Evolutionary transitions between beneficial and phytopathogenic Rhodococcus challenge disease management
- Elizabeth A Savory

- Alexandra J Weisberg
- William J Thomas
- Michael I Gordon
- Danielle M Stevens
- Jeff H Chang
- Oregon State University, United States
- United States Department of Agriculture and Agricultural Research Service, United States
Abstract
Understanding how bacteria affect plant health is crucial for developing sustainable crop production systems. We coupled ecological sampling and genome sequencing to characterize the population genetic history of Rhodococcus and the distribution patterns of virulence plasmids in isolates from nurseries. Analysis of chromosome sequences shows that plants host multiple lineages of Rhodococcus, and suggested that these bacteria are transmitted due to independent introductions, reservoir populations, and point source outbreaks. We demonstrate that isolates lacking virulence genes promote beneficial plant growth, and that the acquisition of a virulence plasmid is sufficient to transition beneficial symbionts to phytopathogens. This evolutionary transition, along with the distribution patterns of plasmids, reveals the impact of horizontal gene transfer in rapidly generating new pathogenic lineages and provides an alternative explanation for pathogen transmission patterns. Results also uncovered a misdiagnosed epidemic that implicated beneficial Rhodococcus bacteria as pathogens of pistachio. The misdiagnosis perpetuated the unnecessary removal of trees and exacerbated economic losses.
https://doi.org/10.7554/eLife.30925.001eLife digest
All organisms live in a world teeming with bacteria. Some bacteria are beneficial and, for example, provide their hosts with nutrients. Others cause harm, for example, by stealing nutrients and causing disease. Many bacteria can also gain DNA from other bacteria, and the genes encoded within the new DNA can help them to live with other organisms. This can start the bacteria on an evolutionary path to becoming beneficial or harmful.
Rhodococcus are bacteria that live in association with many species of plants, including trees. Most are harmless but some cause disease. Plants infected with harmful Rhodococcus can show deformed growth, which causes major losses to the nursery industry.
Savory, Fuller, Weisberg et al. set out to understand how disease-causing Rhodococcus are introduced into nurseries, if they are transferred between nurseries, whether they persist in nurseries, and how to limit their spread. It turns out that harmless Rhodococcus are beneficial to plants. However, if these harmless bacteria gain a certain DNA molecule – called a virulence plasmid – they can convert into harmful bacteria. Further analysis showed that some nurseries repeatedly acquired the harmful bacteria. The pattern of affected nurseries suggested that some might have purchased diseased plants from a common provider. In other cases, the sources remained a mystery.
Savory et al. also report that, contrary to previous findings, there is no evidence to support the diagnosis that Rhodococcus without a virulence plasmid are responsible for an unusual growth problem that has plagued the pistachio industry. In recent years, this incorrect diagnosis led to trees being unnecessarily destroyed, worsening the economic losses.
These findings suggest that genes moving between bacteria can dramatically change how those bacteria interact with the organisms in which they live. It needs to be shown whether this is an exceptional process, unique to only certain groups of bacteria, or if it is more widespread in nature. These findings could inform future disease management strategies to better protect agricultural systems.
https://doi.org/10.7554/eLife.30925.002Introduction
Symbioses are persistent and intimate interactions between organisms. In pathogenic interactions, one partner benefits at the expense of the other. In mutualistic symbioses, specific partners interact and reciprocally benefit. Associative symbioses are a variation of mutualism in which there is lower specificity between interacting partners (Drogue et al., 2012). In agricultural systems, practices are employed to limit pathogens, to introduce nitrogen-fixing mutualistic rhizobia and to restore associative symbionts such as plant growth-promoting bacteria (PGPB). PGPB can directly promote the growth of plants and protect against pathogens (Barea et al., 2005; Pieterse et al., 2014).
The beneficial or parasitic outcomes of symbioses, especially those involving environmentally acquired partners, are often not guaranteed. The health of the host, location of the symbiont on the host, or unregulated proliferation of the symbiont can lead to alternative outcomes (Lin and Koskella, 2015). The genotype of the symbiont is also a critical factor, as horizontal gene transfer (HGT) can lead to the acquisition of new genes that innovate genomes, driving evolutionary transitions and establishing new lineages of beneficial or pathogenic symbionts (Soucy et al., 2015). In some pathogenic symbionts, however, HGT does not bestow the genome with innovative functions, nor do these genomes exhibit substantive changes. Rather, the few horizontally acquired genes encode products that reprogram core genes, thereby co-opting the genome for virulence (Letek et al., 2010).
Rhodococcus is a genus of Gram-positive bacteria with members that persist in a variety of terrestrial and aquatic ecosystems (de Carvalho et al., 2014; Larkin et al., 2005). Rhodococcus includes taxonomic groups with members that have been repeatedly recovered from leaf and root tissues of various species of plants (Bai et al., 2015; Bodenhausen et al., 2013; Bulgarelli et al., 2012; Hong et al., 2015, 2016; Lebeis et al., 2015; Lundberg et al., 2012; Qin et al., 2009, 2011; Salam et al., 2017). It has been suggested that hosts enrich for members of Rhodococcus because of the beneficial traits of the bacteria (Hong et al., 2015, 2016).
Plant-associated Rhodococcus species are better known as pathogens (Putnam and Miller, 2007). Two clades of Rhodococcus include members that can cause disease in over 100 genera of plants (Creason et al., 2014a2014a; Putnam and Miller, 2007). Herbaceous plants are the most commonly affected whereas woody plants are less frequently infected. Disease symptoms include leafy galls, witches’-brooms, and other disfiguring growths. Pathogenic isolates of Rhodococcus, typified by the most-studied isolate D188, require three virulence loci that are most frequently found clustered on virulence plasmids (Crespi et al., 1992; Stes et al., 2011). These plasmids are approximately 200 kb in length and are linear replicons (Francis et al., 2012; Creason et al., 2014b). Some of the fas (fasciation) genes are necessary for disease and encode proteins that synthesize and modify cytokinins, which are predicted to be secreted effectors (Crespi et al., 1994; Pertry et al., 2009). The fasR gene, predicted to be a transcriptional regulator, is also necessary for pathogenicity (Temmerman et al., 2000). att (attenuation) mutants are reportedly attenuated in disease and thus implicated in virulence (Crespi et al., 1992; Maes et al., 2001). The vicA gene, which encodes malate synthasean enzyme in the glyoxylate cycle, is the only locus encoded on the chromosome implicated in virulence (Vereecke et al., 2002).
Pathogenic Rhodococcus are particularly problematic in agricultural settings that produce plants for their aesthetic value. A variety of biotic and abiotic stresses, some induced by anthropogenic practices, cause symptoms that are confused with those caused by Rhodococcus (Putnam and Miller, 2007). Furthermore, isolates of Rhodococcus that lack virulence genes are often cultured from symptomatic tissues (Creason et al., 2014b; Nikolaeva et al., 2009). Multiple tests are used to confirm that a plant is infected by pathogenic Rhodococcus. Bacteria must exhibit the proper morphology on selective media and be taxonomically assigned to Rhodococcus. The bacteria must have virulence genes and must cause disease symptoms in susceptible indicator plants. Because the fasR gene and some of the fas genes are necessary for pathogenicity of the bacteria, their detection is sufficient to confirm pathogenicity of Rhodococcus isolates.
In 2011, populations of micropropagated pistachio UCB-1 (Pistacia atlantica × Pistacia integerrima) rootstocks planted in commercial fields began showing an unusual phenotype (Stamler et al., 2015a, 2015b). Aerial phenotypes of ‘pistachio bushy top syndrome’ include shortened internodes, loss of apical dominance, stem galls, and reduced grafting success. Estimates suggest that more than 1 million trees grown on 25,000–30,000 acres were affected. Rhodococcus isolates were cultured from symptomatic plants, and when inoculated onto UCB-1, they caused morphological changes to the hosts (Stamler et al., 2015b). This was the first report of pistachio being susceptible to Rhodococcus. Subsequent release of the genome sequences for PBTS1 and PBTS2, the reported outbreak strains, indicated they lack virulence loci (Stamler et al., 2016). The detection of vicA was used as evidence for pathogenic Rhodococcus bacteria and to guide management practices, the most extreme and costly being the removal of entire orchards. A second incidence of pistachio bushy top syndrome occurred in 2016, resulting in the destruction of 1.5 million nursery trees.
We determined and analyzed genome sequences from over 80 isolates of Rhodococcus, mostly collected from symptomatic herbaceous plants grown in production settings. Analysis of chromosomal sequences shows that plants host multiple lineages of Rhodococcus. Isolates that lack virulence plasmids can promote changes to the architecture of roots, but if a virulence plasmid is acquired, the isolates transition to being pathogenic. The analysis of chromosomal sequences of pathogens revealed the potential for multiple infections and reservoir populations at nursery sites, as well as for point source outbreaks. However, the distribution patterns of virulence plasmids suggested that agricultural systems can be locations that promote evolutionary transitions and the rapid generation of new lineages of pathogens, providing an alternative route for the spread of pathogens. Last, our results challenge previous conclusions that Rhodococcus isolates lacking virulence genes are causative agents of pistachio bushy top syndrome, suggesting that the pistachio syndrome was likely misdiagnosed (Stamler et al., 2015a, 2015b).
Results
Epidemiological links in nurseries
We used a genomic epidemiological approach to study the transmission patterns of Rhodococcus in a plant agricultural system. Sixty isolates were collected hierarchically across space and time. Multiple isolates were cultured from the same symptomatic tissues or from different plants grown at the same production site (Supplementary file 1A). Previously sequenced isolates, many also collected from production sites, were included (Supplementary file 1A; Creason et al., 2014b). The nursery sources of the isolates were anonymized.
Phylogenetic analysis placed the 60 isolates within Rhodococcus Clades I and II, which also included the 15 previously confirmed pathogenic isolates (Figure 1; Creason et al., 2014b). The two clades are sisters to Clades III and IV. Clade III consists of isolates that were cultured from microbiota of the model plantArabidopsis thaliana (Bai et al., 2015; Bodenhausen et al., 2013; Bulgarelli et al., 2012; Lebeis et al., 2015; Lundberg et al., 2012). No member of Clades III or IV have virulence genes. The four clades are distinct, and separated by a long branch, from the other species of Rhodococcus (Figure 1—figure supplement 1). The isolates in the four clades were operationally classified into 17 species, indicated with a lowercase letter, of which 14 have at least one isolate cultured from a plant (Supplementary file 1B).
Figure 1 with 1 supplement see all
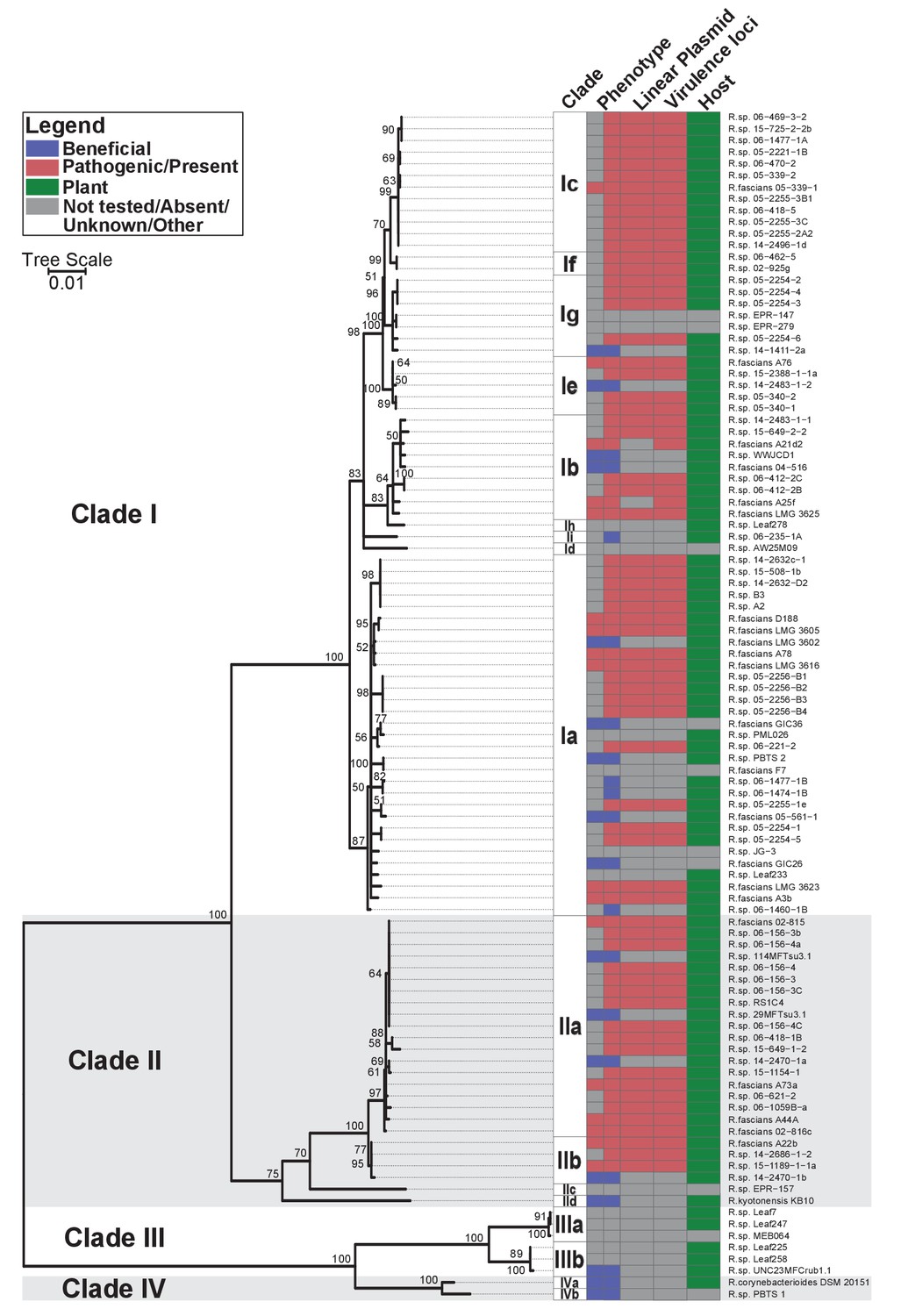
Plant-associated isolates of Rhodococcus form four sister clades.
