Structure of a bacterial ATP synthase
- The Hospital for Sick Children Research Institute, Canada
- The University of Toronto, Canada
- Tokyo Institute of Technology, Japan
- Kyoto-Sangyo University, Japan
Abstract
ATP synthases produce ATP from ADP and inorganic phosphate with energy from a transmembrane proton motive force. Bacterial ATP synthases have been studied extensively because they are the simplest form of the enzyme and because of the relative ease of genetic manipulation of these complexes. We expressed the Bacillus PS3 ATP synthase in Eschericia coli, purified it, and imaged it by cryo-EM, allowing us to build atomic models of the complex in three rotational states. The position of subunit ε shows how it is able to inhibit ATP hydrolysis while allowing ATP synthesis. The architecture of the membrane region shows how the simple bacterial ATP synthase is able to perform the same core functions as the equivalent, but more complicated, mitochondrial complex. The structures reveal the path of transmembrane proton translocation and provide a model for understanding decades of biochemical analysis interrogating the roles of specific residues in the enzyme.
https://doi.org/10.7554/eLife.43128.001Introduction
Adenosine triphosphate (ATP) synthases are multi-subunit protein complexes that use an electrochemical proton motive force across a membrane to make the cell’s supply of ATP from adenosine diphosphate (ADP) and inorganic phosphate (Pi). These enzymes are found in bacteria and chloroplasts as monomers, and in mitochondria as rows of dimers that bend the inner membrane to facilitate formation of the mitochondrial cristae (Davies et al., 2012; Paumard et al., 2002). Proton translocation across the membrane-embedded FO region of the complex occurs via two offset half-channels (Vik and Antonio, 1994; Junge et al., 1997). Studies with Bacillus PS3 ATP synthase in liposomes showed that proton translocation may be driven by ΔpH or ΔΨ alone (Soga et al., 2012). The passage of protons causes rotation of a rotor subcomplex, inducing conformational change in the catalytic F1 region to produce ATP (Walker, 2013) while a peripheral stalk subcomplex holds the F1 region stationary relative to the spinning rotor during catalysis. For the mitochondrial enzyme, X-ray crystallography has been used to determine structures of the soluble F1 region (Abrahams et al., 1994), partial structures of the peripheral stalk subcomplex alone (Dickson et al., 2006) and with the F1 region (Rees et al., 2009), and structures of the F1 region with the membrane-embedded ring of c-subunits attached (Stock et al., 1999; Watt et al., 2010). Recent breakthroughs in electron cryomicroscopy (cryo-EM) allowed the structures of the membrane-embedded FO regions from mitochondrial and chloroplast ATP synthases to be determined to near-atomic resolutions (Guo et al., 2017; Klusch et al., 2017; Srivastava et al., 2018; Hahn et al., 2018).
Compared to their mitochondrial counterparts, bacterial ATP synthases have a simpler subunit composition. The F1 region consists of subunits α3β3γδε, while the FO region is usually formed by three subunits with the stoichiometry ab2c9-15. Chloroplasts and a few bacteria, such as Paracoccus denitrificans, possess two different but homologous copies of subunit b, named subunits b and b’ (Walker, 2013). Each copy of subunit α and β contains a nucleotide binding site. The non-catalytic α subunits each bind to a magnesium ion (Mg2+) and a nucleotide, while the catalytic β subunits can adopt different conformations and bind to Mg-ADP (βDP), Mg-ATP (βTP), or remain empty (βE). Crystal structures of bacterial F1-ATPases and c-rings from the FO regions of several species have been determined (Stocker et al., 2007; Cingolani and Duncan, 2011; Morales-Rios et al., 2015; Shirakihara et al., 2015; Ferguson et al., 2016; Pogoryelov et al., 2009; Preiss et al., 2010; Preiss et al., 2013; Preiss et al., 2015). Structures of intact ATP synthases from E. coli have been determined to overall resolutions of 6 to 7 Å by cryo-EM, with the FO region showing lower quality than the rest of the maps, presumably due to conformational flexibility (Sobti et al., 2016). In structures of both intact ATP synthase (Sobti et al., 2016) and dissociated F1-ATPase (Cingolani and Duncan, 2011; Shirakihara et al., 2015) from bacteria, subunit ε adopts an ‘up’ conformation that inhibits the ATP hydrolysis by the enzyme. In the thermophilic bacterium Bacillus PS3, this subunit ε mediated inhibition is dependent on the concentration of free ATP (Kato et al., 1997; Suzuki et al., 2003; Saita et al., 2010). Low ATP concentrations (e.g. <0.7 mM) promote the inhibitory up conformation while a permissive ‘down’ conformation can be induced by a high concentration of ATP (e.g. >1 mM). This mechanism would allow the Bacillus PS3 ATP synthase to run in reverse, establishing a proton motive force by ATP hydrolysis, when the ATP concentration is sufficient to do so without depleting the cell’s supply of ATP. In E. coli, however, in the absence of a sufficient proton motive force to drive ATP synthesis, inhibition of ATP hydrolysis by subunit ε persists even when the concentration of free ATP is high (Laget and Smith, 1979; Sekiya et al., 2010).
Although bacterial ATP synthases have been subjected to extensive biochemical analysis, high-resolution structural information is lacking for the intact enzyme or the membrane-embedded proton-conducting subunit a and the associated subunit b. We determined structures of intact ATP synthase from Bacillus PS3 in three rotational states by cryo-EM. The structures reached overall resolutions of 3.0, 3.0, and 3.2 Å (Figure 1), allowing construction of nearly complete atomic models for the entire complex. The structures reveal how loops in subunit a of the bacterial enzyme fill the role of additional subunits in the FO region of the mitochondrial enzyme. Most significantly, the structures provide a framework for understanding decades of mutagenesis experiments designed to probe the mechanism of ATP synthases.
Figure 1 with 5 supplements see all
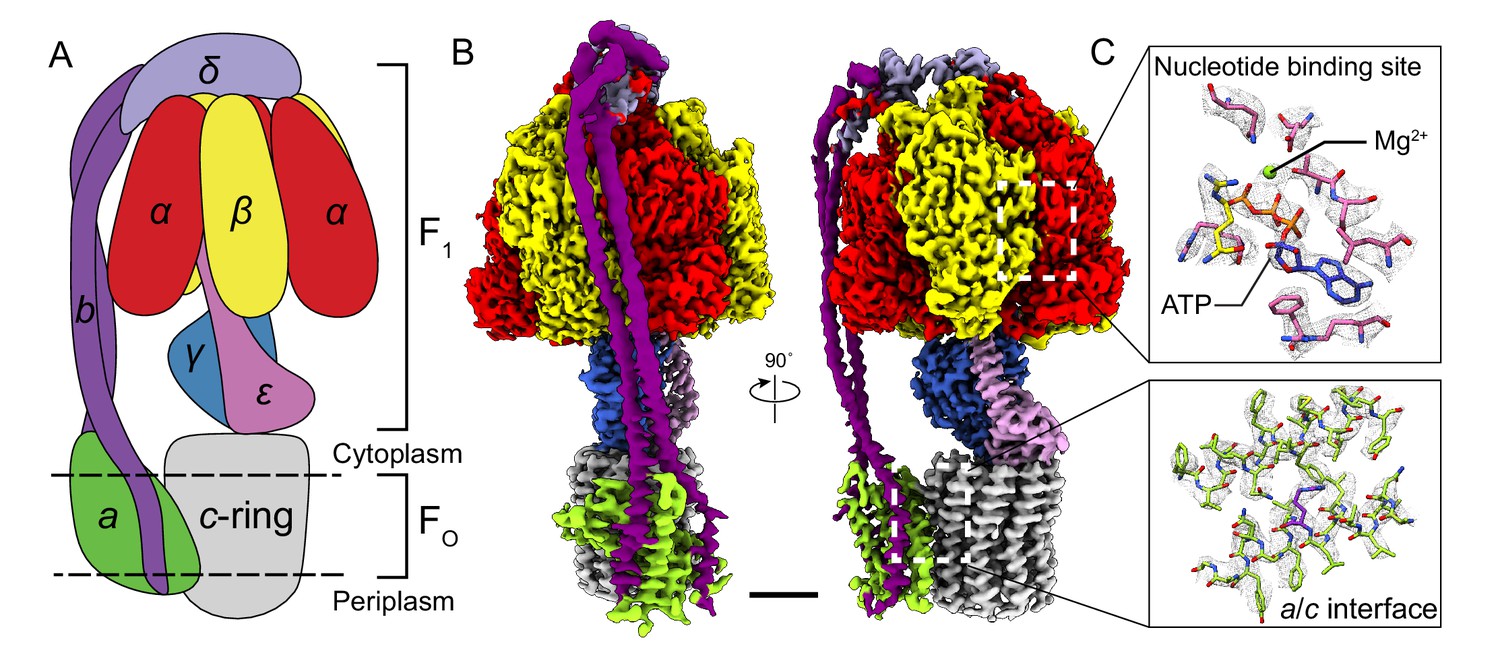
Overall structure of Bacillus PS3 ATP synthase.
