GWAS and functional studies suggest a role for altered DNA repair in the evolution of drug resistance in Mycobacterium tuberculosis
- National Institute of Immunology, India
- Centre for Cellular and Molecular Biology, India
- Department of Zoology, University of Delhi, India
- Centre for Genetic Manipulation of Crop Plants, University of Delhi South Campus, India
- Department of Microbiology and Cell Biology, Indian Institute of Science Bangalore, India
Abstract
The emergence of drug resistance in Mycobacterium tuberculosis (Mtb) is alarming and demands in-depth knowledge for timely diagnosis. We performed genome-wide association analysis using 2237 clinical strains of Mtb to identify novel genetic factors that evoke drug resistance. In addition to the known direct targets, we identified for the first time, a strong association between mutations in DNA repair genes and the multidrug-resistant phenotype. To evaluate the impact of variants identified in the clinical samples in the evolution of drug resistance, we utilized knockouts and complemented strains in Mycobacterium smegmatis and Mtb. Results show that variant mutations compromised the functions of MutY and UvrB. MutY variant showed enhanced survival compared with wild-type (Rv) when the Mtb strains were subjected to multiple rounds of ex vivo antibiotic stress. In an in vivo guinea pig infection model, the MutY variant outcompeted the wild-type strain. We show that novel variant mutations in the DNA repair genes collectively compromise their functions and contribute to better survival under antibiotic/host stress conditions.
Editor's evaluation
This paper provides important evidence implicating polymorphisms in the mycobacterial adenine DNA glycosylase, MutY, in the emergence of antibiotic resistance in Mycobacterium tuberculosis. While the precise mechanism underlying this phenotype requires further investigation, the inference from genome-wide association analyses of sequenced clinical isolates, supported by laboratory experiments and animal infection models, is convincing. This work adds a new locus of interest to the list of polymorphisms associated with tuberculosis drug resistance, and is likely to be relevant to the mycobacterial research field.
https://doi.org/10.7554/eLife.75860.sa0Introduction
The acquisition of drug resistance in Mycobacterium tuberculosis (Mtb) has evoked a perilous situation worldwide (WHO, 2020). Resistance to isoniazid and rifampicin, the first-line drugs, results in multidrug resistant-TB (MDR-TB). The pathogen is defined as extensively drug resistant (XDR) when it becomes resistant to first-line TB drugs, any fluoroquinolones, and at least one additional Group A drug (moxifloxacin, levofloxacin, linezolid, and bedaquiline) (WHO, 2021). Prolonged treatment duration, high drug toxicity, and the expensive drug regimen pose a challenge for treating the MDR and XDR-TB. Moreover, the inadequate treatment of drug-resistant TB leads to the augmentation of resistance to other anti-TB drugs, increasing the probability of transmission of these strains in the population (Alexander and De, 2007; Bastos et al., 2014).
Seven major lineages of Mtb are present across the globe, out of which four lineages: Lineage 1-Indo Oceanic (EAI); Lineage 2- Beijing; Lineage 3- Central Asian (CAS); and Lineage 4- Euro-American (Gagneux et al., 2006) are prevalent in humans. Clinical strains belonging to lineage 2 are more prone to developing drug resistance than lineage 4 strains (Ford et al., 2013). Acquisition of drug resistance in Mtb is majorly attributed to the chromosomal mutations that either modify the antibiotic’s direct target or increase the expression of efflux pumps that helps in decreasing the effective concentration of the drug inside the cell. The expression of drug modifying/degrading enzymes also contributes to the acquisition of drug resistance (Gygli et al., 2017). Despite well-known mechanisms of drug resistance, it is difficult to predict the resistance based on direct target mutations alone, implying the presence of hitherto unknown mechanisms that impart drug resistance. In addition, the diagnosis based on mutations in a particular known target region increases the bias that may have multiple repercussions, such as misdiagnosis and eventual spread of drug resistance (CRyPTIC Consortium and the 100,000 Genomes Project et al., 2018; The CRyPTIC Consortium, 2022b; The CRyPTIC Consortium, 2022a). The current knowledge of the mechanisms and biological triggers involved in the evolution of MDR or XDR-TB is inadequate. This knowledge is crucial for developing new drug targets and improved diagnosis. Multiple efforts have been made to determine the mechanisms for the emergence of MDR and XDR-TB. Genome-wide association studies (GWAS) identified different genes that abet the emergence of drug resistance (Farhat et al., 2013; Hicks et al., 2018; Zhang et al., 2013; Safi et al., 2019). However, only a few genes, such as ponA1, prpR, ald, glpK, and the mutation in the thyA-Rv2765 thyX-hsdS.1 loci, are validated (Farhat et al., 2013; Hicks et al., 2018; Zhang et al., 2013; Safi et al., 2019).
In a quest to identify genetic triggers that aid in the evolution of antibiotic resistance in Mtb, we performed GWAS using global data set of 2237 clinical strains that consist of antibiotic susceptible, MDR, poly-drug resistant (resistant to more than one first-line anti-TB drug other than both isoniazid and rifampicin), pre-XDR, and XDR. Interestingly, we have identified mutations in the multiple DNA repair genes of Mtb associated with the MDR phenotype. Functional validation of the identified mutations in DNA repair enzymes revealed that perturbations in the DNA repair mechanisms result in the enhanced survival of strains in the presence of antibiotics ex vivo and in vivo.
Results
GWAS unveils mutations in the DNA repair genes
To identify the genetic determinants contributing to the development of antibiotic resistance in Mtb, we performed genome-wide association analysis using the whole-genome sequences of clinical strains from nine published studies. After the quality filtering of raw reads, the dataset had 2773 clinical strains from 9 different countries belonging to all 4 lineages (Figure 1a, Figure 1—figure supplement 1; Hicks et al., 2018; Zhang et al., 2013; Casali et al., 2014; Blouin et al., 2012; Shanmugam et al., 2019; Guerra-Assunção et al., 2015; Clark et al., 2013; Bryant et al., 2013; Walker et al., 2013). The dataset chiefly represented Lineage 2 and 4 isolates that are predominant across the globe (Figure 1b). Based on the computational predictions and the phenotypes provided by the previous studies, strains were categorized as susceptible, mono-drug resistant, MDR, Poly-DR, and pre-XDR (Supplementary file 1; Supplementary file 2Manson et al., 2017). We identified ~160,000 single nucleotide polymorphisms (SNPs) and indels after mapping the short reads on the reference Rv genome. A phylogenetic tree constructed using the SNPs shows the proper clustering of lineages (Figure 1b). The total number of SNPs observed for all the strains was comparable, suggesting the absence of genetic drift (Figure 1c).
Figure 1 with 1 supplement see all
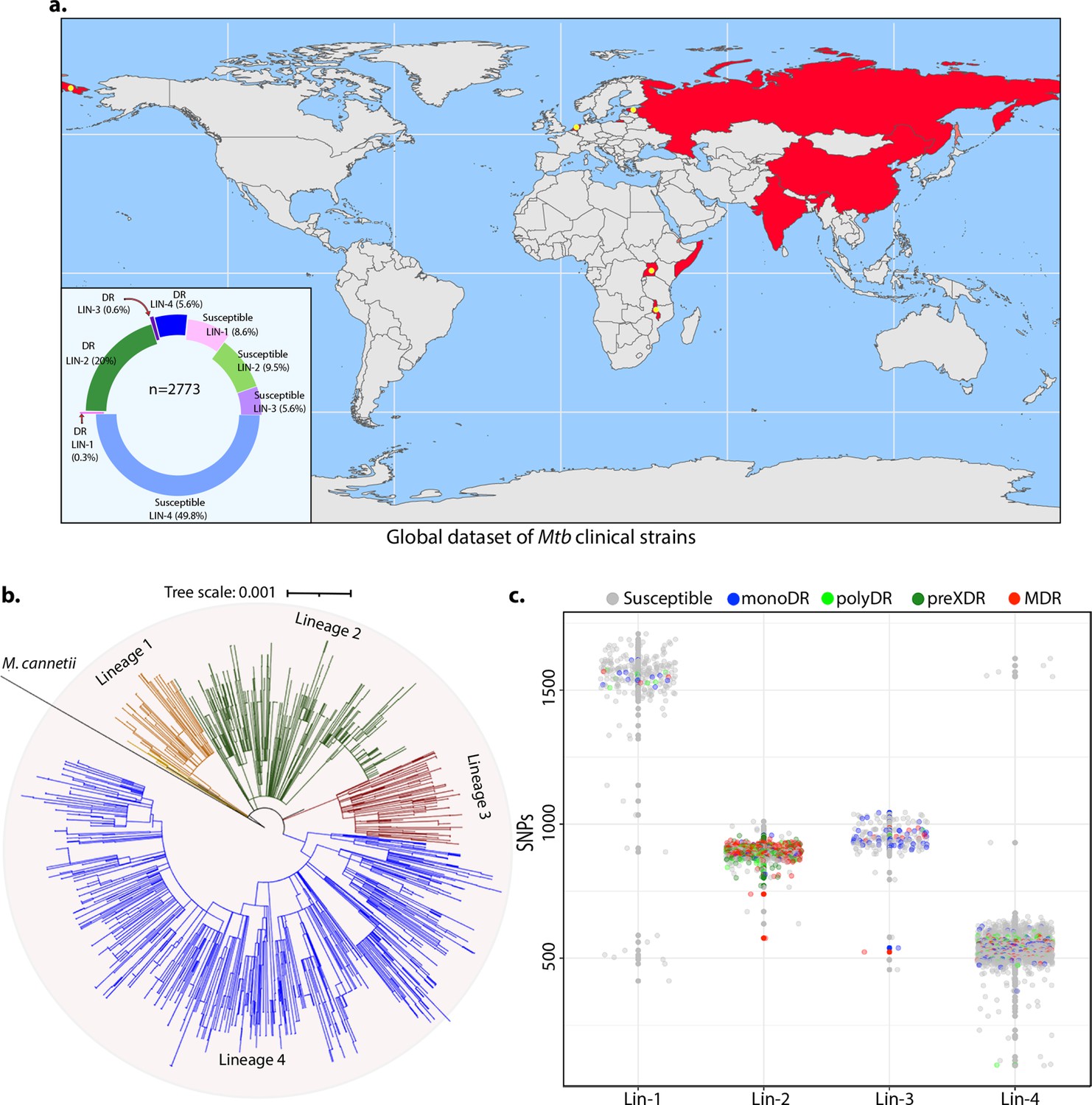
Genome-wide association study unveils mutations in the DNA repair genes.
(a) Geographical distribution of 2773 clinical strains of Mycobacterium tuberculosis (Mtb). The donut plot represents the proportion of susceptible and drug-resistant (DR) strains in each lineage. DR includes mono-DR, poly-DR, multidrug resistant (MDR), and pre-extensively drug resistant (XDR). A detailed breakup of distribution is given in Supplementary file 1. (b) Phylogenetic tree constructed using 1,60,000 single nucleotide polymorphisms (SNPs) using Mycobacterium canetti as an outgroup. (c) Dot-plot showing the number of SNPs identified in each strain. Different colored dots indicate the drug resistance phenotype of strain.
