Analysis of the crystal structure of an active MCM hexamer
- St Jude Children's Research Hospital, United States
Abstract
In a previous Research article (Froelich et al., 2014), we suggested an MCM helicase activation mechanism, but were limited in discussing the ATPase domain because it was absent from the crystal structure. Here we present the crystal structure of a nearly full-length MCM hexamer that is helicase-active and thus has all features essential for unwinding DNA. The structure is a chimera of Sulfolobus solfataricus N-terminal domain and Pyrococcus furiosus ATPase domain. We discuss three major findings: 1) a novel conformation for the A-subdomain that could play a role in MCM regulation; 2) interaction of a universally conserved glutamine in the N-terminal Allosteric Communication Loop with the AAA+ domain helix-2-insert (h2i); and 3) a recessed binding pocket for the MCM ssDNA-binding motif influenced by the h2i. We suggest that during helicase activation, the h2i clamps down on the leading strand to facilitate strand retention and regulate ATP hydrolysis.
https://doi.org/10.7554/eLife.03433.001Introduction
Hexameric MCM rings act as the replicative DNA helicase (Bochman and Schwacha, 2008; Ilves et al., 2010), encircling the leading strand DNA template at the replication fork (Fu et al., 2011). Mcm2-7 complexes are loaded (reviewed in Remus and Diffley, 2009) to encircle double-stranded DNA (dsDNA) via a ‘gate’ between Mcm2 and Mcm5 (Bochman and Schwacha, 2007, 2008; Costa et al., 2011) to yield a double hexamer (Evrin et al., 2009; Remus et al., 2009) that does not unwind DNA. During helicase activation, the Dbf4-dependent Cdc7 kinase (DDK) and cyclin-dependent kinases (CDKs) drive recruitment of Cdc45 and the GINS complex (Labib, 2010). These factors stimulate the Mcm2-7 ATPase and helicase (Ilves et al., 2010) and with Mcm2-7 form the CMG complex (Cdc45-Mcm2-7-GINS), the active replicative helicase (Moyer et al., 2006; Bochman and Schwacha, 2008; Ilves et al., 2010). Following activation, two Mcm2-7 helicases encircle single-stranded DNA (ssDNA) and translocate independently (Yardimci et al., 2010), 3ʹ→5ʹ, on the leading strand DNA template (Fu et al., 2011) with the ATPase domain leading (McGeoch et al., 2005).
Our previous crystal structure of the Pyrococcus furiosus MCM N-terminal domain (PfMCMN) bound to ssDNA revealed an MCM single-stranded binding motif (MSSB) that binds ssDNA (Froelich et al., 2014). Our discussion of an MSSB role in helicase activation invoked action of the AAA+ (reviewed in Duderstadt and Berger, 2013) ATPase domain to translocate DNA, but we could not discuss specifically how the MSSB was affected by the AAA+ domain because it was not present in the PfMCMN:ssDNA structure. Now, we present the crystal structure of a helicase-active MCM hexamer to reveal a novel conformation for the A-subdomain that could play a role in MCM regulation and how the AAA+ helix-2-insert is tethered to the N-terminal domain to create a recessed binding pocket for the MSSB.
Results and discussion
We identified a chimera of the N-terminal domain of Sulfolobus solfataricus (Sso) and the AAA+ domain of PfMCM, Sso-PfMCM (Figure 1A) with a robust DNA unwinding activity (Figure 1B; Figure 1—figure supplement 1). We present an analysis of the crystal structure of the Sso-PfMCM hexamer bound to Mg/ADP (Table 1).
Figure 1 with 3 supplements see all
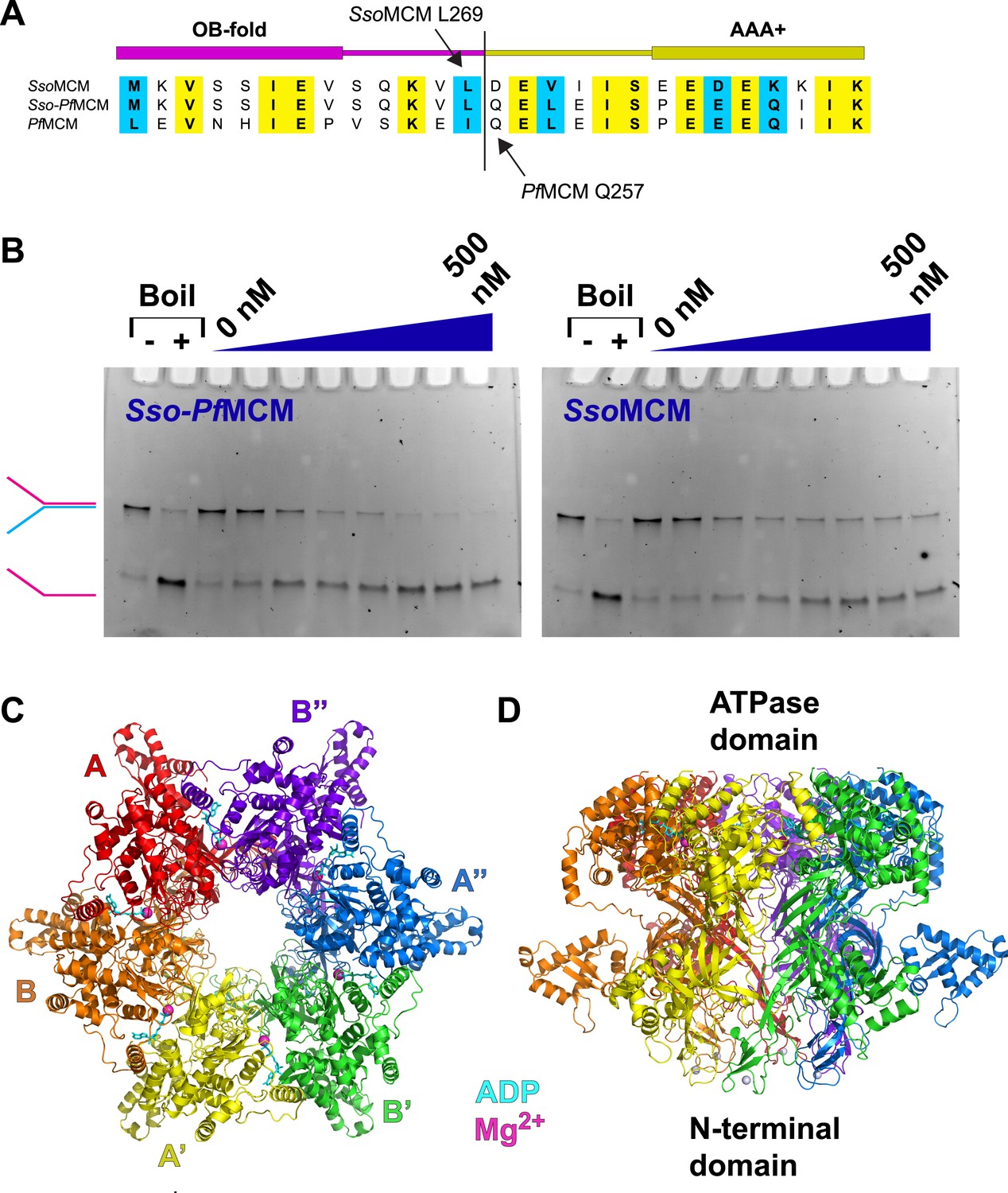
Properties of Sso-PfMCM.
