STING mediates immune responses in the closest living relatives of animals
- Department of Microbiology, University of Texas Southwestern Medical Center, United States
- McDermott Center Bioinformatics Lab, University of Texas Southwestern Medical Center, United States
- Howard Hughes Medical Institute, and Department of Molecular and Cell Biology, University of California, Berkeley, United States
Abstract
Animals have evolved unique repertoires of innate immune genes and pathways that provide their first line of defense against pathogens. To reconstruct the ancestry of animal innate immunity, we have developed the choanoflagellate Monosiga brevicollis, one of the closest living relatives of animals, as a model for studying mechanisms underlying pathogen recognition and immune response. We found that M. brevicollis is killed by exposure to Pseudomonas aeruginosa bacteria. Moreover, M. brevicollis expresses STING, which, in animals, activates innate immune pathways in response to cyclic dinucleotides during pathogen sensing. M. brevicollis STING increases the susceptibility of M. brevicollis to P. aeruginosa-induced cell death and is required for responding to the cyclic dinucleotide 2’3’ cGAMP. Furthermore, similar to animals, autophagic signaling in M. brevicollis is induced by 2’3’ cGAMP in a STING-dependent manner. This study provides evidence for a pre-animal role for STING in antibacterial immunity and establishes M. brevicollis as a model system for the study of immune responses.
Editor's evaluation
This is a very exciting story on the ancient role that STING plays in microbial infections.
https://doi.org/10.7554/eLife.70436.sa0Introduction
Innate immunity is the first line of defense against pathogens for all animals, in which it is crucial for distinguishing between self and non-self, recognizing and responding to pathogens, and repairing cellular damage. Some mechanisms of animal immunity have likely been present since the last common eukaryotic ancestor, including RNAi, production of antimicrobial peptides, and the production of nitric oxide (Shabalina and Koonin, 2008; Richter and Levin, 2019). However, many gene families that play critical roles in animal innate immune responses are unique to animals (Richter et al., 2018).
Comparing animals with their closest relatives, the choanoflagellates, can provide unique insights into the ancestry of animal immunity and reveal other key features of the first animal, the ‘Urmetazoan’ (King et al., 2008; Brunet and King, 2017; Richter and King, 2013). Choanoflagellates are microbial eukaryotes that live in diverse aquatic environments and survive by capturing and phagocytosing environmental bacteria (Leadbeater, 2015) using their ‘collar complex’, an apical flagellum surrounded by actin-filled microvilli (Figure 1A; Leadbeater, 2015; Dayel and King, 2014). Several innate immune pathway genes once considered to be animal-specific are present in choanoflagellates, including cGAS and STING, both of which are crucial for innate responses to cytosolic DNA in animals (Figure 1—figure supplement 1; Richter et al., 2018; Wu et al., 2014; Levin et al., 2014). Although the phylogenetic distribution of these gene families reveals that they first evolved before animal origins, their functions in choanoflagellates and their contributions to the early evolution of animal innate immunity are unknown.
Figure 1 with 1 supplement see all

P. aeruginosa has pathogenic effects on M. brevicollis.
(A) Immunofluorescence illuminates the diagnostic cellular architecture of M. brevicollis, including an apical flagellum (f) made of microtubules, surrounded by an actin-filled microvilli feeding collar (co). Staining for tubulin (green) also highlights cortical microtubules that run along the periphery of the cell body, and staining for F-actin (magenta) highlights basal filopodia (fp). DNA staining (blue) highlights the nucleus (n). (B) M. brevicollis exhibits truncated flagella after exposure to P. aeruginosa. M. brevicollis were exposed to E. coli or P. aeruginosa for 24 hr, and then fixed and immunostained. Arrows point to flagella. Green: anti-tubulin antibody (flagella and cell body), magenta: phalloidin (collar), blue: Hoechst (bacterial and choanoflagellate nuclei). Scale bars represent 10 μm. Flagellar length was quantified using Fiji, and statistical analysis (unpaired t-tests) was performed in GraphPad software. (C) Exposure to P. aeruginosa, but not other Gammaproteobacteria, results in M. brevicollis cell death. Bacteria were added to M. brevicollis culture at an MOI of 1.5 (at Hours = 0), and M. brevicollis cell density was quantified at indicated time points. Data represent mean ± SD for three biological replicates. Statistical analysis (multiple unpaired t-tests) was performed in GraphPad software; p-values shown are from comparisons between Flavobacterium and P. aeruginosa. (D–F) M. brevicollis does not ingest P. aeruginosa bacteria. (D,E) M. brevicollis were fed either fluorescent E. coli (D) or P. aeruginosa (E) for 1 hr, and then visualized by DIC (D,E, left) and green fluorescence (D, E, right). Fluorescent food vacuoles were observed in choanoflagellates fed E. coli, but not P. aeruginosa. (F) M. brevicollis was exposed to GFP-expressing E. coli, V. parahaemolyticus, C. jejuni, or P. aeruginosa (MOI = 50) for 1 hr, and then imaged by DIC and green fluorescence to quantify number of cells with internalized bacteria. Choanoflagellate cells with ≥1 GFP+ food vacuole were scored as GFP+, and cells without any GFP+ food vacuoles were scored as GFP–. Data represent cells quantified over three biological replicates. (G,H) P. aeruginosa does not broadly inhibit M. brevicollis phagocytosis. (G) Internalization of 0.2 μm fluorescent beads was used to quantify phagocytic activity after exposure to E. coli or P. aeruginosa bacteria. Although cells did not phagocytose P. aeruginosa, cells exposed to E. coli and P. aeruginosa had similar phagocytic uptake of beads. Data represent n = 600 cells from three biological replicates. Statistical analyses (multiple unpaired t-tests) were performed in GraphPad software. (H) Exposure to P. aeruginosa does not inhibit phagocytic uptake of E. coli. Internalization of fluorescent E. coli or P. aeruginosa bacteria was quantified after exposure to unlabeled P. aeruginosa (PAO1 strain). Data represent n = 200 cells from two biological replicates. Statistical analysis (unpaired t-test) was performed in GraphPad software. (I) Secreted P. aeruginosa molecules are sufficient to induce M. brevicollis cell death. 5 % (vol/vol) bacterial conditioned medium was added to M. brevicollis culture (at Hours = 0), and M. brevicollis cell density was quantified at indicated time points. Data represent mean ± SD for three biological replicates. Statistical analysis (multiple unpaired t-tests) was performed in GraphPad software, and p-values shown are from comparisons between Flavobacterium and P. aeruginosa. (J) Sustained exposure to secreted P. aeruginosa molecules is required to induce M. brevicollis cell death. P. aeruginosa or Flavobacterium conditioned medium (5% vol/vol) was added to stationary-phase M. brevicollis cultures. After indicated times, cultures were washed and resuspended in fresh media. M. brevicollis cell density was quantified after 24 hr. The % survival is a measure of the cell density of P. aeruginosa-treated cells relative to Flavobacterium-treated controls. Data represent mean ± SD for three biological replicates.
STING (stimulator of interferon genes) is a signaling protein that activates innate immune responses to cytosolic DNA during bacterial or viral infection (Ablasser and Chen, 2019; Ablasser et al., 2013; Ahn and Barber, 2019). Although STING homologs are conserved in diverse invertebrate and vertebrate animals (reviewed in Margolis et al., 2017; Wu et al., 2014; Margolis et al., 2017; Kranzusch et al., 2015), mechanisms of STING activation are best understood in mammals. In mammals, STING is activated by binding 2’3’ cGAMP, an endogenous cyclic dinucleotide produced by the sensor cGAS (cyclic GMP-AMP synthase) upon detecting cytosolic DNA (Ishikawa and Barber, 2008; Sun et al., 2013; Burdette et al., 2011; Diner et al., 2013; Gao et al., 2013; Ablasser et al., 2013). In addition, cyclic dinucleotides produced by bacteria can also activate STING (Burdette et al., 2011; Moretti et al., 2017). Importantly, STING domain-containing systems are present in bacteria where they may contribute to anti-phage defense (Cohen et al., 2019; Morehouse et al., 2020), raising the possibility that eukaryotic STING-like proteins were acquired from lateral gene transfer (Burroughs et al., 2020). Comparative genomics suggests that STING domains arose at least three independent times in eukaryotes, including once in the stem lineage leading to Choanozoa, the clade containing animals and choanoflagellates (Burroughs et al., 2020).
Choanoflagellates have already served as powerful models for studying the origin of animal multicellularity and cell differentiation (Levin et al., 2014; Alegado et al., 2012; Brunet et al., 2021; Dayel et al., 2011; Woznica et al., 2017; Brunet et al., 2019; Laundon et al., 2019) and are ideally positioned to yield insights into the evolution of animal immune pathways. Therefore, we sought to establish the choanoflagellate Monosiga brevicollis as a model for studying pathogen recognition and immune responses. Here, we report that Pseudomonas aeruginosa bacteria are pathogenic for M. brevicollis. Through our study of interactions between P. aeruginosa and M. brevicollis, we determined that STING functions in the choanoflagellate antibacterial response. In addition, we demonstrate that STING is necessary for mediating responses to the STING agonist 2’3’ cGAMP in vivo, and that 2’3’ cGAMP induces STING-dependent autophagic signaling. Our results demonstrate that key features of STING-mediated immune responses are conserved in M. brevicollis, thereby expanding our understanding of the pre-metazoan ancestry of STING signaling.
Results
P. aeruginosa has pathogenic effects on M. brevicollis
One impediment to studying immune responses in choanoflagellates has been the lack of known choanoflagellate pathogens. While bacteria are obligate prey and can regulate mating, multicellular development, and cell contractility in choanoflagellates, to our knowledge no bacteria with pathogenic effects have been described (Alegado et al., 2012; Woznica et al., 2017; Brunet et al., 2019; Woznica et al., 2016; Ireland et al., 2020; Hake, 2021). For this study, we focused on the choanoflagellate Monosiga brevicollis, which has a sequenced genome (King et al., 2008) and grows robustly under laboratory conditions in co-culture with Flavobacterium prey bacteria (Brunet and King, 2017). To identify potential pathogens of choanoflagellates, we screened select bacteria – including environmental isolates and known animal pathogens and commensals (Table 1) – to test whether any of these induced M. brevicollis behavioral changes or reduced cell survival.
Table 1
Bacteria screened for pathogenic effects.
| Bacterium | Pathogenic effects | Reference or details | Source |
|---|---|---|---|
| Aeromonas hyrophila | – | Environmental isolate | This study |
| Bacillus aquimaris | – | Environmental isolate | This study |
| Bacillus badius | – | Mouse isolate | Julie Pfeiffer |
| Bacillus cereus | – | Environmental isolate | This study |
| Bacillus indicus | – | Environmental isolate | This study |
| Bacillus marisflavi | – | Environmental isolate | This study |
| Bacillus pumilus | – | Mouse isolate | Julie Pfeiffer |
| Bacillus safensis | – | Mouse isolate | Julie Pfeiffer |
| Bacillus subtilus | – | ATCC 6633 | Julie Pfeiffer |
| Bacteroides acidifaciens | – | Mouse isolate | Julie Pfeiffer |
| Burkholderia multivorans | – | ATCC 17616 | David Greenberg |
| Campylobacter jejuni GFP | – | DRH3123 | David Hendrixson |
| Deinococcus sp. | – | Environmental isolate | This study |
| Enterococcus cloacae | – | Mouse isolate | Julie Pfeiffer |
| Enterococcus faecium | – | Mouse isolate | Julie Pfeiffer |
| Escherichia coli BW25113 | – | Datsenko and Wanner, 2000 | David Greenberg |
| Escherichia coli DH5a GFP | – | David Hendrixson | |
| Escherichia coli ECC-1470 | – | Leimbach et al., 2015 | Julie Pfeiffer |
| Escherichia coli K12 | – | ATCC 10798 | Julie Pfeiffer |
| Flavobacterium sp. | – | King et al., 2008 | Isolated from ATCC PRA-258 |
| Lactobacillus johnsonii | – | Mouse isolate | Julie Pfeiffer |
| Pseudoalteromonas sp. | – | Environmental isolate | This study |
| Pseudomonas aeruginosa PA-14 | + | Rahme et al., 1995 | Andrew Koh |
| Pseudomonas aeruginosa PAO1 | + | ATCC 15692 | David Greenberg |
| Pseudomonas aeruginosa PAO1-GFP | + | Bloemberg et al., 1997 | David Greenberg |
| Pseudomonas granadensis | – | Environmental isolate | This study |
| Staphylococcus aureus | – | ATCC 23235 | Julie Pfeiffer |
| Staphylococcus sp. | – | Mouse isolate | Julie Pfeiffer |
| Vibrio alginolyticus | – | Environmental isolate | Kim Orth |
| Vibrio furnissii | – | Environmental isolate | This study |
| Vibrio parahaemolyticus | – | Environmental isolate | This study |
| Vibrio parahaemolyticus RimD-GFP | – | Ritchie et al., 2012 | Kim Orth |
| Vibrio ruber | – | Environmental isolate | This study |
| Vibrio sp. | – | Environmental isolate | This study |
After co-culturing M. brevicollis with bacteria for 24 hr, only the gammaproteobacterium Pseudomonas aeruginosa, a ubiquitous environmental bacterium and opportunistic pathogen of diverse eukaryotes (Moradali et al., 2017; Mahajan-Miklos et al., 1999; Pukatzki et al., 2002; Rahme et al., 1997), altered the behavior and growth dynamics of M. brevicollis. Under standard laboratory conditions, M. brevicollis is a highly motile flagellate and swims up in the water column (Video 1, Supplementary file 1). However, after 12–14 hr in the presence of P. aeruginosa strains PAO1 and PA14, a large proportion of M. brevicollis cells settled to the bottom of the culture dish (Video 1, Supplementary file 1). Immunofluorescence staining revealed that M. brevicollis cells exposed to P. aeruginosa had truncated flagella compared to cells exposed to E. coli or other bacteria that did not induce cell settling (Figure 1B). To determine the effects of P. aeruginosa on cell viability, we added P. aeruginosa strain PAO1 or control gammaproteobacteria to M. brevicollis and monitored cell density over the course of 72 hr (Figure 1C). While M. brevicollis continued to proliferate in the presence of control gammaproteobacteria, exposure to P. aeruginosa PAO1 resulted in cell death.
