Homeostatic control of cell wall hydrolysis by the WalRK two-component signaling pathway in Bacillus subtilis
- Harvard Medical School, United States
Abstract
Bacterial cells are encased in a peptidoglycan (PG) exoskeleton that protects them from osmotic lysis and specifies their distinct shapes. Cell wall hydrolases are required to enlarge this covalently closed macromolecule during growth, but how these autolytic enzymes are regulated remains poorly understood. Bacillus subtilis encodes two functionally redundant D,L-endopeptidases (CwlO and LytE) that cleave peptide crosslinks to allow expansion of the PG meshwork during growth. Here, we provide evidence that the essential and broadly conserved WalR-WalK two component regulatory system continuously monitors changes in the activity of these hydrolases by sensing the cleavage products generated by these enzymes and modulating their levels and activity in response. The WalR-WalK pathway is conserved among many Gram-positive pathogens where it controls transcription of distinct sets of PG hydrolases. Cell wall remodeling in these bacteria may be subject to homeostatic control mechanisms similar to the one reported here.
Introduction
The cell wall peptidoglycan (PG) is composed of long glycan strands cross-linked together by short peptides. This three-dimensional exoskeleton specifies shape and protects the cell from osmotic rupture. For cells to grow they must enlarge this covalently closed macromolecule and this requires both the synthesis of new material and hydrolysis of the PG meshwork to allow for its expansion. How the cell maintains the appropriate levels of these potentially autolytic enzymes remains unclear. Here, we report that the WalR-WalK (WalRK) two-component signaling pathway functions in the homeostatic control of the cell wall hydrolases required for expansion of the PG during growth.
The WalRK two-component system (TCS) was discovered over two decades ago and is among the most broadly conserved TCS in Firmicutes (Fabret and Hoch, 1998; Dubrac et al., 2008a; Takada et al., 2018). WalK (also referred to as YycG, VicK, or MicA) is a membrane-anchored sensor kinase and WalR (also called YycF, VicR, or MicB) is a DNA binding response regulator of the OmpR family (Fabret and Hoch, 1998; Dubrac et al., 2008a; Dubrac and Msadek, 2008b; Okajima et al., 2008). In most bacteria that encode this TCS, two additional genes, walH (yycH) and walI (yycI), reside in an operon with them. In B. subtilis, WalH and WalI are negative regulators of the WalK sensor kinase and the three integral membrane proteins assemble into a multimeric complex (Szurmant et al., 2005; Szurmant et al., 2007; Szurmant et al., 2008). The WalRK system is essential in most Firmicutes, making it an attractive antibiotic target (Barrett and Hoch, 1998; Gotoh et al., 2010). Accordingly, the WalRK pathway has been extensively studied in B. subtilis as well as several important Gram-positive pathogens. In all cases where it has been examined, the WalR regulon contains genes encoding cell wall hydrolases (Bisicchia et al., 2007; Howell et al., 2003; Ahn and Burne, 2007; Ng et al., 2005; Liu, 2006; Dubrac et al., 2007). Furthermore, cells engineered to constitutively express a subset of these enzymes can bypass the essentiality of the signaling pathway (Ng et al., 2003; Delaune et al., 2011; Takada et al., 2018). These findings have led to the view that the essential role of WalRK is to coordinate cell wall metabolism with growth. However, despite two decades of research, what the WalK sensor kinases senses and how this pathway functions in cell wall homeostasis have remained mysterious.
In B. subtilis, phosphorylated WalR (WalR ~P) controls the synthesis of several cell wall hydrolases; among them are two enzymes (CwlO and LytE) that are critical for cell wall elongation. Cells lacking either PG hydrolase are viable but depletion of one in the absence of the other causes a cessation of growth followed by lysis (Bisicchia et al., 2007; Hashimoto et al., 2012). Both enzymes are D,L-endopeptidases and cleave the peptide bond between the second (γ-D-Glu) and third (mDAP) amino acid in the stem peptide of PG (Ishikawa et al., 1998; Yamaguchi et al., 2004). CwlO is controlled by a membrane complex composed of the non-canonical ABC transporter FtsEX and two integral membrane proteins SweC and SweD (Meisner et al., 2013; Domínguez-Cuevas et al., 2012; Brunet et al., 2019). LytE is a secreted enzyme with LysM domains that direct it to the lateral cell wall (Margot et al., 1998; Ishikawa et al., 1998; Buist et al., 2008). How the level and activity of these essential elongation hydrolases are regulated remains incompletely understood. Here, we report that the WalK sensor kinase monitors the activity of CwlO and LytE by sensing the cleavage products generated by them. In response, WalK controls WalR-dependent changes in the expression and activity of these enzymes. Thus, this essential two component system functions in the homeostatic control of PG hydrolysis required for growth. This represents the first homeostatic pathway for cell wall hydrolysis in bacteria and we propose that cell wall remodeling in related Gram-positive pathogens is subject to similar regulatory control.
Results
LytE levels increase in the absence of CwlO maintaining cell envelope integrity
In the course of characterizing LytE protein levels in various mutant backgrounds, we discovered that LytE levels increase approximately 2-fold in the absence of CwlO (Figure 1A). To determine whether this increase was due to changes in lytE transcription, we fused the lytE promoter to lacZ and compared ß-galactosidase activity in wild-type and cells lacking CwlO. As can be seen in Figure 1B, transcription from the PlytE promoter increased ~2 fold in the ∆cwlO mutant. A similar increase in PlytE transcription was observed in cells lacking the FtsEX complex, which is required for CwlO activity (Figure 1B) (Meisner et al., 2013). Furthermore, a point mutation in the Walker A motif in FtsE, predicted to impair ATP binding (Yang et al., 2011; Meisner et al., 2013) but not CwlO association with FtsX (Brunet et al., 2019) also resulted in increased lytE transcription (Figure 1—figure supplement 1). From these experiments we conclude that cells lacking CwlO activity increase expression of lytE. We also observed a modest but reproducible increase in lytE transcription in cells lacking LytE (Figure 1B), suggesting that B. subtilis increases lytE expression in response to reduction in D,L-endopeptidase activity in general.
Figure 1 with 2 supplements see all

B. subtilis increases lytE expression in the absence of CwlO activity to maintain cell envelope integrity.
(A) Immunoblot analysis of LytE produced under the control of its native promoter or under IPTG control. The indicated strains (∆lytE, wild-type (wt), ∆cwlO, and ∆cwlO ∆lytE P(IPTG)-lytE (LytE = wt)) were grown in CH medium with or without 500 µM IPTG and harvested at an OD600 ~0.4. SigA protein levels were analyzed to control for loading. (B) Bar graph showing β-galactosidase activity from a lytE promoter (PlytE) fusion to lacZ in wild-type (wt), ∆cwlO, ∆ftsEX, and ∆lytE strains. Activity was assayed in exponentially growing cultures in LB. Error bars represent standard deviation from three biological replicates. Asterisks indicate p-values calculated using Welch’s unequal variances t-test compared to wildtype (*<0.02, **<0.005). (C) Representative fluorescent images of strains from (A) harboring cytoplasmic mCherry grown under identical conditions as in (A). Membranes were visualized with TMA-DPH (top), and merged images of cytoplasmic mCherry and phase-contrast are shown (bottom). Scale bar indicates 2 μm. (D) Bar graph showing the percentage of cells with envelope integrity defects in wild-type (wt), ∆cwlO, and a ∆cwlO mutant in which LytE levels are held at levels equivalent to wild-type (LytE = wt). Cells without cytoplasmic fluorescence and/or that stained with propidium iodide were scored as lysed or PI positive. >500 cells were scored per strain. The images and immunoblots in this figure were representatives from three independent experiments.
-
Figure 1—source data 1
Figure 1 B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig1-data1-v2.xlsx
To investigate whether the ~2 fold change in LytE levels in the ∆cwlO mutant has any physiological consequences, we used a strain lacking both cwlO and lytE that contained an IPTG-regulated allele of lytE. First, we determined the inducer concentration that resulted in LytE levels equivalent to wild-type (Figure 1A) and then examined the cells by fluorescence microscopy (Figure 1C, Figure 1—figure supplement 2). As reported previously, cells lacking cwlO were shorter and fatter than wild-type and these morphological phenotypes were largely homogenous throughout the population (Hashimoto et al., 2012; Meisner et al., 2013; Brunet et al., 2019). Furthermore, based on cytoplasmic mCherry fluorescence (Figure 1C) and propidium iodide staining (Figure 1D), the ∆cwlO mutant cells had intact membranes. By contrast, cells lacking cwlO in which LytE was artificially maintained at wild-type levels had heterogeneous morphologies with >20% lysis or membrane permeability defects (Figure 1C and D, Figure 1—figure supplement 2). These results indicate that the increase in LytE levels allows the ∆cwlO mutant to maintain membrane integrity and a homogenous morphology. Thus, these data suggest that cells lacking CwlO compensate for the reduction in D,L-endopeptidase activity by increasing expression of a second D,L-endopeptidase, LytE.
B. subtilis modulates lytE transcription in response to changes in D,L-endopeptidase activity
The experiments described above indicate that B. subtilis increases lytE expression in the absence of CwlO activity. We next investigated whether cells decrease lytE transcription in the presence of too much D,L-endopeptidase activity. Because CwlO is regulated post-translationally by FtsEX (Domínguez-Cuevas et al., 2012; Meisner et al., 2013), we used LytE to generate high D,L-endopeptidase activity. We introduced a strong IPTG-inducible promoter fusion to lytE and a catalytic mutant (C247S) into a strain harboring our PlytE-lacZ reporter and monitored ß-galactosidase activity after induction. Strikingly, cells with increased levels of wild-type LytE had reduced lytE transcription, while cells over-expressing the catalytic mutant had PlytE promoter activity similar to wild-type (Figure 2A and B).