(A) Cartoon of ATP synthase. (B) Cryo-EM map of ATP synthase with subunits coloured the same as the cartoon. (C) Example map density that allowed construction of an atomic model. Scale bar, 30 Å.
Results and discussion
Structure determination and overall architecture
Subunits of Bacillus PS3 ATP synthase, including subunit β bearing an N-terminal 10 × His tag, were expressed from a plasmid in E. coli strain DK8, which lacks endogenous ATP synthase (Klionsky et al., 1984; Suzuki et al., 2002). The complex was extracted from membranes with detergent, purified by metal-affinity chromatography, and subjected to cryo-EM analysis (Figure 1—figure supplement 1). Three conformations corresponding to different rotational states of the enzyme were identified by ab-initio 3D classification and refined to high resolution. The 3D classes contain 45, 35, and 20% of particle images and the overall resolutions of the corresponding cryo-EM maps were 3.0, 3.0, and 3.2 Å, respectively (Figure 1—figure supplements 2 and 3). Estimation of local resolution suggests that the F1 regions of the maps, which are larger than the FO regions and appear to dominate the image alignment process, are mostly at between 2.5 and 3.5 Å resolution, whereas the FO regions were limited to lower resolution (Figure 1—figure supplement 3). Focused refinement (Bai et al., 2015) of the FO region and peripheral stalk subunits ab2c10 and δ (corresponding to the subunit OSCP in mitochondrial ATP synthase) improved the resolution of the FO regions considerably for all three classes but not enough to resolve density for most of the amino acid side chains. An improved map of the FO region was obtained by focused refinement of the membrane-embedded region only, excluding the soluble portion of subunit b with particle images from all three classes (Figure 1—figure supplement 2). Overall, amino acid side chain detail can be seen for subunits α3, β3, γ, δ, ε, a, c10-ring, and the transmembrane α-helices of b2 (Figure 1—figure supplement 4). The soluble region of the two b-subunits was modeled as poly-alanine (Supplementary file 1).
The general architecture of the enzyme resembles E. coli ATP synthase (Sobti et al., 2016) and the more distantly related spinach chloroplast enzyme (Hahn et al., 2018) but with striking differences. As observed previously in a Bacillus PS3 F1-ATPase crystal structure (PDB 4XD7) (Shirakihara et al., 2015), the three catalytic β subunits adopt ‘open’, ‘closed’, and ‘open’ conformations, different from the ‘half-closed’, ‘closed’, and ‘open’ conformations seen in the auto-inhibited E. coli F1-ATPase (Cingolani and Duncan, 2011), and the ‘closed’, ‘closed’, and ‘open’ conformations seen in chloroplast ATP synthase (Hahn et al., 2018) and most mitochondrial ATP synthase structures (Abrahams et al., 1994; Stock et al., 1999). This difference, with the half-closed βDP of the E. coli enzyme appearing as open in the Bacillus PS3 enzyme, suggests species-specific differences in inhibition by subunit ε (Figure 1B, pink density), which inserts into the α/β interface and forces βDP into the open conformation.
Thermophilic proteins achieve stability at high temperature through mechanisms that include an increased number of ionic interactions, shorter loops between secondary structure elements, and tighter packing of hydrophobic regions (Jaenicke and Böhm, 1998; Kumar and Nussinov, 2001; Szilágyi and Závodszky, 2000). Comparison of individual subunit structures from the F1 regions of ATP synthases from thermophiles (Bacillus PS3 and Caldalaklibacillus thermarum [PDB 5HKK] (Ferguson et al., 2016)) and mesophiles (E. coli [PDB 3OAA] (Cingolani and Duncan, 2011), Paracoccus denitrificans [5DN6] (Morales-Rios et al., 2015), and Spinacia oleracea chloroplast [PDB 6FKF] (Hahn et al., 2018)) did not show clear evidence of tighter packing or shorter loops in the complexes from thermophiles. However, there are more ionic interactions in the F1-ATPase structures from thermophiles than from mesophiles, suggesting that these interactions may play a role in stabilizing the complexes.
In the FO region, one copy of subunit b is positioned at a location equivalent to that of the mitochondrial subunit b, while the second copy occupies the position of yeast subunit 8 (mammalian A6L) on the other side of subunit a (Figure 1B). Despite the different c-ring sizes (10 c-subunits in Bacillus PS3 versus 14 in spinach chloroplasts), the backbone positions of subunits ab2 from Bacillus PS3 overlap with subunits abb’ from spinach chloroplast ATP synthase (Hahn et al., 2018) (Figure 1—figure supplement 5A). Comparison of the atomic model of the FO region from Bacillus PS3 and the backbone model of the E. coli complex from cryo-EM at ~7 Å resolution (PDB 5T4O) (Sobti et al., 2016) showed significant structural differences in transmembrane α-helices of subunit b relative to subunit a (Figure 1—figure supplement 5B). Rather than reflecting true differences between E. coli and Bacillus PS3 ATP synthase structures, these deviations are likely due to the lower resolution of the E. coli maps.
Flexibility in the peripheral and central stalks
As expected, the most striking difference between the three rotational states of the Bacillus PS3 structure is the angular position of the rotor (subunits γεc10) (Figure 2A, Video 1). The structure of the ATP synthase, with three αβ pairs in the F1 region and 10 c-subunits in the FO region, results in symmetry mismatch between the 120° steps of the F1 motor and 36° steps of the FO motor. The 120° steps of the F1 motor gives an average rotational step of 3.3 c-subunits, with the closest integer steps being 3, 4 and 3 c-subunits. By comparing the positions of equivalent c-subunits in different rotational states, the observed rotational step sizes in the three rotational states of the ATP synthase appear to be almost exactly 3, 4 and 3 c-subunits (Figure 2B). At the present resolution, the structures of subunit a and the c-ring do not appear to differ between rotary states. Similar integer step sizes were found in yeast ATP synthase (Vinothkumar et al., 2016) and V-ATPase (Zhao et al., 2015), which also contain 10 c-subunits. However, non-integer steps were seen in the chloroplast (14 c-subunits) (Hahn et al., 2018) and bovine (8 c-subunits) (Zhou et al., 2015) ATP synthases, indicating that the c-subunit steps between the rotational states of rotary ATPases likely depends on the number of c-subunits.