We hypothesized that the probability of finding the genetic determinants contributing to drug resistance would be higher in the strains resistant to more than two antibiotics. Thus, we performed GWAS using 1815 drug-susceptible and 422 drug-resistant strains (Figure 1—figure supplement 1, Figure 2—figure supplements 1–4 & Supplementary file 2). We employed a genome association and prediction integrated tool (GAPIT) software, with stringent false discovery rate (FDR) adjusted p-value (Gao et al., 2016; Zegeye et al., 2014). After setting the adjusted p-value cut-off at 10–5, we identified 188 mutations, including 24 intergenic regions correlated with multidrug resistance (Supplementary file 3; Supplementary file 4; Supplementary file 5; Supplementary file 6). The effect of identified SNPs on the development of MDR/XDR reveals positive or negative contributions (Figure 2a). We have identified known first- and second-line drug resistance target genes (Figure 2b & Table 1). Although we identified multiple mutations in rpoB, only p.Leu452Pro and p.Val496Met were above the cut-off. Notably, mutations in the rrs, katG (p.Ser315Thr), embB (p.Gly406Ser), pncA (p.His71Arg), gyrA (p.Ala90Val), and recently reported genetic determinants such as folC (p.Ser150Gly), and pks were part of the 164 genes, validating our approach (Figure 2b & Table 1, Supplementary file 4; Supplementary file 6). Also, we identified the known compensatory mutations in the fabG1 upstream region, eis-Rv2417c, and oxyR-ahpC loci (Coll et al., 2018; Supplementary file 6). Importantly, the above mutations were absent in the mono-DR and drug-susceptible strains (Figure 2—figure supplement 6).
Table 1
Mutations identified in the direct targets of antibiotics.
| Antibiotic | Gene | Mutations identified |
|---|---|---|
| Rifampicin | rpoB | Leu452Pro, Val496Meth |
| Isoniazid | katG | Ser315Thr |
| Ethambutol | embB | Gly406Ser |
| Ofloxacin | gyrA | Ala90Val, Ser91Pro |
| Kanamycin | rrs | 7 independent mutations |
| Pyrazinamide | pncA | His71Arg |
| Ethionamide | ethA | Met95Arg, Pro160(frame-shift) |
| Streptomycin | gidB | Leu35 (frame-shift) |
| Cycloserine | ald | Thr427Pro |
Figure 2 with 6 supplements see all
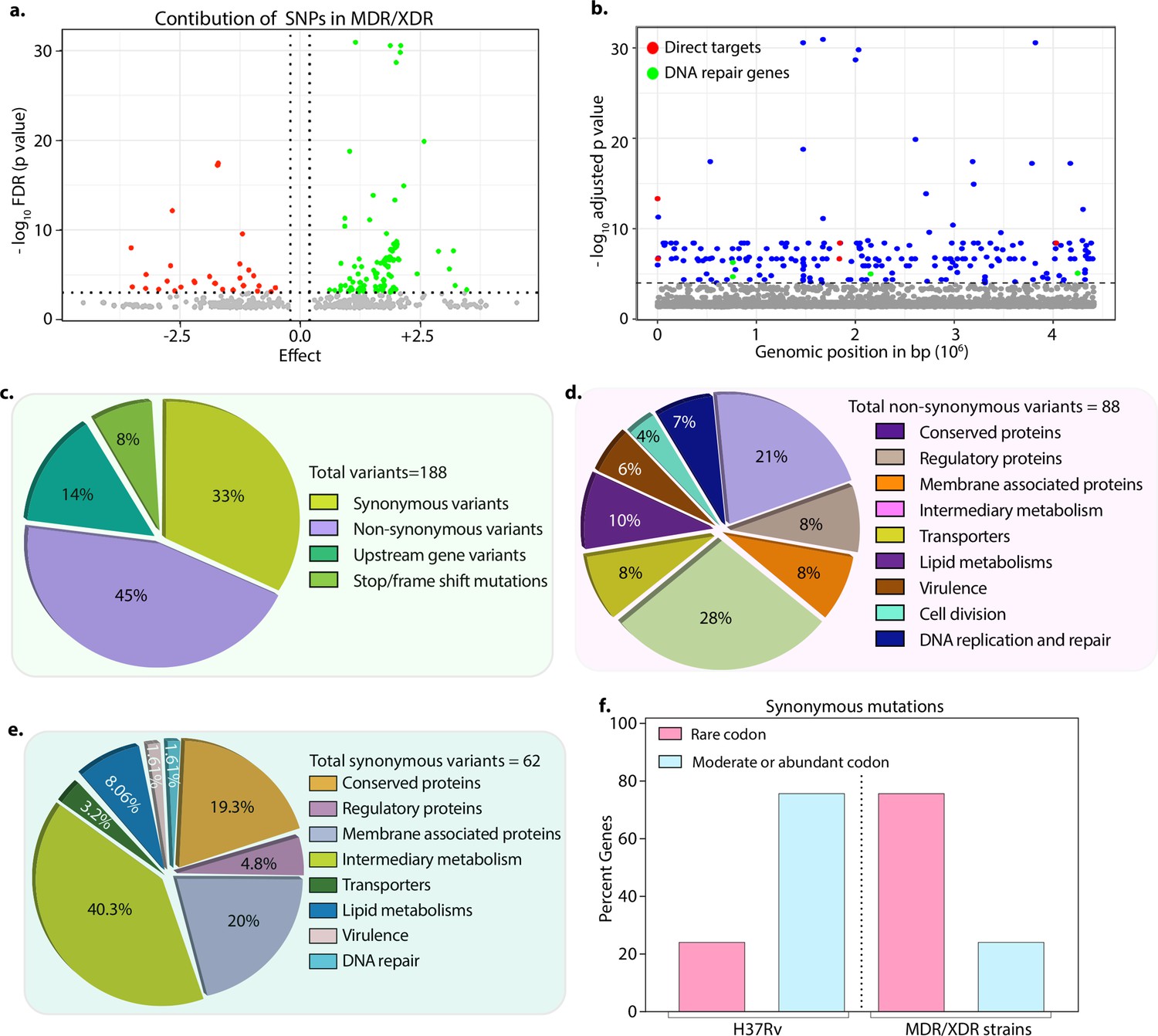
Drug-resistant strains carry mutations in the DNA repair genes.
(a) Volcano plot represents the effect of identified single nucleotide polymorphisms (SNPs) on the development of multidrug resistant/extensively drug resistant TB (MDR/XDR-TB). The positive effect (green dots) shows that the identified SNPs would aid in MDR/XDR development. The negative effect (red dots) shows that the SNPs would restrain the development of MDR/XDR. (b) Manhattan plot representing the association between the genes and drug resistance phenotype. A total of 188 genes that include intergenic regions were identified above the 10–5 cut-off value through association studies. Blue dots represent mutation in the lipid metabolism, membrane proteins, intermediary metabolism genes, and others. Green dots represent mutation in the direct targets for the first- and second-line antibiotics. Red dots represent mutations associated with the DNA repair genes. A detailed list of associated genes is provided in Supplementary file 3; Supplementary file 4. (c–e) Pie chart represents the total (c), non-synonymous (d), and synonymous (e) SNPs identified in the genes that belong to different categories. (f) Bar plot represents the percentage of synonymous mutations in the genes that resulted in abundant/moderate codon usage to the rare codon compared to H37Rv.
-
Figure 2—source data 1
Mutations identified in genes that belong to different categories.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig2-data1-v3.xlsx
Among the 188 genes, 45% of the mutations resulted in non-synonymous changes, whereas 33% resulted in synonymous changes, 14% in the upstream regions of the genes, and 8% in the stop/frameshift mutations (Figure 2c–e & -- Supplementary file 3; Supplementary file 4; Supplementary file 5; Supplementary file 6). While the non-synonymous and the stop/frameshift mutations most likely affect the functions of the proteins, the intergenic region mutations may impact gene expression. We identified mutations in genes involved in lipid metabolism, intermediary metabolism and respiration, membrane transporters, cell wall and cell processes, membrane-associated proteins, and others (https://mycobrowser.epfl.ch/) (Figure 2c–e). Synonymous changes may alter the mRNA stability or stall the translation process by changing an abundant codon to a rare codon (Brandis and Hughes, 2016; Kristofich et al., 2018; Plotkin and Kudla, 2011). Analysis of the synonymous mutations for the codon bias revealed that in >50% of events, codons were converted from moderate/abundant to rare codons (compare 75% in MDR/XDR with 25% in H37Rv) (Figure 2f, Supplementary file 7).
In addition to the mutations described above, we identified novel mutations in base excision repair (BER), nucleotide excision repair (NER), and homologous recombination (HR) pathway genes, mutY, uvrA, uvrB, and recF that are associated with the MDR and XDR-TB (Figure 2b & Table 2). Mutations in the DNA repair pathway genes could contribute to the selection and evolution of antibiotic resistance (Table 2; Figure 2—figure supplement 5). Analysis showed that mutations in the DNA repair genes are distributed specifically in MDR, PDR, and preXDR/XDR strains (Figure 2—figure supplement 6a–d). Furthermore, these strains also harbored mutations in the direct targets of the antibiotics (Figure 2—figure supplement 6i–k). Collectively, in addition to mutations in the direct targets, we identified novel uncharacterized variants, including mutations in the DNA repair genes.
Table 2
Mutations in DNA repair genes associated with drug resistance phenotype.
| Gene | Amino acid change | Wild type | Mutated | False discovery rate-adjusted p-value |
|---|---|---|---|---|
| mutY | Arg262Gln | G | A | 3.83E-09 |
| uvrB | Ala524Val | C | T | 2.15E-07 |
| uvrA | Gln135Lys | C | A | 3.83E-09 |
| RecF | Gly269Gly | G | T | 2.15E-07 |
The mutations in DNA repair genes result in their deficient function
DNA repair pathways, including NER, HR, and BER, guard the genomic integrity (Cole et al., 1998; Singh, 2017). The Mtb MutY is a 302 amino acid (aa) long adenine DNA glycosylase encoded by rv3589. We identified Arg262Gln mutation at the C-terminal region of the MutY. Oxidative damage to DNA results in the formation of 7,8-dihydro 8-oxoguanine (8-oxoG). If left unrepaired by MutM (fpg), it results in 8-oxoG:A (mostly) or 8-oxoG:G base pairing. MutY removes A or G paired against 8-oxoG, allowing MutM to correct the mistake. The absence of repair leads to G:C to T:A or C:G mutations in the genome (Figure 3a; Kurthkoti et al., 2010). The analysis of the mutation spectrum in the drug-resistant clinical strains harboring mutY-R262Q mutation and closely related drug-susceptible strains showed a bias toward C→A, A→G, and C→T mutations (Figure 2—figure supplement 6l). To decipher the biological role of the identified variant, we cloned Mtb mutY and performed site-directed mutagenesis to generate the mutant allele. The wild type and mutant mutY genes were subcloned into an integrative Mtb shuttle vector. Constructs were electroporated into Mycobacterium smegmatis mutY mutant strain (msmΔmutY) to generate msmΔmutY::mutY and msmΔmutY::mutY-R262Q strains. We performed mutation frequency analysis to evaluate the impact of Arg262Gln mutation on its DNA repair function. In accordance with the published data, deletion resulted in a 4.32-fold increase in the mutation frequency (Figure 3b; Kurthkoti et al., 2010). While complementation with wild-type mutY rescued the phenotype, complementation with mutY-R262Q failed to do so (Figure 3b and c).