(A) Sequence alignment showing the construction of the Sso-PfMCM chimera. The N-terminal domain of SsoMCM (residues 1–269, top sequence) was fused to the AAA+ domain of PfMCM (starting at residue 257, bottom sequence) to yield the chimera (middle sequence). (B) The Sso-PfMCM chimera shows enhanced unwinding activity when compared to wild-type SsoMCM. Helicase reactions were performed at 69°C for 60 min with a Y-shaped DNA substrate with a 5ʹ-fluorescein label on one strand. Unwinding reactions were in the presence of 4 mM ATP and contained 0, 50, 100, 150, 200, 300, 400, or 500 nM protein. Views of the Sso-PfMCM hexamer crystal structure parallel (C) and perpendicular (D) to the central channel with each subunit uniquely colored. The magnesium ions are magenta spheres, and ADP molecules are shown as cyan stick. (C) View down the crystallographic threefold axis with the unique and symmetry-derived chains labeled. The ATPase domains are projected out of the page. (D) View perpendicular to the channel axis. The ATPase domains are located at the top, and the N-terminal domains are located at the bottom. The Zn ions are light grey spheres at the bottom.
Table 1
Data collection and refinement statistics
| Sso-PfMCM:MgADP Hexamer | PfMCMAAA:MgADP Double-octamer | |
|---|---|---|
| Data collection | ||
| Space group | P63 | P1 |
| Cell dimensions | ||
| a, b, c (Å) | 118.902, 118.902, 199.317 | 124.956, 127.082, 128.025 |
| α, β, γ (°) | 90, 90, 120 | 71.852, 72.819, 80.392 |
| Resolution (Å) | 50–2.70 (2.80–2.70) | 50–3.80 (3.94–3.80) |
| Rsym | 0.107 (0.750) | 0.169 (0.429) |
| I/σI | 14.8 (1.79) | 8.3 (2.41) |
| Completeness (%) | 99.8 (98.3) | 98.9 (97.0) |
| Redundancy | 6.8 (5.0) | 3.3 (2.6) |
| Refinement | ||
| Resolution (Å) | 50–2.70 (2.80–2.70) | 50–3.80 (3.94–3.80) |
| No. reflections | 39,044/1976 (2042/123) | 69,126/3486 (6206/357) |
| Rwork/Rfree | 0.263/0.295 (0.360/0.353) | 0.301/0.314 (0.367/0.368) |
| No. atoms | ||
| Protein | 9432 | 2429 (1/16 of ASU) |
| ADP | 54 | 27 (1/16 of ASU) |
| ions | 10 | 1 (1/16 of ASU) |
| Water | 0 | 0 |
| B-factors | ||
| Protein | 60 | 91 |
| ADP | 118 | 72 |
| ions | 75 | 81 |
| Water | N/A | N/A |
| R.m.s. deviations | ||
| Bond lengths (Å) | 0.010 | 0.010 |
| Bond angles (°) | 1.488 | 1.597 |
MCM:Mg/ADP hexamer crystal structure
The Sso-PfMCM hexamer (Figure 1C–D; Video 1, 0:00) forms a ring with a channel large enough to accommodate double-stranded DNA (dsDNA) (see ‘Materials and methods’). The Sso-PfMCM structure therefore could mechanistically represent the structure of the MCM hexamer prior to loading (no DNA), after it loads to encircle dsDNA, or after its activation (encircling ssDNA). The two-tiered structure is consistent with electron microscopy studies (Chong et al., 2000; Pape et al., 2003; Gomez-Llorente et al., 2005; Costa et al., 2006; Bochman and Schwacha, 2007; Remus et al., 2009; Costa et al., 2011). The N-terminal tier has three subdomains, A–C (Fletcher et al., 2003; Liu et al., 2008; Froelich et al., 2014) with the A-subdomains in a different orientation (Figure 2A; Video 1, 0:20; see below) than observed previously. The relative positions of MCMN and MCMAAA differ considerably compared to monomeric and filament MCM crystal structures (Brewster et al., 2008; Bae et al., 2009; Slaymaker et al., 2013) (Figure 1—figure supplement 2). This difference is needed to prevent serious clashes that would occur among the ATPase domains.
Video 1
Crystal structure details for Sso-PfMCM.
The video illustrates the arrangement of the subunits in the hexamer and the positions of the subdomains. The A-subdomain conformation is animated to transform to that observed in other crystal structures of MCMN to illustrate how they differ. The different A-subdomain conformations correlate with the conformation of a proline (P104) at the junction between the A- and C-subdomains. The relative position of the mcm5-bob1 mutation is noted. Several central channel modules are highlighted, including the ps1β, h2i, β-turn, MSSB, and the interaction of ACL Q198 with the h2i. The ATPase site is compared to that of papillomavirus E1 (Enemark and Joshua-Tor, 2006), and several key residues are highlighted for MCM. The MSSB location is shown in a surface representation to illustrate that it sits at a recessed binding pocket where the ssDNA (green) of the aligned PfMCMN:ssDNA crystal structure (Froelich et al., 2014) would position snugly.
