Rap2 and TNIK control Plexin-dependent tiled synaptic innervation in C. elegans
- The University of British Columbia, Canada
- National Institute for Physiological Sciences, Japan
Abstract
During development, neurons form synapses with their fate-determined targets. While we begin to elucidate the mechanisms by which extracellular ligand-receptor interactions enhance synapse specificity by inhibiting synaptogenesis, our knowledge about their intracellular mechanisms remains limited. Here we show that Rap2 GTPase (rap-2) and its effector, TNIK (mig-15), act genetically downstream of Plexin (plx-1) to restrict presynaptic assembly and to form tiled synaptic innervation in C. elegans. Both constitutively GTP- and GDP-forms of rap-2 mutants exhibit synaptic tiling defects as plx-1 mutants, suggesting that cycling of the RAP-2 nucleotide state is critical for synapse inhibition. Consistently, PLX-1 suppresses local RAP-2 activity. Excessive ectopic synapse formation in mig-15 mutants causes a severe synaptic tiling defect. Conversely, overexpression of mig-15 strongly inhibited synapse formation, suggesting that mig-15 is a negative regulator of synapse formation. These results reveal that subcellular regulation of small GTPase activity by Plexin shapes proper synapse patterning in vivo.
https://doi.org/10.7554/eLife.38801.001eLife digest
Genes do more than just direct the color of our hair or eyes. They produce proteins that are involved in almost every process in the body. In humans, the majority of active genes can be found in the brain, where they help it to develop and work properly – effectively controlling how we move and behave.
The brain’s functional units, the nerve cells or neurons, communicate with each other by releasing messenger molecules in the gap between them, the synapse. These molecules are then picked up from specific receptor proteins of the receiving neuron.
In the nervous system, neurons only form synapses with the cells they need to connect with, even though they are surrounded by many more cells. This implies that they use specific mechanisms to stop neurons from forming synapses with incorrect target cells. This is important, because if too many synapses were present or if synapses formed with incorrect target cells, it would compromise the information flow in the nervous system. This would ultimately lead to various neurological conditions, including Autism Spectrum Disorder.
In 2013, researchers found that in the roundworm Caenorhabditis elegans, a receptor protein called Plexin, is located at the surface of the neurons and can inhibit the formation of nearby synapses. Now, Chen et al. – including one author involved in the previous research – wanted to find out what genes Plexin manipulates when it stops synapses from growing. Knowing what each of those genes does can help us understand how neurons can inhibit synapses.
The results revealed that Plexin appears to regulate two genes, Rap2 and TNIK. Plexin reduced the activity of Rap2 in the neuron that released the messenger, which hindered the formation of synapses. The gene TNIK and its protein on the other hand, have the ability to modify other proteins and could so inhibit the growth of synapses. When TNIK was experimentally removed, the number of synapses increased, but when its activity was increased, the number of synapses was strongly reduced.
These findings could help scientists understand how mutations in Rap2 or TNIK can lead to various neurological conditions. A next step will be to test if these genes also affect the formation of synapses in other species such as mice, which have a more complex nervous system that is structurally and functionally more similar to that of humans.
https://doi.org/10.7554/eLife.38801.002Introduction
During nervous system development, various instructive and repulsive signaling cues cooperatively direct neurons to form chemical synapses with their appropriate targets. Studies have identified some molecules and elucidated their downstream mechanisms that instruct synaptogenesis such as FGF, Ephrin/Eph, Ig-family of cell adhesion molecules (IgCAMs) and synaptic cell adhesion molecules (SynCAMs) (Shen and Bargmann, 2003; Shen et al., 2004; Dabrowski and Umemori, 2016; Dabrowski et al., 2015; Feng et al., 2000; Kayser et al., 2008; Terauchi et al., 2010; Yamagata et al., 2003). Several axon guidance cues and their receptors also play critical roles to inhibit synapse formation (Inaki et al., 2007; Klassen and Shen, 2007; Poon et al., 2008). Semaphorins (Sema) and their receptors, Plexins, are two conserved families of molecules that have a well-established function to repel axons during development (Kolodkin et al., 1993, 1992; Negishi et al., 2005a; Tran et al., 2007) and play prominent roles contributing to immune system, cardiovascular development and cancer regulation (Epstein et al., 2015; Neufeld et al., 2005; Takamatsu and Kumanogoh, 2012).
In addition to its function as a long-range axon guidance cue during neuronal development, Sema/Plexin signaling plays a critical role as a negative regulator of synapse formation. The role of Sema/Plexin signaling to inhibit synapse formation was first observed in Drosophila, where ectopic expression of Sema2a causes elimination of specific neuromuscular junctions (Matthes et al., 1995). In mammals, Sema3E/PlexinD1 specifies sensory-motor connections (Pecho-Vrieseling et al., 2009). Secreted Sema3F locally inhibits spine development through its receptors PlexinA3 and Neuropilin-2 in hippocampal granule cells (Tran et al., 2009). Sema5A/PlexinA2 signaling inhibits excitatory synapse formation in dentate granule cells (Duan et al., 2014). Sema5B diminishes synaptic connections in cultured hippocampal neurons (O'Connor et al., 2009). However, little is known about the intracellular mechanisms through which Sema/Plexin signaling inhibits synapse formation.
The cytoplasmic domain of Plexin contains a GAP (GTPase-activating protein) domain that inactivates small GTPases (Oinuma et al., 2004; Rohm et al., 2000). Upon activation by Semaphorins, Plexins repel axon outgrowth by inhibiting R-Ras (Negishi et al., 2005b; Hota and Buck, 2012; Tasaka et al., 2012). Recent biochemical and structural analyses demonstrated that the GAP domain of mammalian PlexinA3 is specific for Rap GTPases, which belong to the Ras family of GTPases and regulate the actin cytoskeleton (Wang et al., 2012, 2013). PlexinA3 dimerization by Semaphorin activates its GAP domain, thereby inhibiting Rap1 from inducing neurite retraction. Drosophila PlexA and zebrafish PlexinA1 promote remodeling of epithelial cells by inhibiting Rap1 GTPase during wound healing (Yoo et al., 2016). Another Rap GTPase, Rap2, can inhibit neurite outgrowth (Kawabe et al., 2010). Similar to Sema/Plexin signaling, Rap GTPases regulate synapse formation and function. Rap2 negatively regulate spine number in cultured hippocampal neurons (Fu et al., 2007). Rap1 and Rap2 regulate synaptic activity by removing AMPA receptors from spines during long-term depression and depotentiation, respectively (Zhu et al., 2002, Zhu et al., 2005). While the GAP domain of Plexin is critical to inhibit synapse formation (Duan et al., 2014; Mizumoto and Shen, 2013a), we still do not know whether Plexin regulates synapse patterning via Rap GTPases at presynaptic sites.
In Caenorhabditis elegans, Sema/Plexin signaling functions in vulva formation and male ray development (Dalpé et al., 2005, 2004; Fujii et al., 2002; Ikegami et al., 2004; Liu et al., 2005; Nakao et al., 2007; Nukazuka et al., 2008, 2011). Using this model system, we previously reported that Sema/Plexin signaling in the nervous system mediates a critical inter-axonal interaction for the tiled synaptic innervation of two DA-class cholinergic motor neurons (DA8 and DA9) (Mizumoto and Shen, 2013a). Cell bodies of nine DA neurons in C. elegans reside in the ventral nerve cord, sending dendrites ventrally and axons dorsally to form en passant synapses onto the dorsal body wall muscles. Even though axons of DA neurons show significant overlap, each motor neuron forms synapses onto muscles within specific sub-axonal domains, which do not overlap with those from neighboring DA neurons. This unique synaptic innervation creates tiled synaptic patterns along the nerve cord (White et al., 1986).
Tiled synaptic innervation occurs within most motor neuron classes and may contribute to the sinusoidal locomotion pattern of C. elegans (White et al., 1986). Using a combination of two fluorescent proteins (GFP and mCherry) fused with the presynaptic vesicle protein, RAB-3, and two tissue specific promoters (Figure 1) (Mizumoto and Shen, 2013a), we can visualize this synaptic tiling between DA8 and DA9 neurons. We reported that PLX-1 localizes at the anterior edge of the DA9 synaptic domain in axon-axon interactions in a Semaphorin-dependent manner, where it locally inhibits formation of the presynaptic specialization via its GAP domain. Loss of Semas or plx-1 causes anterior expansion of DA9 synaptic domain and posterior expansion of DA8 synaptic domain. This result indicates loss of inter-axonal interactions between DA8 and DA9 neurons. Consistently, Tran et al., also observed excess dendritic spine formation, specifically within the region close to the cell body, in the plexin knockout mouse (Tran et al., 2009). These findings suggest a conserved mechanism by which Sema/Plexin locally inhibits synapse formation.
Figure 1 with 3 supplements see all
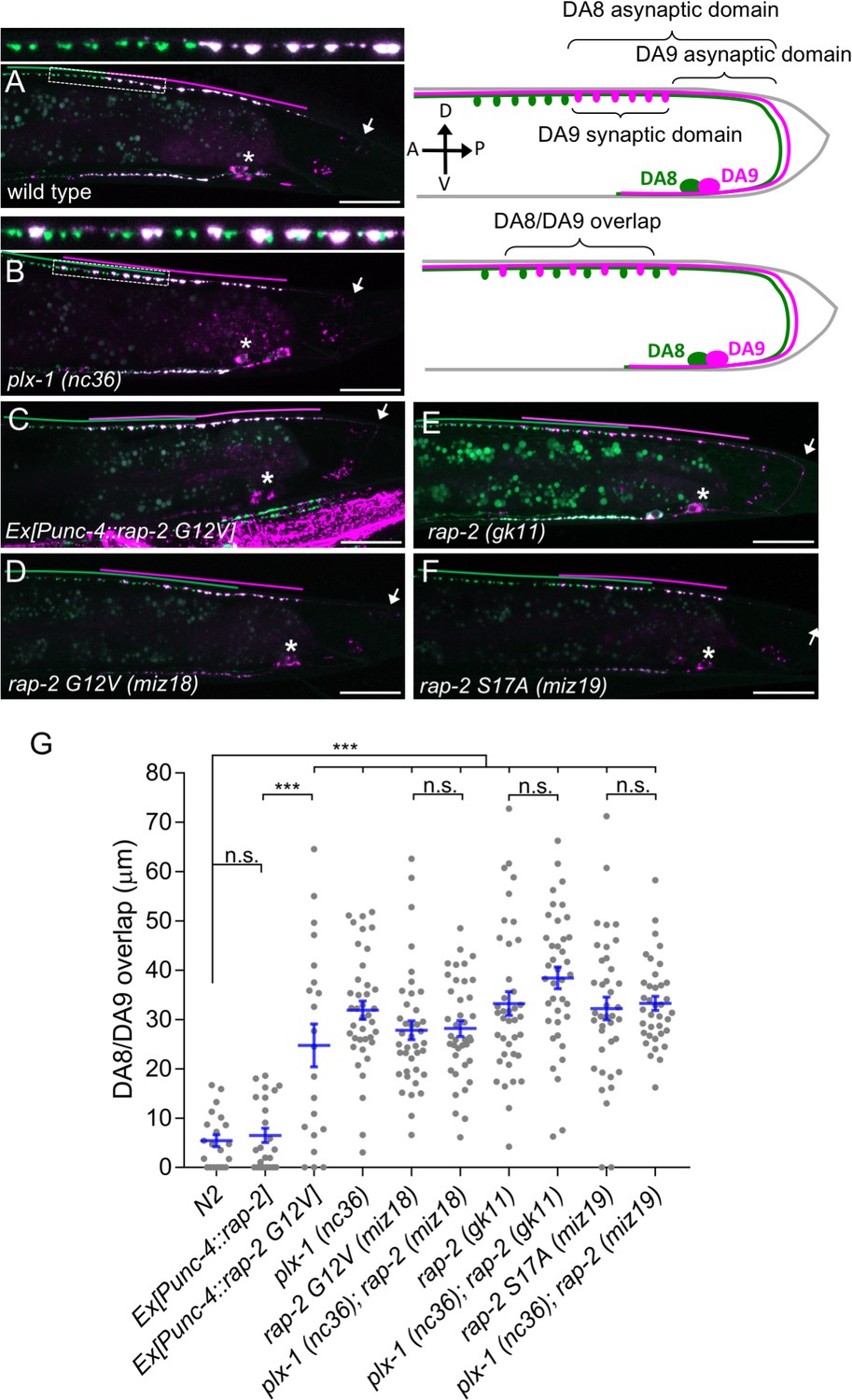
Gain- and loss-of-function rap-2 mutants show synaptic tiling defects.