Figure 3 with 2 supplements see all
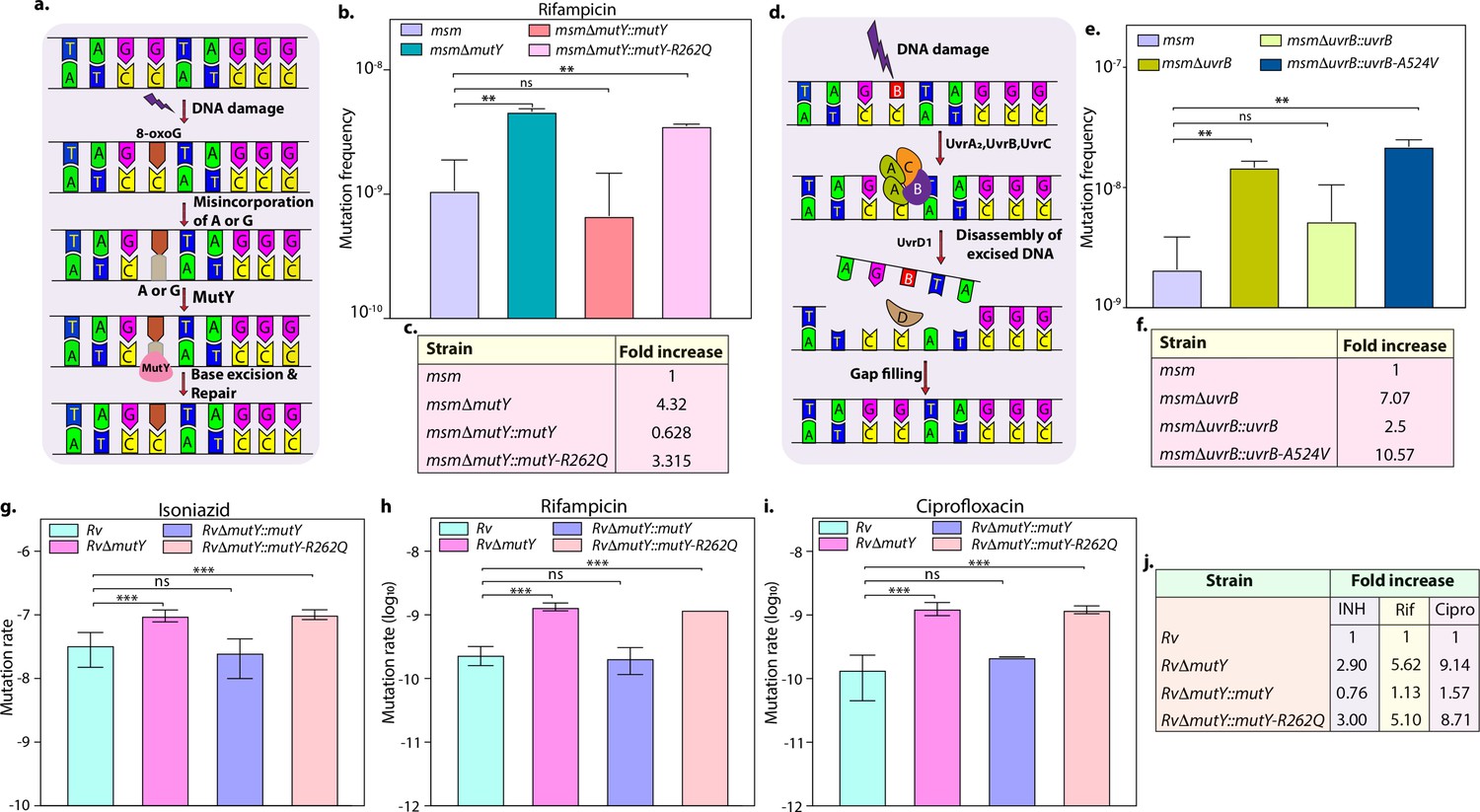
Variants identified in DNA repair genes abrogate their function.
(a) A schematic representation of the base excision repair pathway that operates in mycobacteria. Oxidative damage can result in the conversion of G to 8-oxo-G. If MutM (Fpg) does not repair 8-oxo-G before replication, often an A is inserted against 8-oxo-G during replication. Under these conditions, MutM must avoid repair of 8-oxo-G until MutY removes the erroneously incorporated A. The predominant target of MutY is 8-oxo-G:A pair where it removes A; thus, the action of MutY provides another opportunity to incorporate C (the correct base) against 8-oxo-G. Now the DNA becomes a target for MutM again, leading to the removal of 8-oxo-G and allowing incorporation of G. (b) Mutation frequency was calculated using msm, msmΔmutY, msmΔmutY::mutY, msmΔmutY::mutY-R262Q. (c) Fold increase in the mutation frequency with respect to wild-type msm. (d) A schematic representation of the nucleotide excision repair pathway showing the recognition and initiation of repair by UvrA-UvrB and UvrC. (e) Mutation frequency of msm, msmΔuvrB, msmΔuvrB::uvrB, and msmΔuvrB::uvrB-A524V. (f) Fold increase in the mutation frequency with respect to wild-type msm. Two biologically independent experiment sets were performed. Each biological experiment was performed in a biological sextet. Data represent one set of experiments. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01. (g, h & i) Mutation rate was calculated for different strains in the presence of isoniazid (g), rifampicin (h), or ciprofloxacin (i). (j) Table showing the fold increase in the mutation rate in comparison with wild-type Rv. The experiment was performed using six independent colonies. Data represent mean and standard deviation. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
-
Figure 3—source data 1
Mutation rate analysis in the presence of different drugs.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig3-data1-v3.xlsx
Next, we investigated the role of mutation in the NER pathway gene UvrB. UvrA, UvrB, and UvrC recognize and initiate the NER pathway upon DNA damage. UvrB, a 698 aa long DNA helicase encoded by rv1633, plays a pivotal role in the NER pathway by interacting with the UvrA and UvrC (Figure 3d; Kurthkoti et al., 2008). UvrB harbors N and C-terminal helicase domain, interaction domain, YAD/RRR motif, and UVR domain. Identified UvrB variant, Ala524Val, mapped to the C-terminal helicase like domain (Theis et al., 2000). To evaluate the functional significance of the mutation of uvrB, Mtb uvrB and uvrB-A524V genes were cloned into an integrative vector. The absence of uvrB led to higher mutation frequency, which could be rescued upon complementation with the wild type but not with the variant (Figure 3e and f).
Subsequently, we sought to extend our investigations in Mtb. Toward this, we generated the gene replacement mutant of mutY in laboratory strain Mtb H37Rv (Rv), wherein the mutY at native loci was disrupted with a hygromycin resistance cassette. Replacement at the native loci was confirmed by performing multiple PCRs (Figure 3—figure supplement 1b–c). Complementation constructs harboring mutY or mutY-R262Q were electroporated in the RvΔmutY to generate RvΔmutY::mutY and RvΔmutY::mutY-R262Q. Western blot analysis showed comparable expression of the MutY or MutY-R262Q (Figure 3—figure supplement 1d). We determined the mutation rates in the presence of isoniazid, rifampicin, and ciprofloxacin (Figure 3g–j). The fold increase in the mutation rates relative to Rv for RvΔmutY, RvΔmutY:mutY, and RvΔmutY::mutY-R262Q were 2.90, 0.76, and 3.0 in the presence of isoniazid; 5.62, 1.13, and 5.10 in the presence of rifampicin; and 9.14, 1.57, and 8.71 in the presence of ciprofloxacin, respectively (Figure 3j). Also, we have determined the mutation frequencies in the presence of isoniazid and rifampicin (Figure 3—figure supplement 2). Results are in line with the mutation rate experiments presented in Figure 3. Together these data suggest that variants of mutY and uvrB compromise their function.
The variant of mutY resists antibiotic killing
The killing kinetics in the presence and absence of isoniazid, rifampicin, ciprofloxacin, and ethambutol was performed to evaluate the effect of different drugs on the survival of RvΔmutY or RvΔmutY::mutY-R262Q (Figure 4a). In the absence of antibiotics, the growth kinetics of Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q were similar (Figure 4b). In the presence of isoniazid, ~2 log-fold decreases in bacterial survival was observed on day 3 in Rv and RvΔmutY::mutY; however, in RvΔmutY and RvΔmutY::mutY-R262Q, the difference was limited to ~1.5 log-fold (Figure 4c). A similar trend was apparent on days 6 and 9, suggesting an ~fivefold increase in the survival of RvΔmutY and RvΔmutY::mutY-R262Q compared with Rv and RvΔmutY::mutY (Figure 4c). Interestingly, in the presence of ethambutol, we did not observe any significant difference (Figure 4d). In the presence of rifampicin and ciprofloxacin, we observed an ~10-fold increase in the survival of RvΔmutY and RvΔmutY::mutY-R262Q compared with Rv and RvΔmutY::mutY (Figure 4e–f). Thus, results suggest that the absence of mutY or the presence of mutY variant aids in subverting the antibiotic stress.
