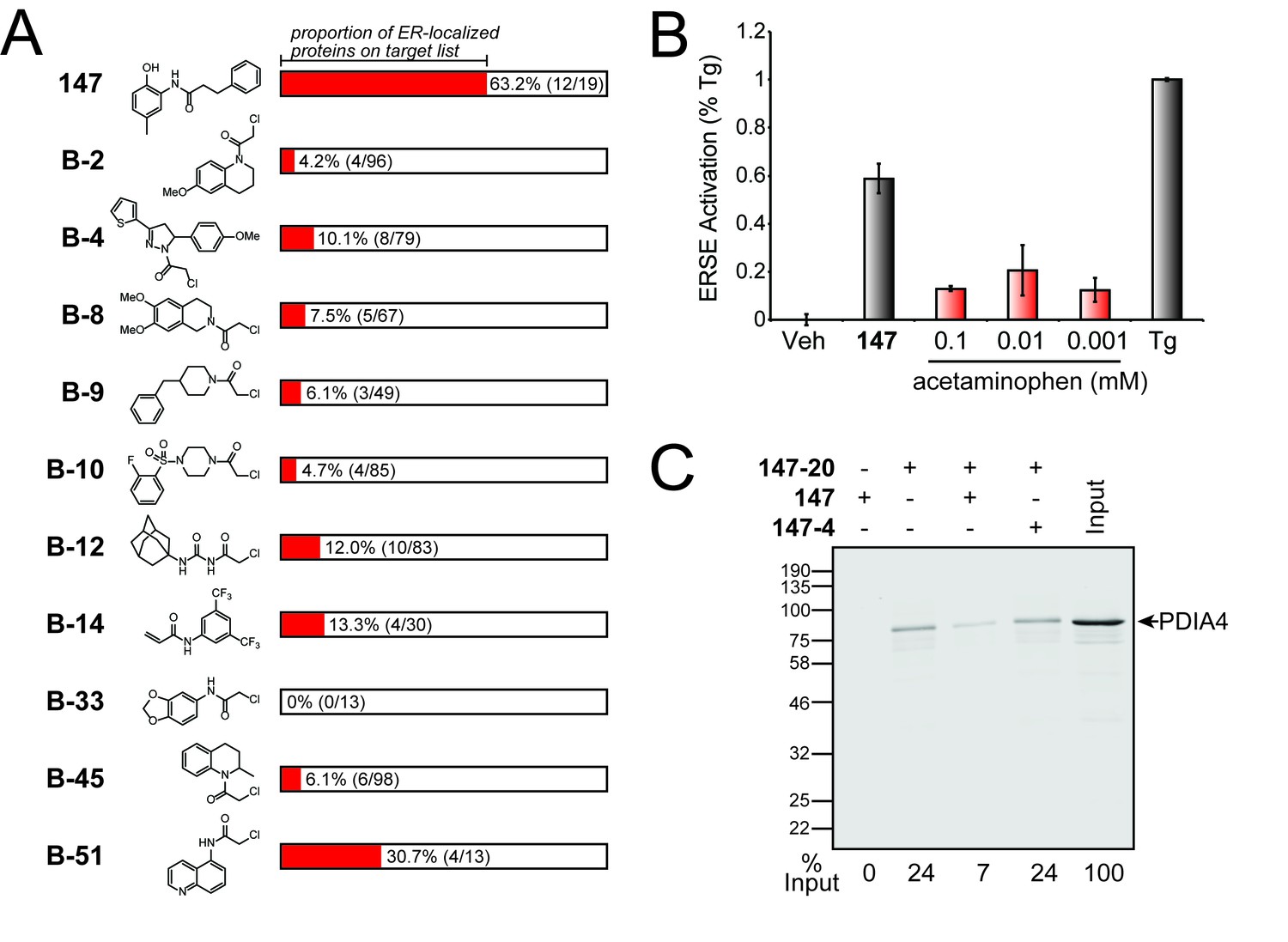
Comparison of S-reactive electrophiles demonstrating that compound 147 selectively modifies ER-localized proteins.
(A) Proportion of ER-localized proteins covalently labeled by 147 or fragment electrophiles reported in (Backus et al., 2016). The ER-localized proportion for each electrophile indicated in red. The numbers in parentheses indicate the number of ER-localized proteins in a given electrophile’s target list on the left and the total number of proteins in the target list on the right. The electrophiles from (Backus et al., 2016) are denoted with a B- followed by the compound numbers used in that work. (B) Bar graph showing activation of the ERSE.FLuc reporter in HEK293T cells treated with 147 (10 µM), thapsigargin (Tg; 0.5 µM) or the indicated dose of acetaminophen for 18 hr. Error bars show SEM for 3 independent experiments. (C) Immunoblot of PDI4 in 147–20 affinity purified proteins from HEK293T cells treated with 147 (10 µM), or 147–20 (10 µM), or the combination of 147–20 (10 µM) and 147 (50 µM), or the combination of 147–20 (10 µM) and 147–4 (50 µM) for 18 hr. The relative recovery of PDIA4 under these different conditions is indicated below the immunoblot.
Moreover, modification of ER-localized proteins and subsequent activation of UPR signaling pathways is also not a general feature of compounds known to form quinone methides. For example, celastrol—a triterpenoid natural product that harbors a quinone methide—activates many stress-responsive signaling pathways, including the heat shock response and the antioxidant response, both of which are largely cytosolic stress responses (Trott et al., 2008). Notably, celastrol does not activate the UPR ERSE.Fluc reporter in cells (Cooley et al., 2014). Furthermore, other compounds known to form reactive quinone methides such as eugenol, also did not significantly activate the ERSE.Fluc reporter (Figure 4B and Figure 4—figure supplement 1). Thus, the reaction preference of 147 for protein targets in the ER (Figure 4A) and subsequent selective activation of the ATF6 UPR signaling pathway is a singular feature of the reactivity and specificity of this compound.