Figure 2
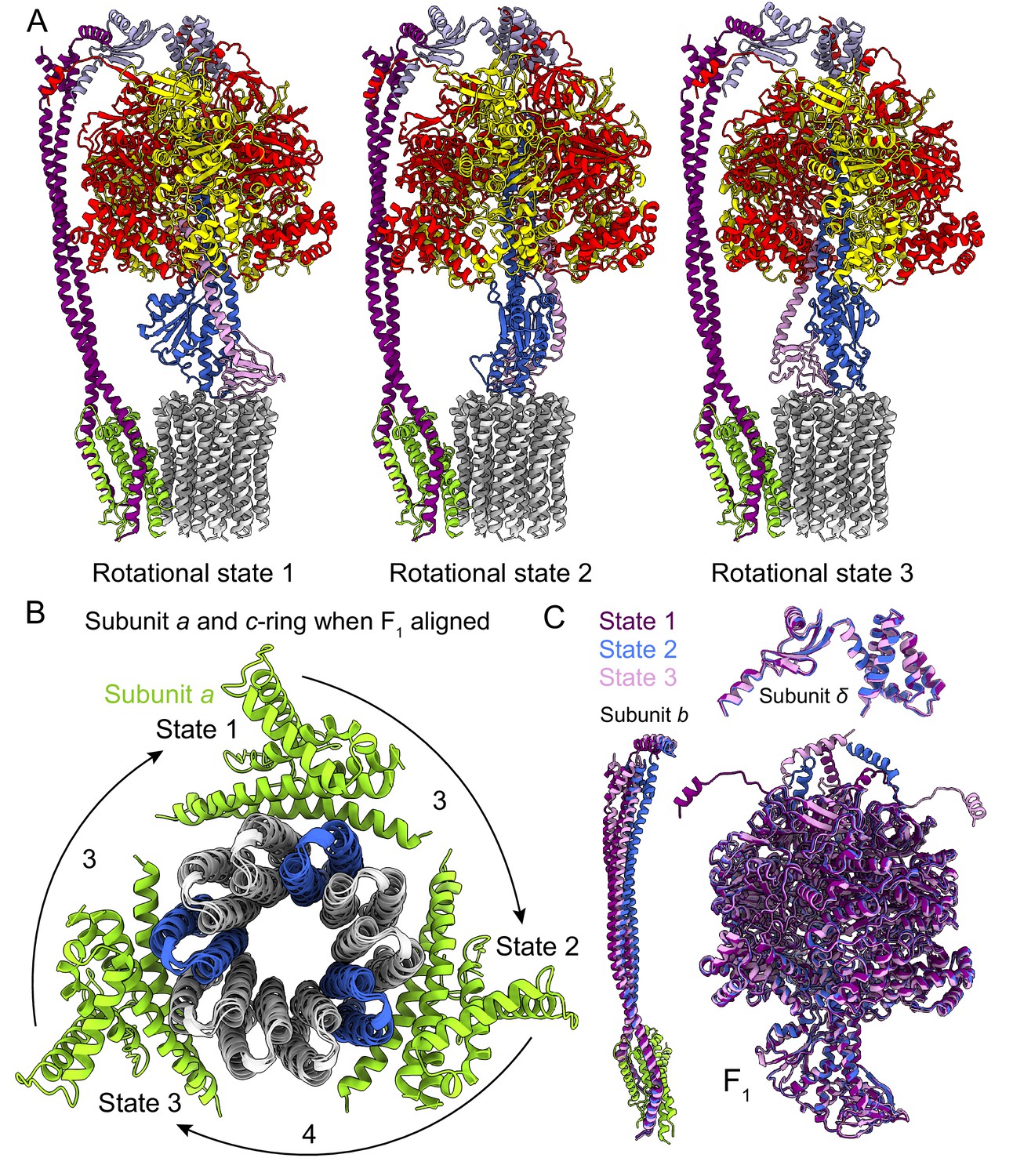
Rotational states of ATP synthase.
(A) Atomic models of the three rotational states of Bacillus PS3 ATP synthase with subunits coloured the same as in Figure 1. (B) Top view of the c-ring and subunit a of the three rotational states from the cytoplasm when the F1 regions of the three states are aligned. Rotation steps of the complex between states are ~3, 4, and 3 c-subunits. (C) Comparison of the atomic models of subunits b, δ, and other F1 region subunits in the different rotational states. The b subunits appear to be the most flexible part of the enzyme.
Video 1
Atomic models of the Bacillus PS3 ATP synthase in three rotational states.
https://doi.org/10.7554/eLife.43128.009The unequal number of c-subunit steps between rotational states or the different interactions made by the three αβ pairs with the b2δ peripheral stalk could lead to a variable rotation speed for the c-ring in the active enzyme, analogous to kinetic limping in kinesin motors (Asbury et al., 2003). Alternatively, flexibility in the enzyme could maintain a constant rotational velocity. Indeed, flexibility is thought to be important for the smooth transmission of power between the F1 and FO regions, which often have mismatched symmetries (Wang and Oster, 1998; Pänke et al., 2001; Mitome et al., 2004). Earlier studies suggested that the central stalk (subunits γ and ε in bacteria) is the main region responsible for the transient storage of torsional energy in rotary ATPases (Sielaff et al., 2008; Wächter et al., 2011; Ernst et al., 2012; Okazaki and Hummer, 2015). Comparison of the three rotational states of the Bacillus PS3 enzyme also shows that C-terminal water-soluble part of subunit b displays the most significant conformational variability between states, while the subunits in the F1 region show little flexibility beyond the catalytic states of the αβ pairs (Figure 2C; Video 1). The structure of the yeast ATP synthase FO dimer (Guo et al., 2017), which lacked the the F1 region and an intact peripheral stalk, showed that the c-ring and subunit a are held together by hydrophobic interactions rather than by the peripheral stalk. In Bacillus PS3 ATP synthase, the peripheral stalk is structurally simpler and more flexible than in yeast mitochondria (Srivastava et al., 2018), suggesting that the bacterial subunits a and the c-ring are also held together by hydrophobic interactions and not the peripheral stalk. Given that these structures represent resting states of the bacterial ATP synthase, additional subunits, such as those in the central stalk, may show flexibility while under strain during rotation.
Nucleotide binding in the F1 region and inhibition by subunit ε
The structure of the F1 region of the intact Bacillus PS3 ATP synthase and the earlier crystal structure of the dissociated F1-ATPase (PDB 4XD7) (Shirakihara et al., 2015) both show that the three catalytic β-subunits (βE, βTP, and βDP) adopt ‘open’, ‘closed’, and ‘open’ conformations, respectively (Figure 3A). In the crystal structure, which was prepared in the presence of CyDTA (trans-1,2-Diaminocyclohexane-N, N, N′, N′-tetraacetic acid monohydrate) as a chelating agent, there was no nucleotide in the three noncatalytic sites of the three α-subunits and the only nucleotide in a catalytic site was an ADP molecule without a Mg2+ ion in the βTP site. In contrast, all three non-catalytic sites in the cryo-EM map are occupied by Mg-ATP, while a Mg-ADP molecule and a weak density tentatively assigned to phosphate are found in the βTP site and by the p-loop of βE, respectively. The presence of physiological Mg2+ ions and nucleotide occupancy (Nalin and Cross, 1982) in the cryo-EM map suggest that it shows a snapshot of the enzyme in the middle of its physiological catalytic cycle.
Figure 3

Inhibition of ATP hydrolysis by subunit ε.
(A) Comparison of βDP (blue) with βE (pink, top) and βTP (light purple, bottom). βDP is forced to adopt an open conformation by subunit ε (dashed box). (B) Comparison of subunits γ (pink) and ε (blue) of ATP synthases from Bacillus PS3 (top) and E. coli (bottom, PDB 3OAA (Cingolani and Duncan, 2011)). The dashed box shows additional interaction between subunits ε and γ in the E. coli complex. (C) Cross-section through the catalytic F1 region of the Bacillus PS3 ATP synthase. Subunit ε (pink) in the rotor is blocked from rotating in the direction of ATP hydrolysis (clockwise) by βTP but is free to rotate in the direction of ATP synthesis (counterclockwise).