Figure 2 with 1 supplement see all
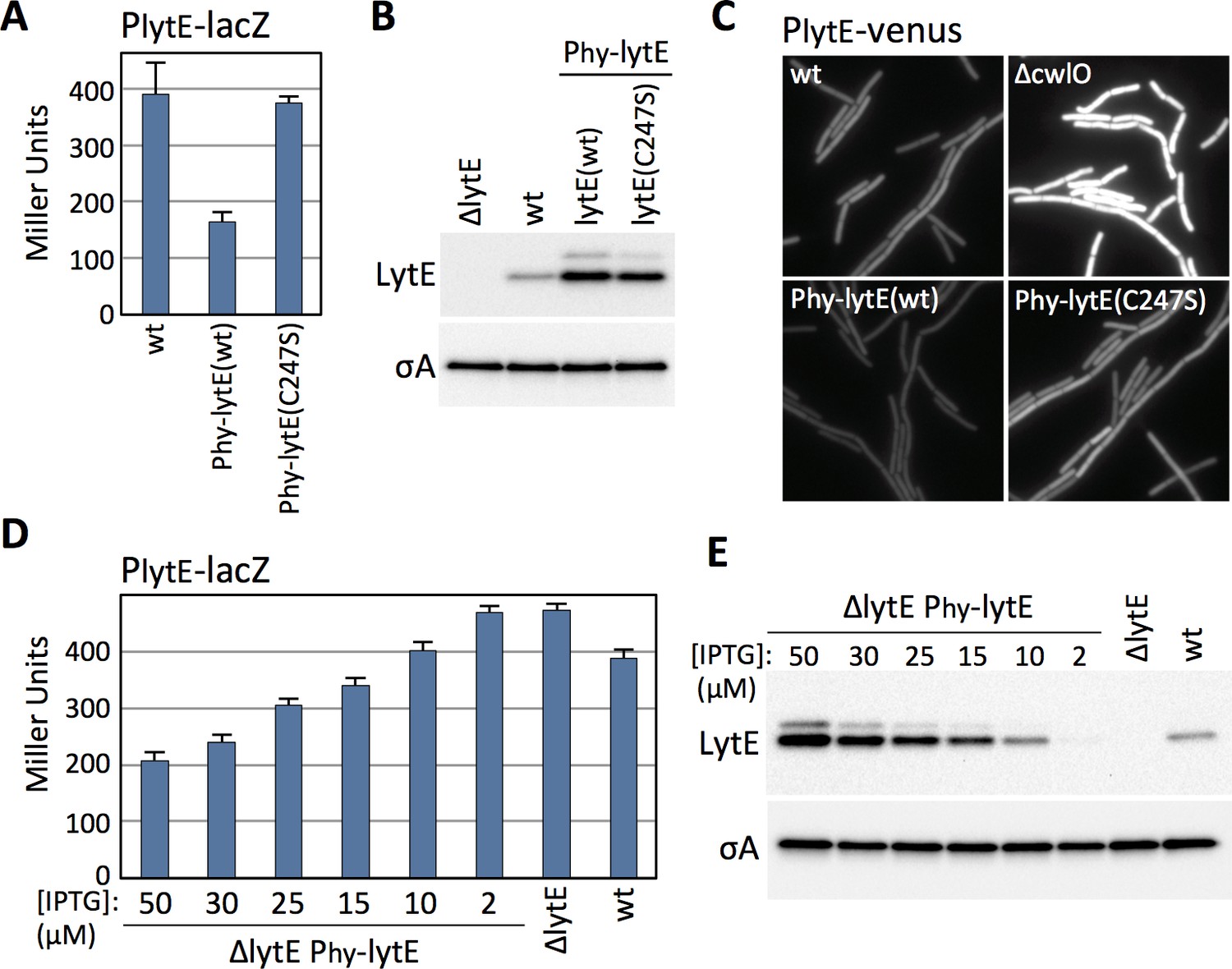
B. subtilis modulates lytE transcription in response to changes in D,L-endopeptidase activity.
(A) Bar graph showing β-galactosidase activity from the PlytE-lacZ reporter in wild-type (wt), and strains harboring the lytE gene or a catalytic mutant (C247S) under the control of the strong IPTG-inducible promoter Phyperspank (Phy) and with an optimized ribosome binding site. Activity was assayed in exponentially growing cultures 60 min after the addition of 50 µM IPTG. Error bars represent standard deviation from three biological replicates. (B) Immunoblot analysis of LytE in the same strains as in (A), 60 min after induction with 50 µM IPTG. SigA protein levels were analyzed to control for loading. (C) Representative images of cytoplasmic Venus fluorescence from the PlytE-venus reporter in the indicated strains visualized 30 min after addition of 50 μM IPTG. (D) Bar graph showing β-galactosidase activity from PlytE-lacZ in the indicated strains. Cells were grown in LB medium or LB medium supplemented with the indicated concentrations of IPTG. β-galactosidase activity was assayed 60 min after induction of lytE. Error bars represent standard deviation from three biological replicates. (E) Immunoblot analysis of LytE and SigA in the strains used in (D). All representative images and immunoblots in this figure are from one of three independent experiments.
-
Figure 2—source data 1
Figure 2A B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig2-data1-v2.xlsx
-
Figure 2—source data 2
Figure 2D B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig2-data2-v2.xlsx
Immunoblot analysis and lacZ reporters are population-based assays. To address whether the changes in lytE transcription in response to high or low D,L-endopeptidase activity were homogenous throughout the population, we built a lytE promoter fusion to the gene encoding the yellow fluorescent protein variant Venus (PlytE-venus). The fluorescent reporter was introduced into strains with high or low D,L-endopeptidase activity and then monitored during exponential growth by fluorescence microscopy. As can be seen in Figure 2C, all cells lacking cwlO had increased Venus fluorescence while all cells over-expressing lytE had reduced fluorescence. Collectively, these results indicate that B. subtilis modulates lytE expression in response to both an increase and decrease in D,L-endopeptidase activity.
The experiments presented thus far indicate that B. subtilis can increase or decrease lytE transcription in cells lacking cwlO or over-expressing lytE. However, it seemed unlikely that B. subtilis evolved a mechanism to compensate for gene deletion and over-expression. To investigate whether B. subtilis modulates lytE transcription in response to more physiological changes in D,L-endopeptidase activity, we used an IPTG-regulated allele of lytE and grew cells at different inducer concentrations to produce a range of LytE levels that were both above and below wild-type levels (Figure 2E). As can been seen in Figure 2D, we found that PlytE transcription inversely correlated with the amount of D,L-endopeptidase produced. Importantly, using the same IPTG-regulated promoter fused to gfp, we found that at IPTG concentrations similar to those used to express lytE all cells in the population had equivalent GFP fluorescence (Figure 2—figure supplement 1). Thus, the graded response to the changes in LytE levels observed in the ensemble assays reflect similar changes in D,L-endopeptidase levels in all cells in the population.
Modulation of lytE expression requires WalR and not SigI
The lytE gene is has two promoters that influence each other (Tseng et al., 2011; Salzberg et al., 2013) (Figure 3A). One is recognized by the alternative sigma factor Sigma I (SigI) and the other is controlled by Sigma A (SigA) but requires the phosphorylated form of the response regulator WalR (Salzberg et al., 2013). Cells lacking SigI have reduced PlytE-lacZ expression and reduced LytE protein levels, while cells lacking the anti-SigI factor RsgI have increased lytE transcription and increased LytE protein levels (Figure 3B and C). Similarly, a point mutation in one of the two WalR binding sites in the lytE promoter (box2*) (Salzberg et al., 2013) abolishes lytE transcription and LytE protein levels (Figure 3B and C). Furthermore, cells lacking WalH, a negative regulator of the WalRK two-component system (Szurmant et al., 2005), results in increased lytE transcription and LytE protein levels (Figure 3B and C). Consistent with these observations and the synthetic lethal relationship between lytE and cwlO, cells lacking SigI or harboring a point mutation in the WalR binding site in the lytE promoter are inviable when CwlO is depleted (Salzberg et al., 2013) (Figure 3D). To investigate whether the RsgI-SigI or the WalRK signaling pathway is involved in the response to changes in D,L-endopeptidase activity, we monitored the PlytE-lacZ response to increased LytE levels in ∆rsgI and ∆walH mutants in which SigI and WalR were constitutively active. As can be seen in Figure 3E, over-expression of lytE in the ∆rsgI mutant resulted in reduced lytE transcription, while in the ∆walH mutant lytE transcription was unchanged. These data suggest that the WalRK signaling pathway is responsible for mediating the observed response to changes in D,L-endopeptidase activity.
Figure 3

Modulation of lytE transcription in response to changes in D,L-endopeptidase activity requires WalR.
(A) Schematic diagram of the lytE gene with its two promoters recognized by SigI and SigA. Phosphorylated WalR binds to two sites (box 1 and 2) flanking the SigA promoter. (B) Bar graph showing β-galactosidase activity from the PlytE-lacZ reporter in the indicated strains. Activity was assayed in exponentially growing cultures in LB medium. Error bars represent standard deviation from three biological replicates. (C) Immunoblot analysis of LytE protein levels in the strains used in (B). SigA levels were analyzed to control for loading. Immunoblots from one of two biological replicates are shown. (D) Spot dilutions of the indicated strains in the presence and absence of cwlO expression. Strains were grown in the presence of IPTG (500 µM) to OD600 ~2.0. The cultures were washed twice without inducer, normalized to an OD600 = 1.5, 10-fold serially diluted, and spotted (5 µL) onto LB agar plates with or without 500 µM IPTG. Representative plates from one of two biological replicates are shown. (E) Bar graph showing β-galactosidase activity from the PlytE-lacZ reporter in indicated strains. Strains harboring the strong IPTG-inducible promoter fusion to lytE (Phy-lytE) were induced for 60 min with 50 µM IPTG. Error bars represent standard deviation from three biological replicates.
-
Figure 3—source data 1
Figure 3B B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig3-data1-v2.xlsx
-
Figure 3—source data 2
Figure 3E B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig3-data2-v2.xlsx
The WalRK signaling pathway responds to changes in D,L-endopeptidase activity
To further test whether the WalRK pathway responds to changes in D,L-endopeptidase activity, we generated transcriptional reporters for two well-characterized genes (yocH and iseA) that are specifically regulated by WalR and not SigI (Bisicchia et al., 2007). yocH is positively regulated by phosphorylated WalR (WalR ~P) while iseA is negatively regulated by WalR ~P (Figure 4A). We fused both promoters to lacZ and separately to venus. To validate these reporters, we monitored their activity in strains lacking WalH or WalI, two negative regulators of WalK (Szurmant et al., 2005; Szurmant et al., 2007). In the absence of either, WalR activity is high and PyocH-venus transcription increased, while PiseA-venus transcription decreased (Figure 4—figure supplement 1A and B). Furthermore, and as expected, depletion of WalRK resulted in strong de-repression of PiseA-venus (Figure 4—figure supplement 1C).
