Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability
- University of Utah, United States
Abstract
The nonsense-mediated mRNA decay (NMD) pathway functions to degrade both abnormal and wild-type mRNAs. NMD is essential for viability in most organisms, but the molecular basis for this requirement is unknown. Here we show that a single, conserved NMD target, the mRNA coding for the stress response factor growth arrest and DNA-damage inducible 45 (GADD45) can account for lethality in Drosophila lacking core NMD genes. Moreover, depletion of Gadd45 in mammalian cells rescues the cell survival defects associated with NMD knockdown. Our findings demonstrate that degradation of Gadd45 mRNA is the essential NMD function and, surprisingly, that the surveillance of abnormal mRNAs by this pathway is not necessarily required for viability.
https://doi.org/10.7554/eLife.12876.001eLife digest
Messenger RNA (mRNA) molecules act as the templates from which proteins are made, and so control the amount of protein in a cell. Having too much of certain proteins can harm cells. Additionally, some mRNAs contain errors, and so can create faulty proteins that may also harm the cell.
Cells have therefore developed ways to destroy excess or error-ridden mRNAs to avoid a deadly build up of proteins. One such quality control mechanism is called nonsense-mediated decay (NMD). This mechanism is so important that cells that cannot perform nonsense-mediated decay die, although it is not clear exactly what kills the cells.
Now, Nelson et al. have found that fruit flies whose cells are unable to perform nonsense-mediated decay die because a harmful protein called Gadd45 builds up in the cells. In normal cells, nonsense-mediated decay destroys the mRNA that relays the instructions for making Gadd45, which keeps the amount of the Gadd45 protein in the cell low. Further experiments show that removing Gadd45 from cells that lack nonsense-mediated decay saves the flies. Removing Gadd45 from human and mouse cells that are unable to perform nonsense-mediated decay also allows these cells to survive.
These findings imply that the only nonsense-mediated decay function needed for cells to live is the destruction of Gadd45 mRNA. This further implies that most faulty and normal mRNAs that are normally destroyed by nonsense-mediated decay do not cause the cells to die when nonsense-mediated decay is lost.
Learning that creating faulty proteins when nonsense-mediated decay is lost is not necessarily harmful to cells opens new possibilities to treating numerous genetic diseases. In some diseases, cells can only produce faulty forms of a particular protein. Nonsense-mediated decay normally destroys all of these mutant proteins, but it may sometimes be better to have faulty versions of a protein than to have none of it. Safely getting rid of nonsense-mediated decay by also eliminating Gadd45 from cells may therefore be a treatment strategy worth exploring.
https://doi.org/10.7554/eLife.12876.002Introduction
Maintaining proper gene expression is critical for normal development and physiology. In addition to de novo transcription, mRNA stability substantially contributes to forming the landscape of expression in a cell. The nonsense-mediated mRNA decay (NMD) pathway is a trans-acting mechanism that destabilizes mRNAs, and is best known for its well-described role as a quality control system, degrading abnormal mRNAs containing premature termination codons (PTCs) (Celik et al., 2015). NMD also degrades many wild-type endogenous mRNAs and thus is an important aspect of their post-transcriptional (Peccarelli and Kebaara, 2014). Loss of either of the core NMD genes Upf1 (Rent1) or Upf2 causes lethality in most eukaryotes (Kerényi et al., 2008; Medghalchi et al., 2001; Metzstein and Krasnow, 2006; Weischenfeldt et al., 2008; Wittkopp et al., 2009), indicating regulation of mRNA stability by NMD is critical for viability. However, the relative contributions to lethality from ectopic stabilization of PTC-containing mRNAs or endogenous NMD targets in NMD mutants remains unclear (Hwang and Maquat, 2011).
To identify which ectopically stabilized mRNAs are responsible for inducing lethality in NMD mutants, we performed an unbiased genetic suppressor screen seeking to restore viability in a Drosophila NMD mutant. To detect subtle increases in survival, we screened to suppress the lethality of animals mutant for the partially viable, hypomorphic Upf225G allele, of which 10% survive to adulthood (Chapin et al., 2014; Metzstein and Krasnow, 2006). We crossed this allele to heterozygous deficiencies to simultaneously reduce the mRNA abundance of several loci (Figure 1A). Of the 376 deficiencies tested, covering more than half the genome, ~10% suppressed NMD mutant lethality (Figure 1B, Figure 1—figure supplement 1A). The suppression effect could not be explained by a reduction in overall mRNA load, as there was only a weak correlation between the increase in mRNAs expressed from a genomic region upon loss of NMD function and the strength of suppression when that region was removed by a deficiency (Figure 1—figure supplement 1B). Rather, deficiencies that suppressed NMD-mutant lethality clustered in three genomic regions (Figure 1—figure supplement 1A). These findings suggest that NMD mutant lethality is not the result of a global excess of nonspecific mRNAs, but rather is mediated by specific genes residing within the few identified regions.