Bacillus PS3 ATP synthase is found in a conformation that has been proposed to allow ATP synthesis while ATP hydrolysis is auto-inhibited. In this state, subunit ε maintains an up conformation and inserts into the αDPβDP interface, forcing βDP to adopt an open conformation (Figure 3A, lower, dashed box) (Shirakihara et al., 2015). In the crystal structure (PDB 4XD7) (Shirakihara et al., 2015), the C-terminal sequence of subunit ε was modeled as two α-helical segments broken at Ser 106, while the cryo-EM structures show the C-terminal part is in fact entirely α-helical. In comparison, subunit ε from the auto-inhibited E. coli F1-ATPase structure (PDB 3OAA) (Cingolani and Duncan, 2011) maintains its two C-terminal α-helices (Figure 3B), with its βDP adopting a half-closed conformation that binds to Mg-ADP. The C-terminal α-helix of the E. coli subunit ε inserts slightly deeper into the αDPβDP interface but overall in a manner similar to that of the Bacillus PS3 subunit ε. However, the second α-helix in E. coli is offset by a 10-residue loop that allows it to interact with subunit γ. This interaction (Figure 3B, lower, dashed box) may stabilize the up conformation of subunit ε in E. coli, explaining why auto-inhibition in E. coli does not depend on ATP concentration (Laget and Smith, 1979; Sekiya et al., 2010) while in Bacillus PS3 it does. Interestingly, during ATP synthesis, Bacillus PS3 subunit ε was proposed to maintain the up conformation (Suzuki et al., 2003), suggesting that it only blocks ATP hydrolysis but not ATP synthesis. For a canonical ATP synthase, the substrates ADP and Pi bind to an open βE. The βE subsequently transitions to become the closed βDP and then βTP, driven by rotation of the central rotor, producing an ATP molecule that is ultimately released when the closed βTP converts back to an open βE (Abrahams et al., 1994). For the Bacillus PS3 ATP synthase to produce ATP with subunit ε in the up conformation, substrate would need to bind to the βDP site instead of the usual βE site, with an ATP molecule produced on transition to a closed βTP. The cryo-EM maps show that a clash between subunit ε and βTP blocks the central rotor turning in the direction of ATP hydrolysis while it is still free to turn in the direction of ATP synthesis (Figure 3C), which could explain the ability of subunit ε to selectively inhibit ATP hydrolysis (Suzuki et al., 2003).
Subunit organization in the FO region
In the bacterial ATP synthase structure, the FO subunits ab2 display an organization similar to the yeast FO complex (PDB 6B2Z, Figure 4A) (Guo et al., 2017). Subunit a and the first copy of subunit b occupy the same positions as their yeast counterparts, while the second copy of subunit b is found at a position equivalent to subunit 8 in the yeast enzyme, which is known as A6L in mammals. Atomic models for ATP synthase from mitochondria (Guo et al., 2017; Klusch et al., 2017; Srivastava et al., 2018) and chloroplasts (Hahn et al., 2018) support the idea that transmembrane proton translocation in ATP synthases occurs via two offset half-channels formed by subunit a (Vik and Antonio, 1994; Junge et al., 1997). Subunit a from Bacillus PS3 shares 21.0% and 29.1% sequence identity with its yeast and chloroplast homologs, respectively, and the atomic model shows that the folding of these homologs is mostly conserved (Figure 4—figure supplement 1). Multi-sequence alignment of subunit a from different species indicates that bacterial and chloroplast subunit a contain a larger periplasmic loop between α-helices 3 and 4 than found in the mitochondrial subunit (Figure 4A, left; Figure 4—figure supplement 2). The sequence for this loop varies significantly among species, suggesting that it is unlikely to be involved in the core function of proton translocation, despite being proximal to the periplasmic proton half-channel. Yeast and mammalian mitochondrial ATP synthases contain subunit f, which has a transmembrane α-helix adjacent to the transmembrane α-helix 1 of subunit a (Figure 4A, right), anchoring subunit b between α-helices 5 and 6 of subunit a. The location of the loop between α-helices 3 and 4 of the Bacillus PS3 subunit a suggests that it serves a similar structural role, compensating for the lack of subunit f in bacteria. The loop forms an additional interface with subunit b near the periplasmic side of the membrane region and may interact with the N terminus of subunit b in the periplasm as well. Two interfaces are also present between the second copy of subunit b and subunit a, one with the first transmembrane α-helix, and the other with the hairpin of α-helices 3 and 4 (Figure 4A). The structure suggests that two interfaces are necessary for subunits a and b to maintain a stable interaction.
Figure 4 with 3 supplements see all
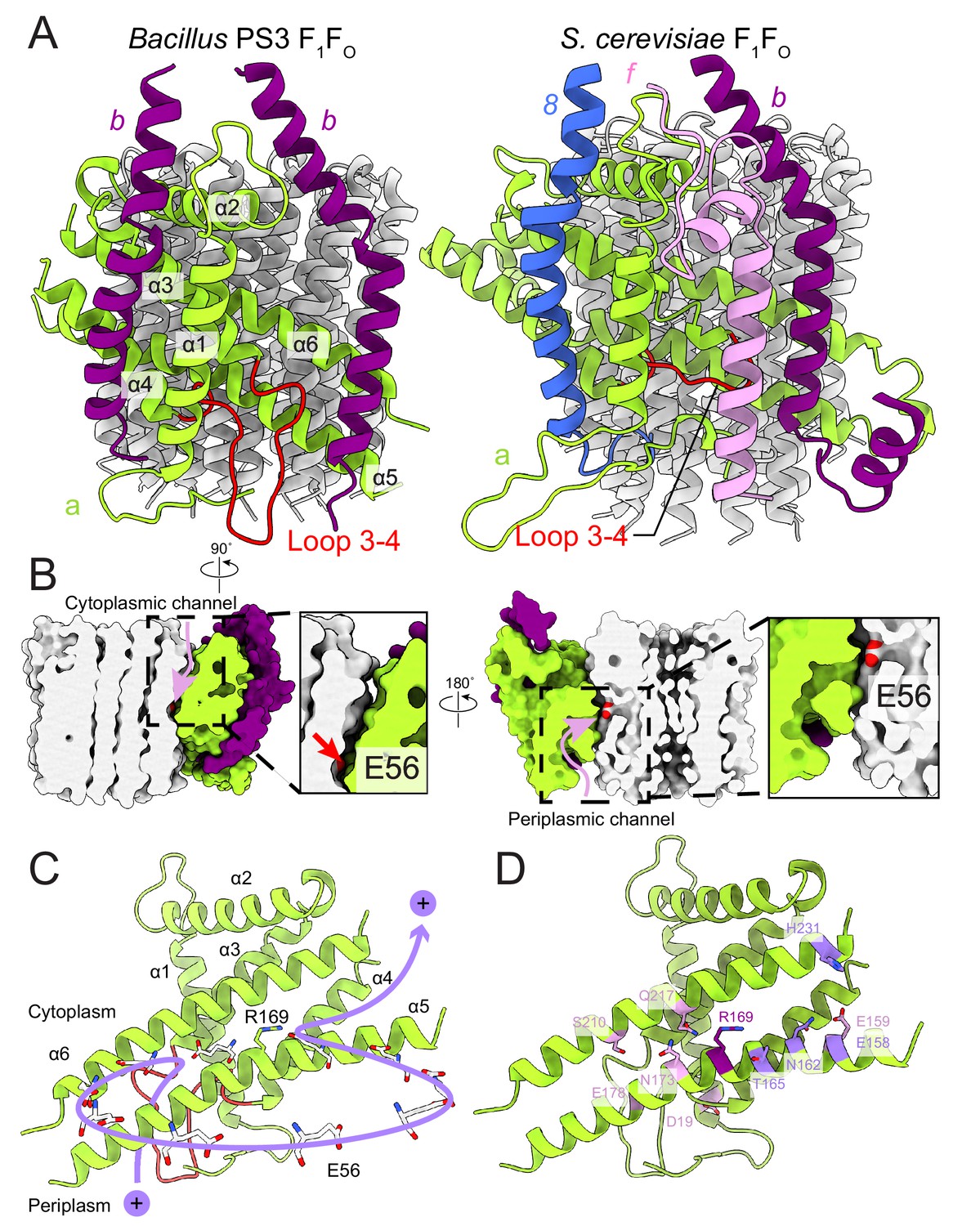
FO region subunits and proton translocation in Bacillus PS3 ATP synthase.
