Distinct effects of complement and of NLRP3- and non-NLRP3 inflammasomes for choroidal neovascularization
- Cutaneous Biology Research Center, Massachusetts General Hospital and Harvard Medical School, United States
- Department of Ophthalmology, University of Halle, Germany
Abstract
NLRP3 inflammasome activation and complement-mediated inflammation have been implicated in promoting choroidal neovascularization (CNV) in age-related macular degeneration (AMD), but central questions regarding their contributions to AMD pathogenesis remain unanswered. Key open questions are (1) whether NLRP3 inflammasome activation mainly in retinal pigment epithelium (RPE) or rather in non-RPE cells promotes CNV, (2) whether inflammasome activation in CNV occurs via NLRP3 or also through NLRP3-independent mechanisms, and (3) whether complement activation induces inflammasome activation in CNV. Here we show in a neovascular AMD mouse model that NLRP3 inflammasome activation in non-RPE cells but not in RPE cells promotes CNV. We demonstrate that both NLRP3-dependent and NLRP3-independent inflammasome activation mechanisms induce CNV. Finally, we find that complement and inflammasomes promote CNV through independent mechanisms. Our findings uncover an unexpected role of non-NLRP3 inflammasomes for CNV and suggest that combination therapies targeting inflammasomes and complement may offer synergistic benefits to inhibit CNV.
Introduction
Inflammasomes are multiprotein complexes that lead to the proteolytic activation of proinflammatory IL-1β and IL-18 through the catalytic activity of caspase-1. Canonical inflammasome activation is initiated by cytosolic pattern recognition receptors (PRRs) when exposed to specific triggers (either microbe-derived pathogen-associated molecular patterns [PAMPs] or host-derived danger-associated molecular patterns [DAMPs]). These PRRs include the family of NOD-like receptors (NLRP1, NLRP3, NLRP6, NLRC4), or AIM2, IFI16, and pyrin. Upon sensing specific stimuli, PRRs oligomerize and form together with the ASC adaptor protein scaffolds that activate caspase-1. This results in autoproteolytic activation of pro-caspase-1 that leads to the release of their effector domains caspase-1 p10 and p20, which are required for the proteolytic processing of pro-IL-1β and pro-IL-18 into their active mature forms that promote inflammation and angiogenesis (Guo et al., 2015; Latz et al., 2013; Schroder and Tschopp, 2010).
Of note, a non-canonical inflammasome has been described in which bacterial lipopolysaccharide (LPS) activates caspase-11 (caspase-4/–5 in humans) to induce pyroptotic cell death (Kayagaki et al., 2011).
Recent studies implicated canonical inflammasome activation in AMD pathogenesis. However, these studies have focused on the NLRP3 inflammasome and did not consider a potential contribution of other PRRs for inflammasome activation in AMD. The reason for this very narrow focus on NLRP3 as a PRR that initiates inflammasome activation in AMD is that various stimuli that are well-established risk factors for AMD, including increased oxidative stress or lipid accumulations, are known activators of the NLRP3 inflammasome (Guo et al., 2015; Latz et al., 2013; Schroder and Tschopp, 2010). This has led to the assumption that AMD risk factors promote NLRP3 inflammasome activation, which further exacerbates AMD pathologies through activation of proinflammatory cytokines, such as IL-1β that is known to stimulate inflammatory angiogenesis (Lavalette et al., 2011).
Indeed, recent studies provided some evidence that NLRP3 inflammasome activation occurs in human AMD and promotes AMD-like pathologies in animal models as well (Doyle et al., 2012; Marneros, 2013; Marneros, 2016; Tarallo et al., 2012). A study by Tarallo et al reported that NLRP3 is expressed in RPE cells in human non-exudative AMD and that targeting the NLRP3 inflammasome or IL-18 could inhibit RPE-degeneration (Tarallo et al., 2012). This study used a model of acute RPE degeneration caused by Alu RNA accumulation or DICER loss as an experimental model for non-exudative AMD, which does not reflect many of the progressive age-dependent pathologies seen in human non-exudative AMD. A study by Doyle et al reported that NLRP3 inflammasome activation occurs in macrophages and results in IL-18 activation that inhibits laser-induced CNV (Doyle et al., 2012). This study used laser-induced CNV as a model for neovascular AMD, which is more of an acute injury model and not reflective of the progressive age-dependent development of CNV seen in human neovascular AMD that occurs without an acute injury (He and Marneros, 2013). Moreover, multiple other groups could not detect an effect of IL-18 on laser-induced CNV (Hirano et al., 2014). We also tested the role of IL-18 in a novel genetic mouse model of neovascular AMD, Vegfahyper mice, in which we observed that increased VEGF-A levels in the RPE resulted in increased inflammasome activation products in RPE/choroids of these mice as well as in spontaneous CNV formation in an age-dependent manner without experimental injury (Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016). Thus, we regard Vegfahyper mice as a more disease-relevant animal model of neovascular AMD than the laser-injury CNV model. We showed that inactivation of Il18 does not significantly affect CNV lesion formation in Vegfahyper mice, whereas inactivation of Nlrp3, Casp1, or Il1r potently inhibits CNV lesion formation in these mice (Marneros, 2013; Marneros, 2016). These findings provide evidence that activation of IL-1β through the NLRP3 inflammasome promotes VEGF-A-induced CNV in mice, whereas IL-18 has no major effect. Notably, inflammasome activation in RPE/choroids of Vegfahyper mice was associated with the accumulation of known triggers of the inflammasome in CNV lesions, including increased markers of oxidative stress and sub-RPE lipid deposits (Marneros, 2013). In this Vegfahyper mouse model of AMD, spontaneous CNV lesion formation can be accurately quantified already at a young age (here quantified at 6 weeks of age). These CNV lesions enlarge with progressive age and can merge with adjacent lesions to form larger confluent areas of CNV (Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016), similarly as observed in human neovascular AMD where multiple adjacent ingrowth sites of neovessels from choriocapillaris through Bruch’s membrane can merge to form larger CNV lesions (Grossniklaus and Green, 2004).
The findings from previous studies regarding the role of IL-18 for AMD have resulted in a controversy about whether inflammasomes play a role in AMD pathogenesis at all. For example, NLRP3 protein was not detected in the RPE of human eyes with AMD in a recent study that tested only a limited number of antibodies, questioning whether inflammasome activation occurs in the RPE at all (Kosmidou et al., 2018). However, this study did not investigate whether actual inflammasome activation occurs in the RPE (cleaved caspase-1 p10 or p20 levels or IL-1β secretion were not assessed, which serve as inflammasome activation markers) (Kosmidou et al., 2018). By contrast, other studies found evidence for inflammasome activation or specifically for NLRP3 inflammasome activation in human RPE cells (Hollborn et al., 2018; Tseng et al., 2013; Wang et al., 2019). However, even if NLRP3 would indeed not be expressed at sufficient levels to be able to activate the inflammasome in the RPE in human eyes with AMD, this would not mean that inflammasome activation does not occur in these cells, as the inflammasome could be activated through other PRRs. Thus, the lack of NLRP3 expression cannot be equated with the lack of inflammasome activation.
Critical unanswered questions are whether NLRP3 inflammasome activation in RPE cells or in non-RPE cells (e.g. macrophages or retinal microglia) promotes AMD pathologies and whether the inflammasome can be activated in AMD not only through NLRP3-dependent but also through NLRP3-independent mechanisms.
Notably, recent in vitro studies suggested that sub-lytic effects of the complement pathway activation product C5b-9 (also called membrane attack complex) can induce NLRP3 inflammasome activation in macrophages (Laudisi et al., 2013; Suresh et al., 2016; Triantafilou et al., 2013). Moreover, the complement activation product C3a has been shown to activate the inflammasome in macrophages in vitro as well (Asgari et al., 2013). As complement-mediated inflammation occurs in AMD and has been linked to AMD disease progression, this raises the question of whether complement activation products may promote AMD pathologies in part by triggering inflammasome activation in AMD lesions (Gehrs et al., 2010). Alternatively, it is possible that complement-mediated inflammation and inflammasome-mediated inflammation are two parallel inflammatory pathways that promote AMD largely independently of each other. Thus, another important unanswered question in the field is whether complement-mediated inflammation promotes AMD pathologies at least in part through inflammasome activation or through inflammasome-independent mechanisms. Vegfahyper mice offer the opportunity to test the roles of complement pathway activation for inflammasome activation and CNV formation, as we observed accumulation of complement C5b-9 in CNV lesions of these mice (Marneros, 2016).
Here our data provide important answers to these key questions: (1) we show that NLRP3 inflammasome activation mainly in non-RPE cells and not in RPE cells promotes CNV; (2) we demonstrate that not only NLRP3-dependent but also NLRP3-independent inflammasome activation promotes CNV; (3) we show that the caspase-11 non-canonical inflammasome does not affect CNV; and (4) we provide evidence that complement pathway activation promotes CNV largely independently of inflammasome activation. These findings have significant translational relevance and suggest that combining therapies that target both the canonical inflammasome as well as the complement pathway likely offer synergistic therapeutic benefits for patients with neovascular AMD.
Results and discussion
NLRP3 inflammasome activation mainly in non-RPE cells but not in RPE cells promotes CNV
We utilized here a genetic mouse model of neovascular AMD, Vegfahyper mice, which forms spontaneous CNV lesions without artificial injury (Marneros, 2013). This model allows us to test effects on both CNV lesion induction (by counting CNV lesion numbers), as well as on CNV lesion growth (measuring CNV lesion area) (Figure 1A–D; Marneros, 2016). This model is more disease-relevant for AMD than the commonly used laser-injury CNV model, as in the laser model we can only assess CNV lesion size after artificial injury but we cannot assess factors that influence CNV lesion induction and CNV lesion numbers. For all CNV quantifications across all experimental mouse groups, we used 6-weeks-old mice, since at this young age early CNV lesions are present, whereas with progressive age CNV lesions increase in size and adjacent CNV lesions can fuse to form confluent larger lesions (Figure 1E; Marneros, 2013). Thus, quantifying CNV lesions in young 6-weeks-old age-matched mice allows us to determine effects on early CNV lesion induction and to quantitate accurately the number of initial CNV lesions present at a defined early stage of the disease process before they become confluent larger lesions. Moreover, we already observed in RPE/choroid lysates of 6-weeks-old Vegfahyper mice a strong increase in the inflammasome activation product caspase-1 p10 (p10 levels serve as a direct correlate of inflammasome activity), linking CNV formation with inflammasome activation in these mice already at this young age (Figure 1F).