Figure 1 with 4 supplements see all
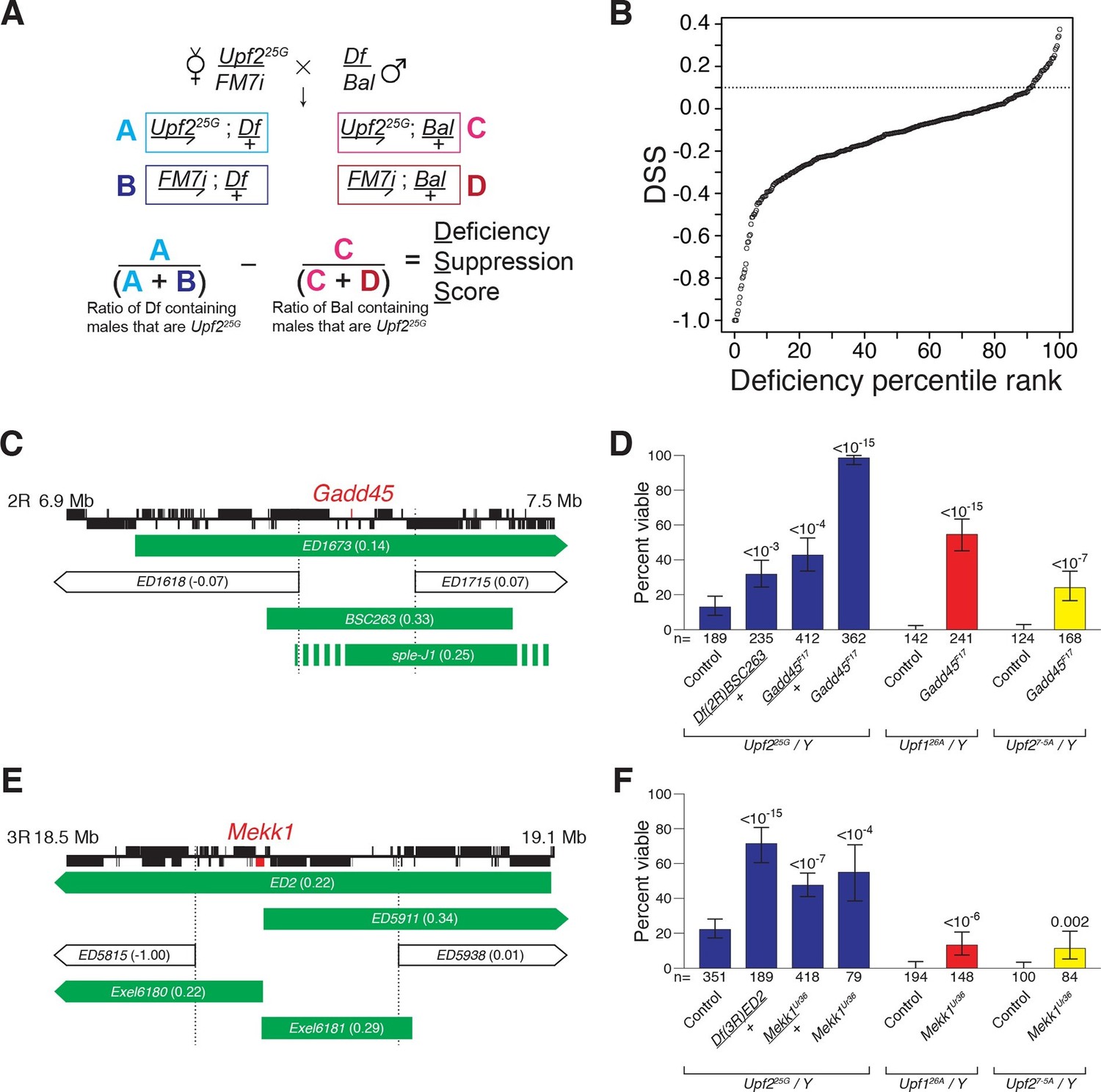
Drosophila suppressor screen identifies the Gadd45 pathway as the inducer of NMD-mutant lethality.
(A) Scheme to screen deficiencies for the suppression of Upf225G partial lethality. The Deficiency Suppression Score (DSS) represents the relative difference in Upf225G viability when crossed to a heterozygous deficiency (Df) compared to when crossed to a balancer (Bal) (See Methods). (B) DSS from 376 screened deficiencies ranked by score. A DSS greater than 0.1 (dotted line) indicates that deficiency suppresses Upf225G lethality. (C and E) Candidate suppressing regions uncovering Gadd45 (C) and Mekk1 (E). DSSs are shown in parenthesis. Dotted lines denote extent of regions deleted by suppressing deficiencies but not non-suppressing deficiencies. Filled blocks on chromosomes indicate predicted gene spans, Gadd45 pathway genes are indicated in red; suppressing deficiencies indicated in green, sple-J1 has undefined breakpoints located within hashed regions. (D and F) NMD mutant adult viability in combination with Gadd45F17 (D) or Mekk1Ur36 (F) mutants. Upf126A and Upf27-5A are null alleles (Frizzell et al., 2012; Metzstein and Krasnow, 2006). p-value compared to controls determined by the test of equal or given proportions indicated. Error bars represent 95% confidence interval of the binomial distribution. n equals total number of animals scored in each cross.
We expected that any specific genes mediating NMD-mutant lethality would have increased expression levels in an NMD mutant and be a direct NMD target. The only gene located within the suppressing regions to fit these criteria is Gadd45 (Figure 1C, Figure 1—figure supplement 2A–C) (Chapin et al., 2014). To determine if NMD targeting of Gadd45 mRNA is critical for viability, we generated a Gadd45 null allele, F17, which completely removes the Gadd45 coding region (Figure 1—figure supplement 3A) and eliminates Gadd45 mRNA expression (Figure 1—figure supplement 2A). As a heterozygote, Gadd45F17 suppressed Upf225Glethality as strongly as the corresponding deficiency identified by our screen (Figure 1D). We found that Gadd45F17 homozygous mutants are fully viable (Figure 1—figure supplement 3B), allowing us to test complete loss of Gadd45 for the suppression of NMD-mutant lethality. Homozygous Gadd45F17 restored full viability to Upf225Gmutants, and remarkably even partially suppressed the complete lethality observed in null Upf1 and Upf2 mutants (Frizzell et al., 2012; Metzstein and Krasnow, 2006) (Figure 1D). Importantly, neither reducing nor eliminating Gadd45 restored NMD function to Upf225G mutants, as measured by the expression of both an endogenous NMD target (Figure 1—figure supplement 4A) and PTC-containing mRNAs (Figure 1—figure supplement 4B).
