The complex relationship of exposure to new Plasmodium infections and incidence of clinical malaria in Papua New Guinea
- Swiss Tropical and Public Health Institute, Switzerland
- University of Basel, Switzerland
- Walter and Eliza Hall Institute of Medical Research, Australia
- Papua New Guinea Institute of Medical Research, Papua New Guinea
- University of Melbourne, Australia
- Burnet Institute, Australia
- ISGlobal, Barcelona Centre for International Health Research, Hospital Clínic-University of Barcelona, Spain
- Institut Pasteur, France
Abstract
The molecular force of blood-stage infection (molFOB) is a quantitative surrogate metric for malaria transmission at population level and for exposure at individual level. Relationships between molFOB, parasite prevalence and clinical incidence were assessed in a treatment-to-reinfection cohort, where P.vivax (Pv) hypnozoites were eliminated in half the children by primaquine (PQ). Discounting relapses, children acquired equal numbers of new P. falciparum (Pf) and Pv blood-stage infections/year (Pf-molFOB = 0–18, Pv-molFOB = 0–23) resulting in comparable spatial and temporal patterns in incidence and prevalence of infections. Including relapses, Pv-molFOB increased >3 fold (relative to PQ-treated children) showing greater heterogeneity at individual (Pv-molFOB = 0–36) and village levels. Pf- and Pv-molFOB were strongly associated with clinical episode risk. Yearly Pf clinical incidence rate (IR = 0.28) was higher than for Pv (IR = 0.12) despite lower Pf-molFOB. These relationships between molFOB, clinical incidence and parasite prevalence reveal a comparable decline in Pf and Pv transmission that is normally hidden by the high burden of Pv relapses.
Clinical trial registration: ClinicalTrials.gov NCT02143934
https://doi.org/10.7554/eLife.23708.001eLife digest
Malaria is caused by five different species of parasites that are transmitted to humans by bites from parasite-carrying mosquitos. Once in human blood, the parasites rapidly multiply. People who live in countries where malaria is common may become infected and never show any symptoms because their immune systems are able to keep parasite numbers low. Repeated infections, or infection with more than one species of malaria parasite also are common. Some species of malaria, including Plasmodium vivax, can hibernate in the liver for weeks or months after the infection and only become active later.
Asymptomatic infections, multi-parasite infections, and reactivating parasites make it hard to measure how often new malaria infections occur. One way scientists can determine if a new infection has occurred is by genotyping the parasites in a person’s blood. Genotyping involves looking for small differences in the parasite DNA. For example, a study in Papua New Guinea, where P. vivax is very common, showed that reactivations of hibernating parasites were more common than new infections.
Now, Hofmann et al. use the same study in Papua New Guinea to compare the frequency and consequences of new infections with P. vivax and another malaria parasite, Plasmodium falciparum. In the study, 466 children from 6 villages were followed for 8 months with tests every 2 to 4 weeks to genotype the parasites in their blood. Some of the children were treated with antimalarial drugs to help wipe out any existing parasites including hibernating ones. While P. vivax was about twice as common in blood samples—likely due to reactivation—genotyping showed that new infections with the two parasites occur at equal rates and often at the same times and locations.
Hofmann et al. also showed that some villages and some children had much higher rates of infection than others. This difference could not fully be explained by use of bednets or other preventive measures. Children were more likely to become ill from P. falciparum than P. vivax even though P. vivax was more common. But children with more frequent infections with P. falciparum seemed better able to manage the parasites and were less likely to develop symptoms that those with infrequent infections. The experiments show that genotyping may help scientists better track new malaria infections and develop better strategies to prevent or treat malaria.
https://doi.org/10.7554/eLife.23708.002Introduction
Renewed emphasis on malaria control has resulted in substantial reductions in overall malaria prevalence and incidence in many endemic countries (World Health Organization, 2015). However, where transmission persists, it is highly heterogeneous even on small spatial scales (Bousema et al., 2012). Individual exposure is further influenced by factors such as use of bednets, attractiveness to mosquitoes, or behavioural differences. In Papua New Guinea (PNG), malaria prevalence has sharply declined in the last decade, largely as a result of two nationwide distributions of long-lasting insecticide treated bednets (LLIN) (Hetzel et al., 2015; Hetzel et al., 2014; Hetzel et al., 2012). P. vivax and P. falciparum PCR-prevalence in the general population was reduced from 32% and 39% in 2006 to 13% and 18% in 2010 (Koepfli et al., 2015). Already before this decline in malaria prevalence, studies in PNG had reported significant heterogeneity in malaria transmission attributed to local population structure and geographical diversity (Hetzel et al., 2015; Cattani et al., 1986; Genton et al., 1995; Müller et al., 2003; Mueller et al., 2009a).
Prior to the up-scaling of malaria control, P. vivax endemicity in PNG was among the highest worldwide (Hetzel et al., 2015). Clinical immunity to P. vivax was acquired very rapidly in PNG children, and the incidence of P. vivax clinical episodes peaked in children younger than two years with only very few P. vivax clinical episodes reported in children older than 5 years or adults (Genton et al., 2008; Michon et al., 2007; Lin et al., 2010; Betuela et al., 2012). In contrast, the risk for uncomplicated P. falciparum clinical episodes increased during early childhood (Lin et al., 2010) and significant reductions in incidence of clinical episodes or high-density infections were only observed in children aged 5 years and older (Michon et al., 2007). Compared to the incidence of clinical malaria, prevalence of P. falciparum and P. vivax peaked in older age groups, with asymptomatic infections remaining common until adulthood in PNG (Koepfli et al., 2015; Mueller et al., 2009a). Concordant with the species-specific pattern in the burden of clinical episodes, P. vivax prevalence peaked in younger age groups than P. falciparum prevalence (Koepfli et al., 2015; Mueller et al., 2009a).
As malaria transmission declines, it is important to understand the resulting changes in malaria prevalence and clinical incidence patterns, as well as the extent of heterogeneity in transmission within malaria endemic regions so that high-risk areas can be identified and targeted (Mosha et al., 2014). Most attempts to delineate high and low transmission areas made to-date, by both researchers and control programs, have used passive case surveillance or cross-sectional malaria indicator surveys. These surveillance strategies result in clinical incidence and prevalence estimates, both of which are surrogate markers for transmission. A more accurate understanding of the relationship between exposure to new infections and malaria prevalence or clinical incidence is needed to determine how accurately these surrogate markers represent heterogeneity in transmission at local scales. In addition, quantifying clinical incidence in relation to exposure to blood-stage infections can increase our insight into the development and maintenance of immunity to malaria in a setting of sustained malaria control (Battle et al., 2015; Cameron et al., 2015).
The molecular force of blood-stage infection (molFOB) describes the number of new genotypes observed in consecutive blood samples from cohort participants over time (Mueller et al., 2012; Koepfli et al., 2013). Genotyping of highly polymorphic markers detects superinfecting parasite clones in asymptomatic (but parasitaemic) or symptomatic individuals. molFOB thus provides a longitudinal, individual and quantitative measure for exposure to new blood-stage malaria infections (Mueller et al., 2012; Koepfli et al., 2013). For P. falciparum, molFOB is closely linked to the number of infective mosquito bites and therefore is a direct proxy for the actual force of infection (FOI) and thus for transmission in endemic settings (Smith et al., 2010). For P. vivax, clones appearing in the blood-stream can either originate directly from an infective mosquito bite or from a relapsing liver hypnozoite (Koepfli et al., 2013). For P. vivax, molFOB is thus a compound measure of exposure to newly acquired infections from mosquito bites and relapsing blood-stage infections.
The usefulness of molFOB as a surrogate marker of individual exposure was validated originally in a cohort of young PNG children 1–4 years of age, in which species-specific molFOB was the most important predictor of clinical incidence for both species (Mueller et al., 2012; Koepfli et al., 2013). Although some spatial heterogeneity of transmission was observed in that study for both species, due to the high level of transmission the species-specific difference in rate of immune acquisition was the predominant feature in that study. While P. vivax molFOB (Pv-molFOB) did not change with age, the incidence of P. vivax clinical episodes decreased significantly with age, with a faster rate of decrease in children with high Pv-molFOB [12]. P. falciparum molFOB (Pf-molFOB) in that cohort was lower compared to Pv-molFOB and the incidence of P. falciparum clinical episodes increased in parallel with an increasing Pf-molFOB in children 1–3 years, reaching a plateau thereafter (Lin et al., 2010; Mueller et al., 2012). These earlier results indicated that (i) immunity to P. vivax is acquired more rapidly in children with higher cumulative exposure, that (ii) this developing immunity led to proportionally fewer clinical P. vivax episodes in older children despite similar exposure to new P. vivax blood-stage infections, and that (iii) higher exposure to P. vivax blood-stage infections, compared to P. falciparum, resulted in a more advanced immunity to P. vivax in this age group compared to P. falciparum (Doolan et al., 2009; Longley et al., 2016). The major challenge to these cross-species comparisons lies within the intrinsic differences of Pf- and Pv-molFOB: whereas Pf-molFOB is a direct marker of mosquito-borne transmission, Pv-molFOB is a composite measure reflecting both newly acquired infections and those caused by relapses of previously acquired infections.
In this study, we extend the analysis of molFOB’s relationship with incidence of clinical malaria episodes to older PNG children and a lower transmission scenario. In addition, given the unique study design that randomized blood-stage only or blood- plus liver-stage treatment at enrolment (Robinson et al., 2015), we are now, for the first time, able to compare the incidence of newly acquired P. falciparum infections with both the incidence of newly acquired P. vivax infections and relapsing P. vivax infections. Advancing on previous studies that investigated each species individually (Mueller et al., 2012; Koepfli et al., 2013), we now provide a comparative analysis of P. falciparum and P. vivax, directly exploring the role of exposure to multiple Plasmodium species in the development of clinical immunity to P. falciparum and P. vivax malaria. We quantify in detail the extent of heterogeneity in molFOB on a small geographical scale and relate this to heterogeneity in clinical episode incidence to investigate effects of small-scale variation in malaria transmission on local malaria epidemiology. By combining in-depth molecular parasitological data with demographic and clinical data, this study thus provides detailed insights into the changing epidemiology of malaria in PNG in response to intense malaria control efforts.
Results
Demographic and parasitological parameters at enrolment
This study was conducted in six villages in Maprik district, East Sepik Province, PNG between August 2009 and May 2010 (Robinson et al., 2015). 524 children aged 5–10 years were enrolled and randomized to receive either chloroquine (CQ), artemeter-lumefantrine (AL) and primaquine (PQ); or CQ, AL and placebo. Demographic parameters of the 466 children that completed the full course of randomized treatment with PQ/CQ/AL (n = 233) or placebo/CQ/AL (n = 233), and were thereafter closely followed for 8 months, were comparable between the six villages (Table 1).