To further explore the origins of the selectivity of 147 and its analogs for modifying ER proteins, we sought to determine whether 147 preferentially partitions to the ER. We treated HEK293T cells with 147 for 15 min and separated membranous organelles including the ER from the cytosol by digitonin extraction (we validated that the digitonin extraction separated the ER and cytosol by blotting the fractions for the ER-localized ERdj3 and cytosolic HSC70; Figure 4—figure supplement 2A). We then used liquid chromatography-mass spectrometry (LC-MS) to quantify the amounts of 147 in the cytosol- and ER-containing fractions based on their peak areas by comparison with a standard curve. We found 2.1 ± 0.2 ng of 147 per million cells in the cytosol fraction compared to 5.6 ± 0.9 ng in the ER fraction (mean ± standard error; Figure 4—figure supplement 2B), corresponding to a ratio of 2.7 ± 0.5. Given that membranous organelles likely constitute a smaller fraction of the total volume of HEK293T cells than the cytosol (even in hepatocytes, membranous organelles are about only 40% of the total cell volume compared to 55% for the cytosol [Weibel et al., 1969]), this likely represents a lower limit for this ratio. This preference is likely due to the hydrophobicity of 147, which would lead to its sequestration in the membranes that surround organelles like the ER.
This result suggests that the bias of 147 for reacting with ER-resident proteins is at least in part the result of its higher concentration in the ER and ER membrane. It is additionally worth noting that the cytochrome P450s that most likely convert 147 to 147-QM (the quinone methide) primarily reside in the ER membrane (Guengerich, 2015). Moreover, the reducing environment of the cytosol would tend to decrease the lifetime of 147-QM (e.g. through reaction with glutathione) relative to the lifetime of 147 in the oxidizing environment of the ER, thereby diminishing the extent of protein modification in the cytosol. The combination of these factors likely drives the preference of 147 to selectively react with ER-resident proteins.
Interestingly, despite preferential labeling of ER-localized PDIs by 147–20, previous work has shown that treatment with 147 does not influence secretion of the endogenous secreted proteome or disulfide-bonded proteins dependent on PDIs for ER folding, such as energetically normal immunoglobulin light chains or fully-assembled immunoglobulins (Plate et al., 2016). This suggests that 147 may label only a fraction of the ER PDIs, allowing the remaining PDI population to be available for facilitating the folding of disulfide-bonded proteins within the ER lumen. To test this, we defined the fraction of PDIA4 labeled with 147–20 in HEK293T cells. We incubated HEK293T cells with 147–20 in the absence or presence of a 5-fold excess of the competitive ATF6 activator 147 or the inactive 147–4 (both lacking the means to attach an affinity handle) and then compared the recovery of PDIA4 in each purification to the total amount of PDIA4 in cell lysates. Only 24% of PDIA4 is covalently modified with 147–20 (Figure 4C). Conjugate formation by 147–20 is nearly eliminated when a 5-fold excess of the parent compound 147 is co-added, but not when the inactive compound 147–4 is co-added in excess. This suggests that while 147 prefers ER PDIs, it does not covalently modify the entire PDI population in the ER, providing an explanation for why treatment with 147 does not influence secretion of the endogenous secreted proteome or specific proteins dependent on PDIs for their efficient folding.
shRNA-depletion of PDIs perturbs 147-dependent ATF6 activation
In the absence of stress, ATF6 is maintained in transport-incompetent monomeric and oligomeric conformations by intra- and intermolecular disulfide bonds (Figure 5A) (Nadanaka et al., 2007; Nadanaka et al., 2006; Higa et al., 2014). ER stress promotes the reduction of ATF6 disulfides, resulting in the formation of a reduced ATF6 monomer that traffics to the Golgi for proteolytic activation by the S1 and S2 proteases (Nadanaka et al., 2007; Nadanaka et al., 2006; Higa et al., 2014). Since the maintenance of disulfides in the ER requires PDI activity, the disulfide-mediated regulation of ATF6 also likely involves PDIs. Consistent with this hypothesis, previous results have shown that PDIA5 has a critical role in regulating ER stress-dependent ATF6 activation in cancer cells (Higa et al., 2014). These results suggest that 147-dependent covalent modification of PDIs could promote ATF6 reduction and subsequent trafficking to the Golgi for activation (Figure 5A). Here, we probe this mechanism using a combination of pharmacologic and genetic approaches.