Figure 4
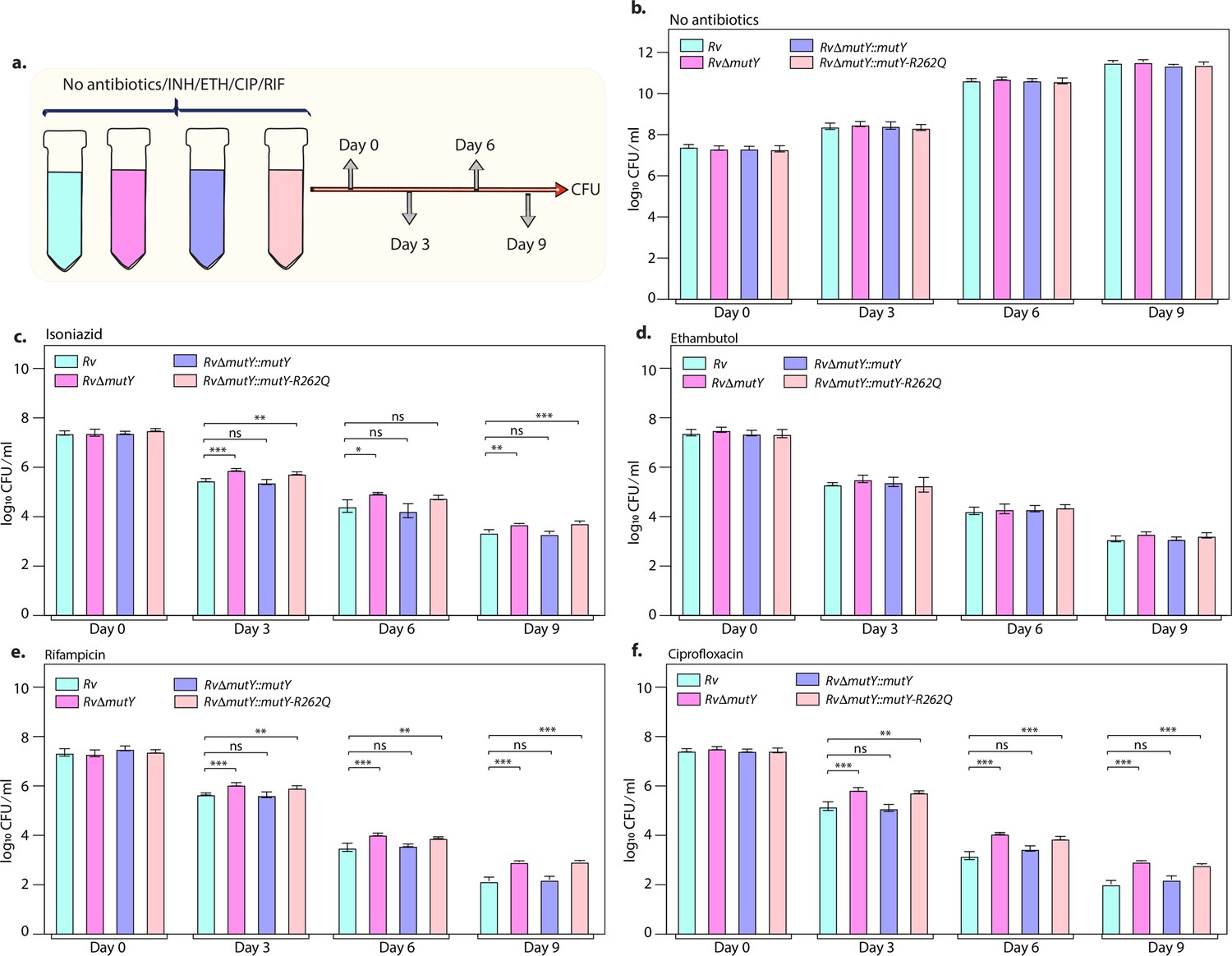
Killing kinetics in the presence of antibiotics show better survival of RvΔmutY and RvΔmutY::mutY R262Q.
(a) Schematic representation of killing kinetics. (b) Growth kinetics in the absence of drugs. (c–f) Growth kinetics in the presence of isoniazid, rifampicin, ciprofloxacin, and ethambutol. Two biologically independent sets of experiments were performed. Each biological experiment was performed in biological triplicates. Data represent one set of experiments. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
-
Figure 4—source data 1
Killing kinetics in the absence and presence of different antibiotics.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig4-data1-v3.xlsx
The variant of mutY confers survival advantage ex vivo
Next, we evaluated the survival of Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q in the peritoneal macrophages. We did not observe any differences in the survival of RvΔmutY or RvΔmutY::mutY-R262Q compared with Rv or RvΔmutY::mutY (Figure 5—figure supplement 1a–b). We speculated that the evolution of a strain to become antibiotic-resistant requires the continued presence of antibiotic and host-directed stress. Therefore, we infected peritoneal macrophages with Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q in the absence or presence of the antibiotics. The bacteria recovered after 120 hr post-infection (p.i.) were cultured in vitro for 5 days and used for the next round of infection. The process was repeated for three rounds, and CFUs were enumerated at 4 and 96 hr p.i. during the fourth (final) round of infection (Figure 5a). CFUs obtained at 4 hr p.i. showed equal load. RvΔmutY and RvΔmutY::mutY-R262Q exhibited better survival in the absence of antibiotics than Rv and RvΔmutY::mutY (Figure 5b–e). There was no additional advantage compared with untreated in the presence of isoniazid (Figure 5f). However, we observed a log-fold advantage for RvΔmutY and RvΔmutY::mutY-R262Q compared with Rv or RvΔmutY::mutY in the presence of rifampicin or ciprofloxacin (Figure 5f).
Figure 5 with 1 supplement see all
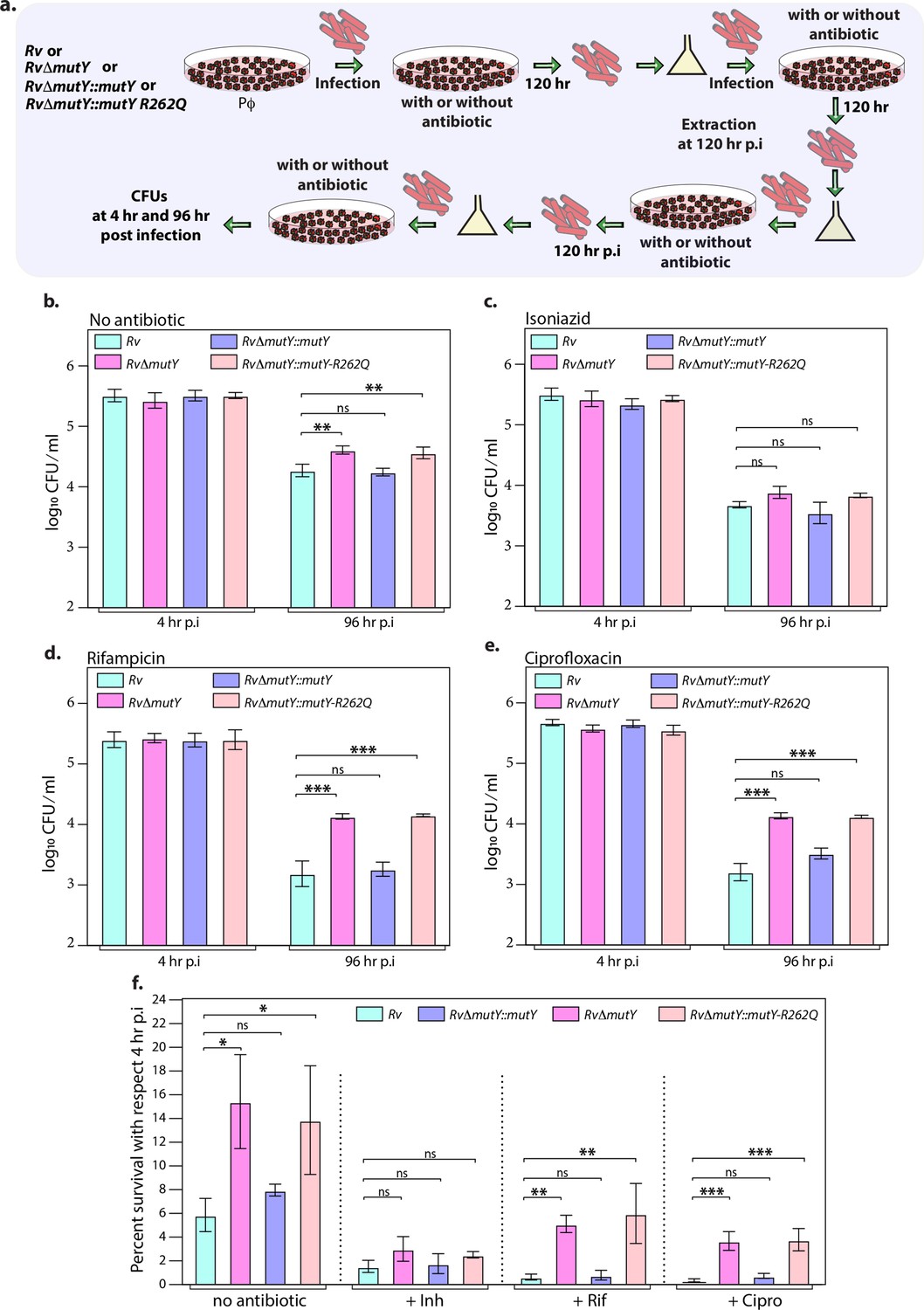
Mutations in the DNA repair genes provide a survival advantage in the presence of antibiotics.
(a) A schematic is representing the ex vivo infection experiment in the presence and absence of different antibiotics. (b–e) Survival of the strains in the peritoneal macrophages at 4 and 96 hr post-infection (p.i.) without and with antibiotics (isoniazid or rifampicin, or ciprofloxacin). (f) Percent survival with respect to 4 hr p.i. was determined for each strain without and with antibiotics (isoniazid or rifampicin or ciprofloxacin). Two biologically independent sets of experiments were performed. Each biological experiment was performed in biological triplicates. Data represent one set of experiments. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
-
Figure 5—source data 1
Survival of different strains in the absence and presence of antibiotics ex vivo.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig5-data1-v3.xlsx
Acquisition of direct target mutations ex vivo in the presence of drugs
We sought to determine if the improved survival of mutY mutant and mutY variant in the above experiment (Figure 5) is due to the acquisition of mutations in the direct target of antibiotics. To identify the mutations, we performed Whole Genome Sequencing (WGS). Genomic DNA extracted from 10 independent colonies (grown in vitro) was mixed in equal proportion prior to library preparation. Only those SNPs present in >20% of the reads were retained for the analysis. Analysis of Rv sequences grown in vitro suggested that the laboratory strain accumulated 100 SNPs compared with the reference strain (data not shown). The sequence of the Rv laboratory strain was used as the reference for the subsequent analysis. WGS data for RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q strains grown in vitro did not show the presence of any mutations in the antibiotic target genes. In a similar vein, 10 independent colonies, each from the 7H11-OADC plates, after the final round of ex vivo infection in the presence or absence of antibiotics, were selected for WGS. Data indicated that in the absence of antibiotics, no direct target mutations were identified in the ex vivo passaged strains (Figure 6a & e). However, in the presence of isoniazid, we found mutations in the katG (Ser315Thr or Ser315Ileu) in the Rv, RvΔmutY but not in RvΔmutY::mutY and RvΔmutY::mutY-R262Q (Figure 6b & e). These findings are in congruence with the ex vivo evolution CFU analysis, wherein we did not observe a significant increase in the survival of RvΔmutY and RvΔmutY::mutY-R262Q in the presence of isoniazid (Figure 5). In the presence of ciprofloxacin and rifampicin, direct target mutations were identified in the gyrA and rpoB (Figure 6c–e). Asp94Glu/Asp94Gly mutations were identified in gyrA, and, His445Tyr/Ser450Leu mutations were identified in rpoB of RvΔmutY and RvΔmutY::mutY-R262Q, respectively. No direct target mutations were identified in the Rv and RvΔmutY::mutY, suggesting that the perturbed DNA repair aids in acquiring the drug resistance-conferring mutations in Mtb (Figure 6c–e & Supplementary file 8; Paritosh, 2022; https://github.com/kumar-paritosh/analysis_of_Mtb_genome).