Table 1
Characteristics of study participants by village.
https://doi.org/10.7554/eLife.23708.003| Village | N | % female | Mean age (±SD) | Mean weight (±SD) | % LLIN use at enrolment* | Mean LLIN use during follow-up† (%, range) | Mean Hb (±SD) |
|---|---|---|---|---|---|---|---|
| Amahup | 119 | 53 | 7.6 (±1.5) | 19.8 (±3.3) | 99 | 99 (50–100) | 11.1 (±1.0) |
| Albinama | 99 | 43 | 7.7 (±1.5) | 20.0 (±3.3) | 95 | 97 (78–100) | 11.7 (±1.8) |
| Balanga | 54 | 59 | 7.8 (±1.6) | 19.8 (±4.3) | 96 | 99 (83–100) | 11.3 (±1.1) |
| Balif | 93 | 51 | 7.8 (±1.5) | 20.3 (±3.3) | 91 | 99 (69–100) | 11.7 (±1.2) |
| Bolumita | 70 | 50 | 7.4 (±1.7) | 19.3 (±2.9) | 77 | 92 (56–100) | 10.7 (±1.0) |
| Numangu | 31 | 55 | 7.4 (±1.6) | 19.2 (±4.6) | 100 | 100 (92–100) | 12.1 (±1.4) |
| Total | 466 | 51 | 7.6 (±1.5) | 19.8 (±3.5) | 93 | 100 (50–100) | 11.4 (±1.4) |
-
* LLIN use in the night preceding enrolment.
† Information on LLIN use in the previous night was collected at each follow-up visit and averaged across follow-up per participant. Mean LLIN use by village was calculated from the averaged individual LLIN use.
-
Hb: Haemoglobin.
P. vivax was the most common infection at enrolment with 48% of children positive by quantitative PCR (qPCR), followed by P. falciparum (24%), P. malariae (15%) and P. ovale (3%; Table 2). 39% of children were not infected with any Plasmodium species at enrolment. The vast majority of P. malariae (75%) and almost all P. ovale infections (93%) occurred in children co-infected with either P. vivax and/or P. falciparum (Table 2). Prevalence of each Plasmodium species varied between villages (P. falciparum, 9–71%; P. vivax, 38–67%; P. malariae, 8–40%; P. ovale, 0–11%; Table 2) and was highest in Bolumita for all species. Accordingly, mixed-species infections were also most prevalent in Bolumita (Table 2). The multiplicity of infection (MOI), that is, the number of parasite genotypes per infection, also varied between villages for both species (mean P. falciparum MOI, 1.1–2.2 clones/infection; mean P. vivax MOI, 1.6–2.9 clones/infection) and children from Bolumita carried more multi-clone infections with P. vivax and P. falciparum than children in other villages (Table 2). Mean P. falciparum parasite density was almost two- to six-fold higher in Bolumita (331 18S rRNA gene copies/µl) than in other villages (56–192 18S rRNA gene copies/µl, Table 2).
Table 2
Plasmodium infection status at enrolment by village.
https://doi.org/10.7554/eLife.23708.004| Village | P. falciparum | P. vivax | P. malariae | P. ovale | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| N pos. | Prevalence by qPCR (CI95) | % mixed* | Mean† density (IQR) | Mean MOI‡ (range) | N pos. | Prevalence by qPCR (CI95) | % mixed* | Mean† density (IQR) | Mean MOI‡ (range) | N pos. | Prevalence by qPCR (CI95) | % mixed* | N pos. | Prevalence by qPCR (CI95) | % mixed* | |
| Albinama | 18 | 18 (11–27) | 72 | 131 (38–189) | 1.4 (1–4) | 54 | 55 (44–65) | 24 | 3 (1–17) | 1.8 (1–7) | 9 | 9 (5–17) | 67 | 5 | 5 (2–12) | 100 |
| Amahup | 14 | 12 (7–19) | 57 | 56 (14–105) | 1.6 (1–5) | 46 | 39 (30–48) | 24 | 3 (1–29) | 2.2 (1–7) | 12 | 10 (6–17) | 83 | 0 | 0 | |
| Balanga | 15 | 28 (17–42) | 67 | 79 (30–848) | 1.7 (1–5) | 23 | 43 (30–57) | 43 | 2 (1–28) | 2.0 (1–7) | 9 | 17 (8–30) | 56 | 2 | 3 (0–14) | 50 |
| Balif | 8 | 9 (4–17) | 63 | 64 (10–325) | 2.0 (1–4) | 35 | 38 (30–48) | 14 | 2 (1–14) | 1.9 (1–6) | 7 | 8 (3–15) | 57 | 0 | 0 | |
| Bolumita | 50 | 71 (59–81) | 80 | 331 (62–1988) | 2.2 (1–8) | 47 | 67 (55–78) | 81 | 3 (2–27) | 2.9 (1–10) | 28 | 40 (29–52) | 89 | 8 | 11 (5–22) | 100 |
| Numangu | 8 | 26 (13–45) | 75 | 192 (30–848) | 1.1 (1–2) | 18 | 58 (39–75) | 28 | 3 (1–25) | 1.6 (1–5) | 4 | 13 (4–31) | 50 | 0 | 0 | |
| Overall | 113 | 24 (20–28) | 73 | 163 (20–1103) | 1.9 (1–8) | 223 | 48 (43–52) | 37 | 3 (1–23) | 2.2 (1–10) | 69 | 15 (12–18) | 75 | 15 | 3 (2–5) | 93 |
| p-value§ | <0.001 | 0.034 | 0.086 | 0.047 | <0.001 | <0.001 | 0.947 | 0.020 | <0.001 | 0.086 | <0.001 | 0.133 | ||||
-
* % of infections by qPCR that are mixed-species infections.
† Geometric mean of species-specific 18S rRNA copy numbers per µl blood.
-
‡ MOI, multiplicity of infection: number of Pf-msp2 and Pv-msp1F3 alleles per infection.
§ Differences between villages were tested for using Chi2 and Fisher’s exact test (prevalence, proportion mixed) or Kruskal-Wallis test (MOI, log10-transformed parasite density).
molFOB and parasite prevalence after randomized radical cure treatment
Children who had received PQ for clearance of P. vivax hypnozoites experienced similar numbers of new blood-stage infections with P. falciparum and P. vivax during follow-up (mean Pf-molFOB = 1.5 CI95 [1.3–1.7] new blood-stage clones/year, Pv-molFOB = 1.6 [1.4–1.9] new blood-stage clones/year, Figure 1A, Figure 1—figure supplement 1). Pf-molFOB in the placebo arm was comparable to the PQ arm (mean Pf-molFOB = 1.4 [1.2–1.6] new blood-stage clones/year), whereas due to the hypnozoite reservoir Pv-molFOB was more than three times higher in the placebo arm compared to the PQ arm (mean Pv-molFOB = 5.4 [4.9–5.8] new blood-stage clones/year). Pv-molFOB in the placebo arm showed a pronounced peak at months 2–3 of follow-up, which likely represents a wave of fast-relapsing hypnozoites in children who did not receive PQ (Figure 1A).
Figure 1 with 2 supplements see all
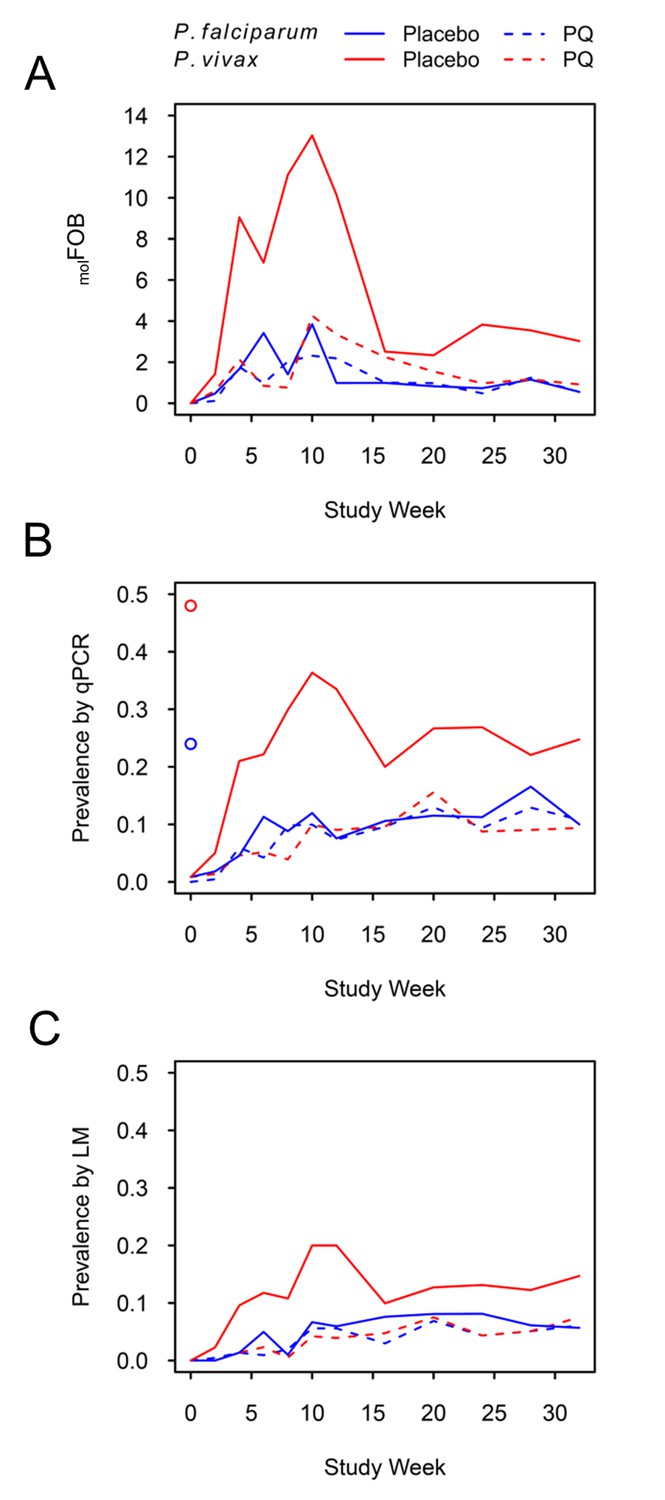
P. falciparum and P. vivax molFOB (A), prevalence by qPCR (B) and LM (C) by week of follow-up.
Blue lines, P. falciparum; red lines, P. vivax; solid lines, placebo arm; dashed lines, PQ arm. Open circles in (B) mark enrolment qPCR prevalence for each species.