In mammals, GADD45 activates the MTK1/MEKK4 kinase in a well-defined stress response pathway (Takekawa and Saito, 1998). Strikingly, the Drosophila MTK1 orthologue, Mekk1, resides within another Upf225G suppressing region (Figure 1E). Similar to Gadd45, we found that Mekk1 null mutants (Inoue et al., 2001) suppressed Upf1 and Upf2 mutant lethality (Figure 1F). This suppression was not as strong as that caused by a loss of Gadd45, revealing that although MEKK1 mediates NMD mutant lethality, it is likely that GADD45 has additional downstream effectors that influence viability. Overall, our findings reveal that increased Gadd45 mRNA stability is the major factor inducing NMD mutant lethality, primarily via increased MEKK1 activity.
Activation of MTK1 in mammals triggers a MAPK signaling cascade that promotes apoptosis (Takekawa and Saito, 1998). Over-expression of Gadd45 in Drosophila also induces apoptosis (Peretz et al., 2007). Interestingly, Drosophila cells lacking NMD function show excess cell death in a variety of tissues (Avery et al., 2011; Frizzell et al., 2012; Metzstein and Krasnow, 2006). To test if increased Gadd45 contributes to this excess death, we used TUNEL staining to examine cell death in wing imaginal discs from Upf225G mutant third instar larvae. This analysis revealed elevated levels of cell death compared to controls (Figure 2A, B, E), and this defect was completely suppressed by Gadd45F17 (Figure 2C–E). To confirm that, this effect was not specific to the Upf2 gene or 25G allele, we examined the wing discs in mutants of another essential NMD gene, Smg5. We found that Smg5 discs also showed elevated TUNEL signal, which was eliminated by loss of Gadd45 (Figure 2—figure supplement 1A–E). These results demonstrate that excess Gadd45 accounts for ectopic cell death in NMD mutant tissues.
Figure 2 with 1 supplement see all
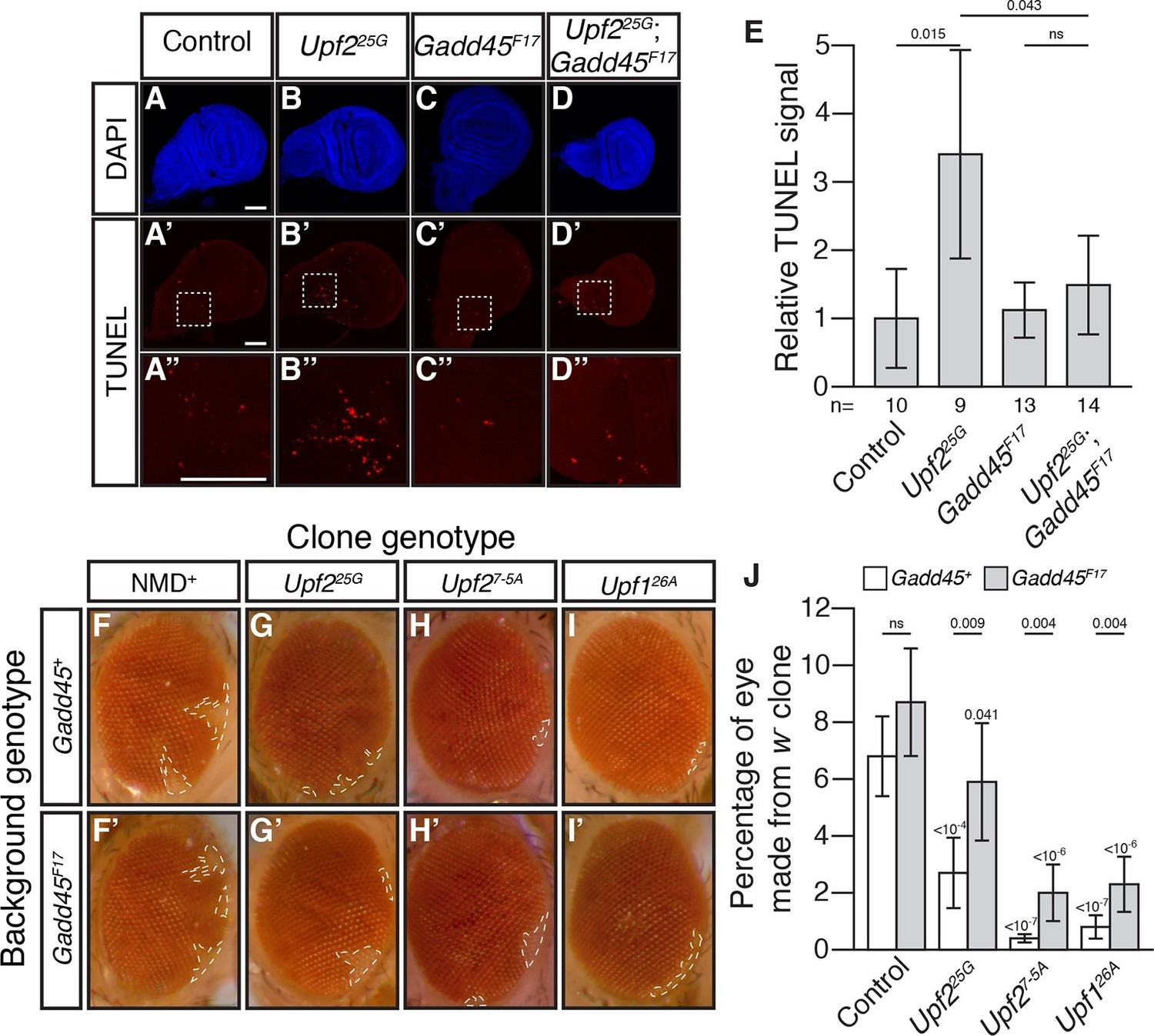
Loss of Gadd45 suppresses NMD-mutant cell death.