Figure 5 with 2 supplements see all
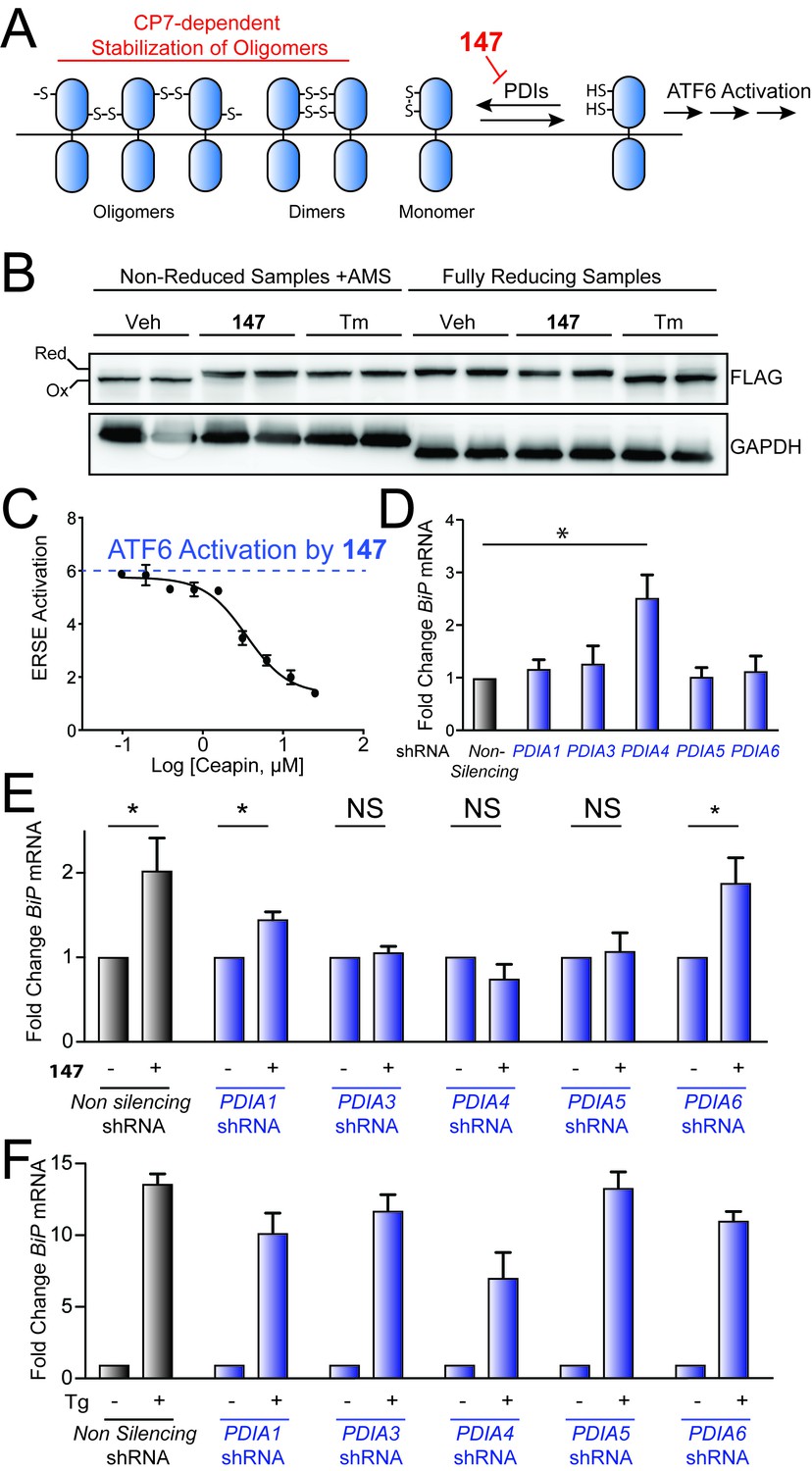
Genetic depletion of select PDIs alters 147-dependent ATF6 activation.
(A) Mechanistic model of 147-dependent ATF6 activation, as described in the main text. The red component of the figure shows how pharmacologic modulation of ATF6 by 147 or CP7 influences ATF6 activity. (B) Immunoblot showing lysates prepared from HeLa cells stably expressing FLAG-tagged ATF6 treated for 6 hr with 147 (10 µM) or tunicamycin (Tm; 1 mg/mL). Lysates were separated by non-reducing or reducing SDS-PAGE prior to immunoblotting. The bands representing oxidized and reduced ATF6 are indicated. Note the increased ATF6 migration observed in reducing gels for Tm-treated cells, reflecting the inhibited N-linked glycosylation of ATF6 in these samples. (C) Graph showing relative activation of the ERSE.Fluc ATF6 reporter in HEK293T cells co-treated for 18 hr with 147 (10 µM) and increasing concentrations of CP7, as indicated. Error bars show SEM for three technical replicates. (D) Bar graph showing BiP expression in HEK293T cells stably expressing non-silencing, PDIA1, PDIA3, PDIA4, PDIA5, or PDIA6 shRNA, as indicated. BiP expression in the PDI-depleted cells is shown relative to cells expressing non-silencing shRNA. Error bars show SEM for 3 independent experiments. *indicates p<0.05. (E) Bar graph showing BiP expression in HEK293T cells expressing non-silencing, PDIA1, PDIA3, PDIA4, PDIA5, or PDIA6 shRNA treated for 6 hr with or without 147 (10 µM). BiP expression levels for samples treated with 147 are normalized to the corresponding vehicle-treated controls. Un-normalized data are shown in Figure 5—figure supplement 1B. Error bars show SEM for 3 independent experiments. *indicates p<0.05. (F) Bar graph showing BiP expression in HEK293T cells expressing non-silencing, PDIA1, PDIA3, PDIA4, PDIA5, or PDIA6 shRNA treated for 6 hr with or without thapsigargin (Tg; 0.5 µM). BiP expression levels for samples treated with Tg are normalized to the corresponding vehicle-treated controls. Un-normalized data are shown in Figure 5—figure supplement 1C Error bars show SEM for 3 independent experiments.