Multi-locus sequence analysis maximum likelihood tree of plant-associated isolates of Rhodococcus. Translated sequences for ftsY, infB, rpoB, rsmA, secY, tsaD, and ychF from 104 members of Rhodococcus were identified using TBLASTN, aligned, and used to generate a multi-locus maximum likelihood tree. Clade designations are based on analysis of average nucleotide identity (Supplementary file 1B). Columns indicate the features of the corresponding isolate. Grey bars indicate not tested, absent, unknown, or other for phenotype, linear plasmid, virulence loci, and host columns, respectively. The left-half of the column corresponding to phenotype indicates a confirmed phenotype, whereas the right-half indicates an inference based on the presence or absence of virulence genes.
Fifty-one of the newly sequenced isolates encode virulence genes. We inspected their genome assemblies as well as those from previously sequenced pathogenic isolates (Creason et al., 2014b). All but four of the 66 genome assemblies had att, fasR, and fas on the same contig as pFi_009. The fas locus is present in a region that is conserved in pFiD188, the virulence plasmid of isolate D188, suggested to be necessary for plasmid replication and maintenance (Francis et al., 2012). The pFi_009 gene is predicted to encode a telomere-associated protein hypothesized to be necessary for the replication of the linear virulence plasmid. The genome assemblies of isolates 06-469-3-2 and 05-2254-6 had contig breaks that disrupted the linkages between virulence and plasmid-associated loci, but these contigs were nonetheless similar in composition and are co-linear to the reference plasmid sequence. The average sequencing coverage of plasmids relative to that of corresponding chromosomes was 1.89 ± 0.56. Only three assemblies had coverages less than 1.0 but all three had virulence loci on the same contig as pFi_009. A21d2 and A25f, previously sequenced, are exceptional because the virulence loci are encoded in their chromosomes (Creason et al., 2014b). Therefore, of the 66 isolates confirmed or inferred to be pathogenic, 64 carry a virulence plasmid. Pathogenic isolates were assigned to eight different species (Figure 1).
We identified single nucleotide polymorphisms (SNPs) for 82 isolates and used these SNPs to define genotypes and to assemble two clade-specific minimum spanning networks (Supplementary files 1C and 1D; Figure 2). The genotypes show pairwise differences in between 220 and 11,714 SNPs. We mapped nursery information onto the network, which provided information on potential transmission patterns (Figure 2). In this figure, the ‘a’ identification is associated with nurseries in which we identified evidence of multiple and independent infections. Plants from nursery N15, indicated with ‘a1’, were infected by five genotypes belonging to Clade I and two belonging to Clade II. Nursery N8 (‘a2’) and several others were also associated with multiple genotypes. However, nursery N8 had a single host plant that was infected by at least three genotypes that represented six of the cultured isolates. The three isolates within one of these genotypes differ by up to 20 pairwise SNPs, whereas the two isolates in one of the other genotypes have no differences (Supplementary file 1C). The third genotype is separated from the other two by 231 and 1414 pairwise SNPs (Figure 2E). Epidemiological link ‘b’ was detected in nursery N15. The corresponding genotype includes seven isolates from Clade IIa, sampled four years apart from Campanula plants (Supplementary file 1A). These isolates are separated by 0–2 pairwise SNPs. Epidemiological links designated as ‘c’ were also made between isolates collected from geographically separated nurseries. Nurseries N1, N12, and N13 (‘c1’) had isolates of the associated genotype (Clade IIb) that are separated by 1–4 pairwise SNPs and were collected up to 13 years apart from Leucanthemum and Geranium plants. Nurseries N7, N11, and N14 (‘c2’) had isolates of the associated genotype (Clade Ia) that were separated by 12–20 pairwise SNPs. The isolates were collected nine years apart, and from Veronica plants.
Figure 2
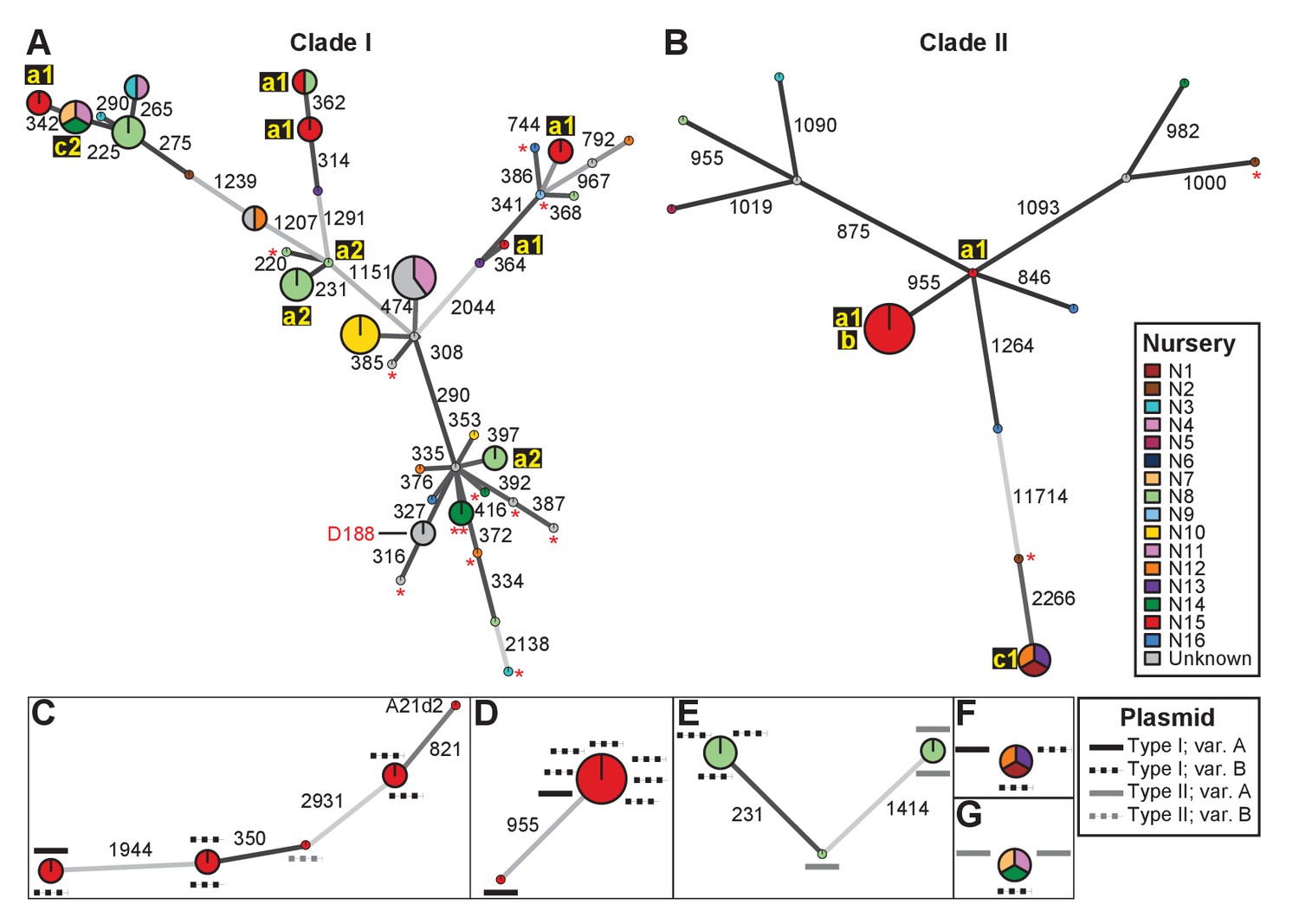
Analysis of SNPs reveals three transmission patterns of pathogenic Rhodococcus.
Minimum spanning networks of isolates of (A) Clade I and (B) Clade II. Each genotype is displayed as a circle, with sizes scaled to represent the number of associated isolates (smallest = 1 isolate). Colors represent the source of the isolates (see key), with coloring proportional to the ratio of isolates from each source. Lower-case letters and numbers (a1, a2, b, c1, and c2) highlight potential transmission patterns; see panels C–G). Asterisks = lacking virulence genes. The genotype that includes D188 is indicated. (C, D) Minimum spanning networks of pathogenic isolates belonging to Clade I (C; ‘a1’) and Clade II (D; ‘a1’ and ‘b’) from nursery N15. A21d2 lacks a virulence plasmid and its virulence loci are present in the chromosome. (E) The minimum spanning network for isolates of pathogenic isolates from nursery N8 (‘a2’). (F) The epidemiological link ‘c1’ between isolates from nurseries N1, N12, and N13. (G) The epidemiological link ‘c2’ between isolates from nurseries N7, N11, and N14. Plasmid types and their variants are mapped onto each of the nodes (see key). Numbers adjacent to connecting lines indicate the number of SNPs that separate each genotype. The lengths of connecting lines are arbitrary; gray lines indicate distances that exceed an arbitrary threshold.
Multiple distribution patterns of plasmids occur in isolates cultured from agricultural settings
The virulence plasmids of 64 plant pathogenic Rhodococcus isolates were categorized on the basis of the phylogenetic analysis of 123 genes that are present in at least 95% of the virulence plasmids, and sub-categorized on the basis of patterns of gene presence/absence (Figure 3; Figure 3—figure supplement 1; Supplementary file 1E). The phylogeny has strong support for two major plasmid types, as well as one unique type only carried by isolate LMG3616 (Figure 3—figure supplement 1). The plasmid sequences within the major clades are conserved and provided few informative nucleotide polymorphisms for sub-categorizing plasmids. The greatest contribution to plasmid diversity is gene gain and loss, and the relative clustering of plasmids based on the presence/absence of genes allowed subgrouping into INDEL variants, as indicated with uppercase letters in Figure 3. At the level of plasmid type, presence/absence categorization was identical to the phylogeny (Figure 3—figure supplement 1). The genes that define each INDEL variant are often present in largely contiguous regions in the plasmid, and could have been acquired as blocks (Francis et al., 2012).
Figure 3 with 2 supplements see all
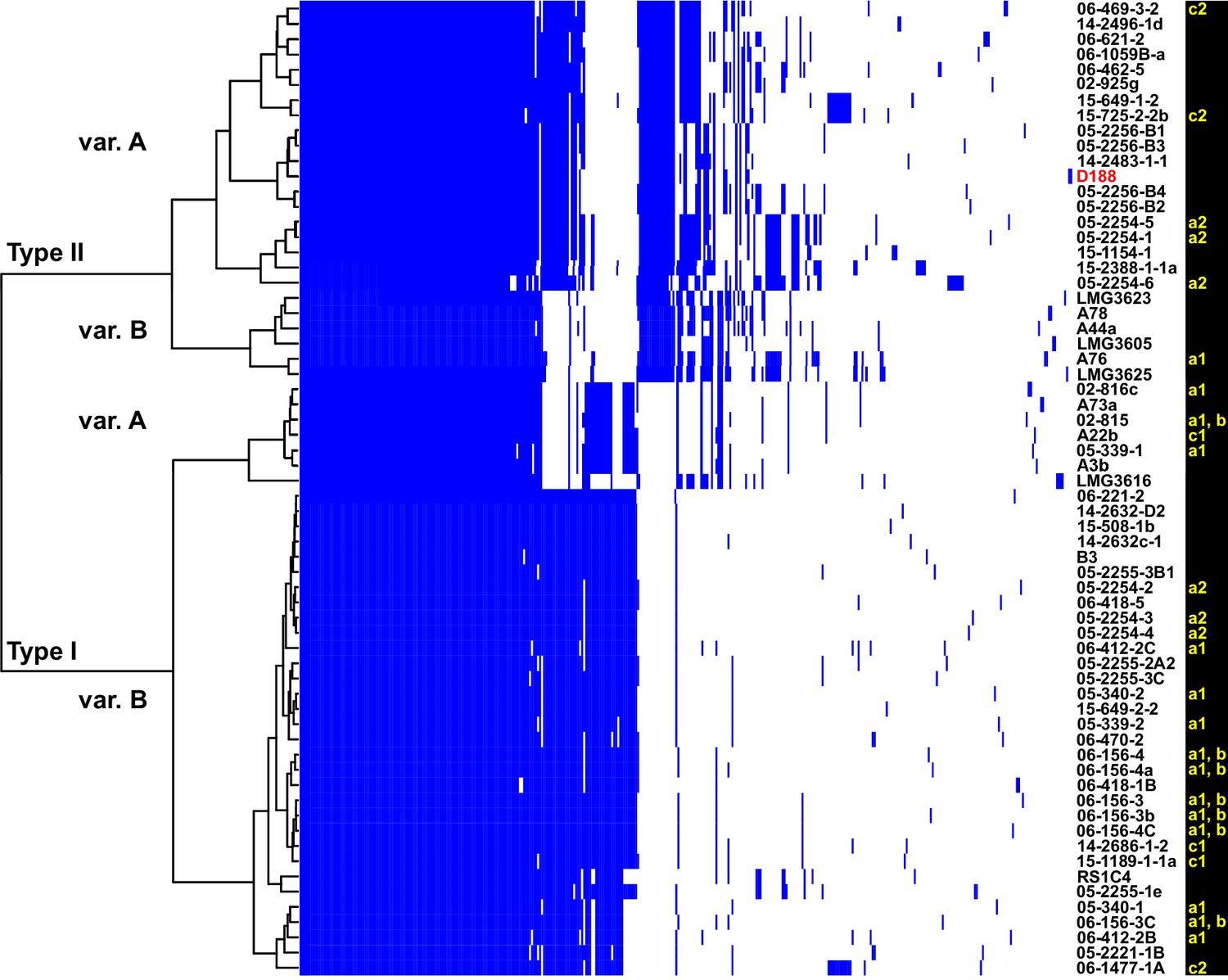
Analysis of plasmid variation reveals multiple patterns in distribution.