Figure 2 with 1 supplement see all
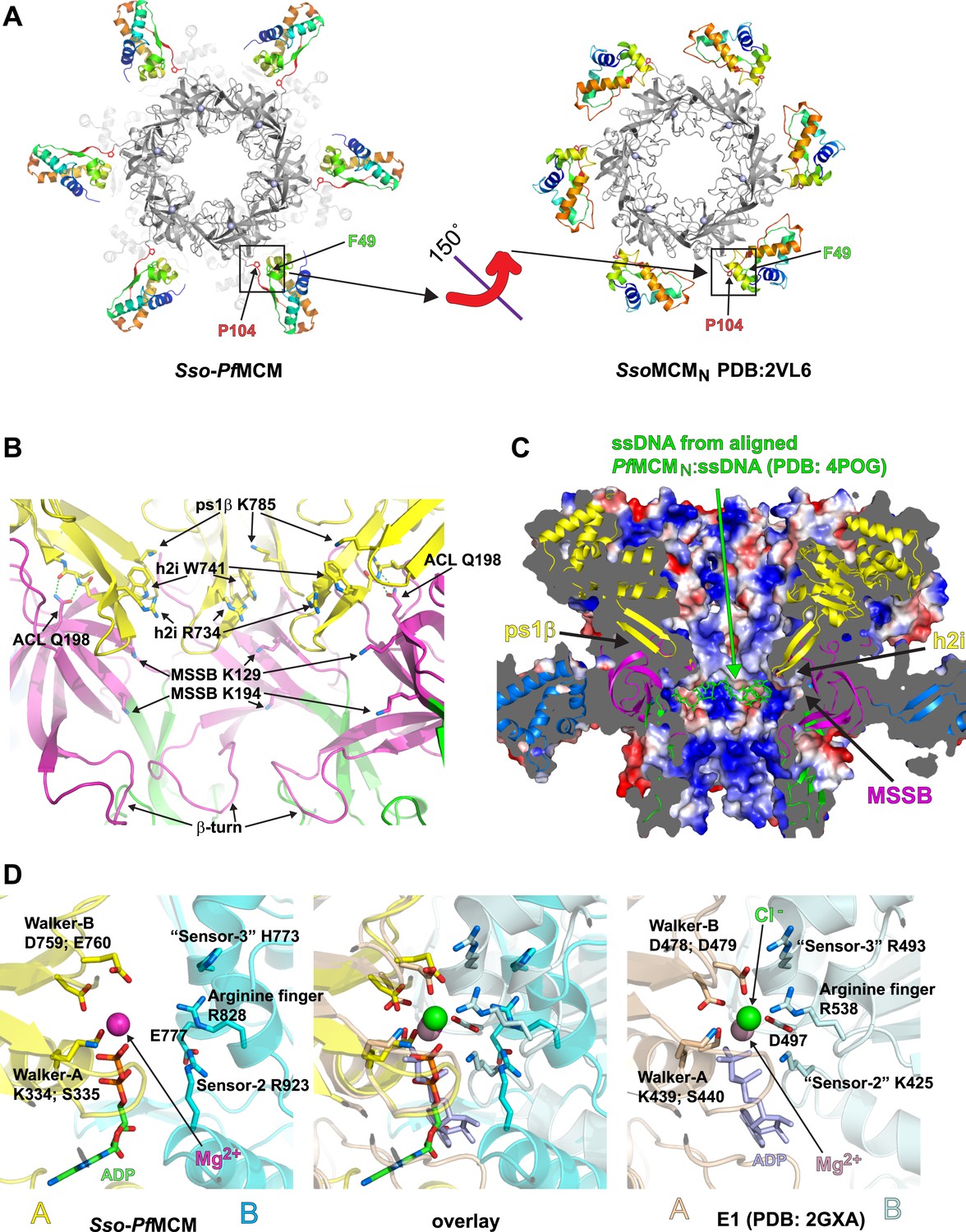
Sso-PfMCM crystal structure details.
(A) The A-subdomains (rainbow) of Sso-PfMCM adopt a unique conformation that is rotated 150° compared to other crystal structures of hexameric MCMN such as SsoMCMN (Liu et al., 2008). The distinct conformations correlate with the conformation of P104 that is located at the junction between the A- and C-subdomains (boxed). In both conformations, P104 packs against the aromatic residue F49. (B) The modules of the central channel. The cartoon is colored with the AAA+ domain in yellow, the Zn-binding B-subdomain in green, and the OB-fold C-subdomain in magenta. The ps1β projects a conserved lysine, K785, into the channel. This lysine packs against W741 of the h2i, which sits adjacent to R734 of the h2i. The MSSB is recessed and sits below the h2i and above the β-turn. The N-terminal domain is tethered to the h2i by a universally conserved glutamine in the ACL, Q198. (C) Surface representation of Sso-PfMCM colored by electrostatic potential. The surface is clipped with a vertical plane through the center to illustrate the central channel features. A cartoon representation of the protein with select modules labeled is colored as in Figure 2B with the helical A-subdomain in blue. The MSSB sits at a recessed pocket. The ssDNA from the aligned PfMCMN:ssDNA structure (Froelich et al., 2014) would be snugly positioned in this pocket. (D) Comparison of the Sso-PfMCM ATPase site (left) with that of E1 (Enemark and Joshua-Tor, 2006, right). The Walker-A and Walker-B residues of one subunit (yellow) are positioned at the left side of the site while three positively charged residues of the adjacent subunit (cyan) line the right side of the site. An acidic residue of the cyan subunit sits below the site. Based on the superposition (middle), we predict that the MCM subunits need to approach each other more closely to generate a competent ATPase site.
A-subdomain is in a different orientation than in previous structures
The A-subdomains of Sso-PfMCM are rotated 150° compared to those in SsoMCMN (Liu et al., 2008) (Figure 2A). The previously identified A-subdomain conformation seems fully possible in our present structure (see Video 1, 0:20), and we suggest that different A-subdomain conformations might play a role in MCM regulation. A similar A-subdomain rotation has been suggested for Methanothermobacter thermautotrophicus (Mt) MCMN based on electron microscopy (Chen et al., 2005) and was suggested to dictate helicase activity (Chen et al., 2005). We suggest the different conformations might regulate interaction with other factors: the A-subdomain might be stabilized against the OB-fold during one cell cycle stage, masking an interaction surface that becomes exposed by a conformational switch. In Sso-PfMCM, the change correlates with a conformer change in P104 at the junction of the A/C-subdomains (phi/psi = −48.5/133.0 in Sso-PfMCM vs −78.0/−14.9 in SsoMCMN, Liu et al., 2008). This proline and an associated aromatic residue, F49 (Figure 2A; Video 1, 0:35), are conserved in Mcm2 and Mcm6 (Video 1, 0:47), suggesting these subunits could be particularly specialized conformational switches. Interestingly, the A-subdomain of Mcm2 interacts with Cdc45 (Costa et al., 2011), providing a potential link between A-subdomain conformation and CMG assembly. While we expect A-subdomain conformations to be more flexible without a proline, proline is not required to attain the conformation in our structure, and the corresponding residue in MtMCM is a serine. As noted previously (Chen et al., 2005), A-subdomain rotation is a conceptual extension of the ‘domain-push’ mechanism described for mcm5-bob1 (Hardy et al., 1997) where the A- and C-subdomain interaction is weakened by bulky side-chains (Fletcher et al., 2003). In our structure, as suggested for MtMCM (Chen et al., 2005), the A/C subdomain interaction is not only weakened, it is broken altogether. Changes in A-subdomain conformation might be driven by MCM phosphorylation, such as phosphorylation of the ScMcm4 N-terminal serine/threonine-rich domain by DDK (Sheu and Stillman, 2006) that serves both inhibitory and facilitating roles in replication (Sheu and Stillman, 2010).