(A and B) Representative image of synaptic tiling of wild type (A) and plx-1 mutant (B) animals. Images show wyIs446 marker to label synapses in DA8 (GFP::RAB-3) and DA9 (GFP::RAB-3+mCherry::RAB-3). Dotted box represents the magnified images from A and B of the synaptic tiling border. Schematics of DA8 (green) and DA9 (magenta) neurons with parameters for analysis shown on the right. (C–F) Representative images of wyIs446 strains with the following genotypes: rap-2(G12V) overexpression in DA neurons (C), rap-2 G12V (miz18) (D), rap-2 null (gk11) (E) and rap-2 S17A (miz19) (F). Synaptic domains from DA8 and DA9 are highlighted with green and magenta lines, respectively. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (G) Quantification of overlap between DA8 and DA9 synaptic domains. Each dot represents a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001.
We previously reported that let-60/KRas gain-of-function mutants showed very mild synaptic tiling defects. Since mammalian Plexin acts as a RapGAP, we hypothesized that Rap GTPase is the major downstream effector of PLX-1 to regulate synaptic tiling of DA neurons. Here, we report that rap-2, a C. elegans ortholog of human Rap2A, and its effector kinase mig-15 (TNIK: Traf2- and Nck-interacting kinase) act genetically downstream of plx-1 to regulate synaptic tiling. PLX-1 delineates the border of synaptic tiling by locally inhibiting RAP-2 along the DA9 axon. We also discovered an unexpected role for mig-15 in inhibiting synapse formation. Our results reveal the mechanism underlying Plexin signaling to form fine synaptic map connectivity.
Results
rap-2 functions downstream of plx-1 to regulate synaptic tiling
Three Rap genes exist in the C. elegans genome (rap-1, rap-2 and rap-3). To delineate which Rap GTPase functions downstream of PLX-1 in synaptic tiling between DA8 and DA9 neurons, we first examined the expression patterns of all three rap genes (Figure 1—figure supplement 1). Among them, only rap-2, an ortholog of mammalian Rap2a, was expressed in motor neurons including DA8 and DA9, while rap-1 and rap-3 were not expressed in these cells (Figure 1—figure supplement 1). In wild type animals, synaptic domains of DA8 and DA9 neurons did not show significant overlap, creating tiled synaptic innervation (Figure 1A and G). In the plx-1(nc36) null mutant, synaptic domains of DA8 and DA9 expanded posteriorly and anteriorly, respectively. As a result, synaptic domains of these neurons overlapped significantly (Figure 1B and G).
Since the intracellular domain of Plexin contains a RapGAP domain, we hypothesized that RAP-2 preferentially exists in a GTP-bound form in the plx-1 mutants. The G12V mutant is widely used as a constitutively GTP-form of small GTPases including mammalian Rap2A and C. elegans RAP-1 (Kawabe et al., 2010; Pellis-van Berkel et al., 2005). Expression of a constitutively GTP-bound form of rap-2(G12V) under the A-type neuron specific promoter, Punc-4, elicited a similar synaptic tiling defect as plx-1 mutants (Figure 1C and G). Expression of wild type rap-2 under the unc-4 promoter did not affect the synaptic tiling pattern, suggesting that G12V mutation but not over-expression of rap-2 caused the synaptic tiling defect (Figure 1G). We then generated rap-2(G12V) mutants using CRISPR/Cas9 genome editing. We observed the same synaptic tiling defects in three independent rap-2(G12V) mutant alleles (miz16, miz17 and miz18) as in plx-1 mutants (Figure 1D and G and Figure 1—figure supplement 2). We found a comparable level of gene expression among all three rap-2(G12V) mutants to wild type rap-2 using RT-qPCR (Figure 1—figure supplement 2). These results confirm that the rap-2(G12V) mutation itself, not changes in gene expression, underlie the synaptic tiling defect in rap-2(G12V) mutants.
Surprisingly, the null mutant of rap-2(gk11) also showed the same synaptic tiling defect, as did the constitutively GTP-form of rap-2(G12V) mutants (Figure 1E and G). This result suggests that synaptic tiling requires both the GTP- and GDP-bound forms of RAP-2. Indeed, the constitutively GDP-bound form of rap-2 mutants (S17A: miz19, miz20) showed the identical synaptic tiling defect as rap-2(G12V) and rap-2(gk11) mutants (Figure 1F and G and Figure 1—figure supplement 2). These results suggest that the cycling between GTP- and GDP-forms of RAP-2 is critical to regulate the spatial patterning of synapses.
Similar to plx-1 mutants, the synaptic tiling defect in all rap-2 mutants is caused by both the posterior expansion of DA8 synaptic domain and the anterior expansion of the DA9 synaptic domain (Figure 1—figure supplement 2). However, none of rap-2(G12V), rap-2(S17A) and rap-2(gk11) mutants enhanced or suppressed the synaptic tiling defect in plx-1 mutants (Figure 1G). These results suggest that plx-1 and rap-2 function in the same genetic pathway. Consistent with the expression patterns of rap-1 and rap-3, neither rap-1(pk2082) nor rap-3(gk3975) null mutants showed significant synaptic tiling defects by themselves and did not enhance the synaptic tiling defect in rap-2(gk11) null mutants (Figure 1—figure supplement 1). All mCherry::RAB-3 puncta co-localized with active zone markers, CLA-1 (Xuan et al., 2017) and UNC-10/Rim (Wu et al., 2013) in the DA9 neurons of plx-1(nc36) and rap-2(gk11) mutants, suggesting that RAB-3 puncta represent bona fide synapses (Figure 1—figure supplement 3). Taken together, these data indicate that plx-1 and rap-2 act in the same genetic pathway for synaptic tiling in DA neurons.
RAP-2 functions cell autonomously in DA neurons
We next determined the cellular location for rap-2 function. Since rap-2(gk11) null mutants showed a synaptic tiling defect, we conducted tissue specific rescue experiments using tissue-specific promoters as previously described (Mizumoto and Shen, 2013a). Expression of rap-2 in the post-synaptic body wall muscle cells under the hlh-1 promoter or in another class of cholinergic motor neurons in the dorsal nerve cord (DB neurons) under the truncated unc-129 promoter did not rescue the synaptic tiling defect in rap-2(gk11) animals (Figure 2A, B and G). However, DA neuron-specific expression using the unc-4c promoter strongly rescued the synaptic tiling defect (Figure 2C and G). DA9-specific expression of rap-2 under the mig-13 promoter partially rescued the synaptic tiling defect (Figure 2E and G). DA9-specific expression of rap-2 rescued the phenotype of anterior expansion of the DA9 synaptic domain but not the posterior expansion of DA8 synaptic domain (Figure 2H and I). These results suggest that rap-2 regulates synapse patterning in a cell-autonomous manner. In contrast to the DA9-specific rescue experiment in rap-2 mutants, DA9-specific expression of plx-1 cDNA was sufficient to rescue synaptic defects in both DA9 and DA8 (Mizumoto and Shen, 2013a) (see Discussion).
Figure 2
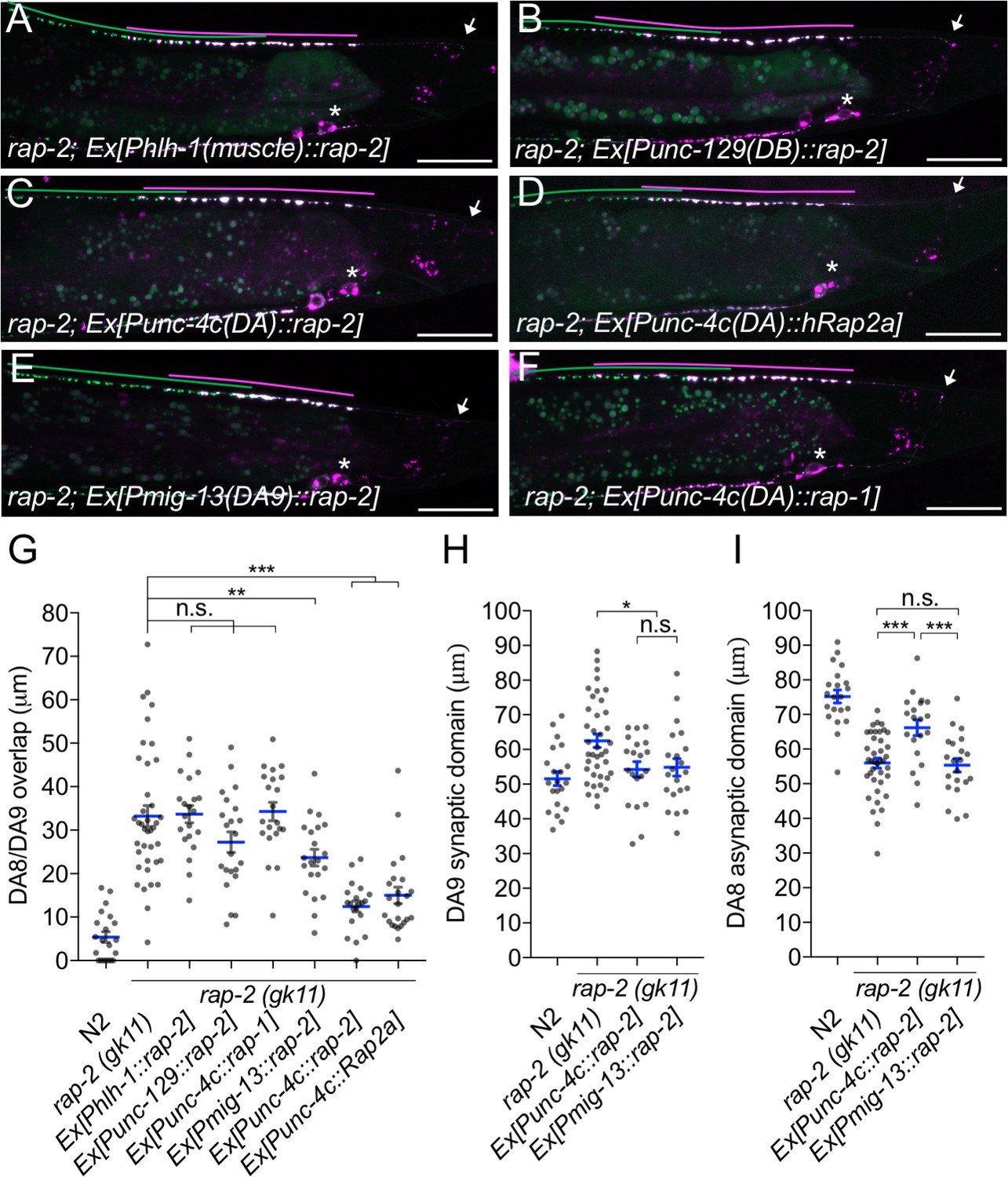
rap-2 functions in DA neurons.