Video 1
Supplementary file 1.
P. aeruginosa influences M. brevicollis motility. Movies depicting M. brevicollis cultures after exposure to E. coli or P. aeruginosa bacteria for 16 hours. In the absence of pathogenic bacteria,M. brevicollis is a highly motile flagellate and swims up in the water column (Movie 1). However, co-culturing M. brevicollis with P. aeruginosa results in reduced motility and cell settling (Movie 2).
Choanoflagellates prey upon bacteria and ingest them through phagocytosis (Leadbeater, 2015; Dayel and King, 2014). However, many bacterial pathogens, including P. aeruginosa, have evolved strategies to prevent or resist phagocytosis by eukaryotic cells (Uribe-Querol and Rosales, 2017; Yoon et al., 2018). Therefore, we examined whether phagocytosis of P. aeruginosa is required to induce cell death. To track phagocytosis, we added GFP-expressing E. coli DH5α (Figure 1D) or P. aeruginosa PAO1 (Figure 1E) to M. brevicollis and monitored the cultures by live imaging. After one hour, while 92 % of M. brevicollis cells incubated with E. coli −GFP had GFP+ food vacuoles, only 3 % of cells incubated with PAO1-GFP had GFP+ food vacuoles (Figure 1F). M. brevicollis also robustly phagocytosed GFP-expressing V. parahaemolyticus and C. jejuni (Figure 1F).
Next, to determine if P. aeruginosa broadly disrupts M. brevicollis phagocytosis, which could induce cell death through starvation, we incubated M. brevicollis with GFP-expressing PAO1 or GFP-expressing E. coli for one hour, and then added 0.2 mm fluorescent beads for an additional 30 min as an independent measure of phagocytic activity. The fraction of M. brevicollis cells with internalized 0.2 mm beads was similar in cultures incubated with E. coli DH5αand PAO1 (Figure 1G). Moreover, exposure to P. aeruginosa did not inhibit phagocytic uptake of E. coli (Figure 1H). These results suggest that exposure to P. aeruginosa does not broadly inhibit phagocytosis.
The above results suggested that the pathogenic effects of P. aeruginosa are induced by factors secreted by extracellular bacteria. In addition, diverse secreted bacterial molecules have been previously shown to influence choanoflagellate cell biology (Alegado et al., 2012; Woznica et al., 2017; Woznica et al., 2016). Therefore, we next investigated the effects of secreted P. aeruginosa molecules on M. brevicollis viability. Exposure of M. brevicollis to conditioned medium from P. aeruginosa PAO1 or diverse non-pathogenic gammaproteobacteria revealed that PAO1 conditioned medium is sufficient to restrict growth and induce cell death (Figure 1I). Similar to live bacteria, exposure to P. aeruginosa conditioned medium led to reduced motility and truncated flagella in M. brevicollis after approximately 8–10 hr.
Because numerous P. aeruginosa secreted virulence factors have been characterized (Moradali et al., 2017; Klockgether and Tümmler, 2017), we screened a battery of isogenic PAO1 strains with deletions in known virulence genes to determine if any of these factors contribute to the pathogenic effects on M. brevicollis (Table 2). All strains tested induced similar levels of M. brevicollis cell death as the parental PAO1 strain, suggesting that none of the deleted virulence genes alone are essential for inducing cytotoxicity in M. brevicollis. The bioactivity in the conditioned media was also found to be heat, protease, and nuclease resistant, indicating that the virulence factors are unlikely to be proteins or nucleic acids (Table 3). In addition, we found that subjecting the conditioned media to methanol extraction followed by liquid chromatography-mass spectrometry resulted in specific fractions that recapitulated the bioactivity of the conditioned media. Although further detailed chemical analysis is required to determine the molecular nature of these factors, these data indicate that secreted P. aeruginosa small molecules are sufficient for inducing cell death in M. brevicollis.
Table 2
P. aeruginosa deletion strains.
| Effects on M. brevicollis | ||||
|---|---|---|---|---|
| Strain name | Gene | Putative ORF function | Truncated Flagellum/Settling | Cell Death |
| MPAO1 | parent to library stain | + | + | |
| PW5035 | pvdE | pyoverdine biosynthesis protein PvdE | + | + |
| PW5034 | pvdE | pyoverdine biosynthesis protein PvdE | + | + |
| PW1059 | exoT | exoenzyme T | + | + |
| PW3078 | toxA | exotoxin A precursor | + | + |
| PW3079 | toxA | exotoxin A precursor | + | + |
| PW4736 | exoY | adenylate cyclase ExoY | + | + |
| PW4737 | exoY | adenylate cyclase ExoY | + | + |
| PW6886 | rhlA | rhamnosyltransferase chain A | + | + |
| PW6887 | rhlA | rhamnosyltransferase chain A | + | + |
| PW7478 | exoS | exoenzyme S | + | + |
| PW7479 | exoS | exoenzyme S | + | + |
| PW7303 | lasB | elastase LasB | + | + |
| PW7302 | lasB | elastase LasB | + | + |
| PW3252 | aprA | alkaline metalloproteinase precursor | + | + |
| PW3253 | aprA | alkaline metalloproteinase precursor | + | + |
| PW4282 | lasA | LasA protease precursor | + | + |
| PW4283 | lasA | LasA protease precursor | + | + |
| RP436 | popB | T3SS translocase | + | + |
| RP576 | exoS, exoT, exoY | T3SS effector molecules | + | + |
Table 3
M. brevicollis response to P. aeruginosa factors.
| Treatment | Cell Death | Interpretation |
|---|---|---|
| Live P. aeruginosa | ++ | |
| P. aeruginosa conditioned media (CM) | ++ | Factor(s) are secreted by P. aeruginosa |
| P. aeruginosa outer membrane vesicles | – | Factor(s) are not present in outer membrane vesicles |
| CM, boiled 20 min | ++ | Factor(s) are not heat labile |
| CM+ proteinase K, followed by 80 C for 30 min | ++ | Factor(s) are not proteins |
| CM+ DNAse and RNAse | ++ | Factor(s) are not nucleic acids |
| CM MeOH extraction | ++ | Factor(s) are organic compounds |
Finally, we investigated whether transient exposure to P. aeruginosa is sufficient to induce cell death in M. brevicollis (Figure 1J). Because choanoflagellates likely experience fluctuations in the chemical composition of aquatic microenvironments on various timescales, we exposed stationary-phase M. brevicollis to P. aeruginosa conditioned media for increasing durations, and then assessed survival relative to Flavobacterium-treated controls after 24 hr. While M. brevicollis cell death was not observed after treatment with P. aeruginosa conditioned media for 4 hr or less, exposures lasting for 6 hr or longer reduced M. brevicollis survival (Figure 1J). These results suggest that cell death pathways are not induced immediately in response P. aeruginosa virulence factors, but are activated after longer exposures to P. aeruginosa conditioned media.
Upregulation of M. brevicollis STING in response to P. aeruginosa
To identify potential genetic pathways activated by M. brevicollis in response to pathogenic bacteria, we performed RNA-seq on M. brevicollis exposed to conditioned medium from either P. aeruginosa or Flavobacterium sp., the non-pathogenic bacterial strain used as a food source (Table 1). We found that 674 genes were up-regulated and 232 genes were downregulated twofold or greater (FDR ≤ 10–4) upon P. aeruginosa exposure compared to cells exposed to Flavobacterium (Figure 2A). The upregulated genes were enriched in biological processes including response to stress, endocytosis, microtubule-based movement, mitochondrial fission, and carbohydrate metabolism. Genes down-regulated in response to P. aeruginosa were enriched in biological processes including RNA modification and metabolism (Figure 2—figure supplement 1A). We also found that the transcription of several genes encoding proteins that function in animal antibacterial innate immunity was upregulated in response to P. aeruginosa, including C-type lectin, glutathione peroxidase, and STING (Figure 2A and B). Using an antibody we raised against the C-terminal portion of M. brevicollis STING (Figure 2—figure supplement 1C,D), we found that STING protein levels are also elevated in response to P. aeruginosa (Figure 2C). Given the importance of STING in animal immunity and its upregulation in response to P. aeruginosa, we pursued its functional relevance in the M. brevicollis pathogen response.
Figure 2 with 1 supplement see all
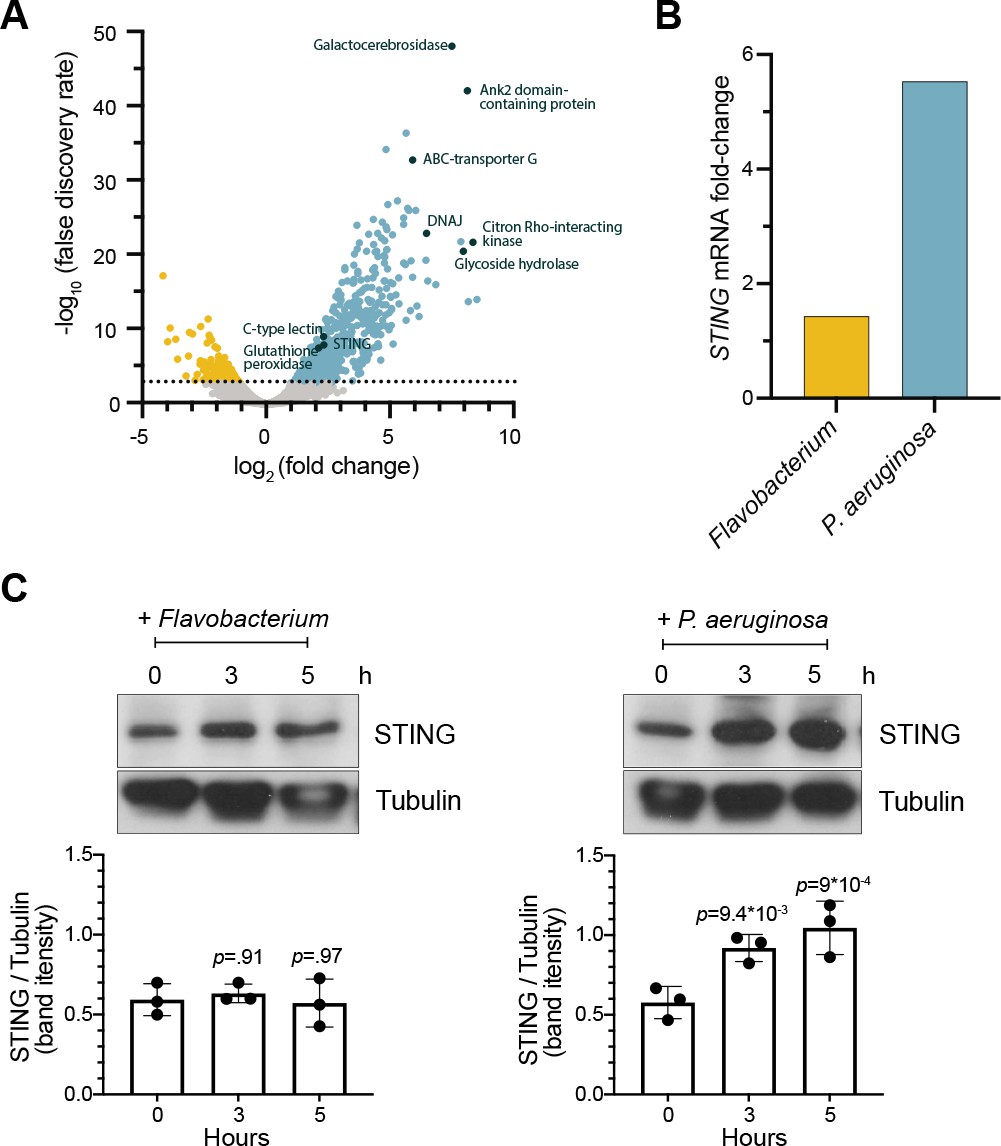
STING is upregulated in M. brevicollis after exposure to P. aeruginosa (A,B) STING transcript levels increase in response to P. aeruginosa.