Figure 1
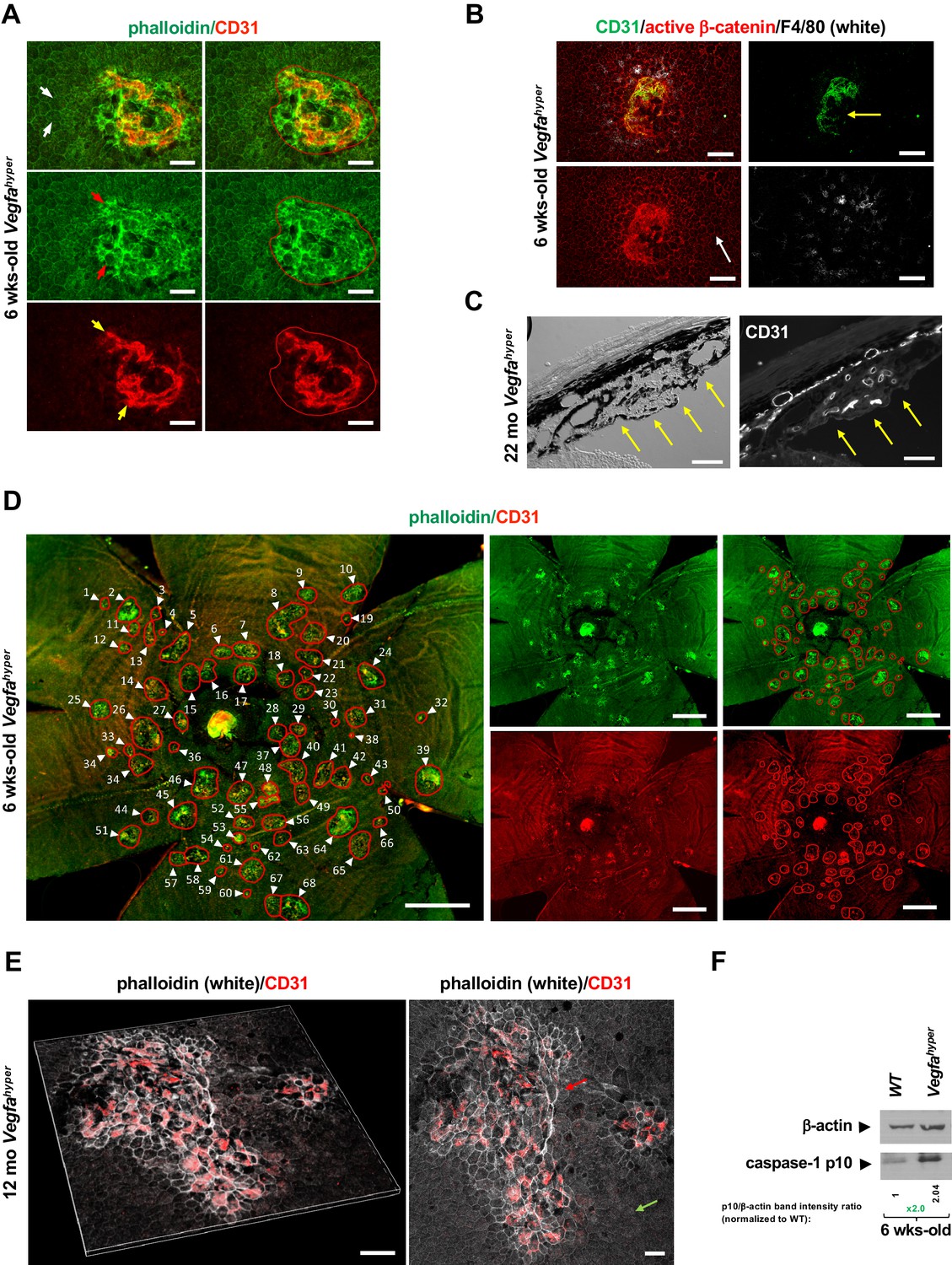
Quantifications of CNV lesions in Vegfahyper mice.
(A) Disrupted honeycomb pattern morphology of RPE cells and sub-RPE protrusions of CD31+ neovessels demarcate a choroidal neovascular lesion in choroidal flat mounts of Vegfahyper mice. A choroidal flat mount of a Vegfahyper mouse is shown. Phalloidin staining highlights RPE cell membranes and shows the regular honeycomb pattern of RPE cells at sites devoid of CNV lesions (white arrows). At the site of a CNV lesion, the typical RPE honeycomb pattern is disrupted (phalloidin, green; red arrows). CD31 (red) staining identifies neovessels of a CNV lesion that protrude into the sub-RPE space (yellow arrows). Disruption of regular RPE cell morphology and CD31 staining of neovessels determine the size of the lesion. A polygon is used to quantify the lesion area. Scale bars, 50 µm. 6-weeks-old mouse. (B) CNV lesion in a 6-weeks-old Vegfahyper mouse shows that RPE disruption occurs only at sites of choroidal neovascularization due to CD31+ neovessels (green) protruding into the sub-RPE space, whereas RPE cells show a normal honeycomb pattern cellular morphology at sites without CNV lesions (white arrow). Labeling for active β-catenin outlines RPE cell membranes (red), similar to phalloidin staining. F4/80+ cells (white) infiltrate sites of CNV lesions. Pigmented cells cover part of the CNV lesion (yellow arrow). Scale bars, 100 µm. (C) Section through a CNV lesion (yellow arrows) in an aged Vegfahyper mouse eye (22-months-old) shows CD31+ neovessels in CNV lesions. Scale bars, 50 µm. (D) Representative image of how CNV lesions were quantified in choroidal flat mounts in mice with the Vegfahyper allele. Measurements of multiple lesions in a choroidal flat mount. A choroidal flat mount of a Vegfahyper mouse is shown in which each CNV lesion is counted (numbered, arrows) and each lesion area shown by a polygon. Phalloidin staining (green) outlines CNV lesions and CD31 immunolabeling (red) detects CD31+ neovessels. Increased phalloidin signal at sites of disruption of the regular RPE morphology occurs only at sites of CD31+ neovessels. Scale bars, 500 µm. 6-weeks-old mouse. (E) Enlarged confluent CNV lesions can be observed in aged Vegfahyper mice. Even at an advanced age, disruption of the honeycomb pattern RPE morphology is strictly localized to the site of CD31+ neovessel protrusions into the sub-RPE space in CNV lesions (red arrow), whereas RPE areas with no CNV maintain their honeycomb pattern (green arrow). The left image shows Z-stack 3D-rendering of CNV lesions in a 12-months-old Vegfahyper mouse eye and the right image shows the projection of this z-stack. CD31+ neovessels of CNV lesions in red; phalloidin in white. Scale bars, 50 µm. (F) RPE/choroid lysates from 6-weeks-old Vegfahyper mice and their WT littermates show that the inflammasome activation product caspase-1 p10 can already be detected at this young age at which CNV lesions were quantified. A > 2 fold increase in p10 levels relative to WT (normalized to β-actin levels) are observed at 6 weeks of age in RPE/choroid lysates of Vegfahyper mice. RPE/choroids of both eyes from four Vegfahyper mice versus four of their WT littermates were pooled. An anti-caspase-1 p10 antibody from Thermo Fisher Scientific was used (PA5-105049) to detect p10.
Notably, the size of CNV lesions can be accurately quantified in this AMD mouse model by immunolabeling of choroidal flat mounts with the endothelial cell marker CD31 that strongly labels neovessels that protrude into the sub-RPE space (Figure 1A–E). These neovessels affect the overlying RPE morphology, which can be visualized by labeling with phalloidin or immunolabeling for active β-catenin, both outlining RPE cell membranes (Figure 1A, B, D and E). Disruption of the typical honeycomb pattern morphology of the RPE occurs in 6-weeks-old Vegfahyper mice only at sites of CD31+ neovessel protrusions into the sub-RPE space (Figure 1D). CNV lesions were quantified in this study by co-labeling for CD31 and with phalloidin in choroidal flat mounts and all CNV lesions showed a strict co-occurrence of disruption of RPE morphology with CD31 neovessel protrusions. Thus, all phalloidin-marked CNV lesions analyzed in this study contained CD31+ neovessels.
At sites devoid of CNV lesions, the RPE and retina appear morphologically normal in 6-weeks-old Vegfahyper mice (Figure 1A and B; Marneros, 2013). By contrast, in aged Vegfahyper mice RPE cells become atrophic and the photoreceptor layer of the retina is attenuated as well, resembling pathologies seen in nonexudative AMD (Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016). However, even in aged Vegfahyper mice co-labeling of choroidal flat mounts for CD31 and with phalloidin shows that the disruption of the honeycomb pattern of RPE morphology co-localizes strictly to the sites of CNV and occurs due to protruding CD31+ neovessels, whereas sites devoid of CNV do not show these RPE changes (Figure 1E).
We previously found that inactivation of Nlrp3 results in reduced CNV lesion numbers in Vegfahyper mice, but we do not know whether this effect is explained by loss of NLRP3 in RPE cells or in non-RPE cells (Marneros, 2013; Marneros, 2016). Controversy exists particularly about whether NLRP3 is expressed in the RPE in sufficient amounts to be functional and to promote AMD pathologies by activating the inflammasome.
To test whether NLRP3 inflammasome activation in RPE cells versus in non-RPE cells promotes CNV, we established mice that have a constitutively active Nlrp3 allele only in the RPE, whereas NLRP3 is inactivated in all other cells of these mice (Figure 2A). For this purpose, we utilized mice that carry a A350V mutation in the Nlrp3 gene (Nlrp3A350V/A350V). This mutation results in uninhibited NLRP3 activation in patients and mice with cryopyrin-associated periodic syndrome (CAPS) (Brydges et al., 2009). In these mice a floxed inverted neoR cassette (neoRfl/fl) in intron 2 inhibits Nlrp3A350V/A350V expression. Thus, in the absence of Cre, the neoR cassette remains in place and prevents expression of Nlrp3A350V/A350V, rendering the mice null for NLRP3. Only in the presence of Cre recombinase is the neoRfl/fl cassette removed, which allows the expression of the mutant Nlrp3A350V/A350V that causes constitutive NLRP3 activation. We crossed floxed neoRfl/fl-Nlrp3A350V/A350V mice with Best1Cre/+ mice (expressing Cre recombinase exclusively in the RPE, also called VMD2Cre/+) and Vegfahyper mice to generate VegfahyperBest1Cre/+Nlrp3A350V/A350V mice (Figure 2A). As Cre recombinase is expressed in Best1Cre/+ mice only in RPE cells and in no other cell types in the eye (Figure 2—figure supplement 1; He et al., 2014; Iacovelli et al., 2011), the constitutively active Nlrp3A350V/A350V mutant allele is exclusively present in the RPE in VegfahyperBest1Cre/+Nlrp3A350V/A350V mice. All other eye tissues (non-RPE cells) of VegfahyperBest1Cre/+Nlrp3A350V/A350V mice do not express Cre and do not remove the neoRfl/fl cassette (Figure 2—figure supplement 1C), resulting in a lack of Nlrp3 expression and NLRP3 inflammasome activity in these Cre-negative non-RPE cells (Brydges et al., 2009). Therefore, comparing VegfahyperBest1Cre/+Nlrp3A350V/A350V mice with VegfahyperNlrp3-/- mice (which are Cre-negative VegfahyperCrenegNlrp3A350V/A350V mice and represent Vegfahyper mice that lack NLRP3 in all cells), allows us to determine whether a constitutively active Nlrp3 allele selectively in the RPE promotes CNV. Only those choroidal flat mounts of VegfahyperBest1Cre/+Nlrp3A350V/A350V mice were included in CNV quantifications that showed uniform Cre expression in the RPE (Figure 2A; Figure 2—figure supplement 1).