Figure 4 with 5 supplements see all
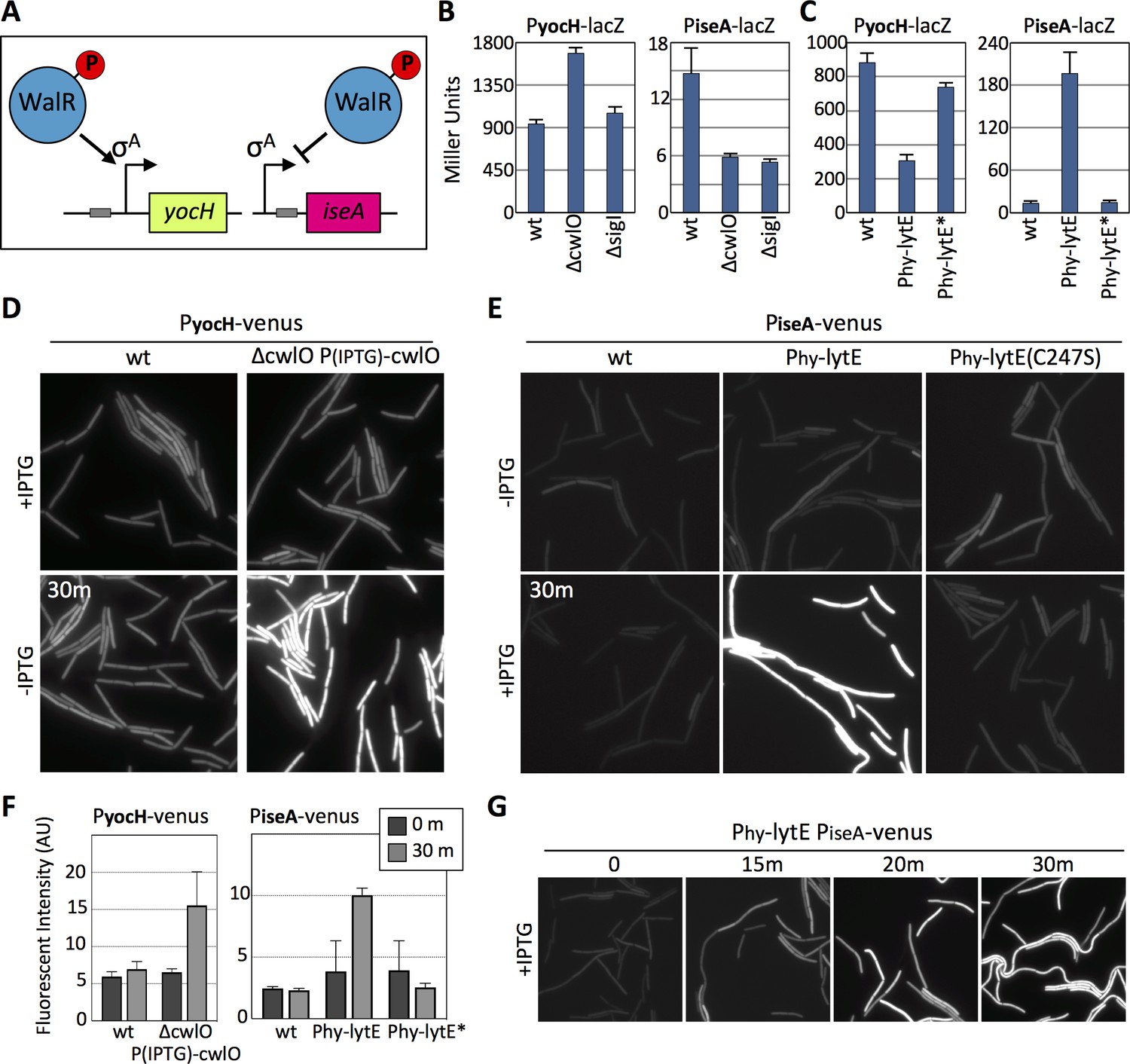
The WalRK signaling pathway responds to changes in D,L-endopeptidase activity.
(A) Schematic diagrams of the yocH and iseA genes. Phosphorylated WalR activates transcription of yocH and represses transcription of iseA. The positions of the WalR binding sites (gray boxes) are shown. (B and C) Bar graph showing β-galactosidase activity from PyocH-lacZ and PiseA-lacZ reporters in the indicated strains. Activity was assayed in exponentially growing cultures in LB medium. Strains harboring strong IPTG-inducible promoter fusion to lytE (Phy-lytE) and the LytE catalytic mutant (C247S) (Phy-lytE*) were induced for 60 min with 50 µM IPTG. Error bars represent standard deviation from three biological replicates. (D) Representative fluorescent images of the indicated strains harboring PyocH-venus. Cells were grown to OD600 ~0.3 in LB medium supplemented with 1 mM IPTG, washed in medium lacking inducer and imaged before and 30 min after growth in LB medium lacking inducer. (E) Representative fluorescent images of the indicated strains harboring PiseA-venus. Strains were grown to OD600 ~0.3 in LB medium and imaged before and 30 min after addition of IPTG (50 µM). Representative images are from one of three independent experiments. (F) Quantification of the average fluorescence intensity, normalized to cell area, of strains shown in (D) and (E).>1000 cells were analyzed from three independent experiments. Dark gray bars (0 m) are from cells prior to IPTG removal for the CwlO depletion experiment monitoring PyocH-venus and prior to IPTG addition for the lytE over-expression experiment monitoring PiseA-venus. Light gray bars (30 m) are from cells 30 min after IPTG removal (left graph) and 30 after IPTG addition (right graph). (G) Representative fluorescent images of PiseA-venus at the indicated times after addition of IPTG (50 µM). PiseA-venus is de-repressed within 15 min after induction of lytE..
-
Figure 4—source data 1
Figure 4B B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Figure 4C B-galactosidase assay Miller Units.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig4-data2-v2.xlsx
-
Figure 4—source data 3
Figure 4F cwlO depletion, cellular fluorescence intensity.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig4-data3-v2.xlsx
-
Figure 4—source data 4
Figure 4F lytE over-expression, cellular fluorescence intensity.
- https://cdn.elifesciences.org/articles/52088/elife-52088-fig4-data4-v2.xlsx
Next, we used these WalR-specific reporters to investigate whether the WalRK pathway responds to changes in D,L-endopeptidase activity. When monitored by ß-galactosidase assay, PyocH transcription increased in the absence of CwlO and decreased when lytE, but not lytE(C247S), was over-expressed (Figure 4B and C). Reciprocally, PiseA transcription was reduced in the absence of CwlO and was strongly de-repressed when lytE was over-expressed (Figure 4B and C). Similar results were obtained with our fluorescent reporters. Within 30 min after shutting off cwlO transcription, PyocH-directed transcription of venus increased (Figure 4D). Furthermore, 15 min after inducing lytE transcription, we could detect de-repression of the PiseA-venus reporter (Figure 4E and G), and after 30 min down-regulation of the PyocH-venus was apparent (Figure 4—figure supplement 2). Quantification of Venus fluorescence (Figure 4F) indicates that our fluorescent and lacZ reporters respond similarly to changes in D,L-endopeptidase levels, although for unknown reasons the magnitude of PiseA de-repression was not as great with the fluorescent reporter. Importantly, these WalR-specific promoters were not affected by a deletion of sigI (Figure 4B and C and Figure 4—figure supplement 3A and B), nor were their dynamics altered in the ∆sigI mutant in response to changes in D,L-endopeptidase activity (Figure 4—figure supplement 3C).
It is noteworthy that both lytE and cwlO are expressed under the control of WalR ~P. Thus, changes in D,L-endopeptidase activity should not only impact the levels of LytE, as shown in Figures 1 and 2, but also the levels of CwlO. Immunoblots to monitor the levels of CwlO in cells over-expressing lytE indicate that this is indeed the case (Figure 4—figure supplement 4A). Furthermore, cells lacking lytE have a modest but reproducible increase in CwlO (Figure 4—figure supplement 4B). Taken together with the data in Figure 3E, these results suggest that the WalRK signaling pathway monitors the extent of D,L endopeptidase activity and modulates lytE and cwlO transcription in response.
Recent studies indicate that the serine/threonine kinase PrkC also controls WalR activity (Libby et al., 2015). PrkC principally regulates WalR in stationary phase and therefore was unlikely to mediate the response to changes in D,L-endopeptidase activity during exponential growth observed here. However, to directly test this we analyzed PiseA and PyocH transcription in wild-type and the ∆prkC mutant before and after inducing lytE (Figure 4—figure supplement 5). As anticipated, the transcriptional responses were similar in the presence and absence of PrkC, ruling out a role for this signaling kinase in responding to D,L-endopeptidase activity.
The WalK sensor kinase responds to changes in D,L-endopeptidase activity in the absence of cell division
Previous work on the WalRK pathway indicates that WalK, but not WalH or WalI, localizes to the divisome and does so in an FtsZ-dependent manner (Fukushima et al., 2008). Furthermore, depletion of FtsZ or other divisome components resulted in a decrease in yocH mRNA levels and an increase in pdaC transcript levels, consistent with a reduction in WalR activity (Fukushima et al., 2008; Fukushima et al., 2011). These studies formed the basis of the prevailing model for WalRK function in which WalK signaling and WalR-dependent expression of cell wall hydrolases are linked to growth via the divisome (Fukushima et al., 2008; Dubrac et al., 2008a; Fukushima et al., 2011). Specifically, it was proposed that in non-dividing cells WalK is held inactive along the lateral membranes by its negative regulators WalH and WalI, while in actively growing and dividing cells, WalK localizes to the septum without its inhibitors where it is competent to activate WalR. Our findings that WalK responds to changes in PG hydrolases that act along the lateral cell wall appear inconsistent with a model in which WalK is only active at the septum. To address this discrepancy, we investigated whether the WalRK pathway could respond to changes in D,L-endopeptidase activity in cells lacking a divisome.
To prevent divisome assembly, we took advantage of the FtsZ inhibitor MciZ (Handler et al., 2008). Cells harboring a xylose-regulated allele of mciZ were grown for 60 min in the presence of inducer to block FtsZ-ring assembly (Figure 5—figure supplement 1). The resulting filaments were then induced to express lytE, and WalRK activity was monitored 30 min later using our PiseA-venus reporter. As can be seen in Figure 5A, lytE over-expression resulted in similar de-repression of PiseA-venus in wild-type and cells lacking a divisome. Reciprocally, depletion of CwlO in filamenting cells for 30 min triggered induction of PyocH-venus to the same extent as CwlO depletion in wild-type cells (Figure 5B). We note that a subset of the filaments in both experiments did not respond to changes in D,L-endopeptidase activity. We suspect this is due to a loss in viability, which is largely consistent with propidium iodide staining (Figure 5—figure supplements 2 and 3). Altogether, our data indicate that the WalRK signaling pathway is capable of responding to changes in PG hydrolase activity in the absence of division, and therefore argue that WalK can function along the lateral membranes.