(A) Volcano plot displaying genes differentially expressed between M. brevicollis exposed to P. aeruginosa PAO1 and Flavobacterium (control) conditioned medium for three hours. Differentially expressed genes are depicted by blue (674 upregulated genes) and yellow (232 downregulated genes) dots (fold change ≥2; FDR ≤ 1e–4). Select genes that are upregulated or may function in innate immunity are labeled. RNA-seq libraries were prepared from four biological replicates. (B) After a 3-hr treatment, STING mRNA levels (determined by RNA-seq) increase 1.42-fold in cells exposed to Flavobacterium conditioned medium and 5.54 fold in cells exposed to P. aeruginosa conditioned medium, compared to untreated controls. (C) STING protein levels increase after exposure to P. aeruginosa. STING levels were examined by immunoblotting at indicated timepoints after exposure to Flavobacterium or P. aeruginosa conditioned medium (5% vol/vol). Tubulin is shown as loading control, and intensity of STING protein bands were quantified relative to tubulin. Statistical analysis (one-way ANOVA, Dunnett’s multiple comparison) was performed in GraphPad software, and p-values shown are calculated using 0 hr timepoint as the control group.
The cyclic dinucleotide 2’3’ cGAMP induces elevated expression of STING in M. brevicollis
The predicted domain architecture of M. brevicollis STING consists of four transmembrane domains followed by a cytosolic STING domain (Figure 3A, Figure 2—figure supplement 1), and likely matches the structure of the ancestral animal STING protein. Vertebrate STING proteins contain a C-terminal tail (CTT; Figure 3A, Figure 2—figure supplement 1) that is required for the induction of interferons (Tanaka and Chen, 2012; Liu et al., 2015; Zhang et al., 2019), and for the activation of other downstream responses, including NFkB (Abe and Barber, 2014) and autophagy (Gui et al., 2019; Yamashiro et al., 2020; Prabakaran et al., 2018) pathways. Both the STING CTT and interferons evolved in vertebrates, and it is currently unclear how choanoflagellate and invertebrate STING proteins mediate downstream immune responses (Margolis et al., 2017; de Oliveira Mann et al., 2019). However, the conservation of putative cyclic dinucleotide-binding residues in M. brevicollis STING (Figure 3B) led us to hypothesize that STING signaling may be induced by cyclic dinucleotides. In addition, because M. brevicollis has a cGAS-like enzyme (Figure 1—figure supplement 1A), it is possible that M. brevicollis produces an endogenous cyclic dinucleotide similar to mammalian 2’3’ cGAMP (Wu et al., 2014; Ahn and Barber, 2019; Watson et al., 2015).
Figure 3 with 1 supplement see all
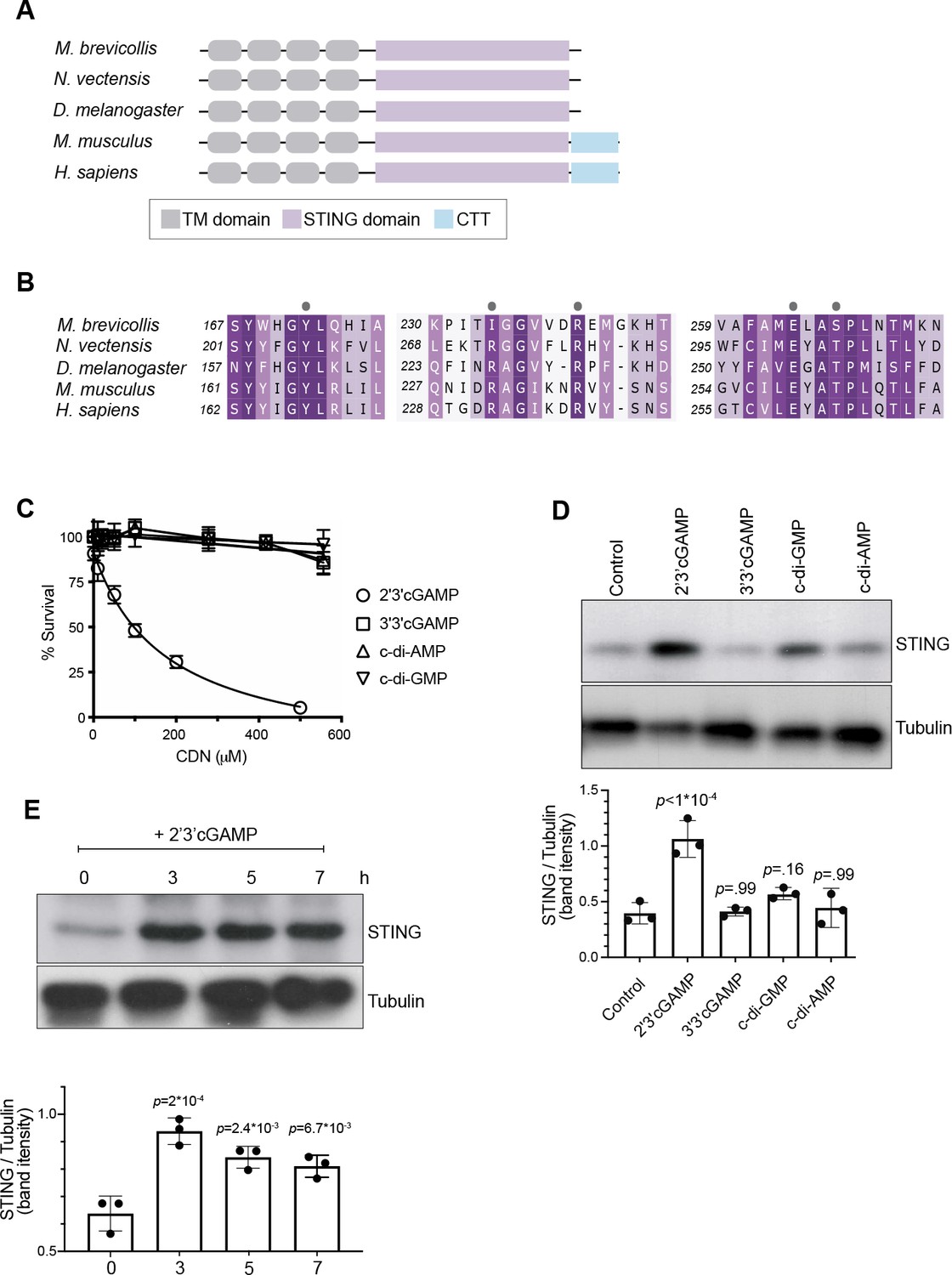
2’3’ cGAMP, but not bacterially produced cyclic dinucleotides, induces elevated levels of STING.
(A) Schematic of choanoflagellate (M. brevicollis), sea anemone (N. vectensis), insect (D. melanogaster), and mammalian (M. musculus and H. sapiens) STING proteins. Transmembrane (TM) domains are depicted in gray, STING cyclic dinucleotide binding domain (CDN) in purple, and C-terminal tail domain (CTT) in blue. (B) Partial protein sequence alignment (generated by Clustal Omega multiple sequence alignment) of M. brevicollis and animal STING proteins, colored by similarity. M. brevicollis STING and human STING are 19.1 % identical and 36.6 % similar at the amino acid level. Key cyclic dinucleotide-interacting residues from human STING structure are indicated by circles. (C) Dose-response curves of M. brevicollis exposed to cyclic dinucleotides for 24 hr reveal that treatment with 2’3’cGAMP, but not 3’3’ cGAMP, c-di-AMP, or c-di-GMP, leads to M. brevicollis cell death in a dose-dependent manner. Data represent mean ± SD for at least three biological replicates. (D) STING protein levels increase after exposure to 2’3’cGAMP, but not bacterially produced cyclic dinucleotides. M. brevicollis STING levels were examined by immunoblotting 5 hr after exposure to 2’3’cGAMP (100 µM), 3’3’cGAMP (200 µM), c-di-GMP (200 µM), or c-di-AMP (200 µM). Tubulin is shown as loading control, and intensity of STING protein bands were quantified relative to tubulin. Shown is a representative blot from three biological replicates. Statistical analysis (one-way ANOVA, Dunnett’s multiple comparison) was performed in GraphPad software. (E) STING protein levels increase and remain elevated after exposure to 100 µM 2’3’cGAMP. Tubulin is shown as loading control, and data are representative of three biological replicates. Statistical analysis (one-way ANOVA, Dunnett’s multiple comparison) was performed in GraphPad software, and p-values shown are calculated using 0 hr timepoint as control group.
To identify potential STING inducers (Watson et al., 2015; Booth et al., 2018), we treated M. brevicollis with purified cyclic dinucleotides, including mammalian cGAMP (2’3’ cGAMP) and 3’3’-linked bacterial cyclic dinucleotides (3’3’ c-di-GMP, 3’3’ c-di-AMP, 3’3’ cGAMP). We first performed dose-response curves to determine if the different cyclic dinucleotides affect the viability of M. brevicollis (Figure 3C). Interestingly, we found that exposure to 2’3’ cGAMP-induced cell death in a dose-dependent manner. In contrast, exposure to 3’3’-linked CDNs cGAMP, c-di-GMP, and c-di-AMP did not alter M. brevicollis survival. Transcriptional profiling of M. brevicollis revealed a robust transcriptional response to 2’3’ cGAMP after three hours (Figure 3—figure supplement 1A). Moreover, transcriptional profiling of M. brevicollis exposed to 2’3’ cGAMP or 3’3’ cGAMP for 3 hr revealed that STING mRNA levels increase in response to 2’3’ cGAMP, but remain unchanged in response to 3’3 cGAMP (Figure 3—figure supplement 1A-C). Therefore, we next treated M. brevicollis with the cyclic dinucleotides for 5 hr, and measured STING protein levels by immunoblot (Figure 3D). Treatment with 2’3’ cGAMP, but not the bacterially produced cyclic dinucleotides, led to elevated levels of STING protein compared to unstimulated cells. A time course of 2’3’ cGAMP treatment revealed that STING protein levels increase as early as 3 hr after exposure to the cyclic dinucleotide and remain elevated for at least 7 hr, approximately one cell cycle (Figure 3E). While we also observed sustained upregulation of STING in the presence of P. aeruginosa, this is markedly different from what has been described in mammals, wherein STING activation results in its translocation to lysosomes and degradation (Prabakaran et al., 2018). In addition, immunostaining for STING in fixed M. brevicollis revealed that the number and intensity of STING puncta increases after exposure to 2’3’ cGAMP (Figure 3—figure supplement 1E,F), although the localization of STING was difficult to assess by immunostaining due to a lack of available subcellular markers. These data suggest that M. brevicollis STING responds to 2’3’ cGAMP, and that this cyclic dinucleotide can be used to further characterize the role of STING in M. brevicollis.
Transfection reveals that STING localizes to the M. brevicollis endoplasmic reticulum
A key barrier to investigating gene function in M. brevicollis has been the absence of transfection and reverse genetics. We found that the transfection protocol recently developed for the choanoflagellate Salpingoeca rosetta (Booth et al., 2018) was not effective in M. brevicollis, but by implementing a number of alterations to optimize reagents and conditions (see Materials and methods) we were able to achieve both reproducible transfection and establishment of stably transformed cell lines in M. brevicollis.
To investigate the subcellular localization of STING, we established a robust transfection protocol for M. brevicollis that would allow the expression of fluorescently labeled STING along with fluorescent subcellular markers for different organelles.