Figure 6
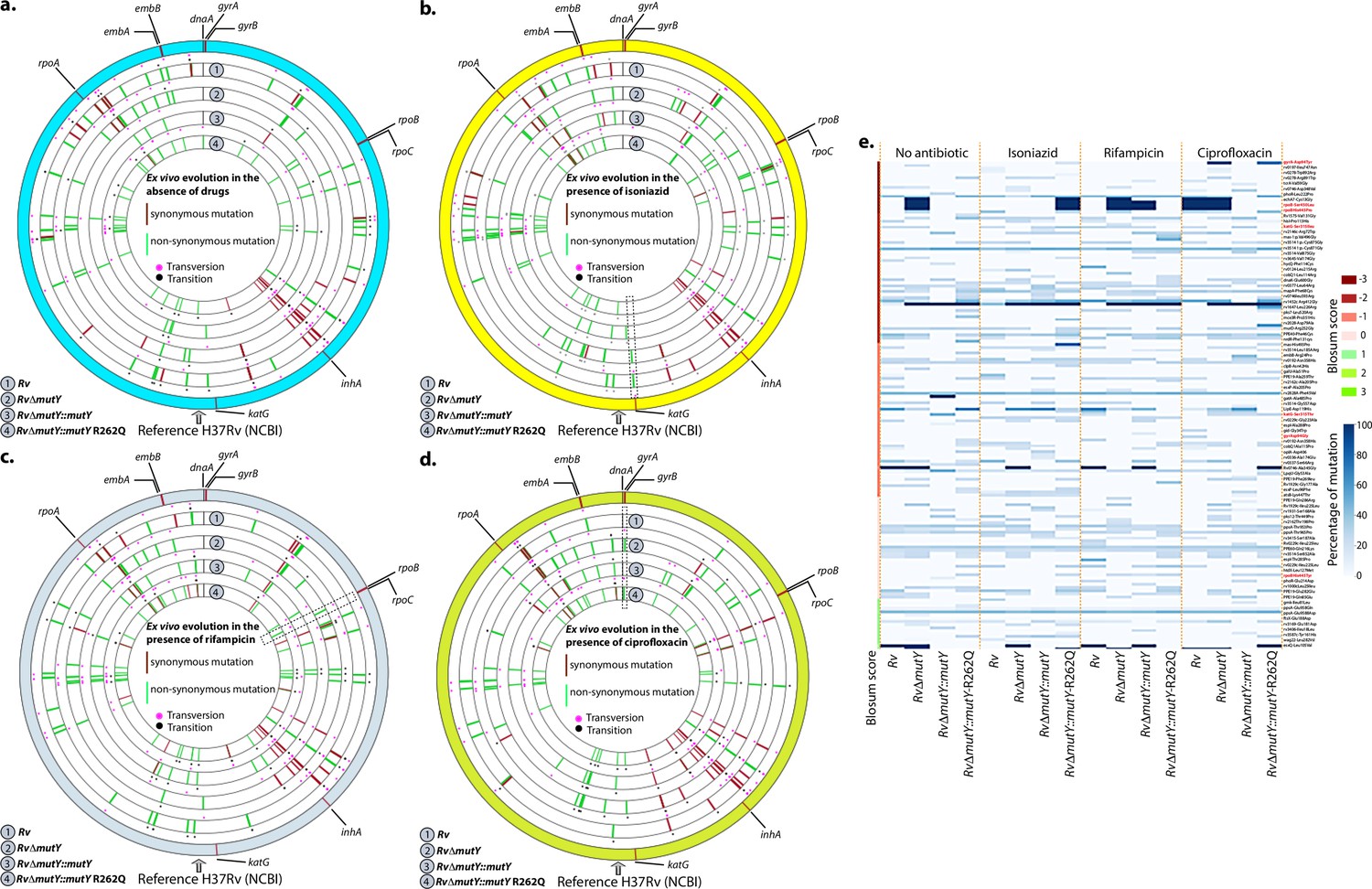
WGS reveals the acquisition of direct target mutations in the ex vivo passaged strains.
(a–d) Circos plot showing the WGS analysis of the strains passaged ex vivo in the absence (a) and in the presence of isoniazid (b), rifampicin (c), and ciprofloxacin (d). The outermost circle represents the reference genome labeled with the known direct target mutations. Circles (from outside to inside) represent Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q genome. (e) Heat map represents the single nucleotide polymorphisms identified in the Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q after ex vivo passage.
Competition experiment ex vivo reveals MutY variant confers a survival advantage
Results above suggested that the mutY variant impacts survival advantage when subjected to antibiotic selection, likely due to its ability to accumulate mutations. We reasoned that, if this is indeed the case, the mutY variant may outcompete the wild-type Rv when both the strains are present together. To test this hypothesis, we infected peritoneal macrophages with a combination of Rv + RvΔmutY or Rv + RvΔmutY::mutY or Rv + RvΔmutY::mutY-R262Q. At 96 hr p.i. host cells were lysed, and Mtb CFUs were enumerated on plain 7H11 or kanamycin (kan) (Rv) or hygromycin (hyg) (RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q) to evaluate the survival rates (Figure 5—figure supplement 1c–d). The percent survival was calculated as (CFUs obtained on kan or hyg/CFUs on kan + hyg) × 100. It is apparent from the data that the survival rates of competing strains were comparable, suggesting that mutY deletion or complementation with variant did not confer a significant advantage (Figure 5—figure supplement 1c). These results indicate that in the absence of prior antibiotic selection, deletion or the presence of mutY variant does not confer an advantage (Figure 5—figure supplement 1c–d).
To test these conclusions, we performed ex vivo co-infection experiment with the strains that were subjected to three rounds of prior selection (Figure 5). Peritoneal macrophages were infected with a combination of Rv + RvΔmutY or Rv + RvΔmutY::mutY or Rv + RvΔmutY::mutY-R262Q (Figure 7a). At 24 hr p.i., cells were either treated or not treated with an antibiotic for the subsequent 72 hr, and total CFUs were enumerated as described above to evaluate the survival rates (Figure 5—figure supplement 1e–f). As expected, there was no difference in the CFUs either at 4 hr p.i. (Figure 7b–e) or 24 hr p.i. (data not shown). At 96 hr p.i., RvΔmutY and RvΔmutY::mutY-R262Q strains showed a distinct advantage over Rv both in the absence or presence of antibiotics. Importantly, RvΔmutY::mutY did not show any advantage over Rv under any conditions. These results suggest that subjecting deletion or variant strains to antibiotic stress in the host helps in evolution of the strains that can outcompete the wild-type strain.
Figure 7

Variant of mutY outcompetes Rv in competition experiment.
(a) Schematic representing the competition experiment performed in peritoneal macrophages. Strains obtained after three rounds of infection in the peritoneal macrophages were used to perform a competition experiment (Figure 4a). (b–e) Percent survival of Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY R262Q in the absence and presence of antibiotics. Two biologically independent experiments, with each experiment performed in technical triplicates. Data represent one of the two biological experiments. Data represent mean and standard deviation. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
-
Figure 7—source data 1
Competition experiment in the presence and absence of different drugs after passage in the peritoneal macrophages.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig7-data1-v3.xlsx
MutY variant exhibits enhanced survival in vivo
Upon entering the host macrophages, Mtb encounters multiple forms of stress that impede its growth. To survive and grow in such a hostile environment, Mtb employs various defense mechanisms (Chai et al., 2020). The treatment regimen with anti-TB drugs imposes a supplementary layer of stress on the pathogen. An auxiliary mechanism the pathogen uses is to accumulate mutations in its genome that improve its ability to combat antibiotic and host-induced stresses. We asked if the variant mutations identified in DNA repair genes provide one such auxiliary mechanism. To test this hypothesis, we performed guinea pig infection experiments using Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q (Figure 8a). CFUs were enumerated after 1 and 56 days post-infection. CFUs obtained on day 1 showed the deposition was equal for wild-type, mutant, and the complemented strains. Gross pathology and histopathology analysis of infected lungs showed the presence of well-formed granulomas (Figure 8b–c). Significantly, 56 hr p.i., RvΔmutY, RvΔmutY::mutY-R262Q strains showed ~fivefold superior survival than Rv RvΔmutY::mutY, suggesting that the variant mutant identified indeed confers advantage (Figure 8d).
Figure 8 with 2 supplements see all
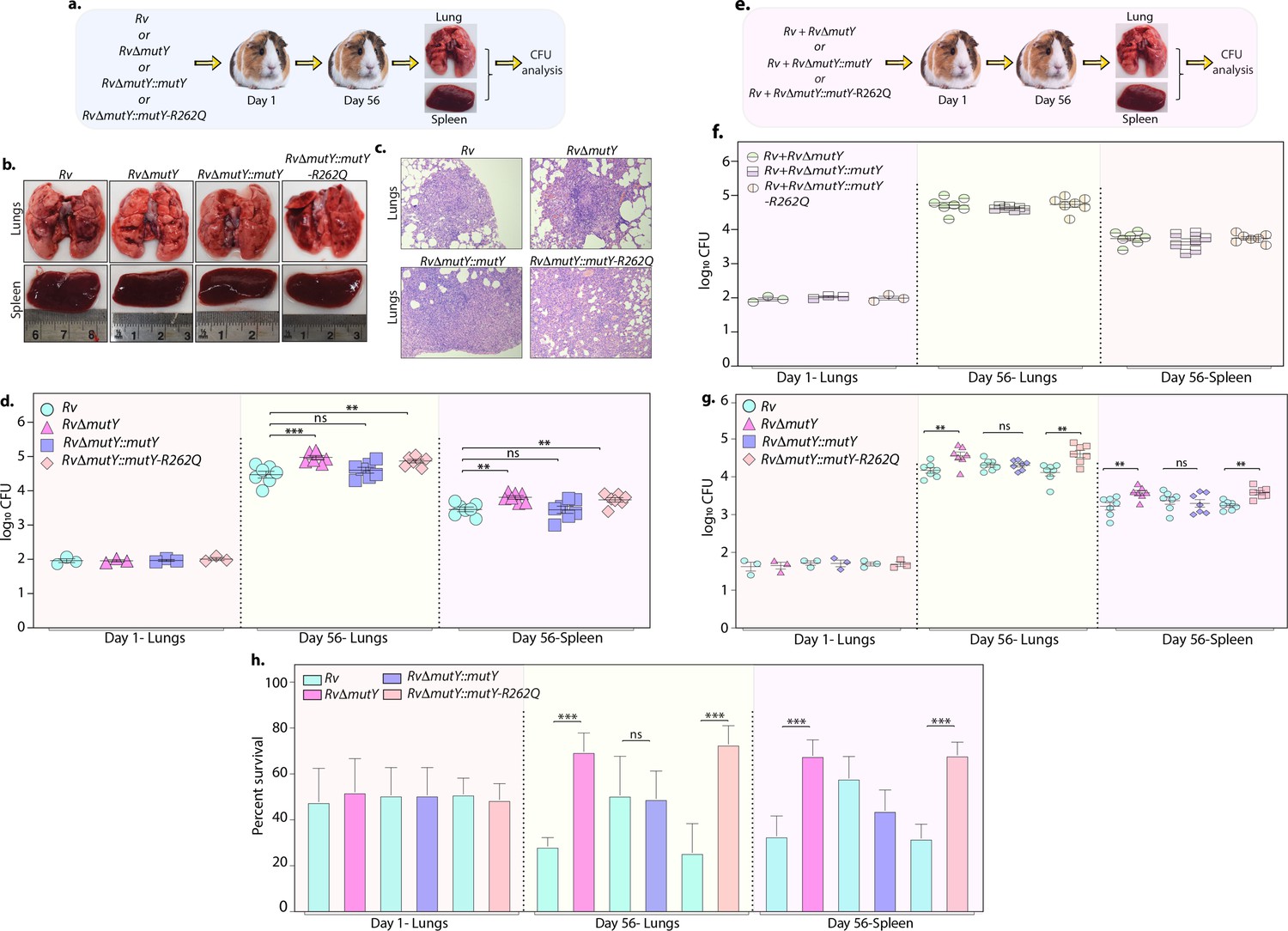
Perturbation of DNA repair results in enhanced survival in vivo.