(A to D) DAPI (blue) and (A’to D’) TUNEL (red) staining in late 3rd instar larval wing discs from control (A); Upf225G (B); Gadd45F17 (C); and Upf225G; Gadd45F17 (D) animals. (A’’ to D’’) are 4x view of outlined section at the base of the blade of the wing disc from A’-D’, respectively. Scale bar represents 100 μm. (E) Relative TUNEL signal in control and mutant wing discs, normalized to control. p-value between indicated samples using a two-sided Student’s t-test are displayed. ns indicates a p-value greater than 0.05. Error bars represent 2 SEM. n equals total number of discs scored. (F to I) w- eye clones in Gadd45+ and Gadd45F17 backgrounds. Dashed lines indicate clone boundaries. (J) Quantification of the fraction of the eye composed of w- cells in control and mutant eyes. p-values indicate differences between Gadd45 mutant and control in the same NMD background (indicated by horizontal bars) or NMD mutant and control in the same Gadd45 background (indicated by value above each individual bar), using a two-sided Student’s t-test. ns indicates a p-value greater than 0.05. Error bars represent 2 SEM. n = 20 eyes for all conditions.
To test if Gadd45-induced cell death is the only cellular defect in NMD mutants, we examined NMD function in the developing eye. NMD is required for proper development of eye cells, as clonal patches of NMD mutant cells in eyes are reduced in size (Frizzell et al., 2012; Metzstein and Krasnow, 2006). We found that Gadd45 is partially responsible for this defect, as the size of eye-cell clones lacking NMD activity in a Gadd45F17 background was increased, although not fully restored (Figure 2F–J). These results indicate that some, but not all, defects associated with loss of NMD are dependent on Gadd45.
Gadd45 is one of the few genes that is directly regulated by NMD in both flies and mammals (Huang et al., 2011; Tani et al., 2012; Viegas et al., 2007), raising the possibility that excess Gadd45 abundance may also contribute to the NMD-mutant lethality observed in mammalian cells (Azzalin and Lingner, 2006; Li et al., 2015; Medghalchi et al., 2001; Weischenfeldt et al., 2008). To test this hypothesis, we analyzed the effects of Gadd45 and Upf1 depletion in mouse NIH-3T3 cells. Gadd45b mRNA (also known as MyD118), which is expressed at least 10-fold higher than any other Gadd45 paralogue in these cells (Yue et al., 2014), was degraded rapidly in a partially Upf1-dependent manner after transcription was blocked with actinomycin D (Figure 3A), and had increased expression during Upf1 knockdown (Figure 3D), confirming it is sensitive to NMD. We found that transfection of 3T3 cells with siRNAs targeting Upf1 resulted in significant reduction in cell counts after 48 hr (Figure 3B), but co-transfection with siRNAs targeting both Upf1 and Gadd45b largely reversed this effect (Figure 3B). The reduction in cell counts was primarily due to increased cell death, as we found that ~25% of cells transfected with Upf1 siRNA were undergoing apoptosis (Figure 3C). Co-transfection of siRNA targeting Gadd45b almost entirely eliminated this increase (Figure 3C), indicating the excess apoptosis observed in Upf1-knockdown cells was mostly due to increased Gadd45 activity. However, while Gadd45b knockdown very greatly suppresses this excess death, it does not as fully rescue cell numbers, suggesting loss of NMD may lead to both Gadd45b-dependent cell death as well as a Gadd45b-independent effect on proliferation. This mirrors the conclusions we made about the partial suppression of cell number defects in the Drosophila eye. Importantly, Upf1 mRNA expression was equivalently reduced and the expression of the mammalian endogenous NMD targets Rassf1 and CRCP (Tani et al., 2012) was equivalently increased in both the single and double knockdown experiments (Figure 3D), indicating that the restoration of viability was not due to a recovery of NMD pathway activity.
Figure 3 with 1 supplement see all
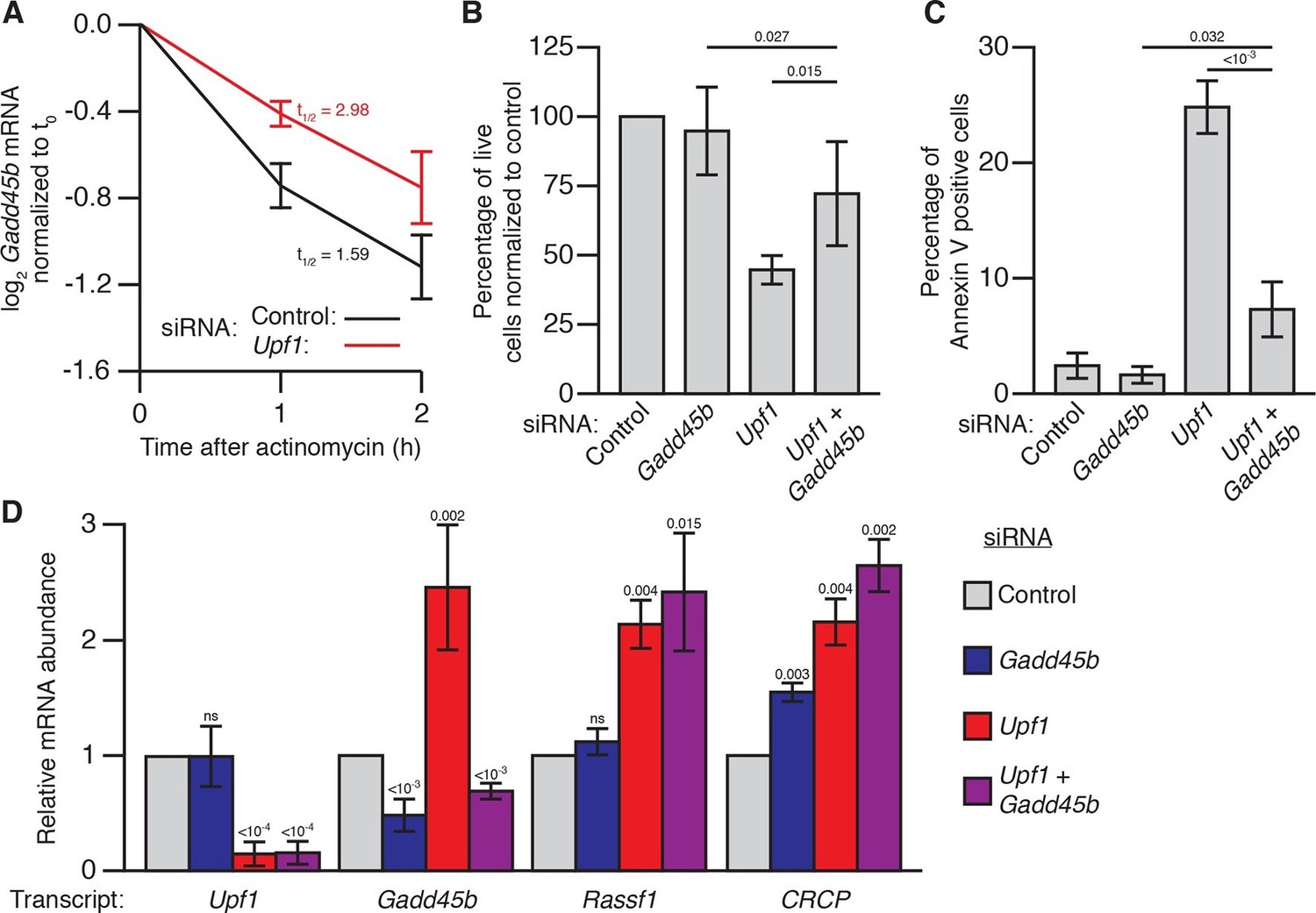
Gadd45b mediates cell lethality in Upf1 siRNA knockdown 3T3 mouse embryonic fibroblasts.