We observed that STING-mTFP protein localized to tubule-like structures around the nucleus (Figure 4A) similar to what was observed by immunostaining with an antibody to STING (Figure 3—figure supplement 1E,F). We then co-transfected STING-mTFP alongside fluorescent reporters marking the endoplasmic reticulum (ER) or mitochondria (Figure 4B,C) and performed live-cell imaging. STING-mTFP co-localized with a fluorescent marker highlighting the ER (Figure 4B). Thus, as in mammalian cells (Ishikawa and Barber, 2008; Dobbs et al., 2015), STING localizes to regions of the ER in M. brevicollis.
Figure 4
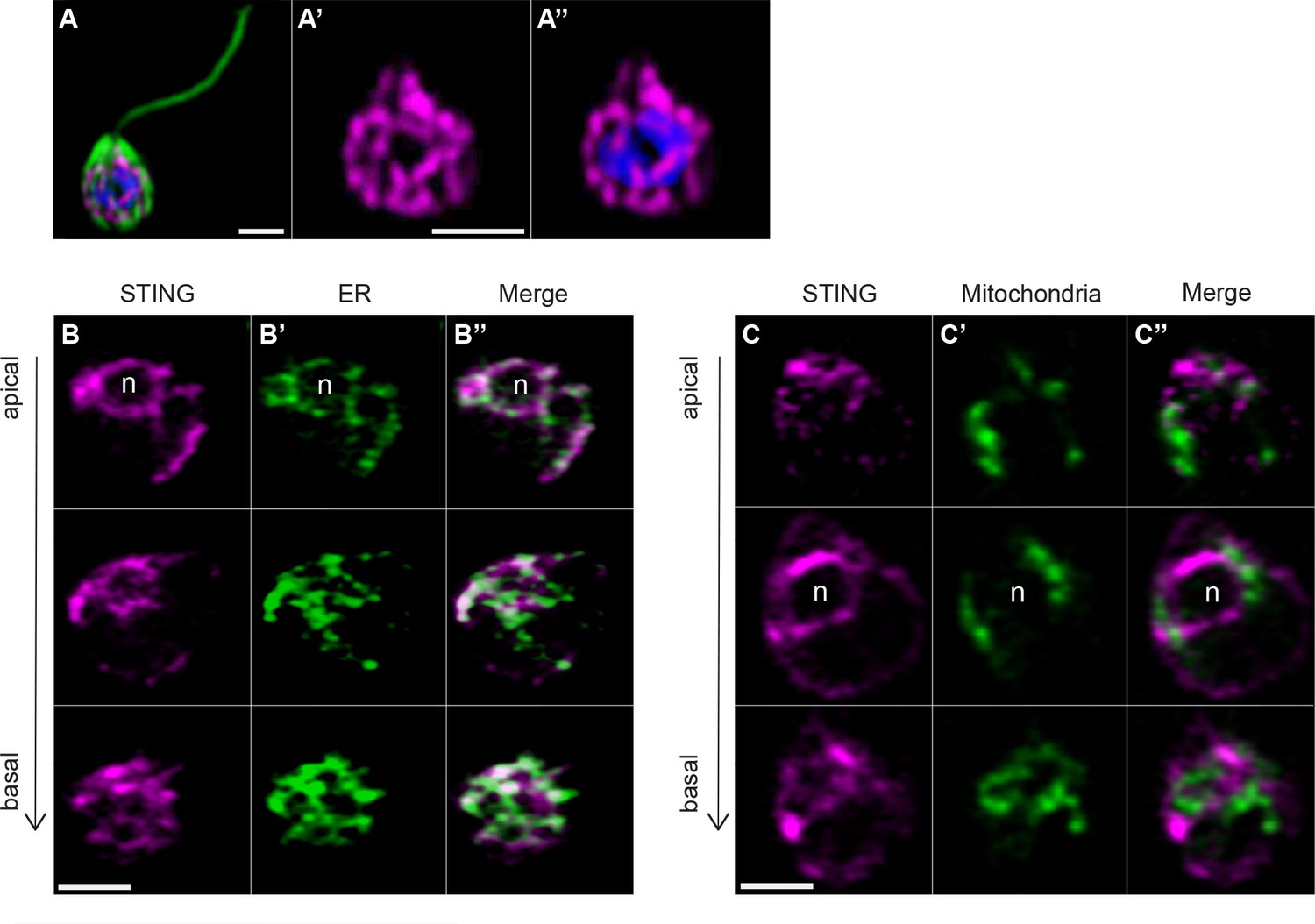
Transfection reveals STING localization to perinuclear and endoplasmic reticulum regions.
(A) STING-mTFP localizes to tubule-like structures around the nucleus in cells stably expressing STING-mTFP. Green: anti-tubulin antibody (flagella and cell body), magenta: anti-STING antibody, blue: Hoechst. Scale bar represents 2 µm. (B,C) Fluorescent markers and live cell imaging reveal that STING is localized to the endoplasmic reticulum (ER). Cells were co-transfected with STING-mTFP and an mCherry fusion protein that localizes either to the endoplasmic reticulum (B; mCherry-HDEL) or mitochondria (C; Cox4-mCherry) (King et al., 2008). Cells were recovered in the presence of Flavobacterium feeding bacteria for 28 hr after co-transfection, and then live cells were visualized with super-resolution microscopy. Each panel shows Z-slice images of a single representative cell. In confocal Z-slice images, cells are oriented with the apical flagella pointing up, and the nucleus is marked by ‘n’ when clearly included in the plane of focus. STING colocalized with the ER marker (B’’), but not the mitochondrial marker (C’’). Scale bar represents 2 µm.
Genetic disruption of STING reveals its role in responding to 2’3’ cGAMP and P. aeruginosa
Disrupting the STING locus using CRISPR/Cas9-mediated genome editing (Figure 5A) enabled us to investigate the function of STING. To overcome low gene editing efficiencies in M. brevicollis, we based our gene editing strategy on a protocol recently developed for S. rosetta that simultaneously edits a gene of interest and confers cycloheximide resistance (Booth and King, 2020). By selecting for cycloheximide resistance and then performing clonal isolation, we were able to isolate a clonal cell line that has a deletion within the STING locus that introduces premature stop codons (Figure 5—figure supplement 1A). We were unable to detect STING protein in STING– cells by immunoblot (Figure 5B). Wild type and STING– cells have similar growth kinetics (Figure 5—figure supplement 1B), suggesting that STING is not required for cell viability under standard laboratory conditions. In addition, overexpression of STING-mTFP did not affect M. brevicollis viability.
Figure 5 with 1 supplement see all
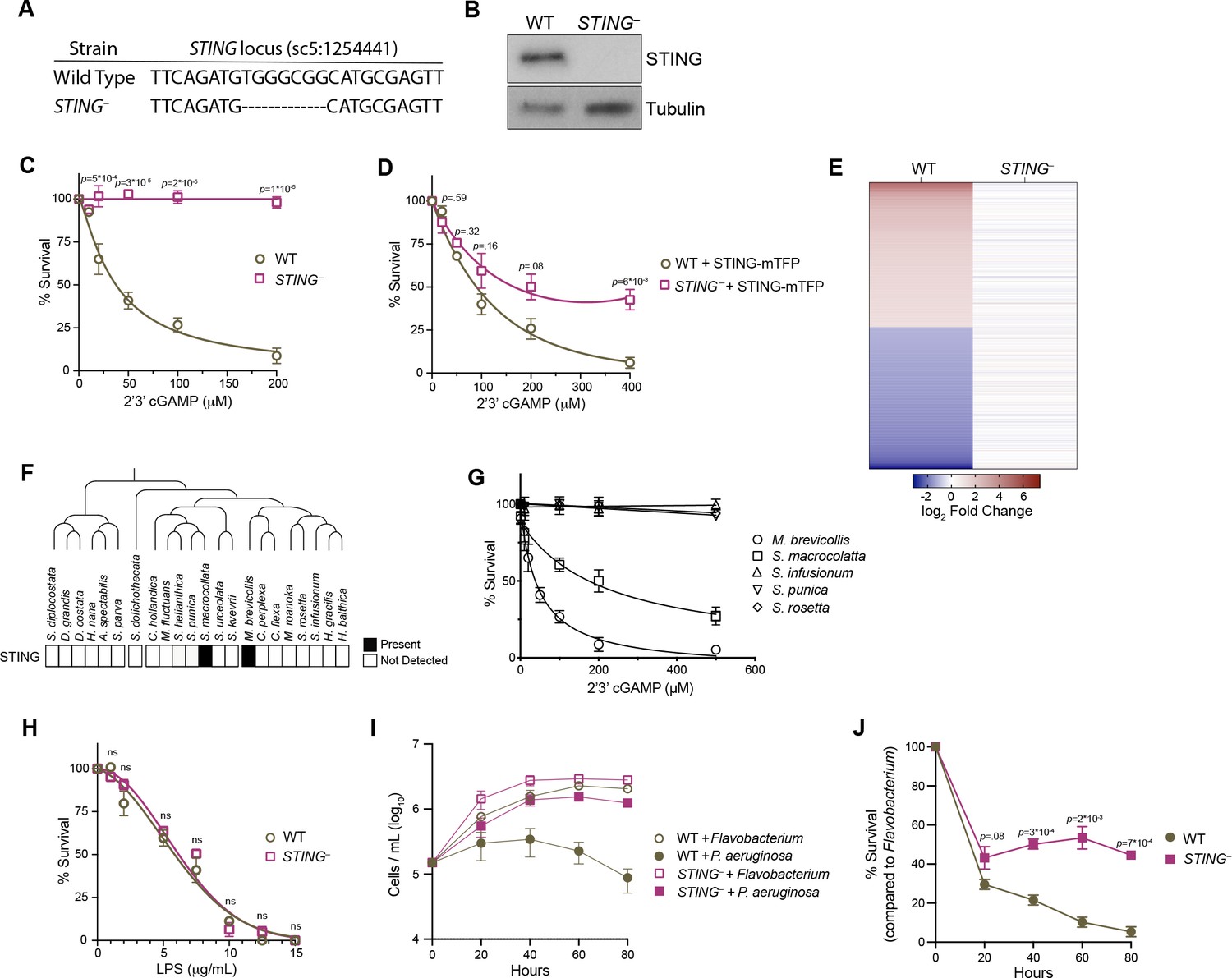
STING mediates responses to 2’3’cGAMP and P. aeruginosa.
(A) The genotypes of wild type and genome-edited STING– strains at the STING locus. (B) STING protein is not detectable by immunoblot in STING– cells. Shown is a representative blot from three biological replicates. (C,D) STING is necessary for 2’3’cGAMP-induced cell death. (C) Wild type and STING– strains were treated with increasing concentrations of 2’3’cGAMP, and survival was quantified after 24 hr. In contrast to wild type cells, 2’3’cGAMP does not induce cell death in STING– cells. Data represent mean ± SD for four biological replicates. (D) Wild type and STING– cells were transfected with STING-mTFP, and treated with puromycin to generate stable clonal strains. Stable expression of STING-mTFP in STING– cells partially rescued the phenotype of 2’3’cGAMP-induced cell death. Data represent mean ± SD for three biological replicates. Statistical analysis (multiple unpaired t-tests) was performed in GraphPad software. (E) Wild type and STING– strains have distinct transcriptional responses to 2’3’ cGAMP. Differential expression analysis was performed on wild type and STING– cells treated with 100 µM 2’3’cGAMP or a vehicle control for 3 hr. A heatmap comparing the log2 fold change of genes identified as differentially expressed (FC ≥2; FDR ≤ 10–4) in wild-type cells after 2’3’ cGAMP treatment, to their log2 fold change in STING– cells after 2’3’ cGAMP treatment. RNA-seq libraries were prepared from two biological replicates. (F) Presence of STING in the transcriptomes of diverse choanoflagellate species. Data from Richter et al., 2018. (G) Effects of 2’3’cGAMP on different choanoflagellate species. Choanoflagellates were grown to late-log phase, and treated with increasing concentrations of 2’3’cGAMP. Survival was quantified after 24 hr. 2’3’cGAMP only affected the survival of M. brevicollis and S. macrocollata, the two sequenced choanoflagellate species with a STING homolog. Data represent mean ± SD for three biological replicates. (H) Wild type and STING– cells have similar survival responses to LPS, suggesting that STING is not required for mediating a response to LPS. Wild type and STING– strains were treated with increasing concentrations of E. coli LPS, and survival was quantified after 24 hr. Data represent mean ± SD for four biological replicates. Statistical analysis (multiple unpaired t-tests) was performed in GraphPad software. (I,J) STING renders M. brevicollis more susceptible to P. aeruginosa-induced growth inhibition. (I) Wild type and STING– cells were exposed to control Flavobacterium or P. aeruginosa conditioned medium (5% vol/vol), and cell densities were quantified at indicated time points. Data represent mean ± SD for three biological replicates. (J) Percent survival calculated from growth curves in (I). Statistical analysis (multiple unpaired t-tests) was performed in GraphPad software.