(a) A schematic representation of the guinea pig infection experiment. (b) Gross histopathology of lungs and spleen of infected guinea pigs. (c) Hematoxylin and eosin staining of infected lung tissue showing the well-formed granuloma. Magnification ×10. (d) Guinea pigs were challenged with Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q via the aerosol route. CFUs were enumerated at day 1 post-infection to determine the lungs’ initial load (n=3). The CFUs at 1 day post-infection (p.i.) are represented for the whole lung. At 56 days p.i., lungs and spleen were isolated to determine survival (CFU / ml, n=7). (e) Outline showing the competition experiment performed in guinea pigs. (f) Total CFU enumeration of Rv + RvΔmutY, Rv + RvΔmutY::mutY, and Rv + RvΔmutY::mutY-R262Q in the lungs and spleen of guinea pigs at 1 day (whole lung, n=3) and 56 days p.i. (CFU/ml, n=7). (g) CFU enumeration of Rv + RvΔmutY, Rv + RvΔmutY::mutY, and Rv + RvΔmutY::mutY-R262Q on kanamycin and hygromycin containing plates. Statistical analysis was performed using two-way ANOVA. Graphpad prism was employed for performing statistical analysis. ***p<0.0001, **p<0.001, and *p<0.01. (h) Survival of each strain at indicated time points in the mixed infection. (i) Percent survival at 1 and 56 days in the competition experiment. Statistical analysis was performed using unpaired t-test. ***p<0.0001, **p<0.001, and *p<0.01.
-
Figure 8—source data 1
Gross histopathology of the infected lungs and spleen isolated from guinea pig.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig8-data1-v3.pdf
-
Figure 8—source data 2
Haematoxylin and eosin staining.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig8-data2-v3.pdf
-
Figure 8—source data 3
Survival of different strains in vivo.
- https://cdn.elifesciences.org/articles/75860/elife-75860-fig8-data3-v3.xlsx
Next, we determined the survival ability of RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q, when competed against wild-type Rv strain (Figure 8e). The lung and spleen homogenates were plated on 7H11 to determine total CFUs (Figure 8f, Figure 8—figure supplement 1). The total CFUs were found to be comparable in both lungs and spleen across all combinations (Figure 8f). Lung and spleen homogenates were plated on either kan (Rv) or hyg (RvΔmutY, RvΔmutY::mutY or RvΔmutY::mutY-R262Q) containing plates to determine survival (Figure 8g). As with independent infections, RvΔmutY, or RvΔmutY::mutY-R262Q, showed fivefold elevated CFUs compared with Rv (Figure 8g). When plotted as percent survival, the data showed that mutY deletion or complementation with the variant confers a survival advantage to the pathogen (Figure 8h). To determine the plausible cause of enhanced survival in vivo, we performed WGS of the strain isolated from guinea pig lungs. Analysis revealed that the specific genes such as cobQ1, smc, espI, and valS were mutated only in RvΔmutY and RvΔmutY::mutY-R262Q but not in Rv and RvΔmutY::mutY. Besides, tcrA and gatA were mutated only in RvΔmutY, whereas rv0746 was mutated exclusively in the RvΔmutY::mutY (Figure 8—figure supplement 2a–b). However, we did not observe any direct antibiotic resistance target mutations; this may be because guinea pigs were not subjected to antibiotic treatment. However, the total number of SNPs observed in all four strains was comparable. Thus the precise mechanistic role of MutY in Mtb pathogenesis needs further investigation. Collectively, results show that variants identified in DNA repair genes abrogate their function and contribute to a better survival in different stress conditions.
Discussion
The WGS of clinical strains provides vital information about many aspects such as, the acquisition and transmission of drug resistance, evolution of compensatory mutations, and evolution of the drug resistance in patients (Cohen et al., 2019). A recent WGS analysis of 10,219 diverse Mtb isolates successfully predicted mutations associated with pyrazinamide resistance (CRyPTIC Consortium and the 100,000 Genomes Project et al., 2018). Large-scale GWAS, employed initially for analyzing human genome data, is an invaluable tool to delineate the mutations that confer antibiotic resistance in bacteria (Power et al., 2017). For example, using the PhyC test on 116 clinical strains of Mtb, ponA1 was identified as one of the targets of independent mutations (Farhat et al., 2013). Analysis of 161 Mtb genomes identified polymorphisms in the intergenic regions that confer resistance to p-aminosalicylic acid (PAS) through overexpression of thyA and thyX (Zhang et al., 2013). GWAS of 498 sequences revealed that mutation in alanine dehydrogenase correlated with the resistance to a second-line drug D-cycloserine (Desjardins et al., 2016). Evaluation of 549 strains led to the identification of prpR, which in an ex vivo infection model confers conditional drug tolerance through regulation of propionate metabolism (Hicks et al., 2018). Largest GWAS involving 6465 clinical isolates uncovered novel resistance-associated mutations in ethA and thyX promoter, associated with ethionamide and PAS resistance, respectively (Coll et al., 2018).
We performed a gene-based GWAS analysis on a large dataset of susceptible and MDR/XDR clinical strains (Figure 1). We identified mutations in the known direct targets of both first- and second-line antibiotics and a few recently reported genetic variants using association analysis (Tables 1 and 2, Table 3). We also identified frameshift and non-synonymous mutations in rv2333c and rv1250, encoding for the transporters known to be differentially expressed in MDR patients (Table 3; Umar et al., 2019). Besides, the analysis captured mutations in genes involved in cell metabolism (Figure 2). This finding supports a recent study in Escherichia coli, where drug treatment led to the acquisition of mutations in the metabolic genes that impart drug resistance (Lopatkin et al., 2021). We believe that these may be compensatory mutations that placate the fitness cost associated with antibiotic resistance, and hence, might be the consequence of antibiotic resistance.
Table 3
Transporters associated with drug resistance phenotype.
| Gene | False discovery rate-adjusted p-value | Variant |
|---|---|---|
| rv1258c | 1.60E-08 | Non-syn |
| mmpL2 | 9.51E-05 | Non-syn |
| rv0987 | 1.60E-08 | Non-syn |
| rv1250 | 3.44E-07 | Non-syn |
| kdpC | 2.15E-07 | Non-syn |
| rv0928 | 2.15E-07 | Non-syn |
| rv2333c | 1.32E-20 | Frameshift |
| mmpL13a | 1.25E-06 | Syn |
| kdpB | 2.15E-07 | Syn |
Most importantly, for the first time, we identified a significant association between mutations in three of the DNA repair pathway genes with drug resistance. This study is in line with the studies on the other pathogens such as Pseudomonas aeruginosa, Helicobacter pylori, Neisseria meningitides, and Salmonella typhimurium, where mutations or deletions in the DNA repair genes were identified in the clinical isolates (Chopra et al., 2003). Moreover, the deletion of ung and udgB (BER pathway genes), independently or together, provides a survival advantage to the bacteria (Naz et al., 2021). It is known that lineage 2 clinical strains have polymorphisms in the DNA repair and replication genes (Mestre et al., 2011; Ebrahimi-Rad et al., 2003). However, we identified mutations in DNA repair genes in lineage 4, suggesting that the phenomenon is not confined to lineage 2 (Figure 3—figure supplement 1a).
The recently published CRYPTIC consortium associates 12,289 Mtb strains with the resistance phenotype to 13 antibiotics does not contain a mutY-Arg262Gln mutation in their data set. This may be due to the different strains used in the study (CRyPTIC Consortium and the 100,000 Genomes Project et al., 2018). Mutation spectrum analysis of the clinical strains harboring the mutY variant and closely related strains showed a trend toward higher C→T, A→G, and C→A mutations, but we could not perform statistical analysis, as the strains harboring the mutY variant were limited. We investigated if the mutations in DNA repair genes are the cause or the consequence of the antibiotic resistance. We functionally validated the identified mutations of two different pathway genes, mutY and uvrB, in Msm using gene replacement mutants. Mutation frequency analysis suggests that GWAS identified Arg262Gln and Ala524Val mutations in mutY and uvrB, respectively, abrogated their functions (Figure 3). The data agrees with the previously published study, wherein WGS analysis of antibiotic susceptible strain isolated from a patient showed a non-synonymous mutation in UvrB (A582V). Notably, the variant strain evolved into XDR-TB over 3.5 years after the first- and second-line drug treatment (Eldholm et al., 2014). Therefore, we propose that mutations identified in DNA repair genes contribute to the evolution of antibiotic resistance (Figure 2—figure supplement 5). Besides, the killing kinetics in the presence of different anti-TB drugs show that RvΔmutY and RvΔmutY::mutY-R262Q exhibit better survival (Figure 4).
Poor adherence of the patients to the antibiotic regimen is the leading cause of the emergence of drug resistance. The continual treatment with antibiotics provides sufficient time to evolve strains into MDR or XDR. We emulated the condition by repeatedly exposing strains to antibiotics in the ex vivo model, followed by growing them in vitro without antibiotics. This led to the improved survival of RvΔmutY and RvΔmutY::mutY-R262Q (Figure 5). WGS of the strains after ex vivo passage shows that the compromised DNA repair helps in acquiring mutations in the direct targets of the antibiotics (Figure 6). We then determined the survival of ex vivo evolved strains in the mixed infection scenario. A competition experiment using the evolved strains showed that RvΔmutY and RvΔmutY::mutY-R262Q could outcompete Rv (Figure 7). Host stress drives the selection of the bacterial population that acquires the ability to withstand adverse conditions. We evaluated the survival of the different strains in guinea pigs. Results suggest that RvΔmutY and RvΔmutY::mutY-R262Q exhibit improved survival. Similarly, we observed that RvΔmutY and RvΔmutY::mutY-R262Q could successfully outcompete Rv in the guinea pig infection model (Figure 8).
Using the GWAS approach and functional validation of the clinical mutations identified in the BER and NER pathways, we established a novel link between the compromised DNA repair and the evolution of antibiotic resistance (Figure 9).