Figure 2 with 1 supplement see all
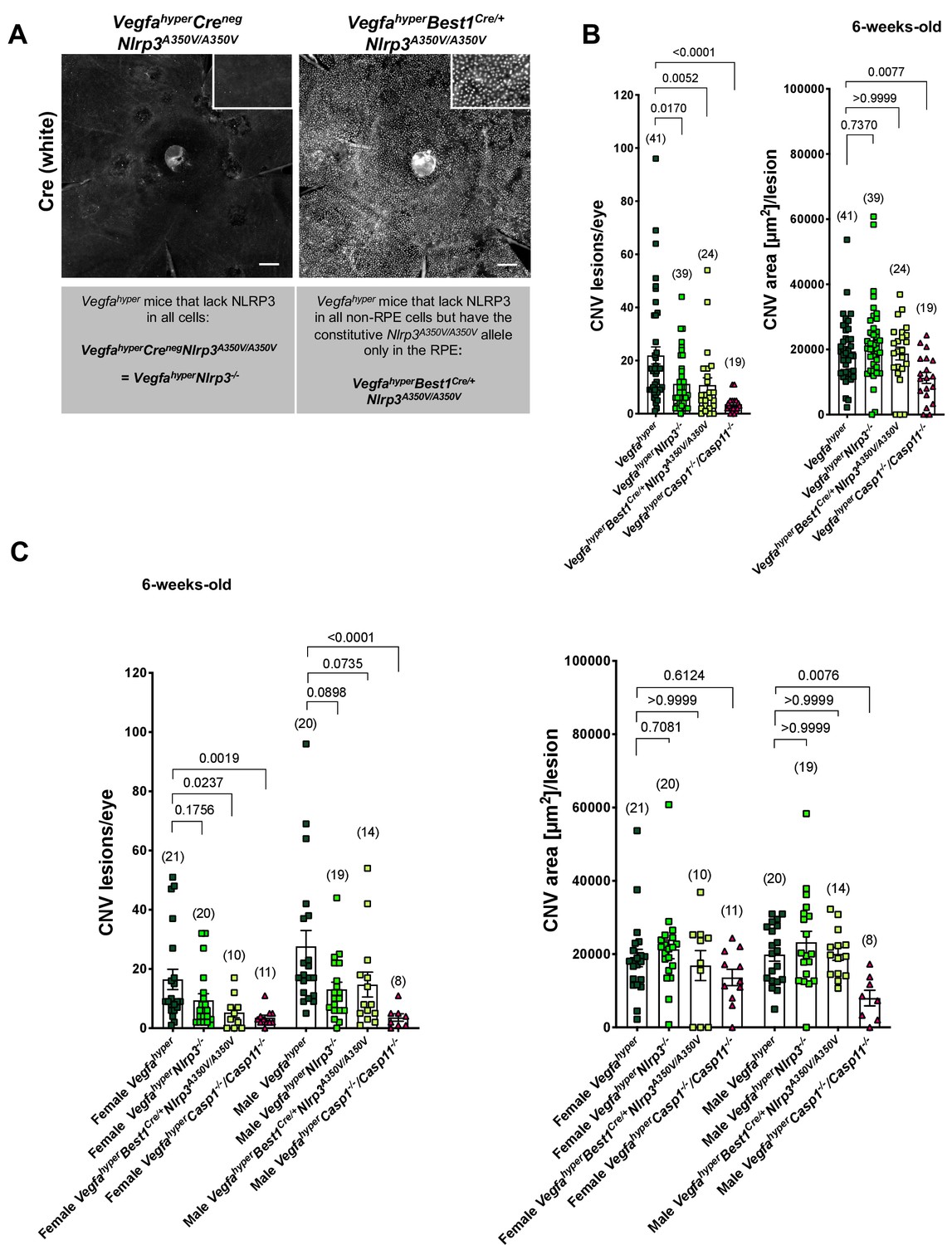
NLRP3 inflammasome activation in non-RPE cells but not in RPE cells promotes CNV.
(A) Nuclear Cre immunostaining (white) in choroidal flat mounts from 6-weeks-old VegfahyperBest1Cre/+Nlrp3A350V/A350V mice confirms uniform Cre expression in the RPE. Inset shows higher magnification image. No Cre staining is seen in VegfahyperCrenegNlrp3A350V/A350V mice ( = VegfahyperNlrp3-/- mice). Scale bars, 200 μm. (B-C). CNV lesion numbers and average CNV lesion area (in μm2/lesion) were measured in 6-weeks-old mice. VegfahyperNlrp3-/- mice (group 2) and VegfahyperBest1Cre/+Nlrp3A350V/A350V mice (group 3) show significantly decreased CNV lesion numbers compared to Vegfahyper mice (group 1), but no significant difference in CNV lesion sizes. CNV lesion sizes and numbers between VegfahyperNlrp3-/- mice and VegfahyperBest1Cre/+Nlrp3A350V/A350V mice are similar. No marked differences in lesion sizes or lesion numbers were observed among male or female groups of VegfahyperNlrp3-/- mice and VegfahyperBest1Cre/+Nlrp3A350V/A350V mice. VegfahyperCasp1-/-Casp11-/- mice (group 4) have fewer CNV lesions than VegfahyperNlrp3-/- mice or VegfahyperBest1Cre/+Nlrp3A350V/A350V mice. Values represent total CNV lesion numbers/eye for each mouse and average lesion area/eye for each mouse. Absolute numbers of mice per group are indicated in parentheses. Graphs show mean ± SEM. P-values are shown and were determined by a Kruskal-Wallis test followed by a Dunn’s test.
Numbers (correlating to lesion induction) and sizes (area, correlating to lesion growth) of CNV lesions were assessed in choroidal flat mounts from 6-weeks-old Vegfahyper mice (group 1), VegfahyperNlrp3-/- mice (with lack of NLRP3 in all cells) (group 2), VegfahyperBest1Cre/+Nlrp3A350V/A350V mice (with the constitutively active Nlrp3A350V/A350V allele in the RPE only while lacking NLRP3 in all non-RPE cells) (group 3), and in VegfahyperCasp1-/-Casp11-/- mice (group 4) that lack all canonical and non-canonical inflammasome activity (Figure 2B and C). We previously reported that 6-weeks-old VegfahyperNlrp3-/- mice have reduced CNV lesion numbers compared to 6-weeks-old Vegfahyper mice, but with similar lesion sizes in both groups (Marneros, 2016). We show now that the strong reduction in CNV lesion numbers compared to Vegfahyper mice (group 1) is similar as seen in VegfahyperNlrp3-/- mice (group 2) also in VegfahyperBest1Cre/+Nlrp3A350V/A350V mice (group 3), and that CNV lesion sizes are not affected (Figure 2B and C). The similar effect on CNV lesion number reduction in VegfahyperBest1Cre/+Nlrp3A350V/A350V mice and VegfahyperNlrp3-/- mice shows that the constitutively active Nlrp3A350V/A350V allele in the RPE does not promote CNV lesion formation, whereas NLRP3 inflammasome inactivation in non-RPE cells potently inhibits CNV lesion induction.
F4/80+ cells in CNV lesions have high levels of caspase-1 protein
These findings suggest that NLRP3 inflammasome activation particularly in non-RPE cells promotes CNV. As caspase-1 is required for inflammasome activation, detection of caspase-1 or its activation products p10/p20 in a specific cell type suggests that inflammasome activation in these cells is more likely than in cells in which no caspase-1 immunolabeling is detected. Thus, in order to test which cell types show significant levels of caspase-1 protein levels, we performed immunolabeling of choroidal flat mounts and eye sections from Vegfahyper mice with several different antibodies that detect either caspase-1 or its activation product p10. We observed strong immunolabeling for caspase-1 or its activation product p10 predominantly in F4/80+ cells in CNV lesions of Vegfahyper mice, whereas no strong immunolabeling was detected in their RPE cells (Figure 3A–C). In the eye, both macrophages as well as retinal microglia express F4/80 (Yu et al., 2020). Notably, we observed that a subset of F4/80+ cells that infiltrate CNV lesions in Vegfahyper mice and showed caspase-1 p10 labeling also expressed the microglia marker P2RY12, whereas P2RY12+ microglia in the retina at sites devoid of CNV lesions did not label for caspase-1 p10 (Figure 3C; Figure 3—figure supplement 1). This suggests that inflammasome activation occurs in both macrophages (F4/80+P2RY12neg) and in activated retinal microglia cells (F4/80+P2RY12+) that infiltrate CNV lesions in these mice. Together with our observation that NLRP3 inflammasome activation mainly in non-RPE cells promotes CNV, these data suggest that inflammasome activation primarily in F4/80+ cells and not in RPE cells drives CNV lesion formation in Vegfahyper mice.
Figure 3 with 1 supplement see all
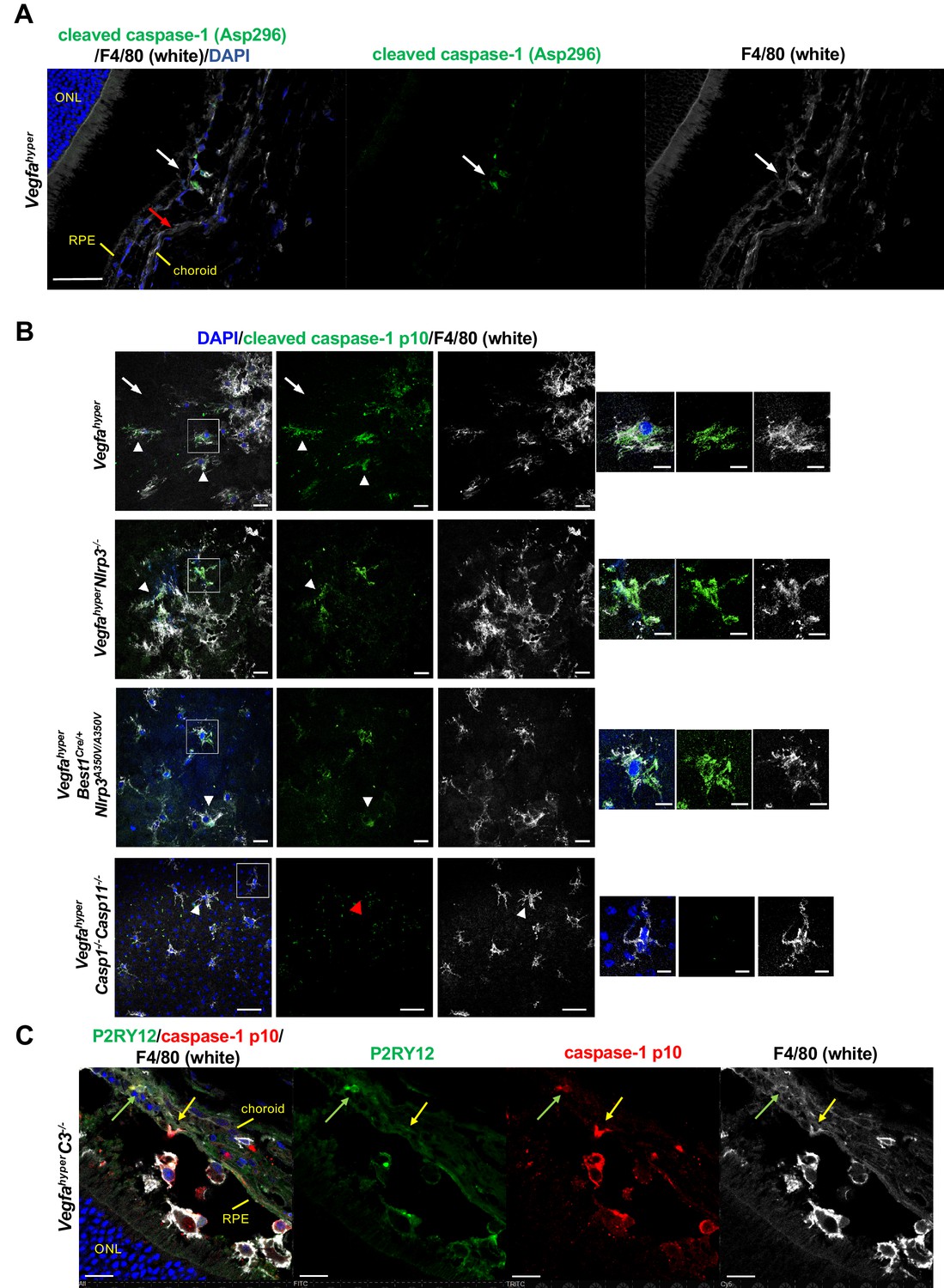
F4/80+ cells in CNV lesions have high levels of caspase-1 protein.