Initially, we tested this model by monitoring the reduction of FLAG-tagged ATF6 (FTATF6) stably expressed in HeLa cells treated with 147 or the ER stressor tunicamycin (Tm). Oxidized and reduced FTATF6 were separated by non-reducing and reducing SDS-PAGE/immunoblotting, as previously described (Nadanaka et al., 2007). In the absence of small molecule treatment, FTATF6 migrates faster in non-reducing gels, consistent with its primarily oxidized status (Figure 5B). Treatment with 147 alters the migration of FTATF6 in non-reducing gels (upward gel shift), consistent with a net reduction of the disulfides in ATF6. Similar results were observed upon Tm treatment, which activates all 3 arms of the UPR. These experiments support the hypothesis that 147-dependent covalent conjugation to PDIs promotes the reduction of ATF6 to increase populations of the reduced ATF6 monomers associated with transcription factor activation (Plate et al., 2016).
A set of small molecules called Ceapins were previously identified to inhibit ER stress-dependent ATF6 activation by stabilizing ATF6 oligomers in the ER and preventing their trafficking to the Golgi for proteolytic activation (Gallagher and Walter, 2016; Gallagher et al., 2016). Based on the proposed model shown in Figure 5A, we predict that stabilization of ATF6 oligomers by Ceapins should impede 147-dependent ATF6 activation by preventing the disruption of the ATF6 oligomers into the reduced monomers required for trafficking. Consistent with this, 147-dependent activation of the ATF6-selective ERSE.Fluc reporter was inhibited by co-addition of increasing concentrations of the active Ceapin, CP7 (Figure 5C; blue dotted line indicates level of 147 activation achieved in the absence of CP7) (Gallagher and Walter, 2016; Gallagher et al., 2016). This indicates that stabilization of ATF6 oligomers disrupts 147-dependent ATF6 activation, consistent with 147 having an important role in promoting ATF6 disulfide reduction and ATF6 dissociation prior to trafficking to the Golgi for proteolytic activation.
Next, we used a genetic approach to define the dependence of 147-mediated ATF6 activation on specific PDIs identified in our quantitative mass spectrometry experiment. We prepared stable pools of HEK293T cells wherein PDIA1, PDIA3, PDIA4, or PDIA6 was shRNA-depleted to extents analogous to the population of PDIAs covalently labeled by 147 (see Figure 4C). We also prepared a stable pool of HEK293T cells shRNA-depleted of PDIA5, which was identified as modified by 147–20 in 2 out of 3 cell types (Table 1—source data 2) and has previously been shown to be important for ATF6 activation (Higa et al., 2014). PDI knockdown in these cells varied from 25–50%, as measured by qPCR (Figure 5—figure supplement 1A), consistent with the population of each PDI predicted to be labeled by 147 (see Figure 4C). Knockdown of PDIA4 increased expression of the ATF6 target gene BiP in untreated cells, indicating that reduced PDIA4 activity activates ATF6 independently of compound addition (Figure 5D). However, knockdown of other PDIs did not significantly influence BiP expression. The addition of 147 did not increase expression of the ATF6-regulated gene BiP, relative to matched untreated controls, in cells in which PDIA3, PDIA4, or PDIA5 were depleted by shRNA (Figure 5E; unnormalized data are shown in Figure 5—figure supplement 1B). In contrast, Tg-dependent induction of BiP was not significantly impaired in these cells upon PDI knockdown (Figure 5F; unnormalized data are shown in Figure 5—figure supplement 1C). Interestingly, shRNA-depletion of PDIA6 did not perturb BiP expression in 147- or Tg-treated cells, suggesting that covalent modification of this PDI is not required for ATF6 activation in these cells. Similarly, PDIA1 depletion only modestly reduced 147-dependent BiP expression, indicating that covalent modification of PDIA1 also does not significantly influence ATF6 activation in these cells. Collectively, these results show that modest depletion (25–50%) of a subset of PDIs selectively inhibits 147-dependent activation of ATF6, suggesting that covalent modification of PDIA3, PDIA4, and/or PDIA5 contribute to 147-dependent ATF6 activation.
The notion that 147 acts through modification of PDIs, some of which are in turn upregulated by 147-mediated ATF6 activation (especially PDIA4, and to a lesser extent PDIA1 and PDIA3) (Plate et al., 2016) raises the question of whether the increased levels of PDIs could diminish the efficacy of 147 as time goes on. ATF6 activity induced by 147 was previously shown to diminish 12 hr after a single treatment of compound in HEK293 cells, suggesting a potential negative feedback loop associated with ATF6 activation (Plate et al., 2016). However, 147 has been used in experiments where it was shown that daily dosing of 147 over weeks could suppress stem cell pluripotency, enhance differentiation, and direct mesodermal cell fate (Kroeger et al., 2018), suggesting that the induction of PDIs does not lead to a substantial feedback attenuation of 147-dependent ATF6 activation when constantly administered. Thus, while the potential for increased PDI levels to suppress 147-dependent ATF6 activation offers an interesting mechanism to modulate 147 activity, further studies will be required to elucidate whether such a negative feedback mechanism is operating.
Discussion
Here, we hypothesize that ATF6 activation by 147 proceeds through a mechanism involving metabolic oxidation of 147 to form a reactive compound, likely a p-quinone methide, that covalently modifies a subset of ER-localized proteins, including PDIs. This targeting of PDIs by 147 appears to disrupt the maintenance of ATF6 disulfides, enabling reduction of disulfides within ATF6, increasing the populations of reduced, trafficking-competent ATF6 monomers. Previous results show that ATF6 reduction is not sufficient to promote the complete trafficking of ATF6 to the Golgi (Nadanaka et al., 2007; Nadanaka et al., 2006). However, increasing populations of reduced, monomeric ATF6 would enhance sensitivity of this transcriptional signaling pathway to normal, homeostatic changes in ER proteostasis, thus increasing basal activity of ATF6 transcriptional signaling (Figure 5A). This mechanism would explain the 30–40% activation of ATF6 afforded by 147 (Plate et al., 2016) and highlights the potential for targeting ATF6 redox to allow for the moderate levels of ATF6 signaling necessary to therapeutically modulate ER proteostasis without impacting global ER function (Plate and Wiseman, 2017).