Rows indicate genes present in (blue) or absent from (white) the plasmids of isolates (listed to the right; D188 is labeled in red for reference). Columns represent individual genes. Type categories were determined on the basis of phylogenetic analysis of the core genes. INDEL variants, delineated by gray and white shading, were determined on the basis of the cladogram. The plasmid carried by LMG3616 is enclosed by dotted lines. The lower-case letters and numbers (a1, a2, b, c1, and c2) listed along the right, relate isolates and their plasmids to the potential transmission patterns indicated in Figure 2.
We next characterized the distribution of plasmids in epidemiologically linked isolates and showed that the patterns of plasmids were inconsistent with those expected of transmission between isolates of a chromosomal genotype (Figures 2C–G and 3). One genotype associated with ‘a1’ (the most left node of network; Figure 2C) includes isolates 05-339-1 and 05-339-2, which differ by seven pairwise SNPs and which carry Type IA and Type IB plasmids, respectively. The genotype associated with ‘b’ includes isolate 02–815, which carries a Type IA plasmid (Figure 2D). This isolate is epidemiologically linked to six isolates (0–2 pairwise SNPs), but the isolates collected four years later carry a Type IB virulence plasmid. Likewise, the genotype associated with ‘c1’ includes isolate A22b, which carries a Type IA virulence plasmid and is linked to two isolates carrying Type IB plasmids (Figure 2F). The most striking deviation is found in the genotype associated with ‘c2’ (Figure 2G). Isolate 06-1477-1A carries a Type IB plasmid and is epidemiologically linked to two isolates that carry a Type IIA plasmid. Even the two Type IIA plasmids are dissimilar as one has a block of genes that, along with other gene INDELs, distinguishes it from the other.
The diverse genotypes of Rhodococcus isolated from nurseries can carry similar plasmid types and these types are not taxonomically restricted. Isolates 06-412-2B of Clade Ib, 05-340-1 of Clade Ie and 06-156-3c of Clade IIa, all of group ‘a1’ detected in nursery N15, have Type IB plasmids that differ by only a few gene INDELs (Figure 2C and D). The phylogeny shows that there are cases in which isolates from the same clade, such as RS1C4 and 06-1059B-a (solid black lines), carry different types of plasmids (Figure 3—figure supplement 2; solid black lines). It was also observed that distantly related Rhodococcus isolates carry the same type of plasmid. For example, 15-649-1-2 of Clade II and LMG 3623 of Clade I both carry Type II plasmids (Figure 3—figure supplement 2; solid red lines).
Two pathogenic isolates were excluded from the analysis because the virulence loci are present in their chromosomes (Creason et al., 2014b). A25f was recovered from nursery N12, whereas A21d2 was recovered from nursery N15 (Supplementary file 1A).
Rhodococcus isolates that lack the virulence gene promote changes in root architecture
In this dataset, we identified nine isolates of Rhodococcus that lack virulence genes. We also identified five such isolates in a previous study, and often fail to detect virulence genes while diagnosing Rhodococcus cultured from diseased plants (Creason et al., 2014b). Others have implied that these are strains that have lost the plasmid (Nikolaeva et al., 2009). The genetic diversity of co-existing isolates described here suggests otherwise (Figure 1; Supplementary files 1A-D). Pathogenic 06-1477-1A and virulence-gene-lacking 06-1477-1B were isolated from a symptomatic Veronica plant, belong to Clades Id and Ia, respectively, and have 1521 pairwise SNPs. Pathogenic isolate 14-2483-1-1 (Clade Ib) was cultured from the same symptomatic plant as virulence-gene-lacking isolate 14-2483-1-2 (Clade Ie), and the two differ by 2820 SNPs. Interestingly, 14-2483-1-2 has the attR and attX virulence genes, as well as 228 nucleotides of the attA virulence gene, on a contig that is dissimilar in sequence and greater in length than the virulence plasmids. The first two coding sequences and the intergenic regions have ≥88% nucleotide identity to corresponding sequences in D188. For attA, only the first 65 nucleotides are identical to its homolog in D188. Results from PCR confirmed that the structure of the locus was not a result of misassembly. Two virulence-gene-lacking isolates, 14-2470-1a and 14-2470-1b, were cultured from a symptomatic plant. These two isolates are in Clades IIa and IIb, respectively, and differ by 13,858 SNPs. Of the isolates cultured and tested from this plant, no pathogenic isolate was detected.
An alternative explanation for the presence of genetically diverse, virulence-gene-lacking isolates of Rhodococcus is that such isolates are beneficial and enriched for by plants. We therefore compared the symbiosis phenotype of seven virulence-plasmid-lacking isolates that represent the four clades against four virulence-gene-carrying isolates, D188, A44a, A25f, and A21d2 (Supplementary file 1F; Creason et al., 2014b; Desomer et al., 1988; Lundberg et al., 2012; Miteva et al., 2004). The virulence-gene-carrying isolates were previously determined to be pathogenic, and were selected on the basis of having variations in the structure and sequence of their virulence loci (Creason et al., 2014b). PBTS1 and PBTS2, implicated as outbreak strains and cultured from the leaf endophytic compartment of pistachio, were also included (Stamler et al., 2015b).
The seven virulence-gene-lacking isolates, as well as PBTS1 and PBTS2, failed to cause disease when inoculated onto the meristems of mature Nicotiana benthamiana. The four pathogenic isolates caused leafy galls (Figure 4A). These four were also the only ones tested that caused significant inhibition of root elongation, thickening of the stem, and terminal arrest at the cotyledon stage (no primary growth or development of lateral roots), when assayed on seedlings of N. benthamiana (Figure 4B and C). We examined plants up to two months after inoculation, and seedlings remained terminally arrested. Most isolates reduced the vertical growth of roots, compared to that of mock-inoculated plants, but inhibition by virulence-gene-lacking isolates was more variable and not as severe as that measured in the roots of pathogen-inoculated seedlings (p-values were <0.0001 except for the treatment with PBTS2 [p-value = 0.1153]). Importantly, seedlings inoculated with virulence-gene-lacking isolates did not show thickening of the stem or terminal arrest at the cotyledon stage, morphological changes associated with disease (Figure 4).
Figure 4
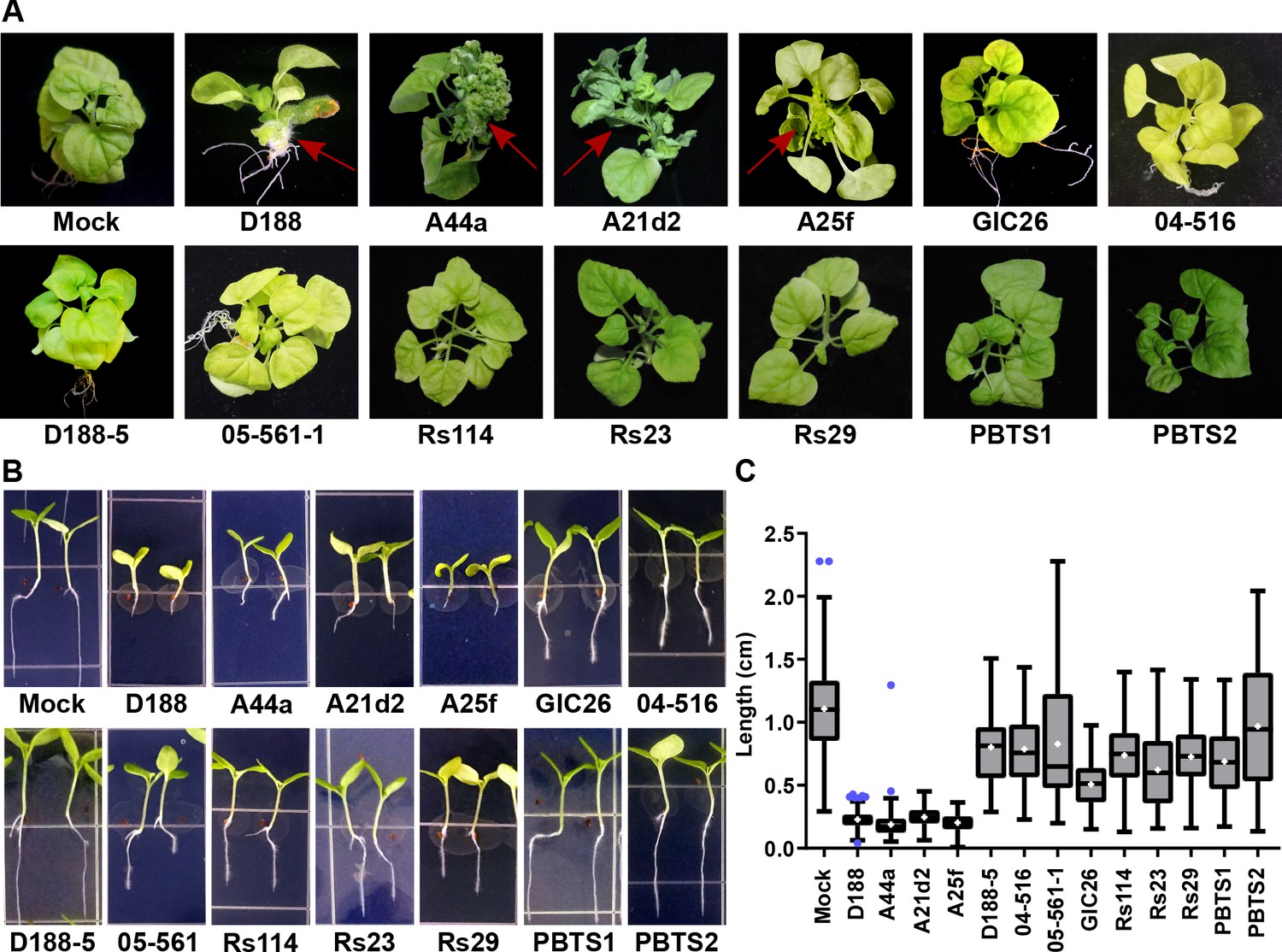
The att, fasR, and fas loci are necessary for the pathogenicity of Rhodococcus.
(A) Representative images of leafy galls on N. benthamiana. Isolates of Rhodococcus were inoculated at the apical meristem. Red arrows indicate the leafy galls. Rs114, Rs23, and Rs29 are abbreviations for isolates 114MFTsu3.1, UNC23MFCrub1.1, and 29MFTsu3.1, respectively. (B) Representative images of the root length of seedlings. Three-day-old N. benthamiana seedlings were inoculated with theindicated isolate of Rhodococcus or water (mock) and grown vertically for seven days under constant light. Isolates D188, A44a, A21d2, and A25f are the only isolates with virulence genes .(C) Quantification of seedling root length. All treatments, except for PBTS2, were significant compared to the mock treatment.
-
Figure 4—source data 1
Lengths of N. benthamiana seedling roots 7 days after inoculation with wild type Rhodococcusisolates.
- https://doi.org/10.7554/eLife.30925.010
Instead, we noticed that all virulence-gene-lacking isolates caused changes to the architecture of the roots (Figure 4B). Relative to mock-inoculated plants, there were proliferations in root hairs and the plants had more lateral roots or earlier development of lateral roots. The former change was quantified in seedlings that were inoculated with members from a subset of the virulence-gene-lacking isolates. There were significant increases in the number of root hairs (averages ranging from 166.1 to 217.3; p-values were all ≤0.0045), compared to mock-inoculated plants (average of 89.8; Figure 5A and B). The isolates varied in their effect, with PBTS2 having the strongest measurable effect (217.3; p-value<0.0001). GIC26 provoked the most visually striking proliferation of root hairs and its extreme effect challenged our ability to count and measure root hairs accurately (Figure 5A; p-value=0.0003). The average length of the root hairs was significantly longer following inoculation with the four isolates of Rhodococcus (averages ranged from 0.02286 to 0.03604 mm vs 0.01186 mm in mock-inoculated plants; p-values were all <0.0001; Figure 5C). Pathogenic isolate D188 was included as a control, but the root hairs of plants that were inoculated with this isolate were too sparse in number to warrant quantification (Figure 5A). Five additional isolates, including 14-2483-1-2 which has part of the att locus, were tested and shownto cause changes to the architecture of the roots of plants (Figure 5—figure supplement 1).