P. vivax prevalence in the PQ arm was comparable to P. falciparum prevalence throughout the study and increased steadily, irrespective of the diagnostic method used (Figure 1B and C). P. vivax prevalence increased more rapidly in the placebo arm until month 3 of follow-up and dropped thereafter, similar to patterns in Pv-molFOB in the same arm. Prevalence as measured by qPCR did not reach pre-treatment levels until the end of the study for any of the four Plasmodium species (Figure 1B, Figure 1—figure supplement 2).
At the end of follow-up, P. vivax prevalence by qPCR in the placebo arm was 25% [19–31%], and therefore more than two-fold higher than in the PQ arm (9% [6–14%]; Figure 1B). Also, throughout follow-up, P. vivax prevalence in the placebo arm was 2–3 fold higher compared to the PQ arm, suggesting that at least 50% of the overall P. vivax prevalence in this cohort can be attributed to the contribution of relapses.
Similarly, throughout and at the end of follow-up P. vivax prevalence in the placebo arm was 2–3 fold higher compared to P. falciparum (irrespective of treatment arm; P. falciparum prevalence at end of follow-up, 10% [8–14%]), which is in agreement with the prevalence pattern at enrolment. Assuming equal transmission from mosquitoes for both species, which was corroborated by a comparable Pf-molFOB and Pv-molFOB in the PQ arm, P. vivax relapses have contributed to a P. vivax prevalence twice as high as that of P. falciparum.
Risk of recurrent blood-stage infections and molFOB during follow-up
In the present study design, recurrent blood-stage infection can either originate from a new transmission event (both arms and all species) or for P. vivax and P. ovale also from a relapse of any previous infection (placebo arm only). After adjusting for the effect of PQ treatment (Robinson et al., 2015), village of residence and infection status by qPCR at enrolment were the main predictors for the risk of recurrent Plasmodium spp. during follow-up (Table 3). Interestingly, in addition to a protective effect against recurrent P. vivax and P. ovale, the risk of recurrent P. falciparum was also reduced by 27% [0–48%] after PQ treatment (p=0.064).
Table 3
Multivariable predictors for time to recurrent blood-stage infection with Plasmodium species by qPCR
https://doi.org/10.7554/eLife.23708.008| Variable | P. vivax | P. falciparum | P. malariae | P. ovale | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AHR* | CI95 | p-value | AHR* | CI95 | p-value | AHR* | CI95 | p-value | AHR* | CI95 | p-value | |
| PQ treatment | 0.18 | 0.13–0.25 | <0.001 | 0.73 | 0.52–1.02 | 0.064 | 0.51 | 0.22–1.19 | 0.121 | 0.31 | 0.12–0.75 | 0.010 |
| Age | 0.95 | 0.87–1.04 | 0.247 | 1.05 | 0.94–1.17 | 0.361 | 0.98 | 0.75–1.29 | 0.905 | 0.96 | 0.74–1.26 | 0.793 |
| LLIN use at enrolment | 0.62 | 0.39–0.98 | 0.043 | 0.84 | 0.49–1.44 | 0.531 | 1.33 | 0.33–6.09 | 0.715 | 0.95 | 0.26–3.43 | 0.936 |
| Hb at enrolment (g/dl) | 0.88 | 0.80–0.98 | 0.019 | 0.90 | 0.80–1.02 | 0.099 | 0.83 | 0.61–1.12 | 0.224 | 0.92 | 0.66–1.28 | 0.634 |
| Village | ||||||||||||
| Albinama (ref) | 1 | 1 | 1 | 1 | ||||||||
| Amahup | 0.45 | 0.29–0.71 | 0.001 | 0.58 | 0.31–1.11 | 0.101 | 0.34 | 0.07–1.79 | 0.205 | 2.83 | 0.29–27.48 | 0.370 |
| Balanga | 2.15 | 1.40–3.31 | <0.001 | 1.81 | 0.99–3.30 | 0.054 | 0.92 | 0.24–3.60 | 0.910 | 7.74 | 0.85–70.45 | 0.070 |
| Balif | 1.00 | 0.66–1.54 | 0.983 | 0.60 | 0.30–1.19 | 0.145 | 0.24 | 0.03–2.07 | 0.193 | 4.60 | 0.51–41.41 | 0.173 |
| Bolumita | 3.34 | 2.09–5.33 | <0.001 | 4.73 | 2.69–8.30 | <0.001 | 1.21 | 0.34–4.31 | 0.770 | 19.43 | 2.19–172.37 | 0.008 |
| Numangu | 0.83 | 0.44–1.59 | 0.583 | 2.29 | 1.17–4.50 | 0.015 | 0.82 | 0.15–4.53 | 0.823 | 3.17 | 0.19–52.41 | 0.420 |
| Infection status at enrolment (by qPCR) | ||||||||||||
| Uninfected (ref) | 1 | 1 | 1 | 1 | ||||||||
| P. vivax | 1.27 | 0.91–1.78 | 0.165 | 1.37 | 0.86–2.20 | 0.186 | 0.92 | 0.20–4.18 | 0.913 | 2.17 | 0.68–6.97 | 0.192 |
| P. falciparum | 1.36 | 0.84–2.19 | 0.205 | 1.56 | 0.86–2.82 | 0.145 | 3.54 | 0.85–14.72 | 0.083 | 1.25 | 0.26–5.90 | 0.779 |
| P. malariae | 0.83 | 0.38–1.85 | 0.655 | 0.99 | 0.38–2.56 | 0.977 | 6.35 | 1.31–30.81 | 0.022 | 1.58 | 0.17–14.30 | 0.676 |
| Mixed P.f. or P.v.† | 1.74 | 1.14–2.65 | 0.010 | 2.08 | 1.25–3.48 | 0.005 | 3.37 | 0.88–12.90 | 0.076 | 2.03 | 0.55–7.53 | 0.287 |
-
* AHRs were modeled using Cox proportional hazard regression.
† Mixed infection including P. falciparum or P. vivax infection in conjunction with one or more other Plasmodium spp.
-
PQ: Primaquine; LLIN: long-lasting insecticide-treated net; Hb: haemoglobin.
The risk of a recurrent infection (measured by qPCR) with P. falciparum, P. vivax and P. ovale varied more than 7-fold between villages, with a higher risk observed in Bolumita (78%, 77%, and 15% with recurrent P. vivax, P. falciparum and P. ovale, respectively) compared to the other villages (recurrent P. vivax, range 25–73%; recurrent P. falciparum, range 12–44%; recurrent P. ovale, range 0–7%). For P. falciparum and P. vivax, a mixed infection at enrolment as measured by qPCR was further associated with up to a two-fold increased risk of recurrent infection (P. falciparum: AHR = 2.08 [1.25–3.48], p=0.005; P. vivax: AHR = 1.74 [1.14–2.65], p=0.010; Table 3), supporting the idea that focal transmission within villages leads to the presence of high-risk and low-risk individuals. For P. malariae, the infection status at enrolment was a stronger predictor of risk of recurrent infection than village of residence. An infection with P. falciparum, P. malariae or a mixed infection at enrolment as measured by qPCR was associated with up to a 6-fold increase in risk of recurrent P. malariae (AHRPf-enrol = 3.54 [0.85–14.72], p=0.083; AHRPm-enrol = 6.35 [1.31–30.81], p=0.022; AHRmixed = 3.37 [0.88–12.90], p=0.076; Table 3).
Reported use of a LLIN during the night previous to enrolment was associated with a reduced risk of recurrent P. vivax and P. falciparum in univariate analyses (Supplementary file 1 - Table 1) but to a lesser extent in multivariable analyses (P. vivax: AHR = 0.62 [0.39–0.98], p=0.043, P. falciparum: AHR = 0.84 [0.49–144], p=0.531). Haemoglobin (Hb) level at enrolment was negatively associated with the risk of recurrent infection with P. vivax (AHR = 0.88 [0.80–0.98], p=0.019) and P. falciparum (AHR = 0.90 [0.80–1.02], p=0.099). Patterns in the risk of recurrent infections with P. falciparum and P. vivax as measured by light microscopy (LM, Supplementary file 2) were similar to those observed for re-infection as measured by qPCR. When based on LM observation (but not as measured by qPCR), increasing age was associated with a reduced risk of recurrent P. vivax (AHR = 0.85 [0.77–0.95], p=0.004; Supplementary file 2) but an increased risk of recurrent P. falciparum (AHR = 1.16 [1.01–1.33], p=0.037; Supplementary file 2).
The incidence of new P. falciparum and P. vivax blood-stage clones detected during follow-up, that is molFOB, was highly variable between individual children and ranged from 0 to 18 new clones/year for P. falciparum (Figure 2A) and 0 to 36 or 23 new blood-stage clones/year for P. vivax in the placebo or PQ arm, respectively (Figure 2B). Mean Pf- and Pv-molFOB varied significantly between villages and were higher in Bolumita (Pf-molFOB = 4.9 new blood-stage clones/year, Pv-molFOBPQ arm=4.4 new blood-stage clones/year, Pv-molFOBplacebo arm=12.1 new blood-stage clones/year; Table 4; Figure 3) than in the other villages (Pf-molFOB, range 0.7–1.8 new blood-stage clones/year; Pv-molFOBPQ arm, range 0.03–2.2 new blood-stage clones/year; Pv-molFOBplacebo arm, range 2.3–7.4 new blood-stage clones/year). In univariate analyses, new P. vivax infections were strongly associated with new P. falciparum infections per sampling interval and vice versa, suggesting concurrent exposure to the two species (Supplementary file 1 – Table 2). However, these effects were reduced when other variables of varying exposure such as village of residence or infection at enrolment were included in the multivariable model (P. vivax: IRRPQ arm=1.32 [0.92–1.89], p=0.134; IRRPlacebo arm=1.10 [0.85–1.42], p=0.466; P. falciparum: IRR = 1.15 [0.97–1.36], p=0.100, Table 4). LLIN use, although strongly associated with lower Pf- and Pv-molFOB in univariate analyses (Supplementary file 1 – Table 2), remained significantly associated in multivariable models only for P. vivax in the placebo arm, where sleeping under a LLIN in the night previous to enrolment was associated with a 38% [9–57%] reduction in Pv-molFOB (p=0.013, Table 4). Each additional year of age was associated with a 14% [0–26%] reduction Pv-molFOB per sampling interval in the PQ arm (p=0.059), while no age effect was observed in the placebo arm or for P. falciparum (Table 4). Hb level at enrolment was negatively associated with Pf- and Pv-molFOB (P. vivax: IRRPQ arm=0.85 [0.72–1.01], p=0.063; IRRPlacebo arm=0.91 [0.85–0.99], p=0.025; P. falciparum: IRR = 0.85 [0.75–0.97], p=0.013), suggesting anaemia in individuals continuously exposed to blood-stage infections.