A remarkable aspect of this mechanism of pharmacologic ATF6 activation is the preferential covalent modification of proteins localized to the ER by 147. This preference likely results from the biased partitioning of 147 to the ER and other membranous compartments of the cell due to its hydrophobic nature as well as the oxidation of 147 by cytochrome P450s localized to the ER membrane. The conversion of 147 to reactive compounds such as a p-quinone methide at the ER membrane limits the ‘sphere of activity’ for this compound, allowing it to selectively modify ER proteins, like PDIs acting on ATF6, similarly localized to the ER membrane. In this scenario, the reactive 147-QM cannot diffuse far from the ER before it hydrolyzes or reacts with small molecule cellular nucleophiles (e.g. glutathione) that prevent the broad modification of cellular proteins localized to other compartments. This is especially true in the cytosol, in which 147-QM likely has a shorter lifetime than in the ER because of the cytosol’s more reducing environment.
This mechanism of activity for 147 is enabled by a coincidence of several structural features of the small molecule. First, the 2-amino-p-cresol substructure of 147 can be oxidized to a p-quinone methide, a highly efficient electrophile for covalent modification of proteins. Second, each of the three primary substructures of 147, the A-ring, B-ring, and three carbon linker (Figure 1A), are largely required for compound activity. The 2-amino-p-cresol substructure of the A-ring is required for 147 oxidation, for example quinone methide formation. However, the dependence on the B-ring and linker for ATF6 activation remains unclear. These substructures could be important for targeting the compound to a specific subset of cytochrome P450s at the ER membrane for metabolic activation and/or directing the reactive, oxidized 147-QM to PDIs for covalent modification, or both. While we continue to define the dependence of 147 activity on these substructures, our results show that the B-ring and linker confer the selectivity of 147 required for compound-dependent ATF6 activation. The third feature of 147 important for ATF6 activation is the unique reactivity of the resulting oxidized product. The reactivity of the metabolically activated 147 is not so high that it is instantly quenched by hydrolysis, nor so low that it can diffuse away from the immediate ER environment and react widely with proteins localized throughout the cell.
Collectively, these features combine to make 147 a precision electrophile that preferentially activates the ATF6 signaling pathway without triggering other pathways activated by similar stimuli, such as the IRE1 and PERK UPR signaling pathways, the heat shock response, or the NRF2 oxidative stress response as we have shown previously by multiplex gene expression profiling, as well as by RNAseq (Plate et al., 2016). ER localization enabling localized generation of electrophiles by P450s and related enzymes that selectively modify protein(s) is a potentially interesting drug development strategy. However, it is important to note that in our previous work we also found that some compounds with similar structures, notably including the 2-amino-p-cresol warhead, are substantially less selective towards activating stress-responsive signaling. The small molecule 132, shown in Figure 5—figure supplement 2, activates ATF6, XBP1, and PERK stress sensors to similar extents (Plate et al., 2016). Given their structural similarity, it is reasonable to expect that 132 is oxidized to a quinone methide followed by proteome reactivity like 147. Thus, it appears that pushing reactivity too far and/or quinone methide concentration too high can result in a broader stress-responsive signaling pathway activation beyond ATF6. The PDIs that 147 modifies could interact with other components of the ER proteostasis network, in addition to ATF6 (Gallagher et al., 2016; Ryno et al., 2014). The selectivity of 147 likely arises from it having a lower propensity to modify the Cys-containing proteome than 132, either because 147 is not as reactive toward protein nucleophiles, or because its quinone methide is at lower concentration, or because it is more rapidly quenched.
Pharmacologic ATF6 activation has substantial potential for ameliorating tissue-specific defects in ER proteostasis implicated in diverse diseases including systemic amyloid diseases, ischemic heart disease, diabetes, and neurodegenerative disorders (Hetz et al., 2015; Plate and Wiseman, 2017). The mechanism of action for our top pharmacologic activator 147 (as defined herein) suggests new potential opportunities to optimize the activity of pharmacologic ATF6 activators for specific tissues. For example, designing 147 analogs with selectivity for a subset of P450s provides a potential opportunity to improve compound activity in tissues specifically expressing this subset of P450s. Similarly, different tissues have different PDI compositions. Thus, improving targeting of reactive compounds for specific PDIs could improve ATF6 activating activity in tissues expressing these PDIs. Interestingly, this type of selectivity for P450s or PDIs provides a potential explanation for how other compounds identified in our original HTS that can be similarly metabolically activated to reactive compounds (e.g. 263) efficiently activate ATF6 in some cell lines (e.g. HEK293T), but not in others (e.g. light chain amyloidosis patient-derived ALMC2 cells) (Plate et al., 2016). Thus, the mechanism defined herein suggests new opportunities to selectively promote ATF6 activation in tissues of interest by further development of these compounds through medicinal chemistry efforts.
Materials and methods
Chemicals
All compounds and reagents were purchased from Sigma-Aldrich, Acros, Alfa Aesar, and EMD Millipore unless otherwise noted and were used without further purification. The ERSE-Firefly luciferase reporter cell line has been reported previously (Plate et al., 2016) and was used without modification.
