Osteoporosis: Finding the genes for fragile bones
Combining transcriptomic data with the analysis of large genome-wide association studies helps identify genes that are likely important for regulating bone mineral density.
- School of Physiology, Pharmacology and Neuroscience, University of Bristol, United Kingdom
Bone fractures are painful and debilitating, particularly as we get older. However, some people are more susceptible than others to breaking bones as they age. Osteoporosis is the most prevalent bone condition of the ageing population (Salari et al., 2021). It is characterized by low bone mass and the deterioration of bone microarchitecture, leading to porous bones that are more likely to break (Trajanoska et al., 2018). But what are the causes of osteoporosis?
Studies involving twins and families have taught us that osteoporosis is heritable (Ralston and Uitterlinden, 2010), which means that genetics play a vital role in determining whether someone is at risk of developing osteoporosis. Learning which genes cause osteoporosis could teach us how the condition develops, opening therapeutic avenues to treat it.
This rationale has inspired researchers to perform genome-wide association studies (GWAS) to identify regions of the genome associated with bone mineral density, which is the trait that is the best indicator of fracture risk (Timpson et al., 2018). It has been 15 years since the first bone mineral density GWAS (Kague et al., 2022). The largest of these studies, published in 2019, involved around 425,000 participants and successfully identified over 500 genomic regions associated with bone mineral density of the heel (Morris et al., 2019).
However, interpreting the associations captured with GWAS can be challenging, because the loci that are identified can contain several genes, reside in non-coding regions near genes, or contain no genes at all. Therefore, despite the remarkable progress made by bone mineral density GWAS, the field needs to bridge a considerable gap that lies between correlation and causation. Filling this gap is essential to understand the underlying biology of osteoporosis and find potential therapeutics for the condition.
Now, in eLife, Charles R Farber and colleagues from the University of Virginia, the University of Colorado and Boston University – including Basel Maher AI-Barghouthi as first author – report on a systematic way to narrow down GWAS signals and shed light on the genes that are likely responsible for variations in human bone mineral density (Al-Barghouthi et al., 2022). To do this, the group leveraged transcriptomic data and computational approaches to systematically screen published bone mineral density GWAS (Figure 1). They identified 512 genes that could be regulators of bone mineral density, including the gene PPP6R3, which had not been associated with bone before.
Figure 1
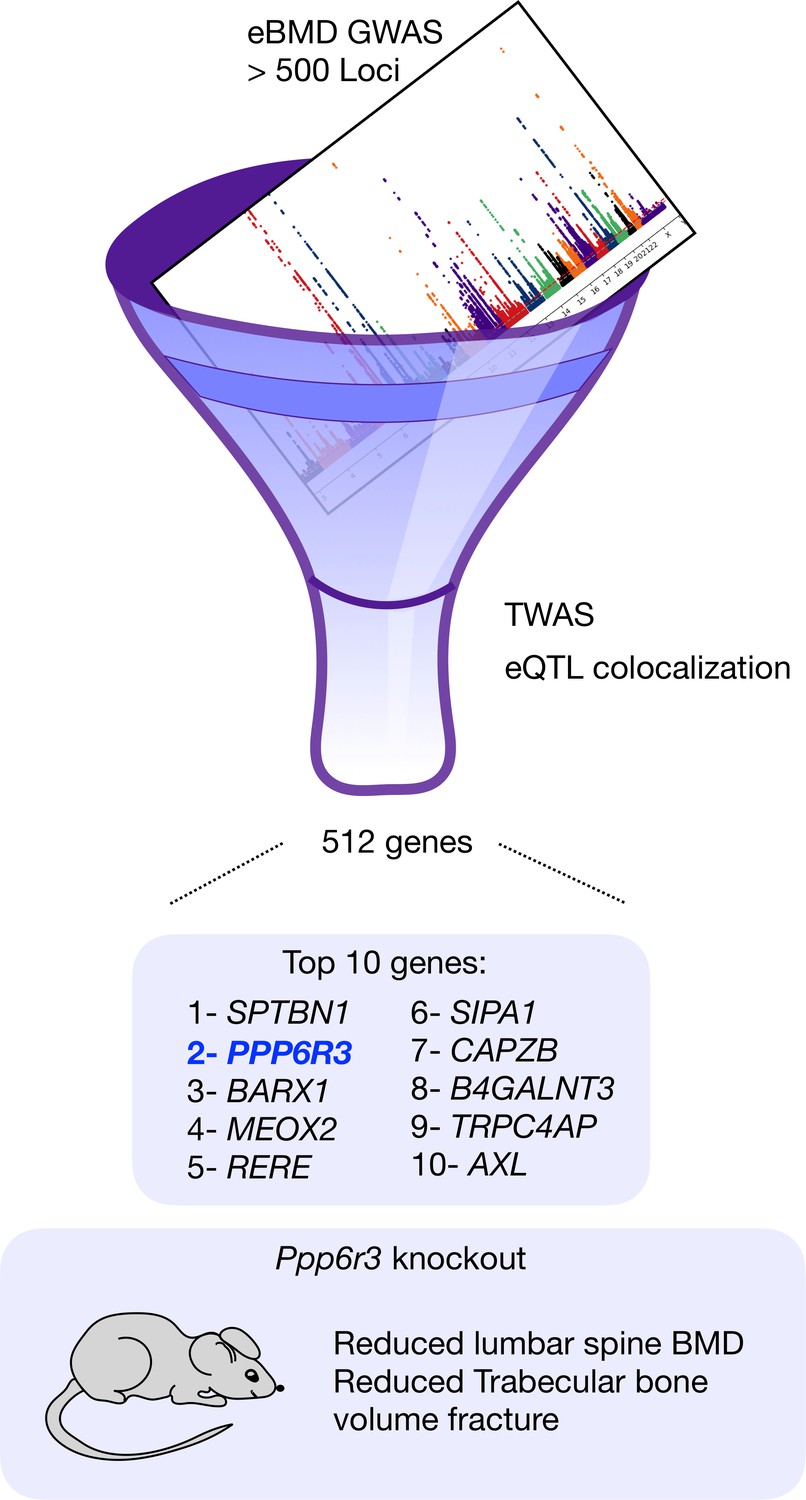
Combining two computational approaches identifies genes that could cause osteoporosis.
