Structural basis of malodour precursor transport in the human axilla
- University of Oxford, United Kingdom
- University of York, United Kingdom
- Unilever Discover, United Kingdom
Abstract
Mammals produce volatile odours that convey different types of societal information. In Homo sapiens, this is now recognised as body odour, a key chemical component of which is the sulphurous thioalcohol, 3-methyl-3-sulfanylhexan-1-ol (3M3SH). Volatile 3M3SH is produced in the underarm as a result of specific microbial activity, which act on the odourless dipeptide-containing malodour precursor molecule, S-Cys-Gly-3M3SH, secreted in the axilla (underarm) during colonisation. The mechanism by which these bacteria recognise S-Cys-Gly-3M3SH and produce body odour is still poorly understood. Here we report the structural and biochemical basis of bacterial transport of S-Cys-Gly-3M3SH by Staphylococcus hominis, which is converted to the sulphurous thioalcohol component 3M3SH in the bacterial cytoplasm, before being released into the environment. Knowledge of the molecular basis of precursor transport, essential for body odour formation, provides a novel opportunity to design specific inhibitors of malodour production in humans.
https://doi.org/10.7554/eLife.34995.001eLife digest
Human body odour contains a number of chemicals, but the most pungent and recognisable are thioalcohols. These molecules are created through a series of chemical reactions that start with an odourless precursor, a compound produced in glands located in our armpits. Then, a type of bacteria called Staphylococcus hominis takes in these molecules and transforms them into smelly thioalcohols. The precise details of how the bacteria do this are not clear.
Now, Minhas, Bawdon et al. show how S. hominis uses a transport protein in its membrane to bring the odourless precursor inside. In the experiments, tools such as X-ray crystallography captured snapshots of this transporter as it was moving the compound into the bacteria. This helped to understand how the bacteria recognize these precursors, as well as the exact structure of these molecules and of their transporters.
The experiments also reveal that bacteria which do not create odour can also ingest the precursors through this same process. This suggests that the odour production is a unique process that happens once these molecules are inside S. hominis.
The findings imply that humans and their body odour-producing bacteria likely evolved together. In other mammals, the bacterial production of bodily smells is linked to the release of pheromones, which are chemicals involved in communication and in selecting sexual partners. It is not clear whether this is also true for humans. Ultimately, learning more about how S. hominis converts precursor molecules into thioalcohols could lead to new ways of nipping body odours in the bud.
https://doi.org/10.7554/eLife.34995.002Introduction
In Homo sapiens the skin of the underarm (axilla) provides a unique niche for bacteria. Through the secretions of various glands, that open onto the skin or into hair follicles, this environment is nutrient-rich and hosts a unique microbial community (Taylor et al., 2003). Esterified fatty acids and other lipids are secreted by the sebaceous gland, while the eccrine glands produce a dilute salt solution with lactate and other organic solutes being secreted (Bovell, 2015). A third exocrine gland, the apocrine, which is more specialized and found only in the axilla, areolae, genitalia and external auditory meatus (ear canal) (Collins, 1989), starts secreting an odourless lipid-rich viscous secretion with the onset of puberty (Figure 1A). The lipid rich secretion is not involved in thermoregulation like the eccrine gland and in mammals has a likely role in scent generation (Collins, 1989).
Figure 1
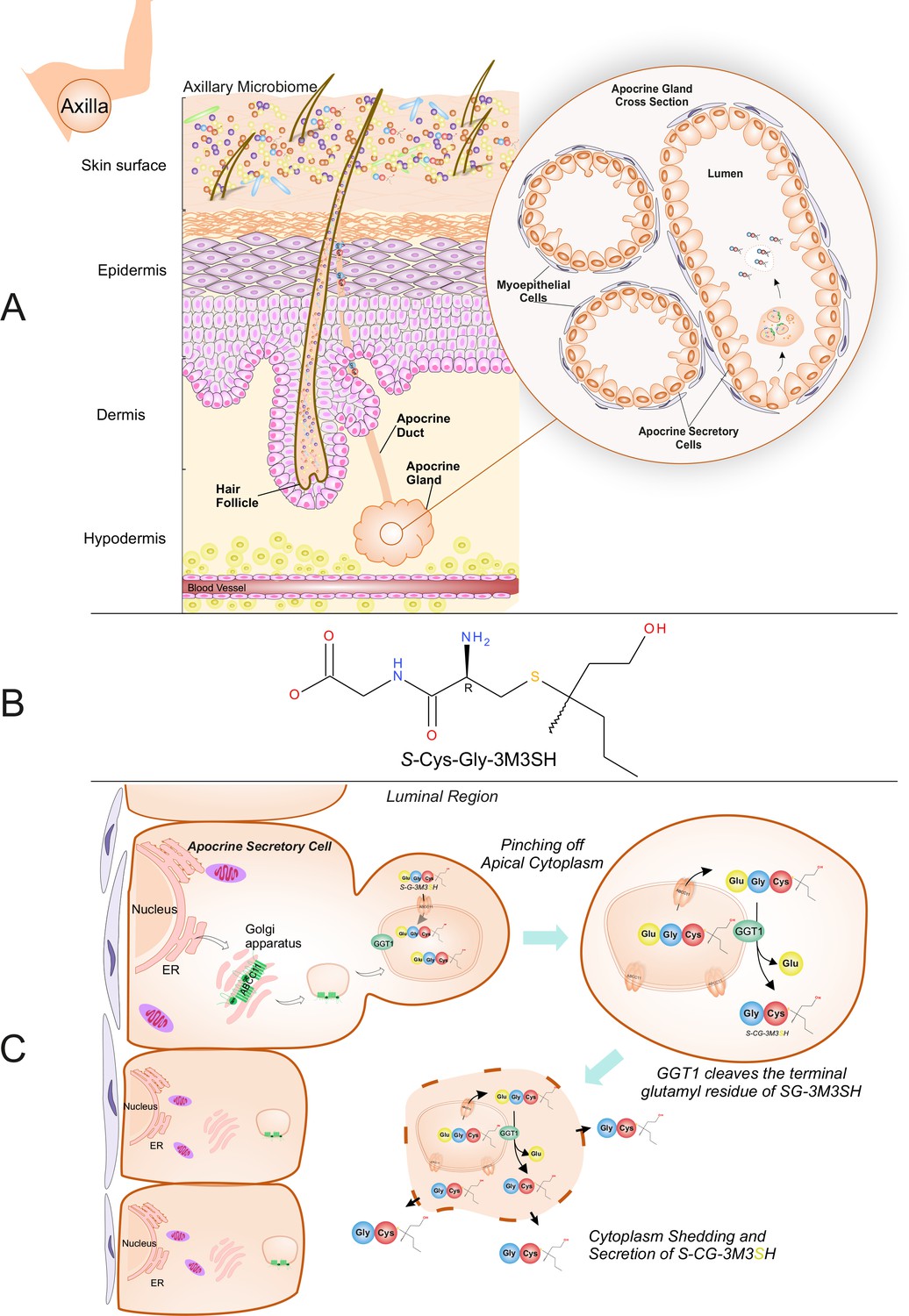
Biochemical route of axillary malodour precursor peptide S-Cys-Gly-3M3SH.
(A) Schematic representation of the axilla. The surface of the skin is composed of the axillary microbiome, typically dominated by either Corynebacterium or Staphylococci. Axillary apocrine glands are subcutaneously located in the hypodermis. Apocrine glands secrete a variety of odourless precursors that are bio-transformed into odorous volatiles by the axillary microbiome. Sulfanylalkanols (e.g. 3-methyl-3-sulfanylhexan-1-ol (3M3SH)) are secreted as cysteinylglycine conjugated peptides (Starkenmann et al., 2005; Emter and Natsch, 2008). Staphylococcus hominis transport and metabolise cysteinylglycine- 3-methyl-3-sulfanylhexan-1-ol (S-Cys-Gly-3M3SH) to liberate the odorous volatile 3M3SH, which is one of the major components of axillary malodour. (B) Chemical structure of the malodour precursor peptide, S-Cys-Gly-3M3SH. (C) Synthesis of S-Cys-Gly-3M3SH. S-Cys-Gly-3M3SH is derived from the glutathione 3-methyl-3-sulfanylhexanol conjugate (SG-3M3SH). SG-3M3SH is actively transported by the ATP-binding cassette transporter sub-family C member 11 (ABCC11) into apocrine secretory vesicles (Baumann et al., 2014b). In the secretory vesicle, S-Gly-3M3SH is bio-transformed by γ-glutamyl transferase 1 (GGT1), which cleaves the terminal glutamyl residue generating S-Cys-Gly-3M3SH. S-Cys-Gly-3M3SH is secreted to the surface of the skin by apocrine secretion, which characteristically involves pinching off the apical cytoplasm and partial shedding releasing intracellular components into the luminal region of the apocrine gland. Upon release the cytoplasm may be recovered and repaired and a new cycle of secretion can occur (Farkaš, 2015).
In humans, the link between the axillary apocrine gland, underarm bacteria and body odour (BO), was first recognised over 50 years ago (Shelley et al., 1953) and early culture-based studies on this relationship found the axillary microbiota to be dominated by Staphylococcus, Corynebacterium and Propionibacterium species, with the Corynebacteria in particular correlating with malodour intensity (Taylor et al., 2003; Leyden et al., 1981). More recent culture-independent meta-taxonomic studies have confirmed that the axilla is dominated by these genera, although the presence of additional taxa not previously indicated by culture-based methods is also evident, notably Gram-positive anaerobic cocci (GPAC) belonging to the Anaerococcus and Peptoniphilus genera (Costello et al., 2009; Egert et al., 2011; Troccaz et al., 2015). Various molecules produced in the axilla have been linked to malodour, including 16-androstene steroids (Gower et al., 1994) and short-chain (C2-C5) volatile fatty acids (VFAs) produced by axillary bacteria via several metabolic routes (James et al., 2004). However, 16-androstenes are now believed to be only minor contributors to axillary malodour (James et al., 2013), and it is accepted that the VFAs most heavily involved in BO are a series of structurally-unusual medium-chain (C6-C10) acids originating from N-acylglutamine precursors present in apocrine sweat (Natsch et al., 2003; Natsch et al., 2006). A third class of molecule, the thioalcohols (also called sulfanylalkanols), have recently been identified as important components of human body odour and a number of these have been detected within the volatiles released from the underarm (Troccaz et al., 2004; Natsch et al., 2004; Hasegawa et al., 2004). They are particularly pungent and can be detected in pg.l−1 quantities, an order of magnitude lower than many other volatile chemicals emanating from the skin (Natsch et al., 2004). The most abundant and pungent of these thioalcohols is 3-methyl-3-sulfanylhexan-1-ol (3M3SH) (Figure 1B), which is a primary causal molecule of axillary malodour and subsequent BO (Troccaz et al., 2004; Hasegawa et al., 2004).