(A) Immunolabeling of eye sections of Vegfahyper mice with an antibody that recognizes endogenous levels of caspase-1 protein only when cleaved at Asp296 that occurs with inflammasome activation but does not detect full-length caspase-1 (Cell Signaling Technology Cat# 89332). F4/80+ cells at the site of a CNV lesion (white) show the presence of cleaved caspase-1 (green, white arrow), whereas this staining is not observed in RPE cells (red arrow) in eyes of Vegfahyper mice. DAPI nuclear staining in blue. Scale bar, 50 μm. Location of the outer nuclear layer of the retina (ONL), choroid, and RPE are shown. (B) Immunolabeling of choroidal flat mounts for caspase-1 p10 (green) shows predominant staining in F4/80+ cells (white, arrowheads) but not in RPE cells (arrows) in CNV lesions. The antibody (sc-22166, Santa Cruz Biotechnology) detects cleaved caspase-1 p10 (inflammasome activation marker) and was raised against a short amino acid sequence containing the neoepitope at Gly315 of caspase-1 of mouse origin. Proteolytic cleavage of the precursor caspase-1 at Gly315 generates the functional caspase-1 subunits, known as p20 and p10 subunits. Choroidal flat mounts from 3-months-old Vegfahyper, VegfahyperNlrp3-/-, and VegfahyperBest1Cre/+Nlrp3A350V/A350V mice are shown. No caspase-1 staining is observed even in aged VegfahyperCasp1-/-Casp11-/- mice (red arrowhead), confirming the specificity of the caspase-1 immunolabeling (a representative flat mount of an 18-months-old mouse is shown). DAPI nuclear staining in blue. Right: Higher magnification images of F4/80+ cells (highlighted in boxes of left images). Scale bars, 20 μm (left) and 10 μm (right). (C) At sites of CNV lesions in mice with the Vegfahyper allele, a subset of infiltrating F4/80+ cells shows immunolabeling for caspase-1 p10 and is also positive for the microglial marker P2RY12 (green arrows). In contrast, other F4/80+p10+ cells show no staining for P2RY12 (yellow arrows). Representative image of a 12-months-old mouse eye with the Vegfahyper allele is shown (here an eye of a VegfahyperC3-/- mouse is shown). Scale bars, 50 μm. DAPI nuclear staining in blue. Location of ONL, choroid, and RPE are shown.
Indeed, F4/80+ cells infiltrate the subretinal space at the site of RPE barrier breakdown in Vegfahyper mice and are spatiotemporally associated with activation of retinal glia cells when CNV lesions form at that site (Marneros, 2013), suggesting that these cells are important for the initiation of CNV lesion formation. Consistent with this hypothesis, we and others found that ablation of macrophages (by using either LysMCre/+iDTR mice treated with diphtheria toxin or using clodronate liposomes) inhibits laser-induced CNV (Espinosa-Heidmann et al., 2003; Marneros, 2013; Sakurai et al., 2003). Thus, the observed strong reduction in CNV lesion formation in Vegfahyper mice that lack inflammasome activity (VegfahyperCasp1-/-Casp11-/- mice) (Figure 2B and C) is likely due to diminished inflammasome-mediated production of proangiogenic cytokines mainly in lesional activated F4/80+ cells, whereas potential low-level inflammasome activation in RPE cells is less likely contributing in a significant manner to CNV lesion formation (Marneros, 2016). Future approaches to target inflammasome components specifically in macrophages/microglia in this AMD mouse model will help clarify the contributions of these cell types for inflammasome-mediated CNV lesion formation.
Progressive inflammasome activation in eyes of Vegfahyper mice occurs through both NLRP3-dependent as well as through NLRP3-independent mechanisms
As AMD pathologies and CNV lesions progress and enlarge in an age-dependent manner in Vegfahyper mice (Figure 1A–E; Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016), as is the case in patients with neovascular AMD, we tested whether inflammasome activation in RPE/choroid lysates of these mice also increases with age progression and correlates with AMD exacerbation. We found in western blots of RPE/choroid lysates of Vegfahyper mice that an increase in caspase-1 inflammasome activation (increased cleaved caspase-1 p10 levels) compared to WT littermates was present already in young mice (increased ratios of p10/β-actin band intensities in RPE/choroid lysates of 6-weeks-old Vegfahyper mice shown in Figure 1F and of 2-months-old Vegfahyper mice shown in Figure 4A). Thus, inflammasome activation is already markedly increased in RPE/choroids of young Vegfahyper mice in which we quantitated CNV lesions. Together with our observation that inactivation of inflammasomes in young Vegfahyper mice (VegfahyperCasp1-/-Casp11-/- mice) potently reduced CNV lesion formation, this demonstrates that inflammasome activation already in young Vegfahyper mice promotes CNV lesion formation. The age-dependent exacerbation of AMD pathologies was also associated with a steady further increase in inflammasome activation in RPE/choroids of Vegfahyper mice with age progression and aged Vegfahyper mice showed high levels of caspase-1 p10 in their RPE/choroids (Figure 4A). A similar relative increase in caspase-1 p10 levels as observed in RPE/choroids of young Vegfahyper mice compared to their WT controls (2-months-old) was also observed in aged (13-months-old) Vegfahyper mice when compared to their age-matched controls (Figure 4A). The specificity of the antibody used to detect caspase-1 and its activation product p10 was confirmed by the absence of a pro-caspase-1 and a caspase-1 p10 band in RPE/choroid lysates from aged VegfahyperCasp1-/-Casp11-/- mice that lack caspase-1 (Figure 4B; ratios of p10/β-actin band intensity quantifications shown).
Figure 4 with 1 supplement see all
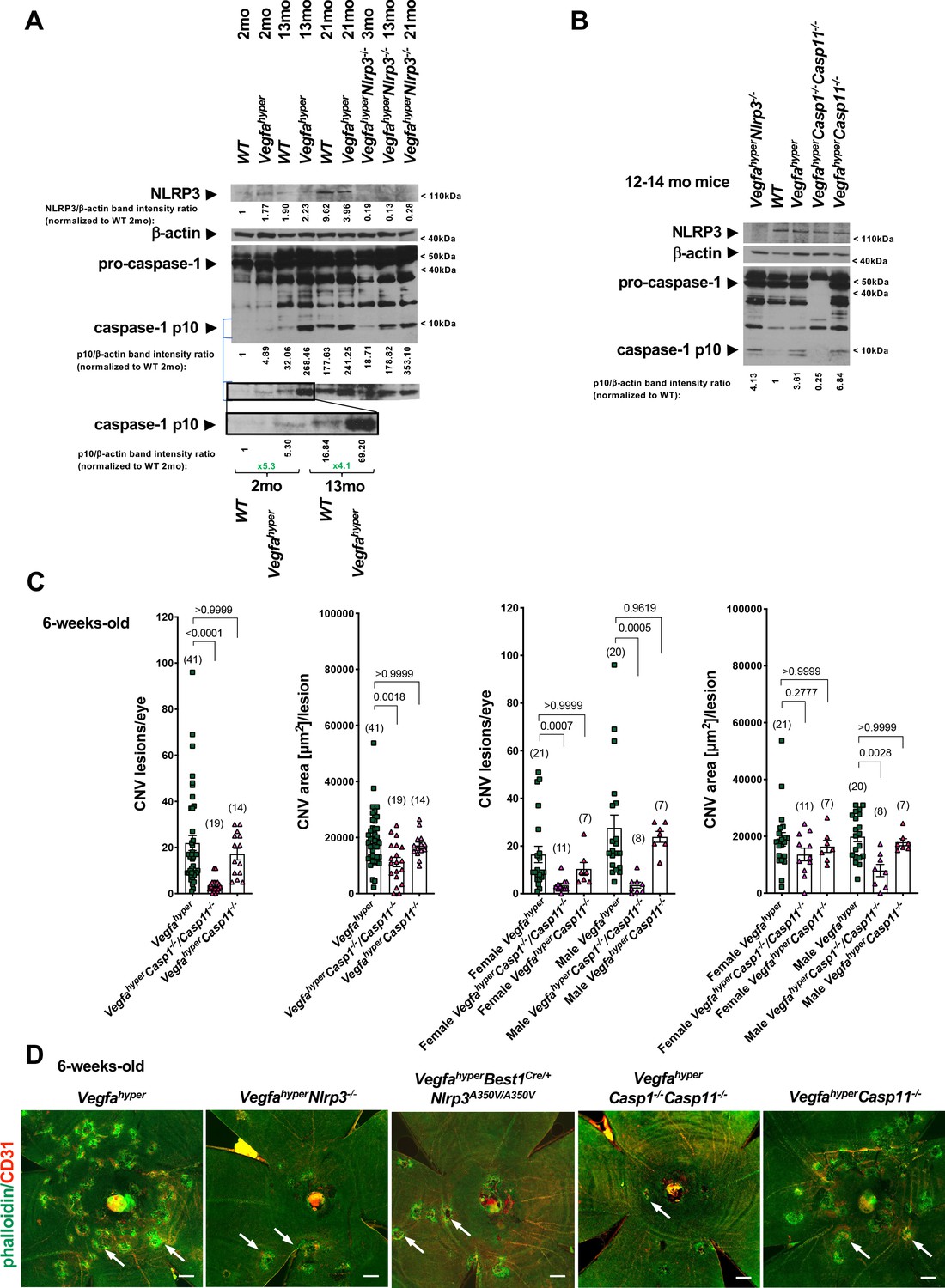
Progressive inflammasome activation in eyes of Vegfahyper mice occurs through both NLRP3-dependent as well as through NLRP3-independent mechanisms.