Figure 5 with 2 supplements see all
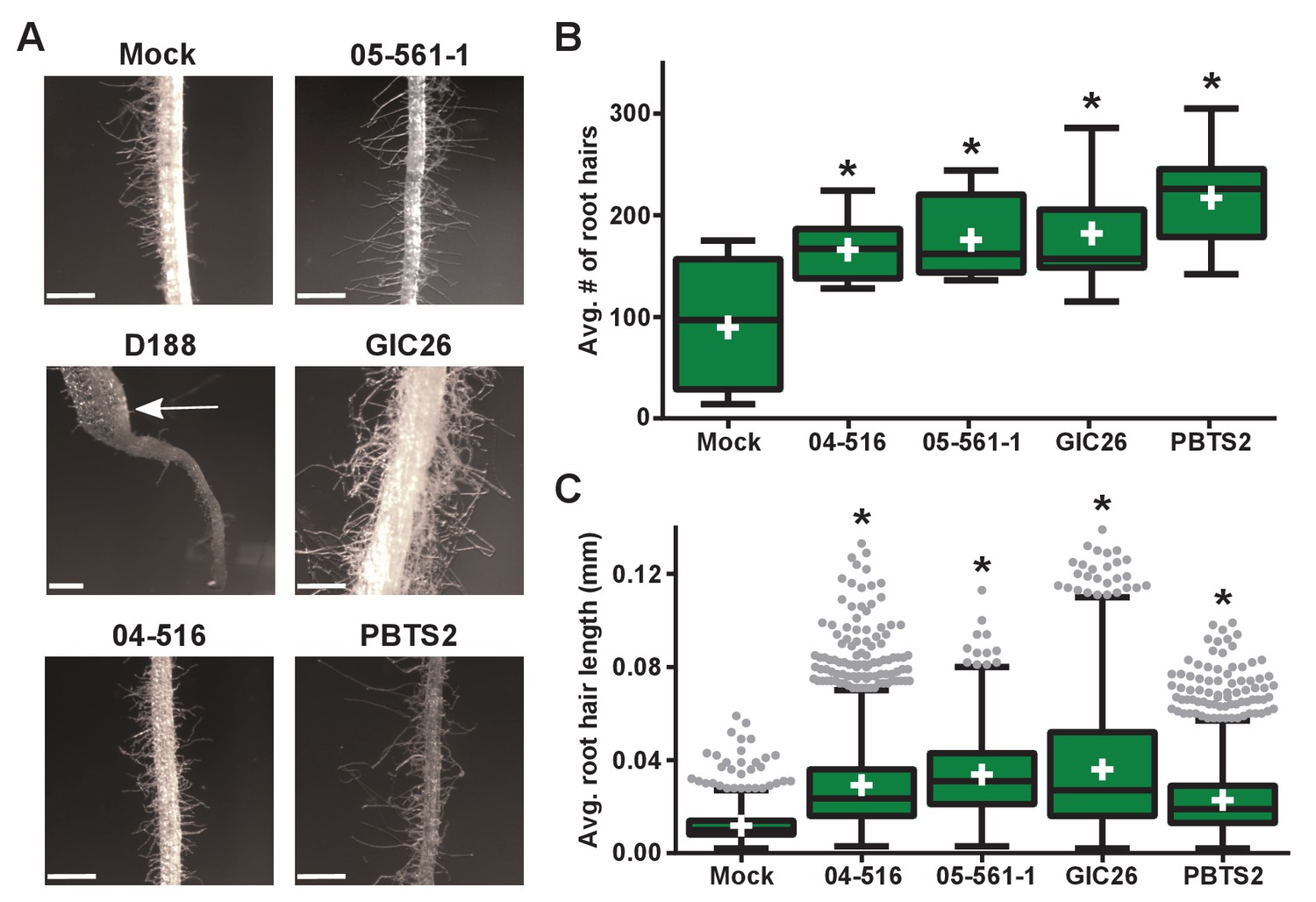
Plant-associated Rhodococcus bacteria cause changes to the root architecture of seedlings.
(A) Representative images of root hairs of N. benthamiana that were inoculated with isolates of Rhodococcus. Images were taken 25 days post inoculation (dpi). The white arrow indicates the thicker stem induced only by isolate D188. Scale bars are 0.5 mm. (B) Quantification of average root hair number at 25 dpi. All root hairs were manually counted for at least five seedlings per treatment. (C) Quantification of root hair lengths at 25 dpi. All root hairs were manually measured for at least five seedlings per treatment. For B and C, data were repeated in two independent biological replicates. * indicates a significant difference compared to the mock treatment.
-
Figure 5—source data 1
Numbers of N. benthamiana seedling root hairs 25 days after inoculation with wild type Rhodococcusisolates.
- https://doi.org/10.7554/eLife.30925.012
-
Figure 5—source data 2
Lengths of N. benthamianaroot hairs 25 days after inoculation with wild type Rhodococcusisolates.
- https://doi.org/10.7554/eLife.30925.013
We analyzed the genome sequences to identify genes that are potentially involved in providing beneficial traits (Figure 5—figure supplement 2; Bruto et al., 2014; Glick, 2012; Sparacino-Watkins et al., 2014). Few homologs or pathways were associated with beneficial traits or an endophytic lifestyle. There was also no obvious correlation with symbiosis phenotype. Some isolates have a homolog predicted to encode 1-aminocyclopropane-1-carboxylate (ACC) deaminase, but this sequence had a strong phylogenetic signal, and is predominantly in members of Clade Ia (Glick, 2014). No complete tryptophan-dependent auxin biosynthetic pathway was identified (Spaepen et al., 2007). The most highly represented classes of carbohydrate active enzymes (CAZYmes) are members of carbohydrate esterase groups CE1 and CE10. CE1 includes xylanases, which degrade hemicellulose (Lombard et al., 2014). CE10 consists of cholinoesterases, a group of enzymes that act on non-carbohydrate sources. In addition, we identified cutinases (C5) and pectate lyases (PL22). These classes of enzymes may contribute to the endophytic lifestyle of Rhodococcus. There are 108 genes that are enriched in the genomes of isolates from the four plant-associated clades (Supplementary file 1G). Forty genes are annotated as hypothetical and many others lack sufficient information in their annotations. AntiSMASH analysis identified anywhere from 1 to ~ 20 loci that may be involved in the production of secondary metabolites (Figure 5—figure supplement 2; Blin et al., 2017). Few had sufficient similarities to previously characterized loci to allow the inference of the identity of the metabolite. Whether and how the functions of these genes contribute to the plant-associated lifestyle are unknown.
The virulence plasmid is sufficient to transition Rhodococcus from being potentially beneficial to being pathogenic
The pFiD188Δatt virulence plasmid was successfully conjugated into a subset of the Rhodococcus isolates that originally lacked virulence genes. This plasmid variant encodes fasR and fasA-F, but has a kanamycin resistance gene that disrupts attR, attX, and attA-G (Maes et al., 2001). Regardless, plasmid pFiD188Δatt is sufficient for isolate D188 to cause disease in mature plants and seedlings, and plants that were treated with a strain containing this plasmid were no different from those infected with D188 (p-value>0.9999; Figure 6—figure supplement 1; Maes et al., 2001). Despite repeated attempts, we were not able to conjugate the plasmid into isolates outside of Clade I successfully. Each of the pFiD188Δatt-carrying isolates caused leafy galls on plants (Figure 6A). In addition, these isolates were no longer capable of causing increases in the growth of root hairs, unlike their corresponding near-isogenic genotypes (Figure 6B). Instead, the isolates carrying pFiD188Δatt, when compared to their near-isogenic plasmid-lacking genotypes, caused disease symptoms and significantly inhibited the growth of seedlings (Figure 6B–C; p-values were all <0.0001).
Figure 6 with 3 supplements see all
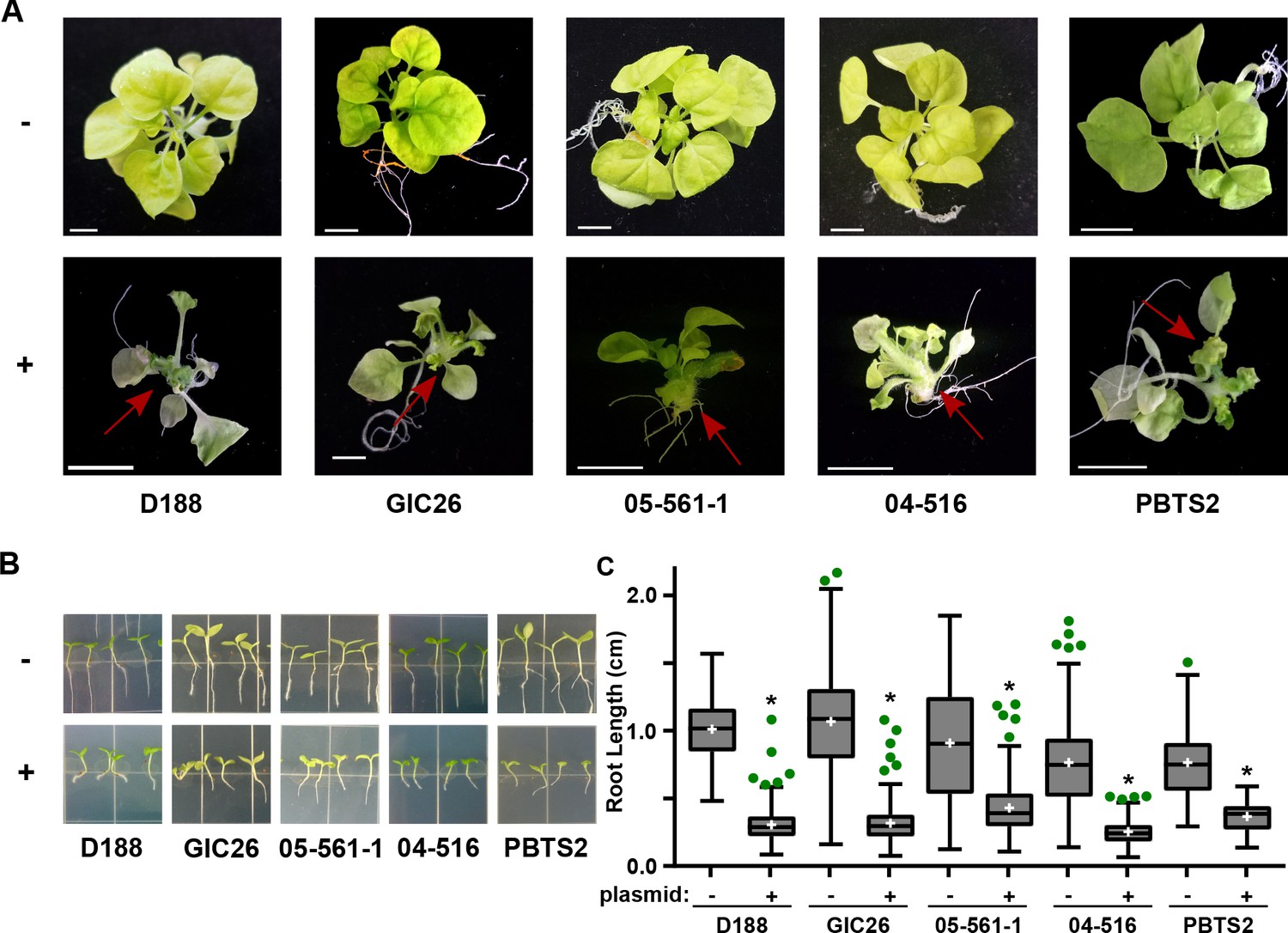
Plasmid pFiD188 with functional fasR and fas is sufficient to transition Rhodococcus isolates to phytopathogens.
(A) Representative images of leafy galls on N. benthamiana. Red arrows indicate leafy galls. Images for GIC26, 04–516, and PBTS2 are repeated from Figure 1. (B) Representative images of the root lengths of seedlings. Three-day-old N. benthamiana seedlings were inoculated with the indicated isolate of Rhodococcus or water (mock) and grown vertically for seven days under constant light. (C) Quantification of the root lengths of N. benthamiana seedlings. In all panels, -/+ indicates absence or presence of pFiD188Δatt. * indicates a significant difference compared to plants treated with the corresponding genotype lacking the plasmid.
-
Figure 6—source data 1
Lengths of N. benthamiana seedling roots 7 days after inoculation with Rhodococcusisolates +/- pFiD188Δatt.
- https://doi.org/10.7554/eLife.30925.022
The inverse transition was also demonstrated. We isolated a variant of D188, D188ΔpFiD188, which lacks the virulence plasmid. When inoculated onto roots of N. benthamiana seedlings, D188ΔpFiD188 caused changes to the architecture of root systems and no longer caused disease to mature plants (Figure 6B; Figure 6—figure supplement 2). We had to generate a new plasmid-lacking strain because the previously generated D188-5 is compromised in in vitro growth (Figure 1; Figure 6C; Figure 6—figure supplement 2; Desomer et al., 1988). Analysis of its genome sequence revealed a significant deletion of 25.4 kb from the chromosome (Supplementary file 1H). Most of the affected 25 genes have annotated functions implicated in housekeeping functions. Sequencing of D188ΔpFiD188 confirmed that it only lacked the virulence plasmid. This isolate also grew similarly to D188 and had no measurable fitness defects (Figure 6C; Figure 6—figure supplement 2).
Only three loci on pFiD188 have been implicated in virulence. We have not been able to repeat results showing that the deletion mutant of att is attenuated in virulence (Figure 6—figure supplement 1; Crespi et al., 1992; Maes et al., 2001). But when constitutively expressing attR, a homolog of the LysR transcriptional regulator necessary for att gene expression, D188 caused unusual leafy galls on N. benthamiana (Figure 6—figure supplement 3). Unlike normal galls that terminate primary growth, those caused by the attR-overexpressing strain regained meristematic activity. When inoculated onto roots, the symptoms were more variable, but nonetheless similar to those caused by D188. The effects were significantly different relative to those seen in mock-inoculated seedlings (p-value<0.0001) or in those inoculated with D188 (p-value<0.0001). The fas locus is predicted to be necessary for Rhodococcus to produce and secrete a mix of cytokinins (Pertry et al., 2009). Approximately 0.1 μM of the synthetic cytokinin 6-benzylaminopurine (BA) was equivalent to a starting inoculum of only ~2.5×103 colony-forming units (cfu) of D188 (Figure 7A). However, regardless of the amount of BA in the medium, the exogenously applied cytokinins only inhibited root elongation and did not provoke the thickening of stems or arrest plant growth.