Cell culture and transfections
Request a detailed protocolHEK293T-Rex (ATCC), HEK293T (ATCC), and HepG2 (ATCC) were cultured in high-glucose Dulbecco’s Modified Eagle’s Medium (DMEM) supplemented with glutamine, penicillin/streptomycin and 10% fetal bovine serum (FBS). Cells were routinely tested for mycoplasma every 6 months. We did not further authenticate the cell lines. HEK293T-Rex cells containing the ERSE-FLuc reporters were created by transfection with ERSE-FLuc pcDNA3.1 by calcium phosphate followed by culturing in geneticin sulfate (G-418, 500 μg/mL). All cells were cultured under typical tissue culture conditions (37°C, 5% CO2).
Measurement of ATF6 activity using the ERSE.Fluc reporter
Request a detailed protocolHEK293T-Rex cells incorporating the ERSE.FLuc reporter were plated at 100 μL/well from suspensions of 200,000 cells/mL in black clear-bottom 96-well plates (Corning) and incubated at 37°C overnight. The following day, cells were treated with 10 µM of compound for 18 hr at 37°C or were pre-treated with the necessary compound as described before addition of compounds for 18 hr at 37°C. The plates were equilibrated to room temperature, then 100 μL of Firefly luciferase assay reagent-1 (Targeting Systems) were added to each well. Luminescence activity was measured 10 min after reagent addition with an EnVision Multilabel Reader (PerkinElmer) using a 100 ms integration time.
SDS-PAGE-based analysis of proteins modified by 147
Request a detailed protocolAffinity-precipitated samples (see below) for SDS-PAGE were normalized for protein concentration using the BCA assay (Thermo Fisher). Protein samples were boiled for 5 min in Laemmli buffer + 100 mM DTT before loading onto a 10% SDS-PAGE gel. The gel was fixed in 40% methanol/10% acetic acid solution. The gel was then stained with 0.1% Coomassie Blue R250 in 10% acetic acid/50% methanol solution. Prominent bands were excised and the proteins were extracted, digested and analyzed by mass spectrometry.
Immunoblotting
Request a detailed protocolFor immunoblotting, cells were lysed in 50 mM Tris buffer, pH 7.5 containing 0.1% TritonX (Fisher Scientific) and supplemented with protease inhibitor cocktail (Roche). Protein lysate concentrations were normalized by BCA assay (Thermo Fisher). Lysates were boiled for 5 min in Laemmli buffer with 100 mM DTT before loading onto SDS-PAGE gels. Proteins were transferred from gel slabs to nitrocellulose and blotted using mouse anti-FLAG M2 antibody (Sigma) or rabbit anti-PDIA4 antibody (Protein Tech) and visualized on the Odyssey Infrared Imaging System (Li-Cor Biosciences).
Affinity precipitation of proteins covalently modified by 147
Request a detailed protocolHEK293T-Rex cells in 10 cm plates were treated for 16 hr with 147 (10 μM), or 147–20 (10 μM), or the combination of 147–20 (10 μM) and 147 (50 μM), or 147–20 (10 μM) in combination with 147–4 (50 μM) at 37°C. Lysates were prepared in radioimmunoprecipitation assay (RIPA) buffer (150 mM NaCl, 50 mM Tris pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, and 0.1% SDS) with fresh protease inhibitor cocktail (Roche, Indianapolis, IN) and centrifuged for 20 min at 10,000 × g. Protein concentrations of supernatants were determined by the BCA assay (Thermo Fisher). For each sample, 100 μg of lysate were reacted with click reagents to give final concentrations as follows: 100 µM of diazo biotin-azide (Click Chemistry Tools, Scottsdale, AZ), 800 µM copper (II) sulfate, 1.6 mM BTTAA ligand (2-(4-((bis((1-tert-butyl-1H-1,2,3-triazol-4-yl)methyl)amino)methyl)−1 H-1,2,3-triazol-1-yl)acetic acid) (Albert Einstein College), and 5 mM sodium ascorbate. The reaction was placed on a shaker at 1000 rpm at 30°C for 2 hr. The proteins were then precipitated from the reaction mixture by adding an equal volume of 3:1 chloroform/methanol. The pellet was washed three times with 1:1 chloroform/methanol. The precipitate was suspended in 500 µL of 6 M urea with 25 mM ammonium bicarbonate and 140 µL of 10% SDS was added to this mixture to help solubilize the protein. 50 µL of high capacity streptavidin beads were washed with PBS and mixed with the protein solution in 6 mL of phosphate-buffered saline (PBS). This suspension was placed on a rotator or a shaker and agitated for 2 hr. The beads were centrifuged and washed five times with PBS and 1% SDS. The protein was eluted from the beads by two washes of 50 mM sodium dithionite in 1% SDS for 1 hr and then precipitated by chloroform/methanol precipitation as described above.