(A) Relative Gadd45b mRNA expression measured by qRT-PCR in NIH-3T3 cells after 48 hr of control (black) or Upf1 (red) siRNA treatment and 0 to 2 hr of actinomycin D treatment, normalized to expression prior to actinomycin treatment. The half-life calculated for each decay curve is indicated. (B) Relative viable cell count of Upf1 and Gadd45b single and double siRNA treatment normalized to control siRNA. p-values display two-sided Student’s t-test between indicated conditions. (C) Quantification of apoptosis as measured by annexin V staining. p-values display two-sided Student’s t-test between indicated conditions. (D) Relative mRNA expression of Upf1, Gadd45b, and two mammalian endogenous NMD targets, Rassf1 and CRCP (Tani et al., 2012) measured by qRT-PCR in Gadd45b and Upf1 single and double siRNA knockdown cells, normalized to expression in the control siRNA condition. p-values display one-sided Student’s t-test for each condition compared to control. Error bars represent 2 SEM.
To extend our analysis to other mammalian cells, we analyzed the role of Gadd45 mediating the effects of loss of NMD in HEK293 cells. We found, similarly to 3T3 cells, that siRNA knockdown of UPF1 in HEK293 cells led to increased GADD45A expression and reduced cell numbers compared to control siRNA (Figure 3—figure supplement 1A,B). Although transfection of siRNA targeting GADD45A alone slightly reduced HEK293 cell numbers, co-transfection with UPF1 siRNA did not further reduce cell count (Figure 3—figure supplement 1B), and UPF1 expression was equivalently reduced in the single and double knockdown conditions (Figure 3—figure supplement 1C). These results suggest that UPF1 knockdown is no longer detrimental to HEK293 cell viability in the absence of GADD45A expression. We conclude that increased expression of mammalian Gadd45 genes contributes to lethality in NMD-deficient mouse and human cells, as Gadd45 does in Drosophila.
Deconvoluting the contributions to organismal viability of the PTC-surveillance versus gene-regulatory functions of NMD has been historically difficult (Hwang and Maquat, 2011). Here, we show that viability can be restored to Drosophila lacking core NMD factors when a single endogenous NMD target, Gadd45, is eliminated, and that the requirement for the regulation of Gadd45 by NMD is evolutionarily conserved from flies to mammals. Although our data suggest that up-regulation of Gadd45 is a major factor contributing to lethality when NMD activity is lost, it is likely that other NMD targets also contribute to the observed lethality. In particular, viability is not restored to 100% in null Upf1; or Upf2; Gadd45 double mutants. In addition, loss of Gadd45 suppresses programmed cell death caused by defects in NMD, but not additional cell cycle defects, as implied by the incomplete suppression in the Drosophila eye and mammalian cell culture. Such defects in the cell cycle may be particularly pronounced during the development of certain tissue, or specific developmental stages. Indeed, NMD has been reported to have differing stage and tissue- specific activities (Bao et al., 2015; Bruno et al., 2011; Colak et al., 2013; Li et al., 2015). Whether this is due to a role in surveillance or another specific target remains unclear, but examination of the effects of loss of NMD in Gadd45 mutants should allow exploration of these possibilities.
The benefit for such a mechanism regulating Gadd45 expression may lie in a function of NMD in restricting viral growth (Balistreri et al., 2014). Because viruses encode trans-acting factors to inhibit NMD (Mocquet et al., 2012), the resulting accumulation of GADD45 in infected cells may act as a “molecular tripwire” that rapidly elicits a stress response and cell death. This outcome suggests that regulating responses to infection may underlie a conserved essential function of NMD. Intriguingly, restriction of pathogens via NMD extends to plants (Garcia et al., 2014), where NMD mutant lethality in A. thaliana, which do not encode Gadd45 orthologues, may be caused by the overexpression of a subset of immune-related intracellular nucleotide-binding leucine-rich repeat receptors, some of which are endogenous NMD targets (Gloggnitzer et al., 2014). In contrast, eukaryotes that do not rely on the activation of programmed cell death to protect against viruses, such as S. cerevisiae, S. pombe, and C. elegans, do not require NMD for viability (Hodgkin et al., 1989; Leeds et al., 1991; Mendell et al., 2000). Together these observations suggest a potential novel role for NMD and Gadd45 in immune responses, triggering the death of infected cells during pathogenic challenges.