Figure 2
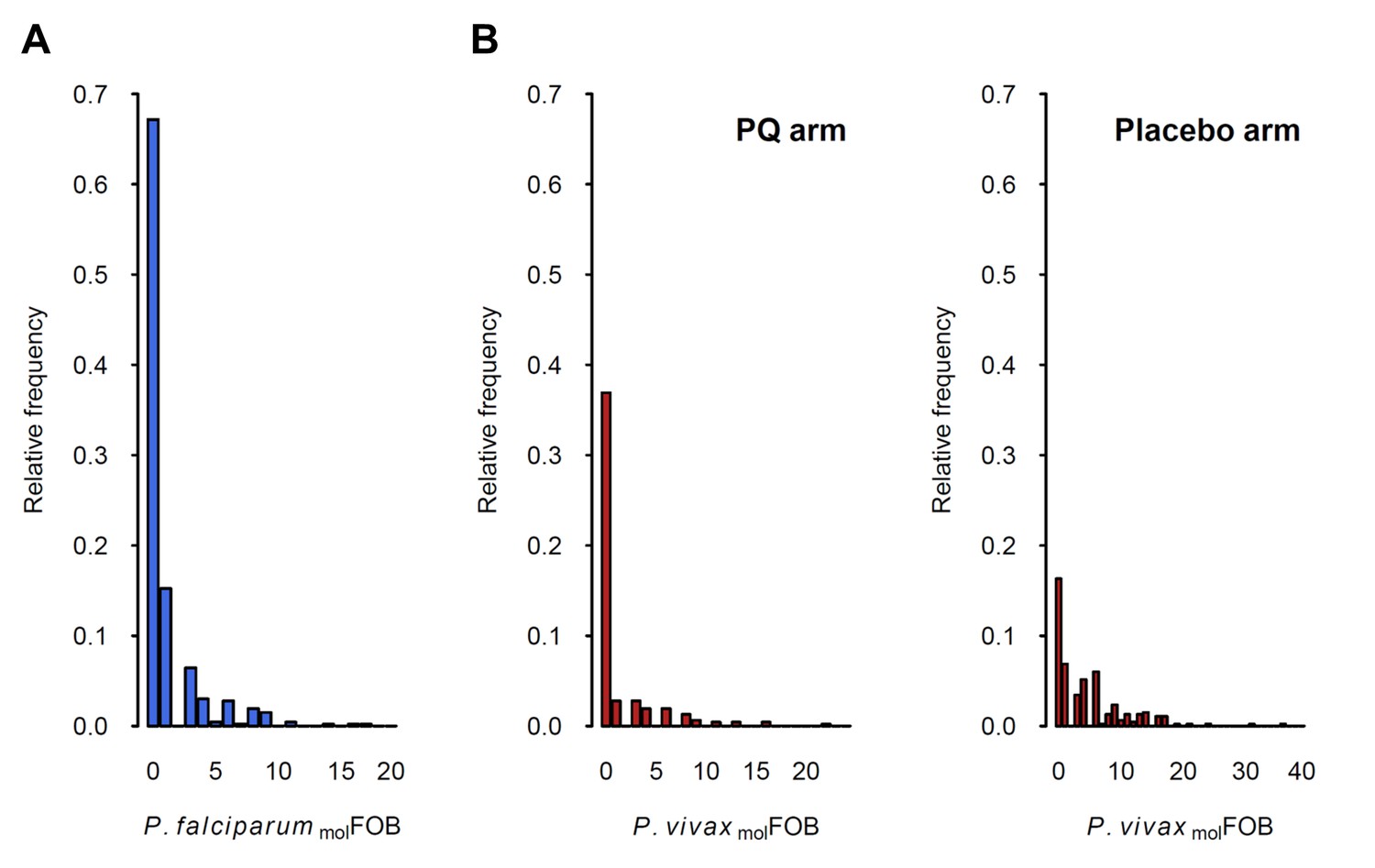
Distribution of P.falciparum molFOB (A) and P. vivax molFOB by treatment arm (B).
Relative frequencies among the 466 children are shown.
Figure 3
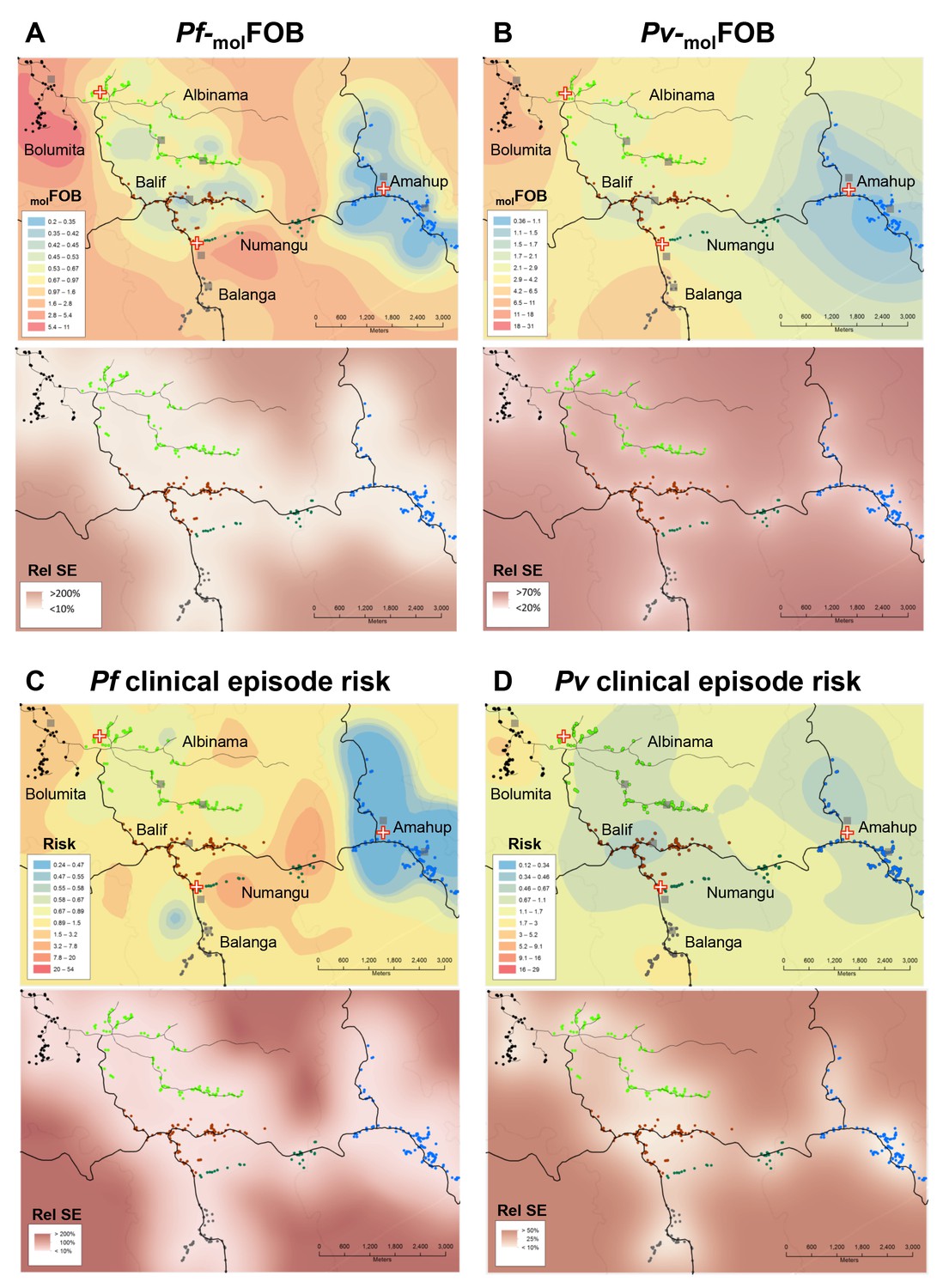
Heterogeneity in molFOB (A, B) and clinical episode risk (C, D) of P.falciparum (A, C) and P. vivax (B, D).
Upper panels show the kriging fit of model predictions of molFOB and clinical episode risk of children in both treatment arms. Lower panels show the standard error relative to the kriging estimate. Dots represent study participants’ houses and are color-coded according to village. Black lines: vehicle-accessible road; dark grey lines: vehicle-inaccessible road; light grey lines: river; red/white cross: health center or aid post; grey square: school or enrolment location. Maps were prepared using ArcGIS 10.2 (Esri, USA).
Table 4
Multivariable predictors of Pv- and Pf-molFOB per follow-up interval.