Figure 9
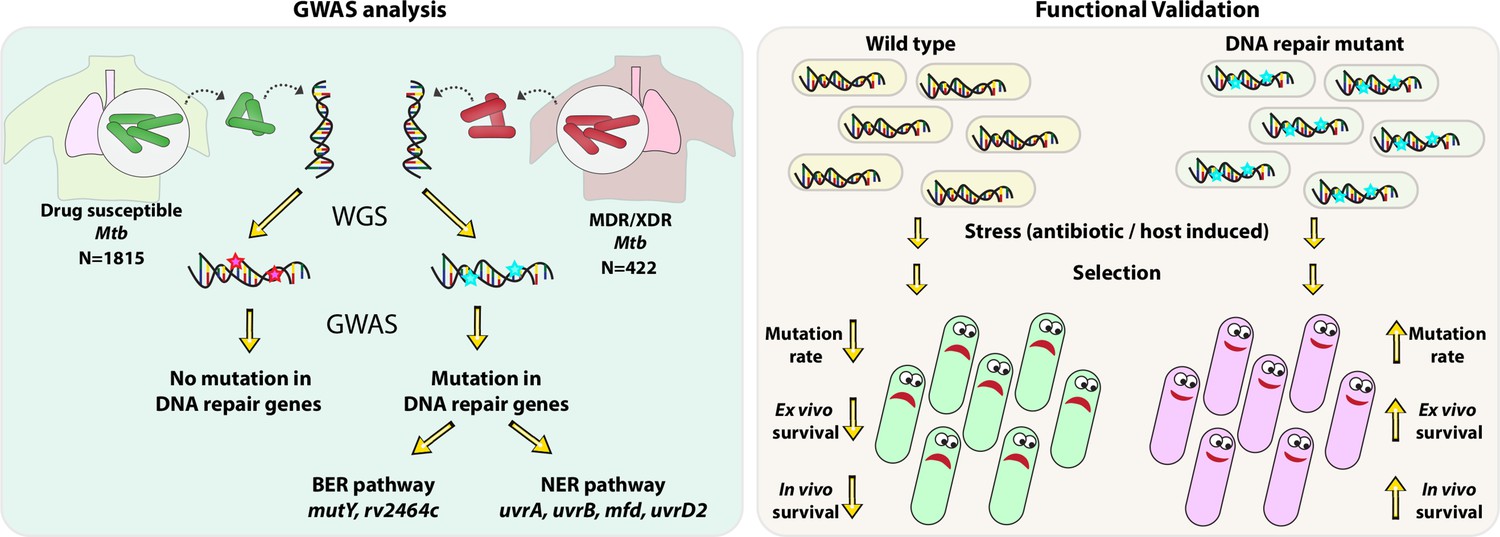
Model.
Model depicts the analysis and subsequent validation. Genome-wide association study (GWAS) revealed mutation in three DNA repair pathway genes in multidrug resistant/extensively drug resistant (MDR/XDR) strains. Based on GWAS, we proposed that mutations in DNA repair genes are associated with the evolution of antibiotic resistance in Mycobacterium tuberculosis (Mtb). Functional validation was performed using the gene replacement mutants of base excision repair (BER) and nucleotide excision repair (NER) pathway genes in Mycobacterium smegmatis and Mtb. In vitro, ex vivo, and in vivo experiments show that compromised DNA repair pathway leads to the enhanced survival of bacteria.
Collectively, the data presented here suggest that the loss of function of DNA repair genes helps acquire drug resistance in the presence of anti-TB drugs. We propose that the evolution of MDR or XDR-TB is likely a consequence of the loss of function of DNA repair genes. The presence of mutations in DNA repair genes can be an early-stage diagnostic marker for the evolution of the strain into MDR/XDR-TB. Molecular diagnosis of DNA repair gene mutations at the onset of infection should help design better therapies to impede the evolution of these strains into MDR or XDR. These findings indicate that bacteria having compromised DNA repair can contribute to the accumulation of mutations, providing an advantage to the bacilli when subjected to antibiotic treatment.
Materials and methods
Sequence retrieval and variant calling
View detailed protocolAccession numbers for 2773 clinical strains of Mtb were obtained from 10 previous studies (Supplementary file 1), representing all 4 lineages from 9 countries. Sequence data were retrieved from EBA (https://www.ebi.ac.uk/) and NCBI databases (https://www.ncbi.nlm.nih.gov/) and quality filtered using Trimmometic software (Bolger et al., 2014). Adapter and E. coli sequence contaminations were removed, the sequences were analyzed at a sliding window of 4 bp, and those with an average phread value of 15 were clipped. Parameters for the trimmometic-based QC were set as (Leading:3, slidingwindow:4:15 trailing:3 minlen:60). Reads with a filtered length of <60 bp were removed. The filtered SE/PE reads were mapped on the H37Rv reference genome (Accession number –ASM19595) using the Burrows Wheeler Alignment (BWA) mem algorithm (Li and Durbin, 2009). GATK pipeline was used for sorting, PCR duplicate removal, and realignment of sequences (Van der Auwera et al., 2013). Variants (SNP and small InDels) were predicted in a batch mode with Platypus script (Rimmer et al., 2014). SNPs with read depth <5 or mapping quality of <20 were marked as missing. SNPs with missing calls in >40% of the accessions were removed from the analysis. Finally, ~160,000 SNPs from 2773 accessions with a variant dataset with an MAF of >1% were selected for the final analysis. The.vcf file was converted to.hmp file with Tassel (Bradbury et al., 2007). A phylogenetic tree was generated using ~160,000 SNPs using SNPhylo (Lee et al., 2014).
Association analysis
View detailed protocolGenome-wide association analysis was carried out with 1815 susceptible strains and 422 MDR/XDR strains (Supplementary file 2). A GAPIT based on a compressed mix linear model was performed under the R environment (Lipka et al., 2012). VanRaden algorithm was employed for the calculation of the Kinship matrix. The kinship matrix assessed the relatedness among the strains included in the association panel. Principal components in GAPIT were used to classify the population structure. An association mapping analysis was carried out by combining the population structure analysis and the relative kinship matrix. The association mapping analysis obtained p-values, R2, and marker effect values. The FDR adjusted p-values in the GAPIT software are highly stringent as it corrects the effects of each marker based on the population structure (Q) as well as kinship (K) values and often lead to overcorrection (Gao et al., 2016; Zegeye et al., 2014). We selected a corrected p-value of 10–5 as the threshold for selecting associated genes. The associated SNPs were annotated using the snpEff v4.11 (Cingolani et al., 2012). A snpeff database was generated using the H37Rv (ASM19595v2, was used as a reference for mapping). The corresponding .gff file and the SNPs were annotated based on their position on the genome. Dot-plot, Manhattan’s plot, and volcano plot were generated using R scripts.
Generation of gene replacement mutant in Mtb
Request a detailed protocolThe upstream (5’ flank) and downstream region (3’ flank) of the mutY were amplified using the Rv genomic DNA. Flanks were digested with an appropriate restriction enzyme and ligated with oriE + lambda cos and hygromycin resistance cassette (Jain et al., 2014). The allelic exchange substrate was digested to generate linearized substrate and electroporated in the recombineering proficient Rv strain harboring pNit-ET plasmid (van Kessel and Hatfull, 2007). Colonies were screened post 3 weeks electroporation for gene replacement mutant.
Generation of complementation constructs and western blot analysis
Request a detailed protocolWild type allele of mutY or uvrB was PCR amplified using Rv genomic DNA. The PCR product and the vector pSTL-giles were digested with NdeI and HindIII to generate pSTL-giles-mutY or pSTL-giles-uvrB. Subsequently, the PCR product was ligated with the vector pSTL-giles. SapI-based cloning was employed for the generation of pSTL-giles-mutY-R262Q or pSTL-giles-uvrB-A524V. Constructs, pSTL-giles-mutY and pSTL-giles-mutY-R262Q, were electroporated in the msmΔmutY or RvΔmutY to generate msmΔmutY::mutY and msmΔmutY::mutY-R262Q or RvΔmutY::mutY and RvΔmutY::mutY-R262Q. Constructs, pSTL-giles-uvrB or pSTL-giles-uvrB-A524V, were electroporated in msmΔuvrB to generate msmΔuvrB::uvrB or msmΔuvrB::uvrB- A524V (oligonucleotides used in the study are given in the Supplementary file 9). 50 ml cultures of Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q were inoculated in 7H9-ADC medium at A600 ~0.1 and grown till A600 ~0.8. Cells were pelleted in 50-ml falcon tubes at 4000 rpm for 10 min and resuspended in the lysis buffer containing protease inhibitors. Cells were transferred in the bead beating tubes containing zirconium beads. Bead beating was performed for eight cycles. The cell lysate was centrifuged twice at 13,000 rpm for 45 min at 4°C. Protein was estimated using the Bradford assay reagent. 50 μg of Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q was loaded on two independent 10% SDS-PAGE and transferred to nitrocellulose membrane. 5% BSA prepared in 1XPBST20 was used for blocking the membrane for 2 hr. Membranes were incubated overnight at 4°C with α-FLAG (1:5000), andα-GroEL-1 (1:10000), respectively. Membranes were washed using 1XPBST20 (thrice) and incubated with anti-rabbit secondary antibody DARPO (1:10000) for 1 hr at room temperature (RT; 25°C). Membranes were washed thrice with 1XPBST20, and a blot was developed using a chemiluminescence reagent.