Restoring the expression of PTC-containing alleles via NMD inhibition has been proposed as a promising therapy for a wide range of recessive genetic diseases (Keeling et al., 2014). Translation of stable PTC-containing mRNAs would produce truncated proteins that may be partially functional and alleviate disease symptoms normally caused by complete loss of the protein. However, the essential function for NMD in viability has raised the concern that these therapies may have prohibitive side effects. Our findings reveal a molecular basis for dealing with this obstacle by suggesting that inhibiting both the NMD and Gadd45 pathways (Tornatore et al., 2014) in combination could provide an effective and safe treatment for patients with debilitating genetic disorders.
Materials and methods
Fly genetics
Request a detailed protocolDrosophila melanogaster stocks were raised on standard cornmeal/dextrose food at 25°. The NMD mutant alleles Upf225G, Upf27-5A, and Upf126A (Frizzell et al., 2012; Metzstein and Krasnow, 2006) are on y w FRT19A chromosomes. These alleles were balanced over FM7i, P{ActGFP}JMR3 (Reichhart and Ferrandon, 1998). Smg5G115 and Smg5C391 are null alleles of Smg5 (J.O.N., D. Förster, S. Luschnig, and M.M.M., unpublished) and will be described in detail later. The Smg5 alleles are balanced over CyO, P{Dfd:eYFP w+} (Le et al., 2006). Other alleles used were P{w[+mC]=EPg}HP20647 (Staudt et al., 2005), Mekk1Ur36 (Inoue et al., 2001) recombined on FRT82B by D. Ryoo, ey-FLP (Newsome et al., 2000), pcm14 (Waldron et al., 2015), Adhn4 (Chia et al., 1987) and DHR783 (Fisk and Thummel, 1998). Control chromosomes were y w FRT19A (for Upf1 and Upf2) and FRT82B (for Mekk1) (Xu and Rubin, 1993). For all experiments using Gadd45F17 we used the Gadd45E8 precise excision as a control.
For viability assays, we mated flies for 3 days and collected all progeny each day for 10 days, starting 10 days after the cross was initiated. The total numbers of F1 mutant and balancer males were scored, and the ratio of mutant males to balancer males was used to determine mutant animal viability. To control for balancer viability within each experiment, we normalized the ratio of mutant to balancer animals to a ratio of the appropriate control chromosome to balancer animals produced from a parallel cross.
Deficiency suppressor screen
Request a detailed protocolWe screened autosomal deficiencies from the DrosDel collection (Ryder et al., 2007). All deficiencies scored can be found in Supplementary file 1. Deficiencies on chromosome 2 were balanced over CyO, and deficiencies on chromosome 3 were balanced over TM6C. We mated males from each deficiency stock to y w Upf225G FRT19A/FM7i, P{ActGFP}JMR3 females and scored all F1 males for the presence or absence of each balancer. For any given deficiency tested, the percentage of Deficiency / + males that are Upf225G mutants, less the percentage of Balancer / + males that are Upf225G mutants was calculated, producing a Deficiency Suppression Score (DSS), which represents the effect of an individual deficiency on the increase or decrease in Upf225G viability, while controlling for each deficiency’s general influence on viability. A DSS greater than 0.1 indicates suppression of lethality. Supplemental deficiencies used were from the Exelixis collection (Parks et al., 2004) and Df(2R)sple-J1 (Heitzler et al., 1993). Deficiency mapping to the Drosophila genome was performed using the 5.1 genome release.
RNA-seq data sets were acquired from Chapin et al. (2014) (archives SRR896609, SRR896616, SRR503415, and SRR503416) and aligned using Bowtie and TopHat alignment with standard remapping parameters to the 5.1 Drosophila genome release. SAMtools accessory scripts were used to retrieve read counts for deficiency and control regions. All read counts were normalized to reads per million within each data set. Average normalized reads in Upf225G samples were normalized to the relative reads of 74 ribosomal proteins in Upf225G samples compared to control samples. Total normalized reads within the regions removed by each deficiency were averaged between biological replicates, and the difference between the Upf225Gand control samples was divided by one million to determine percent increase in genomic load across each deficiency region.
Generation of Gadd45 mutants
Request a detailed protocolWe produced P-element excision lines from the P{w[+mC]=EPg}HP20647 P-element insertion line crossed to a Δ2–3 transposase stock. We mated F1 males containing the P-element and transposase on a CyO balancer to w; Tft / CyO females. Cy+ Tft white-eyed F2 males were then individually mated to w; Tft / CyO females. We then collected Tft+, Cy males and females to create an isogenic stock from each individually mated F2 male. To identify precise excisions we used the primers Gadd45_F1 / Gadd45_R1 flanking the P-element insert site to amplify a region across the excised P-element. Lines that failed to amplify with these primers were candidate imprecise excisions, which we then tested with Gadd45_F1 / Gadd45_R3 primers for deletions. Any detected deletions were subsequently sequenced using these same primers. Primer sequences are found in Supplementary file 2.
Induction and analysis of eye clones
Request a detailed protocolWe generated eye clones with the FLP/FRT system using the ey-FLP driver (Newsome et al., 2000) to induce recombination. We imaged eyes on a Leica MZ125 stereo microscope with a Retiga-2000R camera (QImaging, Canada) with QCapture 3.1.2 software (QImaging). We focused images using the ImageJ stack focuser plugin and quantified relative eye clone size using the ImageJ analyzer tools. A total of 20 eyes from 20 individual animals were scored for each condition.
Cell death assays
Request a detailed protocolFor TUNEL assays, third instar larval wing discs were dissected as described in Sullivan et al. (Sullivan et al., 2000). TUNEL staining was performed using the Apoptag Red in situ Apoptosis Detection Kit (Chimicon International Inc., Billerica, MA) according to Chakraborty et al. (Chakraborty et al., 2015). We DAPI stained wing discs (1:5000) for 5 min prior to mounting. Confocal images were acquired using a Zeiss LSM710 laser scanning confocal microscope (Carl Zeiss AG, Germany). 3-dimensional datasets were acquired with a Plan-Apochromat 20X/0.8 lens, 1.34 μm z-step, using the Zeiss ZEN software. To measure TUNEL signal intensity z-projections images were summed with ImageJ. Background signal was removed by using the ImageJ MaxEntropy auto-threshold. Relative total TUNEL signal intensity was calculated using the ImageJ analyzer tools to measure the total pixel intensity within the wing discs of TUNEL images and normalized to the average intensity in control conditions.