(A–F) Representative images of rap-2; wyIs446 animals expressing Phlh-1::rap-2 (A), Punc-129::rap-2 (B), Punc-4c::rap-2 (C), Punc-4c::Rap2a (human) (D), Pmig-13::rap-2 (E) and Punc-4c;;rap-1 (F). Synaptic domains of DA8 and DA9 are highlighted with green and magenta lines, respectively. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (G–I) Quantification of DA8/DA9 overlap (G), DA9 synaptic domain (H) and DA8 asynaptic domain (I). Each dot represents a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001; **p<0.01; *p<0.05.
We also observed that expression of human Rap2a in DA neurons rescued the synaptic tiling defect of rap-2 mutants, suggesting the function of rap-2 in synapse patterning is conserved across species (Figure 2D and G). Previous work suggested a partial functional redundancy between rap-1 and rap-2 in C. elegans (Pellis-van Berkel et al., 2005). However, we found that rap-1 expression in DA neurons did not rescue the synaptic tiling defect of rap-2 mutants, suggesting functional diversity between rap-1 and rap-2 (Figure 2F and G). Taken together, we conclude that rap-2 functions cell autonomously in DA neurons to regulate synaptic tiling.
RAP-2 activity is spatially regulated by PLX-1
Previously, we demonstrated that PLX-1::GFP is localized at the anterior edge of the DA9 synaptic domain, where it negatively regulates synapse formation through its cytoplasmic GAP domain (Figure 3A and E) (Mizumoto and Shen, 2013a). In the rap-2(gk11) mutant background, we observed no change in PLX-1::GFP localization but did observe ectopic synapses in the axonal region anterior to the PLX-1::GFP domain (Figure 3B and F). This result is consistent with our hypothesis that rap-2 acts downstream of plx-1 to regulate synaptic tiling. Together with our finding that synaptic tiling requires both GTP- and GDP-bound forms of RAP-2, we speculate that PLX-1 acting at the anterior edge of the DA9 synaptic domain regulates the spatial activity of RAP-2 along the axon.
Figure 3
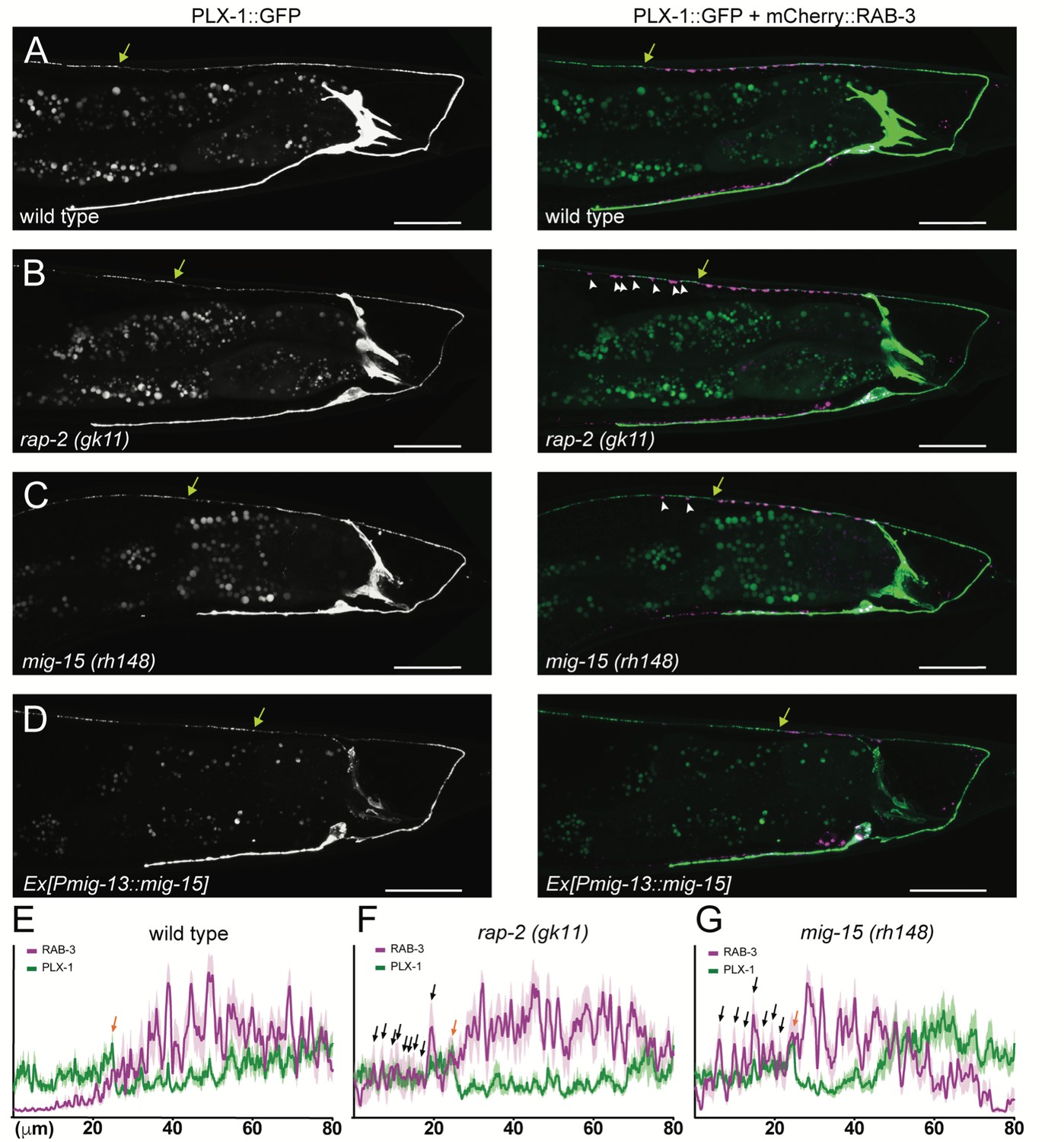
PLX-1::GFP localization is not affected in rap-2 and mig-15 mutants.
(A–D) Representative image of PLX-1::GFP alone (top) and PLX-1::GFP with mCherry::RAB-3 (middle) labeled with wyIs320 in DA9 of wildtype (A), rap-2 (gk11) (B) and mig-15(rh148) (C). Bracket indicates the PLX-1::GFP patch localized at the anterior edge of the DA9 synaptic domain. Arrowheads indicate ectopic synapses formed anterior to the PLX-1::GFP patch. Scale bars: 20 μm. (E–G) Quantification of the normalized mCherry::RAB-3 signal (magenta) and PLX-1::GFP signal (green) in the dorsal axon of DA9 in wildtype (E), rap-2 (gk11) (F) and mig-15(rh148) (G). Animal were aligned according to the PLX-1::GFP patch at the anterior edge of the DA9 synaptic domain (orange arrow). Light colors indicate SEM.
We then sought to determine the spatial distribution of GTP-RAP-2 in DA9 axon. We conducted Fluorescence Lifetime Imaging Microscopy (FLIM)-based FRET (Förster Resonance Energy Transfer) measurements using EGFP-Rap2A (human) and mRFP-RalGDS(RBD: Ras Binding Domain)-mRFP (Yasuda et al., 2006). As RalGDS-RBD specifically binds to GTP-Rap2 but not GDP-Rap2 (Ohba et al., 2000), FRET from EGFP-Rap2A to mRFP-RalGDS(RBD)-mRFP can be used as a readout of Rap2 activity. We detected FRET signal as a change of GFP fluorescence lifetime (Figure 4A). In HeLa cells, we observed a shorter lifetime of constitutively bound GTP construct EGFP-Rap2A(G12V) compared to GDP-bound EGFP-Rap2A(S17A), indicating that the FRET sensor can detect the nucleotide state of Rap2A (Figure 4B and C). Due to the low expression of C. elegans RAP-2 constructs in HeLa cells, we were not able to test whether the mammalian FRET sensor can detect C. elegans RAP-2 activity (data not shown).
Figure 4
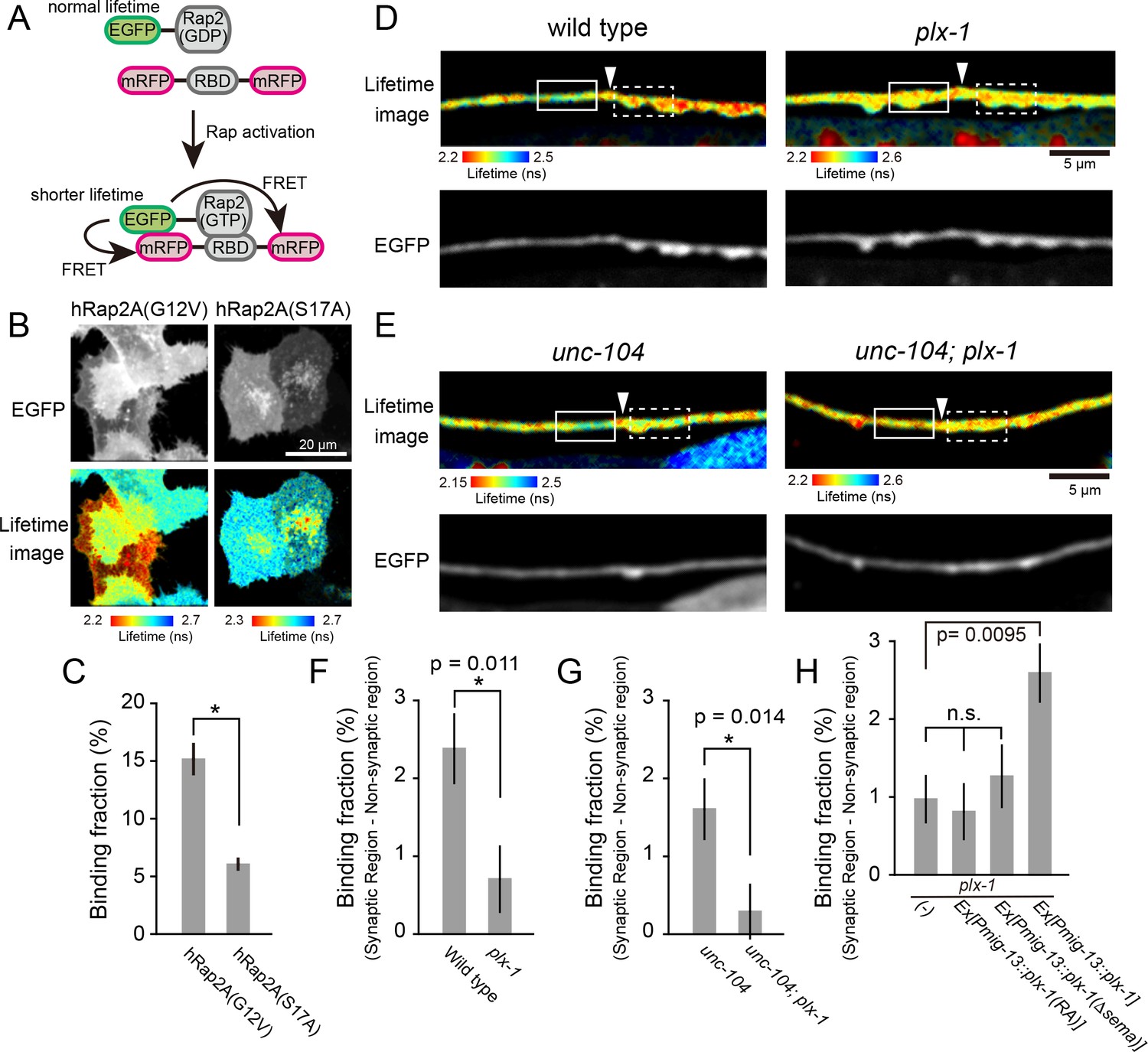
PLX-1 locally inhibits Rap2 activity in DA9.