Thioalcohols are known to be released as a peptide-conjugated precursor from the apocrine gland, which secretes directly into the hair follicle (Figure 1A). The biosynthetic origin of the thioalcohol precursors is currently poorly understood, but they are thought to be secreted into the axilla via the ABC transporter ABCC11 as odourless glutathione conjugates (Baumann et al., 2014a). The glutathione conjugated form (SG-3M3SH) has never been detected in axillary sweat; however, the L-cysteinylglycine dipeptide conjugated form, S-[1-(2-hydroxyethyl)−1-methylbutyl]-L-cysteinylglycine (S-Cys-Gly-3M3SH) (Figure 1B), is readily detectable (Starkenmann et al., 2005) and recently a γ-glutamyl transferase (GGT1) has been demonstrated to localise to the apocrine sweat gland and convert SG-3M3SH to S-Cys-Gly-3M3SH (Baumann et al., 2014a) (Figure 1C). We and others have demonstrated that S-Cys-Gly-3M3SH can be taken up and metabolised by a highly limited range of staphylococcal species present in the axilla, namely Staphylococcus hominis, Staphylococcus haemolyticus and Staphylocooccus lugdunensis, to liberate 3M3SH, that subsequently exits the cell through an undefined mechanism (Troccaz et al., 2004; Starkenmann et al., 2005; Bawdon et al., 2015). A strong correlation between S. hominis and body odour production has recently been established using culture-independent taxonomic studies of the axillary microbiota (Troccaz et al., 2015), confirming the biological significance of this low-abundance species within the axilla in BO production in humans. However, the mechanism by which the S-Cys-Gly-3M3SH precursor molecule is taken up into S. hominis and related thioalcohol producers is currently unknown, hampering efforts to identify novel mechanisms to disrupt this process and control odour production in humans.
Here we identify the biochemical and structural basis for the recognition and specific transport of the malodour precursor peptide, S-Cys-Gly-3M3SH, into S. hominis. We first show that S-Cys-Gly-3M3SH is actively taken up by S. hominis through a specific secondary active transporter, STAH0001_1446 (SH1446), a member of the proton coupled oligopeptide transporter (POT) family (Hagting et al., 1994; Paulsen and Skurray, 1994). Through a combination of structural, biochemical and cell based assays we reveal how this peptide transporter is able to recognise a thioalcohol-conjugated peptide and further, show that transport is coupled to the inwardly directed proton electrochemical gradient. Our results suggest that SH1446 is a clear target for inhibitor design to prevent malodour production, thus providing further molecular insights into the role of the skin microbiota in human scent generation.
Results
POT transporters are essential for transport of thiol-conjugated dipeptides in E. coli
To elucidate how thiol-conjugated dipeptides are transported by bacteria, we first examined the model organism Escherichia coli, which is able to recognise and metabolise the dipeptide version of the non-physiological malodour precursor S-benzyl-L-cysteine, namely, S-benzyl-L-cysteinylglycine (James et al., 2013). Resting cells of E. coli were indeed able to remove S-benzyl-L-cysteinylglycine from solution and break it down to release the thiol active product, benzyl mercaptan (Figure 2—figure supplement 1A,B). Following transport into the cell, the dipeptide must be cleaved to release the benzyl mercaptan, most likely through a cysteine-S-conjugate β-lyase-type activity, for which a number have been described in E. coli, including MetC and MalY (Dwivedi et al., 1982; Zdych et al., 1995; Awano et al., 2003). Interestingly, TnaA which is a tryptophanase known to have L-cysteine desulfhydrase activity (Awano et al., 2005) is also capable of releasing benzyl mercaptan.
Individual disruption of metC and malY did not decrease the amount of benzyl mercaptan produced by E. coli, while disruption of tnaA led to an almost complete loss of biotransformation (Figure 2—figure supplement 1C). This suggests that E. coli takes up the S-benzyl-L-cysteinylglycine, whereupon a host dipeptidase cleaves the glycine residue prior to cleavage of the S-benzyl-cysteine by TnaA to release the benzyl mercaptan.
To discover the genetic basis of the transport and metabolism of these thiol-conjugated dipeptides, we exploited the knock out (KO) library of single gene disruptions in E. coli K-12 (Baba et al., 2006). We hypothesised that the dipeptide moiety might direct the molecule through a peptide transporter. In E. coli the main peptide systems are relatively well characterised, the two main transport systems belong to the ATP binding cassette (ABC) family and the proton dependent oligopeptide transporter (POT) family (Figure 2B). The E. coli Opp and Dpp ABC transporters have been extensively studied in E. coli and the closely related bacterium Salmonella typhimurium (Smith et al., 1999; Klepsch et al., 2011; Abouhamad et al., 1991; Goodell and Higgins, 1987; Tame et al., 1995) and an E. coli BW25113 strain with non-functional Opp and Dpp system (ΔDB1), was used to assess the role of these transporters in the production of benzyl mercaptan. Surprisingly, benzyl mercaptan production was similar to the wild-type strain, suggesting that neither ABC transporter was important for Cys-Gly-conjugate transport (Figure 2A). Having ruled out transport via the ABC peptide transporters, we then broadened our search to include additional peptide transporters that have been described in E. coli. These were the four members of the proton dependent oligopeptide transporter, or POT family, encoded by dtpA-D, that have poorly defined physiological roles during growth on peptides (Harder et al., 2008; Prabhala et al., 2014; Casagrande et al., 2009), the CstA protein, orthologues of which have been demonstrated to be peptide transporters (Garai et al., 2016) and the Sap system, which is implicated in uptake of cationic antimicrobial peptides (Groisman et al., 1992). Most of these strains had similar levels of benzyl mercaptan production to the wild-type, with the exception of dtpB which had an overall 50% drop in thiol production (Figure 2A). These data suggest that transporters belonging to the POT family are most likely responsible for Cys-Gly-conjugate transport, revealing a new biological role for bacterial POT transporters in the uptake of Cys-Gly-(S) conjugates.
Figure 2 with 5 supplements see all
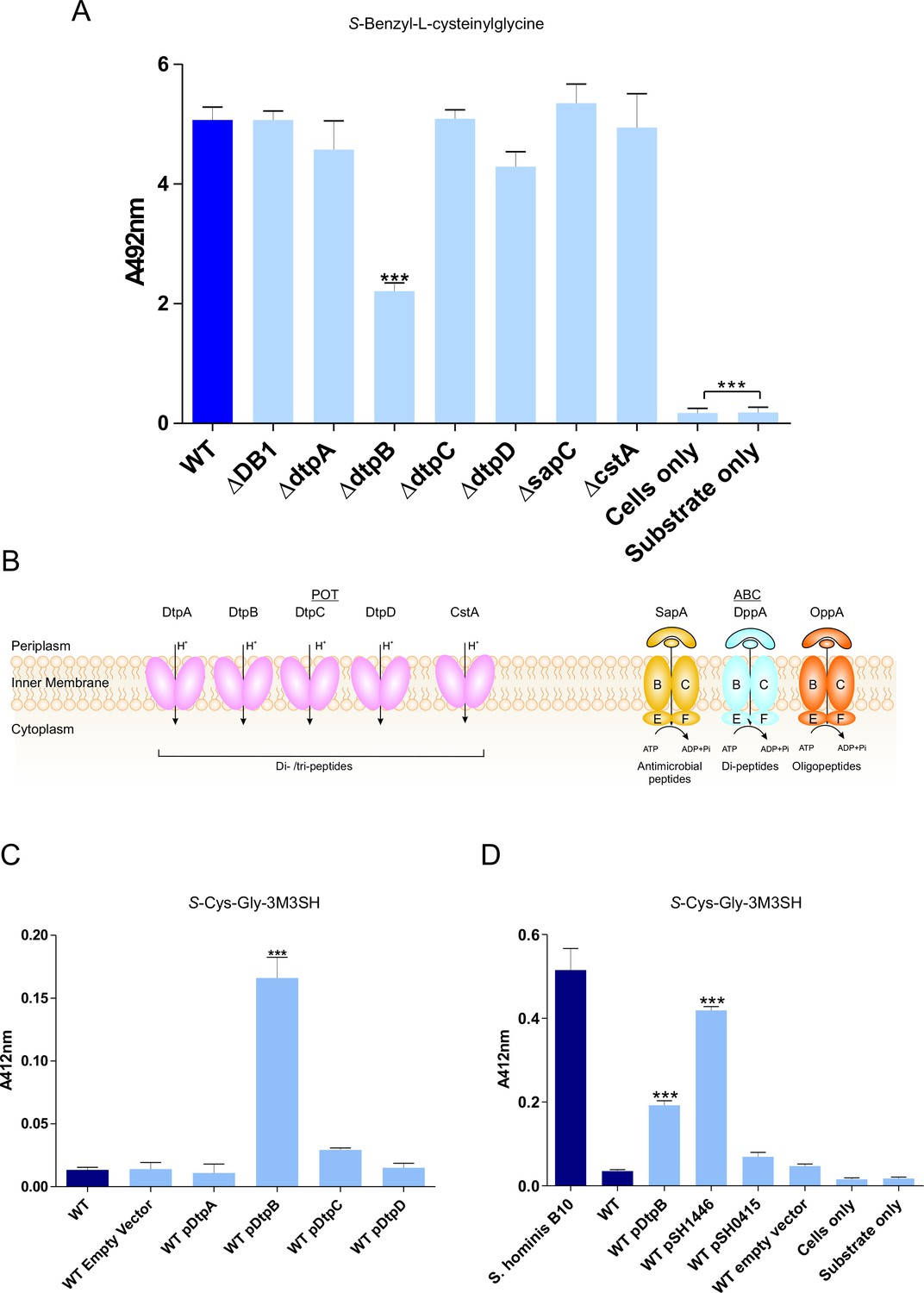
Biotransformation of both model (S-Benzyl-L-cysteinylglycine) and physiological substrate S-Cys-Gly-3M3SH by model organism E.coli K-12.
(A) Biotransformation of S-Benzyl-L-cysteinylglycine by E. coli strains containing deletion in known or predicted peptide transporters. Strain ΔDB1 is an oppA dppA double mutant. Thioalcohol yield (A492) was quantified for each strain using MTS/PMS following 4 hr incubation. (B) Schematic overview of the main peptide transport systems in E. coli. (C) Biotransformation of S-Cys-Gly-3M3SH in E. coli K-12 overexpressing Dtp POT transporter proteins. (D) S-Cys-Gly-3M3SH biotransformation by E. coli K-12 overexpressing S. hominis STAHO0001_1446 (pSH1446) and STAHO0001_0415 (pSH0415). E. coli overexpressing dtpB (pDtpB) is shown for reference. Thioalcohol yield (A412) was quantified using DTNB after 24 hr incubation. Cultures with overexpressing plasmids were pre-induced with 0.0001% L-arabinose overnight. E. coli expressing pBADcLIC2005 with no gene insertion was included as a negative control (Empty vector). Error bars: ±SD (biological triplicates); *p<0.05, **p<0.01, ***p<0.001; One-way ANOVA followed by Dunnett’s Multiple Comparison test.
Identification of a POT transporter from S. hominis that confers malodour production to E. coli
Having established that E. coli has the ability to transport and cleave a Cys-Gly-(S) conjugated substrate, we reasoned that overexpression of a POT transporter might confer the ability to recognise the physiological malodour precursor S-Cys-Gly-3M3SH. The four dtp genes were cloned into a pBAD vector commonly used for overexpression of membrane transporters (Mulligan et al., 2009; Mulligan et al., 2012) and transformed into the wild-type E. coli background. The ability of the resulting strains to convert S-Cys-Gly-3M3SH to 3M3SH was subsequently measured (Bawdon et al., 2015). Consistent with the reduction in benzyl mercaptan production reported in Figure 2A, the dtpB gene alone was able to confer a malodour production (BO+) phenotype to E. coli, which was not seen with any of the other dtp genes (Figure 2C). The same pattern was observed with S-benzyl-Cys-Gly as a substrate (Figure 2—figure supplement 2B), suggesting that DtpB has a unique ability to transport these unusual conjugated peptides.