For annexin V staining, we collected media (including floating cells) from siRNA treated cells. We spun down media at 950g for 4 min to pellet cells, and then aspirated remaining media. Concurrently, we trypsinized siRNA-treated cells still on plates and added them to the same respective tube as previously spun-down media. Following the Alexa Fluor 488 Annexin V/Dead Cell Apoptosis Kit (Abcam, UK) protocol, we stained for apoptotic cells. We visualized cells on an Olympus IX51 microscope (Olympus, Japan) with 20X objective. We collected bright field as well as fluorescent images using a FITC filter with a QImaging QICam Fast1394 camera and QCaptureP software (QImaging). We analyzed cells by counting all cells within a bright field image as well as the annexin V positive cells from the same image. The number of annexin V positive cells was divided by total cell number to generate the fraction of apoptotic cells for each treatment. >3000 total cells were counted across three biological replicates for each treatment.
Cell culture experiments
Request a detailed protocolWe cultured mouse NIH-3T3 cells (ATCC) or HEK293 cells (ATCC) in DMEM (Thermo-Fisher, Waltham, MA) supplemented with 10% fetal bovine serum and glutamine. For siRNA experiments, we transfected cells using RNAiMax and 24 pmol of negative control siRNA (Qiagen, Netherlands), Upf1 siRNA (Qiagen), or Gadd45b siRNA (Sigma-Aldrich) for 3T3 cell experiments, or negative control siRNA (Qiagen), UPF1 siRNA (Qiagen), or GADD45A siRNA (Sigma-Aldrich, St. Louis, MO) for HEK293 experiments. For double siRNA-treated cells, we used 24 pmol of each Upf1 and Gadd45b siRNA for 3T3 experiments or UPF1 and GADD45A siRNA for HEK293 experiments.
For actinomycin experiments, we incubated cells with siRNA for 48 hr before changing the media and then incubated with 2 μg/mL actinomycin (Sigma-Aldrich) for 1 or 2 hr. mRNA half-life was determined by fitting an exponential decay curve to the relative expression at each time point (Tani et al., 2012). t1/2 was calculated based on the average expression at each time point, and the mean t1/2 for each condition is represented.
For cell counting experiments, we trypsinized cells, incubated a small aliquot with Trypan Blue at a final concentration of 0.04% in complete media, and counted Trypan Blue negative cells. RNA was collected from the remaining cells, and relative mRNA levels were measured as described below.
RNA isolation and quantitative RT-PCR
Request a detailed protocolFor Drosophila qRT-PCR analyses, we collected 5–10 adult animals frozen in liquid nitrogen. We isolated total RNA using TRIzol reagent (Invitrogen) and phase-lock tubes (5-Prime), and the RNeasy mini kit (Qiagen). We used on-column RNase-free DNase treatment (Qiagen) to reduce genomic contamination. We determined RNA concentration by spectrophotometer and normalized concentration for reverse transcription. For reverse transcription, we used random decamers and MMLV8 reverse transcriptase (Retroscript Kit, Thermo-FIsher). We performed qRT-PCR analysis using the SYBR Green qPCR Supermix (Bio-Rad, Hercules, CA) and the Bio-Rad iCycler thermocycler. All experimental reactions were performed using three technical replicates and a minimum of three biological replicates per condition, and the expression level of all experimental assays was normalized to RpL32 mRNA expression.
For cell culture qRT-PCR analyses, we collected RNA following the Zymo Research Quick RNA MiniPrep kits protocol, and synthesized cDNA using MMLV reverse transcriptase (NEB, Ipswich, MA) with a template of 1 µg of total RNA and priming with a T18 oligo. We measured relative mRNA levels by qRT-PCR using the Masterplex ep realplex (Eppendorf, Germany) with SYBR green fluorescent dye. Each sample was measured with technical triplicates and three biological replicates, and target mRNA levels were normalized to those of ribosomal protein 19 (Rpl19) mRNA.
For all qRT-PCR analyses we also measured samples that had been made without reverse transcriptase to ensure that signal was not due to genomic DNA. Primer sequences can be found in Supplementary file 2.
3’ UTR cloning and sensitivity assay
Request a detailed protocolWe cloned the UAS-GFP::Gadd45 3’ UTR and control UAS-GFP::Act5C 3’ UTR constructs using the primers G45_3U_X1_F / G45_3U_S1_R or Act5C_X1_F / Act5C_S1_R (Supplementary file 2) to amplify the Gadd45 and Act5C 3’ UTRs, respectively, from genomic DNA. PCR fragments were inserted into the Zero Blunt TOPO vector (Thermo-Fisher), sequenced to assure fidelity, and digested and cloned into a pUAST-attB GFP vector using standard cloning procedures to replace the SV40 3’ UTR. Plasmids were injected by BestGene (Chino Hills, CA) into a stock containing the VK00027 attP site (Venken et al., 2006) for phiC31 directed integration. We used previously described UAS-GFP::SV40 3’ UTR animals (Metzstein and Krasnow, 2006). For imaging, wandering late L3 larvae were collected and examined using a Leica MZ 16F microscope and the Leica DFC340 FX camera with the Leica Application Suite v3.3.0 software.