(A) Schematic of Rap2 FRET sensor system. Binding of mRFP- RalGDS(RBD)-mRFP to EGFP-Rap2 induces FRET from EGFP to mRFP, leading to decreased EGFP fluorescence lifetime. (B) Representative fluorescence (top) and fluorescence lifetime images (bottom) of HeLa cells expressing Rap2 FRET sensor (mRFP-RalGDS(RBD)-mRFP) with either constitutively GTP(G12V)- or GDP(S17A)- forms of human Rap2A. (C) Quantification of the binding fraction of Rap2 mutants in HeLa cells. Binding fractions measured from fluorescence decay curves of individual cells (G12V, n = 25; S17A, n = 25), as described previously (Murakoshi et al., 2011). Data shown as mean ± SEM. Asterisks denote statistical significance (p<0.05, student’s t-test). (D) Representative images of fluorescence lifetime (top) and fluorescence (bottom) of mizIs19 in wild type (left) and plx-1 mutants (right). White arrowheads indicate the position of the putative synaptic tiling border, as judged by the slight dorsal shift of the DA9 axon, as reported previously (Mizumoto and Shen, 2013a). (E) Representative images of fluorescence lifetime (top) and fluorescence (bottom) of mizIs19 in unc-104 (left) and unc-104; plx-1 double mutants (right). (F) Quantification of the difference in binding fraction of GTP-Rap2a and RalGDS(RBD) between synaptic region (dotted boxes in D) and anterior asynaptic region (solid boxes in D). The binding fraction measured in synaptic region (dotted rectangle) was subtracted by that in non-synaptic region (solid rectangle). Five micrometers along the axon line from the synaptic tiling border were used for quantification. Data presented as mean ± SEM (wild type, n = 22; plx-1, n = 17). (G) Quantification of the difference in binding fraction of GTP-Rap2A and RalGDS(RBD) between synaptic region (dotted boxes in E) and anterior asynaptic region (solid boxes in E). The binding fraction measured in synaptic region (dotted rectangle) subtracted from that in non-synaptic region (solid rectangle). Five micrometers along the axon line from the synaptic tiling border were used for quantification. Data are presented as mean ± SEM (unc-104, n = 21; unc-104; plx-1, n = 23). (H) Quantification of the difference in binding fraction of GTP-Rap2a and RalGDS(RBD) between synaptic region and anterior asynaptic region in plx-1 mutants and plx-1 mutants expressing rescuing constructs. Five micrometers along the axon line from the synaptic tiling border were used for quantification. Data are presented as mean ± SEM (no array, n = 35; Ex[Pmig-13::plx-1(RA)], n = 39; Ex[Pmig-13::plx-1(Δsema)], n = 41; Ex[Pmig-13::plx-1], n = 51).
We then expressed EGFP-Rap2A and mRFP-RalGDS(RBD)-mRFP FRET sensors in DA9 neurons in C. elegans. As human Rap2a rescued the synaptic tiling defect of rap-2(gk11) mutants (Figure 2G), we reasoned that the activity pattern of human Rap2A should recapitulate that of endogenous RAP-2. We indeed observed lower Rap2A activity at the anterior edge of the DA9 synaptic domain compared to within the synaptic domain (Figure 4D and F). This observation is consistent with the localization of PLX-1::GFP at the anterior edge of DA9 synaptic domain (Figure 3A) (Mizumoto and Shen, 2013a). Local inhibition of Rap2a activity was strongly diminished in the plx-1 mutant background (Figure 4D and F). Higher Rap2 activity in the synaptic region could simply indicate the presence of synapses within the synaptic domain, rather than Rap2 inactivation by Plexin at the anterior edge of the synaptic domain. To exclude this possibility, we examined Rap2 activity in unc-104/Kif1A mutants, which show no synapses are formed in DA9 axon (Ou et al., 2010). We showed previously that PLX-1::GFP localization to the synaptic tiling border was independent of synapses, since it was unaffected in unc-104/Kif1A mutants (Mizumoto and Shen, 2013a). In unc-104 mutants, we observed the same local inhibition of Rap2A activity at the putative synaptic tiling border, but not in unc-104; plx-1 double mutants (Figure 4E and G), indicating that Plexin controls local Rap2 activity independent of synapses.
To understand that this local Rap2 inactivation at the synaptic tiling border depends on the localized RapGAP activity of PLX-1, we examined the rescue activity of two PLX-1 mutant constructs, PLX-1(RA) and PLX-1(ΔSema), neither of which rescued the synaptic tiling defect of plx-1 mutants (Mizumoto and Shen, 2013a). PLX-1(RA) is a GAP-deficient mutant but localizes normally at the anterior edge of the DA9 synaptic domain. PLX-1(ΔSema) contains intact GAP domain but cannot be activated by the endogenous ligand and shows diffused localization due to deletion of the extracellular SEMA domain (Mizumoto and Shen, 2013a). We observed no local Rap2 inactivation in plx-1 mutant animals expressing these mutant PLX-1 constructs in DA9, while expression of wild type PLX-1 cDNA rescued local Rap2 inactivation at the anterior edge of the DA9 synaptic domain (Figure 4H).
While we do not fully exclude the possibility that PLX-1 indirectly regulates local Rap2 activity, these data taken together with the biochemical evidence that mammalian Plexin acts as RapGAP (Wang et al., 2013, 2012) strongly suggests that Plexin localized at the anterior edge of the DA9 synaptic domain locally inactivates Rap2 GTPase to delineate the synaptic tiling border in DA9.
mig-15(TNIK) regulates synaptic tiling
In mammals, TNIK (Traf2 and Nck1-interacting kinase) acts with Rap2 to regulate neurite extension, AMPA receptor trafficking in hippocampal neurons and microvilli formation in intestinal cells (Hussain et al., 2010; Kawabe et al., 2010; Gloerich et al., 2012). In C. elegans, mig-15 is the sole ortholog of mammalian TNIK and its paralog MINK1 (Misshapen-like kinase 1), which also is an effector of Rap GTPase (Nonaka et al., 2008 ). mig-15 can regulate various cellular processes, such as axon guidance and cell migration (Chapman et al., 2008; Poinat et al., 2002; Shakir et al., 2006; Teulière et al., 2011). We found that mig-15(rh148) hypomorphic mutants showed a severe synaptic tiling defect (Figure 5A and D). Similar to plx-1 and rap-2 mutants, the synaptic tiling defect followed the anterior expansion of the DA9 synaptic domain and the posterior expansion of the DA8 synaptic domain (Figure 5E and F). All RAB-3 puncta in mig-15(rh148) mutants co-localized with active zone markers, CLA-1 and UNC-10 (Figure 1—figure supplement 3), suggesting that these RAB-3 puncta represent bona fide synapses. We observed axon guidance defects (23%, n = 100) or ectopic branch formation (56%, n = 100) in DA9 of mig-15 mutant animals (Figure 5—figure supplement 1). These were excluded from our analysis of synaptic tiling phenotypes. While only half of the mig-15 mutant animals showed axon guidance defects or ectopic branch formation, the synaptic tiling defect of mig-15 mutants was almost fully penetrant (Figure 5D). We did not observe significant synaptic tiling defects in cdh-4(rh310) mutants (DA8/DA9 overlap: 4.6 ± 1.02 μm, n = 21), which can exhibit an axon defasciculation phenotype in the dorsal nerve cord neurons (Schmitz et al., 2008). These data suggest that the synaptic tiling defect in the mig-15 mutants is not a secondary effect of axon outgrowth and guidance.
Figure 5 with 2 supplements see all
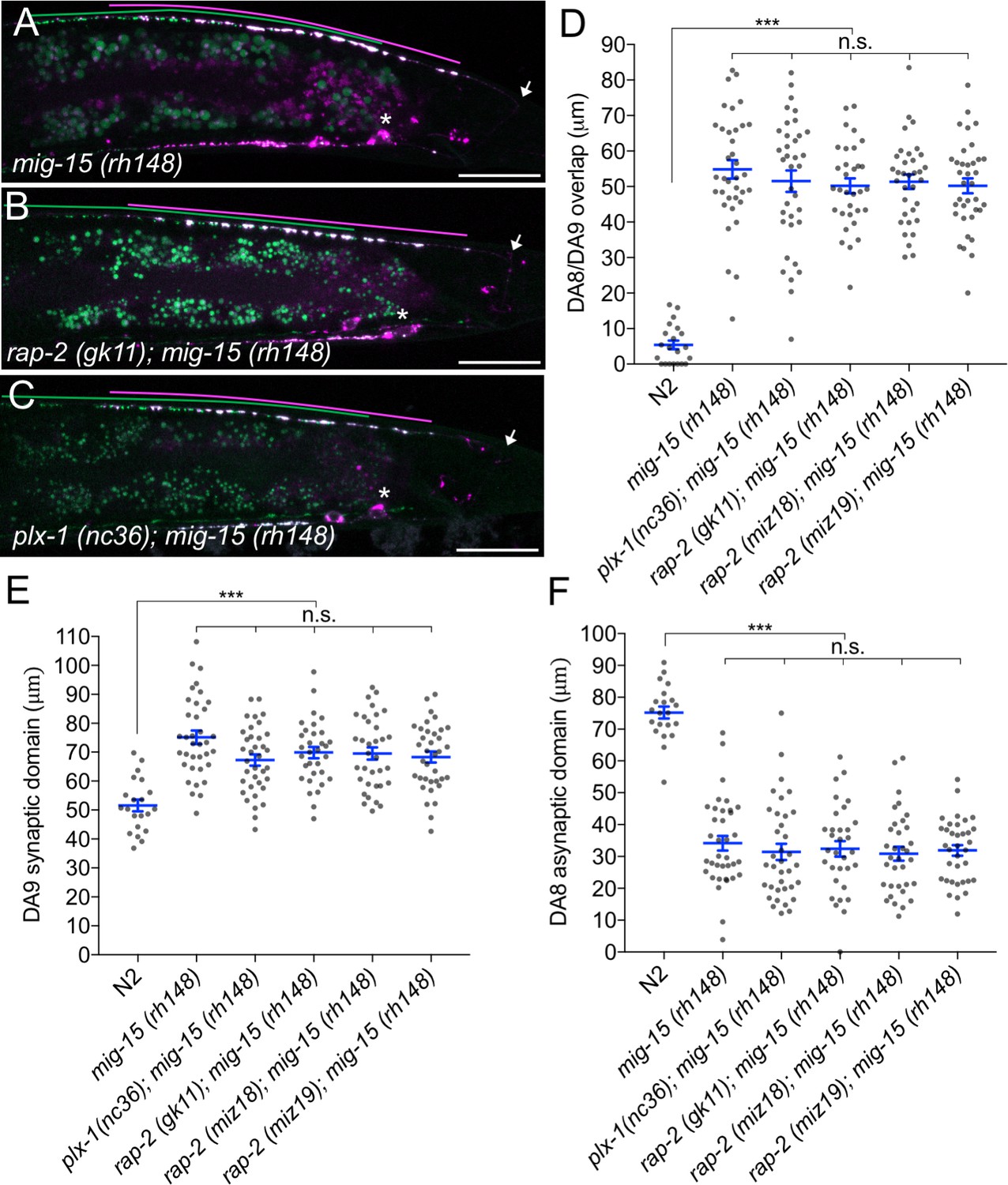
mig-15(TNIK) mutants show a severe synaptic tiling defect.