Figure 7 with 1 supplement see all
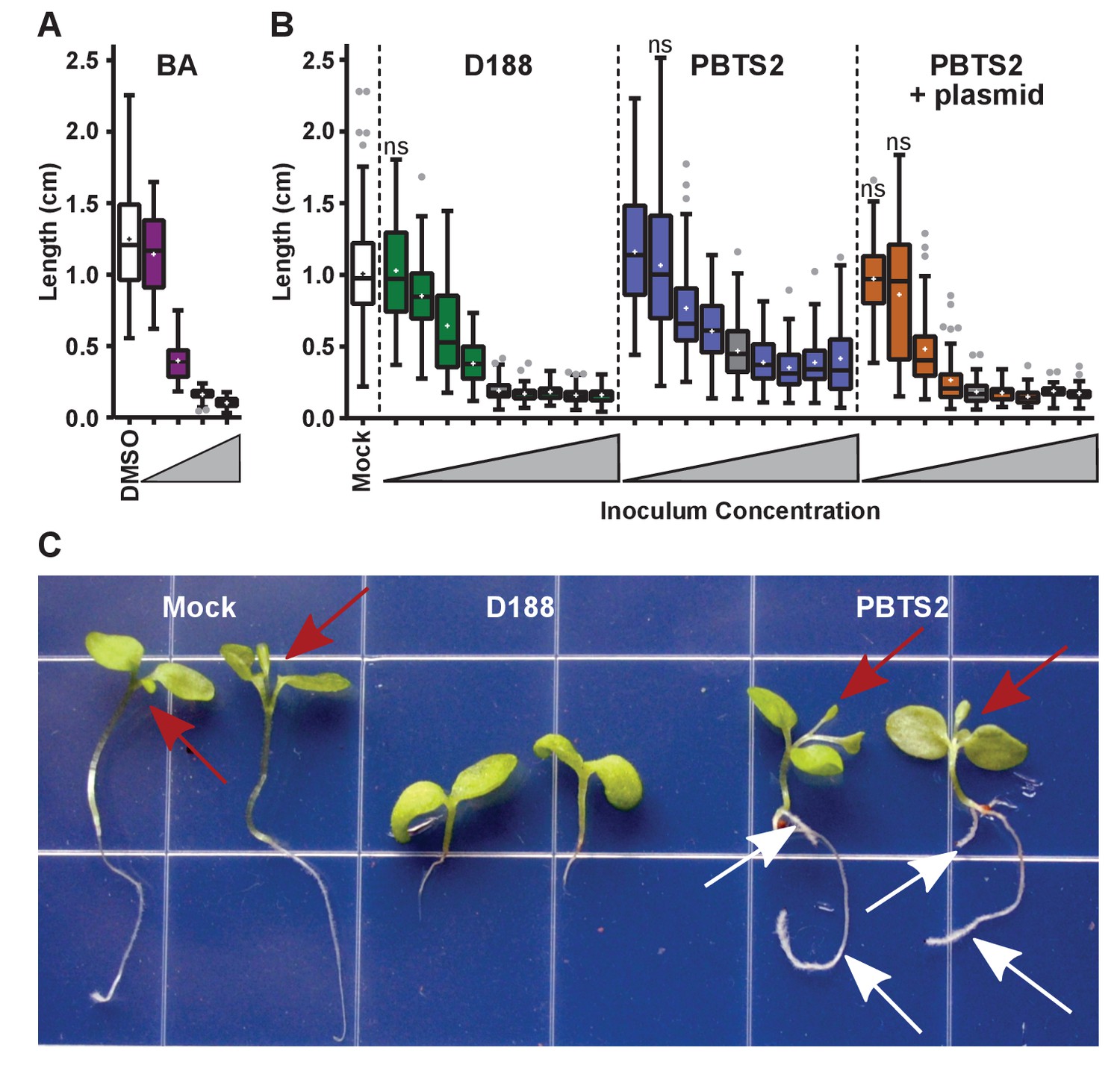
Rhodococcus has a dose-dependent effect on the root elongation of N. benthamiana seedlings.
(A) Quantification of the seedling root lengths of plants grown in exogenously applied cytokinin (6-benyzlaminopurine; BA). Three-day-old N. benthamiana seedlings were transferred to media supplemented with BA (0.01, 0.1, 1.0, and 10 µM) or dimethyl sulfoxide (DMSO). (B) Quantification of the root lengths of seedlings inoculated with increasing doses of Rhodococcus. Three-day-old N. benthamiana seedlings were inoculated with isolates D188, PBTS2, or PBTS2 + pFiD188Δatt, with doses ranging from 2.5 × 102 to 1.0 × 1012 colony-forming units (cfu). The sample shaded in gray highlights the inoculum of OD600 = 0.5 (1x = 2.5 × 1010 cfu) used in all other assays. Inocula below this decrease in 100-fold intervals. Inocula above increase at 2x, 4x, 10x, and 20x. All treatments are significantly different from mock unless otherwise noted with ‘ns’. (C) Representative image of morphological changes in seedlings. Seedlings inoculated with Rhodococcus D188 or PBTS2 (5 × 1011 cfu; 10x typical amount) or water (mock) were photographed. Red arrows indicate true leaves. White arrows indicate lateral roots and the proliferation of root hairs.
-
Figure 7—source data 1
- https://doi.org/10.7554/eLife.30925.024
PBTS1 and PBTS2 are not outbreak strains
Our results show that PBTS1 and PBTS2 are not pathogenic on N. benthamiana (Figures 4 and 5). We next tested whether altering the dose influences the outcome of interaction between N. benthamiana and PBTS2. As inoculum levels of PBTS2 were increased, there was a greater reduction in root length (Figure 7B), but the effect was never to the same robustness and degree as that measured in seedlings infected with D188. In addition, PBTS2 did not cause thickening of stems or terminal arrest in the growth of the plant. At 28 days post-inoculation (dpi), the leaves of seedlings inoculated with the highest tested levels of PBTS2 had developed to the same stage as those of mock-inoculated seedlings, whereas D188-inoculated seedlings remained arrested in growth (Figure 7C). The roots of seedlings inoculated with PBTS2 also formed lateral roots. As inoculum levels of PBTS2 were decreased, there was less reduction of root length, and at the lowest dose tested, roots were significantly longer (1.161 cm; p-value = 0.0043) than those of mock-treated plants (1.007 cm; Figure 7B). A pathogenic PBTS2 strain carrying pFiD188Δatt showed a dose effect similar to that seen for D188 (Figure 7B; p-values were all >0.3584 for all within-dose comparisons).
To exclude the possibility that these results are due to incompatibility between PBTS1 and PBTS2 and N. benthamiana, other species of plants were tested. We used pea, an indicator species for confirming pathogenic Rhodococcus, and UCB-1 pistachio, reportedly the host of the epidemic. Both plant species failed to show disease symptoms, regardless of whether isolates were tested individually or in combination (Figure 7—figure supplement 1). Nine additional pistachio isolates that lack virulence genes also failed to cause disease. Even pathogenic isolates D188 and A44a failed to cause disease symptoms in UCB-1 pistachio.
Another aspect of the diagnosis of pistachio bushy top syndrome was the use of vicA to confirm pathogenic Rhodococcus (Stamler et al., 2015a, 2015b). Primers designed for fasA and fasD specifically amplified a product of expected size from pathogenic isolates D188, A44a, and A25f, and failed to amplify a product from any of the tested beneficial strains (Figure 8A; Supplementary file 1I; Nikolaeva et al., 2012; Serdani et al., 2013). The primers for fasA, and fasD failed to yield a product from A21d2 because this isolate carries an analog of the fas locus (Creason et al., 2014b). The molecular detection of fasR using a loop-mediated isothermal amplification (LAMP)-based assay specifically distinguished all tested pathogenic isolates from beneficial isolates.
Figure 8
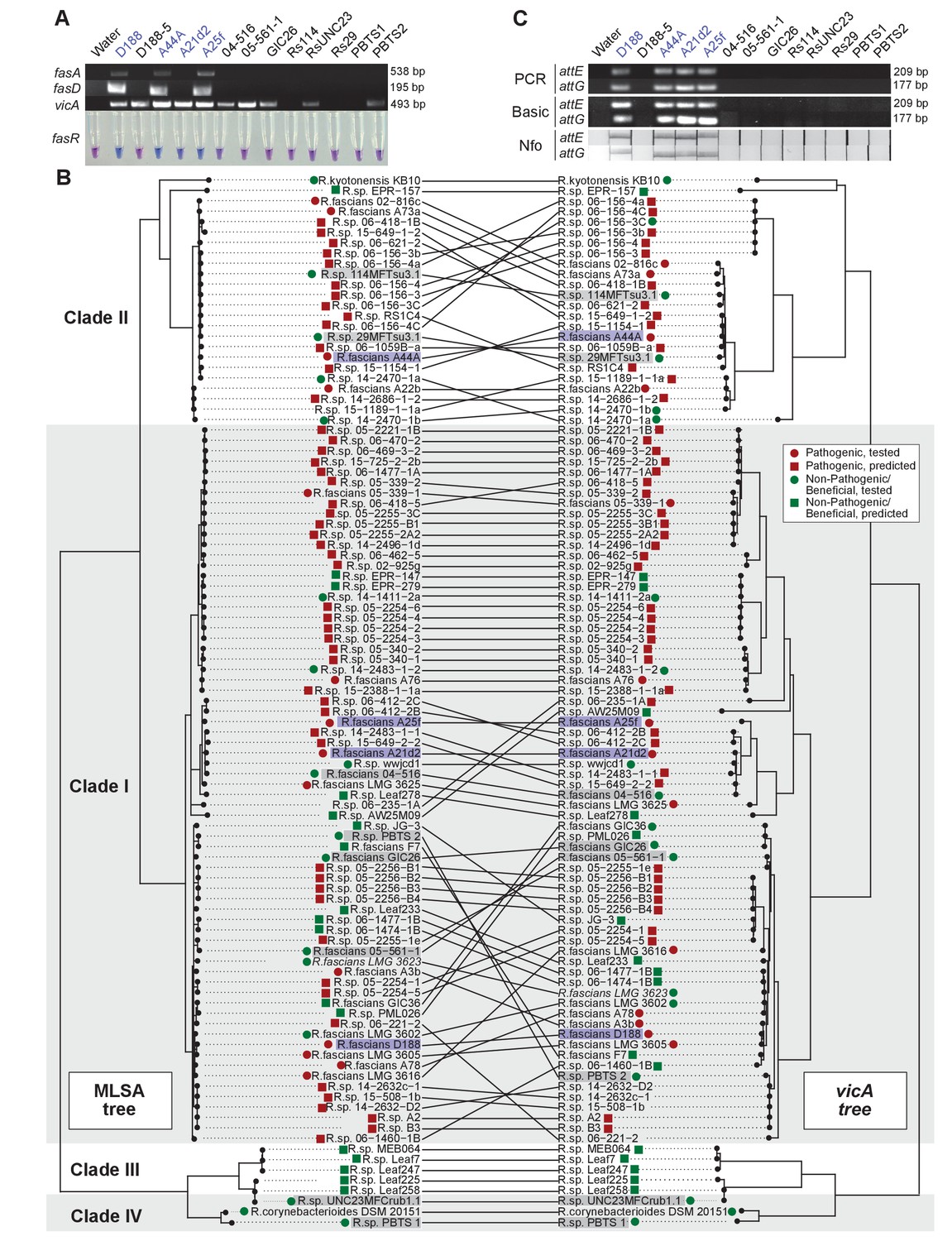
Use of the vicA gene does not discriminate pathogenic Rhodococcus.
(A) (Top) Using locus-specific primers and DNA from the listed isolates, fragments of fasA, fasD and vicA were PCR amplified. Pathogenic isolates are labeled in blue; virulence-gene-lacking isolates are labeled in black. Products were resolved on a 1% TAE agarose gel. Amplicon sizes are listed to the right of the gel images. (Bottom) LAMP assay detection of fasR from the same DNA samples. A positive result is visualized by a blue color. Negative results are light purple. (B) Congruency between species (left) and vicA trees (right). Clades other than I–IV are not shown. Highlighted isolates are the same as in A, pathogenic isolates are highlighted in blue, virulence-gene-lacking isolates are highlighted in gray. (C) Standard endpoint PCR, RPA Basic, and RPA nfo were used to detect attE or attG from DNA extracted from isolates of Rhodococcus. Pathogenic isolates are labeled in blue. For PCR and RPA basic, product sizes are list to the right of the figure. For RPA nfo, the presence of the test band is indicative of a positive reaction; the control bands for all strips were confirmed (not shown).
The detection of vicA did not follow a pattern consistent with the pathogenicity phenotype (Figure 8A). It has a high false-positive rate and detected several, but not all, beneficial isolates. Our repeated attempts to amplify vicA from PBTS1 were unsuccessful; there are four and six mismatches between the two primers used for PCR and the vicA sequence from PBTS1. Homologs of vicA are predicted to be present in nearly all members of the Actinobacteria, including in all 407 Rhodococcus isolates for which genome sequences are available. The topologies of the Rhodococcus genus and the vicA trees are largely congruent, indicating that this locus is mostly vertically inherited, but with some evidence of recombination (Figure 8B).
To address the need for on-site molecular tools to distinguish pathogenic from beneficial genotypes, we used a new molecular detection method that is rapid, robust, and sensitive (Piepenburg et al., 2006). We targeted attE and attG because they are the most unique relative to all sequences in the databases and are conserved among the pathogenic isolates that we have examined (Creason et al., 2014b). The use of the primers for attE and attG in standard PCR and recombinase polymerase amplification (RPA) basic successfully amplified products of expected size from DNA of pathogenic strains, including A21d2 and A25f (Figure 8C). No products were detected when DNAs from beneficial strains were used as templates. An additional oligonucleotide probe that anneals within the amplified fragment was designed for RPA nfo, and when coupled with modified amplification primers, this probe was successful in detecting a product via lateral flow. This method was specific and discriminated between pathogenic and beneficial Rhodococcus. Moreover, RPA nfo can be completed in the absence of specialized equipment, and can yield results in just 30 min.