Analysis of dHR783 and Adhn4 PTC allele stability
Request a detailed protocolWe collected adult F1 Upf2+; Gadd45E8/+, Upf225G; Gadd45E8/+, and Upf225G; Gadd45F17/+males that were also heterozygous for either the dHR783 or Adhn4. The Adhn4allele is a PTC-containing allele and has been demonstrated to be a direct NMD target based on cleavage by Smg6 (Gatfield and Izaurralde, 2004). The dHR783 allele is also a PTC-containing allele and thus is presumably degraded by NMD (Fisk and Thummel, 1998). At least three biological replicates were collected for each condition. We isolated RNA and generated cDNA as described in methods above and used this cDNA as a template for PCR amplification of the dHR78 transcript with the DRH78_F3 / DHR78_R3 primers and the Adh transcript with the Adh_F and Adh_R primers (Supplementary file 2), which flank the nonsense mutation in the respective transcripts. To compare the relative abundance of the dHR783 allele to the wild-type allele, PCR products were Sanger sequenced, and the relative peak intensity for a T (dHR783 allele) compared to a C (wild-type allele) at nucleotide 1063 was compared. To compare the relative abundance of the Adhn4 allele to the wild-type allele, PCR products were digested with PvuII (a site disrupted by the n4 mutation), separated on a 1% agarose gel and stained with ethidium bromide. The relative intensity of the cut and uncut bands was determined using ImageJ and normalized for fragment length. All samples were ran on the same gel and compared under identical conditions. All ratios were normalized to the ratio in the Upf225G; Gadd45E8/+condition.
Statistical analysis
Request a detailed protocolAll figures displaying viability assays represent a proportion of animals of the indicated genotypes that survive to adulthood; error bars for these figures represent the 95% confidence interval of the binomial distribution, and the Test of qual or Given Proportions was used to determine significance difference in these proportions between genotypes. All other figures represent the mean value of multiple replicates have error bars depicting ± 2 SEM, which is a close approximation of the 95% confidence interval (Krzywinski and Altman, 2013). For tests between two variable measures, a two-sided paired Student’s t-test was used to determine significance difference between mean value data. For most qPCR experiments, data was compared to a normalized control, set to a constant of 1, so these tests were performed with a one-sided Student’s t-test.
Data availability
-
D.melanogaster Nonsense-Mediated mRNA Decay studyhttp://trace.ncbi.nlm.nih.gov/Traces/sra/.
References
-
In Vivo Determination of Direct Targets of the Nonsense-Mediated Decay Pathway in DrosophilaG3: Genes|Genomes|Genetics 4:485–496.https://doi.org/10.1534/g3.113.009357
-
Adhn4 of Drosophila melanogaster is a nonsense mutationNucleic Acids Research 15:3931.
-
Genetic and cytogenetic analysis of the 43A-E region containing the segment polarity gene costa and the cellular polarity genes prickle and spiny-legs in Drosophila melanogasterGenetics 135:105–115.
-
A new kind of informational suppression in the nematode Caenorhabditis elegansGenetics 123:301–313.
-
Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the questionCurrent Opinion in Genetics & Development 21:422–430.https://doi.org/10.1016/j.gde.2011.03.008
-
Therapeutics based on stop codon readthroughAnnual Review of Genomics and Human Genetics 15:371–394.https://doi.org/10.1146/annurev-genom-091212-153527
-
Inter-kingdom conservation of mechanism of nonsense-mediated mRNA decayThe EMBO Journal 27:1585–1595.https://doi.org/10.1038/emboj.2008.88
-
Rent1, a trans-effector of nonsense-mediated mRNA decay, is essential for mammalian embryonic viabilityHuman Molecular Genetics 10:99–105.https://doi.org/10.1093/hmg/10.2.99
-
Novel Upf2p Orthologues Suggest a Functional Link between Translation Initiation and Nonsense Surveillance ComplexesMolecular and Cellular Biology 20:8944–8957.https://doi.org/10.1128/MCB.20.23.8944-8957.2000
-
Analysis of Drosophila photoreceptor axon guidance in eye-specific mosaicsDevelopment 127:851–860.
-
Regulation of Natural mRNAs by the Nonsense-Mediated mRNA Decay PathwayEukaryotic Cell 13:1126–1135.https://doi.org/10.1128/EC.00090-14
-
Drosophila protocols, New York, NY, Cold Spring Harbor Laboratory PressDrosophila protocols, New York, NY, Cold Spring Harbor Laboratory Press.
-
Nonsense-mediated mRNA decay effectors are essential for zebrafish embryonic development and survivalMolecular and Cellular Biology 29:3517–3528.https://doi.org/10.1128/MCB.00177-09
-
Analysis of genetic mosaics in developing and adult Drosophila tissuesDevelopment 117:1223–1237.
Article and author information
Author details
Funding
National Institutes of Health (1R01GM084011)
- Jonathan O Nelson
- Mark M Metzstein
March of Dimes Foundation (5-FY07-664)
- Mark M Metzstein
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank the Metzstein and Thummel labs for helpful discussion; Shawn Rynearson, Esther Ellison, Zev Kronenberg, and EJ Osborne for assistance with collection and analysis of the deficiency suppressor screen; Kate Sanders for assistance isolating the Gadd45F17 allele; Don Ryoo, Stefan Luschnig, Sarah Newbury, and Carl Thummel for providing Drosophila lines; Kim Frizzell for assistance imaging Drosophila eyes; Ria Chakraborty and Kent Golic for assistance with TUNEL; Chase Bryan for assistance with confocal microscopy; and Carl Thummel, Gillian Stanfield, and Nels Elde for helpful comments on the manuscript. Fly stocks were obtained from the Bloomington Drosophila Stock Center. Exelixis deficiencies were provided by Exelixis, Inc. This work was supported by National Institutes of Health (NIH) grant 1R01GM084011 (to MMM) and a March of Dimes Award 5-FY07-664 (to MMM).
Copyright
© 2016, Nelson et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,988
- views
-
- 891
- downloads
-
- 71
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 71
- citations for umbrella DOI https://doi.org/10.7554/eLife.12876
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Degradation of Gadd45 mRNA by nonsense-mediated decay is essential for viability
eLife 5:e12876.
https://doi.org/10.7554/eLife.12876




{kind=link}
{kind=link}
{kind=link}