Homing Genetic Elements: Mobile DNAs and switching mating types in yeast
The gene that allows budding yeast cells to switch their mating type evolved from a newly discovered family of genes named weird HO.
- Department of Biological Science, University of Buffalo, The State University of New York, United States
Biologists working in evolution are often surprised when they find a ‘missing link’ with features that could not have been predicted from the features of its predecessors and successors. Such findings demonstrate the wonderful meandering path that evolution can take, with fitness rather than simplicity determining the route. One recent example is the discovery of a missing link in the evolutionary trajectory of a gene called HO that is involved in mating in Saccharomyces cerevisiae and some other species of budding yeast.
Individual yeast cells in these species belong to one of two mating types, known as ‘a’ and ‘α’, and mating occurs when a cell of one type fuses with a cell of the other type. However, if a cell is isolated from potential mating partners, it can 'switch' to the other type and self-mate with its own daughter cell. Switching is triggered by the HO gene, which encodes an endonuclease that specifically cuts the genomic locus (called MAT) that determines the mating type of the cell (Lee and Haber, 2015). This cut is repaired by copying a silent copy of the gene for the other mating type and inserting it into the MAT locus.
Switching between mating types is thought to have evolved in two stages (Hanson and Wolfe, 2017). First, the three loci that carry the mating-type genes (the active MAT locus and the two loci with the silent genes) came together in one genome through gene duplications and genome rearrangements. Although this allowed switching to occur, the process was inefficient. Second, the HO gene emerged and increased the frequency of switching. However, the evolutionary history of this gene is still unclear (Haber and Wolfe, 2005; Keeling and Roger, 1995).
It has been suggested that the HO gene evolved from mobile DNA elements called homing inteins, which ‘home’ into the coding sequence of a specific host gene. Specifically, a homing intein in yeast called VDE has a similar sequence to HO and is thought to be the evolutionary ancestor of this gene. The protein encoded by the VDE gene is initially expressed within its host protein; it then extracts itself and goes on to cut copies of the host gene that lack the intein sequence. This cut triggers a repair reaction similar to the one induced by the endonuclease encoded by HO, and results in VDE being copied from an occupied gene to an empty host gene. Thus, both VDE and HO code for proteins that initiate DNA repair reactions that change the genetic sequence of a cell. However, there are differences: HO is free-standing and does not self-splice, whereas VDE is embedded in a host gene; moreover, the protein produced by HO also contains a zinc finger domain that binds to DNA.
Now, in eLife, Kenneth Wolfe and co-workers from University College Dublin and Université Montpellier – including Aisling Coughlan as first author – report the discovery of a gene family which represents a missing link between VDE and HO (Coughlan et al., 2020). Similar to HO, the genes in the new family – named WHO for weird HO – have an intein-like cutting domain fused to a zinc finger domain (Figure 1A). Coughlan et al. discovered WHO genes in the genomes of yeast species that belong to the Torulaspora and Lachancea genera, and found them clustered next to a gene called FBA1. This arrangement suggests that WHO genes are mobile DNA elements that have inserted themselves at the end of the FBA1 gene. Furthermore, fragments of FBA1 can be found interspersed among the WHO genes, suggesting that FBA1 is cut and partly duplicated during insertion.