Central channel modules
Several modules are directed into the central channel where they could interact with encircled DNA (Figure 2B; Video 1, 1:19). The pre-sensor-1-β-hairpin (ps1β) projects a universally conserved lysine, K785, that is essential for unwinding by SsoMCM (McGeoch et al., 2005). The helix-2-insert (h2i), required for helicase activity in MtMCM (Jenkinson and Chong, 2006), prominently directs R734 and W741 into the central channel. These residues are conserved in a family-specific fashion for Mcms (Video 1, 1:32). The h2i and ps1β are located further from the N-terminal domain than predicted by monomeric crystal structures (Figure 1—figure supplement 2) due to interdomain differences (see above). The h2i projects further into the channel than the ps1β and appears to divide the AAA+ and N-terminal DNA-binding regions. As viewed in Figure 2B, the ps1β and the h2i direct their putative DNA-binding residues above the h2i, while the MSSB is below the h2i. The h2i creates a DNA-binding pocket at the MSSB where ssDNA was observed previously (Froelich et al., 2014). In SsoMCM, alanine mutants of lysine residues in this pocket (K129A and K194A) show severe DNA-binding and unwinding defects (Pucci et al., 2004). While the MSSB pocket of Sso-PfMCM appears poised to bind ssDNA in the fashion observed previously (Figure 2C), some remodeling of h2i side-chains or the ssDNA would be necessary to avoid clashes, particularly involving F737.
Allosteric Communication Loop
In each subunit of our Sso-PfMCM hexamer crystal structure, residues 198–212 comprise a conserved loop of the OB-fold that projects towards the h2i of the same subunit and the ps1β of an adjacent subunit. This loop has been termed the ‘Allosteric Communication Loop’ (ACL) (Barry et al., 2009) due to its predicted proximity to the AAA+ domain and its observed influence on unwinding (Sakakibara et al., 2008; Barry et al., 2009). Although the ATPase domain alone is sufficient to generate DNA unwinding in SsoMCM (Barry et al., 2007; Pucci et al., 2007) and in Aeropyrum pernix MCM (Atanassova and Grainge, 2008), several mutants located on this loop show unwinding defects (Sakakibara et al., 2008; Barry et al., 2009). In our hexamer structure, the proximity of the ACL to the h2i and ps1β, highly significant AAA+ modules (see above), strongly supports an ACL role in N- and C-terminal domain communication. The ACL position near the ps1β of an adjacent subunit is also consistent with previous studies (Barry et al., 2009).
We tested the DNA unwinding activity of PfMCMAAA to compare with that of the Sso-PfMCM chimera to explore interdomain communication. We found PfMCMAAA had a negligible unwinding activity (Figure 1—figure supplement 3). The inactivity of PfMCMAAA could result from attributes identified in the PfMCMAAA double-octamer crystal structure (see ‘Materials and methods’). Specifically, the non-hexameric ring architecture or the alternative topology for the helix-2-insert region, both of which we consider artifacts of the N-terminal truncation, could fully explain the lack of unwinding by this domain. Thus, although SsoMCMN does enhance the unwinding activity of PfMCMAAA, this could simply be due to enforcing a hexameric architecture or by disallowing the unusual h2i topology in favor of the canonical topology.
We next examined the Sso-PfMCM structure for interdomain interactions that could be conserved in native proteins. The structure reveals a fully conserved glutamine, Q198, of the ACL interacts with the main-chain amide atoms of the h2i (Figure 2B; Video 1, 1:50). This interaction could occur in any MCM protein because the constituent atoms are fully conserved. The Q198:h2i interaction is not required for DNA unwinding because no unwinding defects are observed for the Q198A mutant (Figure 1—figure supplement 1), and in the corresponding alanine mutant of MtMCM (Sakakibara et al., 2008). We suggest that the glutamine-h2i interaction functions prior to unwinding to lock the h2i in a holding position. The movement of DNA proposed previously to facilitate initial strand separation (Froelich et al., 2014) could occur by ATP-hydrolysis-driven inward movement of the h2i bound to DNA (Figure 2—figure supplement 1). After the h2i modules reach the position observed in our structure, they would be locked in place by interaction with Q198. This would tighten the grasp on one strand while the opposing strand exits the open Mcm2/5 gate (Bochman and Schwacha, 2007; Costa et al., 2011). With the h2i modules fixed in this position, the ATPase sites would be unable to adopt a productive hydrolysis conformation because each ATPase site is fundamentally tied to h2i position, potentially to prevent further ATPase activity during an important activation event. ATP hydrolysis inhibition by fixed h2i is directly analogous to inhibition of ATP hydrolysis in ϕ12 P4, an RNA-translocating hexamer, by cross-linked RNA-binding loops (Kainov et al., 2008).