(A) Comparison of the FO regions from Bacillus PS3 (left) and S. cerevisiae (right). (B) Cross sections through a surface representation of the FO region (simulated with rolling of a 1.4 Å sphere (Goddard et al., 2018)) show the cytoplasmic (left) and periplasmic (right) proton half-channels. (C) Proton translocation pathway of Bacillus PS3 ATP synthase. During ATP synthesis, a proton enters the complex via the periplasmic half-channel, passing between α-helices 5 and 6 of subunit a to bind to the Glu 56 residue of a subunit c. The proton then rotates with the c-ring until it reaches the cytoplasmic half-channel formed between subunit a and the c-ring. In the cytoplasmic half-channel, the proton is released from the Glu residue due to its interaction with the positively charged Arg 169 of subunit a. A Glu 56 residue from each protomer of the c-ring is shown. (D) Subunit a of Bacillus PS3 ATP synthase. Arg 169 is in purple, important residues for proton translocation identified by mutagenesis in E. coli ATP synthase are in pink, and other residues that appears to contribute to proton transfer in the cytosolic proton half-channel are in light purple.
Proton translocation through the FO region
The Bacillus PS3 ATP synthase structure implies a path for proton translocation through the bacterial complex involving two half-channels similar to the paths described for the mitochondrial and chloroplast enzymes. The cytoplasmic half-channel consists of an aqueous cavity at the interface of subunit a and the c-ring (Figure 4B, left). The periplasmic half-channel is formed from a cavity between α-helices 1, 3, 4 and 5 of subunit a, and reaches the c-ring via a gap between α-helices 5 and 6 (Figure 4B, right). In the atomic model, both channels are visible when modeling the surface with a 1.4 Å sphere that mimics a water molecule (Goddard et al., 2018) (Figure 4B). The channels are wide and hydrophilic, suggesting that water molecules could pass freely through each of the channels before accessing the conserved Glu 56 of the c-subunits. During ATP synthesis, protons travel to the middle of the c-ring via the periplasmic half-channel and bind to the Glu 56 residue of a subunit c (Figure 4C). Protonation of the glutamate allows rotation of the ring counter-clockwise, when viewed from F1 toward FO, delivering the subunit c into the hydrophobic lipid bilayer. Protonation of the remaining nine subunits in the c-ring returns the first glutamate to subunit a, now into the cytoplasmic half-channel, where it releases its proton to the cytoplasm due to interaction with the positively charged Arg 169 of subunit a. The proposed channels are consistent with a series of experiments probing water accessibility of residues in the E. coli ATP synthase subunit a by mutating them to cysteines and testing their accessibility by Ag+ ions (Angevine et al., 2003; Angevine and Fillingame, 2003; Angevine et al., 2007; Moore et al., 2008). Residues that are close to the c-ring, such as S202, S206, N214, and I249 (A161, T165, N173, and G214 in Bacillus PS3) are among the most accessible by Ag+ 52, suggesting that the half-channels do not exclude Ag+. Therefore, it is likely that Na+ ions, which are similar in size to Ag+ ions, are also not excluded by subunit a. It is also known that the c-rings from Na+- and H+-driven ATP synthases have different affinities for Na+ and H (Krah et al., 2010; Leone et al., 2015; Mayer et al., 2012; Schlegel et al., 2012), and that the c-ring of the Na+-driven ATP synthase from llyobacter tartaricus does not bind K+ or Cs+ ions (Leone et al., 2015). Together, these results suggest that ion selectivity in ATP synthases is probably determined by the c-ring, not subunit a.
In eukaryotes, subunit a is encoded by the mitochondrial genome, limiting genetic interrogation of the roles of different residues. In contrast, numerous mutagenesis studies have been performed on bacterial subunits a and b, with E. coli ATP synthase being the most frequently studied (Vik et al., 2000; Cain, 2000). A single G9D mutation in the E. coli subunit b (positionally equivalent to Y13D in Bacillus PS3), results in assembled but non-functional ATP synthase (Porter et al., 1985), while multiple N-terminal mutations in subunit b can either disrupt enzyme assembly or ATP hydrolysis (Hardy et al., 2003). In Bacillus PS3, Tyr 13 is part of the transmembrane α-helix of subunit b and is adjacent to Gly 188 of subunit a (Figure 4—figure supplement 3, dashed box). In E. coli subunit a, Gly 188 is replaced by a leucine (Leu 229). Therefore, the G9D mutation in E. coli not only introduces a charged residue into a hydrophobic transmembrane α-helix, but also creates a steric clash with Leu 229 of subunit a, explaining why the mutation leads to an inactive enzyme. Remarkably, the single N-terminal membrane-embedded α-helix in each of the two copies of subunit b in the Bacillus PS3 ATP synthase forms different interactions with subunit a (Figure 4A). One surface interacts with transmembrane α-helices 1, 2, 3, and 4 of subunit a while the other interacts with α-helices 5 and 6 and the loop between α-helices 3 and 4 of subunit a. Given that the N-terminal α-helix of subunit b makes interactions with different regions of subunit a, it is not surprising that mutations in this region are often detrimental to the assembly and activity of the complex. Cross-linking experiments suggested that the N terminus of the two copies of subunit b are in close proximity to each other (Dmitriev et al., 1999). However, the atomic model shows that the transmembrane α-helix of the b-subunits are on opposite sides of subunit a, suggesting that the cross-linking results may be due to non-specific interactions of b-subunits from neighboring ATP synthases.