Model predictions from this model were used for mapping molFOB in Figure 3A.
| Variable | P. vivax | P. falciparum | |||||||
|---|---|---|---|---|---|---|---|---|---|
| PQ arm | Placebo arm | Combined arms | |||||||
| IRR* | CI95 | p-value | IRR* | CI95 | p-value | IRR* | CI95 | p-value | |
| PQ treatment | n.a.† | n.a. | n.a. | n.a. | n.a. | n.a. | 0.89 | 0.65–1.22 | 0.474 |
| New P. falc. infections in interval‡ | 1.32 | 0.92–1.89 | 0.134 | 1.10 | 0.85–1.42 | 0.466 | n.a. | n.a. | n.a. |
| New P. vivax infections in interval‡ | n.a. | n.a. | n.a. | n.a. | n.a. | n.a. | 1.15 | 0.97–1.36 | 0.100 |
| Age | 0.86 | 0.74–1.01 | 0.059 | 0.95 | 0.87–1.04 | 0.305 | 1.03 | 0.92–1.14 | 0.640 |
| LLIN use at enrolment | 0.96 | 0.51–1.79 | 0.897 | 0.62 | 0.43–0.91 | 0.013 | 1.07 | 0.7–1.62 | 0.755 |
| Hb at enrolment (g/dl) | 0.85 | 0.72–1.01 | 0.063 | 0.91 | 0.85–0.99 | 0.025 | 0.85 | 0.75–0.97 | 0.013 |
| Village | |||||||||
| Albinama (ref) | 1 | 1 | 1 | ||||||
| Amahup | 0.02 | 0–0.11 | <0.001 | 0.56 | 0.34–0.91 | 0.020 | 0.52 | 0.25–1.07 | 0.074 |
| Balif | 0.85 | 0.4–1.8 | 0.664 | 1.74 | 1.16–2.61 | 0.007 | 1.81 | 0.98–3.35 | 0.059 |
| Balanga | 0.28 | 0.1–0.82 | 0.020 | 1.13 | 0.73–1.73 | 0.590 | 0.75 | 0.37–1.52 | 0.423 |
| Bolumita | 1.52 | 0.73–3.17 | 0.268 | 2.67 | 1.83–3.9 | <0.001 | 6.05 | 3.32–11.05 | <0.001 |
| Numangu | 0.5 | 0.15–1.68 | 0.264 | 0.76 | 0.4–1.43 | 0.394 | 2.8 | 1.39–5.64 | 0.004 |
| Study Day | |||||||||
| Day 0–35 (ref) | 1 | 1 | 1 | ||||||
| Day 36–80 | 1.37 | 0.54–3.48 | 0.509 | 1.99 | 1.39–2.84 | <0.001 | 2.42 | 1.44–4.07 | 0.001 |
| Day 81–175 | 1.34 | 0.57–3.12 | 0.503 | 0.89 | 0.61–1.3 | 0.538 | 1.13 | 0.7–1.84 | 0.616 |
| Day > 175 | 0.65 | 0.25–1.69 | 0.374 | 0.56 | 0.38–0.83 | 0.004 | 0.87 | 0.48–1.56 | 0.643 |
-
*IRRs were modeled per sampling interval using negative binomial generalized estimating equations allowing for repeated visits with log-link and an exchangeable correlation structure.
† n.a., not applicable.
-
‡molFOB in the follow-up interval (time-varying covariate).
PQ: Primaquine; LLIN: long-lasting insecticide-treated net; Hb: haemoglobin.
Patterns in the risk of P. vivax and P. falciparum clinical episodes
A total of 98 clinical malaria episodes, here defined as fever plus presence of LM-detectable parasites, were observed during the study period. Of these, 64 (65%) exceeded the previously established pyrogenic thresholds of 2500 and 500 parasites/µl per LM for P. falciparum and P. vivax, respectively (Mueller et al., 2009b). P. falciparum was the most common cause of clinical malaria episodes (P. falciparum, 64 clinical episodes; P. vivax, 31 clinical episodes; mixed P. falciparum/P. vivax by LM, 3 clinical episodes), despite lower incidence of new P. falciparum blood-stage clones compared with P. vivax (P. falciparum, 342 new P. falciparum blood-stage clones; P. vivax, 849 new blood-stage clones). Including clinical episodes with mixed infection as determined by LM in the estimates for both species, clinical incidence rate (IR) was 0.28 [0.21–0.35] P. falciparum episodes/year and 0.12 [0.08–0.17] P. vivax episodes/year. At least one new blood-stage clone was detected in 70% (47/67) of samples from P. falciparum and 71% (24/34) of samples from P. vivax clinical episodes. Of these clinical episodes with new blood-stage clones, 96% (45/47) and 83% (20/24) carried only the new but no persistent P. falciparum and P. vivax clones, respectively.
P. vivax clinical episodes occurred mainly in the placebo arm shortly after directly observed treatment (DOT) (Robinson et al., 2015), the time of peak Pv-molFOB due to relapsing hypnozoites (Figure 1A). On an individual level, Pv-molFOB was positively associated with the risk of clinical episodes and each additional blood-stage P. vivax clone increased the risk of experiencing a P. vivax clinical episode slightly (AHR = 1.07 [1.04–1.09], p<0.001; Table 5). No significant differences in P. vivax clinical episode risk were observed between villages after adjusting for individual molFOB. The risk for a P. vivax clinical episode decreased significantly with age (AHR = 0.62 [0.46–0.84], p=0.002; Table 5). This was paralleled by a decrease in P. vivax densities with age (by qPCR, exp(β)=0.90 [0.83–0.98], p=0.016; Supplementary file 3) indicative of more advanced immunity against P. vivax and thus better control of P. vivax densities in older children.
Table 5
Multivariable predictors for time to P. vivax and P. falciparum clinical episodes.
Model predictions from this model were used for mapping the relative risk of clinical malaria episodes in Figure 3C and D.
| Variable | P. vivax | P. falciparum | ||||
|---|---|---|---|---|---|---|
| AHR* | CI95 | p-value | AHR* | CI95 | p-value | |
| PQ treatment | 0.76 | 0.34–1.68 | 0.497 | 1.79 | 1.05–3.03 | 0.031 |
| P. vivax molFOB‡ | 1.07 | 1.04–1.09 | <0.001 | n.a. | n.a. | n.a. |
| P. falciparum molFOB‡ | n.a. | n.a. | n.a. | 1.15 | 1.11–1.21 | <0.001 |
| Age | 0.62 | 0.46–0.84 | 0.002 | 0.98 | 0.85–1.13 | 0. 799 |
| LLIN use at enrolment | 0.84 | 0.24–2.88 | 0.778 | 0.44 | 0.22–0.87 | 0.018 |
| Hb at enrolment (g/dl) | 0.95 | 0.74–0.67 | 0.668 | 0.85 | 0.71–1.01 | 0.070 |
| Village | ||||||
| Albinama (ref) | 1 | 1 | ||||
| Amahup | 0.89 | 0.23–3.46 | 0.871 | 0.65 | 0.20–2.08 | 0.465 |
| Balif | 1.48 | 0.45–4.86 | 0.518 | 1.26 | 0.50–3.14 | 0.626 |
| Balanga | 0.85 | 0.21–3.53 | 0.827 | 1.39 | 0.59–3.30 | 0.455 |
| Bolumita | 0.99 | 0.24–4.03 | 0.987 | 1.32 | 0.58–3.03 | 0.508 |
| Numangu | 1.00 | 0.23–4.31 | 0.997 | 4.29 | 2.06–8.97 | <0.001 |
| Infection status at enrolment (by qPCR) | ||||||
| Uninfected (ref) | 1 | 1 | ||||
| P. vivax | 0.77 | 0.29–2.07 | 0.608 | 1.64 | 0.91–2.95 | 0.101 |
| P. falciparum | 1.74 | 0.59–5.11 | 0.316 | 0.97 | 0.34–2.77 | 0.954 |
| Mixed P.f. or P.v. | 1.59 | 0.56–4.50 | 0.381 | 1.24 | 0.57–2.68 | 0.582 |
-
* AHRs were modeled using multiple failure Cox proportional hazard regression.
† n.a., not applicable
-
‡ Average molFOB until the time of failure (time-varying covariate).
PQ: Primaquine; LLIN: long-lasting insecticide-treated net; Hb: haemoglobin.
Patterns in the occurrence of P. falciparum clinical episodes during follow-up were more complex. Between-village variation in P. falciparum clinical episode risk remained significant even after adjusting for individual exposure. This effect was mainly apparent in Numangu, where children were at three- to six-fold higher risk for clinical episodes than children in other villages (Numangu AHR = 4.29 [2.06–8.97], other villages range AHR = 0.65 [0.20–2.08] to 1.39 [0.59–3.30]; Table 5). Overall, Pv-molFOB was positively associated with the risk of clinical episodes and each additional P. falciparum blood-stage clone slightly increased the risk for P. falciparum clinical episodes (AHR = 1.15 [1.11–1.21], p<0.001; Table 5); however, relative to the number of new blood-stage clones, P. falciparum clinical episodes were less frequent in highly exposed children compared to low-exposed children (Figure 4). One clinical episode per three new blood-stage clones was detected in the least exposed children (Pf-molFOB <4 new blood-stage clones/year), but only one clinical episode per 15 blood-stage clones in the highest exposed children (Pf-molFOB >9 new blood-stage clones/year, Fisher’s exact test p<0.001). Age was not associated with the risk of P. falciparum clinical episodes. LLIN use at enrolment was associated with a 56% [13–78%] reduced risk of P. falciparum clinical episodes (p=0.018; Table 5).
Figure 4
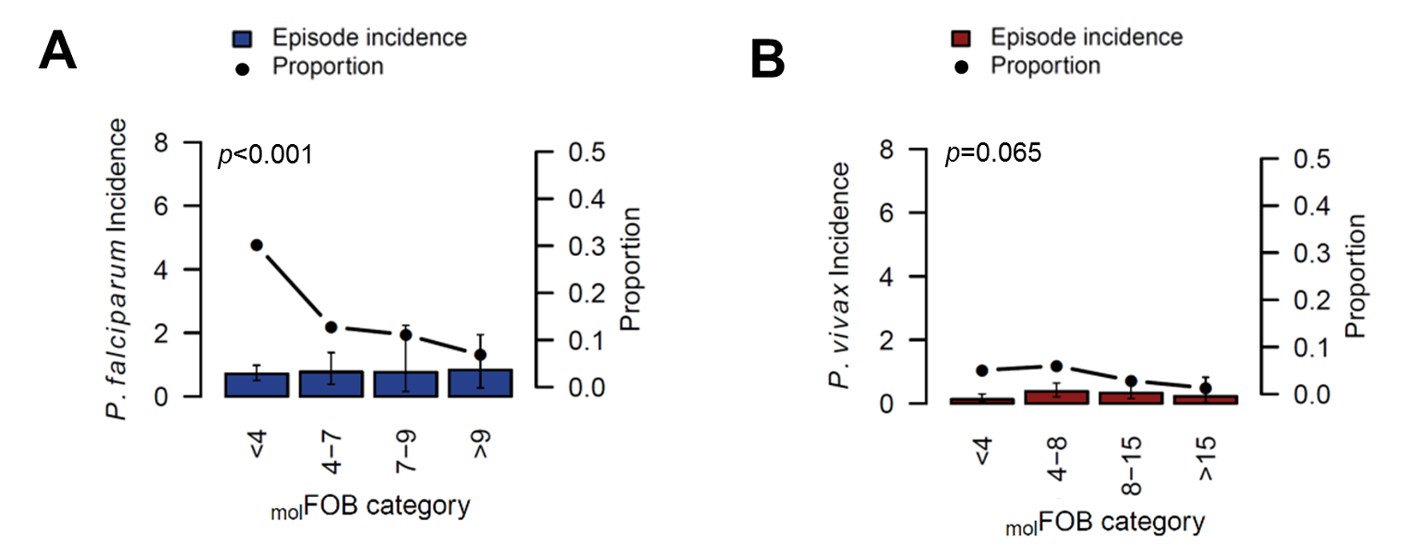
The incidence of P.falciparum (A) and P. vivax (B) clinical episodes relative to molFOB.