Figure 5 with 3 supplements see all
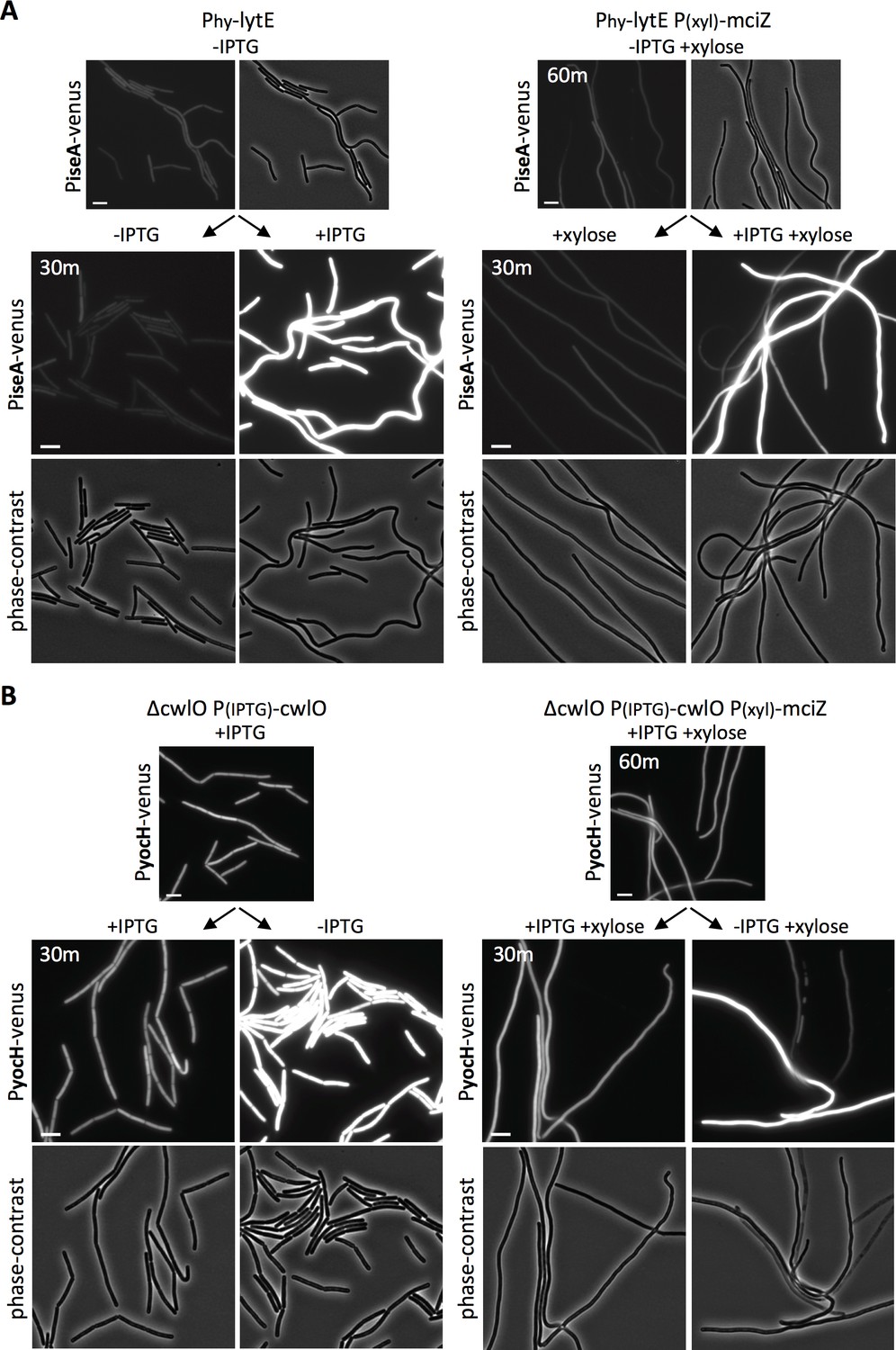
WalRK responds to changes in D,L endopeptidase activity in cells inhibited for divisome assembly.
(A) Representative fluorescence and phase-contrast images of cells harboring PiseA-venus before and 30 min after induction of lytE with 50 µM IPTG. Images on the right are from a strain that also contained the FtsZ inhibitor MciZ under xylose control (P(xyl)-mciZ) and were grown in the presence of 10 mM xylose for 60 min prior to IPTG addition. (B) Representative fluorescence and phase-contrast images of cells harboring PyocH-venus before and 30 min after removal of IPTG to shut off cwlO transcription. Images on the right are from a strain that also contained P(xyl)-mciZ and was grown in the presence of 10 mM xylose for 40 min, prior to IPTG removal. A subset of the division-inhibited cells did not de-repress PiseA-venus (A) or induce PyocH-venus (B); this could be due to loss of viability (see Figure 5—figure supplements 2 and 3). Representative images are from one of three independent experiments. Scale bar indicates 5 µm.
WalK responds to changes in D,L-endopeptidase activity in the absence of the extracellular domains of the WalH and WalI regulators
The two WalK inhibitors, WalH and WalI, are single-pass integral membrane proteins with large extracellular domains (ECDs) (Szurmant et al., 2005) (Figure 6A - schematic). Both regulators reside in a membrane complex with WalK (Szurmant et al., 2007) and could therefore function in signal recognition. Previous work from Szurmant and Hoch showed that basal WalRK activity was unaffected by deletions of the ECDs of WalH and WalI (Szurmant et al., 2008). To investigate whether either of the extracellular domains were required for WalRK signaling in response to changes in D,L-endopeptidase activity, we sought to test strains in which these domains were deleted. We generated xylose-regulated alleles of full-length walH and walI and deletion variants (walH(∆61–455) and walI(∆36–280)) that were identical to those used previously (Szurmant et al., 2008). We introduced these alleles at an ectopic genomic locus in strains lacking walH or walI that harbored the PiseA-venus reporter. As expected, in the absence of xylose, the ∆walH and ∆walI strains had virtually undetectable PiseA-venus expression, indicative of high WalRK activity and strong repression of the PiseA promoter (Figure 6A, Figure 6—figure supplement 1). Similar to what was reported previously, expression of both the full-length and truncated alleles of walH and walI restored basal PiseA-venus transcription (Figure 6A, Figure 6—figure supplement 1). Importantly, cells harboring the full-length and truncated alleles of walH and walI responded similarly to wild-type when lytE was over-expressed (Figure 6A, Figure 6—figure supplement 1). In all cases, PiseA-venus transcription was de-repressed, indicative of a reduction in WalRK signaling. These data indicate that the extracellular domains of WalH and WalI are not individually required for WalRK signaling in response to changes in D,L-endopeptidase activity. To test whether cells lacking the extracellular domains of both WalH and WalI can respond to lytE over-expression, we built stains harboring xylose-regulated alleles of both walH and walI or their deletion variants. As can be seen in Figure 6B, in both strains PiseA-venus transcription was de-repressed upon over-expression of lytE, although the response was more modest in the strain harboring the deletion variants. Collectively, these data argue that the extracellular domains of WalH and WalI are not necessary for WalRK signaling in response to changes in D,L-endopeptidase activity and suggest that the extracellular domain of WalK functions as the sensor.
Figure 6 with 1 supplement see all
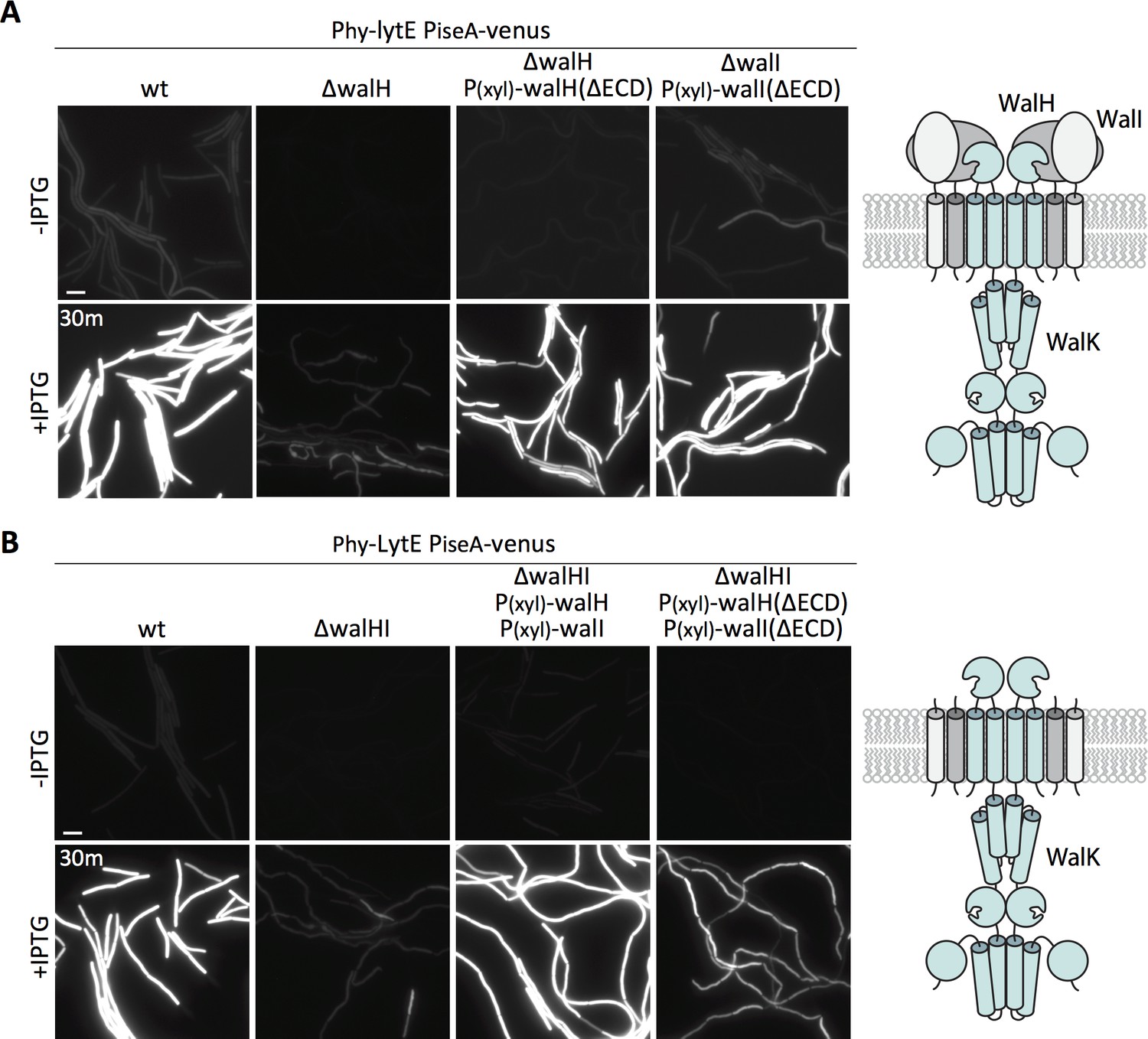
The extracellular domains of WalH and WalI are not required for WalK to respond to changes in D,L-endopeptidase activity.
(A) Representative fluorescence images of the indicated strains harboring PiseA-venus and a strong IPTG-inducible promoter fused to lytE (Phy-lytE). Strains were grown to OD600 ~0.3 in LB medium and imaged before and 30 min after addition of 50 µM IPTG. The medium was supplemented with 10 mM xylose for the strains harboring xylose-regulated alleles of walH or walI with deletions of their extracellular domains (ΔECD). Schematic diagram of the putative WalK/WalH/WalI membrane complex is shown to the right. (B) Representative images of PiseA-venus expression before and 30 min after lytE over-expression in strain lacking both walH and walI and complemented by xylose-induced full-length genes or ΔECD deletion variants. Schematic diagram of the putative membrane complex with WalH and WalI lacking their ECDs is shown on the right. Representative images are from one of three independent experiments. Scale bar indicates 5 µm.