In E. coli, Arg 210 of subunit a (Arg 169 in Bacillus PS3) tolerates the fewest mutations (Cain and Simoni, 1989; Hatch et al., 1995; Lightowlers et al., 1987; Ishmukhametov et al., 2008). Recent structures of rotary ATPases suggest that the importance of this residue derives from its role in releasing protons bound to the Glu residues of the c-subunits as they enter the cytoplasmic half-channel, as well as preventing short-circuiting of the proton path by protons flowing between half-channels without rotation of the c-ring (Morales-Rios et al., 2015; Zhao et al., 2015; Zhou et al., 2015; Allegretti et al., 2015; Mazhab-Jafari et al., 2016). Other residues in the E. coli subunit a identified by mutation as being functionally important include Glu 196 (Glu 159 in Bacillus PS3) (Vik et al., 1988; Lightowlers et al., 1988), Glu 219 (Glu 178) (Vik et al., 1988; Lightowlers et al., 1988; Eya et al., 1991), His 245 (Ser 210) (Lightowlers et al., 1987; Cain and Simoni, 1986; Cain and Simoni, 1988), Asp 44 (Asp 19) (Howitt et al., 1990), Asn 214 (Asn 173) (Cain and Simoni, 1989), and Gln 252 (Gln 217) (Eya et al., 1991; Hartzog and Cain, 1993) (Figure 4D). When mapped to the Bacillus PS3 structure, only Glu 196 (Glu 159 in Bacillus PS3) is close to the cytoplasmic half-channel. Extensive mutations of E. coli Glu 196 showed that enzyme activity depends on the charge and polarity of the residue with Glu > Asp > Gln = Ser = His > Asn > Ala > Lys (Vik et al., 1988). Therefore, the negative surface charge from Glu 196 (Glu 159) near the cytoplasmic half-channel facilitates proton transport across the lipid bilayer. The atomic model of subunit a also suggests that other residues such as Bacillus PS3 Thr 165, Asn 162, Glu 158, Tyr 228, and His 231, which are close to the cytoplasmic half-channel, may contribute to channel formation. Many functional residues identified by mutagenesis are clustered around the periplasmic half-channel. In the atomic model of the Bacillus PS3 subunit a, Asp 19 and Glu 178 are close to the periplasm, while Ser 210, Asn 173, and Gln 217 are deeper inside the membrane. Among these residues, Glu 178 and Ser 210 are considered to be more important to enzyme function than Asn 173 and Gln 217, as mutations of corresponding residues in E. coli are more likely to abolish the proton translocation by the complex (Vik et al., 2000). The Glu 219/His 245 residue pair in E. coli (Cain and Simoni, 1988) also occur in the S. cerevisiae (His 185/Glu 223) and human (His 168/Glu 203) mitochondrial ATP synthases (Figure 4—figure supplement 2). These residues do not appear to be close enough to form a hydrogen bond in the S. cerevisae FO dimer structure (Guo et al., 2017). In Bacillus PS3 subunit a, the His residue is replaced by a serine (Ser 210) that similarly does not appear to close enough to Glu 178 to form a hydrogen bond. Interestingly, although many of these functional residues appear important, their mutation to amino acids that cannot be protonated or deprotonated often does not completely abolish proton translocation (Vik et al., 2000). The atomic model of Bacillus PS3 subunit a shows that the proton half-channels are wide enough for water molecules to pass through freely. This observation suggests that the function of these conserved polar and charged residues is not the direct transfer of protons during translocation. Rather, their presence may help maintain a hydrophilic environment for water-filled proton channels. This role allows different species to use unique sets of polar and charged residues to form their proton half-channels. This variability suggests a remarkably flexible proton translocation mechanism for this highly efficient macromolecular machine.
Materials and methods
Protein expression and purification
Request a detailed protocolE. coli strain DK8, in which the genes encoding endogenous ATP synthase subunits were deleted (Klionsky et al., 1984), was transformed with plasmid pTR-ASDS (Suzuki et al., 2002) encoding Bacillus PS3 ATP synthase with a 10 × His tag at the N terminus of subunit β. Transformed E. coli cells were grown in 2 × TY medium at 37°C for 20 hr before being harvested by centrifugation at 5400 g. Cell pellets were resuspended in lysis buffer (50 mM Tris-HCl pH 7.4, 5 mM MgCl2, 10% [w/v] glycerol, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF) and lysed with three passes through an EmulsiFlex-C3 homogenizer (Avestin) at 15 to 20 kbar. All protein preparation steps were performed at 4°C unless otherwise stated. Cell debris was removed at 12,250 g for 20 min, and the cell membrane fraction was collected by centrifugation at 184,000 g for 1 hr. Membranes were washed twice with lysis buffer before being resuspended in solubilization buffer (50 mM Tris-HCl pH 7.4, 10% [w/v] glycerol, 250 mM sucrose, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 1 mM PMSF) and solubilized by the addition of glycol-diosgenin (GDN) to 1% (w/v) and mixing for 1 hr at room temperature. Insoluble material was removed by centrifugation at 184,000 g for 45 min and solubilized membranes were loaded onto a 5 ml HisTrap HP column (GE Healthcare) equilibrated with buffer A (solubilization buffer with 20 mM imidazole, 300 mM sodium chloride, and 0.02% [w/v] GDN). The column was washed with five column volumes of buffer A, and ATP synthase was eluted with three column volumes of buffer B (buffer A with 200 mM imidazole). Fractions containing ATP synthase were pooled and concentrated prior to being loaded onto a Superose 6 increase 10/300 column (GE Healthcare) equilibrated with gel filtration buffer (20 mM Tris-HCl pH 7.4, 5 mM MgCl2, 10% [w/v] glycerol, 150 mM sodium chloride, 5 mM 6-aminocaproic acid, 5 mM benzamidine, 0.02% [w/v] GDN). The peak corresponding to Bacillus PS3 ATP synthase was pooled and concentrated to ~10 mg/ml prior to storage at −80°C.
Cryo-EM and image analysis
Request a detailed protocolPrior to grid freezing, glycerol was removed from samples with a Zeba spin desalting column (Thermo Fisher Scientific). Purified ATP synthase (2.5 μL) was applied to homemade nanofabricated EM grids (Marr et al., 2014) consisting of a holey layer of gold (Russo and Passmore, 2014; Meyerson et al., 2014) that had been glow-discharged in air for 2 min. Grids were then blotted on both sides in a FEI Vitrobot mark III for 26 s at 4°C and ~100% RH before freezing in a liquid ethane/propane mixture (Tivol et al., 2008). Cryo-EM data were collected with a Titan Krios G3 electron microscope (Thermo Fisher Scientific) operated at 300 kV equipped with a Falcon 3EC direct detector device camera automated with EPU software. Data were recorded as 60 s movies at 2 s per frame with an exposure rate of 0.8 electron/pixel/s, and a calibrated pixel size of 1.06 Å.
All image processing steps were performed in cryoSPARC v2 (Punjani et al., 2017) unless otherwise stated. 10,940 movies were collected. Movie frames were aligned with an implementation of alignframes_lmbfgs within cryoSPARC v2 (Rubinstein and Brubaker, 2015) and CTF parameters were estimated from the average of aligned frames with CTFFIND4 (Rohou and Grigorieff, 2015). 1,866,804 single particle images were selected from the aligned frames with Relion 2.1 (Fernandez-Leiro and Scheres, 2017) and beam-induced motion of individual particles corrected with an improved implementation of alignparts_lmbfgs within cryoSPARC v2 (Rubinstein and Brubaker, 2015). A subset of 1,238,140 particle images were selected by 2D classification in cryoSPARC v2. After initial rounds of ab-initio 3D classification and heterogeneous refinement, three classes corresponding to three main rotational states of the enzyme were identified, containing 405,432, 314,448, and 175,694 particles images (Figure 1—figure supplement 2). These 3D classes were refined with non-uniform refinement to overall resolutions of 3.0 Å, 3.0 Å and 3.2 Å, respectively, with the F1 region reaching higher resolution than the FO region of the complex as seen from estimation of local resolution (Figure 1—figure supplement 3). Masked refinement with signal subtraction (focused refinement) (Bai et al., 2015) around subunits ab2c10δ excluding the detergent micelle improved the map quality of the membrane-embedded region as well as the peripheral stalk for all three classes. The membrane-embedded region (subunits ac10 and transmembrane α-helices of the b-subunits) was improved further by focused refinement with particle images from all three classes, yielding a map at 3.3 Å resolution. All Fourier shell correlation (FSC) curves were calculated with independently refined half-maps and resolution was assessed at the 0.143 criterion with correction for the effects of masking maps. For illustration purposes, composite maps for each of the three rotational states were generated by combining the F1 region of the maps from non-uniform refinement, the peripheral stalk region from the maps obtained with focused refinement of subunits ab2c10δ, and the map from focused refinement of the membrane-embedded region. Specifically, each map was multiplied by a mask surrounding the region of interest and the resulting maps were adjusted to similar absolute grey scale by multiplying with a constant with relion_image_handler before being merged with the maximum function volume operation in UCSF Chimera (Pettersen et al., 2004). These composite maps were not used for model refinement.