(A–C) Representative images of synaptic tiling marker (wyIs446) in mig-15(rh148) (A), rap-2(gk11); mig-15(rh148) (B) and plx-1(nc36); mig-15(rh148) (C) mutants. Synaptic domains of DA8 and DA9 are highlighted with green and magenta lines, respectively. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (D–F) Quantification of overlap between DA8 and DA9 synaptic domains (D), DA9 synaptic domain (E) and DA8 asynaptic domain (F) in respective mutant backgrounds. Each dot represents measurement from a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001.
The other two nonsense alleles (rh326: Q439Stop, rh80: W898Stop) also showed identical synaptic tiling defects as mig-15(rh148) (Figure 5—figure supplement 2). mig-15(rh80) has a nonsense mutation within the highly conserved CNH (citron/NIK homology) domain, which is required to interact with Rap2 in both mammals and C. elegans (Taira et al., 2004). This suggests a physical interaction between RAP-2 and MIG-15 for synaptic tiling. plx-1 or rap-2 mutants did not enhance the synaptic tiling defect in mig-15 mutants (Figure 5B–F). This result is consistent with our hypothesis that mig-15 acts in the same genetic pathway as plx-1 and rap-2. The PLX-1::GFP patch at the putative synaptic tiling border was unaffected in mig-15 mutants, even though the position of the PLX-1::GFP patch has shifted slightly posteriorly compared with wild type (Figure 3C and G). Taken together, these results suggest that mig-15 acts downstream of plx-1 to regulate synaptic tiling.
Interestingly, the degree of overlap between DA8 and DA9 synaptic domains was even larger in mig-15 mutants than those observed in plx-1 and rap-2 mutants (compare Figures 1G and 5D), suggesting that mig-15 also acts downstream of additional signaling pathways (see discussion).
mig-15 functions in DA neurons
We then determined in which cells mig-15 functions by conducting tissue specific rescue experiments. Since several mig-15 isoforms (wormbase and data not shown) exist, we used the mig-15 genomic sequence for the rescue experiments. Expression of mig-15 under the DA neuron specific promoter (Punc-4c) strongly rescued the synaptic tiling defect of mig-15(rh148) mutants (Figure 6A and F), consistent with our hypothesis that mig-15 acts in the same genetic pathway as plx-1 and rap-2.
Figure 6
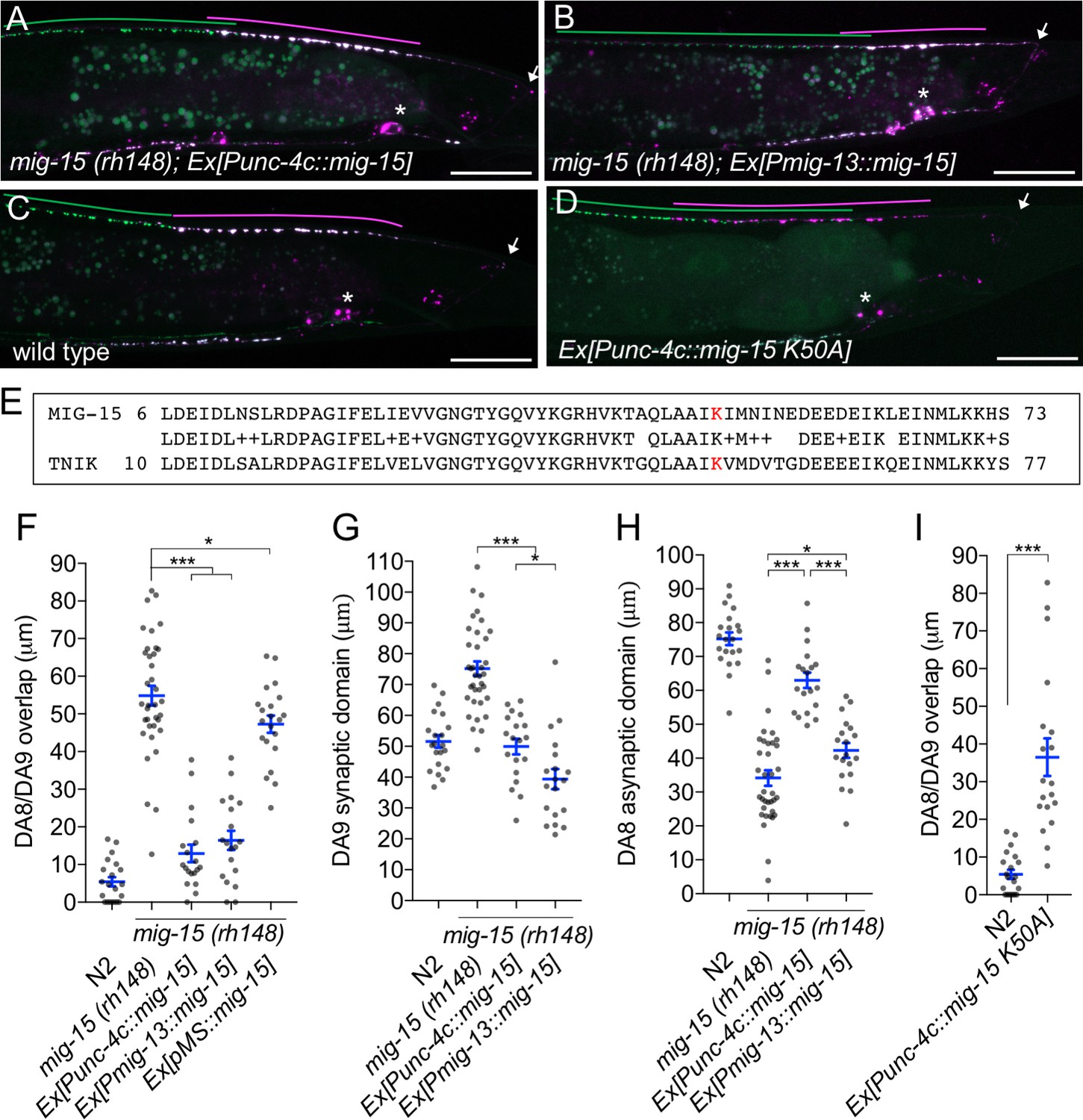
mig-15 functions in DA neurons.
(A–D) Representative images of synaptic tiling marker (wyIs446) in the following backgrounds: mig-15(rh148); Ex[Punc-4c::mig-15] (A), mig-15(rh148); Ex[Pmig-13::mig-15] (B), wild type (C) and wild type animals expressing dominant-negative mig-15(K50A) in DA neurons (D). Synaptic domains of DA8 and DA9 are highlighted with green and magenta lines, respectively. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (E) Amino acid alignment of amino-terminal region of MIG-15 and TNIK. A kinase-dead mutation in TNIK (K54A) and corresponding mutation in MIG-15 (K50A) are highlighted in red. (F–I) Quantification of overlap between DA8 and DA9 synaptic domains (F and I), DA9 synaptic domain (G), DA8 asynaptic domain (H) in respective mutant backgrounds. Each dot represents measurements from a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001; *p<0.05.
Expression of mig-15 in both DA8 and DA9 rescued both posterior expansion of the DA8 synaptic domain and anterior expansion of the DA9 synaptic domain (Figure 6G and H). DA9 specific expression of mig-15 under the mig-13 promoter rescued anterior expansion of the DA9 synaptic domain, suggesting that mig-15 functions cell autonomously in DA9 (Figure 6B and G). We observed that Pmig-13::mig-15 weakly rescued the posterior expansion of the DA8 synaptic domain (Figure 6H). This is likely due to the leaky expression of mig-15 in DA8, as the mig-15 genomic fragment without promoter showed slight rescue of the synaptic tiling defect in mig-15(rh148) mutants (Figure 6F). Kinase dead TNIK mutants act as a dominant-negative (Mahmoudi et al., 2009). In DA neurons, expression of mutant mig-15(kd), which carries the same mutation at the corresponding amino acid of the dominant-negative TNIK (Figure 6E), in DA neurons caused a severe synaptic tiling defect (Figure 6C, D and I). Based on these results, we conclude that mig-15 functions cell autonomously in DA neurons.
mig-15 inhibits synapse formation
We observed that DA9-specific expression of mig-15 under the mig-13 promoter in mig-15 mutants often exhibited a shorter synaptic domain compared to wild type (Figure 6B and G). So, we speculated that an excess amount of mig-15 inhibits synapse formation. We tested the effect of mig-15 overexpression in the wild type background. Strikingly, DA9-specific mig-15 overexpression in wild type (mig-15(OE)) significantly reduced synapse number compared to wild type (Figure 7A, C and D and Figure 7—figure supplement 1). This reduction occurred without affecting the overall morphology of the DA9 neuron (Figure 5—figure supplement 1). Conversely, DA9 synapse number was significantly increased in the mig-15(rh148) mutants (Figure 7B and D and Figure 7—figure supplement 1). mig-15 overexpression also significantly reduced synapse number in DD-type GABAergic motor neurons (Figure 7—figure supplement 2). These results indicate that mig-15 is a negative regulator of synapse formation. Further, pan-neuronal expression of mig-15 under the rab-3 promoter caused severe uncoordinated locomotion in wildtype animals (Figure 7—figure supplement 3). These locomotor defects occurred concomitant with significantly reduced GFP::RAB-3 intensity in the dorsal nerve cord in mig-15 over-expressing animals and without causing significant axon guidance defects (Figure 7—figure supplement 3). Taken together, these data indicate that reduced synapse number by mig-15 overexpression disrupted proper functioning of the motor circuit. Importantly, we observed no significant increase in synapse numbers in plx-1 or rap-2 mutants (Figure 7—figure supplement 1), suggesting that the role of mig-15 in negatively regulating synapse number is independent of its role in PLX-1/RAP-2 -mediated synaptic tiling (Figure 8G).