The WalK extracellular domain is required to respond to changes in D,L-endopeptidase activity
WalK contains a 148 amino acid extracellular loop that is homologous to Per-Arnt-Sim (PAS)-like domains, also known as an sCache domain (Upadhyay et al., 2016) (Figure 6A – schematic). These domains are a common feature of sensor kinases and in some cases have been shown to bind signaling ligands (Chang et al., 2010; Henry and Crosson, 2011; Upadhyay et al., 2016). Previous studies indicate that deletion of the extracellular sCache domain of WalK (WalK∆sCache) results in constitutive signaling and high WalR activity (Fukushima et al., 2011) and we confirmed this using both PyocH-venus and PiseA-venus reporters (Figure 7A). To determine whether this domain is required to respond to changes in D,L-endopeptidase activity, we monitored PiseA-venus transcription after over-expression of lytE. PiseA transcription was de-repressed in both wild-type cells and the ∆walK mutant complemented with full-length walK, indicative of a decrease in WalRK signaling (Figure 7B). However, the ∆walK mutant harboring the walK(∆sCache) variant was unresponsive to lytE over-expression (Figure 7B). Furthermore, the walK(∆sCache) variant failed to respond to depletion of CwlO (Figure 7—figure supplement 1).
Figure 7 with 1 supplement see all
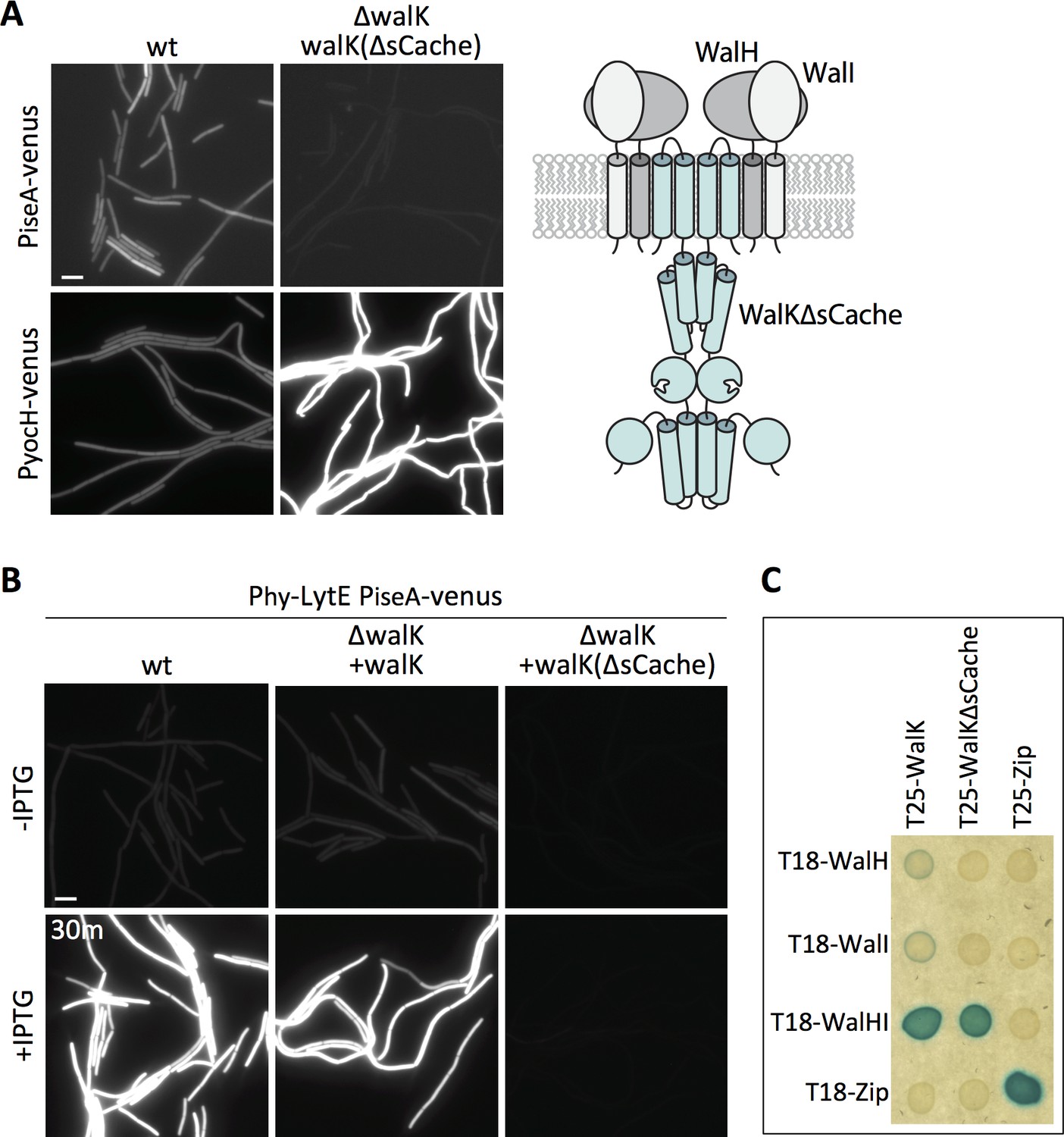
Deletion of the extracellular sCache domain of WalK renders the sensor kinase unresponsive to changes in D,L-endopeptidase activity.
(A) Representative fluorescence images of the indicated strains harboring PiseA-venus or PyocH-venus reporters. Cells were grown to OD600 ~0.4 in LB medium. Strains in which WalK lacks its extracellular sensory domain (∆sCache) have high WalRK activity, leading to strong repression of PiseA-venus and elevated transcription of PyocH-venus. Schematic model of the signaling complex with WalK lacking its extracellular domain is shown on the right. (B) Representative fluorescence images of the indicated strains harboring PiseA-venus and Phy-lytE. Strains were grown to OD600 ~0.3 in LB medium and imaged before and 30 min after addition of 50 µM IPTG. Representative images are from one of three independent experiments. Scale bar indicates 5 µm. (C) WalK and WalK∆sCache interact with WalH and WalI in the bacterial adenylate cyclase two-hybrid (BACTH) assay. The BTH101 E. coli reporter strain containing plasmids expressing the indicated protein fusions to the complementing (T18 and T25) domains of the Bordetella adenylate cyclase. The T18-WalHI plasmid contains a T18-WalH fusion and untagged WalI. Cells were grown to early stationary phase in LB at 37°C, normalized to OD600 = 0.2, and 3 µL of each was spotted on LB agar plates supplemented with X-Gal and IPTG. Plates were incubated overnight at 30°C. The ‘Zip’ fusions, composed of the leucine zipper domain of GCN4, served as positive and negative controls.
These data are consistent with the idea that the sCache domain of WalK is necessary for sensing changes in D,L-endopeptidase activity. However, previous in vivo formaldehyde crosslinking experiments suggested that the interaction between WalK and WalH/WalI requires the extracellular domain of WalK (Fukushima et al., 2011). If correct, the constitutive signaling in the walK(∆sCache) mutant and the failure to respond to changes in D,L-endopeptidase activity could be due to the loss of inhibition by WalH and WalI. To more directly test the interactions among these proteins, we used the Bacterial Adenylate Cyclase Two Hybrid (BACTH) system (Ladant and Ullmann, 1999; Karimova et al., 2005). We generated fusions with complementary fragments (T18 and T25) of Bordetella pertussis adenylate cyclase to WalK, WalK(∆sCache), WalH, and WalI. The fusions were co-transformed into E. coli and assayed for interaction on LB agar supplemented with X-Gal (see Materials and methods). As can be seen in Figure 6C, a strong interaction was only observed when T25-WalK and T18-WalH were co-expressed with untagged WalI, indicating that all three proteins are required for stable interaction. Importantly, we observed a similarly strong interaction using T25-WalK(∆sCache) (Figure 6C). These data argue that the WalK variant lacking its ECD stably interacts with WalH and WalI in vivo. Collectively, these results and those from Figure 5 suggest that the extracellular sCache domain of WalK functions as the sensing domain that monitors changes in D,L-endopeptidase activity.
The WalK response is specific to D,L-endopeptidase crosslink cleavage
The data presented thus far indicate that the WalK sensor kinase responds to changes in cell wall hydrolysis. We envisioned two possible models for what WalK might be sensing. Since CwlO and LytE are both D,L-endopeptidases, WalK could specifically monitor D,L-endopeptidase cleavage products. Alternatively, WalK could be sensing some aspect of the cell wall that is affected by PG hydrolysis in general. For example, WalK could monitor the presence of intact crosslinks or the extent of tension on the PG meshwork. To help distinguish between these two models, we investigated whether WalK responds to PG hydrolases that cleave distinct bonds in the PG meshwork (Figure 8B). To target these enzymes to the lateral cell wall, we fused the catalytic domains of each PG hydrolase to the N-terminal LysM domains of LytE (M3) (Figure 8A) that direct it to the lateral wall (Buist et al., 2008; Hashimoto et al., 2012; Hashimoto et al., 2018). Each chimera was expressed under the control of the same IPTG-regulated promoter used to over-express lytE, and WalK signaling was monitored 30 min after induction using the PiseA-venus reporter. Consistent with the idea that WalK monitors D,L-endopeptidase activity, the chimeras that contained D,L-endopeptidase domains from PG hydrolases that function in cell separation (LytF and CwlS from B. subtilis) (Ohnishi et al., 1999; Yamamoto et al., 2003; Fukushima et al., 2006) de-repressed PiseA transcription (Figure 8C, Figure 8—figure supplement 1B). By contrast, WalK signaling was not impacted by expression of chimeras containing D,D-endopeptidase domains (MepM and MepS from E. coli) (Singh et al., 2012), L,D-endopeptidase domains (LytH and CwlK from B. subtilis) (Horsburgh et al., 2003; Fukushima et al., 2007), the amidase domain from B. subtilis YrvJ (Wendrich and Marahiel, 1997), or the glucosaminidase domain from B. subtilis LytD (Margot et al., 1994) (Figure 8C, Figure 8—figure supplement 1B). Over-expression of most of the chimeras did not cause discernable morphological defects raising the possibility that the fusions were not active enough to alter WalRK signaling, however, we note that the MepM D,D-endopeptidase chimera was able to suppress the lethality of ∆cwlO ∆lytE double mutant (Hashimoto et al., 2018) but even this fusion did not impact PiseA transcription (Figure 8—figure supplement 1A). Collectively, these data favor the model that WalK specifically monitors D,L-endopeptidase activity.