Model building and refinement
Request a detailed protocolAtomic models for subunits α3β3γεδ from all three rotational states were built with Coot (Emsley and Cowtan, 2004) into the maps of the intact complex from non-uniform refinement using PDB 4XD7 (Shirakihara et al., 2015) and PDB 6FKF (Hahn et al., 2018) as initial models for subunits α3β3γε and subunit δ, respectively. Subunits ac10 and the membrane-embedded regions of subunits b2 were built de novo in the 3.3 Å map of the membrane-embedded region of the complex from focused refinement. Backbone models of the soluble region of subunits b2 for all three conformations were built with the maps from focused refinement of the peripheral stalk. Models were refined into their respective maps with phenix.real_space_refine (Adams et al., 2010) using secondary structure and geometric restraints followed by manual adjustments in Coot (Supplementary file 1). The quality of the models was evaluated by MolProbity (Chen et al., 2010) and EMRinger (Barad et al., 2015). To generate full models for all three rotational states, the model of subunits ac10 and the membrane region of subunit b2 were fit into the full maps of each conformation as three rigid bodies (a, c10, and b2 membrane region) with phenix.real_space_refine. For classes 1 and 3, the backbone models of the soluble region of subunit b2 did not fit the full maps well, and thus the fit was improved by molecular dynamics flexible fitting (MDFF) (Trabuco et al., 2008). The final composite model for each rotational state was generated by combining the models of subunits α3β3γεδ, the rigid body refined subunits ac10 and membrane region of b2, and the backbone model of the soluble region of b2. Figures and movie were generated with Chimera (Pettersen et al., 2004) and ChimeraX (Goddard et al., 2018).
Data availability
CryoEM maps have been deposited in EMDB and atomic models in PDB.
-
Electron Microscopy Data BankID EMD-9333. Intact class 1.
-
Electron Microscopy Data BankID EMD-9334. Intact class 2.
-
Electron Microscopy Data BankID EMD-9335. Intact class 3.
-
Electron Microscopy Data BankID EMD-9336. Focused Fo/stalk class 1.
-
Electron Microscopy Data BankID EMD-9337. Focused Fo/stalk class 2.
-
Electron Microscopy Data BankID EMD-9338. Focused Fo/stalk class 3.
-
Electron Microscopy Data BankID EMD-9327. Focused Fo.
References
-
PHENIX: a comprehensive Python-based system for macromolecular structure solutionActa Crystallographica Section D Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
Aqueous access pathways in ATP synthase subunit a. reactivity of cysteine substituted into transmembrane helices 1, 3, and 5The Journal of Biological Chemistry 282:9001–9007.https://doi.org/10.1074/jbc.M610848200
-
Aqueous access channels in subunit a of rotary ATP synthaseThe Journal of Biological Chemistry 278:6066–6074.https://doi.org/10.1074/jbc.M210199200
-
Mutagenic analysis of the F0 stator subunitsJournal of Bioenergetics and Biomembranes 32:365–371.
-
Impaired proton conductivity resulting from mutations in the a subunit of F1F0 ATPase in Escherichia coliThe Journal of Biological Chemistry 261:10043–10050.
-
Interaction between Glu-219 and His-245 within the a subunit of F1F0-ATPase in Escherichia coliThe Journal of Biological Chemistry 263:6606–6612.
-
Proton translocation by the F1F0ATPase of Escherichia coli. mutagenic analysis of the a subunitThe Journal of Biological Chemistry 264:3292–3300.
-
MolProbity: all-atom structure validation for macromolecular crystallographyActa Crystallographica Section D Biological Crystallography 66:12–21.https://doi.org/10.1107/S0907444909042073
-
Structure of the ATP synthase catalytic complex (F(1)) from Escherichia coli in an autoinhibited conformationNature Structural & Molecular Biology 18:701–707.https://doi.org/10.1038/nsmb.2058
-
On the structure of the stator of the mitochondrial ATP synthaseThe EMBO Journal 25:2911–2918.https://doi.org/10.1038/sj.emboj.7601177
-
Structure of the membrane domain of subunit b of the Escherichia coli F0F1 ATP synthaseThe Journal of Biological Chemistry 274:15598–15604.
-
Coot: model-building tools for molecular graphicsActa crystallographica. Section D, Biological crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Elastic deformations of the rotary double motor of single FoF1-ATP synthases detected in real time by förster resonance energy transferBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1817:1722–1731.https://doi.org/10.1016/j.bbabio.2012.03.034
-
Role of the carboxyl terminal region of H(+)-ATPase (F0F1) a subunit from Escherichia coliArchives of Biochemistry and Biophysics 284:71–77.https://doi.org/10.1016/0003-9861(91)90265-K
-
A pipeline approach to single-particle processing in RELIONActa Crystallographica Section D Structural Biology 73:496–502.https://doi.org/10.1107/S2059798316019276
-
Mutagenesis studies of the F1F0 ATP synthase b subunit membrane domainJournal of Bioenergetics and Biomembranes 35:389–397.
-
Mutational analysis of the function of the a-subunit of the F0F1-APPase of Escherichia coliBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1015:264–268.https://doi.org/10.1016/0005-2728(90)90030-8
-
ATP synthesis without R210 of subunit a in the Escherichia coli ATP synthaseBiochimica et biophysica acta 1777:32–38.https://doi.org/10.1016/j.bbabio.2007.11.004
-
The stability of proteins in extreme environmentsCurrent Opinion in Structural Biology 8:738–748.https://doi.org/10.1016/S0959-440X(98)80094-8
-
ATP synthase: an electrochemical transducer with rotatory mechanicsTrends in Biochemical Sciences 22:420–423.https://doi.org/10.1016/S0968-0004(97)01129-8
-
Thermophilic F1-ATPase is activated without dissociation of an endogenous inhibitor, epsilon subunitThe Journal of Biological Chemistry 272:24906–24912.
-
In vivo evidence for the role of the epsilon subunit as an inhibitor of the proton-translocating ATPase of Escherichia coliJournal of Bacteriology 160:1055–1060.
-
Structural and energetic basis for H+ versus na+ binding selectivity in ATP synthase fo rotorsBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1797:763–772.https://doi.org/10.1016/j.bbabio.2010.04.014
-
How do thermophilic proteins deal with heat?Cellular and Molecular Life Sciences 58:1216–1233.https://doi.org/10.1007/PL00000935
-
Inhibitory properties of endogenous subunit epsilon in the Escherichia coli F1 ATPaseArchives of Biochemistry and Biophysics 197:83–89.https://doi.org/10.1016/0003-9861(79)90222-4
-
The proton pore in the Escherichia coli F0F1-ATPase: a requirement for arginine at position 210 of the a-subunitBiochimica Et Biophysica Acta (BBA) - Bioenergetics 894:399–406.https://doi.org/10.1016/0005-2728(87)90118-6
-
The proton pore in the Escherichia coli F0F1-ATPase: substitution of glutamate by glutamine at position 219 of the alpha-subunit prevents F0-mediated proton permeabilityBiochimica Et Biophysica Acta (BBA) - Bioenergetics 933:241–248.https://doi.org/10.1016/0005-2728(88)90031-X
-
Fabrication of carbon films with ∼ 500nm holes for cryo-EM with a direct detector deviceJournal of Structural Biology 185:42–47.https://doi.org/10.1016/j.jsb.2013.11.002
-
A c subunit with four transmembrane helices and one ion (Na+)-binding site in an archaeal ATP synthase: implications for c ring function and structureThe Journal of Biological Chemistry 287:39327–39337.https://doi.org/10.1074/jbc.M112.411223
-
The cytoplasmic loops of subunit a of Escherichia coli ATP synthase may participate in the proton translocating mechanismThe Journal of Biological Chemistry 283:13044–13052.https://doi.org/10.1074/jbc.M800900200
-
Adenine nucleotide binding sites on beef heart F1-ATPase. Specificity of cooperative interactions between catalytic sitesThe Journal of Biological Chemistry 257:8055–8060.