(A) RPE/choroid lysates of Vegfahyper mice show increased levels of the inflammasome activation marker cleaved caspase-1 p10 compared to age-matched WT mice both at 2 months of age as well as at 13 months of age. Caspase-1 p10 levels increase with progressive age in eyes of both WT and Vegfahyper mice and maintain their relative difference with age. Caspase-1 p10 levels are already markedly increased in RPE/choroid lysates from 2-months-old Vegfahyper mice when compared to their WT littermates (~5 fold). They are further increased in 13-months-old mice with a similar relative increase in Vegfahyper mice when compared to their WT littermates (~4 fold). NLRP3 immunoblotting confirms the presence of NLRP3 in RPE/choroid lysates of Vegfahyper mice already at 2 months of age and shows the lack of NLRP3 protein in VegfahyperNlrp3-/- mice, validating the specificity of the NLRP3 antibody. Lack of NLRP3 does not prevent inflammasome activation in RPE/choroid lysates and VegfahyperNlrp3-/- mice show caspase-1 p10 levels in their eyes that increase with progressive age, similarly as observed in Vegfahyper mice. Four RPE/choroid tissues from four independent mice were pooled for each of the groups indicated. Age of groups are indicated. Ratios of p10/β-actin or NLRP3/β-actin band intensities normalized to 2-months-old WT values are shown. Blue lines indicate p10 bands from the same western blot after re-probing with secondary antibody and longer exposure of the film. Boxed area is magnified and p10 bands are quantified, demonstrating that p10 levels are already increased in 2-months-old Vegfahyper mouse groups. (B) RPE/choroid lysates of Vegfahyper mice, VegfahyperNlrp3-/- mice and VegfahyperCasp11-/- mice show increased levels of caspase-1 p10 compared to those of WT mice. No pro-caspase-1 or p10 were observed in lysates of VegfahyperCasp1-/-Casp11-/- mice, confirming the specificity of the antibody used (Santa Cruz Biotechnology Cat# sc-514). WT mice, Vegfahyper mice and VegfahyperCasp1-/-Casp11-/- mice express NLRP3. Lysates of RPE/choroid tissues from 12 to 14 months-old mice (four RPE/choroids pooled for each group). Ratios of p10/β-actin band intensities normalized to WT values are shown. (C) VegfahyperCasp1-/-Casp11-/- mice show significantly reduced CNV lesion numbers compared to Vegfahyper mice. Significantly smaller CNV lesion sizes were observed in male but not in female VegfahyperCasp1-/-Casp11-/- mice compared to male and female Vegfahyper mice respectively. Comparing VegfahyperCasp11-/- with Vegfahyper mice shows no significant difference in either CNV lesion numbers or their sizes. 6-weeks-old mice. Values represent total CNV lesion numbers/eye for each mouse and average lesion area/eye for each mouse. Absolute numbers of mice per group are indicated in parentheses. Graphs show mean ± SEM. P-values are shown and were determined with a Kruskal-Wallis test followed by a Dunn’s test. (D) Representative choroidal flat mount images show multiple CD31+ CNV lesions (white arrows) in 6-weeks-old Vegfahyper and VegfahyperCasp11-/- mice and much fewer lesions in VegfahyperNlrp3-/-, VegfahyperBest1Cre/+Nlrp3A350V/A350V, and VegfahyperCasp1-/-Casp11-/- mice. Staining for phalloidin in green (outlining CNV lesions prominently) and for CD31 in red. Scale bars, 200 μm. Phalloidin-marked loss of honeycomb morphology of RPE occurs only at sites of CD31+ neovessel protrusion into the sub-RPE space. This can also be seen when assessing red and green channels of these images separately (shown in Figure 4—figure supplement 1).
Our finding that inactivation of Nlrp3 reduces CNV lesion numbers in Vegfahyper mice (VegfahyperNlrp3-/- mice) demonstrates that NLRP3 inflammasome activation promotes CNV lesion formation, but it does not answer the question whether other types of inflammasomes that are activated through different PRRs other than NLRP3 also contribute to CNV lesion formation. Previous studies focused mainly on a role of the NLRP3 inflammasome for AMD pathogenesis but did not examine whether NLRP3-independent inflammasome activation mechanisms may contribute to the progression of AMD pathologies. Thus, we tested whether inflammasome activation in RPE/choroid lysates of Vegfahyper mice can be attributed mainly to NLRP3-mediated inflammasome activation or whether other NLRP3-independent mechanisms can also promote inflammasome activation in CNV. Western blots detected NLRP3 protein already in RPE/choroid lysates from young WT and Vegfahyper mice, whereas NLRP3 protein was absent in RPE/choroid lysates from VegfahyperNlrp3-/- mice (Figure 4A and B). Moreover, RPE/choroid lysates from VegfahyperNlrp3-/- mice showed the presence of a cleaved caspase-1 p10 band despite the absence of NLRP3 protein, demonstrating that inflammasome activation in eyes of Vegfahyper mice is not dependent on NLRP3 and can also be mediated through other inflammasome activators/PRRs in the absence of NLRP3 (Figure 4A and B). Caspase-1 activation increased in VegfahyperNlrp3-/- mice in an age-dependent manner similarly as in Vegfahyper mice (Figure 4A). This is also consistent with our observation that strong caspase-1 immunolabeling occurred in F4/80+ cells in CNV lesions of VegfahyperNlrp3-/- mice to a similar extent as seen in Vegfahyper mice (Figure 3B). Together with the observation of reduced CNV lesion formation in VegfahyperNlrp3-/- mice compared to Vegfahyper mice, these data suggest that inflammasome activation in RPE/choroid tissues of Vegfahyper mice can occur through both NLRP3-dependent and NLRP3-independent mechanisms. These findings are in accordance with our observation that Vegfahyper mice that lack caspase-1/caspase-11 (and, thus, lack all canonical and non-canonical inflammasomes) exhibit a stronger reduction in CNV lesion numbers compared to Vegfahyper mice that lack only NLRP3 (Figure 2B and C; Marneros, 2016). In fact, CNV lesions were fewer and smaller in size (decreased lesion formation as well as a decreased lesion growth) in VegfahyperCasp1-/-Casp11-/- mice when compared to VegfahyperNlrp3-/- mice or VegfahyperBest1Cre/+Nlrp3A350V/A350V mice (Figure 2B and C).
Non-canonical inflammasome activation does not affect CNV lesions in Vegfahyper mice
Mice that lack Casp1 (thus, lacking all canonical inflammasomes as caspase-1 is a required component of all inflammasomes) have also an incidental inactivating mutation in Casp11 (they are therefore double KOs for both caspase-1 and caspase-11 [referred to as VegfahyperCasp1-/-Casp11-/-mice; Kayagaki et al., 2011]. We show that these VegfahyperCasp1-/-Casp11-/- mice have indeed no caspase-1 protein [thus, no inflammasome activity], as no pro-caspase-1 nor any caspase-1 p10 can be detected in western blots of RPE/choroid lysates from these mice [Figure 4B]). To rule out a significant contribution of caspase-11 to inflammasome activation or CNV lesion formation in eyes of Vegfahyper mice, we generated VegfahyperCasp11-/- mice, which have a functional Casp1 gene (thus, can have canonical inflammasome activation) but lack Casp11 (Kayagaki et al., 2011; Li et al., 2007). We show in this western blot that, indeed, lack of caspase-11 does not reduce inflammasome activation (caspase-1 p10 levels are not diminished in RPE/choroid lysates of VegfahyperCasp11-/- mice) (Figure 4B). Consistent with this finding, lack of caspase-11 does also not affect CNV lesion numbers or sizes (Figure 4C and D; Figure 4—figure supplement 1). Thus, caspase-11-independent caspase-1 inflammasome activation that is triggered by both NLRP3-dependent and NLRP3-independent mechanisms contributes to CNV induction in Vegfahyper mice. Notably, lack of caspase-1/caspase-11 potently reduces not only CNV lesion numbers but also CNV lesion sizes (Figure 4C).
Collectively, our findings resolve important open questions in the field by demonstrating that (1) NLRP3 inflammasome activation predominantly in non-RPE cells and not in RPE cells promotes VEGF-A-induced CNV and that (2) NLRP3-independent and caspase-11-independent inflammasome activation mechanisms contribute to CNV lesion formation as well.
Targeting complement-mediated inflammation inhibits CNV without preventing inflammasome activation
Complement factor H (CFH) has been shown to inhibit CD47-mediated elimination of mononuclear phagocytes that maintains homeostasis in the subretinal space, and AMD-associated CFH variants may through this mechanism cause an accumulation of subretinal mononuclear phagocytes that promote AMD pathologies (Calippe et al., 2017). This study provides further evidence that links activated phagocytes to the progression of AMD pathologies. Moreover, several human genetic studies implicated a disease-promoting role of complement-mediated inflammation for neovascular AMD (Gehrs et al., 2010). Complement activation pathways converge on the critical complement component C3, leading to the generation of complement activation products C3a, C5a and the membrane attack complex C5b-9, which have been shown to accumulate in AMD lesions in patients (Johnson et al., 2000; Mullins et al., 2014; Nozaki et al., 2006). Indeed, we also observed accumulation of C5b-9 in CNV lesions of Vegfahyper mice (Marneros, 2016), suggesting that complement pathway activation may promote CNV in this neovascular AMD mouse model and that Vegfahyper mice provide an opportunity to assess the role of complement-mediated inflammation for the manifestation of AMD-like pathologies. Complement C3 is found in the retina particularly along Bruch’s membrane and we also observed strong immunolabeling for C3 in CNV lesions of Vegfahyper mice (Figure 5A), whereas no C3 was detected in CNV lesions of VegfahyperC3-/- mice (Figure 5B). Consistent with a requirement of C3 for the formation of C5b-9, immunolabeling detected C5b-9 in CNV lesions of Vegfahyper mice but not in CNV lesions of VegfahyperC3-/- mice (Figure 5B; Figure 5—figure supplement 1).
Figure 5 with 2 supplements see all
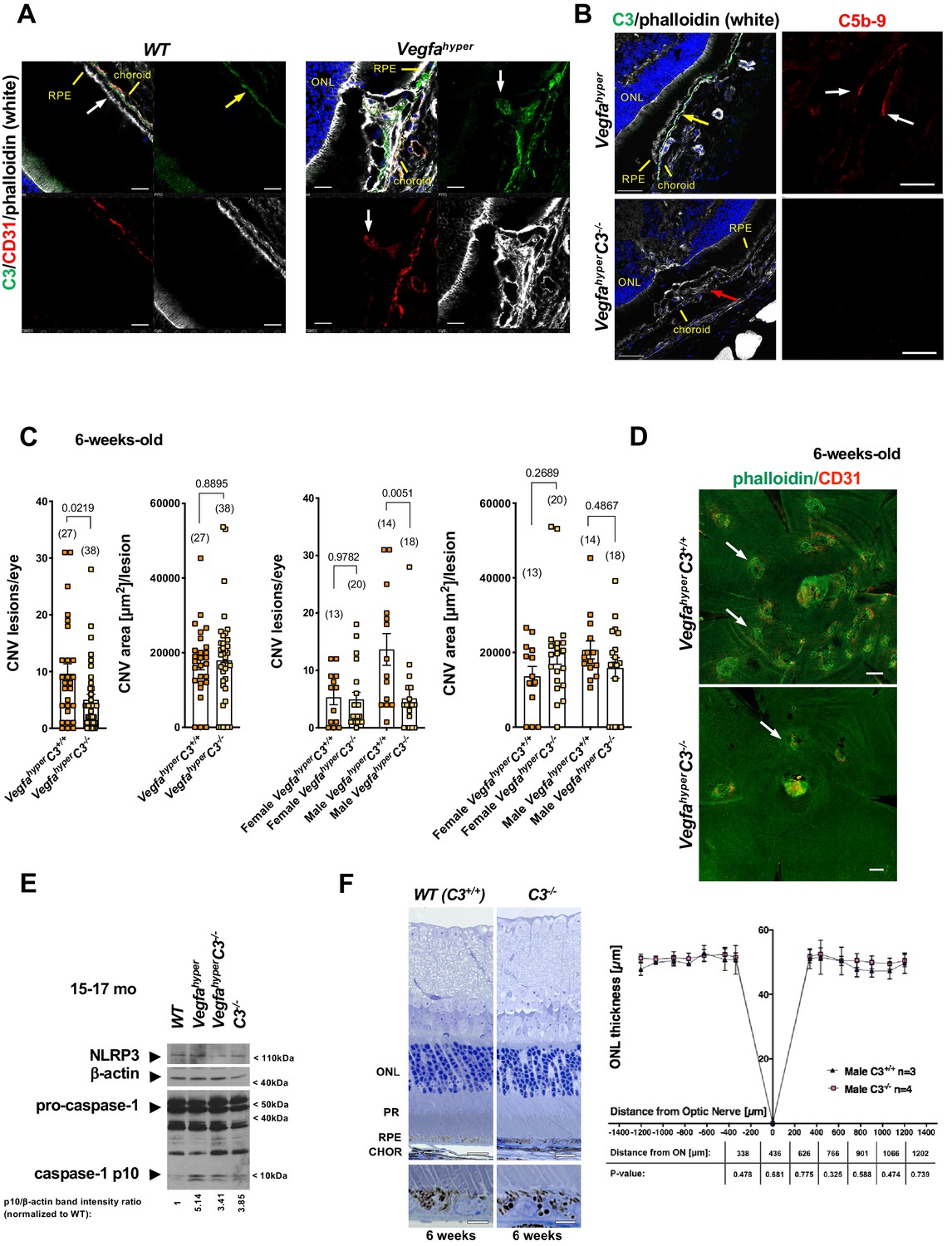
Targeting complement-mediated inflammation inhibits CNV without preventing inflammasome activation.