The molecular basis for S-Cys-Gly-3M3H transport into malodour producing staphylococci is unknown, and the genetic intractability of these poorly studied coagulase negative Staphylococcal species makes it difficult to study using genetic approaches. We therefore used an alternative approach exploiting our discovery that expression of a transporter in trans could produce a BO+ phenotype to E. coli. Given our discovery that E. coli POT family peptide transporters can transport S-Cys-Gly-3M3SH, we examined the genome of S. hominis SK1119, as S. hominis are the most abundant malodour producing Staphylococcal species in the underarm microbiota. Unlike E. coli, S. hominis contains only a single POT family member DtpT (SH1446), orthologues of which were seen in all sequenced staphylococci (Figure 2—figure supplement 3A). The POT peptide systems remain largely uncharacterised in staphyoloccci, although in the well-studied pathogen S. aureus, the single POT transporter encoded by dtpT is the sole route for dipeptide uptake in this species (Hiron et al., 2007). We also identified a second system, encoded by STAHO0001_0415 (SH0415), which is annotated as an MFS transporter and only present in S. hominis and S. haemolyticus, two known thioalcohol producers, and which is genetically linked to genes involved in peptide and amino acid metabolism (Figure 2—figure supplement 3B). Both transporters were cloned into pBADcLIC2005 and transformed into E. coli for functional analysis. When levels of 3M3SH production were measured in these strains, uptake was only enhanced by the presence of SH1446, with SH0415 having little effect on 3M3SH activity (Figure 2D). In contrast, with S-benzyl-cysteinlyglycine as a substrate, there was no increase in production of benzyl mercaptan with SH1446 but a significant increase was observed with SH0415 (Figure 2—figure supplement 2B). Together these data strongly support the conclusion that the S-Cys-Gly-3M3SH transporter in S. hominis is the POT transporter STAH0001_1446 (SH1446).
To further demonstrate the importance of SH1446 in the physiological transport of S-Cys-Gly-3M3SH we used a biochemical approach to examine whether peptides could inhibit 3M3SH production by competing with S-Cys-Gly-3M3SH uptake. Firstly, we used our E. coli system where 3M3SH production is seen when SH1446 is expressed in trans and we observed clear inhibition of 3M3SH production with both di- and tri-Ala peptides (Figure 2—figure supplement 4) but not with L-Ala alone nor tetra-Ala. The purified TnaA enzyme that cleaves Cys-3M3SH was not inhibited by any of these peptides (Figure 2—figure supplement 5) , supporting the hypothesis that the whole cell inhibition is due to inhibition of peptide transport. Similarly, we measured the ability of the same peptides to inhibit 3M3SH production directly in S. hominis (Figure 2—figure supplement 4) and observed a comparable profile. It should be noted that a significant affect was observed for 25 mM tetra-Ala in S. hominis wild-type, however this affect is due to the acidic pH that was required to dissolve the peptide which appears to inhibit metabolism in S. hominis but not E. coli.
The S. hominis POT transporter, PepTSh transports S-Cys-Gly-3M3SH
POT transporters belong to the Major Facilitator Superfamily (MFS) and use an inwardly directed proton electrochemical gradient to drive the concentrative uptake of di- and tri-peptides across the cell membrane for metabolic assimilation (Daniel and Kottra, 2004). To establish SH1446 as the protein responsible for S-Cys-Gly-3M3SH recognition and transport, the gene was cloned and overexpressed in E. coli for functional and structural studies. The purified protein, hereafter referred to as PepTSh (Peptide Transporter Staphylococcus hominis) was purified and reconstituted into liposomes. Transport was studied using a previously employed pyranine based reporter assay, which measures transport via changes to the internal pH of the liposomes (Parker et al., 2014) (Figure 3A). Acidification of the lumen of the liposome was observed in the presence of a membrane potential (ΔΨ, negative inside), and addition of external di- or tri-peptide substrate, demonstrating that PepTSh is a proton coupled peptide transporter. Importantly we also observed acidification in the presence of the S-Cys-Gly-3M3SH precursor peptide, confirming our earlier identification of PepTSh as the transporter for this molecule in S. hominis.
Figure 3
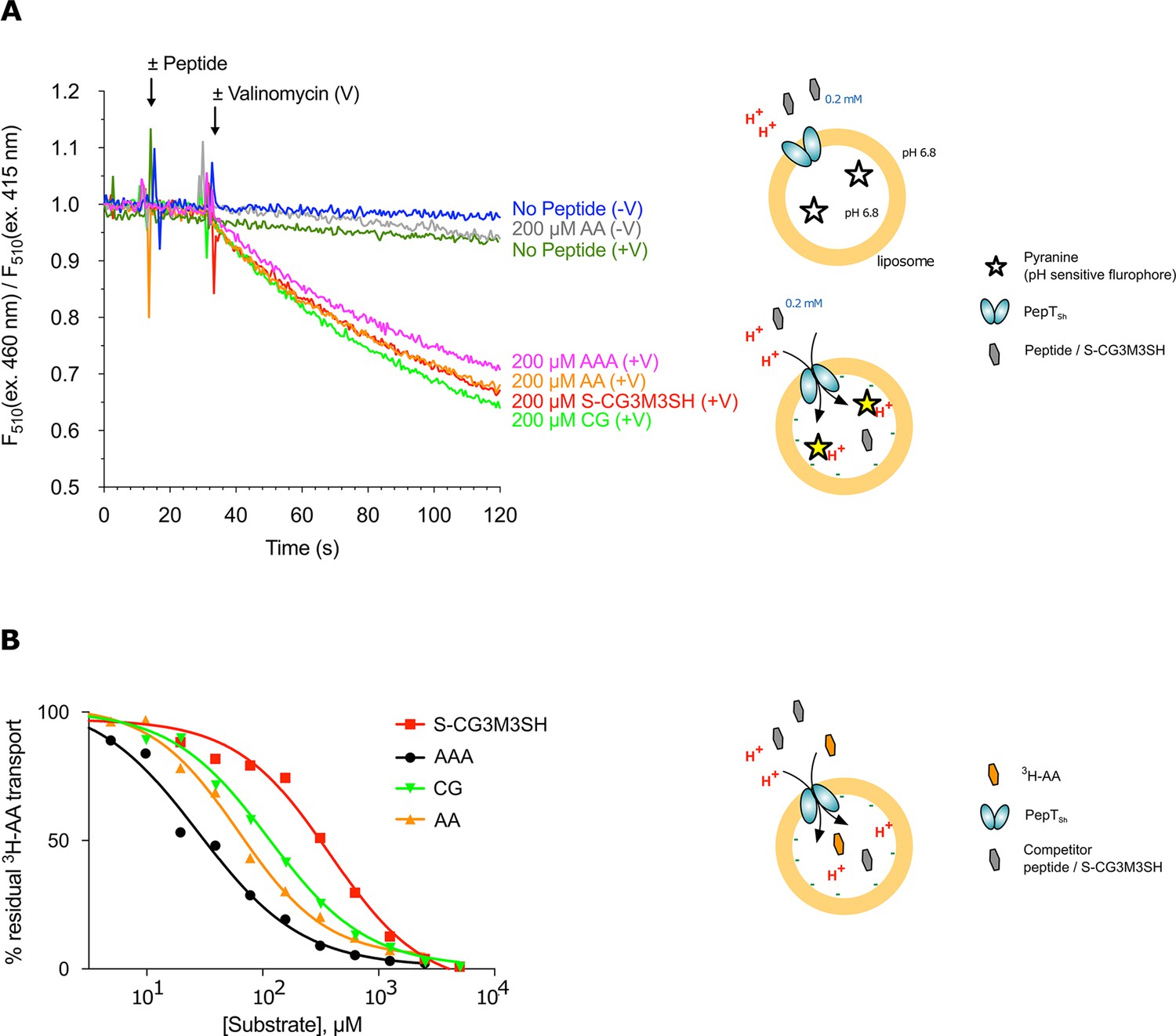
Functional characterisation of PepTSh.
(A) Monitoring proton coupled peptide uptake using a pH sensitive dye. PepTSh was reconstituted into liposomes loaded with pyranine and a high concentration of potassium ions. The external solution contains either peptide, Ala-Ala (orange), Ala-Ala-Ala (purple), Cys-Gly (Green) or S-Cys-Gly-3M3SH (red) and a low potassium concentration. On addition of valinomycin (V) the membrane becomes highly permeable to potassium, generating a hyperpolarised membrane potential (negative inside), which drives the uptake of protons and ligand, protonating the pyranine dye and altering its fluorescent properties. A change in the ratio of fluorescence demonstrates active transport of the ligand. (B) Representative IC50 curves showing the relative affinity of the different ligands shown in A for PepTSh. Assays were set up to measure the ability of competitor peptides to reduce uptake of tritiated di-Alanine (AA) peptide.
To understand the recognition of S-Cys-Gly-3M3SH compared to natural peptides, we determined IC50 values using competition with 3H-di-alanine. The IC50 value for tri-alanine, di-alanine and Cys-Gly were 23.7 ± 3.4, 72.2 ± 9.7 and 115 ± 22.2 μM, respectively (Figure 3B), indicating that PepTSh is able to recognise tri-Ala more favourably than di-Ala. However, for S-Cys-Gly-3M3SH the IC50 value was 362 ± 21.8 μM, three-fold higher than the Cys-Gly peptide. Our data therefore show that while PepTSh can transport the modified peptide, the addition of the thioalcohol group on the cysteine side chain has substantially impaired the recognition and transport versus natural peptides.
Crystal structure of PepTSh bound to S-Cys-Gly-3M3SH
To gain further insight into the mechanism of substrate binding we successfully obtained a high-resolution co-crystal structure of PepTSh at 2.5 Å resolution, bound to S-Cys-Gly-3M3SH using the in meso crystallisation method (Figure 4A, Table 1 and Figure 4—figure supplement 1). PepTSh adopts the canonical MFS fold with helices H1-H6 forming the N-terminal bundle and H7-H12 the C-terminal bundle. PepTSh was crystallised in an inward open state, observed previously for other members of the POT family (Newstead, 2015), wherein the extracellular gate, consisting of helices H1-2 and H7-8 from the N- and C-terminal bundles respectively, is closed and the intracellular gate, consisting of helices H4-5 and H10-11 is open. Similar to other bacterial POT family members, PepTSh contains two additional helices inserted between the N- and C-terminal bundles, termed HA and HB (Figure 4B and Figure 4—figure supplement 2). Unlike previous POT structures however, PepTSh also contains a well-structured β-hairpin motif between helices H7 and H8 on the extracellular side of the membrane, extending out in line with the lipid head groups. Interestingly we observed a number of monoolein lipid molecules that cluster around both the β- hairpin and the cavity created between helices HA and HB with the transporter domain. The role of helices HA and HB in prokaryotic POT family transporters still remains unclear. However, the observation of lipid molecules bound between these helices and the transport domain suggests a possible role in stabilising these transporters in the membrane, which may be an adaption specific to prokaryotic members of the POT family (Newstead et al., 2011).