Discussion
Whole-genome-enabled epidemiological studies have revealed local, global, and historical patterns for the transmission of human pathogens and have informed on health care (Comas et al., 2013; Croucher et al., 2011; Harris et al., 2010, 2013; Mutreja et al., 2011; Parkhill and Wren, 2011; Walker et al., 2013). Ours is a case study for using genomic epidemiology to uncover and explain the transmission patterns of phytopathogens in agricultural systems. The investigation of plasmid distribution highlighted the significant role of HGT in shaping the population structure of pathogenic bacteria and revealed challenges in modeling their transmission. Our analysis of chromosomal SNPs suggested that nurseries experience multiple and independent introductions of pathogenic Rhodococcus, exemplified by ‘a’ isolates observed in nurseries N15 and N8 (Figure 2). The link between isolates collected across time (‘b’) is indicative of a reservoir population that has a pathogenic genotype. The epidemiological links (‘c1’ and ‘c2’) of isolates from different nurseries support the possibility of point source outbreaks and suggest that the sources have reservoir populations.
However, the distribution of plasmids also indicated that alternative processes may be occurring (Figures 3 and 9). First, two different plasmid types are associated with ‘c2’. This is not expected from an outbreak and is more consistent with different members of a lineage acquiring plasmids separately. Second, different plasmid variants are carried by isolates that are defined by genotypes associated with ‘a1’, ‘b’, and ‘c1’ (Figure 2). These are not expected patterns and probably reflect separate acquisitions of plasmids by different members of a lineage or the rapid and independent evolution of plasmids. Third, genetically distinct lineages of Rhodococcus at nursery N15 carry the same variant of plasmid. This is best explained by multiple lineages' acquiring plasmids from a common donor population. Nurseries often produce a large variety of perennials and clonally propagated plants that are frequently handled and intensely managed in multiple production settings, and are often in regions that produce many different agricultural commodities. These are prime locations for different genotypes of Rhodococcus to interact and for plasmids to be transferred, switched, and evolved.
Figure 9
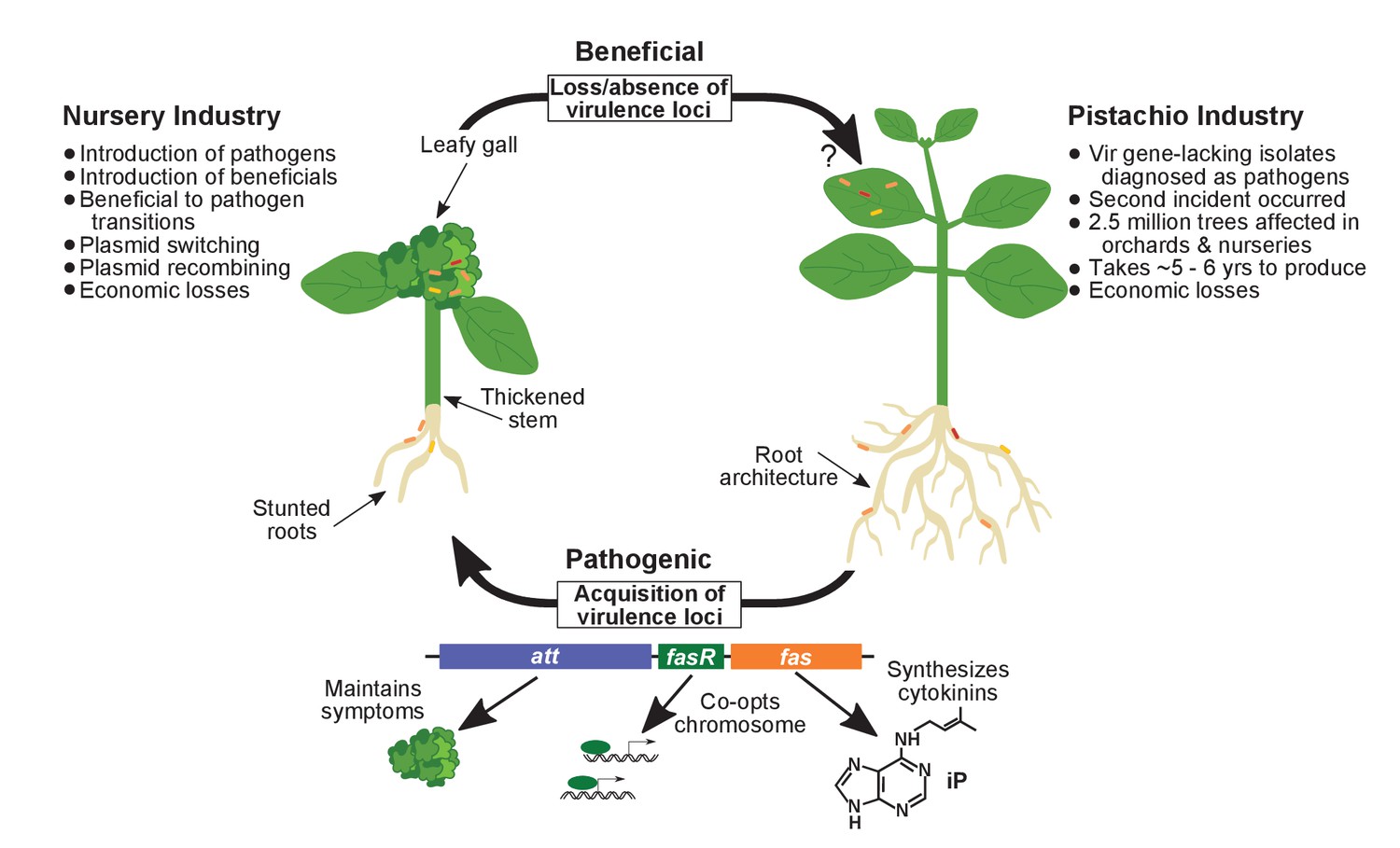
Model of the evolutionary transition in Rhodococcus and effects on agricultural sectors.
The presence or absence of the virulence plasmid in eight species of Rhodococcusdetermines whether the bacterium is beneficial (right), and promotes growth in root architecture, or pathogenic (left), and causes leafy galls and inhibits primary growth. The virulence plasmid carries three loci identified as necessary for pathogenicity and their predicted functions are described. Horizontal gene transfer and evolutionary transitions affect Rhodococcus and impact the nursery industry (as indicated on the left of the figure). Virulence-gene-lacking isolates of Rhodococcus were diagnosed as outbreak strains and the probable misdiagnosis could have had detrimental impacts on the pistachio industry (as indicated on the right of the figure).
We also demonstrated that plants in agricultural systems are hosts to isolates of Rhodococcus that are probably beneficial associative symbionts (Supplementary file 1A; Vacheron et al., 2013). Root hairs are extensions that increase the surface area of roots, forming an extensive interface between plant and soil. Virulence-gene-lacking isolates of Rhodococcus caused significant increases in the number and length of root hairs, which may enable plants to be more efficient in the uptake of water and dissolved nutrients (Figure 5). This potentially beneficial growth-promoting phenotype is consistent with the finding of a number of reports identifying Rhodococcus within endophytic compartments and the rhizosphere of plants, and with the suggestions that the bacteria are enriched for by plants because of their beneficial traits (Bai et al., 2015; Bodenhausen et al., 2013; Bulgarelli et al., 2012; Hong et al., 2015, 2016; Lundberg et al., 2012; Qin et al., 2009, 2011; Salam et al., 2017). The associative symbionts are genetically diverse and represent 14 different species circumscribed by four sister clades (Figure 1; Supplementary file 1B). The mechanism by which isolates of Rhodococcus cause growth changes to plants is unknown, as genome mining efforts suggest that these traits are potentially novel (Figure 5—figure supplement 2).
Acquisition of a virulence plasmid by isolates representing eight species of Clades I and II is sufficient to drive an evolutionary transition (Figures 1 and 6). Loss of the plasmid reverted Rhodococcus to being beneficial, consistent with the hypothesis that virulence genes function irrespective of genomic background (Figure 6—figure supplement 2). In addition, we could not identify any chromosome-located genes that are enriched in pathogenic isolates, in comparison to non-pathogenic isolates, that could be potential candidate virulence genes. Evolutionary transitions, such as the switch from being a free-living, non-pathogenic lineage to being a pathogenic lineage, have beendetected frequently (for example, by Bruto et al., 2017). The transition described for Rhodococcus is seamless and the mutualist to pathogen transition has beendescribed only rarely (Sachs et al., 2011). It is possible that focus on characterizing binary outcomes in symbioses have obscured the true fluidity of symbioses. Similar transitions may have occurred in Agrobacterium/Rhizobium, a group of bacteria that express plasmid-mediated traits of significance to plant agriculture (de Lajudie et al., 1999; Glaeser et al., 2016; Hao et al., 2012; Lacroix and Citovsky, 2016; Wang et al., 2006).
Transitions and the rapid generation of new lineages of pathogens could occur in agricultural systems, where plants are frequently host to multiple Rhodococcus isolates of different genotypes. Pathogenic isolate 05-2254-6, which is associated with ‘a2’, is most similar to virulence-gene-lacking isolate, 14-1411-2a, also collected from N8. The 220 pairwise SNPs that differ between these two isolates exceeded the threshold used to define genotypes, but the two isolates are nonetheless closely related (Figures 1 and 2; Supplementary file 1C). This is not unique, as several pathogenic genotypes are related to virulence-gene-lacking genotypes, as indicated by their intermingling in the phylogeny and their connectivity in the network. The patterns involving distinct genotypes or plasmid types, such as ‘a1’ and ‘c2’, respectively, are also consistent with evolutionary transitions (Figures 2 and 3). An example that is similar to ‘a1’ is the genotype of D188 and LMG3605, both of which have different plasmid variants.
The limited genetic diversity in virulence plasmids within our dataset was unexpected and suggests that their common ancestor evolved recently (Figure 3—figure supplement 1). This is in dramatic contrast to the virulence plasmids of Agrobacterium species of bacteria, where the Ti and Ri plasmids form different types that can be easily distinguished on the basis of the phylogeny of a single, conserved virulence gene (Fuller et al., 2017). It is also remarkable that the virulence plasmid, which carries only three virulence loci, is sufficient for pathogenicity across eight genetically diverse species of Rhodococcus (Figures 1 and 9; Creason et al., 2014b; Francis et al., 2012; Letek et al., 2010). FasR is a predicted transcriptional regulator, and is probably key for reprogramming the genome to transition bacteria to pathogens. The roles of the other two loci are still unclear. Results presented here suggest that att contributes to the maintenance of disease symptoms, but the mechanism is unknown (Figure 6—figure supplement 3). The Fas-produced mix of cytokinins are predicted to be secreted into plants and necessary to cause disease symptoms, but the existing data are not consistent with this hypothesis (Pertry et al., 2009). Exceedingly low amounts of cytokinins are detected in culture-grown bacteria and plants have a variety of cytokinin-buffering mechanisms (Creason et al., 2014b; Kieber and Schaller, 2014; Pertry et al., 2009, 2010). A miniscule amount of starting bacterial inoculum is sufficient to provoke disease symptoms, and exogenous applications of cytokinins fail to phenocopy the effects of pathogenic Rhodococcus (Figure 7). Last, the cytokinin mixture model is challenged by the revelation that the fitness of D188-5, a key isolate used to develop the model, is severely compromised by a 25-kb deletion (Figure 6—figure supplement 2; Supplementary file 1H; Desomer et al., 1988; Maes et al., 2001; Pertry et al., 2009, 2010; Stes et al., 2011; Temmerman et al., 2001).
This study highlights the importance of understanding the genetic and phenotypic characteristics of an organism and the consequences of prematurely drawing conclusions from incomplete data. There is no evidence to suggest that HGT or evolutionary transitions confounded the diagnosis of pistachio, and we could not detect pathogenic Rhodococcus from symptomatic tissues of pistachio. We were unsuccessful in repeating results showing that PBTS1 and PBTS2, or other isolates cultured from pistachio, cause disease symptoms on plants (Figure 4; Figure 7—figure supplement 1). We could not amplify virulence genes from PBTS1 or PBTS2, but when a virulence plasmid was introduced into PBTS2, it was sufficient to transition PBTS2 to a pathogen of a plant species that is demonstrably susceptible to Rhodococcus (Figures 6 and 7B). Whether pistachio is even a host for pathogenic Rhodococcus is unresolved (Figure 7—figure supplement 1). Nevertheless, we recognize the insurmountable challenge in showing that there is no possibility that pathogenic Rhodococcus causes pistachio bushy top syndrome.
The results from this work prompted us to examine previous studies retrospectively (Figure 9; [Stamler et al., 2015a, 2015b]). There was a targeted search for Rhodococcus, the justification for which is unfounded because the symptoms on pistachio are unlike any produced by pathogenic Rhodococcus in any of the 100+ known hosts (Putnam and Miller, 2007). Bacteria were inexplicably cultured from asymptomatic leaves distal to symptomatic stem tissues. The key study did not include control strains or reproduce galling and graft failure, the most defining disease symptoms observed in field settings. A high inoculum of Rhodococcus was used and high doses of even beneficial bacteria can have costs (Figure 7). For example, some human diseases are caused by dysbiosis in which an imbalance of gut microbiotia causes disease (Bloom et al., 2011). In addition, hosts often employ mechanisms to regulate or ensure nonpersistent interactions with beneficial bacteria (Gutjahr and Parniske, 2013; Magori et al., 2009; Nyholm and McFall-Ngai, 2004; Reid et al., 2011; Wopereis et al., 2000).