Mean clinical episode incidence is shown as bars (left axis) and proportion of clinical episode incidence divided by molFOB as connected dots (right axis). Error bars represent 95% CIs. p-values refer to the differences between groups in the proportion of clinical episodes and new infections, assessed by Chi2 or Fisher’s exact test.
A higher risk for P. falciparum clinical episodes in children that had received PQ treatment for clearance of P. vivax liver stages was observed (AHR = 1.79 [1.05–3.03], p=0.031; Table 5), suggesting a potential protective effect of P. vivax infections against P. falciparum clinical episodes. Analysis to further explore this revealed that a concurrent or recent infection (i.e., at the same or preceding follow-up visit) with P. vivax reduced the odds of a P. falciparum clinical episode by 65% [22-85%] (p=0.011; Table 6). Further indications for a potential interaction between the two species was also observed when analyzing P. falciparum parasite densities, which were reduced by 55% [19–75%] (p=0.008; Supplementary file 3) in mixed P. falciparum/P. vivax infections compared to P. falciparum single infections, indicative of suppression of one of the species in mixed infections.
Table 6
Multivariable predictors for odds of P. falciparum clinical episodes
https://doi.org/10.7554/eLife.23708.015| P. falciparum episode | |||
|---|---|---|---|
| Variable | OR* | CI95 | p-value |
| PQ treatment | 1.42 | 0.80–2.52 | 0.226 |
| P. vivax qPCR positive† | 0.35 | 0.15–0.78 | 0.011 |
| P. falciparum molFOB‡ | 1.21 | 1.10–1.34 | <0.001 |
| Age | 0.93 | 0.80–1.09 | 0.370 |
| LLIN at enrolment | 0.37 | 0.16–0.83 | 0.016 |
| Hb (g/dl) at enrolment | 0.88 | 0.70–1.11 | 0.292 |
| Village | |||
| Albinama (ref) | 1 | ||
| Amahup | 0.41 | 0.12–1.39 | 0.154 |
| Balif | 0.9 | 0.26–3.08 | 0.870 |
| Balanga | 1.19 | 0.42–3.39 | 0.747 |
| Bolumita | 1.48 | 0.42–5.17 | 0.540 |
| Numangu | 4.17 | 1.64–10.58 | 0.003 |
| Study Day | |||
| Day 0–80 | 1 | ||
| Day 81–175 | 0.99 | 0.51–1.91 | 0.972 |
| Day > 175 | 0.83 | 0.39–1.75 | 0.629 |
-
* ORs were modeled using a binomial generalized estimating equation with logit link function using an exchangeable correlation structure.
† Determined as P. vivax positive at the same or previous sampling visit.
-
‡molFOB in the follow-up interval (time-varying covariate).
PQ: Primaquine; LLIN: long-lasting insecticide-treated net; Hb: haemoglobin.
Discussion
In the present study, we describe striking heterogeneity in malaria transmission not only between closely neighboring communities in Maprik district, PNG, but also substantial differences in exposure between individual children from the same village. On village level this heterogeneity is apparent both when using traditional markers such as prevalence of infection, as well as when using the novel `reference standard` marker of individual exposure molFOB. The increased resolution provided by molFOB further allows quantifying heterogeneity in exposure to new blood-stage infections between individual children. Extending an earlier study in a neighboring area in which younger children had been enrolled, and that had identified molFOB as the most important predictor of malaria clinical episodes (Mueller et al., 2012; Koepfli et al., 2013), we confirmed that molFOB remains significantly associated with the risk for clinical episodes, but other factors such as age (P. vivax), or a mixed Pf/Pv infection and village factors not captured by any of the other parameters assessed (P. falciparum) have a stronger effect on the risk for clinical malaria (Table 5 and 6).
Malaria transmission is often estimated by investigating the more accessible human host rather than the mosquito vector (Tusting et al., 2014). Because P. falciparum blood-stage infections are a direct outcome of mosquito-to-human transmission, infection parameters assessed in the human blood closely reflect P. falciparum transmission. In contrast, relapses arising from dormant hypnozoites contribute substantially to P. vivax blood-stage infections (Robinson et al., 2015), thus complicating the assessment of mosquito-to-human P. vivax transmission via infection parameters measured in the human blood. The unique design of this study, that combined clearance of hypnozoites in half of the study participants with subsequent measurement of Pv-molFOB, allowed us to identify the burden of P. vivax infections due to mosquito-to-human transmission (in hypnozoite-cleared individuals) and compare it to the total burden of P. vivax infections. We found a highly similar incidence and comparable temporal and spatial heterogeneity of P. falciparum and P. vivax infection acquired through renewed exposure to infected mosquito bites. In children that experienced the full burden of relapses we found two-fold higher P. vivax infection prevalence and 4-times higher incidence (molFOB) compared to P. falciparum. This first quantitative comparative assessment of P. falciparum and P. vivax transmission using non-entomological molecular parameters thus indicates that the observed differences in epidemiology between the two species are largely due to the high burden of relapsing P. vivax blood-stage infections (Robinson et al., 2015). Our molecular results thus support recent entomological data from Dreikikir district, 50 km from Maprik in East Sepik Province (Reimer et al., 2016) as well as earlier studies in East Sepik (Hii et al., 2001) that found similar sporozoite rates for P. falciparum and P. vivax.
Our previous analysis of this cohort had investigated the contribution of the hypnozoite reservoir to P. vivax infection and disease in order to inform strategies for achieving a sustained reduction of the P. vivax burden in PNG (Robinson et al., 2015). Here, we now describe the post-treatment re-infection dynamics in higher temporal detail. Through a detailed comparison of these patterns for P. vivax and P. falciparum in PQ and placebo-treated children we further elucidate the contribution of relapses to P. vivax prevalence and clinical incidence, which are the most commonly used parameters for planning and monitoring of malaria control strategies. P. vivax relapses accounted for more than half of the observed P. vivax prevalence in this cohort, which is lower than what was previously estimated as the contribution of relapses towards Pv-molFOB by comparison of treatment arms (77%, Robinson et al., 2015). This difference can be accounted for by the higher number of P. vivax multiple clone infections that will accumulate more rapidly in the placebo-arm, where additional parasite clones from relapses and/or new infections may overlap with without a corresponding change in overall prevalence.
While we previously described a sustained effect of PQ treatment with significant reductions in Pv-molFOB observed up to eight months post treatment (Robinson et al., 2015), here, we describe temporal variation in relapse rate with a rapid and wave-like recurrence of P. vivax in children from the placebo arm, who had retained their hypnozoites (Figure 1A). The concurrent, modest peaks in Pf-molFOB and Pv-molFOB in the PQ arm represent seasonal variation in transmission, which is highest in December and January in the study area (Mueller et al., 2012); corresponding to weeks 8–14 of follow-up). A much higher peak and subsequent drop in appearance of new clones within three months after blood-stage only treatment, which was mirrored by a corresponding peak and drop in P. vivax prevalence (Figure 1B andC), suggests that the incidence of relapse infections in the blood was not constant during follow-up. P. vivax infections are often observed following treatment of P. falciparum malaria (Douglas et al., 2011), and it can thus been hypothesized that the frequency of relapses may be temporarily increased after blood-stage antimalarial treatment (White and Imwong, 2012). It is thus conceivable that the blood-stage antimalarial at baseline either triggered P. vivax relapses directly, or indirectly by allowing more hypnozoites to establish blood-stage infections in parasite-free hosts. Alternatively, blood-stage P. vivax infections from hypnozoites relapsing shortly after baseline treatment (during a period when antimalarial drugs were present at sub-curative levels) may be suppressed to sub-detectable densities until complete waning of drug levels, resulting in simultaneous proliferation and detection of many new blood-stage clones within the first weeks after treatment (Douglas et al., 2011; Tarning et al., 2014). More detailed modeling of the dynamics of individual P. vivax blood-stage infections and their association with potential triggers such as treatment or febrile illness will be required to determine the existence and importance of proposed relapse-triggers.
Malaria transmission showed high micro-spatial heterogeneity with more than 10-fold differences in Pf- and Pv-molFOB (in the PQ arm) between villages despite an overall high LLIN use by the study participants (during follow-up; village average use,>90%; individual use,>50%). Individual LLIN use at enrolment was nevertheless associated with a reduced risk of recurrent P. falciparum and P. vivax in univariate analyses. However, after adjustment for other related variables (i.e., village of residence or infection status) this association became non-significant. Children living in Bolumita, where both P. falciparum and P. vivax molFOB and prevalence were highest, had a modestly lower LLIN use (mean during follow-up, 92%; at enrolment, 77%) compared to children from other villages (mean during follow-up, 97–100%; at enrolment, 91–100%). It is conceivable that LLIN use in the Bolumita community may be less effective in reducing malaria transmission (Killeen et al., 2007; Smith et al., 2009). Potential differences between villages in mosquito density, behavior, sporozoite rate, proximity of house or play areas to mosquito breeding sites, or human behavioral factors (related to LLIN use or other risk factors) are however likely to be more important determinants for exposure to infective bites. Small-scale variations in vector species and distribution between and within villages in PNG have been described previously (Cattani et al., 1986; Reimer et al., 2016; Charlwood et al., 1986; Hii et al., 1997; Burkot et al., 1988) and likely account in a large part for the micro-geographic heterogeneity in malariological parameters observed in this and other studies.
Assessing the incidence of new infections from consecutive blood samples using molecular methods (as is necessary to determine molFOB), is complicated by fluctuating densities of clonal parasitemia that may temporarily fall below the limit of detection of the genotyping PCR, leading to imperfect detectability of clones (Bretscher et al., 2010; Felger et al., 2012; Koepfli et al., 2011). For P. falciparum, periodical sequestration of clones and absence from the peripheral blood at time of sampling may further contribute to imperfect detectability. For P. vivax, generally low parasite densities aggravate the problem of imperfect detectability, and dis- and re-appearance of clones may be a result of imperfect detectability or relapsing hypnozoites. The overall estimates of molFOB presented here may thus be biased. Accurately assessing the effects of this imperfect detectability on parameters estimated from longitudinal genotyping data, such as molFOB, requires complex mathematical modeling (Bretscher et al., 2010; Felger et al., 2012; Sama et al., 2005; Sama et al., 2006). However, although clonal detectability has been shown to decrease with age (Felger et al., 2012; Sama et al., 2006) and MOI (Koepfli et al., 2011), it is unlikely to vary substantially within our cohort’s age range and transmission setting. Hence the observed differences in molFOB are likely to accurately reflect the relative differences in individual exposure as well as in population transmission levels within the study area.