Figure 8 with 2 supplements see all
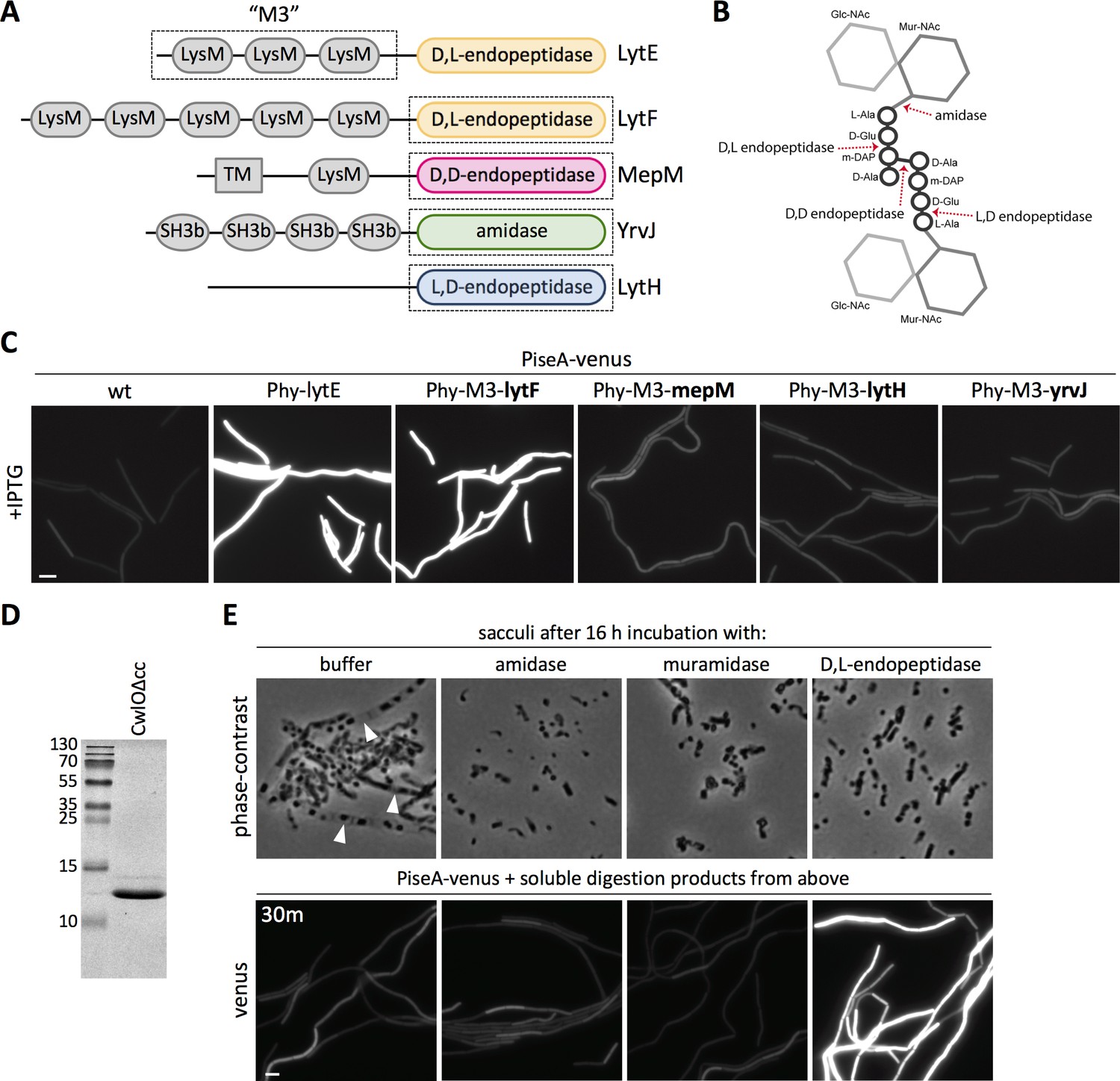
The WalRK pathway specifically responds to D,L-endopeptidase-cleaved peptidoglycan.
(A) Schematic diagram of the cell wall hydrolases used to make LytE chimeras. The three amino-terminal LysM domains of LytE (M3) were fused to the carboxyl-terminal catalytic domains of LytF, MepM, LytH, and YrvJ. (B) Schematic of a single PG crosslink and the cleavage sites of the cell wall hydrolase domains used in (A); the amino acids in the crossbridge are represented by black circles and the di-saccharide with gray hexagons. (C) Representative fluorescence images of the indicated strains harboring PiseA-venus reporter and a strong IPTG-inducible promoter (Phy) fused to lytE or the chimeras. Cells were grown to OD600 ~0.3 in LB medium and imaged 30 min after addition of IPTG. 50 µM IPTG was used to induce LytE and the D,L-endopeptidase chimera and 500 µM IPTG was used for all the other chimeras. (D) Coomassie-stained gel of purified CwlO lacking its coiled-coil domain (CwlOΔcc). (E) Phase-contrast images of purified B. subtilis sacculi incubated overnight at 37°C with buffer or purified CwlO∆cc, the amidase LytA, or the muramidase mutanolysin (top panels). Translucent sacculi (carets) surrounding phase-dark insoluble aggregates are only visible after incubation with buffer. Representative fluorescence images of B. subtilis cells harboring PiseA-venus after 30 min incubation with the soluble material generated from overnight incubation of sacculi under the indicated conditions (bottom panels). Representative images are from one of three independent experiments. Scale bar indicates 5 µm.
To directly test whether D,L-endopeptidase cleavage products inhibit WalRK signaling, we sought to investigate whether soluble PG cleavage products generated by a D,L-endopeptidase in vitro could de-repress the PiseA-venus reporter. To this end, we incubated purified B. subtilis sacculi at 37°C for 16 hr with a commercial muramidase (mutanolysin from Streptomyces globisporus), a purified amidase (LytA from Streptococcus pneumoniae) (Flores-Kim et al., 2019), or the constitutively active D,L-endopeptidase domain from B. subtilis CwlO that lacks its regulatory coiled-coil domain (CwlO∆cc) (Figure 8D). Phase-contrast microscopy was used to assess cell wall hydrolysis (Figure 8E - top panels). Translucent sacculi containing phase-dark aggregates (Liechti et al., 2014) were readily detectable in buffer-treated sacculi, while only the liberated phase-dark aggregates were present in the sacculi incubated with the PG hydrolases. The soluble material was collected from each digestion and added to cells harboring the PiseA-venus reporter. Notably, only the cleavage products generated by D,L-endopeptidase digestion reduced WalRK activity and de-repressed PiseA-venus (Figure 8E - bottom panels). Furthermore, the soluble material from sacculi incubated with iodoacetamide-inactivated CwlO∆cc failed to reduce WalRK activity (Figure 8—figure supplement 2). These results indicate that WalK specifically responds to D,L-endopeptidase cleavage products.
Discussion
Altogether, our data support a model in which the WalR-WalK two-component signaling pathway functions in homeostatic control of the cell wall elongation hydrolases LytE and CwlO, and does so by sensing and responding to PG cleavage products generated by these enzymes (Figure 9). When D,L-endopeptidase activity is low, the concentration of these cleavage products drops leading to an increase in WalK kinase activity and a concomitant increase in WalR ~P. WalR ~P increases transcription of lytE and cwlO, restoring homeostasis (Figure 9 – left panel). Reciprocally, when there is elevated D,L-endopeptidase activity, the concentration of cleavage products increases. High concentrations of these putative allosteric inhibitors reduce WalK activity, resulting in a decrease in lytE and cwlO transcription (Figure 9 – right panel).
Figure 9 with 2 supplements see all
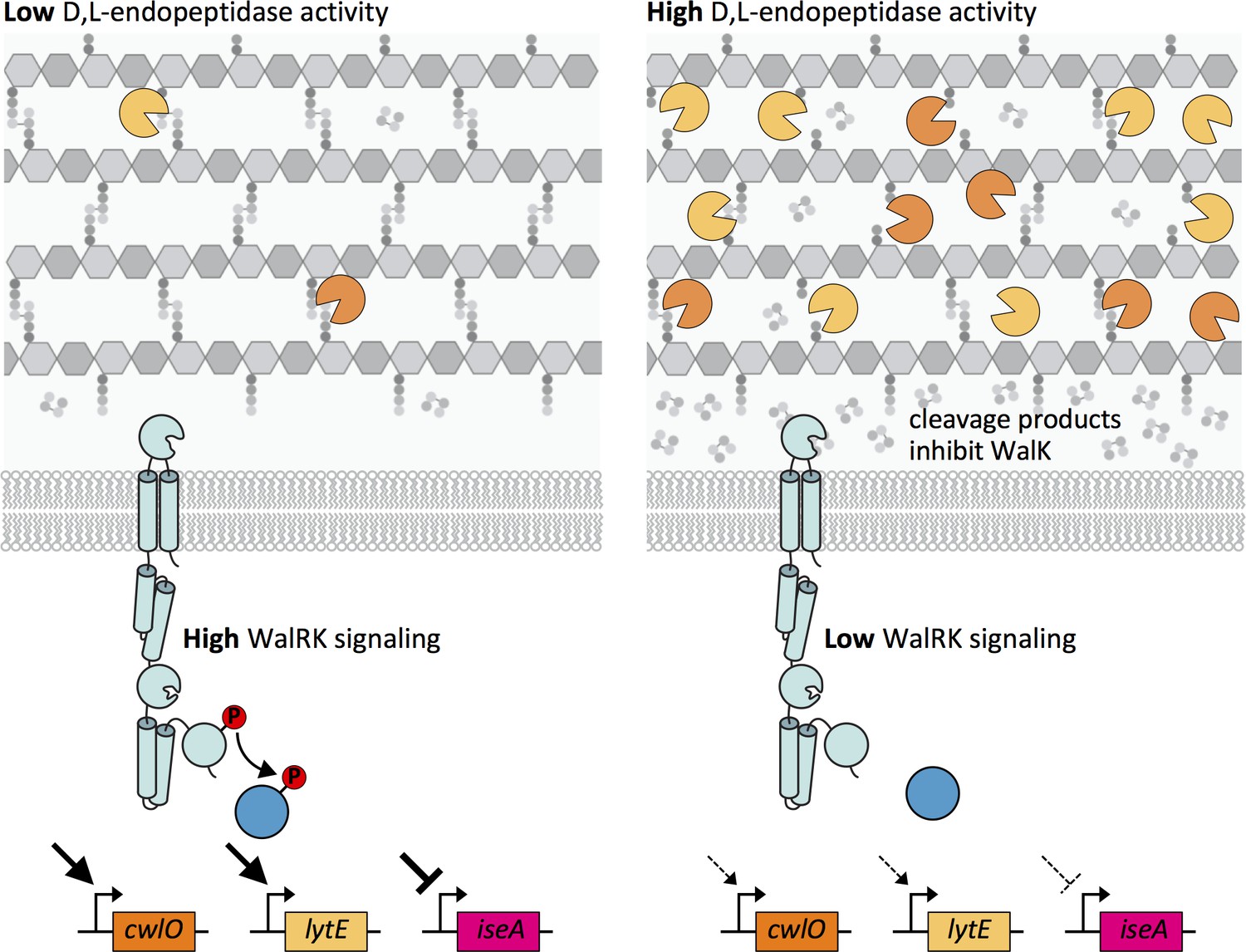
Model depicting the homeostatic control of cell wall elongation hydrolases by the WalRK two-component system.