ATPase active site
The ATPase active site structurally resembles the ATPase site of the AAA+ helicase E1 (Enemark and Joshua-Tor, 2006) (Figure 2D, Video 1, 2:04) with Walker-A/B residues (Abrahams et al., 1994; Neuwald et al., 1999) of one subunit, and three positively charged residues of the adjacent subunit. The three positive residues consist of sensor-2 (Neuwald et al., 1999), the arginine finger (Neuwald et al., 1999), and residues that we classify as sensor-3 (Enemark and Joshua-Tor, 2006). Although not typical for AAA+ proteins, the placement of sensor-2 in an ATPase site among Walker-A/B residues of the neighboring subunit (in trans) was predicted for MCM proteins in defining AAA+ Clade 7 (Erzberger and Berger, 2006). Biochemical experiments (Moreau et al., 2007) and MCM structure-based predictions (Bae et al., 2009) are also consistent with this arrangement. Based on comparison of the ATPase site with the tight ‘ATP-like’ configuration of E1 (Enemark and Joshua-Tor, 2006), we expect MCM subunits must approach more closely to generate a competent ATPase site. In this state, the h2i and ps1β are predicted to move upward in the view shown in Figure 2—figure supplement 1 to increase the distance between the ACL and the ps1β as identified by DEER-spectroscopy (Barry et al., 2009). ATP-hydrolysis would drive the h2i/ps1β downward to translocate one ssDNA strand with expected polarity and orientation (McGeoch et al., 2005) while the complementary strand is excluded from the ring (Fu et al., 2011). Our present structural findings cannot differentiate several mechanistic details such as hydrolysis order or timing (reviewed in Singleton et al., 2007). We speculate that the MCM helicase unwinds DNA with helically-arranged h2i/ps1β modules analogous to E1 (Enemark and Joshua-Tor, 2006) and Rho (Thomsen and Berger, 2009), but the six non-identical subunits of Eukaryotic Mcm2-7 could operate asymmetrically during unwinding. Indeed, the ATPase modules of the different Mcm2-7 subunits show distinct roles and specialization during different functional stages (Coster et al., 2014; Kang et al., 2014). Elucidation of how the AAA+ domain interacts with DNA in an unwinding conformation will help reveal more details of the MCM unwinding mechanism.
Materials and methods
Cloning, mutagenesis, expression, and purification
Request a detailed protocolThe chimera protein construct consists of SsoMCMN, (SsoMCM aa 1–269) fused to PfMCMAAA (PfMCM aa 257–361/729–966 = aa 257–966 with its intein, aa 362–728, removed). It corresponds to a full-length MCM protein lacking the short (aa 967–1049) C-terminal helix-turn-helix domain (Aravind and Koonin, 1999) that is dispensible for unwinding activity in MtMCM (Jenkinson and Chong, 2006) and in SsoMCM (Barry et al., 2007). All expression constructs were prepared as N-terminal His6-SUMO fusions. The original SUMO vector was the generous gift of Dr Christopher D Lima (Mossessova and Lima, 2000). The PfMCM gene contains an intein, aa 362–728 (Yoshimochi et al., 2008) in the helix-2-insert region of ATPase domain. We genetically removed the intein by sequentially cloning two fragments (amplified from P. furiosus genomic DNA, ATCC) incorporating a silent NotI mutation at the junction. This plasmid served as the PCR template to generate constructs of Sso-PfMCM (SsoMCM aa 1–269/PfMCM aa 257–361/729–966, pJM001.3) and PfMCMAAA (aa 263–361/729–966, pEE021.1 = crystallized construct; and PfMCM aa 252–361/729–1049 = construct of unwinding experiments). The Sso-PfMCM chimera construct was generated by overlap extension of PCR fragments encoding the N-terminal domain of SsoMCM (amplified from S. solfataricus genomic DNA, ATCC) and PfMCMAAA that had 93 bases of overlap at the junction. The full-length SsoMCM expression construct (pEE045.1) was prepared via PCR amplification of the full SsoMCM gene. The Q198A mutant (pJM005.5) was generated by site-directed mutagenesis of pJM001.3. DNA sequencing verified the integrity of the coding region of all constructs. Proteins were purified as described previously (Froelich et al., 2014), including removal of the SUMO tag by digestion by Ulp1 protease (the Ulp1 protease plasmid was the generous gift of Dr Christopher D Lima) (Mossessova and Lima, 2000).
Crystallization, data-collection, structure-solution, and refinement
Request a detailed protocolPrior to crystallization, purified Sso-PfMCM was dialyzed into buffer containing 25 mM HEPES, pH 7.6; 10 mM NaCl; and 5 mM Mg(OAc)2. Crystals of Sso-PfMCM with Mg/ADP grew by hanging drop by mixing 2 μl of protein:ADP solution (10.8 mg/ml Sso-PfMCM; 5 mM ADP) and 2 μl of well solution (100 mM HEPES, pH 7.6; 350 mM MgCl2; 3% (wt/vol) PEG 3350). Crystals were cryoprotected by quickly passing through a 1:3 ethylene glycol:well solution and flash frozen in liquid nitrogen. Data were collected at SER-CAT beamline 22-ID. Data were collected at 1.0 Å wavelength in 0.25° oscillations for 112.5° at a temperature of 100 K. All data were scaled and integrated using the HKL-2000 software package (Otwinowski and Minor, 1997) to 2.70 Å resolution.
The structure was solved in space group P63 by the program Phaser (McCoy et al., 2007), which placed two copies of PfMCMAAA (see below) and two copies of a monomer of SsoMCMN, PDB 2VL6 (Liu et al., 2008), in a single hexamer on a crystallographic threefold axis. Overall, the unit cell contains two nearly sixfold symmetric hexamers offset by a strong NCS translation of [1/3, 2/3, 1/2] (Patterson peak height 50% of origin). Initial electron density maps revealed a clear misplacement of the helical A-subdomain, which was corrected by Phaser (McCoy et al., 2007) by using separate search models for the A-subdomain and the B/C-subdomains. The initial electron density map was greatly improved by multi-crystal electron density averaging in the AAA+ domain region with the program Dmmulti (Cowtan, 1994) by implementing twofold averaging of Sso-PfMCM and 16-fold averaging of PfMCMAAA. The model was refined at various stages with CNS (Brunger et al., 1998; Brunger, 2007), phenix (Afonine et al., 2012), and refmac5 (Vagin et al., 2004) and manually improved with Coot (Emsley and Cowtan, 2004). The final refinement was carried out in CNS (Brunger et al., 1998; Brunger, 2007). A Ramachandran plot calculated by Procheck (Laskowski et al., 1993) indicated the following statistics: core: 917 (86.8%); allowed: 124 (11.7%); generously allowed: 15 (1.4%); disallowed: 0 (0%). Figures were prepared with PyMOL (Schrodinger, 2010), Molscript (Kraulis, 1991), and Raster3D (Merritt and Bacon, 1997).