Figure 4 with 4 supplements see all
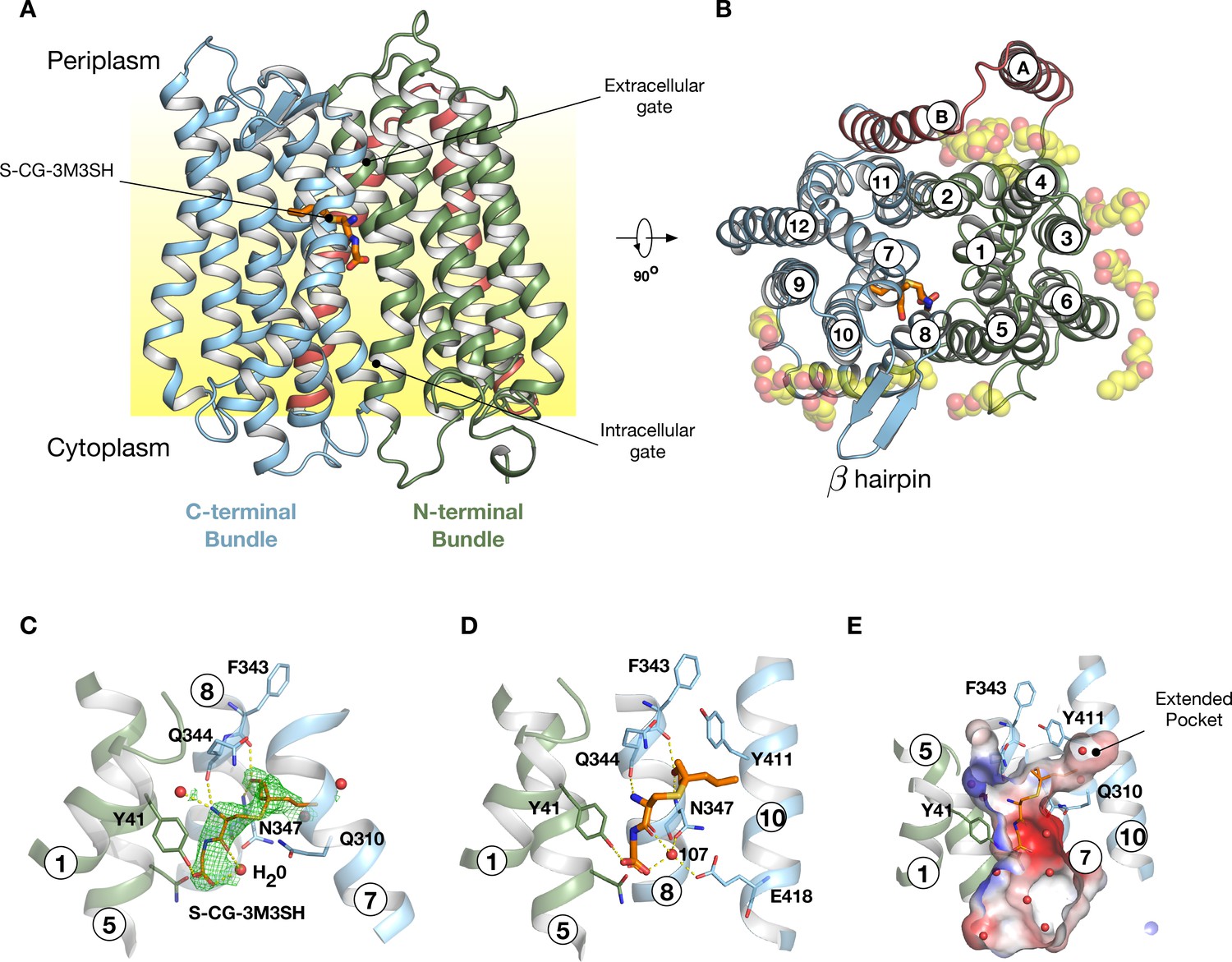
Crystal structure of PepTSh.
(A) Crystal structure of PepTSh shown in the plane of the membrane. Transmembrane helices making up the N-terminal bundle, H1-H6, are coloured green and pack against the C-terminal bundle, helices H7-H12. The bound S-Cys-Gly-3M3SH peptide is shown in sticks. (B) View of A rotated 90 °. The additional helices, HA and HB, can be clearly observed packing to one side of the transporter. The bound lipids are shown as spheres. (C) Zoomed in view of the binding site showing the bound S-Cys-Gly-3M3SH ligand with the mFo-DFc difference electron density map contoured at 3 σ in green mesh. (D) Equivalent view of the binding site showing the location of ordered water molecules in the binding site. (E) View showing the electrostatic surface of the binding site of the transporter with the hydrophobic pocket accommodating the acyl chain of the ligand.
Table 1
Data collection and refinement statistics.
Statistics for the highest-resolution shell are shown in parentheses.
| PepTSh (PDB: 6EXS) | |
|---|---|
| Wavelength | 0.8729 |
| Resolution range | 45.1–2.5 (2.589–2.5) |
| Space group | P 1 21 1 |
| Unit cell | 60.04 55.576 100.866 90 104.749 90 |
| Total reflections | 151797 (14474) |
| Unique reflections | 22501 (2204) |
| Multiplicity | 6.7 (6.6) |
| Completeness (%) | 99.43 (99.73) |
| Mean I/sigma (I) | 9.76 (1.48) |
| Wilson B-factor | 43.87 |
| R-merge | 0.159 (1.28) |
| R-meas | 0.172 (1.39) |
| R-pim | 0.07 (0.54) |
| CC1/2 | 0.997 (0.59) |
| CC* | 0.999 (0.86) |
| Reflections used in refinement | 22504 (2204) |
| Reflections used for R-free | 1137 (110) |
| R-work | 0.21 (0.21) |
| R-free | 0.23 (0.24) |
| CC (work) | 0.88 (0.69) |
| CC (free) | 0.85 (0.52) |
| Number of non-hydrogen atoms | 4063 |
| macromolecules | 3614 |
| ligands | 384 |
| solvent | 65 |
| Protein residues | 487 |
| RMS(bonds) | 0.01 |
| RMS(angles) | 1.04 |
| Ramachandran favored (%) | 98.35 |
| Ramachandran allowed (%) | 1.65 |
| Ramachandran outliers (%) | 0 |
| Rotamer outliers (%) | 1.47 |
| Clashscore | 2.59 |
| Average B-factor | 57.0 |
| macromolecules | 54.72 |
| ligands | 80.53 |
| solvent | 51.2 |
Following extensive screening and co-crystallisation we observed clear mFo-DFc difference electron density for the S-Cys-Gly-3M3SH ligand in the peptide binding site (Figure 4C and Figure 4—figure supplement 3). The Cys-Gly part of the precursor is positioned such that the carboxy termini of the peptide faces towards the intracellular side of the binding site. The terminal carboxyl group makes two hydrogen bond interactions, one with Tyr41 on H1 and a second interaction to a water molecule (W107), which links the carboxyl group with Asn347 on H8. The carbonyl group of the CG peptide also makes a hydrogen bond to this water molecule, creating a well-coordinated interaction network between the peptide and Asn347 (Figure 4D). The terminal amino group makes two further polar interactions, with a water molecule that sits next to Tyr41 and also to the carbonyl group of Gln344, which sits further up on H8. In contrast, the thioalcohol moiety points towards the extracellular gate, with the sulphur atom sitting close to Gln310 on H7, whereas the alcohol group extends towards helix H8 making interactions with backbone carbonyl and amide groups of Phe343 and Asn347, respectively. The acyl group extends into a large pocket nestled between helices H7 and H10 (Figure 4E). This long pocket (~10 Å) extends from the central peptide binding site into the C-terminal bundle of the transporter and is both hydrophobic and polar in character.
Previously, we showed that a related POT family transporter, PepTSt, is able to bind peptides in different orientations in the binding site, with a di-peptide adopting a horizontal position and a tri-peptide a vertical one (Lyons et al., 2014) and suggested this flexibility may be responsible for the accomodation of larger peptide ligands (Newstead, 2017). Here we see that the S-Cys-Gly-3M3SH peptide adopts a more vertical orientation, similar to the tri-alanine position in PepTSt. However, there are significant differences in the interactions. A structural overlay of the S-Cys-Gly-3M3SH peptide shows that it sits much further up in the binding site compared to the natural peptides captured in PepTSt (Figure 4—figure supplement 4A & B), most likely as a result of the requirement to accommodate the thioalcohol group in the extended pocket. This has a knock-on effect with respect to the position of the amino and carboxy termini. In PepTSt, the amino terminal group of the di-peptide directly interacts with helix H8 and H10, whereas the tri-peptide makes a much weaker interaction through a carbonyl group. In contrast, in PepTSh this interaction is achieved through a water mediated hydrogen bond (Figure 4D).
The role of water in mediating interactions between peptides and their binding sites has been previously observed in the ABC transporter binding domains (Tame et al., 1995), albeit for side chain independent binding. A similar mechanism may be employed by PepTSh to facilitate transport of modified peptides by replacing direct interactions between the peptide and the transporter when these are sterically restricted, as we observe with S-Cys-Gly-3M3SH. The interaction of the amino termini of peptides with both the mammalian and bacterial POT family transporters has been identified as facilitating high affinity binding (Weitz et al., 2007; Meredith et al., 2000). The reduced IC50 value for the modified peptide in PepTSh (Figure 3B), may therefore be a combination of steric restraints resulting in the accommodation of the extended side chain group, and the subsequent knock on effects on the position of the interactions between the amino and carboxy groups with conserved side chains in the binding site.
Functional characterisation of PepTSh binding site
S-Cys-Gly-3M3SH is the first human derived peptide with a modified side chain to be captured in a POT family transporter, and contains biochemical features similar to physiological peptides (Cys-Gly), but also incorporates the non-peptide thioalcohol group (3M3SH). We therefore wished to explore whether S-Cys-Gly-3M3SH is recognised using a similar mechanism to physiological peptides. Several mutations in the binding site that have previously been shown to play an important role in peptide recognition within the bacterial POT family were therefore generated (Newstead, 2017). Tyrosine 41 forms part of a conserved E33xxERFxYY41 motif on TM1 (Solcan et al., 2012), which plays a role in proton coupling and influences peptide uptake in several POT family members (Doki et al., 2013; Ernst et al., 2009; Guettou et al., 2014; Lyons et al., 2014). In PepTSh Tyr41 hydrogen bonds with the carboxyl terminus of the terminal glycine amino acid in S-Cys-Gly-3M3SH (Figure 4C,D). Mutating Tyr41 to phenylalanine or alanine severely reduced transport of di-alanine compared to WT protein (Figure 5A). A similar negative effect on transport was observed in the related POT family transporter PepTSt, from Streptococcus thermophilus (Solcan et al., 2012), suggesting a common role for the conserved tyrosine in peptide recognition within the POT family. We then used the same variants in our whole cell assay for 3M3SH production. Interestingly, while both Tyr41Phe and Tyr41Ala variants had reduced activity, at about 50% of the wild-type protein (Figure 5B), the phenotype was not as severe as that observed in the liposome based assay. We interpret this result to indicate that recognition of S-Cys-Gly-3M3SH is less dependent on this residue than di-Ala.