The results from a genome-wide association study (GWAS) that identified over 500 genomic regions that were associated with low bone mineral density in the heel were filtered using a combination of two techniques – transcriptome-wide association studies and expression quantitative trait loci (eQTL) colocalization – and this resulted in the identification of 512 genes that could be causing osteoporosis. The 10 most significant genes are shown. The gene with the second highest score, PPP6R3, had not previously been linked to bone. Al-Barghouthi et al. went on to show that mice in which this gene had been knocked out had reduced bone mineral density in their vertebrae.
Al-Barghouthi et al. used data from the Genotype-Tissue Expression (GTEx) project, which is an ongoing public resource containing gene expression data from 54 healthy tissue sites across nearly 1000 individuals whose genomes have been sequenced (GTEx Consortium, 2013). GTEx data allows researchers to infer how DNA variations in the proximity of a gene can influence fluctuations in gene expression, what is known as expression quantitative trait loci (eQTL). The team used this information to determine which of the genomic regions identified in the 2019 study on the heel (Morris et al., 2019) are most likely to have significant effects on bone mineral density based on the eQTLs they contain. This computational approach is known as a transcriptome wide association study or TWAS. Next, Al-Barghouthi et al. performed eQTL colocalization, a statistical technique that can determine whether an eQTL variant found in a specific region of the genome is driving the GWAS signal in that region. This approach allows researchers to identify specific sequences or genes that are likely responsible for a trait. Combining these two approaches, the team identified 512 genes that could potentially cause osteoporosis.
To confirm that their method was capturing genes regulating bone mineral density, Al-Barghouthi et al. asked whether the genes they had identified were enriched for known bone genes. They found that about 13% of the genes they had captured were known bone genes, over what would be expected by chance. Identified genes were also enriched for ontologies associated with bone (i.e. ossification, skeletal system development, and osteoblast differentiation). Additionally, Al-Barghouthi et al. attempted to validate their data using information on bone mineral density from knockout mice available through the International Mouse Phenotype Consortium (Dickinson et al., 2016; Swan et al., 2020). This consortium had available knockouts for 142 out of the 512 genes. Of these, 64 mutants led to alteration of whole-body bone mineral density. An exciting finding was that a large percentage (76.5%) of genes that cause changes in bone mineral density in mice knockouts did not belong to a ‘known bone gene’, which gives us a hint about the complexity of the biological mechanisms that regulate bone mineral density.
Finally, Al-Barghouthi et al. tightened their analysis to capture genes with the strongest evidence for causality. PPP6R3 was the second top gene identified and the first without functional evidence for bone mineral density. Surprisingly, the associated locus that PPP6R3 belongs to is very well known, as it harbors a well-studied bone gene called LRP5. The question therefore arises: is PPP6R3 a new gene in bone mineral density regulation, or is the GWAS signal associated with LRP5? To answer this question, Al-Barghouthi et al. generated mice knockouts for Ppp6r3, the mouse version of the gene, and then measured their bone mineral density at the femur and the lumbar spine. They found that in addition to showing lower bone mineral density at the spine, the Ppp6r3 knockout mice demonstrated deterioration of bone microarchitecture and increased bone turnover, which is commonly observed in models of osteoporosis.
One limitation of the work of Al-Barghouthi et al. is that the GTEx project does not contain expression data from bone tissues. However, due to the complex nature of bone mineral density, new genes can still be identified using their approach. Of course, questions still remain regarding the direct or indirect effect of genes on bone mineral density. For example, how does PPP6R3 regulate osteoblast or osteoclast activity? Another important finding of the latest work is that many of the genes potentially causing osteoporosis are in gene-rich regions of the genome. Indeed, as PPP6R3 and LRP5 demonstrate, a single associated genomic region may harbor more than one causative gene.
While a follow up from this work is necessary to test the causality of other identified genes using animal models, it is equally important to explore if and how PPP6R3 contributes to fracture risk and osteoporosis. In the future, approaches like the one used by Al-Barghouthi et al. will allow researchers to slowly learn more about the genetic causes of osteoporosis, which could lead to better treatments.
References
-
The genotype-tissue expression (GTEx) projectNature Genetics 45:580–585.https://doi.org/10.1038/ng.2653
-
The genetic overlap between osteoporosis and craniosynostosisFrontiers in Endocrinology 13:1020821.https://doi.org/10.3389/fendo.2022.1020821
-
The global prevalence of osteoporosis in the world: a comprehensive systematic review and meta-analysisJournal of Orthopaedic Surgery and Research 16:609.https://doi.org/10.1186/s13018-021-02772-0
-
Genetic architecture: the shape of the genetic contribution to human traits and diseaseNature Reviews Genetics 19:110–124.https://doi.org/10.1038/nrg.2017.101
Article and author information
Author details
Publication history
Copyright
© 2022, Kague
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,016
- views
-
- 88
- downloads
-
- 1
- citation
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 1
- citation for umbrella DOI https://doi.org/10.7554/eLife.85161
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Osteoporosis: Finding the genes for fragile bones
eLife 11:e85161.
https://doi.org/10.7554/eLife.85161




{kind=link}