To investigate the connection between 2’3’ cGAMP and STING signaling in M. brevicollis, we exposed STING– cells to increasing concentrations of 2’3’ cGAMP. In contrast to wild-type M. brevicollis, STING– cells are resistant to 2’3’ cGAMP-induced cell death (Figure 5C). The 2’3’ cGAMP resistance phenotype could be partially reversed by stably expressing STING within the STING– mutant background (Figure 5D). In addition, STING– cells fail to induce a strong transcriptional response to 2’3’ cGAMP compared to wild-type cells (Figure 5E, Figure 3—figure supplement 1A, Figure 5—figure supplement 1C,D). While 371 genes are differentially expressed in wild-type cells after exposure to 2’3’ cGAMP for 3 hr, only 28 genes are differentially expressed in STING– cells (FC ≥3; FDR ≤ 10–4). Thus, 2’3’ cGAMP induces a STING-dependent transcriptional response in M. brevicollis.
Interestingly, of the 22 choanoflagellate species with sequenced transcriptomes (Richter et al., 2018; Brunet et al., 2019), only M. brevicollis and Salpingoeca macrocollata, express homologs of both STING and cGAS (Figure 1—figure supplement 1A, Figure 5F, Figure 5—figure supplement 1E). Therefore, we were curious whether other choanoflagellate species are able to respond to 2’3’ cGAMP in the absence of a putative STING protein. We exposed four other choanoflagellate species (Salpingoeca infusionum, S. macrocollata, S. rosetta, and Salpingoeca punica) to increasing 2’3’ cGAMP concentrations, and quantified survival after 24 hours (Figure 5G). Of these additional species, only S. macrocollata had impaired survival in the presence of 2’3’ cGAMP. Thus, it is possible that STING also responds to 2’3’ cGAMP in S. macrocollata.
We next asked whether STING– cells have altered responses to other immune agonists. Although M. brevicollis is continuously co-cultured with feeding bacteria, we observed that treatment with high concentrations of E. coli lipopolysaccharides induces cell death (Figure 5H). As LPS is not known to activate STING signaling, we treated wild type and STING– cells with LPS to probe the specificity of STING-mediated immune responses in M. brevicollis. The survival responses of wild type and STING– cells to LPS were indistinguishable (Figure 5H), suggesting that there are separable pathways for responding to 2’3’ cGAMP and LPS. We also examined the survival of STING– cells exposed to P. aeruginosa conditioned medium (Figure 5I and J). In growth curve experiments, P. aeruginosa hindered the growth rate and stationary phase cell density of STING– cells compared to Flavobacterium (Figure 5I and J). However, STING– cells were still able to divide in the presence of P. aeruginosa, whereas wild-type cell growth was completely restricted (Figure 5I and J). These results indicate that wild-type cells are more susceptible to P. aeruginosa than STING– cells, although it is unclear how STING contributes to P. aeruginosa-induced growth restriction and cell death.
2’3’ cGAMP-induces autophagic signaling via STING
One downstream consequence of STING signaling in animals is the initiation of autophagy (Moretti et al., 2017; Gui et al., 2019; Yamashiro et al., 2020; Watson et al., 2015; Liu et al., 2018). Based on viral infection studies in D. melanogaster (Liu et al., 2018) and experiments expressing invertebrate STING in mammalian cells (Gui et al., 2019), it has been proposed that the induction of autophagy may be an interferon-independent ancestral function of STING. Although M. brevicollis lacks many effectors required for immune responses downstream of STING in animals (including TBK1 and NF-kB; Figure S1A), autophagy machinery is well conserved in M. brevicollis. Therefore, we asked if one outcome of 2’3’ cGAMP exposure in M. brevicollis is the induction of autophagy.
The evolutionarily conserved protein Atg8/LC3 is a ubiquitin-like protein that can be used to monitor autophagy (Klionsky, 2021; Tanida et al., 2005). During autophagosome formation, unmodified Atg8, called Atg8-I, is conjugated to phosphatidylethanolamine. Lipidated Atg8, called Atg8-II, remains associated with growing autophagosomes. As such, two indicators of autophagy are elevated Atg8-II levels relative to Atg8-I and increased formation of Atg8+ autophagosome puncta. Because antibodies are not available to detect endogenous M. brevicollis autophagy markers or cargo receptors, we generated wild type and STING– cell lines stably expressing mCherry-Atg8. Stable expression of mCherry-Atg8 under the control of the constitutive pEFL promoter did not alter the relative susceptibilities of these cell lines to 2’3’cGAMP (Figure 6—figure supplement 1A). By immunoblot, mCherry-Atg8-II can be distinguished from mCherry-Atg8-I based on its enhanced gel mobility. When we exposed both cell lines to 2’3’ cGAMP for three hours, we observed increased levels of Atg8-II relative to Atg8-I by immunoblot in wild type, but not STING– cells (Figure 6A). These results suggest that treatment with 2’3’ cGAMP induces autophagic signaling in a STING-dependent manner; however, making this conclusion requires evidence of autophagy induction through inhibitor studies. To confirm autophagy induction, we treated cells with chloroquine, a lysosomotropic agent which inhibits autophagy by blocking endosomal acidification, thereby preventing amphisome formation and Atg8-II turnover (Klionsky, 2021). Exposing wild-type cells pretreated with chloroquine to 2’3’ cGAMP for 3 hr resulted in increased levels of Atg8-II relative to Atg8-I, suggesting that 2’3’ cGAMP treatment indeed induces the autophagic pathway (Figure 6B, Figure 6—figure supplement 1B). In cells pretreated with chloroquine, STING levels did not markedly increase after exposure to 2’3’ cGAMP (Figure 6—figure supplement 1B), raising the possibility that the autophagic pathway is important for regulating STING protein levels. We next examined whether 2’3’ cGAMP induces Atg8+ puncta formation by treating wild type and STING– cells with 2’3’ cGAMP for 3 hr, and observing mCherry foci by microscopy (Figure 6C–F). Quantifying images revealed that Atg8+ puncta accumulate after 2’3’ cGAMP treatment in wild type, but not STING– cells (Figure 6G). Overall, these results suggest that M. brevicollis responds to 2’3’ cGAMP through STING-dependent induction of the autophagy pathway.
Figure 6 with 1 supplement see all

STING mediates 2’3’cGAMP-induced autophagic pathway.
(A) 2’3’cGAMP-induced Atg8 lipidation requires STING. WT and STING– cells stably expressing mCherry-Atg8 were treated with a vehicle control or 100 µM 2’3’cGAMP for 3 hr, followed by immunoblotting. The band intensity of Atg8-I (unmodified Atg8) and Atg8-II (lipidated Atg8) were quantified for each sample. Relative levels of Atg8 lipidation were assessed by dividing the band intensities of Atg8-II/Atg8-I. Tubulin is shown as loading control. Immunoblot is representative of three biological replicates. (B) 2’3’cGAMP induces Atg8 lipidation in chloroquine-treated wild type cells. WT cells stably expressing mCherry-Atg8 were first incubated with 40 mM chloroquine for 6 hr, and then treated with a vehicle control or 100 µM 2’3’cGAMP for 3 hr in the presence of chloroquine, followed by immunoblotting. For each sample, relative levels of Atg8 lipidation were assessed by dividing the band intensities of Atg8-II/Atg8-I. Tubulin is shown as loading control. Immunoblot is representative of three biological replicates. For a representative immunoblot and quantification of Atg8-II/Atg8-I levels in chloroquine-treated STING– cells, refer to Figure 6—figure supplement 1B. (C–G) STING is required for 2’3’cGAMP-induced autophagosome formation. (C–F) WT and STING– cells stably expressing mCherry-Atg8 were treated with a vehicle control or 100 µM 2’3’cGAMP for 3 hr, and then fixed and immunostained. (C,D) Representative confocal images of wild-type cells show that Atg8 puncta accumulate after 2’3’cGAMP treatment. Magenta: mCherry-Atg8; Green: anti-tubulin antibody (flagella and cell body). (E,F) Representative confocal images of STING– cells show that Atg8 remains evenly distributed in the cytoplasm after 2’3’cGAMP treatment. (G) The number of Atg8 puncta/cell was quantified for WT and STING– cells treated with a vehicle control or 2’3’cGAMP for 3 hr. Data represent cells quantified from two biological replicates (n = 150 cells per treatment group). Statistical analyses (unpaired two-tailed t-tests) were performed in GraphPad software. (H) Treatment with lysosomotropic agents that inhibit autophagy rescue 2’3’ cGAMP-induced cell death in wild-type cells. WT and STING– cells were pre-treated with 40 mM chloroquine, 10 mM NH4Cl, or a vehicle control for 6 hr. Cells were then exposed to either 100 µM 2’3’cGAMP or a vehicle control for 18 hr before quantifying survival. Data represent mean ± SD for three biological replicates. Statistical analyses (multiple unpaired t-tests) were performed in GraphPad software.
Finally, we asked whether STING-mediated autophagic pathway induction affects survival after exposure to 2’3’ cGAMP (Figure 6H). To determine if inhibiting autophagy impacts 2’3’ cGAMP-induced cell death, we examined the survival responses of wild type and STING– M. brevicollis to 2’3’ cGAMP after pretreatment with lysosomotropic agents chloroquine or NH4Cl. Chloroquine or NH4Cl pretreatment rescued 2’3’ cGAMP-induced cell death in wild type M. brevicollis (Figure 6H), whereas the survival of STING– cells, which are already resistant to 2’3’ cGAMP-induced cell death, was not affected. Therefore, we hypothesize that 2’3’ cGAMP induces cell death in M. brevicollis by overstimulating STING-mediated autophagic signaling.
Discussion
Investigating choanoflagellate immune responses has the potential to inform the ancestry of animal immune pathways. In this study, we screened diverse bacteria to identify a choanoflagellate pathogen, and determined that M. brevicollis is killed by sustained exposure to P. aeruginosa bacteria. We found that STING, a crucial component of animal innate responses to cytosolic DNA, is upregulated in M. brevicollis after exposure to P. aeruginosa or the STING ligand 2’3’ cGAMP. The application of newly developed transgenic and genetic tools for M. brevicollis revealed that, similar to mammalian STING, M. brevicollis STING localizes to perinuclear endoplasmic reticulum regions. In addition, STING mediates responses to P. aeruginosa bacteria, and is required for inducing transcriptional changes and autophagic signaling in response to 2’3’ cGAMP. These data reveal that STING plays conserved roles in choanoflagellate immune responses, and provide insight into the evolution of STING signaling on the animal stem lineage.
The discovery that M. brevicollis STING mediates immune responses raises a number of interesting questions about the full extent of its physiological roles in choanoflagellates. For example, while our results demonstrate that M. brevicollis STING responds to exogenous 2’3’ cGAMP, the endogenous triggers of STING activation in M. brevicollis remain to be determined. M. brevicollis has a putative cGAS homolog, suggesting that STING may respond to an endogenously produced cyclic dinucleotide similar to 2’3’ cGAMP. Determining the enzymatic activities of M. brevicollis cGAS and identifying the endogenous trigger of M. brevicollis STING will be critical steps towards elucidating mechanisms of STING activation. Although cGAS and STING are rare among sequenced choanoflagellate species, both species with STING homologs, M. brevicollis and S. macrocollata, also harbor a cGAS homolog (Figure 1—figure supplement 1A), suggesting the presence of an intact choanoflagellate cGAS-STING pathway.
Our results suggest that M. brevicollis has distinct responses to 2’3’ cGAMP versus 3’3’-linked cyclic dinucleotides produced by bacteria (Figure 3C and D, Figure 3—figure supplement 1). In contrast to 2’3’ cGAMP, the purified bacterial cyclic dinucleotides tested (3’3’ cGAMP, c-di-AMP, c-di-GMP) do not lead to STING upregulation or induce cell death in M. brevicollis. However, 3’3’ cGAMP induces a robust transcriptional response in M. brevicollis, indicating that STING, or a different cyclic dinucleotide receptor (McFarland et al., 2017), responds to these bacterial molecules. One hypothesis is that M. brevicollis STING, similar to animal STING proteins (Kranzusch et al., 2015), may have different binding affinities for 2’3’ and 3’3’-linked cyclic dinucleotides. It is also possible that bacterial cyclic dinucleotides activate additional pathways that influence survival in M. brevicollis. As bacterivores, choanoflagellates likely benefit from a fine-tuned response to bacterial cyclic dinucleotides that enables them to interpret higher and lower concentrations in their environment. Elucidating mechanisms of STING activation in M. brevicollis could help reveal how STING proteins in animals evolved to respond to both bacterially produced and endogenous cyclic dinucleotides.