Figure 5
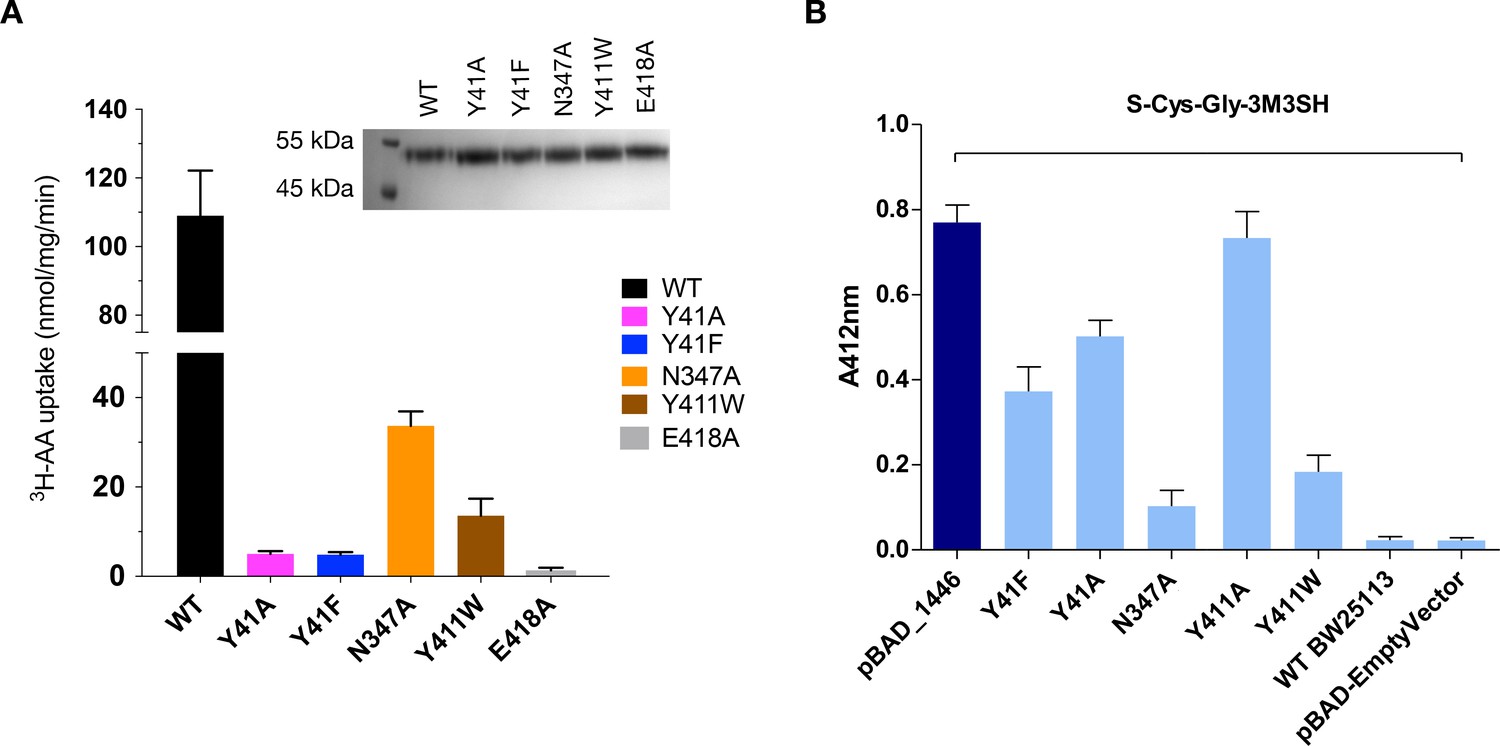
Functional characterisation of binding site residues.
(A) Bar chart showing the di-alanine uptake for the WT and mutant variants of PepTSh. Inset – SDS-PAGE analysis of the reconstituted proteins used in the assay. (B) Biotransformation of S-Cys-Gly-3M3SH by E. coli K-12 (BW25113) overexpressing WT PepTSt (pBAD_1446), or variants indicated, compared to background uptake in non-transformed cells (WT BW25113) and cells harbouring empty plasmid (pBAD-EmptyVector). Error bars: ±SD (biological triplicates).
We next wanted to test the role of other residues we observed sitting close to the S-Cys-Gly-3M3SH ligand. In particular, we were interested in the contribution made by the Asn347 and Glu418, which coordinate a water molecule (W107) linking both the carboxyl and carbonyl groups of the ligand to the binding site (Figure 4B). The equivalent residues in homologous POT family transporters have been implicated in both peptide recognition (Lyons et al., 2014) and gating (Solcan et al., 2012), and in the case of Glu418 may facilitate the coupling between peptide recognition, proton movement and transport (Parker et al., 2017). Although a Glu418Ala variant had no activity in our assays, consistent with its proposed role in coordinating the intracellular gate (Fowler et al., 2015), we observed reduced uptake of di-Ala in the Asn347Ala variant (Figure 5A). Interestingly, this reduction was far more severe in the in vivo assay (Figure 5B), indicating that this interaction is more important for transport of the S-Cys-Gly-3M3SH peptide than di-Ala. This may reflect the requirement of Asn347 to coordinate the water molecule linking the peptide to the intracellular gate side chain Glu418, which in the case of the di-peptide may interact directly as in PepTSt, lessening the requirement for the Asn347 interaction.
The co-crystal structure also identified the acyl chain portion of the thioalcohol group was accommodated within an extended pocket, formed near the extracellular side of the binding site (Figure 4E). To test the importance of this pocket on S-Cys-Gly-3M3SH transport, we made a Tyr411Trp variant, which would block the cavity. Uptake was noticeably reduced, but still measurable at ~19% compared to WT (Figure 5A). The same mutation resulted in a similar large decrease in the biotransformation of S-Cys-Gly-3M3SH, while a Tyr411Ala mutation had wild-type biotransformation rates (Figure 5B). It is clear that the hydrophobic pocket in PepTSh plays an important role in facilitating the recognition of the thioalcohol group in the binding pocket and, as discussed below, is a potential site for inhibitor design.
Taken together the current data lend further support to our model of an adaptable binding site within the POT family that can accommodate chemically diverse peptides and their derivatives through the use of both side chain and water mediated interactions with conserved residues and different specificity pockets to accommodate the different chemical side chains.
Discussion
The production of malodour by humans is an evolutionary ancient olfactory process that likely was involved in mate selection and evolutionary processes that relied on chemical signalling (Stoddart, 1990). Evidence for the microbial origins of chemical signalling molecules is also seen in other animals such as foxes, deer, cats and beetles (Wyatt, 2003), but the details of this process in humans is less well understood. The thioalcohol 3M3SH, a major component of body odour, is produced in the human axilla as a glutathione-conjugated precursor and is processed to a unique dipeptide structure (Figure 1). The discovery that axilla-isolated strains of S. hominis, S. lugdunensis and S. heamolyticus are able to produce 3M3SH prompted the search for the molecular basis for this process (Bawdon et al., 2015). While S-Cys-Gly-3M3SH is a highly modified and uniquely human dipeptide, we reasoned that this moiety might dictate the uptake mechanism into malodour producing bacteria and here we identify a peptide transporter, PepTSh (SH1446), from the POT family of secondary active transporters as being responsible for this process. Each of these strains have a POT family transporter that is closely related to PepTSh (Figure 4—figure supplement 2), and we would predict they will also be able to uptake S-Cys-Gly-3M3SH.
The genetic intractability of S. hominis dictated that we use a biochemical approach to demonstrate that SH1446 is likely the sole route of uptake of S-Cys-Gly-3M3SH. First, this staphylococci has a similar repertoire of peptide transporters in S. aureus, in that it has a single POT transporter and one Opp ABC transporter, and previous work in S. aureus has demonstrated that the POT transporter, DtpT, is essential for growth on dipeptides and some tripeptides, while the ABC transporter (Opp3) has a function in transporting longer oligopeptides (Hiron et al., 2007). The use of L-Ala containing peptides as inhibitors in this study demonstrates an inhibitory effect on 3M3SH production that is consistent with the properties of a POT family transporter. Several attempts were made to genetically disrupt SH1446 in S. hominis, using tools developed for S. aureus and S. epidermidis, but these were unsuccessful, most likely due to the presence of multiple species-specific restriction modification (RM) systems which render this organism, as with many other coagulase negative (CoNS) species, genetically intractable.
The specific role for POT transporters in the transport of these unusually modified dipeptides is supported from the inability of the E. coli ABC transporters for oligopeptides and dipeptides, Opp and Dpp, to recognise and transport S-Cys-Gly-3M3SH. Given that S-Cys-Gly-3M3SH is effectively a dipeptide with a highly extended R-group on the first amino acid, it is noteworthy that only the promiscuous nature of the POT binding site is able to recognise this ligand. For OppA and DppA, the substrate binding proteins that initially recognise peptides transported by the ABC systems, the binding pocket is well defined and makes general main chain-main chain interactions to ensure that the protein can bind peptides in a sequence independent manner (Tame et al., 1995; Sleigh et al., 1997; Sleigh et al., 1999). However, the pocket has evolved to accommodate regular amino acid side chains and the unusually large structure of the thioalcohol attachment would likely be sterically incompatible with recognition by either OppA or DppA. The data presented here further support the idea that POT transporters have an unusually promiscuous binding site that may play an evolutionary advantageous role in accepting modified peptides as substrates.
Recently, the human POT family transporters, PepT1 and PepT2, have gained additional attention due to their role in the absorption and retention of several important classes of drug molecule (Smith et al., 2013), including peptide modified pro-drugs. Similar to S-Cys-Gly-3M3SH, these pro-drugs are drug molecules conjugated to a peptide or amino acid (Brandsch, 2013). Pro-drugs are able to compete with natural peptides for transport via PepT1 and PepT2, and are therefore actively transported into the body (Ganapathy et al., 1998; Gupta et al., 2013; Terada and Inui, 2012). The promiscuous nature of the POT family binding site has been shown to derive, in part, from their ability to recognise peptides in different conformations and through the availability of different specificity pockets, which are able to accommodate chemically different classes of peptides (Newstead, 2017). Our results here provide the first evidence that specificity pockets can also accommodate exotic chemical extensions. The observation that water plays a role in facilitating the interaction between the S-Cys-Gly-3M3SH peptide and PepTSh may also indicate a broader role for water in facilitating the recognition of xenobiotic peptides. In particular, the observation that water may bridge interactions between the binding site and either the amino or carboxy group of the ligand would greatly increase the number of conformations a peptide could adopt in the transporter and still trigger proton coupled transport.
Finally, we can ask the questions, why do these bacteria transport S-Cys-Gly-3M3SH and can this process be inhibited to provide effective malodour control? The presumed biochemical pathway for breakdown of the S-Cys-Gly-3M3SH is outlined in Figure 6. The firststep is the likely removal of the glycine by a dipeptidase and then the lyase reaction that cleaves the carbon-sulphur bond to release pyruvate and ammonia in addition to 3M3SH. The route of export of 3M3SH is unknown, but currently assumed to be diffusion across the bacterial inner membrane. The bacterium will gain a nutritional benefit from the catabolism of S-Cys-Gly-3M3SH via the release of both nitrogen and carbon as glycine, pyruvate and ammonia (Figure 6), which may have been the evolutionary trigger for the adaptation to catabolise this chemical. Asian individuals with single nucleotide polymorphisms (SNPs) in ABCC11, the transporters involved in the secretion of S-Cys-Gly-3M3SH (Figure 1) have been studied (Nakano et al., 2009; Baumann et al., 2014b). Recently, a microbiological investigation of individuals with wild-type alleles of ABCC11 compared to heterozygous and double mutant genotypes revealed that the proportion of Staphylococcus sp. is higher in the underarms of humans that produce the precursor (Harker et al., 2014). While these studies are limited in that they do not provide species level identification, they are at least consistent with the idea that the production of the precursor positively correlates with the presence of Staphylococcus sp. in the underarm. The discovery of the transporter and molecular understanding of the mode of S-Cys-Gly-3M3SH binding now suggests a specific route to develop inhibitors of malodour production by directly inhibiting the transporter SH1446 (Grice, 2014). Importantly, such intervention would not impact on the overall axillar microbiome, but instead specifically target the reduction in body odour production.