The WalK sensor kinase and its two regulators WalH and WalI (not shown) sense and respond to the extent of D,L-endopeptidase cleavage. (Left panel) When the CwlO and LytE activities (orange and yellow scissors) are low, the concentration of their cleavage products is low and WalK kinase activity is high generating high levels of phosphorylated WalR. WalR ~P activates transcription of lytE and cwlO and represses transcription of iseA, encoding a secreted inhibitor of LytE. Thus, increased WalRK signaling returns D,L-endopeptidase activity to its homeostatic set point (not shown). (Right panel) When D,L-endopeptidase activity is high, the concentration of cleavage products generated by these enzymes increases and inhibits WalK signaling leading to a reduction in WalR ~P. Low WalR ~P reduces transcription of lytE and cwlO and de-represses iseA expression. CwlO levels are rapidly reduced due to its short half-life (Figure 9—figure supplement 1) and LytE activity is rapidly inhibited by IseA (Figure 9—figure supplement 2A). Thus, inhibition of WalK signaling by high concentrations of cleavage products efficiently restores homeostatic levels of the elongation D,L-endopeptidases.
In this model, transcriptional activation of lytE and cwlO rapidly boosts D,L-endopeptidase activity when the levels of these enzymes drop. However, this pathway can also rapidly reduce D,L-endopeptidase activity when it gets too high. The iseA gene, which is repressed by WalR ~P, encodes a secreted inhibitor of D,L-endopeptidases (Bisicchia et al., 2007; Salzberg and Helmann, 2007; Yamamoto et al., 2008). It was given its name (inhibitor of cell separation A) because of its ability to inhibit cell separation D,L-endopeptidases when over-expressed on a multi-copy plasmid (Yamamoto et al., 2008). However, we have found that LytE is likely to be the primary target of IseA. Specifically, we found that the levels of IseA needed to inhibit LytE had no impact on cell separation (Figure 9—figure supplement 1A and B). Furthermore, we have also found that CwlO has a half-life of ~7 min (Figure 9—figure supplement 2A). Accordingly, when D,L-endopeptidase activity is too high, a drop in WalRK signaling not only causes a reduction in lytE and cwlO transcription, but also inhibition of LytE activity via de-repression of iseA and a reduction in CwlO levels through degradation or shedding of the enzyme into the medium (Figure 9—figure supplement 2B). Thus, the WalRK signaling pathway is able to adjust the levels and activity of CwlO and LytE on the time-scale of a cell cycle to maintain homeostatic control of these essential enzymes during growth.
Previous work on the WalRK signaling pathway led to the model that WalK localization to the septal ring, in the absence of its negative regulators WalH and WalI, couples its activity to growth via the divisome (Fukushima et al., 2008; Dubrac et al., 2008a). Support for this model comes from experiments in which depletion of FtsZ or other divisome components results in a decrease in yocH mRNA levels and an increase in pdaC mRNA levels, suggesting that WalK-dependent phosphorylation of WalR requires an intact divisome (Fukushima et al., 2008; Fukushima et al., 2011). One caveat of these experiments is that yocH and pdaC transcript levels were monitored 3 hr after depletion of the divisome components, raising the possibility that the reduction in WalR activity could be indirect. We note that we did not detect changes in PiseA- and PyocH-directed Venus fluorescence after inhibiting FtsZ for 70–90 min (Figure 5, Figure 5—figure supplement 2, Figure 5—figure supplement 3). However, RT-PCR is likely to be more sensitive than our fluorescence-based assay. It is therefore possible that WalRK functions in the homeostatic control of the elongation hydrolases as described here, and separately acts at the divisome to boost CwlO and LytE levels during cytokinesis in anticipation of cell wall growth following division. Time-lapse microscopy in microfluidic devices like the mother machine (Wang et al., 2010) will enable a direct test of this model in the future.
We have demonstrated that the soluble cleavage products generated by D,L-endopeptidase-digestion can inhibit WalK signaling and could therefore function as allosteric inhibitors of the kinase. Although the specific cleavage product that inhibits WalK is currently unknown, we favor the idea that the cross-linked tetra- and/or penta-peptides liberated by cleavage of peptide crosslinks (Figures 8B and 9) are the WalK ligand because cleavage of PG crossbridges is required for expansion of the PG meshwork during growth (Bisicchia et al., 2007; Meisner et al., 2013; Hashimoto et al., 2012; Hashimoto et al., 2018). To generate these products, both γ-D-Glu-mDAP bonds must be cleaved in the peptide crossbridge. Concerted cleavage of identical bonds in peptide crosslinks has been proposed previously (Wong et al., 2015), however it has not been rigorously tested. Alternatively, it is also possible that di- or tri-peptides released upon cleavage of un-cross-linked stem peptides or even short glycan strands with attached di-peptides could serve as the signal. Future experiments will be directed at defining the specific cleavage product that is sensed by WalK and ultimately the ligand-binding site in the sensor domain.
As indicated above, our data are most consistent with a model in which WalK is inhibited when it binds its signal and active when un-liganded (Figure 9). Specifically, we showed that D,L-endopeptidase cleavage products cause de-repression of PiseA, indicative of low levels of WalR ~P and low WalK kinase activity. Reciprocally, we found that depletion of CwlO, and therefore low concentrations of cleavage products, leads to high PyocH transcription, indicative of high levels of WalR ~P and high WalK kinase activity. Another, closely related, two-component system in B. subtilis is the PhoR-PhoP system, involved in sensing and responding to phosphate limitation (Seki et al., 1987; Botella et al., 2014). The PhoP response regulator activates genes involved in phosphate scavenging and inhibits the synthesis of the phosphate-rich surface polymers called wall teichoic acid (WTA). Interestingly, work from the Devine lab suggests that the sensor kinase PhoR is active when un-liganded and inhibited when bound by intermediates in the WTA biosynthetic pathway (Botella et al., 2014). In this case, the signal is thought to be sensed by the intracellular PAS domain of PhoR. Nonetheless, it is interesting that in both cases these kinases appear to be regulated by allosteric inhibition.
Cell growth requires an intimate balance between cell wall synthesis and its hydrolysis. The homeostatic control pathway we have defined here ensures that exponentially growing cells maintain a defined amount of D,L-endopeptidase activity for cell wall elongation. However, under different growth conditions, for example entry into stationary phase, the cell is likely to require different levels of hydrolase activity. We propose that the cell could adapt by modulating the homeostatic ‘set point’ of the WalRK signaling pathway. This set point is likely determined by the levels of the WalK-WalH-WalI sensor kinase complex; the affinity of WalK for its putative allosteric inhibitor; and the activities of the histidine kinase and response regulator. A link between one or more of these determinants and PG synthesis could ensure coordination. Since the wal operon is not under auto-regulatory control (Fabret and Hoch, 1998), altering the set point is unlikely to occur through changes in the levels in the WalK-WalH-WalI complex. Instead, B. subtilis could modulate WalK's affinity for D,L-endopeptidase cleavage products or adjust WalK kinase activity or WalR-dependent transcription. For example, the conserved extracellular domains of WalH and WalI could function in modulating ligand affinity in response to an extracellular signal generated during PG synthesis, or in response to changes in the cell envelope itself. Similarly, the conserved intracellular PAS domain of WalK could alter kinase activity in response to a cytoplasmic signal. For example, this domain could sense intermediates in the PG precursor (lipid II) pathway, analogous to the signal sensed by the PhoR PAS domain. Finally, WalR activity is known to be modulated by PrkC-dependent phosphorylation during entry into stationary phase (Libby et al., 2015), which is thought to respond to extracellular muropeptides. By changing the homeostatic set point for hydrolase activity, extracellular signals could directly coordinate hydrolase activity with PG synthesis while the intracellular signal could link hydrolase activity to flux through the precursor synthesis pathway. Identifying these potential allosteric modulators and defining how they impact the homeostatic control of LytE and CwlO are important challenges for the future.
The WalRK two-component system is the most broadly conserved TCS in the Firmicutes and can be found in important human pathogens including Staphylococcus aureus, Listeria monocytogenes, Bacillus anthracis, Streptococcus pneumoniae, and Streptococcus mutans (Dubrac et al., 2008a). In most cases, this pathway is essential and, in the organisms in which it has been investigated, the WalR regulon contains cell wall hydrolases although not necessarily D,L-endopeptidases (Martin et al., 1999; Howell et al., 2003; Dubrac and Msadek, 2004; Ng et al., 2005; Liu, 2006; Bisicchia et al., 2007; Ahn and Burne, 2007; Delaune et al., 2011). Furthermore, as is the case with B. subtilis (Takada et al., 2018), the essentiality of this pathway can be bypassed by engineering these bacteria to express a subset of the PG hydrolytic enzymes in the WalR regulon (Ng et al., 2003; Delaune et al., 2011). Based on these similarities, we hypothesize that the WalRK pathway functions in homeostatic control of cell wall hydrolysis in these pathogens and uses distinct cleavage products to monitor PG hydrolase activity.
It is noteworthy that the WalK sensor kinases in Streptococci and Lactococci lack an extracellular sCache domain (Lange et al., 1999; Dubrac et al., 2008a; Wang et al., 2013). Accordingly, if there is homeostatic control of cell wall hydrolases in these bacteria they must use a different sensing mechanism or employ a distinct signaling pathway. Intriguingly, the StkP serine/theronine kinase in S. pneumoniae has been implicated in modulating cell wall hydrolysis and has been suggested to function in concert with WalRK pathway in this organism (Fleurie et al., 2012; Stamsås et al., 2017). Analysis of the WalK signaling in diverse Gram-positive pathogens will establish the similarities and differences in the regulatory logic we have uncovered here. Finally, the cleavage products of the PG hydrolases in the WalR regulons could potently inhibit WalK signaling and therefore offer the potential for therapeutic development.