While it is clear that 2’3’ cGAMP stimulates STING-dependent transcriptional responses and autophagic signaling in M. brevicollis (Figures 5 and 6), the signaling pathways downstream of STING in choanoflagellates are unknown. Much of what is known about STING signaling comes from mammals and involves the extended CTT domain of STING, which M. brevicollis lacks, and immune genes that are restricted to vertebrates. Two pathways downstream of STING activation that are conserved in invertebrates, and as such are proposed ancestral functions of STING, are autophagy and NF-kB signaling. Here, we observed that STING is required for induction of the autophagy pathway in response to 2’3’ cGAMP in M. brevicollis, indicating that the role of STING in regulating autophagy predates animal origins. While exposure to 2’3’ cGAMP leads to NF-kB activation in the sea anemone N. vectensis (Margolis, 2021) and in insects (Cai et al., 2020; Martin et al., 2018; Goto et al., 2018; Hua et al., 2018), neither M. brevicollis nor S. macrocollata, the two choanoflagellate species with STING, possess a NF-kB homolog (Richter et al., 2018). Nonetheless, 2’3’ cGAMP activates an extensive transcriptional response downstream of STING in M. brevicollis, although the specific signaling pathways remain to be identified. This does not negate the hypothesis that STING signaling led to NF-kB activation in the Urmetazoan, but strongly suggests that additional pathways exist downstream of STING activation in choanoflagellates, and potentially in animals.
Choanoflagellates forage on diverse environmental bacteria for sustenance, yet how they recognize and respond to pathogens is a mystery. Our finding that P. aeruginosa has pathogenic effects on M. brevicollis (Figure 1) provides a much-needed framework for uncovering mechanisms of pathogen recognition and antibacterial immunity in choanoflagellates. Profiling the host transcriptional response to P. aeruginosa has allowed us to identify choanoflagellate genes that may be involved in recognizing (C-type lectins) and combating (polysaccharide lyases, antimicrobial peptides) bacteria; yet, it has also revealed the immense complexity of this interaction, with more than 600 M. brevicollis genes differentially expressed in response P. aeruginosa. Thus, identifying specific P. aeruginosa virulence factors will be critical for understanding why P. aeruginosa – but not other bacteria – have pathogenic effects on M. brevicollis, and facilitate the characterization of mechanisms underlying choanoflagellate pathogen responses.
With the establishment of molecular genetic techniques in choanoflagellates – first for S. rosetta (Booth et al., 2018; Booth and King, 2020), and here for M. brevicollis – we now have the opportunity to explore the functions of candidate immune genes. Identifying additional choanoflagellate pathogens, particularly viral pathogens, will also be key to delineating immune response pathways. Finally, as choanoflagellates are at least as genetically diverse as animals (Richter et al., 2018), expanding studies of immune responses to diverse choanoflagellate species will be essential for reconstructing the evolution of immune pathways in animals.
Materials and methods
Culturing choanoflagellates
Request a detailed protocolAll strains of M. brevicollis were co-cultured with Flavobacterium sp. bacteria (King et al., 2008) (American Type Culture Collection [ATCC], Manassas, VA; Cat. No. PRA-258) in a seawater based media enriched with glycerol, yeast extract, peptone and cereal grass (details in Media Recipes). Cells were grown either at room temperature, or at 16 °C in a wine cooler (Koldfront). All M. brevicollis cell lines were verified by 18 S sequencing and RNA-seq. Choanoflagellate cell lines S. rosetta, S. macrocollata, S. punica, and S. infusionum were verified by 18 S sequencing.
Bacterial effects on M. brevicollis
Isolating environmental bacteria
Request a detailed protocolEnvironmental bacterial species were isolated from water samples from Woods Hole, MA, St. Petersburg, FL, and Dallas, TX. Water samples were streaked onto Sea Water Complete media or LB plates, and grown at 30 ° C or 37 ° C. After isolating individual colonies, partial 16 S sequencing using 16 S universal primers (27 F: 5′-AGAGTTTGATCCTGGCTCAG-3′, 1492 R: 5′-TACGGYTACCTTGTTACGACTT-3′) was used to determine the identity of the bacterial isolates.
Screening for pathogenic effects
Request a detailed protocolM. brevicollis was grown for 30 hr, and feeding bacteria were reduced through one round of centrifugation and resuspension in artificial seawater (ASW). Cells were counted on a hemocytometer and diluted to 5 × 106 cells/mL in High Nutrient Medium, and plated into 24-well plates.
For each bacterium, a single colony was inoculated into LB and grown shaking overnight at either 30 ° C (environmental isolates) or 37 ° C (mouse isolates). Bacterial cells were pelleted by centrifugation for 5 minutes at 4000 x g, and resuspended in artificial seawater (ASW) to an OD~1.
Each bacterial species was added to M. brevicollis culture at two concentrations (10 mL/mL and 50 mL/mL) in duplicate. M. brevicollis was then monitored at regular intervals for changes in behavior and growth.
Growth curves in the presence of bacteria
Request a detailed protocolAll bacteria were grown shaking at 30 ° C in Sea Water Complete media or LB (to optical density of 0.8). For each bacterial strain, CFU plating was used to estimate the number of bacterial cells/ mL under these growth conditions. To prepare bacterial conditioned media, bacterial cells were pelleted by centrifugation for 10 minutes at 4000 x g, and supernatant was passed through a 0.22 mm sterilizing filter.
M. brevicollis was grown for 30 hr, and bacteria were washed away through two consecutive rounds of centrifugation and resuspension in artificial seawater (ASW). Cells were counted on a hemocytometer and diluted to 1.0 × 106 cells/mL (growth curves with live bacteria) or 1.5 × 105 cells/mL (growth curves with conditioned medium) in High Nutrient Medium. To test the effects of live bacteria, 1.5 × 106 bacterial cells were added per 1 mL of M. brevicollis culture. To test the effects of bacterial conditioned media, 50 ml of bacterial conditioned media was added per 1 mL of M. brevicollis culture. For each growth curve biological replicate, cells were plated into 24-well plates, and two wells were counted per time point as technical replicates. At least three biological replicates are represented in each graph.
Bacterial internalization
Request a detailed protocolFluorescent E. coli and P. aeruginosa were grown shaking at 30 ° C in LB to an optical density of OD600 = 0.8. Fluorescent C. jejuni was grown from freezer stocks in microaerobic conditions on Mueller-Hinton agar. For each bacterial strain, CFU plating was used to estimate the number of bacterial cells/ mL under these growth conditions.
M. brevicollis was grown for 30 hr, and feeding bacteria were washed away with one round of centrifugation and resuspension in artificial seawater (ASW). Cells were counted and diluted to 1.5 × 105 cells/mL in ASW. 1.5 × 107 bacterial cells were added to 2 mL M. brevicollis culture (MOI = 50), and co-incubated at room temperature for 1 hr with gentle mixing at regular intervals to avoid settling. To quantify bead internalization, M. brevicollis was co-incubated with bacteria for 1 hr (as described above), at which point ~1 × 1010 beads (0.2 mm diameter, resuspended in 1 % BSA to prevent clumping) were added to the conical for an additional 30 min.
Prior to imaging, 200 mL aliquots were transferred to eight-well glass bottom chambers (Ibidi Cat. No 80827). Live imaging was performed on a Zeiss Axio Observer widefield microscope using a 63 x objective. Images were processed and analyzed using Fij (Schindelin et al., 2012).
P. aeruginosa deletion mutants
Request a detailed protocolP. aeruginosa deletion strains were acquired from the Seattle PAO1 transposon mutant library (NIH P30 DK089507). Strains RP436 and RP576 (PMID 15731071) were acquired from Russell Vance. The effects of both live bacteria and bacterial conditioned medium were tested for all acquired strains at a range of PFU/mL (live bacteria) or percent volume (conditioned medium).
Conditioned media treatments
Request a detailed protocolP. aeruginosa was grown shaking at 30 ° C in LB for 24 hr, and bacterial conditioned media was prepared by pelleting bacterial cells by centrifugation for 10 min at 4000 x g, and filtering supernatant twice through 0.22 mm sterilizing filters. To determine if the bioactivity of the conditioned media is present in secreted outer membrane vesicles (OMVs), OMVs were depleted from the conditioned media by ultracentrifugation as described in Woznica et al., 2016. To determine if the bioactivity of the conditioned media is heat labile, conditioned media was incubated at 100 ° C for 20 min. To determine if the bioactivity of the conditioned media is due to a protein, conditioned media was incubated with 200 µg/mL proteinase K (New England Biolabs) at 37 ° C for 12 hr, after which the enzyme was deactivated by heating the conditioned media to 80 ° C for 30 min. To determine if the bioactivity of the conditioned media is due to a nucleic acid, the conditioned media boiled for 20 min at 100 ° C, lyophilized, and resuspended in 40 mM Tris-Cl (pH = 8, 10 mM MgCl2, 1 mM CaCl2) for digestion with RNase A (100 μg mL−1; Sigma), DNase I (100 μg mL−1; Sigma), or Phosphodiesterase I from Crotalus adamanteus venom (2.5mU; Sigma). For digestion with Nuclease P1 from Penicillium citrinum (2.5mU; Sigma), lyophilized conditioned media was resuspended in 40 mM Tris-Cl (pH = 6, 2 mM ZnCl2). All nucleic acid digestions took place for 2.5 hr at 37 °C. To test whether the bioactivity of the conditioned media is in the methanolic extract, conditioned media was lyophilized and resuspended in methanol. The methanolic suspension was centrifuged at 8000 rpm for 5 min, and the methanol layer recovered and dried.
Immune agonist dose-response curves
Request a detailed protocolM. brevicollis was grown to late-log phase, and feeding bacteria were reduced through one round of centrifugation and resuspension in artificial seawater (ASW). Cells were counted on a hemocytometer and diluted to 1.0 × 106 cells/mL (growth curves with live bacteria) in High Nutrient Medium, and aliquoted into 96-well (100 µL/well) or 24-well (1 mL/well) plates. Immune agonists were added at indicated concentrations in technical duplicate, and cells were counted again after 24 hr. % survival is a calculation of: [mean experimental (cells/mL) / mean control (cells/mL)]. Each dose-response curve is representative of at least three biological replicates.
RNA-Seq
Growth of choanoflagellate cultures
Request a detailed protocolAfter thawing new cultures, growth curves were conducted to determine the seeding density and time required to harvest cells at late-log phase growth. To grow large numbers of cells for RNA-seq, cells were seeded one to two days prior to the experiment in either three-layer flasks (Falcon; Corning, Oneonta, NY, USA; Cat. No. 14-826-95) or 75 cm2 flasks (Falcon; Corning, Oneonta, NY, USA; Cat. No. 13-680-65), and grown at room temperature. Bacteria were washed away from choanoflagellate cells through two rounds of centrifugation and resuspension in artificial seawater (ASW). To count the cell density, cells were diluted 100-fold in 200 µl of ASW, and fixed with 1 µL of 16 % paraformaldehyde. Cells were counted on a hemocytometer, and the remaining cells were diluted to a final concentration of 4 × 106 choanoflagellate cells/mL. The resuspended cells were divided into 2.5 mL aliquots and plated in six-well plates prior to treatment. After treatment, cells were transferred to a 15 mL conical and pelleted by centrifugation at 2400 x g for 5 min, flash frozen with liquid nitrogen, and stored at –80 °C.
RNA isolation
Request a detailed protocolTotal RNA was isolated from cell pellets with the RNAqueous kit (Ambion, Thermo Fisher Scientific). Double the amount of lysis buffer was used to increase RNA yield and decrease degradation, and RNA was eluted in minimal volumes in each of the two elution steps (40 µL and 15 µL). RNA was precipitated in LiCl to remove contaminating genomic DNA. Total RNA concentration and quality was evaluated using the Agilent Bioanalyzer 2,100 system and RNA Nano Chip kit (Cat No. 5067–1511).
Library preparation, sequencing, and analysis
Request a detailed protocolLibraries were prepared and sequenced by the UTSW Genomics Sequencing Core. RNA libraries were generated with the Illumina TruSeq Stranded mRNA Library prep kit (Cat No. 20020594), using a starting total RNA input of 2–3 µg. To remove contaminating bacterial RNA, samples were first poly-A selected using oligo-dT attached magnetic beads. Following purification, the mRNA was fragmented at 94 °C for 4 min, and cleaved RNA fragments were synthesized into cDNA. After an end repair step, UMI adapters (synthesized by IDT) were ligated to the cDNA, and the products were twice purified using AMPure XP beads before amplification.