Figure 6

Overview of transport and intracellular biotransformation of S-Cys-Gly-3M3SH in Staphylococcus hominis.
1. Cys-Gly-(S)−3M3SH is actively transported across the membrane by the di/tri peptide transporter PepTSh along with the movement of protons. 2. A dipeptidase cleaves the terminal glycine residue of S-Cys-Gly-3M3SH producing Cys-(S)−3M3SH. 3. The thiol is liberated from Cys-(S)−3M3SH by a C-S β-lyase that also releases ammonia and pyruvate, the volatile thiol (3M3SH) then diffuses or is exported out the cell. 4.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Ggene (Escherichia coli) | dtpA | BW25113 | b1634, ECK1630 | POT transporter family |
| Gene (Escherichia coli) | dtpB | BW25113 | b3496, ECK3481 | POT transporter family |
| Gene (Escherichia coli) | dtpC | BW25113 | b4130, ECK4124 | POT transporter family |
| Gene (Escherichia coli) | dtpD | BW25113 | b0709, ECK0698 | POT transporter family |
| Gene (Escherichia coli) | tnaA | BW25113 | b3708, ECK3701 | Tryptophanase |
| Gene (Staphylococcus hominis) | SH1446 | SK119 | STAHO0001_1446 (GenBank: EEK12089.1) | POT transporter family |
| Gene (Staphylococcus hominis) | SH0415 | SK119 | STAHO0001_0415 (GenBank: EEK11635.1) | MFS transporter family |
| Strain, strain background (Escherichia coli) | BW25113 | Baba et al., 2006 | F-, Δ(araD-araB)567, ΔlacZ4787(::rrnB-3), lambda-, rph-1, Δ(rhaD-rhaB)568, hsdR514 | |
| Strain, strain background (Escherichia coli) | JW1237 | Baba et al., 2006 | ΔoppC::kanR | |
| Strain, strain background (Escherichia coli) | JW3511 | Baba et al., 2006 | ΔdppC::kanR | |
| Strain, strain background (Escherichia coli) | JW1626 | Baba et al., 2006 | ΔdtpA::kanR | |
| Strain, strain background (Escherichia coli) | JW3463 | Baba et al., 2006 | ΔdtpB::kanR | |
| Strain, strain background (Escherichia coli) | JW4091 | Baba et al., 2006 | ΔdtpC::kanR | |
| Strain, strain background (Escherichia coli) | JW0699 | Baba et al., 2006 | ΔdtpD::kanR | |
| Strain, strain background (Escherichia coli) | JW3686 | Baba et al., 2006 | ΔtnaA::kanR | |
| Strain, strain background (Escherichia coli) | JW2975 | Baba et al., 2006 | ΔmetC::kanR | |
| Strain, strain background (Escherichia coli) | JW1614 | Baba et al., 2006 | ΔmalY::kanR | |
| Strain, strain background (Escherichia coli) | JW1285 | Baba et al., 2006 | ΔsapC::kanR | |
| Strain, strain background (Escherichia coli) | JW0590 | Baba et al., 2006 | ΔcstA::kanR | |
| Strain, strain background (Escherichia coli) | ΔoppC | This study. KanR removed from parent strain | ΔoppC | |
| Strain, strain background (Escherichia coli) | ΔdppC | This study. KanR removed from parent strain | ΔdppC | |
| Strain, strain background (Escherichia coli) | ΔdtpA | This study. KanR removed from parent strain | ΔdtpA | |
| Strain, strain background (Escherichia coli) | ΔdtpB | This study. KanR removed from parent strain | ΔdtpB | |
| Strain, strain background (Escherichia coli) | ΔdtpC | This study. KanR removed from parent strain | ΔdtpC | |
| Strain, strain background (Escherichia coli) | ΔdtpD | This study. KanR removed from parent strain | ΔdtpD | |
| Strain, strain background (Escherichia coli) | ΔtnaA | This study. KanR removed from parent strain | ΔtnaA | |
| Strain, strain background (Escherichia coli) | ΔDB1 | This study. KanR removed from parent strain | ΔoppC ΔdppC | |
| Strain, strain background (Staphylococcus hominis) | SK119 | Taxonomy ID: 629742 (Accession: PRJNA34087) | ||
| Strain, strain background | C43(DE3) | Miroux and Walker, 1996 | ||
| Strain, strain background | pWaldo-GFPe | Drew et al., 2001 | ||
| Genetic reagent (Escherichia coli) | pBADcLIC2005 | Geertsma and Poolman, 2007 | pBAD vector modified with LIC cassette, PBAD | |
| Genetic reagent (Escherichia coli) | pKD46 | Datsenko and Wanner, 2000 | Contains KanR cassette | |
| Genetic reagent (Escherichia coli) | pCP20 | Cherepanov and Wackernagel, 1995 | FLP+, pSC101 origin, λ cI857+, λ pR Repts, AmpR, CmR | |
| Recombinant DNA reagent | pBADcLIC-dtpA | This study. Cloned using primers in Supplementary file 1 | ||
| Recombinant DNA reagent | pBADcLIC-dtpB | This study. Cloned using primers in Supplementary file 1 | ||
| Recombinant DNA reagent | pBADcLIC-dtpC | This study. Cloned using primers in Supplementary file 1 | ||
| Recombinant DNA reagent | pBADcLIC-dtpD | This study. Cloned using primers in Supplementary file 1 | ||
| Recombinant DNA reagent | pBADcLIC-SH0415 | This study. Cloned using primers in Supplementary file 1 | ||
| Recombinant DNA reagent | pBADcLIC-SH1446 | This study. Cloned using primers in Supplementary file 1 | ||
| Peptide, recombinant protein | SH1446 | This study. Purified from E. coli BW25113 containing pBADcLIC-SH1446 | ||
| Peptide, recombinant protein | TnaA | This study. Purified from E. coli BW25113 containing pBADcLIC-TnaA | ||
| Chemical compound, drug | S-Cys-Gly-3M3SH | Peakdale Molecular (UK) | Custom made malodour substrates | |
| Chemical compound, drug | L-Ala | Sigma-Aldrich (St. Louis, MO, USA) | A7627 | |
| Chemical compound, drug | L-Ala-Ala | Sigma-Aldrich (St. Louis, MO, USA) | A9502 | |
| Chemical compound, drug | L-Ala-Ala-Ala | Sigma-Aldrich (St. Louis, MO, USA) | A9627 | |
| Chemical compound, drug | L-Ala-Ala-Ala-Ala | Cambridge Biosciences | H-1260.0250 | |
| Chemical compound, drug | n-Decyl-β-D-maltopyranoside | Anatrace | D322LA | |
| Chemical compound, drug | n-Dodecyl-β-D-maltoside | Anatrace | D310LA | |
| Chemical compound, drug | Nickel-NTA Superflow resin | ThermoFisher | 88221 |
Bacterial strains, media and plasmids
Request a detailed protocolEscherichia coli K-12 was used as the model organism throughout this study. Escherichia coli K-12 single gene deletions were obtained from the in-house E. coli Keio collection at University of York. Bacterial strains and plasmids used in this study are listed in Supplementary file 1 and 2. All strains were cultured in LB (1% tryptone, 0.5% yeast extract, 1% NaCl) unless otherwise stated, Antibiotics were used at the following concentrations Ampicillin 100 μg/mL and Kanamycin 50 μg/mL; L-arabinose concentrations are indicated. PCR, plasmid extractions, and transformations were carried out as per standard protocols. For routine cloning, genomic DNA was extracted from BW25113 and high fidelity KOD polymerase was used (Novagen). Correct clones were confirmed by DNA sequencing. Specific details of plasmid constructions are detailed below.
Construction of multiple mutants in single keio deletions
Request a detailed protocolMultiple gene knockouts were created in single keio deletion strains. To create the double KO strain DB1 (ΔoppC ΔdppC), the KanR cassette was removed from a target gene using pCP20 (encoding Flp recombinase) at 30°C, loss of antibiotic cassette was confirmed by PCR and sequencing. The second gene to be disrupted was PCR amplified with 100 bp homology including the FRT flanked KanR cassette from the single gene deletion strain in the keio collection. Targeted gene disruptions were created using λ Red recombination as previously described (Datsenko and Wanner, 2000).
Plasmid constructions and protein purification: pBAD-dtpA, dtpB, dtpC, dtpC, tnaA
Request a detailed protocolOverexpression of the E. coli POT transporters were cloned into pBADcLIC2005 vector under the control of arabinose. Full-length genes were amplified from E. coli BW25113 genomic DNA using high fidelity KOD polymerase (Merck) using the corresponding primers listed in Supplementary file 3. In silico analysis of the available Staphylococcus hominis SK1119 genome identified STAH0001_1446 (SH1446) as a POT family member which orthologues present in all sequenced staphylococci and a MFS transporter STAH0001_0415 (SH0415) present in only S. hominis and S. haemolyticus. SH0415 and SH1446 genes were amplified from S. hominis genomic DNA using high fidelity KOD polymerase (Merck) using primers listed in Supplementary file 3. All PCR products were purified using Wizard SV Gel and PCR Clean-Up System (Promega), cloned into pBADcLIC2005 vector using standard ligation-independent cloning (LIC) techniques. All fragments were confirmed by PCR and DNA sequencing (GATC Biotech).
Site directed mutagenesis of SH1446
Request a detailed protocolThe wildtype STAHO0001_1446 (SH1446) cloned onto the expression plasmid pBADcLIC2005 was used to individually generate the Y411W, Y411A, Y41A, Y41F, N167A and N347A mutants. To introduce the mutation, high fidelity inverse PCR was performed using divergent primers with one per pair being mutagenic (Supplementary file 3). The resultant products were then circularised by performing a blunt end ligation and transformed into E. coli BW25113. The mutant sequences were verified by DNA sequencing (GATC Biotech).
Malodour precursor biotransformation assay malodour precursor substrates
Request a detailed protocolS-Benzyl-L-cysteinylglycine and (S)-(1-(2-hydroxyethyl)−1-methylbutyl)-L-cysteinylglycine (S-Cys-Gly-3M3SH) (Peakdale Molecular) were prepared at a stock concentration of 10 mM in M9 salts (6 g Na2HPO4 g/L, 3 g KH2PO4 g/L, 0.5 g NaCl g/L) and stored at 4°C.
S-Benzyl-L-cysteinylglycine and S-Cys-Gly-3M3SH biotransformation assays using resting cells
Request a detailed protocolE. coli were grown overnight in LB, cells were harvested by centrifugation at 3, 500 rpm 10 min and resuspended in sterile M9 salts. Where appropriate, E. coli strains harbouring inducible genes on the pBADcLIC2005 plasmid were pre-induced during overnight growth with 0.0001% L-arabinose. To a sterile Eppendorf the following was added; 2 mM substrate (S-Benzyl-L-cysteinylglycine or S-Cys-Gly-3M3SH), cells to an OD650nm of 5 and M9 salts up to 200 μL. Cells only (no added substrate) and substrate only (no added cells) reactions were also included in every individual experiment. The reactions were incubated at 37°C for up to 48 hr with samples taken at appropriate time points. Liberated thiols were quantified using either MTS/PMS or DTNB labelling as detailed below.