Figure 7 with 3 supplements see all
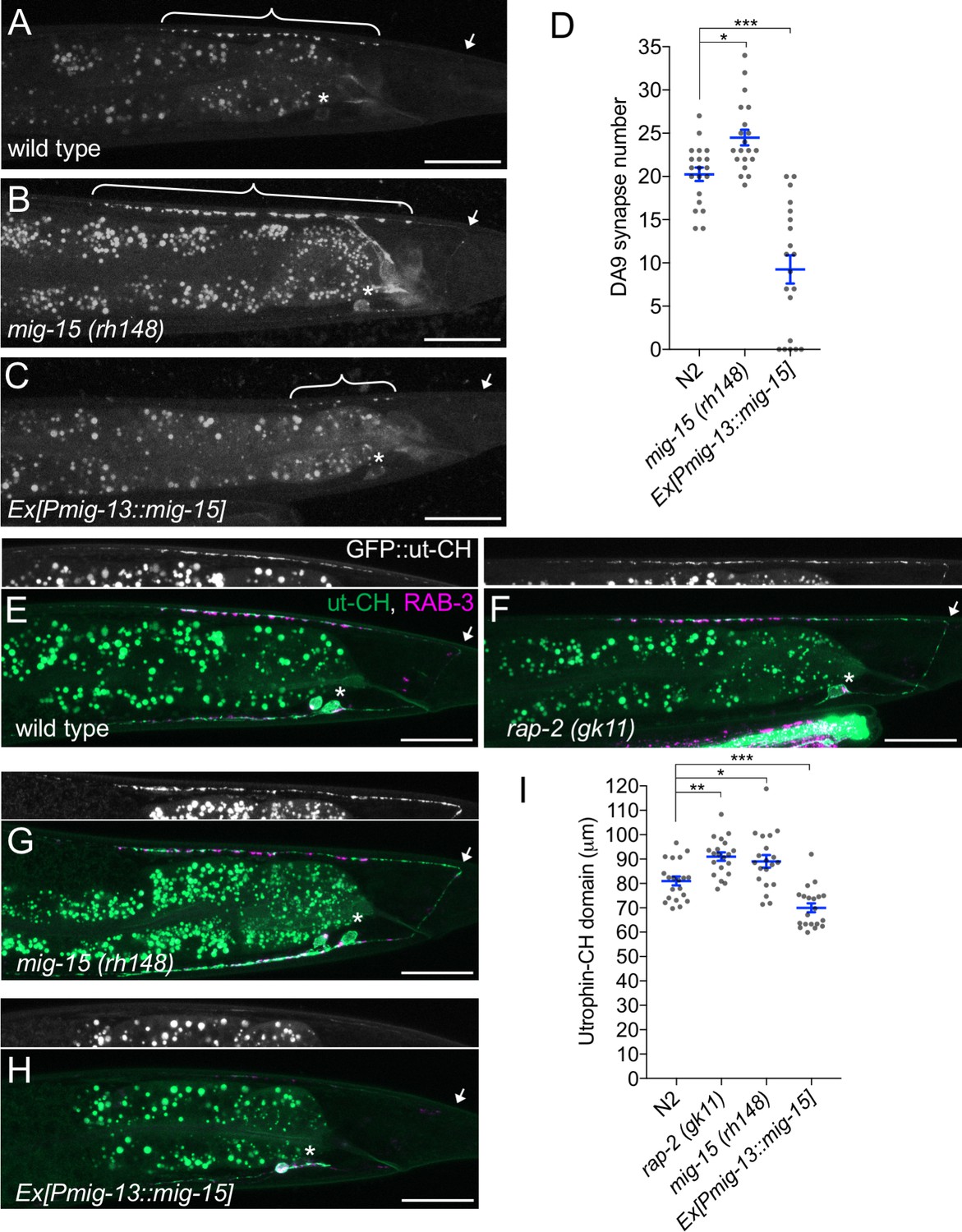
mig-15 negatively regulates synapse number.
(A–C) Representative images of DA9 presynaptic marker, GFP::RAB-3 (wyIs85), in wild type (A), mig-15(rh148) (B) and mig-15 overexpressing animals (C). Brackets represent DA9 synaptic domain. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (D) Quantification of DA9 synapse number. Each dot represents measurements from a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001; *p<0.05. (E–H) Representative images of synaptic branched F-actin labeled with GFP::ut-CH (wyIs329) in wild type (E), rap-2(gk11) (F), mig-15(rh148) (G) and mig-15 overexpressing animals (H). (I) Quantification of the length of GFP::ut-CH. Distance from the dorsal commissure to the most anterior and brightest GFP spot was measured. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001; **p<0.01, *p<0.05.
Figure 8

PLX-1/RAP-2 signaling coordinates synapse number and synaptic tiling border.
(A–C) Representative images of synaptic tiling marker (wyIs446) overexpressing mig-15 in DA9 from wild type (A), plx-1(nc36) (B) and rap-2(gk11) (C). Synaptic domains of DA8 and DA9 highlighted with green and magenta lines, respectively. Asterisks: DA9 cell body. Arrows: dorsal commissure of DA9. Scale bars: 20 μm. (D) Schematic illustration of synapse distribution in wild type (left), animals overexpressing mig-15 in DA9 (middle) and synaptic tiling mutants overexpressing mig-15 in DA9 (right). Arrows indicate the synaptic tiling border. Colored arrows represent expanded or shortened synaptic domain from DA8 (green) and DA9 (magenta). (E) Quantification of the DA8 asynaptic domain. (F) Quantification of the DA9 synaptic domain. Each dot represents measurements from a single animal. Blue bars indicate mean ± SEM. n.s.: not significant; ***p<0.001. (G) A model of synaptic tiling. PLX-1/RAP-2 signaling controls synaptic tiling via MIG-15, while MIG-15 also plays crucial roles in inhibiting the number of synapses. Solid lines indicate putative direct regulations, dotted lines represent indirect or unknown mode of regulations.
Rap GTPase and TNIK are well-known actin cytoskeleton regulators (Lin et al., 2010, 2008; Taira et al., 2004). Previous studies demonstrated that presynaptic development requires ARP2/3-dependent branched F-actin (Chia et al., 2012, 2014). Branched F-actin visualized by GFP::ut-CH (utrophin calponin homology domain) is enriched within the DA9 synaptic domain (Figure 7E) (Chia et al., 2012; Mizumoto and Shen, 2013a). We predicted that mig-15 negatively regulates synapse formation by re-organizing branched F-actin at the anterior edge of the synaptic domain. Consistently, we observed longer synaptic F-actin distribution in rap-2(gk11) and mig-15(rh148) mutants (Figure 7F, G and I). While GFP::ut-CH was observed in the posterior asynaptic axonal region or in the dendrite of DA9, synapse formation is likely inhibited by Wnt and Netrin signaling as previously reported (Klassen and Shen, 2007; Poon et al., 2008). Conversely, overexpression of mig-15 in DA9 significantly decreased the length of synaptic F-actin (Figure 7H and I). Overexpression of mig-15 also appeared to decrease the overall amount of synaptic F-actin (Figure 7H). This result suggests that mig-15 inhibits synapse formation by negatively regulating the formation of synaptic F-actin.
plx-1 and rap-2 coordinate synaptic tiling border positioning and synapse number
mig-15(OE) reduced the number of synapses in DA9. As a result, the length of the DA9 synaptic domain was significantly reduced in mig-15(OE) animals than in wild type (Figure 8A and E). Yet, synaptic tiling is maintained without a significant gap between DA8 and DA9 synaptic domains in mig-15(OE) animals, suggesting that the position of synaptic tiling border shifted posteriorly in mig-15(OE) animals. Indeed, the length of the posterior asynaptic domain of DA8 was significantly shorter in mig-15(OE) animals, indicating that the DA8 synaptic domain expanded posteriorly (Figure 8D). The PLX-1::GFP patch at the anterior edge of the DA9 synaptic domain also shifted posteriorly in mig-15(OE) animals with reduced synapse number (Figure 3D). These results strongly suggest that the PLX-1/RAP-2 signaling pathway may specify the position of the synaptic tiling border according to the available number of synapses in each DA neuron (Figure 8G). We propose that synaptic tiling is a mechanism to maintain a uniform distribution of synapses from one class of motor neuron in the nerve cord. Consistently, we found that DA8 synaptic domain did not shift posteriorly when mig-15 was overexpressed in DA9 of the synaptic tiling mutants, plx-1 or rap-2 (Figure 8B–E). This result suggests that DA8 no longer senses the reduction of DA9 synapse number in the synaptic tiling mutants. Synapse number was not different between mig-15(OE) and in rap-2(gk11); mig-15(OE) animals (Figure 7—figure supplement 1), suggesting that mig-15 is not dependent on the Plexin/Rap2 signaling pathway to inhibit synapse number (Figure 8G).
In summary, we demonstrate that synaptic tiling maintains a uniform distribution of synapses from one class of motor neurons along the nerve cord. Further, our results indicate plx-1 and rap-2 play critical roles in this process by coordinating the position of the synaptic tiling border.
Discussion
While much is known about the morphogenic triggers for axon guidance and patterned synapse formation, the downstream sequelae of these intracellular effectors have remained unclear. We discovered the role of Rap2 GTPase and TNIK in synapse pattern formation in C. elegans. Since Sema/Plexin signaling processes to inhibit synapse formation are well conserved across species, we propose that Sema/Plexin in mammals also utilize Rap2 and TNIK to regulate synapse patterning.
Cell autonomous and non-autonomous functions of Sema/Plexin signaling components
Previously we showed that both smp-1 and plx-1 are necessary and sufficient in DA9, which suggests that smp-1 and plx-1 act cell-autonomously in DA9 and non-autonomously in DA8 to regulate synaptic tiling. We proposed that Sema/PLX-1 in DA9 send a retrograde signal to DA8 through an unidentified signaling molecule (X) to induce the synaptic tiling pattern in DA8 (Mizumoto and Shen, 2013a). However, we found that both rap-2 and mig-15 act cell autonomously, since our DA9-specific rescue experiment only rescued the DA9 phenotype, but not the DA8 phenotype. This conclusion is further supported since the synaptic tiling defects of these mutants were fully rescued when both neurons express functional rap-2 cDNA or mig-15 genomic DNA. We propose that each neuron utilizes a different set of cell surface proteins but share common intracellular mechanisms to specify synapse patterning. Diverse signaling and cell adhesion molecules, such as atrial natriuretic peptide receptor (NPR) and GPCRs, regulate Rap activity (Gloerich and Bos, 2011; Birukova et al., 2008; Weissman et al., 2004). Screening for these potential Rap regulators should identify novel molecules that interact with Sema/Plexin and act in DA9.