Figure 1
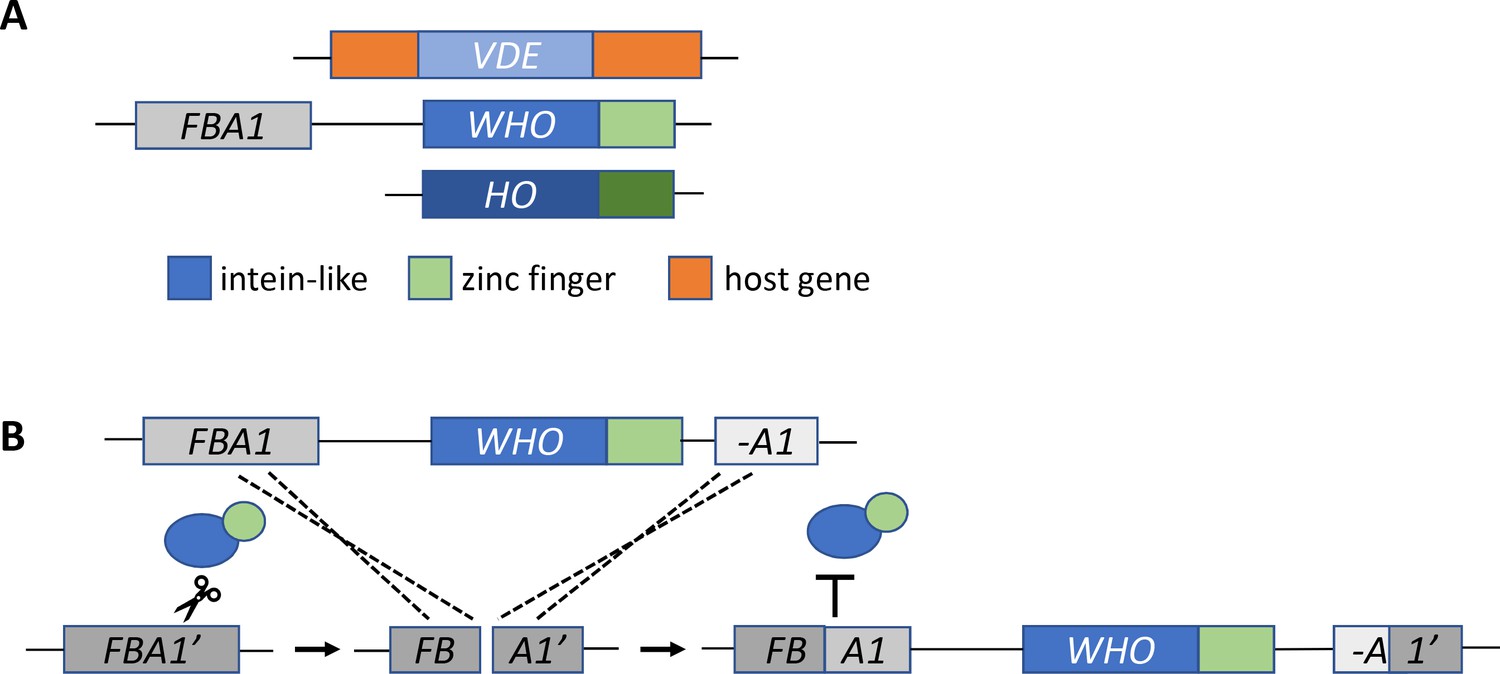
The missing link in the evolution of HO.
(A) The HO gene is structurally similar to two other mobile DNA elements, VDE and WHO. All three genes have an intein-like domain (blue rectangle) that includes an endonuclease domain: in HO this domain cuts DNA at the MAT locus. The WHO and HO genes also have a zinc finger domain (green). The WHO gene is also located next to its target, the FBA1 gene. (B) Coughlan et al. propose that the WHO gene homes into the FBA1 gene as follows. First, the enzyme encoded by the WHO gene (blue and green ovals) cuts a target gene, represented as FBA1’ (dark gray box), which is not flanked by WHO. Next, the break is repaired using a template of the FBA1 gene (medium gray) which is adjacent to the WHO gene and a fragment of FBA1 (A1, light gray). The template sequence (including the WHO gene) is copied and inserted into the cleaved site of the target gene. Recombination between the target FBA1’ gene and the template FBA1 gene creates a new FBA1 gene (dark and medium gray) and FBA1 fragment (light gray and dark gray), which sit on either side of the WHO gene. The newly recombined FBA1 gene is no longer susceptible to cleavage by the enzyme encoded by WHO (represented by the T symbol).
To find out whether the cutting domain in the WHO family of genes targets FBA1, Coughlan et al. studied a gene called WHO6 that comes from the species Torulaspora delbrueckii. The experiments were carried out in S. cerevisiae cells that contained a copy of the FBA1 gene from T. delbrueckii. If the enzyme encoded by WHO6 cuts the TdFBA1 target gene, cell division will be halted until the DNA is repaired. Coughlan et al. found that when WHO6 was overexpressed, cells that contained the TdFBA1 gene were less likely to survive. The only cells that survived were those in which the TdFBA1 gene had been modified to make it resistant to further cutting (Figure 1B).