(3-(4,5-dimethylthiazol-2-yl)−5-(3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium) (MTS) and phenazine methosulfate (PMS)-mediated chemical labelling of benzyl-mercaptan thioalcohol
Request a detailed protocolLiberated benzyl mercaptan was labelled as follows, 500 μl from each reaction was taken at the appropriate time point and centrifuged at 13 000 r.p.m. for 2 min. To chemically label benzyl-mercaptan, 50 µl of labelling reagent, consisting of 4.09 mM 3-(4,5-dimethylthiazol-2-yl)−5- (3-carboxymethoxyphenyl)−2-(4-sulfophenyl)−2H-tetrazolium (MTS) (Promega) and 0.2 mM phenazine methosulfate (PMS) (Sigma) (Goodwin et al., 1995), dissolved in dH2O, was aliquoted into either a 96 well microplate or 0.5 ml Eppendorf tubes. For reactions using a 96 well microplate, 200 µl reaction supernatant was added to the pre- aliquoted 50 µl MTS/PMS. The plate was sealed with a semi-permeable film lid, incubated at room temperature for 50 min and the A492nm recorded on a Biotek PowerWave XS microplate spectrophotometer. The data was captured by KC junior and exported to Microsoft Excel. For reactions in Eppendorf tubes, 20 µl - 50 µl reaction supernatant was added to 50 µl MTS/PMS. The reaction was incubated at room temperature for 50 min. 20 µl was subsequently removed and diluted in 980 µl dH2O in a 1.5 ml disposable plastic cuvette. The A492nm was recorded.
5,5'-dithiobis-(2-nitrobenzoic acid) (DTNB)-mediated chemical labelling of 3M3SH
Request a detailed protocolTo quantify free 3M3SH, a 75 µl volume from each reaction (isolated as described above) was taken at the appropriate time point and centrifuged at 13, 000 rpm for 2 min. 950 µl labelling solution, consisting of: 800 µl dH2O, 100 µl UltraPure Tris (pH 8.0) and 50 µl DTNB (Ellman, 1958) stock solution (50 mM sodium acetate, 2 mM DTNB, dissolved in dH2O) was added to a 1.5 ml disposable cuvette. 50 µl reaction supernatant was added to the cuvette, mixed by pipetting and incubated at room temperature for 5 min. The A412nm was recorded on a Jenway 6305 spectrophotometer. (Van Horn, 2003). Where appropriate, E. coli strains harbouring inducible genes on the pBADcLIC2005 plasmid were pre-induced during overnight growth with 0.0001% L-arabinose. In all experiments cells only (no added substrate) and substrate only (no added cells) were also included.
Protein expression and purification
Request a detailed protocolThe gene encoding S. hominis SH1446 was cloned into a C-terminal octa histidine GFP fusion vector (pWaldo-GFPe) (Drew et al., 2006) and subsequently transformed into Escherichia coli C43(DE3) cells (Miroux and Walker, 1996). Mutations were introduced using site directed mutagenesis followed by DpnI digestion. An overnight starter culture was used to inoculate 4 L of Terrific Broth (TB). Cells were left to grow at 37°C and aerated by shaking at 250 rpm. Overexpression of the fusion protein was induced when the culture reached an OD600 of ~0.6, by adding isopropyl-β-D-thiogalactopyranoside (IPTG) to a final concentration of 0.4 mM. Following induction, the temperature was reduced to 25°C. Cells were harvested 16 hr later, and resuspended in phosphate buffered saline (PBS) containing deoxyribonuclease I from bovine pancreas, and stored at −80°C.Unless stated, the following steps were carried out at 4°C. Cells were thawed and disrupted at 32 kpsi (Constant Systems, UK). Cellular debris was removed by centrifugation at 30,000 x g for 30 min, and membranes were isolated by ultracentrifugation at 230,000 x g for 120 min. Membranes were resuspended in PBS using a dounce homogeniser, flash frozen, and stored at −80°C. Thawed membranes were diluted with 1x PBS (10 mL per gram of membrane), 20 mM imidazole (pH 8.0), 150 mM NaCl, and 1% n-Dodecyl-β-D-maltoside (DDM) and left to solubilise for 60 min, stirring. Non-solubilised membranes were removed by ultracentrifugation at 230,000 g for 60 min. Nickel-NTA resin (ThermoFisher Scientific) was added to the sample, using a ratio of 1 mL resin per gram of membrane, and left to bind, stirring, for approximately 90 min. The resin was then packed into a glass econo-column (BioRad, USA) under gravity. The resin was washed with 20 column volumes (CV) of wash buffer (1x PBS, 20 mM imidazole (pH 8.0), 150 mM NaCl and 0.05% DDM). The resin was further washed with 10CV of wash buffer containing 30 mM imidazole. The bound GFP-SH1446 fusion protein was eluted with wash buffer containing 250 mM imidazole. The eluent was combined with an equivalent concentration of hexa histidine tagged TEV protease and dialysed overnight against 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.03% DDM. The cleaved protein was filtered using a 0.22 μm filter (Millipore, USA) and passed through a Ni-NTA 5 mL HisTrap column (GE Healthcare) to remove uncleaved GFP-SH1446 protein and TEV protease. The flow through was collected and concentrated to 300 μl using a 50 kDa MWCO concentrator (Vivapsin 20, Sartorius AG) and applied to a size exclusion column (Superdex 200 10/30, GE Healthcare) pre-equilibrated with 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.03% DDM. Fractions containing purified SH1446 protein were pooled and concentrated to a final concentration of 15–20 mg.ml−1.
For expression and purification of TnaA, overnight cultures were grown in LB at 37°C, 200 rpm. 1 L of LB was inoculated 1:50 with the starter culture and grown to an OD600nm ~0.6–0.8, target protein expression was induced with 0.001% arabinose. Following induction the culture was further incubated at 37°C for ~8 hr. Cells were harvested by centrifugation at 4,500 rpm for 15 min and resuspended in 35 mL resuspension buffer (50 mM Kpi, 20% Glycerol, 200 mM NaCl and 10 mL Imidazole, pH 7.8) and stored at −80°C. All subsequent steps were carried out at 4°C, for protein purification cells were thawed and lysed using sonication with 3 s pulsed and 7 s pause for 3mins. Lysates were clarified by centrifugation 25,000 rpm ~30 min. The clarified supernatant was loaded onto a 5 mL HisTrap column (GE Healthcare) and affinity purified on an AKTA Start (GE Healthcare) using a standard protocol as per manufacturer’s instructions.
Protein crystallisation
Request a detailed protocolCrystals of SH1446 were obtained in-meso phase. Briefly, concentrated protein was mixed with 9.9 monoacylglycerol (Monoolein, Sigma-Aldrich) at a protein:lipid ratio of 2:3, in a mechanical syringe mixer at 20°C (Ai and Caffrey, 2000). 50 nL of mesophase and 800 nL of precipitant solution was dispensed on to a 96-well glass plate using an in meso robot (Gryphon, Art Robbins Instruments). SH1446 S-Cys-Gly-3M3SH complex was crystallised in precipitant solution containing 26–27% (v/v) PEG 200, 220 mM (NH4)2HPO4, 110 mM sodium citrate (pH 5.0) and 5 mM S-Cys-Gly-3M3SH (Peakdale Molecular) and left overnight at 4°C, prior to crystallisation. Crystals appeared after 2–3 days, and grew to its maximum size after 10 days. Crystallisation wells were accessed using a tungsten-carbide glasscutter, and crystals were directly mounted onto 50–100 μm microloops (MiTeGen) and flash frozen at 100 K.
X-ray diffraction data were collected at ID23-2 at the European Synchrotron Radiation Facility on a Pilatus3 2M detector with an exposure setting of 0.05 s with a 0.2° oscillation range over 360° Diffraction data was indexed and integrated in XDS (Kabsch, 2010) and merged in Aimless (Evans, 2011).
Structure solution and refinement
Request a detailed protocolThe ligand bound complex was solved by molecular replacement in PHASER (McCoy et al., 2007) using a modified search model of the bacterial peptide transporter from Streptococcus thermophilus, PepTSt (Lyons et al., 2014), PDB: 4D2B. Iterative rounds of manual model building in COOT (Emsley et al., 2010) followed by refinement in autoBUSTER (Bricogne G. 2017) followed by refinement in Phenix (Adams et al., 2010), resulted in a final model, which was validated using the Molprobity server (Chen et al., 2010). Additional difference density was observed in the binding site and matched the stereo chemical properties and size of S-CG3M3SH. Ligand restraints were generated using the Grade Web Server (http://grade.globalphasing.org). Figures were prepared using PyMOL (Schrödinger, LLC).
Reconstitution into proteoliposomes and transport assays
Request a detailed protocolPurified SH1446 was detergent exchanged into n-Decyl-β-D-maltopyranoside (DM, Anatrace) using a size exclusion column (Superdex 200 10/30, GE Healthcare) pre-equilibrated with 20 mM Tris-HCl (pH 7.5), 150 mM NaCl, and 0.3% DM. Lipids (POPE:POPG in a 3:1 ratio) were mixed with the eluted protein at a ratio of 60:1. The protein:lipid mixture was rapidly diluted with 50 mM KPO4 (pH 7.0) buffer to below the CMC value of DM. Proteoliposomes were harvested by ultracentrifugation (234,000 xg) for 120 min. The pellet was resuspended in 50 mM KPO4 and dialysed overnight against 3L of 50 mM KPO4 (pH 7.0). Proteoliposomes were recovered and subjected to three rounds of freeze thawing before storage at −80°C.
Proteoliposomes were thawed and harvested by ultracentrifugation at 150,000 g. The resulting pellet was carefully resuspended in INSIDE buffer (120 mM KCl, 2 mM MgSO4, 5 mM HEPES pH 6.8) and 1 mM pyranine (trisodium 8-hydroxypyrene-1,3,6-trisulfonate). Proteoliposomes underwent eleven freeze-thaw cycles in liquid nitrogen before the sample was extruded through a 0.4 μm polycarbonate membrane and harvested by ultracentrifugation as before. Excess pyranine dye was removed with a Microspin G-25 column (GE Healthcare) pre-equilibrated with INSIDE buffer without pyranine. For the assay, proteoliposomes were diluted into OUTSIDE buffer (120 mM NaCl, 5 mM HEPES (pH 6.8), 2 mM MgSO4).
A Cary Eclipse fluorescence spectrophotometer (Agilent Technologies) equipped with a cuvette and magnetic flea was used to carry out the transport assay. Dual fluorescence excitation was set to 460/415 nm with emission at 510 nm. Transport was initiated with 1 μM valinomycin, the resulting data was exported and analysed using Prism (GraphPad Software). Transport data was normalised to one to facilitate comparison with data collected from multiple conditions.