Crystallographic datasets for crystals grown in the presence of several nucleotide cofactors (ADP, AMP-PNP, ATP-γS, ADP-AlFx) were collected. All were strongly isomorphic with crystals grown with Mg/ADP, and no evidence of a γ-PO4 or its analog was ever detected in resulting electron density maps. We therefore conclude that all crystal datasets adopt a highly similar structure most consistent with an ADP-bound state and that the γ-PO4/analog hydrolyzes over the course of crystallization or is crystallographically disordered.
The crystal structure of PfMCMAAA was pivotal in obtaining the crystal structure of Sso-PfMCM (above), and we therefore include the details of its structure determination. However, the resolution of PfMCMAAA, (3.80 Å) limits the overall detail of the structure. Crystals of PfMCMAAA with Mg/ADP grew by hanging drop by mixing 2 μl protein/Mg/ADP (6 mg/ml; 5 mM ADP; 50 mM MgCl2; 18 mM HEPES, pH 7.6; 180 mM NaCl; 4.5 mM β-mercaptoethanol) with 2 μl well solution (50 mM sodium cacodylate, pH 6.0; 50 mM magnesium acetate; 30% MPD; 5% glycerol). A Crystal was flash frozen in liquid nitrogen and data were collected at SER-CAT beamline 22-BM at 1.0 Å wavelength in 0.5° oscillations for 360° at a temperature of 100 K. The data were scaled and integrated using the HKL-2000 software package (Otwinowski and Minor, 1997) to 3.80 Å resolution. A weak molecular replacement solution was obtained with the program Phaser (McCoy et al., 2007), which placed 16 monomers of the AAA+ portion of PDB 4FDG (Slaymaker et al., 2013) as a double-octamer. The MCM complex is not presumed to adopt an octameric assembly in vivo, but we note that a hypothetical hexamer generated by removing two adjacent subunits from the octameric ring would correspond to an open hexameric ring with an opening large enough to permit entry of B-form dsDNA. Initial electron density maps were greatly improved by 16-fold NCS-averaging and solvent flattening with the program Resolve (Terwilliger, 2000, 2004), which revealed obvious side-chain positions. The sequence was assigned to the structure based upon the location of selenium positions (7 per subunit) for a selenomethionine derivative in an NCS-averaged anomalous difference Fourier map generated with the Resolve-improved phases, and by alignment with the 1.90 Å resolution structure of a monomeric MCM homolog (Bae et al., 2009). The selenomethionine derivative was expressed in B834(DE3) cells (EMD Millipore, Darmstadt, Germany) in LeMaster's media (Hendrickson et al., 1990), and anomalous difference data were collected at SER-CAT beamline 22-ID at 0.97915 Å wavelength in 0.5° oscillations for 260° at a temperature of 100 K. The data were scaled and integrated using the HKL-2000 software package (Otwinowski and Minor, 1997) to 4.0 Å resolution. The anomalous signal was too weak to generate starting phases, but readily identified the selenium positions by an NCS-averaged anomalous difference fourier map generated with the Resolve-improved molecular replacement phases (see above). The structure was refined with a strict 16-fold NCS protocol in CNS (Brunger et al., 1998; Brunger, 2007) and manually improved with Coot (Emsley and Cowtan, 2004). Following refinement of the higher resolution structure of Sso-PfMCM (above), the coordinates were updated, and the h2i region was rebuilt. The structure was subjected to coordinate and group B-factor refinement with strict 16-fold NCS in CNS (Brunger et al., 1998; Brunger, 2007) to yield the final model. The h2i is folded differently than in other AAA+ proteins to mediate a β-sheet interface with the ps1β of a subunit in the other octamer. Twofold symmetric dimers are arranged around an eightfold symmetry axis to yield approximate D8-symmetry. A total of 16 of these β-sheet interfaces occur around the ring. This h2i structure is not compatible with the position of the N-terminal domain seen in Sso-PfMCM, and it is therefore almost certainly an artifact of removing the N-terminal domain in the PfMCMAAA construct. The precise sequence registry for these h2i residues is not clearly defined, and the residues have been modeled as poly-alanine in our best assessment of the polypeptide direction. A Ramachandran plot calculated by Procheck (Laskowski et al., 1993) indicated the following statistics: core: 213 (74.7%); allowed: 56 (19.6%); generously allowed: 8 (2.8%); disallowed: 8 (2.8%).
Helicase assay
Request a detailed protocolAll unwinding experiments were performed with a Y-shaped DNA substrate with a 55-mer double-stranded region, a 50-mer poly-dT 3ʹ-arm, and a 30-mer 5ʹ-arm with a fluorescein label at the 5ʹ- end. The substrate was prepared by annealing a 5ʹ-fluorescein-labeled oligonucleotide (5ʹ-TTGAACCACCCCCTTGTTAAATCACTTCTACTTGCATGCCTGCAGGTCGACTCTAGAGGATCCCCGGGTACCGAGCTCGAATTCG–3ʹ with an unlabeled oligonucleotide (5ʹ- CGAATTCGAGCTCGGTACCCGGGGATCCTCTAGAGTCGACCTGCAGGCATGCAAGTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTTT-3ʹ), Sigma–Aldrich, St. Louis, MO). The 85-mer oligonucleotide was identical to a previously published substrate that had been annealed to M13 plasmid ssDNA to study SsoMCM unwinding (Pucci et al., 2004). Helicase activity assays were prepared in 20 μl reaction mixtures with 25 mM HEPES (pH = 7.6), 100 mM Na(OAc), 5 mM Mg(OAc)2, 4 mM ATP, and 3.7 nM fluorescein-labeled DNA substrate. For protein concentration titrations, the protein concentration ranged from 0 to 500 nM (monomer), and reactions were incubated at 69°C for 60 min. For time-course experiments, 500 nM protein was incubated at 69°C for 1 to 90 min. Prior to the addition of ATP to initiate unwinding, time points were incubated at 69°C for 5 min to allow for thermal equilibration. For all samples, reactions were stopped by the addition of 5 μl of loading buffer containing 40% (vol/vol) glycerol, 5% (wt/vol) sodium dodecyl sulfate (SDS), and 50 mM ethylenediaminetetraacetic acid (EDTA), and a 20 μl aliquot was loaded on a 4–20% 1× TBE gradient PAGE gel (Biorad, Berkeley, CA) and run at 150 V for 90 min. Gel imaging was performed with a Fuji LAS-4000 using a 15-min exposure time and a SYBR-Green filter.