Evaluating the impact of malaria control efforts requires monitoring changes in malariological metrics over extended periods of time. Drawing comparisons between studies performed at different times in different age groups is particularly challenging because of the interplay of past and current exposure to infective bites and the resulting anti-malarial immunity in the study population of a certain age. In our cohort, fewer P. vivax clinical episodes than P. falciparum clinical episodes were detected despite a higher incidence of P. vivax blood-stage infections, which is consistent with earlier studies in children of similar age (Michon et al., 2007). The very low incidence of clinical P. vivax episodes in our cohort, at 0.16 clinical episodes/year (placebo arm [Robinson et al., 2015]), contrasts drastically with that of 2.46 P. vivax clinical episodes/year observed in an earlier observational cohort of younger children aged 1–4 years from the same area (Lin et al., 2010). The 3-fold difference in Pv-molFOB between the two cohorts seems modest when compared to the 15-fold difference in the incidence of clinical Pv episodes (Koepfli et al., 2013). This suggests that the much lower incidence of P. vivax clinical illness in 5–10 years old children of this study is more likely explained by an advanced state of immunity to P. vivax compared to the younger children of the earlier cohort than the drop in P. vivax transmission. Consistently, age emerged as the strongest factor associated with protection against P. vivax clinical episodes, Pv-molFOB and P. vivax parasite density. Like in the previous cohort of younger children from neighboring villages (Lin et al., 2010) the incidence clinical P. vivax clinical episodes dropped significantly with age. Unlike in the previous cohort of younger children (Koepfli et al., 2013), in this cohort we additionally observed a drop in Pv-molFOB as well as P. vivax densities with age (Table 4 and Supplementary file 3). This is a further indication of the substantial clinical immunity to P. vivax acquired during years of past exposure in the children of this study, which is still ongoing after the age of five.
In sharp contrast to the age-dependent decline of P. vivax clinical episode incidence, no age-dependent decrease in the incidence of clinical episodes was observed for P. falciparum. In a previous cohort study conducted in 2004 in 5–14 year old children in an area from PNG with substantially higher transmission levels (mean incidence risk 5.0 versus 0.8 infections/year, Michon et al., 2007; Robinson et al., 2015), the risk of moderate- to high-density P. falciparum infections decreased significantly with age (Michon et al., 2007). Clinical immunity to P. falciparum in children of this earlier cohort was not only significantly further advanced compared to children of this cohort, but in addition, transmission was more homogeneous in the area of that study. As a consequence age was a much better marker of life-time exposure and thus immune status compared to the present cohort.
In this cohort, exposure to P. falciparum infections was highly heterogeneous between study participants. Mathematical modeling suggests that at heterogeneous transmission, changes of parasite prevalence and clinical episode incidence with age are less pronounced compared to settings with homogeneous transmission (Ross and Smith, 2010). Although no age trends were observed for P. falciparum in this cohort, when children were stratified into groups ranging from low to high exposure we found that the proportion of P. falciparum clinical episodes relative to new infections decreased with increasing exposure. This could either reflect the development of clinical immunity in highly exposed children, or premunition, a proposed mechanism by which established infections help to control superinfections by immunological cross-protection (Sergent and Parrot, 1935; Smith et al., 1999). In settings of decreasing and heterogeneous transmission, age alone may therefore not be a suitable marker of immunity to P. falciparum. Instead, combining age and molFOB to estimate cumulative life-time exposure may provide a more accurate surrogate measure of the extent of acquired clinical immunity. With P. falciparum transmission declining in PNG due to successful malaria control strategies (Koepfli et al., 2015), it is conceivable that immunity against P. falciparum will develop more slowly, shifting the burden of disease towards older age groups or towards more complex, non-linear age patterns. This delay in immune acquisition is however more than compensated by the overall much lower incidence of clinical malaria clinical episodes: in cohort studies in children younger than 4 years from Maprik district, clinical P. falciparum incidence had dropped from 2.56 clinical episodes/year before (observational cohort, [Lin et al., 2010; Mueller et al., 2012]) to 0.67 clinical episodes/year immediately after the free LLIN distribution campaign (placebo arm, [Betuela et al., 2012]).
Finally, given that four Plasmodium species co-exist in PNG, there has long been considerable interest in potential mechanisms of cross-species immunity and mixed species interactions (Mueller et al., 2009a; Bruce et al., 2000; Smith et al., 2001; Mehlotra et al., 2000). However, there is so far no consistent evidence for the presence or absence of cross-protection among Plasmodium species. P. vivax and P. falciparum infections in our study were concentrated in the same children and villages (Figure 3), likely due to overlapping focal transmission for P. falciparum and P. vivax and thus potentially high co-infection rates in mosquitoes. Contrary to an earlier cohort in younger PNG children that found a decreased risk of P. falciparum clinical episodes after PQ radical cure (Betuela et al., 2012), we found indications for an increased risk of P. falciparum illness after clearance of P. vivax hypnozoites using PQ. Our data suggests that in individuals with substantial clinical immunity against P. vivax, a concurrent P. vivax infection may provide protection against P. falciparum clinical episodes by limiting P. falciparum densities (Table 6, Supplementary file 3). However, the comparably small number of clinical episodes in this and the earlier contrasting study does not allow an in-depth analysis of causal relationships and therefore does not allow firm conclusions on the potential effects and mechanisms of cross-species interactions in mixed infections.
In conclusion, this study provides detailed insight into the changing epidemiology of malaria in PNG children under sustained malaria control, by using molFOB as a powerful measure to quantitatively investigate patterns of new mosquito-derived P. falciparum and P. vivax infections versus those for P. vivax relapsing infections, as well as spatial and age trends in exposure to these infections. Striking heterogeneity in malaria transmission between villages as well as in individual exposure to new P. falciparum and P. vivax infections persisted in our study area despite very high use of LLINs. This presents a significant challenge for on-going malaria control efforts. The comparable patterns of new mosquito-derived P. falciparum and P. vivax infections indicate that sustained use of LLINs does result in a comparable reduction in transmission of both species. The higher incidence and prevalence of P. vivax infections observed in our data is thus directly linked to its ability to cause relapsing infections, highlighting the crucial role of hypnozoites for P. vivax epidemiology and the need to effectively intervene against these hidden stages. Together, these insights provide a crucial link to evaluate the level of P. vivax mosquito-based transmission against that of P. falciparum and serve to calibrate other standard malaria indicators such as parasite prevalence or incidence of clinical episodes and to ultimately inform new approaches to surveillance and response systems.
Materials and methods
Study design and participants
Request a detailed protocolThis study was conducted in six villages in the Albinama and Balif areas, Maprik district, East Sepik Province, PNG between August 2009 and May 2010. The area is serviced by the Albinama health sub-center, Balif aid post and a network of health workers in all study villages. The study design has been described in detail elsewhere (Robinson et al., 2015). Briefly, 524 children aged 5–10 years whose parents provided written informed consent for their participation were enrolled and randomized to receive either chloroquine (CQ, days 1–3, total dose 25 mg/kg), artemeter-lumefantrine (Coartem, AL, days 11–13, 2 mg/kg A, 12 mg/kg L) and primaquine (PQ, days 1–20, 0.5 mg/kg/day); or CQ (days 1–3), AL (days 11–13), and placebo (days 1–20) over 20 days of directly observed treatment (DOT1-20) in a double-blinded manner. Children were actively visited and examined for signs and symptoms of malaria fortnightly at their schools for 8 months. In addition, passive surveillance was provided by the local health centre, aid post and village health workers throughout the study period. Finger-prick blood samples (250 µl) were collected at fortnightly active-follow-up visits in the first 12 weeks and monthly thereafter, as well as from symptomatic children detected during active or passive morbidity surveillance. Symptomatic children were tested for malaria infection with rapid diagnostic test (RDT, CareStartMalaria pLDH/HRP2 Combo, AccessBio, USA), and only RDT and or LM-confirmed Plasmodium infections of any density were treated with a 3 day course of AL.
Household, village and health facility location data was collected using a handheld GPS receiver (Garmin GPSmap62sc) and maps were prepared using ArcGIS 10.2 (Esri, USA).
The study received ethical clearance from the PNG IMR Institutional Review Board (0908), the PNG Medical Advisory Committee (09.11), the Ethics Committee of Basel 237/11 and was conducted in full concordance with the Declaration of Helsinki. The study was registered on ClinicalTrials.gov (NCT02143934).
Laboratory methods
Request a detailed protocolAll blood samples were examined by LM and qPCR for detection and speciation of Plasmodium infections as described earlier (Robinson et al., 2015). Each blood slide was read independently by two skilled microscopists and re-read by an expert microscopist in case of discrepancies in positivity, speciation or density (≥2 x log10 difference). Thick blood films were examined by LM for 200 fields (1000x magnification) before being declared parasite-negative. Parasite density was converted from the number of parasites per 200–500 white blood cells (WBC) to parasites/µl assuming 8000 WBC/µl (WHO malaria microscopy training guide) and calculated as the geometric mean of all positive reads.
DNA was extracted from the red blood cell pellet using the FavorPrep 96-well genomic DNA extraction kit (Favorgen). Samples carrying any Plasmodium spp. infection were identified using a generic qPCR (Wampfler et al., 2013) and positives were subsequently tested in species-specific qPCRs (Rosanas-Urgell et al., 2010; Wampfler et al., 2013). All qPCRs targeted the small subunit (18S) ribosomal RNA gene and were performed as simplex (P. vivax and P. falciparum) or duplex qPCR (P. malariae, P. ovale). The concentration of target copies per µl of DNA was determined relative to a dilution row of standard plasmid as previously described (Rosanas-Urgell et al., 2010). The qPCR limit of detection (LOD) was determined using a standard plasmid dilution row and defined as the last point with more than 50% of replicates positive. The LOD was 2 target copies/µl DNA, equaling 4 target copies/reaction, for all qPCRs. All samples that crossed the fluorescence threshold were scored as positive for species-specific qPCRs. In all samples positive in P. falciparum and/or P. vivax qPCRs, individual parasite clones were distinguished by genotyping the length-polymorphic Pf-msp2 or Pv-msp1F3 marker genes using capillary electrophoresis for highly precise fragment sizing (Koepfli et al., 2013; Koepfli et al., 2011; Falk et al., 2006; Schoepflin et al., 2009). MOI was determined by counting the number of detected Pf-msp2 or Pv-msp1F3 alleles per sample. molFOB was calculated from the number of new parasite clones detected per child or per sampling interval in the peripheral blood, divided by the individual time at risk or length of the interval. A new infection was defined as a Pf-msp2 or Pv-msp1F3 allele not present in the two preceding genotyping-positive samples collected during active or passive surveillance (Figure 1—figure supplement 1). Imperfect diagnostic detectability was not further adjusted for.