Results based on molecular detection were similarly tenuous. The use of the vicA locus was misleading and led to conclusions regarding the pathogenicity of Rhodococcus (Figure 8). The reported amplification of fragments of fasA and fasD (GenBank accession numbers KP274062 and KP274064) from PBTS1 as well as fasD (KP274067) from PBTS2 cannot be reconciled with the absence of the genes from the genome sequences (Stamler et al., 2015b, 2016). We used a different technology to re-sequence independently prepared DNA from PBTS2 (Supplementary file 1F). The assembly is co-linear with the publically available sequence and lacks the virulence plasmid and virulence genes (Stamler et al., 2016). The possibility that virulence plasmids are unstable in populations grown outside of plant environments is not supported by the data. DNA extracted from bacteria grown in culture was used as a template in both PCR and whole-genome sequencing (Stamler et al., 2015b, 2016). We successfully sequenced plasmids from 64 culture-grown pathogenic isolates, which had undergone multiple transfers, and repeatedly and successfully detected pathogenic isolates cultured from symptomatic tissues (Creason et al., 2014b).
Some of the results of molecular detection were clearly artifacts. The two fragments reported to correspond to fasD are identical in sequence, each consisting of two short fragments of 147 and 194 nucleotides long that are artificially joined together by 280 ‘Ns’. In our dataset, A21d2 is the only sequenced pathogenic isolate that lacks fasA, but it also lacks a homolog of fasD. It is thus not expected that PBTS2 should have only fasD but no fasA (Stamler et al., 2015b). The most egregious artifact was the reported amplification of vicA from PBTS1 (GenBank accession number KP274063), which we could not reproduce (Figure 8). When the sequence of the reportedly amplified fragment was used as a query to BLAST search the PBTS1 genome sequence directly, we failed to identify a homologous region. When used in searches against publically available databases, the top hits other than KP274063 were NM-J PBTS (KR153287; 100% identity), DMS3-9 (KJ677035; 97% identity), D188 complete genome (CP015235; 96% identity), and PBTS2 complete genome (CP015220; 96% identity).
Another study posited that the virulence plasmids are unstable in populations persisting within plant environments (Nikolaeva et al., 2009). Genome sequences showed instead that Rhodococcus isolates are from genetically distinct lineages. This observation further emphasizes the need to use appropriate molecular diagnostic tools that discriminate pathogenic from non-pathogenic bacteria. LAMP to detect fasR or RPA nfo to detect attE or attG are both sensitive methods that have the additional benefit of being rapid and less dependent on specialized equipment (Figure 8; Serdani et al., 2013).
The data are consistent with the possibility that previous conclusions rest on a misdiagnosis of pistachio bushy top syndrome (Stamler et al., 2015a, 2015b). If so, Rhodococcus was implicated as pathogenic, irrespective of genotype and in disregard of the genetic and phenotypic diversity of the genus. This mindset conflates all bacteria as causative agents of disease and demotes the importance of bacteria in promoting the health of their hosts (Figure 9). This potential misdiagnosis of pistachio bushy top syndrome could be responsible for catastrophic effects. An estimated 2.5 million trees have been destroyed, resulting in tremendous economic loss. Efforts to identify the true cause were decelerated. Accusations regarding the source of Rhodococcus have introduced conflict into an industry struggling with a considerable and unfamiliar problem. Considering the data described here, previous conclusions should, at the very least, be tempered and recommendations for managing plants in which Rhodococcus bacteria have been detected, reexamined. Ideally, these findings will renew efforts to identify the true nature of the syndrome afflicting UCB-1 pistachio rootstocks.
Actinobacteria are prominent members of plant-associated communities, but the mechanisms and evolution of traits that are important for Gram-positive bacteria to reside in microbial communities and influence plant health are not well understood (Figure 9). Members of the Rhodococcus genus are excellent models for addressing this knowledge gap. A wealth of associated resources, such as an extensive and diverse collection of genotypes, associated genome sequences, and genetic tools have been developed for Rhodococcus. The members of this taxon can also interact with and benefit genetically tractable plant species. Last, the members of Rhodococcus are model organisms for characterizing evolutionary transitions between alternative symbiotic states.
Materials and methods
Bacterial isolates and growth conditions
Request a detailed protocolRhodococcus isolates used in this study are listed in Supplementary files 1A and F. Bacteria were maintained on solid LB medium at 28oC or grown overnight in LB medium at 28oC with shaking. Rhodococcus conjugations were done as previously described (Desomer et al., 1988). Escherichia coli was grown on LB medium at 37oC. When appropriate, the medium was amended with 50 µg/ml of antibiotic kanamycin for Rhodococcus or E. coli. For growth curves, cultures of overnight-grown Rhodococcus were pelleted, washed, and resuspended at OD600 = 0.5 in a final volume of 200 µl of LB medium in 96-well flat-bottomed plates. The bacteria were grown for a period of 14 hr at 28oC with shaking in a Tecan Spark 10 m plate reader. Optical density (OD600) measurements were taken every hour. Three technical replicates were included for each isolate, and the experiment was repeated at least three times with similar results.
Genome sequencing, assembly, and annotation
Request a detailed protocolThe Wizard genomic prep kit (Promega, Fitchburg, WI, USA) was used to extract genomic DNA from Rhodococcus. Directions for Gram-positive bacteria were followed. DNA was quantified with a Nanodrop spectrophotometer and adjusted to 50 ng/µl. Total genomic DNA was used to prepare Nextera XT libraries, and the resulting multiplexed libraries were sequenced on an Illumina HiSeq 3000 to generate 250mer paired end sequencing reads (Center for Genome Research and Biocomputing [CGRB], Oregon State University). Reads were processed as follows. FastQC was used to assess sequencing reads for quality (Andrews, 2014). BBduk v.35.82, with the parameters ‘ktrim = r k = 23 mink = 9 hdist = 1 minlength = 100 tpe tbo’, was used to remove adapter sequences (Bushnell, 2014). SPAdes v. 3.1.1, with the parameters ‘--careful -k 21,33,55,77,99’ was used to correct errors and to de novo assemble the reads into contigs (Bankevich et al., 2012). Blobtools was used to assess assemblies and guide elimination of contigs likely to be derived from contaminating bacteria (based on combined GC content, coverage, and contig annotation) (Kumar et al., 2013). Prokka was used to annotate the assembled genome sequences (Seemann, 2014).
Phylogenetic analyses
Request a detailed protocolSequences for the maximum likelihood multi-locus sequence analysis (MLSA) tree were acquired using the autoMLSA tool (Davis Ii et al., 2016). The sequences for genes, ftsY (ABG98302.1), infH (ABG98417.1), rpoB (ABG93773.1), rsmA (ABG97450.1), secY (ABG97930.1), tsaD (ABG97962.1), and ychF (ABG97656.1) from the genome sequence of Rhodococcus jostii RHA1 were translated and used as queries in TBLASTN v. 2.2.31 searches against the assembled genome sequences and the NCBI nt database, masked to Rhodococcus (Adékambi et al., 2011; accessed 12/2016). Of those from NCBI nt, eight strains lacking all seven sequences and/or duplicate results were removed from the analysis. The sequences were aligned using MAFFT v. 6.864b with default settings (Katoh and Standley, 2013). A RAxML accessory script was used to determine the best-fitting protein model for each protein sequence alignment. Phylogenetic trees (100 ML searches, ‘autoMRE’ criterion bootstrap replicates) were generated using RAxML v. 8.1.17 with a partitioned alignment of the MLSA protein sequences (Stamatakis, 2014).
A similar analysis using R. fascians D188 vicA (AMY55488.1) as a query was used to acquire and assemble a phylogeny of 162 malate synthase gene sequences from the NCBI nr database masked to Rhodococcus (accessed 04/2017). A cophylo plot of the MLSA and vicA trees was generated using the R package phytools (Revell, 2012).
For the genes present in 95% of the virulence plasmids, sequences were concatenated using the R package EvobiR SuperMatrix function prior to constructing phylogenies (Blackmon and Adams, 2015).
Only bootstrap values greater than 50 are shown.
Genome analyses and bioinformatics tools
Request a detailed protocolBowtie2 v. 2.2.3, with the option ‘--local’, was used to align reads to the chromosome references sequences of D188 (CP015235.1) or A44a (GCF_000760735.1), based on the clade assignment of the corresponding isolates (Langmead et al., 2009). Alignments were converted to bam format using samtools v. 0.1.18 and read groups were added using Picard tools v. 2.0.1 (Li et al., 2009; Picard Tools, 2015). GATK v. 3.7 HaplotypeCaller and the options ‘-ERC GVCF -ploidy 1’ were used to call variants for each isolate, and the data were then combined using GenotypeGVCFs (McKenna et al., 2010). Variants were filtered using the R package vcfR with depth filtering using quantile probabilities of 0.25 and 0.75 as cutoffs and a minimum of four reads, as well as a missing data cutoff of 20% (Knaus and Grünwald, 2017). Variants were converted into a fasta alignment using bcftools v. 1.3–14-ge0890a1 vcf-to-tab and the perl script vcftab-to-fasta (Li et al., 2009; Chen, 2012). Genotypes were called based on a threshold of 25 SNPs, and bitwise distances, using the R package poppr, were used to assemble minimum spanning networks (Kamvar et al., 2014).
Pairwise average nucleotide identity (ANI) between Rhodococcus isolates was calculated using autoANI (Davis Ii et al., 2016).
Get_homologues v. 20170418 with MCL clustering was used to cluster genes from 206 Rhodococcus genomes into orthologous groups (Contreras-Moreira and Vinuesa, 2013). The parse_pangenome_matrix.pl script of get_homologues was used to identify genes enriched (with a 95% threshold) in genomes of Rhodococcus in the four plant-associated clades.
To identify pathogenicity loci in sequenced isolates, fasR, fas and att were used as queries in TBLASTN searches against genome assemblies. CONTIGuator was used to map assembled contigs to the reference strain D188 to search for sequences corresponding to pFiD188 or pFID188-like plasmids (Galardini et al., 2011). Get_homologues v. 20170418 was used to cluster genes from each of the virulence plasmids as well as the other Rhodococcus linear plasmids (Francis et al., 2012). Plasmids were clustered on the basis of gene presence/absence using binary distances and Ward's method for clustering (ward.D2). dbCAN HMMs 5.0 was downloaded and used with ad hoc scripts to identify CAZYmes from translated genome sequences (Yin et al., 2012). The antiSMASH database was downloaded on 06/2017 and analyzed using antiSMASH ver. 4.0 (Blin et al., 2017). Queries used in TBLASTN searches were gene sequences ascertained from searching the literature (Bruto et al., 2014; Glick, 2012; Sparacino-Watkins et al., 2014).
HISAT2 was used to align sequencing reads from strain D188-5 to the D188 reference genome sequence. Variants (SNPs) were called using freebayes, filtered to those with quality score greater than 20 using vcffilter, and annotated using SNPdat v. 1.0.5 (Doran and Creevey, 2013).
Plant growth conditions, assays, and data analysis
Request a detailed protocolSeedling root inhibition assays were performed as described previously, with the exception that after bacteria were adjusted to OD600 = 0.5, they were sometimes diluted or concentrated (Creason et al., 2014b). At least 100 seedlings were assayed per treatment. Images were taken at 7 days post inoculation (dpi) and data were analyzed. For root hair quantification, a dissecting microscope, equipped with a camera, was used to capture images at 10 and 25 dpi. Root hairs within a 1 cm segment, 1 cm below the stem were quantified using ImageJ (Schneider et al., 2012). For cytokinin inhibition assays, three-day-old germinated seedlings were transplanted to MS (half-strength MS, 0.5M MES) medium containing DMSO (control) or 0.01–10.0 µM 6-benzylaminopurine (BA) and then grown and quantified in the same manner.
Leafy galls were induced using the decapitation method on four-week-old N. benthamiana plants (Creason et al., 2014b). Images were taken 28 dpi.
Pisum sativum ‘Alaska’ seeds were surface-sterilized in 70% ethanol for 1 min, 10% bleach for 10 min, and washed three times with sterile water. Seeds were soaked in sterile water for 60 min and then plated on water agar (15 g agar/L). Plates were incubated at 23°C until radicles were approximately 5 mm. Germinated seeds were soaked in suspension of Rhodococcus isolates (OD600 = 0.2) or 10 mM MgCl2 buffer for 45 min. Ten seeds per treatment were included in each experiment. Inoculated seeds were placed in sterile test tubes containing 5 ml of Hoagland’s nutrient agar. Samples were incubated for 14 days at 23°C with a 16/8 light/dark cycle. Stem number and length were quantified at 14 dpi.
Infection of UCB-1 pistachio was done, with minor modifications, according to previously described protocols (Stamler et al., 2015b). Briefly, control isolates, PBTS1, PBTS2, and a 1:1 mixture of PBTS1 and PBTS2 were suspended in 10 mM MgCl2 (final OD600 = 0.7). Treatment groups consisting of 15 seedlings were spray-inoculated with 200 ml of bacterial suspension. A mock-inoculated control group was sprayed with 200 ml of 10 mM MgCl2. Inoculated seedlings were placed in humidity chambers for 14 days and the plants were maintained in a greenhouse for seven months. Tree height and internode length were measured at 30 day intervals, and the final measurements were recorded at 210 dpi.
Unless indicated, all experiments were repeated at least three times with similar results. For all data sets, outliers were identified using the ROUT method (Q = 1%) and removed. Data were analyzed using One-way or Two-way ANOVA followed by Tukey’s multiple comparisons test (GraphPad Prism v.7, GraphPad Software, La Jolla, CA, USA). Box and whisker plots were generated using the Tukey method; colored dots indicate outliers. Means are indicated by +.
Nucleic acid manipulations
Request a detailed protocolThe attR gene was PCR-amplified from D188 genomic DNA and subcloned downstream to the L5 bacteriophage promoter in vector pJDC165 (Jeff Cirillo, Texas A and M). The L5::attR construct was verified via Sanger sequencing. Rhodococcus competent cells were prepared from overnight-grown 3 ml cultures. Cells were pelleted and washed twice with sterile, cold dH2O, followed by one wash with sterile, cold 10% glycerol. Cells were resuspended in 50 µl 10% glycerol. Plasmid DNA (0.5–1 µg) was added to the cells. After 30 min of incubation on ice, the cells were electroporated in 1 mm gap cuvettes at 2.2 kV. Cells were resuspended in 250 µl SOC medium and incubated at 28°C with shaking for 16 hr prior to plating on LB medium with appropriate antibiotics.