Definition of central channel axis, mathematical analysis
Request a detailed protocolWe conclude that the central channel of our structure of the Sso-PfMCM hexamer is sufficiently large to accommodate dsDNA based on a channel radius minimum of 12.7 Å for its polyalanine model. For comparison, the polyalanine model of BPV E1:ssDNA (encircles ssDNA; Enemark and Joshua-Tor, 2006; Lee et al., 2014) has a minimum radius of 6.1 Å (PDB: 2GXA, hexamer 1, Enemark and Joshua-Tor, 2006); topoisomerase I:dsDNA (tightly encircles dsDNA) has a minimum radius of 7.5 Å (PDB: 1A35, Redinbo et al., 1998); and PCNA (encircles dsDNA but can slide) has a minimum radius of 16.0 Å (PDB: 1PLQ, Krishna et al., 1994). Based on these comparisons, the channel diameter in our structure of Sso-PfMCM is large enough to accommodate dsDNA, but might not slide over dsDNA as readily as PCNA. The details of these calculations are provided below.
Our analysis of the central channel of a ring requires a definition of the channel axis. We define this axis as the rotation axis for least-squares permutation of the subunits. For Sso-PfMCM, this axis coincides with a crystallographic threefold axis. For straightforward analysis, we use the following procedure, which is general, to produce a transformed PDB coordinate file with the channel axis coincident with the [0, 0, Z] axis of the standard PDB coordinate system. First, the coordinates of a full ring molecule were translated to place the center-of-mass on the origin of the standard PDB coordinate system with the program MOLEMAN (Kleywegt, 1997). Second, the least-squares rotation that explicitly permutes the subunits (for example, chains ABCDEF onto chains BCDEFA in a hexamer) was calculated by LSQMAN (Kleywegt, 1996), expressed in polar angles (Omega = O, Phi = P, Chi = C). These polar angles were used to transform the origin-shifted coordinates in two successive polar rotation operations with the program MOLEMAN (Kleywegt, 1997). The first polar rotation was by (0, 0, −P), and the second was by (90, 90, −O). Subsequently, the distance of any atom to the channel axis can be calculated from its X and Y coordinates in the transformed PDB coordinate file as the square root of (X2 + Y2). For Topoisomerase I (PDB id: 1A35, Redinbo et al., 1998), the channel axis was defined as the least-squares helical axis of the bound dsDNA, and this axis was transformed to coincide with the [0, 0, Z] axis of the PDB coordinate system as described above.
Data availability
-
Crystal structure of an active MCM hexamer (SsoPfMCM hexamer)Publicly available at RCSB Protein Data Bank.
-
PfMCM-AAA double-octamerPublicly available at RCSB Protein Data Bank.
-
Crystal structure of papillomavirus E1 hexameric helicase with ssDNA and MgADPPublicly available at RCSB Protein Data Bank.
-
Human topoisomeras I/DNA complexPublicly available at RCSB Protein Data Bank.
-
Crystal structure of the eukaryotic DNA polymerase processivity factor PCNAPublicly available at RCSB Protein Data Bank.
-
Structural analysis of the Sulfolobus solfataricus MCM protein n- terminal domainPublicly available at RCSB Protein Data Bank.
-
Crystal Structure of an Archaeal MCM FilamentPublicly available at RCSB Protein Data Bank.
References
-
Towards automated crystallographic structure refinement with phenix.refineActa Crystallographica Section D, Biological Crystallography 68:352–367.https://doi.org/10.1107/S0907444912001308
-
DNA-binding proteins and evolution of transcription regulation in the archaeaNucleic Acids Research 27:4658–4670.https://doi.org/10.1093/nar/27.23.4658
-
Intersubunit allosteric communication mediated by a conserved loop in the MCM helicaseProceedings of the National Academy of Sciences of USA 106:1051–1056.https://doi.org/10.1073/pnas.0809192106
-
Archaeal MCM has separable processivity, substrate choice and helicase domainsNucleic Acids Research 35:988–998.https://doi.org/10.1093/nar/gkl1117
-
Differences in the single-stranded DNA binding activities of MCM2-7 and MCM467: MCM2 and MCM5 define a slow ATP-dependent stepThe Journal of Biological Chemistry 282:33795–33804.https://doi.org/10.1074/jbc.M703824200
-
The Mcm2-7 complex has in vitro helicase activityMolecular Cell 31:287–293.https://doi.org/10.1016/j.molcel.2008.05.020
-
Crystal structure of a near-full-length archaeal MCM: functional insights for an AAA+ hexameric helicaseProceedings of the National Academy of Sciences of USA 105:20191–20196.https://doi.org/10.1073/pnas.0808037105
-
Version 1.2 of the Crystallography and NMR systemNature Protocols 2:2728–2733.https://doi.org/10.1038/nprot.2007.406
-
Crystallography & NMR system: a new software suite for macromolecular structure determinationActa Crystallographica Section D, Biological Crystallography 54:905–921.https://doi.org/10.1107/S0907444998003254
-
Structural polymorphism of Methanothermobacter thermautotrophicus MCMJournal of Molecular Biology 346:389–394.https://doi.org/10.1016/j.jmb.2004.11.076
-
A double-hexamer archaeal minichromosome maintenance protein is an ATP-dependent DNA helicaseProceedings of the National Academy of Sciences of USA 97:1530–1535.https://doi.org/10.1073/pnas.030539597
-
The structural basis for MCM2-7 helicase activation by GINS and Cdc45Nature Structural & Molecular Biology 18:471–477.https://doi.org/10.1038/nsmb.2004
-
Structural basis of the Methanothermobacter thermautotrophicus MCM helicase activityNucleic Acids Research 34:5829–5838.https://doi.org/10.1093/nar/gkl708
-
‘dm’: An automated procedure for phase improvement by density modificationJoint CCP4 and ESF-EACBM Newsletter on Protein Crystallography 31:34–38.