Statistical analysis
Request a detailed protocolChildren were considered at risk for clinical malaria clinical episodes until the end of the study or until they were censored (on the last visit before two consecutively missed scheduled follow-up visits [Robinson et al., 2015]). For clinical endpoints, time-at-risk (TAR) was not further adjusted for interim missed follow-up visits because the intense active and passive case detection presumably led to detection of all malaria clinical episodes. In contrast, TAR for analysis of molecular data (e.g., molFOB) was reduced by the duration of the missed interval if a child was not seen by the study team for six weeks or more (≥42 days). Children with a TAR of less than 3 months (<84 days) were excluded. This resulted in an analyzed population of 466 children (characterized in Table 1) of which 430 (92.3%) completed the whole follow-up period, with a median of 15 (IQR: 13–17) study contacts and mean TAR of 186 days (IQR 168–223 days).
Time to first Plasmodium infection by qPCR and LM and its association with covariates were modeled using Cox regression, and the proportional hazards assumption was checked using the test based on the Schoenfeld residuals. Multiple failure Cox regression was used to model the time to P. vivax and P. falciparum clinical episodes.
For statistical analysis, a malaria clinical episode was defined as fever (>37.5°C axillary) plus the presence of LM-detectable parasites, irrespective of RDT result or antimalarial treatment during the field visit. Negative binomial generalized estimating equations (GEE) with log link function using an exchangeable correlation structure were used to model incidence of new infections with P. falciparum and P. vivax per sampling interval. For these analyses, the time at risk was restricted to the intervals where molFOB could be estimated (i.e., starting in the third follow-up interval). A binomial GEE with logit link function using an exchangeable correlation structure was used to model the odds of a P. falciparum clinical episode per interval. Gaussian GEEs with log link function using an exchangeable correlation matrix were used to model log-transformed qPCR parasite densities in qPCR-positive samples, measured as 18S rRNA copy numbers/µl blood. In the GEE and Cox models where molFOB was a covariate, it was included as a time-varying covariate. When modelling molFOB (Table 4) and the odds of clinical episodes (Table 6) using GEEs, molFOB was calculated for each follow-up interval and used as predictor. In Cox models investigating the risk of clinical episodes (Table 5), molFOB was calculated based on the new infections up to the time of failure and used as predictor. In exploratory preliminary analyses we tested for a wide variety of interactions between covariates including interactions between all combinations of molFOB, enrolment infection status, age, village and bednet-usage. All analyses were done using STATA v14 and R.
Maps were drawn using Arcgis 10.1 (Esri Inc.). Ordinary kriging was used to generate the contour maps. Semivariograms were used as the mathematical forms used to express autocorrelation. Input variables for the spatial models were (i)molFOB (prediction of independent variable, molFOB) using the model presented in Table 4 (negative binomial GEE) for Pf and Supplementary file 4 for Pv (same as that shown in Table 4 but with primaquine and placebo arms combined), resulting in Figure 3 Panels A and B; (ii) relative risk of clinical episodes as predicted by the model shown in Table 5, resulting in Panels C and D of Figure 3. Relative standard error maps were generated by dividing the absolute standard error map by the model prediction map.
Data availability
-
Data from: The complex relationship of exposure to new Plasmodium infections and incidence of clinical malaria in Papua New GuineaAvailable at Dryad Digital Repository under a CC0 Public Domain Dedication.
-
Data from: Strategies for understanding and reducing the Plasmodium vivax and Plasmodium ovale hypnozoite reservoir in Papua New Guinean children: a randomised placebo-controlled trial and mathematical modelAvailable at Dryad Digital Repository under a CC0 Public Domain Dedication.
References
-
Relapses contribute significantly to the risk of plasmodium vivax infection and disease in papua new guinean children 1-5 years of ageThe Journal of Infectious Diseases 206:1771–1780.https://doi.org/10.1093/infdis/jis580
-
Detectability of Plasmodium falciparum clonesMalaria Journal 9:234.https://doi.org/10.1186/1475-2875-9-234
-
Human malaria transmission studies in the Anopheles punctulatus complex in Papua New Guinea: sporozoite rates, inoculation rates, and sporozoite densitiesThe American Journal of Tropical Medicine and Hygiene 39:135–144.https://doi.org/10.4269/ajtmh.1988.39.135
-
Small-area variations in the epidemiology of malaria in madang provincePapua and New Guinea medical journal 29:11–17.
-
The ecology of the anopheles punctulatus group of mosquitoes from Papua New Guinea: a review of recent workPapua and New Guinea Medical Journal 29:19–26.
-
Acquired immunity to malariaClinical Microbiology Reviews 22:13–36.https://doi.org/10.1128/CMR.00025-08
-
Comparison of PCR-RFLP and Genescan-based genotyping for analyzing infection dynamics of Plasmodium falciparumThe American Journal of Tropical Medicine and Hygiene 74:944–950.
-
The epidemiology of malaria in the wosera area, East Sepik Province, Papua New Guinea for vaccine trials. I. Malariometric indices and immunityAnnals of Tropical Medicine & Parasitology 89:359–376.https://doi.org/10.1080/00034983.1995.11812965
-
Prevalence of malaria across Papua New Guinea after initial roll-out of insecticide-treated mosquito netsTropical Medicine & International Health 20:1745–1755.https://doi.org/10.1111/tmi.12616
-
Area effects of bednet use in a malaria-endemic area in Papua New GuineaTransactions of the Royal Society of Tropical Medicine and Hygiene 95:7–13.https://doi.org/10.1016/S0035-9203(01)90315-3
-
Random distribution of mixed species malaria infections in Papua New GuineaThe American Journal of Tropical Medicine and Hygiene 62:225–231.https://doi.org/10.4269/ajtmh.2000.62.225
-
The risk of malarial infections and disease in Papua New Guinean childrenThe American Journal of Tropical Medicine and Hygiene 76:997–1008.
-
The epidemiology of malaria in Papua New GuineaTrends in Parasitology 19:253–259.https://doi.org/10.1016/S1471-4922(03)00091-6
-
Premunition in plasmodium falciparum infection: insights from the epidemiology of multiple infectionsTransactions of the Royal Society of Tropical Medicine and Hygiene 93:59–64.https://doi.org/10.1016/S0035-9203(99)90329-2
-
Prospective risk of morbidity in relation to malaria infection in an area of high endemicity of multiple species of PlasmodiumThe American Journal of Tropical Medicine and Hygiene 64:262–267.https://doi.org/10.4269/ajtmh.2001.64.262
-
Predicting changing malaria risk after expanded insecticide-treated net coverage in AfricaTrends in Parasitology 25:511–516.https://doi.org/10.1016/j.pt.2009.08.002
-
Population Pharmacokinetics and Antimalarial Pharmacodynamics of Piperaquine in Patients With Plasmodium vivax Malaria in ThailandCPT Pharmacometrics Syst. Pharmacol. 3:e132.https://doi.org/10.1038/psp.2014.29
Article and author information
Author details
Funding
National Health and Medical Research Council (Early Career Fellowship #1052760)
- Stephan Karl
National Institute of Allergy and Infectious Diseases (South West Pacific International Centers of Excellence in malaria research U19 AI089686)
- Inoni Betuela
- Ingrid Felger
- Leanne J Robinson
- Ivo Mueller
Bill and Melinda Gates Foundation (TransEpi consortium)
- Inoni Betuela
- Ingrid Felger
- Leanne J Robinson
- Ivo Mueller
National Health and Medical Research Council (Project Grant #1021544)
- Inoni Betuela
- Ingrid Felger
- Leanne J Robinson
- Ivo Mueller
Fundación Cellex
- Inoni Betuela
- Ivo Mueller
Schweizerischer Nationalfonds zur Förderung der Wissenschaftlichen Forschung (310030-134889 310030-159580)
- Ingrid Felger
- Ivo Mueller
National Health and Medical Research Council (Early Career Fellowship #1016443)
- Leanne J Robinson
National Health and Medical Research Council (Senior Research Fellowship #1043345)
- Ivo Mueller
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We sincerely thank the children, their parents and guardians, school principals, teachers, and communities for their willingness to be involved in this study. We are grateful to the staff at Albinama Health Centre and village-based health workers for their assistance. We also wish to thank the field team, administration and laboratory staff at Maprik branch, as well as the molecular parasitology laboratory staff at Goroka branch of PNG IMR for their efforts in sample collection and processing. We thank Matthew Phillip for assistance with GPS data collection. We thank Amanda Ross for statistical advice, as well as Jessica Brewster and Cristian Koepfli for assistance with qPCR. Funding was obtained from the Swiss National Science Foundation (grant no. 310030–134889 and 310030–159580), the International Centers of Excellence in Malaria Research (grant U19 AI089686), and the TransEpi consortium funded by the Bill and Melinda Gates, the NHMRC (#1021544) and the Cellex Foundation. This work was also made possible through Victorian State Government Operational Infrastructure Support and Australian Government NHMRC IRIISS. LJR was supported by an NHMRC Early Career Fellowship #1016443. SK is supported by an NHMRC Early Career Fellowship #1052760. IM is supported by an NHMRC Senior Research Fellowship (#1043345). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Ethics
Clinical trial registration: ClinicalTrials.gov NCT02143934
Human subjects: The study received ethical clearance from the PNG IMR Institutional Review Board (0908), the PNG Medical Advisory Committee (09.11), the Ethics Committee of Basel 237/11 and was conducted in full concordance with the Declaration of Helsinki. Written informed consent was obtained from the parents/guardians of all children enrolled in the study.
Copyright
© 2017, Hofmann et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,548
- views
-
- 283
- downloads
-
- 42
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 42
- citations for umbrella DOI https://doi.org/10.7554/eLife.23708
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The complex relationship of exposure to new Plasmodium infections and incidence of clinical malaria in Papua New Guinea
eLife 6:e23708.
https://doi.org/10.7554/eLife.23708




{kind=link}
{kind=link}
{kind=link}
{kind=link}