Analysis of mutation frequency and rate
Request a detailed protocolAntibiotic sensitive cultures of msm, msmΔmutY, msmΔmutY::mutY, msmΔmutY::mutY-R262Q, msmΔuvrB, msmΔuvrB::uvrB, and msmΔuvrB::uvrB-A524V were grown in 7H9-ADC medium A600 ~0.6, and 50,000 cells/ml were inoculated in fresh 10-ml medium. Cultures were grown for 6 days in a 37°C incubator at 200 pm. On the seventh day, appropriate dilutions were plated on 7H11-OADC plain plates to determine the load, and 1 ml was plated on rifampicin (50 μg/ml). Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY R262Q strains were inoculated at A600~0.1. Strains were grown to A600 ~0.6 and plated on 7H11-plain or rifampicin (2 μg/ml) or isoniazid (5 μg/ml) plates to calculate the mutation frequency. Six antibiotic-sensitive colonies were grown in a 7H9-ADC medium up to A600 ~0.8, and 50,000 cells/ml were inoculated in a fresh 10-ml 7H9-ADC medium in the presence of 15% sterile Rv culture filtrate. On the 15th day, appropriate dilutions were plated on 7H11-OADC plain plates to determine the load, and 1 ml was plated on rifampicin (2 μg/ml), isoniazid (5 μg/ml), and ciprofloxacin (1.5 μg/ml). The mutation rate was determined as reported previously (Boshoff et al., 2003; David, 1970). Two biologically independent experiment sets were performed to determine mutation frequency. Each experiment was performed in the biological triplicate (mutation frequency) or six triplicate (mutation rate). Data represent one of the two biological sets of experiments. Data represent mean and standard deviation. Statistical analysis (two-way ANOVA) was performed using Graphpad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
Killing kinetics in the presence of anti-TB drugs
Request a detailed protocolRv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q were grown in 7H9-ADC medium up to A600 ~0.8. Cultures were inoculated in the fresh medium at the A600 ~0.6 in 10-ml 7H9-ADC medium. Different antibiotics- rifampicin (2 μg/ml), isoniazid (5 μg/ml), and ciprofloxacin (1.5 μg/ml) were added, and the CFUs were enumerated at day 0, 3, 6, and 9 post on 7H11-OADC plates. Two biologically independent sets of experiments were performed. Each experiment was performed in the biological triplicate. Data represent one of the two biological sets of experiments. Data represent mean and standard deviation. Statistical analysis (two-way ANOVA) was performed using Graphpad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
Survival of strains ex vivo
Request a detailed protocolBalb/c mice were injected with thioglycollate, and 72 hr post-injection, peritoneal macrophages were isolated. One million cells were seeded in each well of a six-well plate. Cells were infected with Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY R262Q independently at an multiplicity of infection (MOI) of 1:5. After 24 hr p.i. cells were treated with rifampicin (1 μg/ml), isoniazid (1 μg/ml), and ciprofloxacin (2.5 μg/ml). Cells were lysed at 120 hr p.i. using 0.05% sodium dodecyl sulfate (SDS), and the bacteria was extracted. Bacteria were washed thrice with PBS to ensure the removal of SDS. Extracted bacteria were cultured in a 7H9-ADC medium without antibiotics for 5–7 days up to A600~0.4. These cultures were used for the next round of infection. The whole process was repeated three times. During the fourth round of infection, CFUs were enumerated at 4 hr p.i. and 96 hr p.i. to determine the survival of different strains. Percent survival was calculated by normalized CFU obtained at 96 hr with respect to the respective CFUs obtained at 4 hr. Two biologically independent experiment sets were performed. Each experiment was performed in biological triplicate. A representative experiment is shown in Figure 5. Similarly, the strains obtained after three rounds of infection from the above experiment were used for the competition experiment. Rv + RvΔmutY, Rv + RvΔmutY::mutY, and Rv + RvΔmutY::mutY-R262Q were mixed in a 1:1 ratio. 24 hr p.i, the cells were either not treated or treated with the antibiotics (isoniazid-1μg/ml, rifampicin- 1 μg/ml, and ciprofloxacin- 2.5 μg/ml) for subsequent 72 hr. CFUs were enumerated at 4 hr and 96 hr p.i. on 7H11-plain (all strains), 7H11-Kan (Rv), and 7H11-Hyg (RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q) plates. Percent survival was calculated as (CFUs on Kan or Hyg plates/[CFUs on Kan +CFUs on Hyg)]×100. Two biologically independent sets of experiments were performed. Each experiment was performed in the biological triplicate. Data represent one of the two biological sets of experiments. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01.
WGS of strains under different conditions
Request a detailed protocolIndependent colonies of Rv (n=10), RvΔmutY (n=10), RvΔmutY::mutY (n=10), and RvΔmutY::mutY-R262Q (n=10) were randomly selected and grown in vitro without any selection for genomic DNA isolation. Rv (n=10), RvΔmutY (n=10), RvΔmutY::mutY (n=10), and RvΔmutY::mutY-R262Q (n=10) were obtained after ex vivo passage in the absence and in the presence of isoniazid, rifampicin, and ciprofloxacin were grown independently for genomic DNA isolation. Similarly, Rv (n=10), RvΔmutY (n=10), RvΔmutY::mutY (n=10), and RvΔmutY::mutY-R262Q (n=10) isolated from guinea pig lungs were grown for the genomic DNA isolation. During the library preparation, genomic DNA isolated from an independent colony of Rv was mixed in equal amounts. The library of the mixed sample was prepared, and the library was sent for WGS after a quality check. A similar procedure was used to prepare each strain’s libraries under different conditions. Variant calling was performed as described in the section-sequence retrieval and variant calling. SNPs less than or equal to 20% cut-off were discarded. A final matrix containing identified SNPs in the genes, mutation percentage, and blosum score was generated. Heat maps and circos plots were generated using custom python scripts.
Guinea pig infection
Request a detailed protocolRv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q cultures were grown up to A600 ~0.8. For preparing single-cell suspension, Rv, RvΔmutY, RvΔmutY::mutY, and RvΔmutY::mutY-R262Q cultures were pelleted at 4000 rpm at RT. After suspending cells in saline, cells were passed through a 26½ gauge needle to obtain a single-cell suspension. 1×108 cells were taken in the 15 ml saline for infection. Female outbred Hartley guinea pigs were challenged using a Madison chamber calibrated to deliver ~100 bacilli/lung through the aersolic route. For determining the deposition of bacteria in the lungs of guinea pigs (n=3), CFUs were enumerated at 1 day post-infection on 7H11-plain plates for each strain. Survival of each strain was determined at 56 days post-infection in the lungs and spleen (n=7). Rv + RvΔmutY, Rv + RvΔmutY::mutY, and Rv + RvΔmutY::mutY R262Q were mixed in a 1:1 ratio, and the guinea pigs (n=10 per strain) were challenged as described above. CFUs were enumerated at 1 and 56 days post-infection on 7H11-plain plates or those containing kanamycin or hygromycin-containing plates. Data represent the SEM. Statistical analysis (two-way ANOVA) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01. Percent survival was calculated as (CFUs on Kan or Hyg plates/[CFUs on Kan + CFUs on Hyg])×100. Data represents Standard deviation and mean. Statistical analysis (Unpaired t-test) was performed using Graph pad prism software. ***p<0.0001, **p<0.001, and *p<0.01. All guinea pig infection experiments were performed at the same time. Guinea pigs were not treated with any antibiotics before or after infection.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting file. Sequencing data have been deposited at NCBI under Bioproject PRJNA885615. Code availability at GitHub: https://github.com/kumar-paritosh/analysis_of_Mtb_genome; (copy archived at swh:1:rev:cf2547e50b00c57dee9b60bca899ed50e617106f).
-
NCBI BioProjectID PRJNA885615. Whole genome sequencing of Mycobacterium tuberculosis strains under different conditions.
-
NCBI BioProjectID PRJNA268900. Clinically prevalent mutations in Mycobacterium tuberculosis alter propionate metabolism and mediate multidrug tolerance.
-
NCBI Sequence Read ArchiveID SRA065095. Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance.
-
ERP000192ID PRJEB2138. Evolution and transmission of drug-resistant tuberculosis in a Russian population.
-
ERP001885ID PRJEB3334. Significance of the identification in the Horn of Africa of an exceptionally deep branching Mycobacterium tuberculosis clade.
-
NCBI BioProjectID 492975. Genome Sequencing of Polydrug-, Multidrug-, and Extensively Drug-Resistant Mycobacterium tuberculosis Strains from South India.
-
ERP001072ID PRJEB2794. Recurrence due to relapse or reinfection with Mycobacterium tuberculosis: a whole-genome sequencing approach in a large, population-based cohort with a high HIV infection prevalence and active follow-up.
-
EBIID PRJEB2424. Elucidating emergence and transmission of multidrug-resistant tuberculosis in treatment experienced patients by whole genome sequencing.
-
DDBJID ERA020628. Whole-genome sequencing to establish relapse or re-infection with Mycobacterium tuberculosis: a retrospective observational study.
-
ERP000276ID PRJEB2221. Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational study.
References
-
The emergence of extensively drug-resistant tuberculosis (TB): TB/HIV coinfection, multidrug-resistant TB and the resulting public health threat from extensively drug-resistant TB, globally and in CanadaThe Canadian Journal of Infectious Diseases & Medical Microbiology = Journal Canadien Des Maladies Infectieuses et de La Microbiologie Medicale 18:289–291.https://doi.org/10.1155/2007/986794
-
Trimmomatic: a flexible trimmer for illumina sequence dataBioinformatics 30:2114–2120.https://doi.org/10.1093/bioinformatics/btu170
-
Host defense mechanisms against Mycobacterium tuberculosisCellular and Molecular Life Sciences 77:1859–1878.https://doi.org/10.1007/s00018-019-03353-5
-
The role of mutators in the emergence of antibiotic-resistant bacteriaDrug Resistance Updates 6:137–145.https://doi.org/10.1016/s1368-7646(03)00041-4
-
Prediction of susceptibility to first-line tuberculosis drugs by DNA sequencingThe New England Journal of Medicine 379:1403–1415.https://doi.org/10.1056/NEJMoa1800474
-
Mutations in putative mutator genes of Mycobacterium tuberculosis strains of the W-Beijing familyEmerging Infectious Diseases 9:838–845.https://doi.org/10.3201/eid0907.020803
-
Antimicrobial resistance in Mycobacterium tuberculosis: mechanistic and evolutionary perspectivesFEMS Microbiology Reviews 41:354–373.https://doi.org/10.1093/femsre/fux011
-
GAPIT: genome association and prediction integrated toolBioinformatics 28:2397–2399.https://doi.org/10.1093/bioinformatics/bts444
-
Synonymous but not the same: the causes and consequences of codon biasNature Reviews. Genetics 12:32–42.https://doi.org/10.1038/nrg2899
-
Microbial genome-wide association studies: lessons from human GWASNature Reviews. Genetics 18:41–50.https://doi.org/10.1038/nrg.2016.132
-
From fastq data to high confidence variant calls: the genome analysis toolkit best practices pipelineCurrent Protocols in Bioinformatics 43:11.https://doi.org/10.1002/0471250953.bi1110s43
-
Recombineering in Mycobacterium tuberculosisNature Methods 4:147–152.https://doi.org/10.1038/nmeth996
-
Whole-Genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational studyThe Lancet. Infectious Diseases 13:137–146.https://doi.org/10.1016/S1473-3099(12)70277-3
-
ReportWHO announces updated definitions of extensively drug-resistant tuberculosisWorld Health Organization.
Article and author information
Author details
Vinay Kumar Nandicoori

Funding
Department of Biotechnology, Ministry of Science and Technology, India (BT/PR13522/COE/34/27/2015)
- Vinay Kumar Nandicoori
Department of Science and Technology, Ministry of Science and Technology, India (JCB/2019/000015)
- Vinay Kumar Nandicoori
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was funded by the Department of Biotechnology, Government of India (BT/PR13522/COE/34/27/2015) and the J.C Bose fellowship (JCB/2019/000015). SN is a Senior Project Associate in the J.C Bose fellowship (JCB/2019/000015). We thank the Tuberculosis Aerosol Challenge Facility at ICGEB and staff for their help in performing animal infection experiments. We are thankful to the bio-containment facility (BSL3) at NII. Vector-pYUB1471 is a kind gift from Prof. William R Jacobs’s laboratory.
Ethics
Animal experiments protocol was approved by the Animal Ethics Committee of the National Institute of Immunology, New Delhi, India. The approval (IAEC#409/16) is as per the guidelines issued by the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), Government of India.
Copyright
© 2023, Naz, Paritosh et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,821
- views
-
- 417
- downloads
-
- 21
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 21
- citations for umbrella DOI https://doi.org/10.7554/eLife.75860
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
GWAS and functional studies suggest a role for altered DNA repair in the evolution of drug resistance in Mycobacterium tuberculosis
eLife 12:e75860.
https://doi.org/10.7554/eLife.75860




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}