TMT-MuDPIT proteomics-based identification of proteins modified by 147
Request a detailed protocolAir-dried pellets from the affinity precipitation were resuspended in 1% RapiGest SF (Waters) in 100 mM HEPES (pH 8.0). Proteins were reduced with 5 mM tris(2-carboxyethyl)phosphine hydrochloride (Thermo Fisher) for 30 min and alkylated with 10 mM iodoacetamide (Sigma Aldrich, St. Louis, MO) for 30 min at ambient temperature and protected from light. Proteins were digested for 18 hr at 37°C with 2 μg trypsin (Promega). After digestion, 20 μg of peptides from each sample were reacted for 1 hr with the appropriate TMT-NHS isobaric reagent (ThermoFisher) in 40% (v/v) anhydrous acetonitrile and quenched with 0.4% NH4HCO3 for 1 hr. Samples with different TMT labels were pooled and acidified with 5% formic acid. Acetonitrile was evaporated on a SpeedVac and debris was removed by centrifugation for 30 min at 18,000 × g. MuDPIT (Multi-Dimensional Protein Identification Technology) microcolumns were prepared as described previously (Ryno et al., 2014). LCMS/MS analysis was performed using a Q Exactive mass spectrometer equipped with an EASY nLC 1000 (Thermo Fisher). MuDPIT experiments were performed by 5 min sequential injections of 0, 20, 50, 80, 100% buffer C (500 mM ammonium acetate in buffer A) and a final step of 90% buffer C/10% buffer B (20% water, 80% acetonitrile, 0.1% fomic acid, v/v/v) and each step followed by a gradient from buffer A (95% water, 5% acetonitrile, 0.1% formic acid) to buffer B. Electrospray ionization was performed directly from the analytical column by applying a voltage of 2.5 kV with an inlet capillary temperature of 275°C. Data-dependent acquisition of MS/MS spectra was performed with the following settings: eluted peptides were scanned from 400 to 1800 m/z with a resolution of 30,000 and the mass spectrometer in a data dependent acquisition mode. The top ten peaks for each full scan were fragmented by HCD using a normalized collision energy of 30%, a 100 ms activation time, a resolution of 7500, and scanned from 100 to 1800 m/z. Dynamic exclusion parameters were 1 repeat count, 30 ms repeat duration, 500 exclusion list size, 120 s exclusion duration, and exclusion width between 0.51 and 1.51. Peptide identification and protein quantification was performed using the Integrated Proteomics Pipeline Suite (IP2, Integrated Proteomics Applications, Inc., San Diego, CA) as described previously (Ryno et al., 2014).
Measurement of 147 concentration in ER versus cytosol
Request a detailed protocolThe concentration of 147 in the ER was determined by treating HEK293T cells for 15 min with 10 µM 147. The cells were scraped off the plate with 1 mM EDTA in TBS and the cellular pellet was lysed with 0.1% digitonin in HEPES (pH 7.5), 100 mM NaCl. The supernatant (cytosol) was analyzed for enrichment of cytosolic proteins (HSC70) and the pellet (ER) was analyzed for ER proteins (ERdj3) by immunoblot. To determine the concentrations of 147 in the ER and cytosol, each enriched fraction was subject to a chloroform: methanol extraction by adding 4× volume of both chloroform and methanol to precipitate the proteins. The organic phase was collected, concentrated, and resuspended in methanol. 147 concentrations were determined using a Waters TQ-XS triple quad mass spectrometer by injecting 5 µL of sample onto a Waters HSS T3 C18 column (2.1 × 10 mm, 1.8 µM) with a flow rate of 0.3 mL per minute and comparing the observed peak areas to a calibration curve.
Measurement of ATF6 oxidation state
Request a detailed protocolThe oxidation state of ATF6 in HeLa cells treated with 147 was analyzed by gel-shift essentially as previously described (Nadanaka et al., 2006). Briefly, cells were infected with adenovirus encoding FLAG-tagged full length inactive ATF6 [ATF6(1–670)] and subsequently treated with respective treatments for 6 hr. Cells were lysed in low-stringency lysis buffer comprising 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, 1% Triton X-100, protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN) and phosphatase inhibitor cocktail (Roche Diagnostics) and 20 µM 4-Acetamido-4'-Maleimidylstilbene-2,2'-Disulfonic Acid, Disodium Salt (AMS) (Thermo Fisher, cat# A485). AMS binds covalently to reduced thiols, typically on cysteine residues, and increases their molecular mass in SDS-PAGE. Thus, proteins that exhibit an upward shift when analyzed under non-reducing conditions compared to reducing are considered to have reduced thiols.
shRNA knockdown of PDIs and subsequent measurement of ATF6 activation by 147
Request a detailed protocolHEK293T-Rex (Invitrogen) cells were cultured in complete DMEM (CellGro) supplemented with 10% FBS (CellGro) and penicillin/streptomycin (CellGro). Lentiviruses encoding a mix of shRNAs that are either non-silencing or directed against PDIA1, PDIA3, PDIA4, PDIA5, or PDIA6 were transduced into HEK293T-Rex with 1–5 mL of virus in media containing 5 mg/mL polybrene (see Supplementary File 1 for lentivirus production methods). Stable cell lines were selected by culturing in puromycin (5 μg/mL) before characterization. The expression of the ATF6 target gene BiP in cells treated with 147 (10 μM) or thapsigargin (0.5 μM) was then measured by qPCR as described previously.
Quantitative RT-PCR
Request a detailed protocolThe relative mRNA expression levels of target genes were measured using quantitative RT-PCR (see Supplementary File 1 for RT-PCR details and sequences of primers used).
Statistical methods
Request a detailed protocolThe data in Figure 2 and Figure 5 were tested for significance using ANOVA with a post-hoc Dunnett’s test. The PANTHER overrepresentation test has been described elsewhere (Mi et al., 2017) and was used as implemented by the PANTHER website (http://pantherdb.org/) using Fisher’s exact test with multiple testing correction.
Synthesis and characterization
Request a detailed protocolDetails of the synthesis and characterization of 147 and its derivatives are reported in Supplementary File 1.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files.
References
-
Increasing the efficacy of bioorthogonal click reactions for bioconjugation: a comparative studyAngewandte Chemie International Edition 50:8051–8056.https://doi.org/10.1002/anie.201101817
-
Quinone methide bioactivation pathway: contribution to toxicity and/or cytoprotection?Current Organic Chemistry 18:61–69.https://doi.org/10.2174/138527281801140121123046
-
Exploiting endogenous cellular process to generate quinone methides in vivoCurrent Organic Chemistry 1818:70–85.