-
A structural framework for replication origin opening by AAA+ initiation factorsCurrent Opinion in Structural Biology 23:144–153.https://doi.org/10.1016/j.sbi.2012.11.012
-
Coot: model-building tools for molecular graphicsActa Crystallographica Section D, Biological Crystallography 60:2126–2132.https://doi.org/10.1107/S0907444904019158
-
Evolutionary relationships and structural mechanisms of AAA+ proteinsAnnual Review of Biophysics and Biomolecular Structure 35:93–114.https://doi.org/10.1146/annurev.biophys.35.040405.101933
-
A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replicationProceedings of the National Academy of Sciences of USA 106:20240–20245.https://doi.org/10.1073/pnas.0911500106
-
The structure and function of MCM from archaeal M. ThermoautotrophicumNature Structural Biology 10:160–167.https://doi.org/10.1038/nsb893
-
Polymorphism and double hexamer structure in the archaeal minichromosome maintenance (MCM) helicase from Methanobacterium thermoautotrophicumThe Journal of Biological Chemistry 280:40909–40915.https://doi.org/10.1074/jbc.M509760200
-
mcm5/cdc46-bob1 bypasses the requirement for the S phase activator Cdc7pProceedings of the National Academy of Sciences of USA 94:3151–3155.https://doi.org/10.1073/pnas.94.7.3151
-
Selenomethionyl proteins produced for analysis by multiwavelength anomalous diffraction (MAD): a vehicle for direct determination of three-dimensional structureThe EMBO Journal 9:1665–1672.
-
Minichromosome maintenance helicase activity is controlled by N- and C-terminal motifs and requires the ATPase domain helix-2 insertProceedings of the National Academy of Sciences of USA 103:7613–7618.https://doi.org/10.1073/pnas.0509297103
-
Structural basis of mechanochemical coupling in a hexameric molecular motorThe Journal of Biological Chemistry 283:3607–3617.https://doi.org/10.1074/jbc.M706366200
-
Use of non-crystallographic symmetry in protein structure refinementActa Crystallographica Section D, Biological Crystallography 52:842–857.https://doi.org/10.1107/S0907444995016477
-
Validation of protein models from Calpha coordinates aloneJournal of Molecular Biology 273:371–376.https://doi.org/10.1006/jmbi.1997.1309
-
Molscript - a program to produce both detailed and schematic plots of protein structuresJournal of Applied Crystallography 24:946–950.https://doi.org/10.1107/S0021889891004399
-
Procheck - a program to check the stereochemical quality of protein structuresJournal of Applied Crystallography 26:283–291.https://doi.org/10.1107/S0021889892009944
-
Dynamic look at DNA unwinding by a replicative helicaseProceedings of the National Academy of Sciences of USA 111:E827–E835.https://doi.org/10.1073/pnas.1322254111
-
Structural analysis of the Sulfolobus solfataricus MCM protein N-terminal domainNucleic Acids Research 36:3235–3243.https://doi.org/10.1093/nar/gkn183
-
Phaser crystallographic softwareJournal of Applied Crystallography 40:658–674.https://doi.org/10.1107/S0021889807021206
-
Organization of the archaeal MCM complex on DNA and implications for the helicase mechanismNature Structural & Molecular Biology 12:756–762.https://doi.org/10.1038/nsmb974
-
Raster3D: photorealistic molecular graphicsMethods in Enzymology 277:505–524.https://doi.org/10.1016/S0076-6879(97)77028-9
-
Isolation of the Cdc45/Mcm2-7/GINS (CMG) complex, a candidate for the eukaryotic DNA replication fork helicaseProceedings of the National Academy of Sciences of USA 103:10236–10241.https://doi.org/10.1073/pnas.0602400103
-
AAA+: a class of chaperone-like ATPases associated with the assembly, operation, and disassembly of protein complexesGenome Research 9:27–43.
-
Processing of X-ray diffraction data collected in oscillation modeMacromolecular Crystallography, Part A 276:307–326.https://doi.org/10.1016/S0076-6879(97)76066-X
-
Modular organization of the Sulfolobus solfataricus mini-chromosome maintenance proteinThe Journal of Biological Chemistry 282:12574–12582.https://doi.org/10.1074/jbc.M610953200
-
Amino acids of the Sulfolobus solfataricus mini-chromosome maintenance-like DNA helicase involved in DNA binding/remodelingThe Journal of Biological Chemistry 279:49222–49228.https://doi.org/10.1074/jbc.M408967200
-
Eukaryotic DNA replication control: lock and load, then fireCurrent Opinion in Cell Biology 21:771–777.https://doi.org/10.1016/j.ceb.2009.08.002
-
Coupling of DNA binding and helicase activity is mediated by a conserved loop in the MCM proteinNucleic Acids Research 36:1309–1320.https://doi.org/10.1093/nar/gkm1160
-
The PyMOL molecular graphics system, Version 1.3r1
-
Structure and mechanism of helicases and nucleic acid translocasesAnnual Review of Biochemistry 76:23–50.https://doi.org/10.1146/annurev.biochem.76.052305.115300
-
Mini-chromosome maintenance complexes form a filament to remodel DNA structure and topologyNucleic Acids Research 41:3446–3456.https://doi.org/10.1093/nar/gkt022
-
Maximum-likelihood density modificationActa Crystallographica Section D, Biological Crystallography 56:965–972.https://doi.org/10.1107/S0907444900005072
-
Using prime-and-switch phasing to reduce model bias in molecular replacementActa Crystallographica Section D, Biological Crystallography 60:2144–2149.https://doi.org/10.1107/S0907444904019535
-
REFMAC5 dictionary: organization of prior chemical knowledge and guidelines for its useActa Crystallographica Section D, Biological Crystallography 60:2184–2195.https://doi.org/10.1107/S0907444904023510
-
The GINS complex from Pyrococcus furiosus stimulates the MCM helicase activityThe Journal of Biological Chemistry 283:1601–1609.https://doi.org/10.1074/jbc.M707654200
Article and author information
Author details
Funding
American Lebanese Syrian Associated Charities (ALSAC)
- Eric J Enemark
National Institute of General Medical Sciences (R01GM098771)
- Eric J Enemark
Cancer Center Support Grant (5 P30 CA021765)
- Eric J Enemark
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Data were collected at Southeast Regional Collaborative Access Team (SER-CAT) 22-ID and 22-BM beamlines at the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at www.ser-cat.org/members.html. We are grateful to SER-CAT staff for experimental support. Use of the Advanced Photon Source was supported by the U. S. Department of Energy, Office of Science, Office of Basic Energy Sciences, under Contract No. W-31-109-Eng-38.
Copyright
© 2014, Miller et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,872
- views
-
- 433
- downloads
-
- 63
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 63
- citations for umbrella DOI https://doi.org/10.7554/eLife.03433
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Analysis of the crystal structure of an active MCM hexamer
eLife 3:e03433.
https://doi.org/10.7554/eLife.03433




{kind=link}
{kind=link}