(A) Left: Complement C3 (yellow arrow, green) is localized particularly along Bruch’s membrane under RPE cells (white arrow) in WT mice. Right: Strong C3 immunolabeling (green) is also observed in CNV lesions of Vegfahyper mice, where it co-localizes with CD31+ neovessel protrusions (red; white arrows). 12-months-old mice. Scale bars, 50 μm. Location of ONL, choroid, and RPE are shown. (B) Left: C3 (green, yellow arrow) is detected in CNV lesions in eyes of Vegfahyper mice but not in CNV lesions in VegfahyperC3-/- mice (CNV lesion shown by a red arrow), confirming loss of C3 protein in C3-/- mice. Right: CNV lesions in Vegfahyper mice show immunolabeling for C5b-9 (red, white arrows), whereas no C5b-9 immunolabeling is detected in CNV lesions in VegfahyperC3-/- mice. 12-months-old mice. Scale bars, 50 μm (left), 20 μm (right). C5b-9 immunolabeling (red) from co-immunolabeling image shown in Figure 5—figure supplement 1. Locations of ONL, choroid, and RPE are shown. (C) VegfahyperC3-/- mice show significantly fewer CNV lesions compared to littermate Vegfahyper mice (VegfahyperC3+/+ mice), but this difference was only observed in male mice. Male Vegfahyper mice have more CNV lesions compared to females, whereas there was no marked difference in lesion numbers for male and female VegfahyperC3-/- mice. Inactivation of C3 did not significantly affect CNV lesion sizes. 6-weeks-old mice. Values represent total CNV lesion numbers/eye for each mouse and average lesion area/eye for each mouse. Absolute numbers of mice per group are indicated in parentheses. Graphs show mean ± SEM. P-values are shown (two-tailed) and were determined with a Mann-Whitney test. (D) Representative choroidal flat mount images of 6-weeks-old male mice show multifocal CD31+ CNV lesions (white arrows) in Vegfahyper mice and significantly fewer CNV lesions in VegfahyperC3-/- mice. Staining with phalloidin in green and immunolabeling for CD31 in red. Scale bars, 200 μm. (E) Lack of complement C3 does not prevent inflammasome activation in eyes of VegfahyperC3-/- mice. Two RPE/choroid tissue lysates were pooled from 15 to 17 months-old mice. Ratios of p10/β-actin band intensities normalized to WT values are shown. (F) C3 deficiency does not cause significant retinal morphological defects at 6 weeks of age. Representative sections through posterior eyes of 6-weeks-old male WT mice and C3-/- mice are shown. Scale bars, top 20 μm, bottom 5 μm. Outer nuclear layer (ONL) thickness was measured at seven distances along the meridian in eyes of 6-weeks-old mice. Values represent average ONL thickness in μm per group. SEMs are shown for each mean. Student’s t-tests were performed for values measured at equal distances from the optic nerve (ON). No significant difference in ONL thickness between WT mice and C3-/- mice was observed. PR: photoreceptors; CHOR: choroid.
Notably, complement activation products C3a and C5b-9 can lead to NLRP3 inflammasome activation in macrophages in vitro (Asgari et al., 2013; Brandstetter et al., 2015; Laudisi et al., 2013; Suresh et al., 2016; Triantafilou et al., 2013). These observations raise the question of whether complement pathway activation promotes CNV through NLRP3 inflammasome activation that leads to the release of pro-inflammatory and proangiogenic IL-1β. However, whether the in vitro observation of complement-mediated inflammasome activation also has a role for CNV lesion formation in vivo or whether complement affects CNV through inflammasome-independent mechanisms is currently not known. Answering this question has important clinical relevance, as an inflammasome-independent effect of complement-mediated inflammation on CNV progression would suggest that combining therapeutic strategies that target both inflammatory pathways would be expected to be more efficient than when only targeting complement activation or inflammasome activation alone. We hypothesize that several factors that accumulate at sites of evolving CNV lesions in Vegfahyper mice and that have been linked to inflammasome activation (such as increased oxidative stress, sub-RPE lipid deposits, and complement activation products) may all promote inflammasome activation and, thereby, increase CNV formation. However, whether complement pathway activation products contribute in a significant way to inflammasome activation at sites of CNV lesion formation in Vegfahyper mice is not known.
Thus, we next tested the effects of inactivation of complement C3 on inflammasome activation and the manifestation of neovascular AMD-like pathologies in Vegfahyper mice by generating Vegfahyper mice that lack complement C3 (VegfahyperC3-/- mice) (Figure 5B; Wessels et al., 1995). We found that 6-weeks-old VegfahyperC3-/- mice had significantly fewer CNV lesions than age-matched Vegfahyper littermates (corresponding to VegfahyperC3+/+ mice without inactivation of C3), but no difference in CNV lesion size was observed (Figure 5C and D). Notably, female Vegfahyper mice tend to have fewer CNV lesions than male Vegfahyper mice (Figures 2C and 4C). However, the potent relative reduction in VEGF-A-induced CNV lesion numbers in mice lacking C3 compared to their Vegfahyper littermates was observed only in male mice but not in female mice (Figure 5C). This is consistent with the previously reported observation that female C57bl6 mice show a reduced ability to promote inflammation via C5b-9, while they show normal complement cascade functionality at the level of C3 (Kotimaa et al., 2016). Thus, the difference in CNV lesion numbers between male and female VegfahyperC3-/- mice also suggests that VEGF-A-induced CNV lesion formation in Vegfahyper mice is likely promoted by terminal complement pathway activation.
Importantly, we observed a > 3 fold increase in caspase-1 p10 levels in RPE/choroid lysates of VegfahyperC3-/- mice (lacking complement C3) compared to age-matched WT mice, demonstrating that lack of C3 does not prevent inflammasome activation in these eyes (Figure 5E; ratios of p10/β-actin band intensity quantifications shown). A moderate decrease in p10 levels in RPE/choroid lysates of VegfahyperC3-/- mice compared to those of age-matched Vegfahyper mice may indicate a minor contribution of complement pathway activation to inflammasome activation in RPE/choroids. However, the findings show that C3 is not required for inflammasome activation in RPE/choroid lysates of Vegfahyper mice and that inflammasome activation can occur through C3-independent parallel activation mechanisms. We confirmed this also by immunolabeling for caspase-1 p10 in eyes of VegfahyperC3-/- mice, where we observed p10 staining in F4/80+ activated macrophages and retinal microglia that infiltrate CNV lesions (Figure 3C). As lack of C3 results in fewer CNV lesions in male VegfahyperC3-/- mice compared to male Vegfahyper mice in a highly significant manner despite inflammasome activation in their RPE/choroids, this suggests that C3 affects CNV lesion formation mainly through inflammasome-independent mechanisms. This conclusion is also supported by the data showing that Vegfahyper mice lacking all inflammasomes (VegfahyperCasp1-/-Casp11-/- mice) have fewer CNV lesions than VegfahyperC3-/- mice (if C3 would be critical for inflammasome activation, then VegfahyperC3-/- mice should have a similarly strong reduction in CNV lesions as VegfahyperCasp1-/-Casp11-/- mice) (Figure 6).
Figure 6
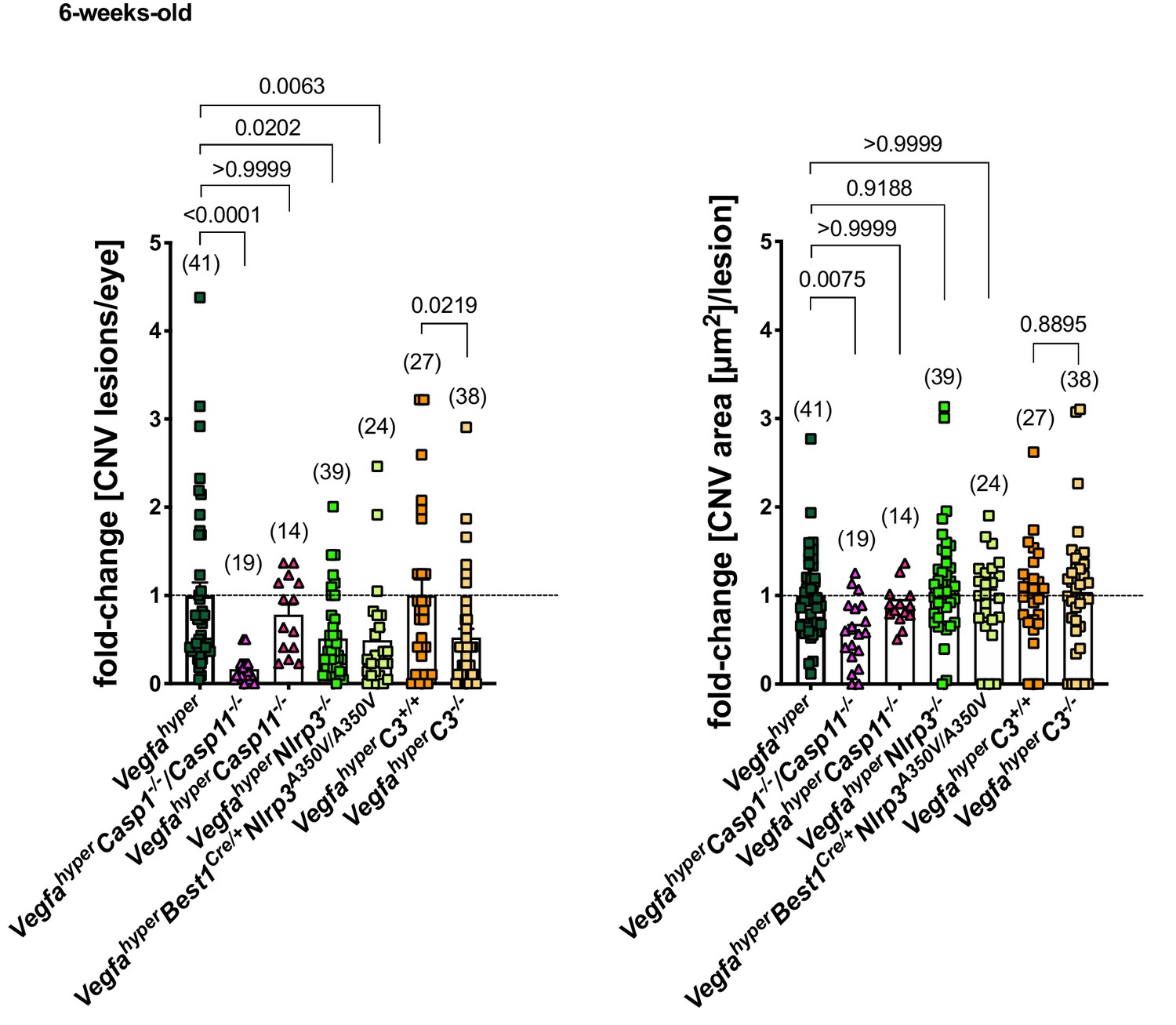
Inactivation of all inflammasomes inhibits CNV lesion formation significantly more than when targeting only NLRP3 or C3.