Competition assays
Request a detailed protocolProteoliposomes were harvested as before, the resulting pellet was resuspended in INSIDE buffer, subjected to three freeze-thaw cycles in liquid nitrogen, extruded through a 0.4 μm polycarbonate membrane and harvested by ultracentrifugation once more. For the assay, proteoliposomes were diluted in external buffer (110 mM NaCl, 10 mM NaPO4 and 2 mM MgSO4) supplemented with 50 μM di-alanine peptide containing trace amounts of tritiated 3H-di-alanine (specific activity 30 Ci/mmol) and the competing substrate at increasing concentrations as indicated. Competition assays were performed at 30°C, samples were taken at specified time points and transport was stopped by addition of 2 mL H2O. Proteoliposomes were immediately filtered onto a 0.22 μm cellulose filter (Merck Millipore) using a vacuum manifold and subsequently washed twice more with 2 mL H2O. The amount of peptide transported into the liposomes was calculated based on the specific activity for each peptide as detailed by the manufacturer and counting efficiency for the radioisotope in Ultima Gold (PerkinElmer) counted in a Wallac scintillation counter (3H, 45% counting efficiency). Transport assays were performed a minimum of three times to generate an overall mean and S.E.M.
Data availability
Request a detailed protocolAtomic coordinates for the atomic models have been deposited in the Protein Data Bank under accession numbers 6EXS.
Data availability
Diffraction data have been deposited in the PDB under the accession code 6EXS. Vectors have been deposited in Addgene (deposit number 75759).
-
Crystal structure of SH1446 in complex with S-Cys-Gly-3M3SHPublicly available at the RCSB Protein Data Bank (accession no: 6EXS).
References
-
PHENIX: a comprehensive Python-based system for macromolecular structure solutionActa Crystallographica Section D Biological Crystallography 66:213–221.https://doi.org/10.1107/S0907444909052925
-
Membrane protein crystallization in Lipidic mesophases: detergent effectsBiophysical Journal 79:394–405.https://doi.org/10.1016/S0006-3495(00)76301-6
-
Effect of cysteine desulfhydrase gene disruption on L-cysteine overproduction in Escherichia coliApplied Microbiology and Biotechnology 62:239–243.https://doi.org/10.1007/s00253-003-1262-2
-
Identification and functional analysis of Escherichia coli cysteine desulfhydrasesApplied and Environmental Microbiology 71:4149–4152.https://doi.org/10.1128/AEM.71.7.4149-4152.2005
-
Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the keio collectionMolecular Systems Biology 2:2006.0008.https://doi.org/10.1038/msb4100050
-
The human eccrine sweat gland: structure, function and disordersJournal of Local and Global Health Science 2015:5.https://doi.org/10.5339/jlghs.2015.5
-
Drug transport via the intestinal peptide transporter PepT1Current Opinion in Pharmacology 13:881–887.https://doi.org/10.1016/j.coph.2013.08.004
-
Projection structure of DtpD (YbgH), a prokaryotic member of the peptide transporter familyJournal of Molecular Biology 394:708–717.https://doi.org/10.1016/j.jmb.2009.09.048
-
MolProbity: all-atom structure validation for macromolecular crystallographyActa Crystallographica Section D Biological Crystallography 66:12–21.https://doi.org/10.1107/S0907444909042073
-
The proton oligopeptide cotransporter family SLC15 in physiology and pharmacologyPflugers Archiv : European Journal of Physiology 447:610–618.https://doi.org/10.1007/s00424-003-1101-4
-
One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR productsProceedings of the National Academy of Sciences 97:6640–6645.https://doi.org/10.1073/pnas.120163297
-
Structural basis for dynamic mechanism of proton-coupled symport by the peptide transporter POTProceedings of the National Academy of Sciences 110:11343–11348.https://doi.org/10.1073/pnas.1301079110
-
A colorimetric method for determining low concentrations of mercaptansArchives of Biochemistry and Biophysics 74:443–450.https://doi.org/10.1016/0003-9861(58)90014-6
-
Features and development of cootActa Crystallographica. Section D, Biological Crystallography 66:486–501.https://doi.org/10.1107/S0907444910007493
-
Ligand binding analyses of the putative peptide transporter YjdL from E. coli display a significant selectivity towards dipeptidesBiochemical and Biophysical Research Communications 389:112–116.https://doi.org/10.1016/j.bbrc.2009.08.098
-
An introduction to data reduction: space-group determination, scaling and intensity statisticsActa Crystallographica Section D Biological Crystallography 67:282–292.https://doi.org/10.1107/S090744491003982X
-
Apocrine secretion: new insights into an old phenomenonBiochimica Et Biophysica Acta (BBA) - General Subjects 1850:1740–1750.https://doi.org/10.1016/j.bbagen.2015.05.003
-
Valacyclovir: a substrate for the intestinal and renal peptide transporters PEPT1 and PEPT2Biochemical and Biophysical Research Communications 246:470–475.https://doi.org/10.1006/bbrc.1998.8628
-
High-throughput cloning and expression in recalcitrant BacteriaNature Methods 4:705–707.https://doi.org/10.1038/nmeth1073
-
Uptake of cell wall peptides by Salmonella typhimurium and Escherichia coliJournal of Bacteriology 169:3861–3865.https://doi.org/10.1128/jb.169.8.3861-3865.1987
-
Microculture tetrazolium assays: a comparison between two new tetrazolium salts, XTT and MTSJournal of Immunological Methods 179:95–103.https://doi.org/10.1016/0022-1759(94)00277-4
-
Comparison of 16-androstene steroid concentrations in sterile apocrine sweat and axillary secretions: interconversions of 16-androstenes by the axillary microflora--a mechanism for axillary odour production in man?The Journal of Steroid Biochemistry and Molecular Biology 48:409–418.https://doi.org/10.1016/0960-0760(94)90082-5
-
The skin microbiome: potential for novel diagnostic and therapeutic approaches to cutaneous diseaseSeminars in Cutaneous Medicine and Surgery 33:98–103.https://doi.org/10.12788/j.sder.0087
-
Selectivity mechanism of a bacterial homolog of the human drug-peptide transporters PepT1 and PepT2Nature Structural & Molecular Biology 21:728–731.https://doi.org/10.1038/nsmb.2860
-
The di- and tripeptide transport protein of Lactococcus lactis. A new type of bacterial peptide transporterThe Journal of Biological Chemistry 269:11391–11399.
-
Identification of new odoriferous compounds in human axillary sweatChemistry & Biodiversity 1:2042–2050.https://doi.org/10.1002/cbdv.200490157
-
Only one of four oligopeptide transport systems mediates nitrogen nutrition in Staphylococcus aureusJournal of Bacteriology 189:5119–5129.https://doi.org/10.1128/JB.00274-07
-
Microbiological and biochemical origins of human axillary odourFEMS Microbiology Ecology 83:527–540.https://doi.org/10.1111/1574-6941.12054
-
Fatty acid metabolism by cutaneous Bacteria and its role in axillary malodourWorld Journal of Microbiology & Biotechnology 20:787–793.https://doi.org/10.1007/s11274-004-5843-8
-
XDSActa Crystallographica. Section D, Biological Crystallography 66:125–132.https://doi.org/10.1107/S0907444909047337
-
Escherichia coli peptide binding protein OppA has a preference for positively charged peptidesJournal of Molecular Biology 414:75–85.https://doi.org/10.1016/j.jmb.2011.09.043
-
The microbiology of the human axilla and its relationship to axillary odorJournal of Investigative Dermatology 77:413–416.https://doi.org/10.1111/1523-1747.ep12494624
-
Phaser crystallographic softwareJournal of Applied Crystallography 40:658–674.https://doi.org/10.1107/S0021889807021206
-
Modified amino acids and peptides as substrates for the intestinal peptide transporter PepT1European Journal of Biochemistry 267:3723–3728.https://doi.org/10.1046/j.1432-1327.2000.01405.x
-
A specific bacterial aminoacylase cleaves odorant precursors secreted in the human axillaJournal of Biological Chemistry 278:5718–5727.https://doi.org/10.1074/jbc.M210142200
-
Molecular insights into proton coupled peptide transport in the PTR family of oligopeptide transportersBiochimica Et Biophysica Acta (BBA) - General Subjects 1850:488–499.https://doi.org/10.1016/j.bbagen.2014.05.011
-
Recent advances in understanding proton coupled peptide transport via the POT familyCurrent Opinion in Structural Biology 45:17–24.https://doi.org/10.1016/j.sbi.2016.10.018
-
Proton movement and coupling in the POT family of peptide transportersProceedings of the National Academy of Sciences 114:13182–13187.https://doi.org/10.1073/pnas.1710727114
-
The POT family of transport proteinsTrends in Biochemical Sciences 19:404.https://doi.org/10.1016/0968-0004(94)90087-6
-
Axillary odor; experimental study of the role of Bacteria, apocrine sweat, and deodorantsA.M.A. Archives of Dermatology and Syphilology 68:430–446.https://doi.org/10.1001/archderm.1953.01540100070012
-
Crystallographic and calorimetric analysis of peptide binding to OppA proteinJournal of Molecular Biology 291:393–415.https://doi.org/10.1006/jmbi.1999.2929
-
Proton-coupled oligopeptide transporter family SLC15: physiological, pharmacological and pathological implicationsMolecular Aspects of Medicine 34:323–336.https://doi.org/10.1016/j.mam.2012.11.003
-
Alternating access mechanism in the POT family of oligopeptide transportersThe EMBO Journal 31:3411–3421.https://doi.org/10.1038/emboj.2012.157
-
BookThe Scented Ape: The Biology and Culture of Human OdourCambridge University Press.
-
Characterization of the microflora of the human axillaInternational Journal of Cosmetic Science 25:137–145.https://doi.org/10.1046/j.1467-2494.2003.00181.x
-
Recent advances in structural biology of peptide transportersCurrent Topics in Membranes 70:257–274.https://doi.org/10.1016/B978-0-12-394316-3.00008-9
-
3-Methyl-3-sulfanylhexan-1-ol as a Major descriptor for the human axilla-sweat odour profileChemistry & Biodiversity 1:1022–1035.https://doi.org/10.1002/cbdv.200490077
-
Functional and structural characterization of a prokaryotic peptide transporter with features similar to mammalian PEPT1Journal of Biological Chemistry 282:2832–2839.https://doi.org/10.1074/jbc.M604866200
-
BookPheromones and Animal Behaviour : Communication by Smell and TasteCambridge University Press.
-
MalY of Escherichia coli is an enzyme with the activity of a beta C-S lyase (cystathionase)Journal of Bacteriology 177:5035–5039.https://doi.org/10.1128/jb.177.17.5035-5039.1995
Article and author information
Author details
Funding
Wellcome (102890/Z/13/Z)
- Simon Newstead
Biotechnology and Biological Sciences Research Council (BB/N006615/1)
- Gavin H Thomas
Biotechnology and Biological Sciences Research Council (BB/L013703/1)
- Gavin H Thomas
- Simon Newstead
Biotechnology and Biological Sciences Research Council (BB/H016201/1)
- Daniel Bawdon
- A Gordon James
- Gavin H Thomas
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank the staff of beamlines I24 Diamond Light Source, UK, and ID23eh2 at the European Synchrotron Radiation Facility (ESRF) for assistance. This work was supported by a Wellcome award (102890/Z/13/Z) to SN and BBSRC LINK funding to GHT (BB/N006615/1) and was initiated by CBMNnet proof of concept funding (BB/L013703/1) to GHT and SN. DB was supported by a BBSRC CASE studentship (BB/H016201/1).
Copyright
© 2018, Minhas et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 13,070
- views
-
- 1,389
- downloads
-
- 50
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 50
- citations for umbrella DOI https://doi.org/10.7554/eLife.34995
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Structural basis of malodour precursor transport in the human axilla
eLife 7:e34995.
https://doi.org/10.7554/eLife.34995




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}