Library quantity was measured using the Quant-iT PicoGreen dsDNA Assay kit by Invitrogen (Cat No. P7589) and a PerkinElmer Victor X3, 2030 Multilabel Reader. Library quality was verified on an Agilent 2,100 Bioanalyzer instrument using Agilent High sensitivity DNA kit (Cat No. 5067–4626) or DNA 1000 kit (Cat No. 5067–1504). Libraries were pooled, and sequenced in different batches on either the Illumina NextSeq 550 system with SE-75 workflow, or the Illumina NovaSeq 6,000 system with S4 flowcell and XP PE-100 workflow, generating 25–40 million reads per sample. Reads were checked for quality using fastqc (v0.11.2) and fastq_screen (v0.4.4), and trimmed using fastq-mcf (ea-utils, v1.1.2–806). Trimmed fastq files were mapped to the Monosiga brevicollis reference genome (NCBI:txid81824) using TopHat (Kim et al., 2013) (v2.0.12). Duplicates were marked using picard-tools (v2.10.10). Read counts were generated using featureCounts (Liao et al., 2014), and differential expression analysis was performed using edgeR (Robinson et al., 2010). Statistical cutoffs of FDR ≤ 10–4 were used to identify significant differentially expressed genes. GO enrichment analysis of differentially expressed genes was performed using DAVID (https://david.ncifcrf.gov/).
RT-qPCR
Request a detailed protocolRNA was isolated as described above, and cDNA was synthesized from total RNA with the High Capacity cDNA Reverse Transcription kit (Applied Biosystems; Thermo Fisher). Real‐time PCR was performed with iTaq Universal SYBR Green Supermix (Biorad; Cat No. 1725121) or SYBR Green PCR master mix (Applied Biosystems; Cat No. 4309155) in either an 7,500 Fast Real-Time PCR System (Applied Biosystems), or a QuantStudio 3 Real-Time PCR System (Applied Biosystems). Ct values were converted into relative gene expression using the ΔΔCt method (Livak and Schmittgen, 2001) and the internal control gene RPL15 (MONBRDRAFT_38309).
Immunoblotting
Request a detailed protocolM. brevicollis was harvested by centrifugation at 5000 x g for 5 min at 4 °C, and resuspended in 100 µL lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 1 mM EDTA, 1 mM ethyleneglycoltetraacetic acid [EGTA], 0.5 % sodium deoxycholate, 1% NP-40) containing protease inhibitor cocktail (Roche) for 10 min at 4 °C. The crude lysate was clarified by centrifugation at 10,000 x g for 10 min at 4 °C, and denatured in Laemmli buffer before SDS-PAGE. Proteins were transferred to an Immobilon-P PVDF membrane (Millipore), and blocked for two hours in PBST (1 x PBS containing 5 % nonfat dry milk and 0.05 % Tween-20). Membranes were incubated with primary antibodies diluted in PBST overnight at 4 °C and washed extensively in PBST. Membranes were incubated with secondary antibodies for 1 hr at room temperature, washed extensively in PBST, and developed using Immobilon Western Chemiluminescent HRP Substrate (Millipore Sigma). Source data files for all western blots are provided as Source Data File 1.
STING antibody production
Request a detailed protocolThe anti-mbreSTING antibody was generated by Pacific Immunology. Rabbits were immunized with a KLH-conjugated peptide corresponding to residues 320–338 of M. brevicollis protein EDQ90889.1 (Cys-KNRSEVLKKMRAEDQYAMP), and serum was affinity purified against the peptide to reduce cross-reactivity and validated using immunoblotting.
Immunofluorescence staining and imaging
Request a detailed protocolDepending on the cell density of the starting culture, between 0.2 and 1 mL of cells were concentrated by centrifugation for 5 min at 2500× g. The cells were resuspended in 200 µl of artificial seawater and applied to poly-l-lysine–coated coverslips (Corning Life Sciences; Cat. No.354085) placed at the bottom of each well of a 24-well cell culture dish. After the cells were allowed to settle on the coverslip for 30 min, 150 µl of the cell solution was gently removed from the side of the dish. All of the subsequent washes and incubations during the staining procedure were performed by adding and removing 200 µl of the indicated buffer.
Cells were fixed in two stages. First, 200 µl cold 6 % acetone diluted in 4 X PBS was added for 5 min at room temperature. Next, 200 µl cold 8 % paraformaldehyde diluted in 4 X PBS was added (yielding a final concentration of 4 % paraformaldehyde), and the fixative mixture was incubated for 15 min at room temperature. After fixation, the coverslip was gently washed three times with 200 µl 4 X PBS.
Cells were permeabilized by incubating in permeabilization buffer (4 X PBS; 3 % [wt/vol] bovine serum albumin (BSA)-fraction V; 0.2 % [vol/vol] Triton X-100) for 30 min. After removing permeabilization buffer, the coverslip was incubated in primary antibody for 1 hour at room temperature, and then washed three times in 4 X PBS. The coverslip was then incubated with secondary antibody for 1 hour at room temperature, and then washed twice in 4 X PBS. The coverslip was next incubated in 4 U/ml Phalloidin (Thermo Fisher Scientific) for 30 min at room temperature, washed once in 4 X PBS. Lastly, the coverslip was incubated in 10 µg/ml Hoechst 33,342 (Thermo Fisher Scientific) for 5 min at room temperature, and then washed once with 4 X PBS.
To prepare a slide for mounting, 10 µl of Pro-Long Gold (Thermo Fisher Scientific) was added to a slide. The coverslip was gently removed from the well with forceps, excess buffer was blotted from the side with a piece of filter paper, and the coverslip was gently placed on the drop of Pro-Long diamond. The mounting media cured overnight before visualization.
Images were acquired on either: (1) a Zeiss LSM 880 Airyscan confocal microscope with a 63 x objective by frame scanning in the superresolution mode (images processed using the automated Airyscan algorithm (Zeiss)), or (2) a Nikon CSU-W1 SoRa spinning disk confocal microscope with a 60 x objective in SR mode (images processed using Imaris).
Live-cell imaging
Request a detailed protocolCells transfected with fluorescent reporter plasmid were prepared for microscopy by transferring 200 µl of cells to a glass-bottom dish or glass-bottom 8-well chamber (Ibidi). Confocal microscopy was performed on a Zeiss Axio Observer LSM 880 with an Fast Airyscan detector and a 63 x/NA1.40 Plan-Apochromatic oil immersion objective (Carl Zeiss AG, Oberkochen, Germany). Confocal stacks were acquired by frame scanning in superresolution mode, and images were processed using the automated Airyscan algorithm (Zeiss).
Transfection of M. brevicollis
Cell culture
Request a detailed protocolOne day prior to transfection, 60 ml of High Nutrient Medium was inoculated with M. brevicollis to a final concentration of 10,000 cells/ml. The culture was split in two, and grown in two 75 cm2 flasks at room temperature, approximately 22 °C (Falcon; Corning, Oneonta, NY, USA; Cat. No. 13-680-65).
Cell washing
Request a detailed protocolAfter 24 hr of growth, bacteria were washed away from M. brevicollis cells through three consecutive rounds of centrifugation and resuspension in artificial seawater (ASW). The culture flasks were combined and vigorously shaken for 30 s, and then transferred to 50 ml conical tubes and spun for 5 min at 2000× g and 22 °C. The supernatant was removed with a serological pipette, and residual media were removed with a fine-tip transfer pipette. The cell pellets were resuspended in a single conical tube in a total volume of 50 ml of ASW, vigorously shaken for 30 s, and then centrifuged for 5 min at 2050× g. The supernatant was removed as before. In a final washing step, the cell pellet was resuspended in 50 mL ASW, shaken vigorously, and centrifuged for 5 min at 2100× g. After the supernatant was removed, the cells were resuspended in a total volume of 400 µl of ASW. To count the cell density, cells were diluted 100-fold in 200 µl of ASW, and fixed with 1 µl of 16 % paraformaldehyde. Cells were counted on a hemocytometer, and the remaining cells were diluted to a final concentration of 5 × 107 choanoflagellate cells/ml. The resuspended cells were divided into 100 µl aliquots with 5 × 106 cells per aliquot to immediately prime cells in the next step.
Cell priming
Request a detailed protocolEach aliquot of M. brevicollis cells was incubated in priming buffer (40 mM HEPES-KOH, pH 7.5; 55 mM lithium citrate; 50 mM l-cysteine; 10 % [wt/vol] PEG 8000; and 2 µM papain) to remove the extracellular material coating the cell. The 100 µl aliquots, which contained 5 × 106 cells, were centrifuged for 5 min at 1700× g. The supernatant was removed, and cells were resuspended in 100 µl of priming buffer and then incubated for 35 min at room temperature. Priming was quenched by adding 4 µl of 50 mg/ml bovine serum albumin-fraction V (Thermo Fisher Scientific, Waltham, MA; Cat. No. BP1600-100) and then centrifuged for 5 min at 1250× g and 22 °C with the centrifuge brake set to a “soft” setting. The supernatant was removed with a fine-tip micropipette, and the cells were resuspended in 25 µl of SG Buffer (Lonza).
Nucleofection
Request a detailed protocolEach transfection reaction was prepared by adding 2 µl of “primed” cells resuspended in SG buffer (Lonza) to a mixture of: 16 µl of SG buffer, 2 µl of 20 µg/µl pUC19, 1 µl of 250 mM ATP (pH 7.5), 1 µl of 100 mg/ml sodium heparin, and ≤7 µl of reporter DNA (volume is dependent on the number of constructs transfected). Each transfection reaction was transferred to one well in 16-well nucleofection strip (Lonza; Cat. No. V4XC-2032). The nucleofection strip was placed in the X-unit (Lonza; Cat. No. AAF-1002F) connected to a Nucleofector 4D core unit (Lonza; Cat. No. AAF-1002B), and the EO100 pulse was applied to each well.
Recovery
Request a detailed protocolA total of 100 µl of cold recovery buffer (10 mM HEPES-KOH, pH 7.5; 0.9 M sorbitol; 8 % [wt/vol] PEG 8000) was added to the cells immediately after pulsation. After 5 min, the whole volume of the transfection reaction plus the recovery buffer was transferred to 2 ml of Low Nutrient Medium in a 12-well plate. The cells were grown for 24–48 hr before being assayed for luminescence or fluorescence.
Puromycin selection
Request a detailed protocolTo generate stably transfected M. brevicollis cell lines, puromycin was added to cells 24 hr after transfection at a final concentration of 300 µg/mL. Cells were monitored over the course of 7–21 days, and fresh High Nutrient Media + 300 µg/mL puromycin was added to the cells as needed.
Genome editing
Request a detailed protocolFor a more detailed description of gRNA and repair oligonucleotide design, refer to Booth et al., 2018.
Design and preparation of gRNAs First, crRNAs were designed by using the extended recognition motif 5’-HNNGRSGGH-3’ (in which the PAM is underlined, N stands for any base, R stands for purine, S stands for G or C, and H stands for any base except G) to search for targets in cDNA sequences (Peng et al., 2014). Next, we confirmed that the RNA sequence did not span exon-exon junctions by aligning the sequence to genomic DNA.
Functional gRNAs were prepared by annealing synthetic crRNA with a synthetic tracrRNA (Booth and King, 2020). To prepare a functional gRNA complex from synthetic RNAs, crRNA and tracrRNA (Integrated DNA Technologies [IDT], Coralville, IA, USA) were resuspended to a final concentration of 200 µM in duplex buffer (30 mM HEPES-KOH, pH 7.5; 100 mM potassium acetate; IDT, Cat. No. 11-0103-01). Equal volumes of crRNA and tracrRNA stocks were mixed together, incubated at 95 °C for 5 min in an aluminum block, and then the entire aluminum block was placed at room temp to slowly cool the RNA to 25 °C. The RNA was stored at –20 °C.
Design and preparation of repair oligonucleotides Repair oligonucleotides for generating knockouts were designed by copying the sequence 50 bases upstream and downstream of the SpCas9 cleavage site. A knockout sequence (5’TTTATTTAATTAAATAAA-3’) was inserted at the cleavage site (Booth and King, 2020).
Dried oligonucleotides (IDT) were resuspended to a concentration of 250 µM in a buffer of 10 mM HEPES-KOH, pH 7.5, incubated at 55 °C for 1 hr, and mixed well by pipetting up and down. The oligonucleotides were stored at –20 °C.
Delivery of gene editing cargoes with nucleofection
The method for delivering SpCas9 RNPs and DNA repair templates into M. brevicollis is as follows:
Cell culture
Request a detailed protocolOne day prior to transfection, 60 ml of High Nutrient Medium was inoculated to a final concentration of M. brevicollis at 10,000 cells/ml. The culture was split in two, and grown in two 75 cm2 flasks at room temperature, approximately 22 °C (Falcon; Corning, Oneonta, NY, USA; Cat. No. 13-680-65).