These results indicate that the WHO6 gene can trigger a repair reaction at the site of FBA1. Presumably, if the FBA1 sequence used to repair the damage is adjacent to a WHO gene, this would allow WHO to home into FBA1 (Figure 1B). This homing mechanism is surprisingly different from that of VDE, highlighting the meandering path of HO evolution. Moreover, it suggests that the structural features of HO, such as the zinc finger, were not originally selected for switching mating types.
The discovery that a WHO-like gene is a stepping stone on the evolutionary path to the HO gene raises a number of questions. For example, how did the preferred cut site shift from FBA1 to MAT? The previous hypothesis was that acquiring the zinc finger domain was connected to the specificity for MAT (Haber and Wolfe, 2005). However, WHO genes do not cut MAT despite having the zinc finger domain. An answer may lie in the repeated homing of WHO genes into the same FBA1 locus, which requires each incoming WHO gene to have a slightly different cutting specificity. Perhaps as new WHO variants were selected, one also happened to cut MAT. In this regard, it would be interesting to determine how the amino acid sequence of the proteins coded by WHO genes impacts the specificity of the cut site.
It is puzzling that the proteins encoded by VDE and HO are active in different cell types (haploid versus diploid), and that their activities are regulated in different ways: HO is only transcribed at certain times, whereas VDE is always transcribed but only localized to the nucleus at certain times (Gimble and Thorner, 1992; Nagai et al., 2003; Bakhrat et al., 2006; Stillman, 2013). How did these changes take place? An intriguing possibility is that the acquisition of the zinc finger domain, which contains a nuclear localization signal, was a key step in the regulation of the HO gene diverging from VDE. Thus, how the zinc finger domain contributed to the evolution of HO may have been different than originally imagined.
Another yeast species called Kluyveromyces lactis lacks the HO gene and instead initiates mating-type switching using two other genes that evolved from different DNA mobile elements (Barsoum et al., 2010; Rajaei et al., 2014). This raises the question of whether mobile DNA elements benefit from triggering the switch between mating type. Alternatively, they may have been captured by yeast because of their ability to re-arrange DNA, which makes them well-suited for switching.
At first glance, switching seems unlikely to benefit a homing element such as WHO, as it allows haploid cells to self-mate, preventing the introduction of new versions of a gene into which the element can home. However, generating a diploid cell does allow the yeast to produce spores which promote the long-term survival of the organism. The benefit of mating-type switching is further supported by the fact that this process has evolved eleven independent times in budding yeast (Krassowski et al., 2019). Perhaps, by promoting the survival of the host cell, the homing element preserves itself.
References
-
Nuclear import of HO endonuclease utilizes two nuclear localization signals and four importins of the ribosomal import systemJournal of Biological Chemistry 281:12218–12226.https://doi.org/10.1074/jbc.M600238200
-
α3, a transposable element that promotes host sexual reproductionGenes & Development 24:33–44.https://doi.org/10.1101/gad.557310
-
BookFunction and Evolution of HO and VDE Endonucleases in FungiIn: Belfort M, Stoddard B. L, Wood D. W, Derbyshire V, editors. Homing Endonucleases and Inteins. Berlin Heidelberg: Springer-Verlag. pp. 161–175.https://doi.org/10.1007/3-540-29474-0
-
Mating-type gene switching in Saccharomyces cerevisiaeMicrobiology Spectrum 3:MDNA3-0013-2014.https://doi.org/10.1128/microbiolspec.MDNA3-0013-2014
-
Dancing the cell cycle two-step: regulation of yeast G1-cell-cycle genes by chromatin structureTrends in Biochemical Sciences 38:467–475.https://doi.org/10.1016/j.tibs.2013.06.009
Article and author information
Author details
Publication history
Copyright
© 2020, Rusche
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,054
- views
-
- 119
- downloads
-
- 3
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 3
- citations for umbrella DOI https://doi.org/10.7554/eLife.58403
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Homing Genetic Elements: Mobile DNAs and switching mating types in yeast
eLife 9:e58403.
https://doi.org/10.7554/eLife.58403




{kind=link}