Cycling of Rap GTPase activity in synaptic tiling
We showed that proper synapse patterning requires both GDP- and GTP-forms of RAP-2. Considering that PLX-1 regulates the spatial distribution of RAP-2 activity and mig-15 acts genetically downstream of plx-1 in synaptic tiling, RAP-2 may also locally regulate MIG-15 (TNIK). While we did not observe a specific subcellular localization of GFP-MIG-15 in DA9 (data not shown), PLX-1 and RAP-2 may instead regulate MIG-15 activity rather than its spatial localization. Further biochemical characterization of MIG-15 regulation by GTP-RAP-2 or GDP-RAP-2 will elucidate the exact functions of RAP-2 in synapse patterning.
Small GTPase activity is regulated by GAP and GEF (Guanine nucleotide exchange factor) proteins. Yet, we did not observe significant synaptic tiling defects in mutants of putative RAP-2 GEFs, which include pxf-1(RAPGEF2/6) and epac-1(RAPGEF3/4/5) (data not shown) (Frische et al., 2007; Pellis-van Berkel et al., 2005). We speculate that multiple RapGEFs act redundantly to activate RAP-2 in synaptic tiling.
mig-15(TNIK) may integrate multiple inhibitory cues during synapse formation
mig-15 mutants show a greater degree of overlap between DA8 and DA9 synaptic domains than plx-1 or rap-2 mutants. This effect partially occurs from excess synaptogenesis in the posterior asynaptic domain of both DA8 and DA9 neurons. Previously, we demonstrated that Wnt morphogens and their receptors, Frizzled, instruct synaptic topographic patterning by locally inhibiting synapse formation. Indeed, synaptic tiling defects in mig-15 mutants was somewhat similar to the combined effect of plx-1 and wnt mutants (Mizumoto and Shen, 2013b). TNIK can act as a positive regulator of the canonical Wnt signaling pathway in colorectal cancer cells (Mahmoudi et al., 2009; Shitashige et al., 2010). While we do not know whether the canonical Wnt signaling pathway contributes to local inhibition of synapse formation, we propose that TNIK integrates multiple signaling pathways for precise synapse pattern formation.
In addition to its role in synapse pattern formation, our data indicate that mig-15 also plays a role as a negative regulator of synapse number. Since neither plx-1 nor rap-2 mutants showed a significant increase in synapse number in DA9, mig-15 may inhibit synapse formation in a different signaling pathway (Figure 8G). As we observed a global reduction of synaptic actin staining in animals over-expressing mig-15, we propose that mig-15 controls synapse number by regulating synaptic F-actin.
The exact mechanisms of synaptic actin regulation by TNIK remain undetermined. TNIK could activate the JNK kinase pathway (Taira et al., 2004; Fu et al., 1999). The MIG-15/JNK-1 signaling pathway inhibits axonal branch formation in s C. elegans ensory neurons (Crawley et al., 2017). In contrast to these well-established roles of MIG-15/TNIK as an activator of the JNK pathway, we did not observe any synaptic tiling defect nor change in synapse number in jnk-1 mutant animals (Figure 5—figure supplement 2 and data not shown). Our results suggest mig-15 does not inhibit synapse formation through the JNK pathway.
Due to the pleiotropic phenotype of the mig-15 mutants, our genetic and phenotypic analyses of mig-15 did not exquisitely reveal the mechanistic relationship between PLX-1 and MIG-15 in synaptic tiling regulation. Further biochemical studies of MIG-15 regulation by Plexin/Rap2 in synaptic tiling will elucidate the molecular mechanisms that underlie the role of MIG-15/TNIK in synapse pattern formation.
Plexin signaling and diseases
Aberrant neuronal wiring underlies many neurological disorders. Not surprisingly, Semaphorin and Plexin genes are associated with various neurodevelopmental disorders and intellectual disabilities, including autism spectrum disorders (ASD) and schizophrenia (Mah et al., 2006; Gene Discovery Project of Johns Hopkins & the Autism Consortium et al., 2009). For example, PLXNB1, SEMA3A, SEMA4D and SEMA6C are significantly upregulated in the prefrontal cortices of schizophrenic patients (Eastwood et al., 2003; Gilabert-Juan et al., 2015). On the other hand, non-synonymous variations in the Sema3D gene had a significant protective effect against developing schizophrenia (Fujii et al., 2011). More recent work showed that loss of Sema5A/PlexA2 signaling induces excess excitatory synapse formation in granule cells, which caused ASD-like behavioural defects in mice (Duan et al., 2014).
Similar to Sema/Plexin signaling, TNIK is also associated with various neurological disorders, including schizophrenia and intellectual disabilities (Anazi et al., 2016; Potkin et al., 2010). TNIK can also physically bind and act with DISC1 (Disrupted in Schizophrenia 1) to regulate synaptic composition (Wang et al., 2011). So, we propose that the Sema/Plexin/Rap2/TNIK signaling pathway plays a critical role to precisely define synaptic connections and its disruption may induce serious neurological disorders.
Interestingly, SNPs in Plexin genes are also associated with extremely high IQ (Spain et al., 2016). Recent work suggests that loss of PlexinA1 confers better motor control in rodents due to increased synaptic connectivity in the corticospinal cord (Gu et al., 2017). Further studies on the Plexin/Rap2/TNIK signaling pathway in synapse map formation, as presented here, will likely reveal the genetic basis of these disorders and conditions.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (C. elegans) | rap-2 | NA | C25D7.7 | |
| Gene (C. elegans) | plx-1 | NA | Y55F3AL.1 | |
| Gene (C. elegans) | mig-15 | NA | ZC504.4 | |
| Strain, strain background (C. elegans) | rap-2(gk11) | C. elegans stock center (CGC) | VC14 | |
| Strain, strain background (C. elegans) | mig-15(rh148) | C. elegans stock center (CGC) | NJ490 | |
| Strain, strain background (C. elegans) | plx-1(nc36) | C. elegans stock center (CGC) | ST36 | |
| Strain, strain background (C. elegans) | rap-2(miz18) | This study | UJ401 | G12V mutant |
| Strain, strain background (C. elegans) | rap-2(miz19) | This study | UJ402 | S17A mutant |
| Strain, strain background (C. elegans) | wyIs446 | This study | TV14517 | synaptic tiling marker |
| Strain, strain background (C. elegans) | mizIs19 | This study | UJ397 | Rap2 FRET sensor strain |
Strains
All C. elegans strains were derived from Bristol N2 and raised on OP50 Escherichia coli-seeded nematode growth medium (NGM) plates at 20C and maintained as described previously (Brenner, 1974). The following mutants were used in this study: unc-104(e1265)II, plx-1(nc36)IV, rap-1(pk2082)IV, rap-3(gk3975)IV, jnk-1(gk7)IV rap-2(gk11)V, rap-2(miz16)V, rap-2(miz17)V, rap-2(miz18)V, rap-2(miz19)V. rap-2(miz20)V, mig-15(rh148)X, mig-15(rh80)X, mig-15(rh326)X.
CRISPR/Cas9 genome editing
Request a detailed protocolrap-2(miz16)V, rap-2(miz17)V, rap-2(miz18)V, rap-2(miz19)V. rap-2(miz20)V were generated using Co-CRISPR method (Kim et al., 2014). unc-22 or dpy-10 co-CRISPR markers were used for selecting candidate animals (Kim et al., 2014; Arribere et al., 2014). Vectors for sgRNA and Cas9 were obtained from Addgene (Plasmid ID: 46169 and 46168, respectively) (Friedland et al., 2013). The rap-2 guide RNA sequence (5’ – gTAGTGGAGGTGTCGGAAAAT-3’) was designed using MIT CRISPR design tool (crispr.mit.edu:8079) and inserted into sgRNA vector using Q5 Site-Directed Mutagenesis kit (NEB). Repair templates with either G12V (miz17 and miz18) and S17A (miz19 and miz20) mutation were generated by PCR with primer sets carrying corresponding mutations (see supplemental Experimental Procesures). Synonymous mutations were also introduced in the sgRNA recognition sequence to avoid Cas9 recruitment to the edited genome. PCR products were cloned into EcoRI site of the pBluescript SK(+) vector. Synthesized double-stranded DNA (GeneArt, Thermo Fisher) was used as a repair template for generating rap-2(miz16) mutant.
Plasmid constructions
Request a detailed protocolC. elegans expression clones were made in a derivative of pPD49.26 (A. Fire), the pSM vector (kind gift from S. McCarroll and C. I. Bargmann). Primer sets used in this study are listed in the Supplemental Experimental Procedures. The following constructs were used and transgenes were generated using standard microinjection method (Mello et al., 1991): wyIs446 (Punc-4::2xGFP-rab-3; Pmig-13::mCherry-rab-3; Podr-1::RFP), wyIs85 (Pitr-1::GFP-rab-3; Podr-1::RFP), wyIs442 (Pflp-13::2xGFP-rab-3; Pplx-2::mCherry-rab-3; Podr-1::RFP), wyIs320 (Pitr-1::plx-1::GFP; Pmig-13::mCherry;;rab-3; Podr-1::GFP), wyIs329 (Pmig-13::GFP-ut-CH; Pmig-13::mCherry::rab-3; Podr-1::GFP), wyIs524 (Punc-4::2xGFP-rab-3; Pmig-13::mCherry-rab-3; Podr-1::RFP), wyIs685 (Pmig-13::mCherry::rab-3; Pmig-13::GFPnovo2::cla-1; Podr-1::GFP) mizIs1 (Pitr-1::GFPnovo2-CAAX; Pvha-6::zif-1; Pitr-1::mCherry::rab-3; Podr-1::GFP), mizIs19 (Pmig-13::eGFP::hRap2a; Pmig-13::mRFP-RalGDS(RBD)-mRFP; Podr-1::GFP), mizIs33 (Prab-3::mig-15; Podr-1::GFP), jsIs682 (Prab-3::GFP::rab-3; lin-15(+)), wyEx5445 (Prap-1::GFP; Punc-4::myr-mCherry; Podr-1::RFP), wyEx5464 (Prap-2::GFP; Punc-4::myr-mCherry; Podr-1::RFP), mizEx194 (Prap-3::GFP; Pmig-13::myr-mCherry; Podr-1::RFP), mizEx165 (Phlh-1::rap-2; Podr-1::GFP), mizEx164 (Punc-129::rap-2; Podr-1::GFP), mizEx174 (Punc-4c::rap-2; Podr-1::GFP), mizEx157 (Pmig-13::rap-2; Podr-1::GFP), mizEx156 (Punc-4c::hRap2a; Podr-1::GFP), mizEx177 (Punc-4c::rap-1; Podr-1::GFP), mizEx151 (Pmig-13::mig-15; Podr-1::GFP), mizEx147 (Punc-4c::mig-15; Podr-1::GFP), mizEx153 (ΔpSMmig-15; Podr-1::GFP), mizEx178 (Punc-4c::mig-15(K50A); Podr-1::GFP), mizEx173 (Punc-4::rap-2(G12V); Podr-1::GFP), mizEx179 (Pflp-13::mig-15; Podr-1::GFP), mizEx170 (Pmig-13::mig-15; Podr-1::GFP), mizEx197 (Pmig-13::mig-15; Podr-1::GFP), mizEx210 (Pmig-13::mig-15; Podr-1::RFP), mizEx257 (Prab-3::GFP; Prab-3::mCherry::rab-3, Podr-1::mScarlet::CAAX), mizEx272 (Podr-1::GFP; Pmig-13::unc-10::TdTomato); mizEx309 (Pmig-13::plx-1(RA); Podr-1::RFP), mizEx312 (Pmig-13::plx-1(Δsema); Podr-1::RFP); mizEx314 (Pmig-13::plx-1; Podr-1::RFP).