Comparison of fold-changes in CNV lesion numbers and sizes in all strains relative to Vegfahyper mice. 6-weeks-old mice. VegfahyperC3+/+ mice are littermates of VegfahyperC3-/- mice that are WT for C3. Graphs show mean ± SEM. Absolute numbers of mice per group are indicated in parentheses. P-values are shown. Kruskal-Wallis test followed by Dunn’s test.
Notably, the observation that CNV lesion induction (CNV numbers) is promoted by complement C3, but not their growth (CNV area) (Figure 5C), was also made when targeting NLRP3 (Figure 2B and C). In contrast, when targeting all inflammasomes (in VegfahyperCasp1-/-Casp11-/- mice), we observed not only a much more potent inhibition of CNV lesion numbers but also of their growth (CNV area) (Figure 2B and C). This suggests that CNV lesion growth is at least in part also affected by NLRP3- and C3-independent mechanisms.
C3 inactivation caused no major morphological abnormalities of the RPE or retina when examined at 6 weeks of age, as assessed by cross-sectional morphology of the RPE and the outer nuclear layer (ONL) as well as by RPE staining of choroidal flat mounts with phalloidin (Figure 5D and F). Choroidal flat mounts of 12-months-old C3-/- mice showed that RPE cells maintain their typical honeycomb pattern morphology even with progressive age (Figure 5—figure supplement 2). Notably, previously published detailed studies showed that aged C3-/- mice developed retinal abnormalities that were not observed in 6-weeks-old mice, confirming our observation that at the time in which we quantified CNV lesions (6 weeks of age) no RPE/retinal abnormalities could be attributed to C3 deficiency (Hoh Kam et al., 2013; Mukai et al., 2018; Yu et al., 2012). While these studies show that complement C3 plays a role in the maintenance of retinal function with age progression, it remains to be shown whether these retinal abnormalities in aged C3-/- mice are a consequence of life-long C3 deficiency and whether targeting C3 or other complement pathway components in aged adults impairs retinal function. Despite the observation of retinal abnormalities in aged C3-/- mice that completely lack complement activity, it is possible that strategies to only attenuate complement-mediated inflammation in aged adults with AMD may have therapeutic benefits while maintaining low-level complement pathway activity to not cause detrimental effects on retinal homeostasis.
Collectively, our data demonstrate that VEGF-A-induced neovascular AMD-like pathologies are promoted through inflammasome activation that occurs predominantly in non-RPE cells, and which is only in part mediated by NLRP3 but can also occur independently of NLRP3, caspase-11, or complement C3 (Figure 6). Thus, therapeutic strategies that combine approaches to inhibit both inflammasome activation and complement-mediated inflammation could result in more potent inhibition of neovascular AMD than either treatment approach alone.
Materials and methods
Animals
For all animal studies, institutional approval by Massachusetts General Hospital was granted. Experiments were performed in compliance with ARRIVAL guidelines. The generation of Vegfahyper mice was previously reported (Miquerol et al., 1999). The increase in VEGF-A levels occurs in these mice as a consequence of the heterozygous insertion of an IRES-NLS-lacZ-SV40pA sequence +202 bp 3’ to the STOP codon into the 3-’UTR of the Vegfa gene locus (Miquerol et al., 1999). The observed ocular abnormalities always co-segregated with the Vegfahyper allele (VegfalacZ/WT) and were not observed in any of the examined control littermate mice (Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016). Vegfahyper mice were crossed with C3-/-, Casp1-/-/Casp11-/-, Casp11-/-, and Nlrp3A350VneoR mice (mice were obtained from JAX laboratories, except Casp1-/-/Casp11-/- that were obtained from the original investigator) (Brydges et al., 2009; Hara et al., 2006; Li et al., 2007; Li et al., 1995; Wessels et al., 1995). Of note, mice lacking Casp1 have an incidental Casp11 mutation (Casp1 and Casp11 are too close in the genome to be segregated by recombination), rendering them double KOs for both caspase-1 and caspase-11 (Kayagaki et al., 2011). In contrast, Casp11 null mice have a functional Casp1 gene. Casp1-/-/Casp11-/- mice and Casp11-/- mice have both been shown to lack functional caspase-11 protein (Kayagaki et al., 2011). Homozygous Nlrp3A350V mice contain a floxed neo cassette in reverse orientation (neoR) in intron 2 leading to the lack of functional Nlrp3 gene expression in the absence of Cre expression (here referred to as Nlrp3-/- mice) (Brydges et al., 2009). VegfahyperNlrp3A350V/A350V mice were crossed with Best1Cre/+ mice (Iacovelli et al., 2011). The specificity of Cre recombinase expression in Best1Cre/+ mice has been extensively analyzed previously, and it was shown both in eye sections as well as in choroidal flat mounts that Cre recombinase expression in the eyes of these mice occurs specifically in RPE cells and not in other cells of the eye (Iacovelli et al., 2011), which we confirmed in our previous study as well (He et al., 2014). Moreover, we and others showed in these previous publications that co-labeling for phalloidin (outlining the typical honeycomb morphology of RPE cells) and with an antibody against Cre recombinase provides an accurate visualization of the numbers of RPE cells that express Cre recombinase. We confirmed these results in extensive studies by combining Cre immunolabeling with markers that accurately visualize RPE cells (e.g. phalloidin staining or active β-catenin immunolabeling, both highlighting RPE cell membranes) (Figure 2A; Figure 2—figure supplement 1; He et al., 2014). Only those choroidal flat mounts of VegfahyperBest1Cre/+Nlrp3A350V/A350V mice were included in CNV quantifications that showed uniform Cre expression in the RPE (Figure 2A; Figure 2—figure supplement 1A and B). We also confirmed that in VegfahyperBest1Cre/+Nlrp3A350V/A350V mice Best1Cre/+-mediated removal of the floxed neoR cassette in intron 2 of the Nlrp3 gene indeed occurs only in RPE cells. RPE cells were isolated from VegfahyperBest1Cre/+Nlrp3A350V/A350V mice with dispase-II treatment (20 mg/ml) (D4693, Sigma) and PCR of isolated genomic DNA with primers flanking the floxed neoR cassette in intron 2 of the Nlrp3 gene showed the presence of a PCR product without the neoR sequence (Nlrp3A350V allele from the subset of RPE cells in which Cre was active) in addition to a PCR product containing the neoR cassette (Nlrp3-/- allele from the subset of RPE cells in which Cre was not active). In contrast, RPE cells from VegfahyperNlrp3-/- mice without the Cre showed only the PCR product containing the neoR cassette (Nlrp3-/- allele). PCR primers used were: 5’-GCTACTTCCATTTGTCACGTCC-3’ (mutant forward), 5’-CACCCTGCATTTTGTTGTTG-3’ (WT forward), 5’-CGTGTAGCGACTGTTGAGGT-3’ (common). Thus, Cre is not only expressed in the RPE of VegfahyperBest1Cre/+Nlrp3A350V/A350V mice but also active and mediates Cre-mediated excision of floxed alleles that lead to the Nlrp3A350V/A350V mutant alleles (Figure 2—figure supplement 1C). Notably, no Cre-mediated excision of the floxed neoR cassette was observed in the retina, lens, or choroid in eyes of VegfahyperBest1Cre/+Nlrp3A350V/A350V mice.
Lack of caspase-1 and NLRP3 protein in Casp1-/-/Casp11-/- mice and Nlrp3-/- mice respectively were confirmed in western blotting experiments of RPE/choroid lysates of these mice with validated anti-caspase-1 or anti-NLRP3 antibodies (Figure 4A and B). Lack of C3 protein in eyes of C3-/- mice was confirmed by immunolabeling with anti-C3 antibodies (Figure 5B). All mice were housed under conventional breeding conditions.
Morphological examination of eyes
Request a detailed protocolFor semithin sections, eyes were fixed in 1.25% paraformaldehyde and 2.5% glutaraldehyde in 0.1 M cacodylate buffer (pH 7.4). After post-fixation in 4% osmium tetroxide, and dehydration steps, tissues were embedded in TAAB epon (Marivac Ltd.). 1 μm-thin sections were used for toluidine blue staining and light microscopy. Outer nuclear layer average thickness of 3 different slides at distances of 338.5, 436.5, 626, 766.5, 901.5, 1066, 1202.5 μm from the optic nerve was determined for each eye using Zeiss Zen blue 2.0 software. Genotype information was concealed when ONL thickness was determined. Results were subsequently assigned to genotypes and differences between mouse strains were determined. P-values were calculated with a two-tailed unpaired Student's t-test. Representative images were obtained from each eye.
Western blotting
Request a detailed protocolFreshly dissected RPE/choroid tissues were lysed in NP-40 lysis buffer (Life Technologies) with 1 mM PMSF and protease inhibitor cocktail using the Qiagen TissueLyser II. After centrifugation, the supernatant was used for protein quantification. RPE/choroid tissues from WT mice, Vegfahyper mice, VegfahyperNlrp3-/- mice, VegfahyperBest1Cre/+Nlrp3A350V/A350V mice, VegfahyperCasp1-/-Casp11-/- mice, VegfahyperCasp11-/- mice, C3-/- mice, and VegfahyperC3-/- mice were used for western blotting. Equal protein loading was assessed using a polyclonal rabbit anti-mouse β-actin antibody (Lab Vision Cat# RB-9421-P0, RRID:AB_720055) at a dilution of 1:1000. NLRP3 was detected using a monoclonal rabbit anti-mouse NLRP3 antibody (Cell Signaling Technology Cat# 15101, RRID:AB_2722591) at a dilution of 1:1000 (Kosmidou et al., 2018). For detection of pro-caspase-1 and activated caspase-1 p10, a polyclonal rabbit anti-mouse antibody (Santa Cruz Biotechnology Cat# sc-514, RRID:AB_2068895) was used at a dilution of 1:200. The specificity of the NLRP3 and caspase-1 p10 antibodies was confirmed by the absence of a signal in RPE/choroid lysates from Vegfahyper mice lacking either NLRP3 or caspase-1. An anti-caspase-1 p10 antibody from Thermo Fisher Scientific (Thermo Fisher Scientific Cat# PA5-105049, RRID:AB_2816522) was used as well. HRP-conjugated secondary antibodies were used and chemiluminescence signal was determined with SuperSignal WestPico chemiluminescent substrate (Thermo Fisher, Waltham, MA). p10, NLRP3, and β-actin band intensities were quantified with Image J.
Immunolabeling and choroidal flat mounting
Request a detailed protocolCNV lesions in mice with the Vegfahyper allele can be accurately quantitated in choroidal flat mounts by co-labeling with phalloidin and immunolabeling for the endothelial cell marker CD31: (1) neovessels in CNV lesions show strongly increased CD31 labeling (allowing distinction from quiescent normal vessels); (2) phalloidin labeling can discern normal RPE cells by highlighting their cell membranes that show a typical honeycomb pattern, whereas this RPE cell morphology is disrupted at the site of CNV lesions by protruding CD31+ neovessels (Ablonczy et al., 2014; Marneros, 2013; Marneros, 2016). Thus, co-labeling for CD31 and with phalloidin provides a sharp demarcation of CNV lesions from surrounding tissue on choroidal flat mounts, allowing for precise measurements of CNV lesion numbers and CNV lesion areas (Figure 1 shows how CNV quantifications were performed).