-
Differential inhibition and inactivation of human CYP1 enzymes by trans-resveratrol: evidence for mechanism-based inactivation of CYP1A2The Journal of Pharmacology and Experimental Therapeutics 299:874–882.
-
Oxidation promotes insertion of the CLIC1 chloride intracellular channel into the membraneEuropean Biophysics Journal 39:129–138.https://doi.org/10.1007/s00249-009-0450-0
-
Proteostasis control by the unfolded protein responseNature Cell Biology 17:829–838.https://doi.org/10.1038/ncb3184
-
Endoplasmic reticulum stress-activated transcription factor ATF6α requires the disulfide isomerase PDIA5 to modulate chemoresistanceMolecular and Cellular Biology 34:1839–1849.https://doi.org/10.1128/MCB.01484-13
-
Cytochrome P450: Structure, Mechanism, and Biochemistry523–785, Human cytochrome P450 enzymes, Cytochrome P450: Structure, Mechanism, and Biochemistry, Switzerland, Springer International.
-
Pd(PhCN)(2)Cl(2)/P(t-Bu)(3): a versatile catalyst for Sonogashira reactions of aryl bromides at room temperatureOrganic Letters 2:1729–1731.
-
XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein responseMolecular and Cellular Biology 23:7448–7459.https://doi.org/10.1128/MCB.23.21.7448-7459.2003
-
The intracellular chloride ion channel protein CLIC1 undergoes a redox-controlled structural transitionJournal of Biological Chemistry 279:9298–9305.https://doi.org/10.1074/jbc.M308444200
-
ERp29, an unusual redox-inactive member of the thioredoxin familyAntioxidants & Redox Signaling 8:325–337.https://doi.org/10.1089/ars.2006.8.325
-
"Inverse Drug Discovery" Strategy To Identify Proteins That Are Targeted by Latent Electrophiles As Exemplified by Aryl FluorosulfatesJournal of the American Chemical Society 140:200–210.https://doi.org/10.1021/jacs.7b08366
-
Role of disulfide bridges formed in the luminal domain of ATF6 in sensing endoplasmic reticulum stressMolecular and Cellular Biology 27:1027–1043.https://doi.org/10.1128/MCB.00408-06
-
Reduction of disulfide bridges in the lumenal domain of ATF6 in response to glucose starvationCell Structure and Function 31:127–134.https://doi.org/10.1247/csf.06024
-
VDAC2-specific cellular functions and the underlying structureBiochimica Et Biophysica Acta (BBA) - Molecular Cell Research 1863:2503–2514.https://doi.org/10.1016/j.bbamcr.2016.04.020
-
How many lives does CLIMP-63 have?Biochemical Society Transactions 43:222–228.https://doi.org/10.1042/BST20140272
-
Metabolism and cytotoxicity of eugenol in isolated rat hepatocytesChemico-Biological Interactions 77:137–147.https://doi.org/10.1016/0009-2797(91)90069-J
-
o-Methoxy-4-alkylphenols that form quinone methides of intermediate reactivity are the most toxic in rat liver slicesChemical Research in Toxicology 8:323–327.https://doi.org/10.1021/tx00045a001
-
Biological and toxicological consequences of quinone methide formationChemico-Biological Interactions 86:129–162.https://doi.org/10.1016/0009-2797(93)90117-H
-
Mutation of ATF6 causes autosomal recessive achromatopsiaHuman Genetics 134:941–950.https://doi.org/10.1007/s00439-015-1571-4
-
Mechanism, specificity, and physiology of signal peptide peptidase (SPP) and SPP-like proteasesBiochimica Et Biophysica Acta (BBA) - Biomembranes 1828:2828–2839.https://doi.org/10.1016/j.bbamem.2013.03.033
-
Bioactivation of 4-methylphenol (p-cresol) via cytochrome P450-mediated aromatic oxidation in human liver microsomesDrug Metabolism and Disposition: The Biological Fate of Chemicals 33:1867–1876.https://doi.org/10.1124/dmd.105.006387
-
Identification of the cis-acting endoplasmic reticulum stress response element responsible for transcriptional induction of mammalian glucose-regulated proteins. involvement of basic leucine zipper transcription factorsThe Journal of Biological Chemistry 273:33741–33749.
Article and author information
Author details
Funding
National Institutes of Health (AG046495)
- R Luke Wiseman
- Jeffery W Kelly
National Institutes of Health (DK107604)
- R Luke Wiseman
National Institutes of Health (HL075573)
- Chris Glembotski
National Institutes of Health (HL104535)
- Chris Glembotski
National Institutes of Health (HL085577)
- Chris Glembotski
Leukemia and Lymphoma Society (5439-16)
- Lars Plate
American Heart Association (17PRES33670796)
- Erik A Blackwood
San Diego State University (Rees-Stealy Research Foundation)
- Erik A Blackwood
San Diego State University (Heart Institute Inamori Foundation)
- Erik A Blackwood
Achievement Rewards for College Scientists Foundation (San Diego chapter)
- Erik A Blackwood
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Copyright
© 2018, Paxman et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,626
- views
-
- 728
- downloads
-
- 116
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 116
- citations for umbrella DOI https://doi.org/10.7554/eLife.37168
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Pharmacologic ATF6 activating compounds are metabolically activated to selectively modify endoplasmic reticulum proteins
eLife 7:e37168.
https://doi.org/10.7554/eLife.37168




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}