Cloning of rap-1, rap-2 and mig-15
Request a detailed protocolcDNAs of rap-1 and rap-2 were obtained from cDNA library prepared from N2 RNA. Trizol (Invitrogen) was used to purify total RNA from N2, and the SuperScript III First-Strand Synthesis System for RT-PCR (Invitrogen) was used for the reverse-transcription. mig-15 genomic DNA was amplified from the N2 genomic DNA purified using GeneJET Genomic DNA Purification Kit (Thermo Scientific). Phusion (NEB) or Q5 (NEB) DNA polymerases were used for all PCR reactions for amplifying cDNA and genomic DNA fragments. Amplified fragments were cloned into the AscI and KpnI sites of pSM vector using SLiCE method (Motohashi, 2015), Gibson assembly (Gibson et al., 2009) or T4 ligase (NEB). List of primers used in this study is available in the Supplemental Experimental Procedures.
Confocal microscopy
Request a detailed protocolImages of fluorescently tagged fusion proteins were captured in live C. elegans using a Zeiss LSM800 confocal microscope (Carl Zeiss, Germany). Worms were immobilized on 2% agarose pad using a mixture of 7.5 mM levamisole (Sigma-Aldrich) and 0.225M BDM (2,3-butanedione monoxime) (Sigma-Aldrich). Images were analyzed with Zen software (Carl Zeiss) or Image J (NIH, USA). Definition of each parameter is as follows (Mizumoto and Shen, 2013a): DA8/9 overlap: a distance between the most anterior DA9 synapse and the most posterior DA8 synapse, DA8 asynaptic domain: a distance from commissure to the most posterior DA8 synapse, DA9 synaptic domain: a distance between the most anterior and posterior DA9 synapses. Middle L4 (judged by the stereotyped shape of developing vulva) animals were used for quantification. Averages were taken from at least 20 samples. For GFP::Utrophin-CH, we measured the length from the posterior end of dorsal axon to the anterior end of GFP::Utrophin-CH domain. For each marker strain, the same imaging setting (laser power, gain pinhole) and image processing were used for comparing different genotypes.
Two-photon FLIM-FRET experiment
Request a detailed protocolExpression vector for cultured cells (pCI-eGFP-hRap2a, pCI-eGFP-RAP-1, pCI-eGFP-RAP-2, pCI-eGFP-RAP-2(G12V), pCI-eGFP-RAP-2(S17A)) were generated by replacing Ras in pCI-eGFP-Ras (Yasuda et al., 2006) with hRap2a and rap-2 cDNAs with XhoI and BamHI. pCI-mRFP-RalGDS-mRFP plasmid is a kind gift from Dr. Yasuda. Rap2 and FRET sensor plasmids were mixed in 1:2 ratio and transfected into HeLa cells using Lipofectamine 3000 (ThermoFisher). FLIM was conducted 24 hr after transfection. For expression of FRET sensor in the DA9 neuron, each fusion protein constructs were cloned into AscI and KpnI sites of the pSM vector containing mig-13 promoter using SLiCE method.
A custom-made two-photon fluorescence lifetime imaging microscope was used as described elsewhere (Murakoshi et al., 2011). Briefly, EGFP-Rap2a was excited with a Ti-sapphire laser (Mai Tai; Spectra-Physics) tuned to 920 nm. The X and Y scanning galvano mirrors (6210 hr; Cambridge Technology) were controlled with ScanImage software (Pologruto et al., 2003). EGFP photon signals were collected an objective lens (60×, 1.0 NA; Olympus) and a photomultiplier tube (H7422-40p; Hamamatsu) placed after a dichroic mirror (FF553-SDi01; Semrock) and emission filter (FF01-510/84; Semrock). A fluorescence lifetime curve was recorded by a time-correlated single-photon-counting board (SPC-150; Becker and Hickl) controlled with the software described previously (Yasuda et al., 2006). For construction of a fluorescence lifetime image, the mean fluorescence lifetime values (τm) in each pixel were translated into a color-coded image. We quantified free EGFP-Rap2a and EGFP-Rap2a undergoing FRET (binding fraction) as described elsewhere (Yasuda et al., 2006). Briefly, we calculated the proportion of EGFP undergoing FRET in individual ROIs using the following formula:
(1)
where τfree and τFRET are the fluorescence lifetime of free EGFP and EGFP undergoing FRET, respectively.
Statistics
Prism (GraphPad) software was used for statistical analysis. One-way ANOVA was done and corrected for multiple comparisons with posthoc Tukey's multiple comparisons tests done between all genotypes. Student’s t-test was used for pairwise comparison. Sample numbers were pre-determined before conducting statistical analyses.
Supplemental experimental procedures
Request a detailed protocolThe method for qRT-PCR and sequences of primers and repair templates used in this study are available in Supplementary file 1.
Data availability
All data generated or analyzed during this study are included in the manuscript and supporting files.
References
-
Epac/Rap and PKA are novel mechanisms of ANP-induced Rac-mediated pulmonary endothelial barrier protectionJournal of Cellular Physiology 215:715–724.https://doi.org/10.1002/jcp.21354
-
Semaphorin signaling in cardiovascular developmentCell Metabolism 21:163–173.https://doi.org/10.1016/j.cmet.2014.12.015
-
Heritable genome editing in C. elegans via a CRISPR-Cas9 systemNature Methods 10:741–743.https://doi.org/10.1038/nmeth.2532
-
TNIK, a novel member of the germinal center kinase family that activates the c-Jun N-terminal kinase pathway and regulates the cytoskeletonThe Journal of Biological Chemistry 274:30729–30766.https://doi.org/10.1074/jbc.274.43.30729
-
Caenorhabditis elegans PlexinA, PLX-1, interacts with transmembrane semaphorins and regulates epidermal morphogenesisDevelopment 129:2053–2063.
-
Possible association of the semaphorin 3D gene (SEMA3D) with schizophreniaJournal of Psychiatric Research 45:47–53.https://doi.org/10.1016/j.jpsychires.2010.05.004
-
Regulating rap small G-proteins in time and spaceTrends in Cell Biology 21:615–623.https://doi.org/10.1016/j.tcb.2011.07.001
-
Rap2A links intestinal cell polarity to brush border formationNature Cell Biology 14:793–801.https://doi.org/10.1038/ncb2537
-
Plexin structures are coming: opportunities for multilevel investigations of semaphorin guidance receptors, their cell signaling mechanisms, and functionsCellular and Molecular Life Sciences 69:3765–3805.https://doi.org/10.1007/s00018-012-1019-0
-
The kinase TNIK is an essential activator of Wnt target genesThe EMBO Journal 28:3329–3340.https://doi.org/10.1038/emboj.2009.285
-
Efficient gene transfer in c.elegans: extrachromosomal maintenance and integration of transforming sequences.The EMBO Journal 10:3959–4029.
-
Plexins: axon guidance and signal transductionCellular and Molecular Life Sciences 62:1363–1371.https://doi.org/10.1007/s00018-005-5018-2
-
R-ras as a key player for signaling pathway of plexinsMolecular Neurobiology 32:217–222.https://doi.org/10.1385/MN:32:3:217
-
MINK is a Rap2 effector for phosphorylation of the postsynaptic scaffold protein TANC1Biochemical and Biophysical Research Communications 377:573–578.https://doi.org/10.1016/j.bbrc.2008.10.038
-
Rap2 as a slowly responding molecular switch in the Rap1 signaling cascadeMolecular and Cellular Biology 20:6074–6083.https://doi.org/10.1128/MCB.20.16.6074-6083.2000
-
Requirement of the Caenorhabditis elegans RapGEF pxf-1 and rap-1 for epithelial integrityMolecular Biology of the Cell 16:106–116.https://doi.org/10.1091/mbc.e04-06-0492
-
ScanImage: flexible software for operating laser scanning microscopesBiomedical Engineering Online 2:13.https://doi.org/10.1186/1475-925X-2-13
-
The Traf2- and Nck-interacting kinase as a putative effector of Rap2 to regulate actin cytoskeletonJournal of Biological Chemistry 279:49488–49496.https://doi.org/10.1074/jbc.M406370200
-
Diverse roles for semaphorin-plexin signaling in the immune systemTrends in Immunology 33:127–135.https://doi.org/10.1016/j.it.2012.01.008
-
Semaphorin regulation of cellular morphologyAnnual Review of Cell and Developmental Biology 23:263–292.https://doi.org/10.1146/annurev.cellbio.22.010605.093554
-
The structure of the nervous system of the nematode Caenorhabditis elegansPhilosophical Transactions of the Royal Society B: Biological Sciences 314:1–340.https://doi.org/10.1098/rstb.1986.0056
-
Synaptic adhesion moleculesCurrent Opinion in Cell Biology 15:621–632.https://doi.org/10.1016/S0955-0674(03)00107-8
-
Plexins function in epithelial repair in both Drosophila and zebrafishNature Communications 7:12282–12335.https://doi.org/10.1038/ncomms12282
Article and author information
Author details
Funding
Human Frontier Science Program (CDA-00004/2014)
- Kota Mizumoto
Canadian Institutes of Health Research (PJT-148667)
- Kota Mizumoto
Natural Sciences and Engineering Research Council of Canada (RGPIN-2015-04022)
- Kota Mizumoto
Canada Research Chairs
- Kota Mizumoto
Michael Smith Foundation for Health Research (16869)
- Kota Mizumoto
Tomizawa Jun-ichi and Keiko Fund of Molecular Biology Society of Japan for Young Scientists
- Kota Mizumoto
Canada Foundation for Innovation (34722)
- Kota Mizumoto
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to Donald Moerman and his lab members for generating rap-3 mutant strain, providing other mutant strains, sharing reagents and for general discussions. We also thank Ryohei Yasuda for FRET sensor plasmids, Hiroshi Kawabe for human Rap2a cDNA, Richard Ikegami for plx-2 promoter construct, Harald Hutter and Peri Kurshan for sharing strains, May Dang-Lawson and Michael Gold for unpublished biochemical experiments, Lisa Fernando for the technical support, all Mizumoto lab members and general discussions, Kang Shen, Shaul Yogev and Maulik Patel for comments on the manuscript. Some strains used in this study were obtained from the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440) and C. elegans gene knock out consortium. Mizumoto Lab is funded by HFSP (CDA-00004/2014), CIHR (PJT-148667), NSERC (RGPIN-2015–04022), CFI-JELF (34722) and Tomizawa Jun-ichi and Keiko Fund for Young Scientist. KM is a recipient of Canada Research Chair and Michael Smith Foundation for Health Research Scholar.
Copyright
© 2018, Chen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,721
- views
-
- 293
- downloads
-
- 26
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 26
- citations for umbrella DOI https://doi.org/10.7554/eLife.38801
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Rap2 and TNIK control Plexin-dependent tiled synaptic innervation in C. elegans
eLife 7:e38801.
https://doi.org/10.7554/eLife.38801




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}