The following protocol was used for choroidal flat mount stainings. Whole eyes were enucleated and fixed in 4% paraformaldehyde overnight at 4°C and then washed in PBS. For choroidal flat mounts anterior segment, lens and retina were removed. RPE/choroid tissues were permeabilized in 0.5% Triton X-100 and blocked with 5% serum in which the secondary antibodies were raised for 30 min. Incubation with primary antibodies in the blocking solution was performed overnight at 4°C. Tissues were then washed in PBS. The following primary antibodies were used at a 1:50 dilution: rat anti-mouse CD31 (BD Biosciences Cat# 550274, RRID:AB_393571) , Alexa-647-conjugated rat anti-mouse F4/80 (BioLegend Cat# 123101, RRID:AB_893504), mouse anti-Cre (Millipore Cat# MAB3120, RRID:AB_2085748), and goat anti-cleaved-caspase-1 p10 (Santa Cruz Biotechnology Cat# sc-22166, RRID:AB_2068884). The sc-22166 antibody from Santa Cruz against cleaved caspase-1 p10 is a goat polyclonal antibody raised against a short amino acid sequence containing the neoepitope at Gly315 of caspase-1 of mouse origin. Proteolytic cleavage of the precursor caspase-1 at glycine residue 317 in humans and 315 in mouse generates the functional caspase-1 subunits, known as p20 and p10 subunits. The specificity of the immunolabeling with this antibody to detect caspase-1-specific peptides was confirmed by the absence of staining in choroidal flat mounts of VegfahyperCasp1-/-Casp11-/- mice that lack caspase-1. Eye sections were also stained with an antibody that recognizes endogenous levels of caspase-1 protein only when cleaved at Asp296 but that does not cross-react with full-length caspase-1 (Cell Signaling Technology Cat# 89332). A monoclonal antibody that detects P2RY12 was obtained from BioLegend (BioLegend Cat# 848001, RRID:AB_2650633).
Cytoskeletal staining was performed with Alexa-488- or Alexa-647-conjugated phalloidin (Life Technologies), which outlines the honeycomb pattern of RPE cells. In addition, RPE cell membranes were identified by immunolabeling for active β-catenin (Cell Signaling Technology Cat# 8814, RRID:AB_11127203). For C3 immunolabeling of eye sections, we used a rabbit polyclonal anti-C3 antibody, whose lack of staining in C3-/- mice confirmed the specificity of this antibody (Abcam Cat# ab11887, RRID:AB_298669). Eye sections were also labeled with rabbit polyclonal anti-C5b-9 antibodies (Abcam Cat# ab58811, RRID:AB_879748). Co-labeling experiments were combined with single-labeling experiments and experiments omitting both either the primary or the secondary antibodies to distinguish immunolabeling from autofluorescence. Secondary Alexa-488, Alexa-555, or Alexa-647 antibodies (at a 1:100 dilution) (Life Technologies) were incubated for 3 hr at room temperature in the dark. DAPI (Life Technologies) was used for the staining of nuclei. Subsequently to immunolabeling, RPE/choroid tissues were radially cut eight times and mounted on a slide for microscopic analysis. For immunolabeling of eye sections, frozen OCT-embedded tissue blocks were cut at 7μm thickness.
Quantification of the size and number of CNV lesions
Request a detailed protocolAllocation, treatment, and handling of mice were the same across all study groups. For all mice randomization was performed and data were collected and processed randomly. Investigators quantifying CNV lesions were blinded to the genotype of the mice. We used 6-weeks-old mice for all CNV quantifications in order to have a uniform age-controlled population of mice in which early stages of CNV lesions are present. Immunolabeling was performed with anti-CD31 antibodies to detect blood vessels and combined with Alexa-488-conjugated phalloidin labeling. Anti-Cre antibody was used to assess the extent of Cre+ RPE in VegfahyperBest1Cre/+Nlrp3A350V/A350V mice. Neovessels in CNV lesions show strong CD31 immunoreactivity allowing for a clear separation of CNV lesions from surrounding tissues (Figure 1). Similarly, phalloidin-Alexa488 labeling outlines the typical honeycomb pattern of RPE cells. At sites of CNV lesions, this pattern is disrupted by protruding CD31+ neovessels, providing an accurate demarcation of CNV lesion areas (Figure 1). The area of each CNV lesion was measured in μm2 using Zen blue 2.0 software. The number of CNV lesions and the average CNV lesion area for each eye was determined. Genotyping information was concealed when lesion sizes and numbers were determined to ensure unbiased analyses. Results were subsequently assigned to genotypes and differences between mouse strains were determined. CNV lesion number and average CNV area per mouse are shown (GraphPad Prism version 8.4.3). Sample size requirements were determined based on our previous CNV quantifications in Vegfahyper mice and those lacking inflammasome components (Marneros, 2016).
Statistics
The Shapiro-Wilk test for normality showed that CNV lesions show no normal distribution. To account for this and to consider comparisons between multiple groups, CNV lesion analyses between multiple experimental groups were performed with a Kruskal-Wallis test followed by Dunn’s test. When only two groups were present (VegfahyperC3+/+ mice versus VegfahyperC3-/- mice) a Mann-Whitney test (two-sided) was performed. P-values<0.05 were considered to be statistically significant. Graphs and analyses were performed with GraphPad Prism version 8.4.3.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Complement component C5a primes retinal pigment epithelial cells for inflammasome activation by Lipofuscin-mediated photooxidative damageJournal of Biological Chemistry 290:31189–31198.https://doi.org/10.1074/jbc.M115.671180
-
Macrophage depletion diminishes lesion size and severity in experimental choroidal neovascularizationInvestigative Opthalmology & Visual Science 44:3586–3592.https://doi.org/10.1167/iovs.03-0038
-
Complement, age-related macular degeneration and a vision of the futureArchives of Ophthalmology 128:349–358.https://doi.org/10.1001/archophthalmol.2010.18
-
Choroidal neovascularizationAmerican Journal of Ophthalmology 137:496–503.https://doi.org/10.1016/j.ajo.2003.09.042
-
Inflammasomes: mechanism of action, role in disease, and therapeuticsNature Medicine 21:677–687.https://doi.org/10.1038/nm.3893
-
Age- and gene-dosage-dependent cre-induced abnormalities in the retinal pigment epitheliumThe American Journal of Pathology 184:1660–1667.https://doi.org/10.1016/j.ajpath.2014.02.007
-
Macrophages are essential for the early wound healing response and the formation of a fibrovascular scarThe American Journal of Pathology 182:2407–2417.https://doi.org/10.1016/j.ajpath.2013.02.032
-
IL-18 is not therapeutic for neovascular age-related macular degenerationNature Medicine 20:1372–1375.https://doi.org/10.1038/nm.3671
-
Complement component C3 plays a critical role in protecting the aging retina in a murine model of age-related macular degenerationThe American Journal of Pathology 183:480–492.https://doi.org/10.1016/j.ajpath.2013.04.008
-
Osmotic and hypoxic induction of the complement factor C9 in cultured human retinal pigment epithelial cells: regulation of VEGF and NLRP3 expressionMolecular Vision 24:518–535.
-
Generation of cre transgenic mice with postnatal RPE-Specific ocular expressionInvestigative Opthalmology & Visual Science 52:1378–1383.https://doi.org/10.1167/iovs.10-6347
-
A potential role for immune complex pathogenesis in drusen formationExperimental Eye Research 70:441–449.https://doi.org/10.1006/exer.1999.0798
-
Activation and regulation of the inflammasomesNature Reviews Immunology 13:397–411.https://doi.org/10.1038/nri3452
-
Cutting edge: the NLRP3 inflammasome links complement-mediated inflammation and IL-1β releaseThe Journal of Immunology 191:1006–1010.https://doi.org/10.4049/jimmunol.1300489
-
Interleukin-1β inhibition prevents choroidal neovascularization and does not exacerbate photoreceptor degenerationThe American Journal of Pathology 178:2416–2423.https://doi.org/10.1016/j.ajpath.2011.01.013
-
Multiple developmental roles of VEGF suggested by a LacZ-tagged alleleDevelopmental Biology 212:307–322.https://doi.org/10.1006/dbio.1999.9355
-
The complement system is critical in maintaining retinal integrity during agingFrontiers in Aging Neuroscience 10:15.https://doi.org/10.3389/fnagi.2018.00015
-
The membrane attack complex in aging human choriocapillaris: relationship to macular degeneration and choroidal thinningThe American Journal of Pathology 184:3142–3153.https://doi.org/10.1016/j.ajpath.2014.07.017
-
Macrophage depletion inhibits experimental choroidal neovascularizationInvestigative Opthalmology & Visual Science 44:3578–3585.https://doi.org/10.1167/iovs.03-0097
-
Complement-mediated 'bystander' damage initiates host NLRP3 inflammasome activationJournal of Cell Science 129:1928–1939.https://doi.org/10.1242/jcs.179291
-
The complement membrane attack complex triggers intracellular Ca2+ fluxes leading to NLRP3 inflammasome activationJournal of Cell Science 126:2903–2913.https://doi.org/10.1242/jcs.124388
-
NLRP3 inflammasome activation in retinal pigment epithelial cells by lysosomal destabilization: implications for Age-Related macular degenerationInvestigative Opthalmology & Visual Science 54:110–120.https://doi.org/10.1167/iovs.12-10655
-
Efficacy of novel selective NLRP3 inhibitors in human and murine retinal pigment epithelial cellsJournal of Molecular Medicine 97:523–532.https://doi.org/10.1007/s00109-019-01753-5
-
A novel role of complement in retinal degenerationInvestigative Opthalmology & Visual Science 53:7684–7692.https://doi.org/10.1167/iovs.12-10069
-
Microglia versus monocytes: distinct roles in degenerative diseases of the retinaTrends in Neurosciences 43:433–449.https://doi.org/10.1016/j.tins.2020.03.012
Article and author information
Author details
Alexander G Marneros

Funding
National Institutes of Health (R21EY027104)
- Joseph O Lamontagne
- Karin Strittmatter
- Alexander G Marneros
BrightFocus Foundation
- Alexander G Marneros
Hans-Böckler-Stiftung (Scholarship)
- Jakob Malsy
Alpha Omega Alpha (Carolyn L. Kucklein Student Research Fellowship)
- Andrea C Alvarado
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by grants from the NIH (R21EY027104) and the BrightFocus foundation to AGM. JM was supported by a scholarship of the Hans-Boeckler-Stiftung. AA was supported by an Alpha Omega Alpha Carolyn L Kucklein Student Research Fellowship. We would like to thank Dr. Mark Vangel (Massachusetts General Hospital) for advice on biostatistical analyses (supported by Harvard Catalyst).
Ethics
Animal experimentation: For all animal studies, institutional approval by Massachusetts General Hospital was granted (IACUC approval 2009N000083). Experiments were performed in compliance with ARRIVAL guidelines.
Copyright
© 2020, Malsy et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,995
- views
-
- 324
- downloads
-
- 22
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 22
- citations for umbrella DOI https://doi.org/10.7554/eLife.60194
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Distinct effects of complement and of NLRP3- and non-NLRP3 inflammasomes for choroidal neovascularization
eLife 9:e60194.
https://doi.org/10.7554/eLife.60194




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}