-
UCSF chimera--a visualization system for exploratory research and analysisJournal of Computational Chemistry 25:1605–1612.https://doi.org/10.1002/jcc.20084
-
High-resolution structure of the rotor ring of a proton-dependent ATP synthaseNature Structural & Molecular Biology 16:1068–1073.https://doi.org/10.1038/nsmb.1678
-
Role of the b subunit of the Escherichia coli proton-translocating ATPase. A mutagenic analysisThe Journal of Biological Chemistry 260:8182–8187.
-
Structure of the mycobacterial ATP synthase fo rotor ring in complex with the anti-TB drug bedaquilineScience Advances 1:e1500106–e1500109.https://doi.org/10.1126/sciadv.1500106
-
Deciphering key features in protein structures with the new ENDscript serverNucleic Acids Research 42:W320–W324.https://doi.org/10.1093/nar/gku316
-
CTFFIND4: fast and accurate defocus estimation from electron micrographsJournal of Structural Biology 192:216–221.https://doi.org/10.1016/j.jsb.2015.08.008
-
Alignment of cryo-EM movies of individual particles by optimization of image translationsJournal of Structural Biology 192:188–195.https://doi.org/10.1016/j.jsb.2015.08.007
-
Activation and stiffness of the inhibited states of F1-ATPase probed by single-molecule manipulationJournal of Biological Chemistry 285:11411–11417.https://doi.org/10.1074/jbc.M109.099143
-
Single molecule behavior of inhibited and active states of Escherichia coli ATP synthase F1 rotationThe Journal of Biological Chemistry 285:42058–42067.https://doi.org/10.1074/jbc.M110.176701
-
Kinetic equivalence of transmembrane pH and electrical potential differences in ATP synthesisJournal of Biological Chemistry 287:9633–9639.https://doi.org/10.1074/jbc.M111.335356
-
F(0) of ATP synthase is a rotary proton channel. obligatory coupling of proton translocation with rotation of c-subunit ringThe Journal of Biological Chemistry 277:13281–13285.https://doi.org/10.1074/jbc.M111210200
-
F0F1-ATPase/synthase is geared to the synthesis mode by conformational rearrangement of epsilon subunit in response to proton motive force and ADP/ATP balanceThe Journal of Biological Chemistry 278:46840–46846.https://doi.org/10.1074/jbc.M307165200
-
An improved cryogen for plunge freezingMicroscopy and Microanalysis 14:375–379.https://doi.org/10.1017/S1431927608080781
-
Mutagenesis of the alpha subunit of the F1Fo-ATPase from Escherichia coli. Mutations at Glu-196, Pro-190, and Ser-199.The Journal of Biological Chemistry 263:6599–6605.
-
A model for the structure of subunit a of the Escherichia coli ATP synthase and its role in proton translocationBiochimica Et Biophysica Acta (BBA) - Bioenergetics 1458:457–466.https://doi.org/10.1016/S0005-2728(00)00094-3
-
A mechanism of proton translocation by F1F0 ATP synthases suggested by double mutants of the a subunitThe Journal of Biological Chemistry 269:30364–30369.
-
The ATP synthase: the understood, the uncertain and the unknownBiochemical Society Transactions 41:1–16.https://doi.org/10.1042/BST20110773
Article and author information
Author details
Funding
Canadian Institutes of Health Research (MOP 81294)
- John L Rubinstein
Canada Research Chairs
- John L Rubinstein
Japan Society for the Promotion of Science (JP18H02409)
- Toshiharu Suzuki
Canada Foundation for Innovation
- John L Rubinstein
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Dr. Samir Benlekbir (the Hospital for Sick Children) for helping with cryo-EM data collection and Prof. Tomitake Tsukihara (Osaka University, Japan) for discussions. This work was supported by Canadian Institutes of Health Research operating grant MOP 81294. Cryo-EM data was collected at the Toronto High-Resolution High-Throughput cryo-EM facility, supported by the Canada Foundation for Innovation and Ontario Research Fund. HG was supported by an Ontario Graduate Scholarship and a University of Toronto Excellence Award. TS was supported by Japan Society for the Promotion of Science Grants-in-Aid for Scientific Research (KAKENHI) Grant JP18H02409. JLR was supported by the Canada Research Chairs program.
Version history
- Received: October 26, 2018
- Accepted: February 2, 2019
- Accepted Manuscript published: February 6, 2019 (version 1)
- Version of Record published: February 15, 2019 (version 2)
Copyright
© 2019, Guo et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 26,643
- views
-
- 1,752
- downloads
-
- 129
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structure of a bacterial ATP synthase
eLife 8:e43128.
https://doi.org/10.7554/eLife.43128
Further reading
-
- Biochemistry and Chemical Biology
- Cell Biology
Mediator of ERBB2-driven Cell Motility 1 (MEMO1) is an evolutionary conserved protein implicated in many biological processes; however, its primary molecular function remains unknown. Importantly, MEMO1 is overexpressed in many types of cancer and was shown to modulate breast cancer metastasis through altered cell motility. To better understand the function of MEMO1 in cancer cells, we analyzed genetic interactions of MEMO1 using gene essentiality data from 1028 cancer cell lines and found multiple iron-related genes exhibiting genetic relationships with MEMO1. We experimentally confirmed several interactions between MEMO1 and iron-related proteins in living cells, most notably, transferrin receptor 2 (TFR2), mitoferrin-2 (SLC25A28), and the global iron response regulator IRP1 (ACO1). These interactions indicate that cells with high MEMO1 expression levels are hypersensitive to the disruptions in iron distribution. Our data also indicate that MEMO1 is involved in ferroptosis and is linked to iron supply to mitochondria. We have found that purified MEMO1 binds iron with high affinity under redox conditions mimicking intracellular environment and solved MEMO1 structures in complex with iron and copper. Our work reveals that the iron coordination mode in MEMO1 is very similar to that of iron-containing extradiol dioxygenases, which also display a similar structural fold. We conclude that MEMO1 is an iron-binding protein that modulates iron homeostasis in cancer cells.
-
- Biochemistry and Chemical Biology
- Structural Biology and Molecular Biophysics
NADPH oxidases (NOX) are transmembrane proteins, widely spread in eukaryotes and prokaryotes, that produce reactive oxygen species (ROS). Eukaryotes use the ROS products for innate immune defense and signaling in critical (patho)physiological processes. Despite the recent structures of human NOX isoforms, the activation of electron transfer remains incompletely understood. SpNOX, a homolog from Streptococcus pneumoniae, can serves as a robust model for exploring electron transfers in the NOX family thanks to its constitutive activity. Crystal structures of SpNOX full-length and dehydrogenase (DH) domain constructs are revealed here. The isolated DH domain acts as a flavin reductase, and both constructs use either NADPH or NADH as substrate. Our findings suggest that hydride transfer from NAD(P)H to FAD is the rate-limiting step in electron transfer. We identify significance of F397 in nicotinamide access to flavin isoalloxazine and confirm flavin binding contributions from both DH and Transmembrane (TM) domains. Comparison with related enzymes suggests that distal access to heme may influence the final electron acceptor, while the relative position of DH and TM does not necessarily correlate with activity, contrary to previous suggestions. It rather suggests requirement of an internal rearrangement, within the DH domain, to switch from a resting to an active state. Thus, SpNOX appears to be a good model of active NOX2, which allows us to propose an explanation for NOX2’s requirement for activation.




{kind=link}
{kind=link}
{kind=link}
{kind=link}