Assembly of Cas9/gRNA RNP
Request a detailed protocolBefore starting transfections, the SpCas9 RNP was assembled. For one reaction, 2 µl of 20 µM SpCas9 (NEB, Cat. No. M0646M) was placed in the bottom of a 0.25 ml PCR tube, and then 2 µl of 100 µM gRNA was slowly pipetted up and down with SpCas9 to gently mix the solutions. The mixed solution was incubated at room temperature for 1 hr, and then placed on ice.
Thaw DNA oligonucleotides
Request a detailed protocolBefore using oligonucleotides in nucleofections, the oligonucleotides were incubated at 55 °C for 1 hr.
Cell washing
Request a detailed protocolAfter 24 hr of growth, bacteria were washed away from M. brevicollis cells through three consecutive rounds of centrifugation and resuspension in artificial seawater (ASW). The culture flasks were combined and vigorously shaken for 30 s, and then transferred to 50 ml conical tubes and spun for 5 min at 2000× g and 22 °C. The supernatant was removed with a serological pipette, and residual media were removed with a fine-tip transfer pipette. The cell pellets were resuspended in a single conical tube in a total volume of 50 ml of ASW, vigorously shaken for 30 s, and then centrifuged for 5 min at 2050× g. The supernatant was removed as before. In a final washing step, the cell pellet was resuspended in 50 mL ASW, shaken vigorously, and centrifuged for 5 min at 2100× g. After the supernatant was removed, the cells were resuspended in a total volume of 400 µl of ASW. To count the cell density, cells were diluted 100-fold in 200 µl of ASW, and fixed with 1 µl of 16 % paraformaldehyde. Cells were counted on a hemocytometer, and the remaining cells were diluted to a final concentration of 5 × 107 choanoflagellate cells/ml. The resuspended cells were divided into 100 µl aliquots with 5 × 106 cells per aliquot to immediately prime cells in the next step.
Cell priming
Request a detailed protocolEach aliquot of M. brevicollis cells was incubated in priming buffer (40 mM HEPES-KOH, pH 7.5; 50 mM lithium citrate; 50 mM l-cysteine; 15 % [wt/vol] PEG 8000; and 2 µM papain) to remove the extracellular material coating the cell. The 100 µl aliquots, which contained 5 × 106 cells, were centrifuged for 5 min at 1700× g and at room temperature. The supernatant was removed, and cells were resuspended in 100 µl of priming buffer and then incubated for 35 min. Priming was quenched by adding 10 µl of 50 mg/ml bovine serum albumin-fraction V (Thermo Fisher Scientific, Waltham, MA; Cat. No. BP1600-100). Cells were then centrifuged for 5 min at 1250× g and 22 °C with the centrifuge brake set to a ‘soft’ setting. The supernatant was removed with a fine-tip micropipette, and the cells were resuspended in 25 µl of SG Buffer (Lonza).
Nucleofection
Request a detailed protocolEach nucleofection reaction was prepared by adding 16 µl of cold SG Buffer to 4 µl of the SpCas9 RNP that was assembled as described above. For reactions that used two different guide RNAs, each gRNA was assembled with SpCas9 separately and 4 µl of each RNP solution were combined at this step. 2 µl of the repair oligonucleotide template was added to the SpCas9 RNP diluted in SG buffer. Finally, 2 µl of primed cells were added to the solution with Cas9 RNP and the repair template. The nucleofection reaction was placed in one well of a 16-well nucleofection strip (Lonza; Cat. No. V4XC-2032). The nucleofection strip was placed in the X-unit (Lonza; Cat. No. AAF-1002F) connected to a Nucleofector 4D core unit (Lonza; Cat. No. AAF-1002B), and the EO100 pulse was applied to each well.
Recovery
Request a detailed protocol100 µl of cold recovery buffer (10 mM HEPES-KOH, pH 7.5; 0.9 M sorbitol; 8 % [wt/vol] PEG 8000) was added to the cells immediately after pulsation. After 5 minutes, the whole volume of the transfection reaction plus the recovery buffer was transferred to 1 ml of High Nutrient Medium in a 12-well plate.
Cycloheximide selection in M. brevicollis
Request a detailed protocolOne day after transfection, 10 µl of 10 µg/ml cycloheximide was added per 1 mL culture of transfected cells. The cells were incubated with cycloheximide for 5 days prior to clonal isolation and genotyping.
Genotyping
Request a detailed protocolCells were harvested for genotyping by spinning 0.5 ml of cells at 4000 g and 22 °C for 5 min. The supernatant was removed and DNA was isolated either by Base-Tris extraction [in which the cell pellet was resuspended in 20 µL base solution (25 mM NaOH, 2 mM EDTA), boiled at 100 °C for 20 min, cooled at 4 °C for 5 min, and neutralized with 20 µL Tris solution (40 mM Tris-HCl, pH 7.5)], or by DNAzol Direct [in which the cell pellet was resuspended in 50 µL and incubated at room temperature for 30 min (Molecular Research Center, Inc [MRC, Inc.], Cincinnati, OH; Cat. No. DN131)]. Three µl of the DNA solution was added to a 25 µl PCR reaction (DreamTaq Green PCR Master Mix, Thermo Fisher Scientific Cat No K1082) and amplified with 34 rounds of thermal cycling.
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (M. brevicollis) | M. brevicollis | ATCC PRA-258 | PMID:18276888 | |
| Genetic reagent, (M. brevicollis) | M. brevicollis STING– | This study | STING–knockout strain; cell line maintained by A. Woznica | |
| Transfected construct (M. brevicollis) | pEFL5’-pac-P2A-STING-mTFP-3'act | This study | Construct to express M, brevicollis STING fused to mTFP; can be obtained from A. Woznica | |
| Transfected construct (M. brevicollis) | pEFL5’-pac-P2A-mCherry-Atg8-–3'act | This study | Construct to express mCherry fused to M. brevicollis Atg8; can be obtained from A. Woznica | |
| Strain, strain background (Flavobacterium) | Flavobacterium sp. | This study | Isolated from MX1 (ATCC PRA-258) culture; can be obtained from A. Woznica | |
| Strain, strain background (Pseudomonas aeruginosa) | PAO1 | ATCC 15692 | PMID:13961373 | |
| Strain, strain background (P. aeruginosa, transgenic strain) | PAO1-GFP | ATCC 15692GFP | PMID:9361441 | |
| Antibody | anti-choano STING (rabbit polyclonal) | This study | Generated by Pacific Immunology; dilution (1:200) for IF, (1:2000) dilution for WB; can be obtained from A. Woznica | |
| Antibody | Anti-mCherry 16D7 (rat monoclonal) | Invitrogen | Cat# M11217 | (1:2000) dilution for WB |
| Antibody | Anti-human Tubulin E7 (Mouse monoclonal) | Developmental Studies Hybridoma Bank | Cat# AB_2315513 | (1:200) dilution for IF |
| Antibody | Alpha-human tubulin (Mouse monoclonal) | Sigma Aldrich | Cat # T64074 | (1:7000) dilution for WB |
| Chemical compound, drug | 2’3’ cGAMP | Cayman Chemical | Cat# 19,887 | |
| Chemical compound, drug | 3’3’ cGAMP | Cayman chemical | Cat# 17,966 | |
| Sequence-based reagent | STING556 gRNA | This study | Guide RNA | TTTCGGGATTCAGATGTGGG |
| Sequence-based reagent | STING locus PCR primers | This study | PCR primers | F: 5’ ATG ATG GTT AAT CTC TCT GAT CTT TCA CAT C 3’ R: 5’ TTA TGG CAT CGC ATA CTG GTC C 3’ |
| Commercial assay or kit | SG Cell Line 4D-NucleofectorTM X Kit S | Lonza, | Cat# V4XC-3032 |
Data availability
Raw sequencing reads and normalized gene counts have been deposited at the NCBI GEO under accession GSE174340.
-
NCBI Gene Expression OmnibusID GSE174340. STING mediates immune responses in a unicellular choanoflagellate.
-
JGIID Monbr1. The genome of the choanoflagellate Monosiga brevicollis and the origin of metazoans.
References
-
STING signaling and host defense against microbial infectionExperimental & Molecular Medicine 51:1–10.https://doi.org/10.1038/s12276-019-0333-0
-
Green fluorescent protein as a marker for Pseudomonas sppApplied and Environmental Microbiology 63:4543–4551.
-
Transfection of choanoflagellates illuminates their cell biology and the ancestry of animal septinsMolecular Biology of the Cell 29:3026–3038.https://doi.org/10.1091/mbc.E18-08-0514
-
The Origin of Animal Multicellularity and Cell DifferentiationDevelopmental Cell 43:124–140.https://doi.org/10.1016/j.devcel.2017.09.016
-
Stimulator of interferon genes (STING) provides insect antiviral immunity by promoting Dredd caspase-mediated NF-κB activationThe Journal of Biological Chemistry 293:11878–11890.https://doi.org/10.1074/jbc.RA117.000194
-
Synergistic Cues from Diverse Bacteria Enhance Multicellular Development in a ChoanoflagellateApplied and Environmental Microbiology 86:e02920-19.https://doi.org/10.1128/AEM.02920-19
-
BookThe Choanoflagellates: Evolution, Ecology, and BiologyBirmingham: Cambridge University Press.https://doi.org/10.1017/CBO9781139051125
-
Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) MethodMethods 25:402–408.
-
Evolutionary Origins of cGAS-STING SignalingTrends in Immunology 38:733–743.https://doi.org/10.1016/j.it.2017.03.004
-
Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and PersistenceFrontiers in Cellular and Infection Microbiology 7:39.https://doi.org/10.3389/fcimb.2017.00039
-
The genomic and cellular foundations of animal originsAnnual Review of Genetics 47:509–537.https://doi.org/10.1146/annurev-genet-111212-133456
-
The origin and evolution of cell-intrinsic antibacterial defenses in eukaryotesCurrent Opinion in Genetics & Development 58–59:111–122.https://doi.org/10.1016/j.gde.2019.09.002
-
Fiji: an open-source platform for biological-image analysisNature Methods 9:676–682.https://doi.org/10.1038/nmeth.2019
-
Origins and evolution of eukaryotic RNA interferenceTrends in Ecology & Evolution 23:578–587.https://doi.org/10.1016/j.tree.2008.06.005
-
Control of Phagocytosis by Microbial PathogensFrontiers in Immunology 8:1368.https://doi.org/10.3389/fimmu.2017.01368
-
Molecular evolutionary and structural analysis of the cytosolic DNA sensor cGAS and STINGNucleic Acids Research 42:8243–8257.https://doi.org/10.1093/nar/gku569
Article and author information
Author details
Funding
Howard Hughes Medical Institute (Hanna Gray Fellows Program)
- Arielle Woznica
Howard Hughes Medical Institute (Faculty Scholars Program)
- Julie K Pfeiffer
Howard Hughes Medical Institute
- Nicole King
Pew Charitable Trusts (Pew Innovation Fund)
- Nicole King
- Julie K Pfeiffer
Burroughs Wellcome Fund (Investigators in the Pathogenesis of Infectious Diseases)
- Julie K Pfeiffer
National Cancer Institute (1P30 CA142543)
- Arielle Woznica
- Julie K Pfeiffer
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Bill Jackson, Tera Levin, Shally Margolis, Russell Vance, and Nan Yan for helpful advice and/or comments on the manuscript, and David Greenberg, David Hendrixson, Andrew Koh, Kim Orth, Russell Vance, and Sebastian Winter for bacterial strains. We thank Neal Alto for use of the widefield microscope, Monika Sigg for use of a reporter construct, and Mya Breitbart for sea water samples. We are grateful for David Booth’s invaluable advice on developing transfection and gene editing protocols.
This work was funded by a Pew Innovation Fund award (JKP and NK), a HHMI Hanna Gray Fellows award (AW), a HHMI Faculty Scholar award (JKP), and a Burroughs Wellcome Fund Investigators in the Pathogenesis of Infectious Diseases (JKP). We acknowledge the assistance of the UT Southwestern Live Cell Imaging Facility, a Shared Resource of the Harold C Simmons Cancer Center, supported in part by an NCI Cancer Center Support Grant, 1P30 CA142543-01.
Copyright
© 2021, Woznica et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,365
- views
-
- 460
- downloads
-
- 54
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 54
- citations for umbrella DOI https://doi.org/10.7554/eLife.70436
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
STING mediates immune responses in the closest living relatives of animals
eLife 10:e70436.
https://doi.org/10.7554/eLife.70436




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}