For PCR, the following were used: 1x ThermoPol reaction buffer (New England Biolab, Ipswich, MA, USA), 200 μM dNTPs, 0.2 μM of each primer, 50 ng genomic DNA template, 0.625 units Taq DNA polymerase (New England Biolab, Ipswich, MA, USA), in a final volume of 25 μl. PCR conditions were 95°C, 3 min; 30 cycles of 95°C for 30 s, 55°C for 30 s, 72°C for 1 min; 72°C for 10 min; 16°C hold. Reactions with water, instead of a DNA template, were used as a negative control.
For LAMP, the reaction mixture was as follows: 0.5 ng DNA template, 1x ThermoPol reaction buffer (New England Biolab, Ipswich, MA, USA), 5 mM MgSO4, 140 µM dNTPs, 146 µM hydoxynaphthol blue (HNB), 1.6 µM each 16FIP and 16BIP primers, 0.2 µM each 16F3 and 16B3 primers, and 12 U Bst polymerase in a final volume of 25 µl. Reactions were incubated at 64°C for 60 min and then cooled to 4°C. Tubes were centrifuged briefly at 8000 rpm.
RPA was done per the manufacturer’s instructions (TwistAmp Basic, TwistDx Limited, Cambridge, UK). Reactions consist of 0.48 μM per primer, 29.5 μl rehydration buffer, 12.2 μl water, and 1.0 μl genomic DNA. A volume of 2.5 μl 280 mM magnesium acetate (MgAc) was added to initiate the reaction. The reaction was incubated at 37°C for 30 min (Fuller et al., 2017). Products were purified using the QIAquick PCR purification kit (Qiagen, Germany), run out on a 2.0% agarose gel, stained with ethidium bromide, and visualized under UV light. Products were verified via Sanger sequencing.
RPA reactions coupled to lateral flow detection were comprised of 0.42 μM forward primer, 0.42 μM biotin-labeled reverse primer, 0.12 μM probe, 29.5 μM rehydration buffer, 12.2 μl water, and 1.0 μl of 25 ng/μl genomic DNA. Reactions were added to a freeze-dried pellet provided by the manufacturer (TwistAmp nfo, TwistDx Limited, Cambridge, UK) with the subsequent addition of 2.5 μl of 280 mM MgAc to initiate the reaction. Following subsequent incubation at 37°C for 30 min, the dual-labelled amplicon was visualized using a lateral flow dipstick (Milenia Biotec GMbH, Germany). One microliter of the RPA product was diluted in 49 μl 1.0x PBST and 10 μl of the dilution were applied to the base of the dipstick, which was subsequently submerged in 100 μl 1.0 PBST at room temperature until the visualization of the positive control band, typically lasting two minutes.
Sequences of primers and probes, and their modifications, are described in Supplementary file 1I.
Data availability
-
Sequencing of plant-associated RhodococcusPublicly available at NCBI BioProject (accession no: PRJNA395383).
-
Analysis of Genome Sequences from Plant Pathogenic Rhodococcus Reveals Genetic Novelties in Virulence LociPublicly available at NCBI Genome (accession no: PRJNA233522).
References
-
FastQC: A Quality Control tool for High Throughput Sequence DataFastQC: A Quality Control tool for High Throughput Sequence Data, http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
-
SPAdes: a new genome assembly algorithm and its applications to single-cell sequencingJournal of Computational Biology 19:455–477.https://doi.org/10.1089/cmb.2012.0021
-
Microbial co-operation in the rhizosphereJournal of Experimental Botany 56:1761–1778.https://doi.org/10.1093/jxb/eri197
-
EvobiR: Tools for comparative analyses and teaching evolutionary biologyEvobiR: Tools for comparative analyses and teaching evolutionary biology, 1.1, https://zenodo.org/record/30938#.Wh5IHdSF5xA, 10.5281/zenodo.30938.
-
antiSMASH 4.0—improvements in chemistry prediction and gene cluster boundary identificationNucleic Acids Research 45:W36–W41.https://doi.org/10.1093/nar/gkx319
-
BBMap short read aligner, and other bioinformatic toolsBBMap short read aligner, and other bioinformatic tools, http://sourceforge.net/projects/bbmap/.
-
vcf_tab_to_fasta_alignment.plvcf_tab_to_fasta_alignment.pl, https://github.com/JinfengChen/vcf-tab-to-fasta.
-
GET_HOMOLOGUES, a versatile software package for scalable and robust microbial pangenome analysisApplied and Environmental Microbiology 79:7696–7701.https://doi.org/10.1128/AEM.02411-13
-
Fasciation induction by the phytopathogen Rhodococcus fascians depends upon a linear plasmid encoding a cytokinin synthase geneThe EMBO journal 11:795–804.
-
Agrobacterium bv. 1 strains isolated from nodules of tropical legumesSystematic and Applied Microbiology 22:119–132.https://doi.org/10.1016/S0723-2020(99)80035-6
-
Conjugative transfer of cadmium resistance plasmids in Rhodococcus fascians strainsJournal of Bacteriology 170:2401–2405.https://doi.org/10.1128/jb.170.5.2401-2405.1988
-
Which specificity in cooperation between phytostimulating rhizobacteria and plants?Research in Microbiology 163:500–510.https://doi.org/10.1016/j.resmic.2012.08.006
-
pFiD188, the linear virulence plasmid of Rhodococcus fascians D188Molecular Plant-Microbe Interactions 25:637–647.https://doi.org/10.1094/MPMI-08-11-0215
-
CONTIGuator: a bacterial genomes finishing tool for structural insights on draft genomesSource Code for Biology and Medicine 6:11.https://doi.org/10.1186/1751-0473-6-11
-
Bacteria with ACC deaminase can promote plant growth and help to feed the worldMicrobiological Research 169:30–39.https://doi.org/10.1016/j.micres.2013.09.009
-
Cell and developmental biology of arbuscular mycorrhiza symbiosisAnnual Review of Cell and Developmental Biology 29:593–617.https://doi.org/10.1146/annurev-cellbio-101512-122413
-
Genome sequence and mutational analysis of plant-growth-promoting bacterium Agrobacterium tumefaciens CCNWGS0286 Isolated from a zinc-lead mine tailingApplied and Environmental Microbiology 78:5384–5394.https://doi.org/10.1128/AEM.01200-12
-
Whole-genome sequencing for analysis of an outbreak of meticillin-resistant Staphylococcus aureus: a descriptive studyThe Lancet Infectious Diseases 13:130–136.https://doi.org/10.1016/S1473-3099(12)70268-2
-
A leaf-inhabiting endophytic bacterium, rhodococcus sp. kb6, enhances sweet potato resistance to black rot disease caused by ceratocystis fimbriataJournal of Microbiology and Biotechnology 26:488–492.https://doi.org/10.4014/jmb.1511.11039
-
MAFFT multiple sequence alignment software version 7: improvements in performance and usabilityMolecular Biology and Evolution 30:772–780.https://doi.org/10.1093/molbev/mst010
-
vcfr: a package to manipulate and visualize variant call format data in RMolecular Ecology Resources 17:44–53.https://doi.org/10.1111/1755-0998.12549
-
Biodegradation and Rhodococcus--masters of catabolic versatilityCurrent Opinion in Biotechnology 16:282–290.https://doi.org/10.1016/j.copbio.2005.04.007
-
The sequence alignment/map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
The carbohydrate-active enzymes database (CAZy) in 2013Nucleic Acids Research 42:D490–D495.https://doi.org/10.1093/nar/gkt1178
-
Too much love, a root regulator associated with the long-distance control of nodulation in Lotus japonicusMolecular Plant-Microbe Interactions 22:259–268.https://doi.org/10.1094/MPMI-22-3-0259
-
Phylogenetic and physiological diversity of microorganisms isolated from a deep greenland glacier ice coreApplied and Environmental Microbiology 70:202–213.https://doi.org/10.1128/AEM.70.1.202-213.2004
-
The winnowing: establishing the squid-vibrio symbiosisNature Reviews Microbiology 2:632–642.https://doi.org/10.1038/nrmicro957
-
Rhodococcus fascians impacts plant development through the dynamic fas-mediated production of a cytokinin mixMolecular Plant-Microbe Interactions 23:1164–1174.https://doi.org/10.1094/MPMI-23-9-1164
-
Induced systemic resistance by beneficial microbesAnnual Review of Phytopathology 52:347–375.https://doi.org/10.1146/annurev-phyto-082712-102340
-
Rhodococcus fascians in Herbaceous PerennialsPlant Disease 91:1064–1076.https://doi.org/10.1094/PDIS-91-9-1064
-
Isolation, diversity, and antimicrobial activity of rare actinobacteria from medicinal plants of tropical rain forests in Xishuangbanna, ChinaApplied and Environmental Microbiology 75:6176–6186.https://doi.org/10.1128/AEM.01034-09
-
Biodiversity, bioactive natural products and biotechnological potential of plant-associated endophytic actinobacteriaApplied Microbiology and Biotechnology 89:457–473.https://doi.org/10.1007/s00253-010-2923-6
-
Molecular mechanisms controlling legume autoregulation of nodulationAnnals of Botany 108:789–795.https://doi.org/10.1093/aob/mcr205
-
Phytools: an R package for phylogenetic comparative biology (and other things)Methods in Ecology and Evolution 3:217–223.https://doi.org/10.1111/j.2041-210X.2011.00169.x
-
Endophytic actinobacteria associated with dracaena cochinchinensis lour.: Isolation, diversity, and their cytotoxic activitiesBioMed Research International 2017:1308563.https://doi.org/10.1155/2017/1308563
-
NIH Image to ImageJ: 25 years of image analysisNature Methods 9:671–675.https://doi.org/10.1038/nmeth.2089
-
Prokka: rapid prokaryotic genome annotationBioinformatics 30:2068–2069.https://doi.org/10.1093/bioinformatics/btu153
-
Horizontal gene transfer: building the web of lifeNature Reviews Genetics 16:472–482.https://doi.org/10.1038/nrg3962
-
Indole-3-acetic acid in microbial and microorganism-plant signalingFEMS Microbiology Reviews 31:425–448.https://doi.org/10.1111/j.1574-6976.2007.00072.x
-
Nitrate and periplasmic nitrate reductasesChem. Soc. Rev. 43:676–706.https://doi.org/10.1039/C3CS60249D
-
A successful bacterial coup d'état: how Rhodococcus fascians redirects plant developmentAnnual Review of Phytopathology 49:69–86.https://doi.org/10.1146/annurev-phyto-072910-095217
-
Leafy gall formation is controlled by fasR, an AraC-type regulatory gene in Rhodococcus fasciansJournal of Bacteriology 182:5832–5840.https://doi.org/10.1128/JB.182.20.5832-5840.2000
-
Picard ToolsPicard Tools, http://broadinstitute.github.io/picard.
-
Plant growth-promoting rhizobacteria and root system functioningFrontiers in Plant Science 4:356.https://doi.org/10.3389/fpls.2013.00356
-
Beneficial microbes affect endogenous mechanisms controlling root developmentTrends in Plant Science 21:218–229.https://doi.org/10.1016/j.tplants.2016.01.013
-
Chromosomal locus that affects pathogenicity of Rhodococcus fasciansJournal of Bacteriology 184:1112–1120.https://doi.org/10.1128/jb.184.4.1112-1120.2002
-
Whole-genome sequencing to delineate Mycobacterium tuberculosis outbreaks: a retrospective observational studyThe Lancet Infectious Diseases 13:137–146.https://doi.org/10.1016/S1473-3099(12)70277-3
-
dbCAN: a web resource for automated carbohydrate-active enzyme annotationNucleic Acids Research 40:W445–W451.https://doi.org/10.1093/nar/gks479
Article and author information
Author details
Alexandra J Weisberg

Niklaus J Grünwald
Funding
National Institute of Food and Agriculture (2014-51181-22384)
- Niklaus J Grünwald
- Melodie L Putnam
- Jeff H Chang
National Institute of Food and Agriculture (2013-67012-21139)
- Elizabeth A Savory
National Institute of Food and Agriculture (2017-67012-26126)
- Alexandra J Weisberg
The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Acknowledgements
This work would not have been possible without the contribution of plants from multiple nurseries, for which we are appreciative. We thank Dr. Edward Davis II for assistance with computational methods, Heidi Lederhos for her assistance in the laboratory, Dr. Zhian Kamvar for assistance with using poppr, Dr. Jeffrey Anderson (Oregon State University) for permitting use of the plate reader, Dr. Jeffery Dangl (University of North Carolina) for providing Rhodococcus spp. isolates 114MFTsu3.1, UNC23MFCrub1.1 and 29MFTsu3.1, Dr. Jeff Cirillo (Texas A and M) for pJDC165, Dr. Jennifer Randall (New Mexico State University) for Rhodococcus spp. isolates PBTS1 and PBTS2, and staff in the Center for Genome Research and Biocomputing at Oregon State University for sequencing services. This work was supported by the National Institute of Food and Agriculture, US Department of Agriculture award 2014-51181-22384, to JHC, MLP, and NJG. EAS was supported by USDA NIFA award 2013-67012-21139. AJW was supported by USDA NIFA award 2017-67012-26126. Last, we thank the Department of Botany and Plant Pathology at Oregon State University for its generous support of SLF and the computing cluster. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Copyright
© 2017, Savory et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 5,241
- views
-
- 660
- downloads
-
- 89
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 89
- citations for umbrella DOI https://doi.org/10.7554/eLife.30925
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Evolutionary transitions between beneficial and phytopathogenic Rhodococcus challenge disease management
eLife 6:e30925.
https://doi.org/10.7554/eLife.30925




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}