Materials and methods
Strains, plasmids, and routine growth conditions
Request a detailed protocolAll Bacillus subtilis strains were derived from the prototrophic strain PY79 (Youngman et al., 1983). Cells were grown in either Luria-Bertani (LB) or casein hydrolysate (CH) medium at 37°C. Unless otherwise indicated, B. subtilis strains were constructed using genomic DNA and a 1-step competence method. Antibiotic concentrations were used at: 100 µg/mL spectinomycin, 5 µg/mL chloramphenicol, 10 µg/mL tetracycline, 10 µg/mL kanamycin, 1 µg/mL erythromycin and 25 µg/mL lincomycin. A list of strains and plasmids used in this study can be found in the Key Resources Table (Supplementary file 2), and oligonucleotide primers can be found in Supplementary file 1.
β-Galactosidase assays
Request a detailed protocolB. subtilis strains were grown in LB medium at 37°C to an OD600 of ~0.5. The optical density was recorded and 1 mL of culture was harvested and assayed for β-galactosidase activity as previously described (Rudner et al., 1999). Briefly, cell pellets were re-suspended in 1 mL Z buffer (40 mM NaH2PO4, 60 mM Na2HPO4, 1 mM MgSO4, 10 mM KCl, and 50 mM β-mercaptoethanol). 250 µL of this suspension was added to 750 µL of Z buffer supplemented with lysozyme (0.25 mg/ml), and the samples were incubated at 37°C for 15 min. The colorimetric reaction was initiated by addition of 200 µL of 2-nitrophenyl-β-D-galactopyranoside (ONPG, 4 mg/ml) in Z buffer and stopped with 500 µL 1M Na2CO3. The absorbance at 420 nm and OD550 of the reactions were recorded, and the β-galactosidase specific activity in Miller Units was calculated according to the formula [A420-1.75x(OD550)] / (time [min] x OD600) x dilution factor x 1000 (Miller, 1972).
Immunoblot analysis
Request a detailed protocolImmunoblot analysis was performed as described previously (Wang et al., 2015). Briefly, the OD600 was recorded for each culture, 1 mL was collected, and the cell pellet re-suspended in lysis buffer (20 mM Tris pH 7.0, 10 mM MgCl2, 1 mM EDTA, 1 mg/mL lysozyme, 10 µg/mL DNase I, 100 µg/mL RNase A, 1 mM PMSF, 1 µg/mL leupeptin, 1 µg/mL pepstatin) to a final OD600 of 10 for equivalent loading. The cells were incubated at 37°C for 10 min followed by addition of an equal volume of sodium dodecyl sulfate (SDS) sample buffer (0.25 M Tris pH 6.8, 4% SDS, 20% glycerol, 10 mM EDTA) containing 10% β-mercaptoethanol. Samples were heated for 15 min at 65°C prior to loading. Proteins were separated by SDS-PAGE on 15% (LytE) or 12.5% (FtsX, CwlO, SigA) polyacrylamide gels, electroblotted onto Immobilon-P membranes (Millipore) and blocked in 5% nonfat milk in phosphate-buffered saline (PBS) with 0.5% Tween-20. The blocked membranes were probed with anti-LytE (1:10,000), anti-SigA (1:10,000) (Fujita and Sadaie, 1998), anti-FtsX (1:10,000) (Meisner et al., 2013), or anti-CwlO (1:10,000) (Meisner et al., 2013) diluted into 3% BSA in 1x PBS with 0.05% Tween-20. Primary antibodies were detected using horseradish peroxidase-conjugated goat anti-rabbit IgG (BioRad) and the Super Signal chemiluminescence reagent as described by the manufacturer (Pierce). Signal was detected using a FluorChem R System (Protein Simple).
LytE purification and antibody production
Request a detailed protocolRecombinant LytE lacking its three N-terminal LysM domains (LytEΔlysMx3) was expressed in E. coli BL21 (DE3) using the PT7-His6-SUMO-lytEΔlysMx3 expression vector (pYB18). Cells were grown in Terrific Broth (Tartof and Hobbs, 1988) supplemented with 100 μg/ml ampicillin at 37°C to OD600 = 0.3. LytEΔlysMx3 expression was induced for 16 hr at 22°C by addition of 0.5 mM IPTG. After induction, cells were collected by centrifugation at 10,000 × g for 10 min. Cell pellets were resuspended in 15 mL Buffer 1 (20 mM Tris pH 7.5, 300 mM NaCl, 5 mM imidazole, 10% glycerol, 0.1 µM Dithiothreitol) and Complete EDTA-free protease inhibitors (Roche) and lysed via passage through a French press. Cell lysates were clarified by centrifugation at 10,000 x g for 10 min at 4°C. Clarified lysates were mixed with 0.5 mL of Ni2+-NTA agarose resin (Qiagen) and incubated for 2 hr at 4°C. The mixture was loaded onto a column (BioRad) and washed with 10 mL Buffer 1.The His6-SUMO-LytEΔlysMx3 fusion protein was eluted with Buffer 2 (20 mM Tris pH 7.5, 300 mM NaCl, 200 mM imidazole, 0.1 µM Dithiothreitol). Eluates were pooled and dialyzed into storage buffer (20 mM Tris pH 7.5, 300 mM NaCl, 10% glycerol, 0.1 µM Dithiothreitol) at 4°C overnight. 10 µL of purified His6-Ulp1 (1.25 mg/ml) was added to the dialysate and was incubated overnight on ice. The reaction was then mixed with 0.5 mL Ni2+-NTA agarose and loaded onto a column. Flow-through fractions containing the cleaved (untagged) LytEΔlysMx3 were collected and used to generate rabbit polyclonal antibodies (Covance).
Fluorescence microscopy
Request a detailed protocolExponentially growing cells were harvested and concentrated by centrifugation at 6800 x g for 1.5 min and re-suspended in 1/10th volume growth medium and then immobilized on 2% (wt/vol) agarose pads containing growth medium. Fluorescence microscopy was performed on a Nikon Ti inverted microscope equipped with a Plan Apo 100x/1.4 Oil Ph3 DM phase contrast objective, an Andor Zyla 4.2 Plus sCMOS camera, and Lumencore SpectraX LED Illumination. Images were acquired using Nikon Elements 4.3 acquisition software. The fluorescent membrane dye TMA-DPH was added to the concentrated cell suspension at 50 µM final. Propidium iodide (PI) was added at a final concentration of 5 μM. Venus and YFP were imaged using a Chroma ET filter cube for YFP (49003) with an exposure time of 800 ms; TMA-DPH was visualized using a Chroma ET filter cube for DAPI (49000) with an exposure time of 300 ms; mCherry and PI were visualized using a Chroma ET filter cube for mCherry (49008) with an exposure time of 800 ms and 500 ms, respectively. Image processing was performed using Metamorph software (version 7.7.0.0) and Oufti (Paintdakhi et al., 2016) was used for quantitative image analysis.
Bacterial two-hybrid assay
Request a detailed protocolThe Bacterial Adenylate Cyclase-based Two Hybrid (BACTH) system was used as previously described (Karimova et al., 1998; Bendezú et al., 2009). Briefly, pairs of proteins were fused to the complementary fragments (T18 and T25) of the Bordetella pertusis adenylate cyclase. Competent BTH101 E. coli cells were co-transformed with the two plasmids containing T18 and T25 protein fusions in one step. Transformants were selected on LB agar plates supplemented with 100 µg/mL ampicillin (Amp100), 50 µg/mL kanamycin (Kan50), 500 µg/mL isopropyl-β-D-thiogalactoside (IPTG500), and 100 µg/mL 5-Bromo-4-chloro-3-indolyl-β-D-galactopyranoside (X-Gal100). Plates were incubated at 30°C and the homogeneity of the colony color among transformants was confirmed. Single colonies were then grown to early stationary phase in LB medium supplemented with Amp100 and Kan50, normalized to OD600 = 0.2, and spotted (3 µL) on LB agar plates containing Amp100, Kan50, IPTG500, X-Gal100. Plates were incubated at 30°C overnight and imaged. Transformations were done in triplicate with selected images representative of three biological replicates.
PG hydrolase over-expression
Request a detailed protocolCultures of exponentially growing cells were diluted to OD600 = 0.01 in LB and grown for ~1 hr at 37°C to an OD600 ~0.05. The cultures were then induced with IPTG (concentrations indicated in the Figure Legends). Fluorescent images were acquired before and at indicated times after induction. For experiments in which cell division was inhibited, cultures of exponentially growing cells were diluted to OD600 = 0.04 and 10 mM xylose was added to induce mciZ. The cultures were then grown for ~1 hr at 37°C to allow cells to filament for ~2 mass doublings before 50 µM IPTG was added to induce lytE expression.
PG hydrolase depletion
Request a detailed protocolCultures were grown in LB supplemented with 1 mM IPTG to mid-exponential phase, washed three times in LB lacking inducer, and diluted to OD600 = 0.05 in LB to initiate depletion. Images were acquired before and at indicated times after depletion as described in the text. For experiments in which cell division was inhibited, cultures were grown in the presence of 1 mM IPTG (to induce cwlO) until mid-exponential phase. Cells were diluted to OD600 = 0.04 with 1 mM IPTG and 10 mM xylose (to induce mciZ). The cultures were then grown for ~1 hr at 37°C to allow cells to filament for ~2 mass doublings. The cells were then washed three times in LB lacking IPTG, and resuspended in LB containing 10 mM xylose. iseA over-expression.
Cultures of exponentially growing cells were diluted to OD600 = 0.02 in CH medium supplemented with 10 mM xylose and grown for ~1.5 hr at 37°C to an OD600 ~0.2. The cultures were then induced with 500 µM IPTG. Fluorescent images were acquired at indicated times after induction.
In vivo protein turnover assay
Request a detailed protocolWild-type B. subtilis was grown in LB medium at 37°C to an OD600 of 0.5. Protein translation was blocked by the addition of both spectinomycin (200 µg/mL, final concentration) and chloramphenicol (10 µg/mL, final concentration). Samples (1 mL of culture) were collected immediately prior to antibiotic treatment and at the indicated times after. Cells were pelleted by centrifugation for 5 min and immediately flash-frozen in liquid nitrogen. The cell pellets were thawed on ice, resuspended in lysis buffer (20 mM Tris pH 7.0, 10 mM MgCl2, 1 mM EDTA, 1 mg/ml lysozyme, 10 µg/ml DNase I, 100 µg/ml RNase A, 1 mM PMSF, 1 µg/ml leupeptin, 1 µg/ml pepstatin), and the suspensions transferred to fresh microfuge tubes to avoid CwlO present in culture medium that non-specifically bound to the plastic tube (Brunet et al., 2019). The lysates were then analyzed by immunoblot as described above.
CwlO∆cc purification
Request a detailed protocolRecombinant CwlO lacking its N-terminal coiled coil domain (Δcc) was expressed in E. coli BL21(DE3) ΔfhuA (New England Biolabs) using the PT7-His6-SUMO-cwlOΔcc expression vector (pJM63). Cells were grown in LB supplemented with 100 µg/mL ampicillin at 37°C to OD600 = 0.5. Cultures were allowed to equilibrate at room temperature for 30 min and then transferred to 30°C. His6-SUMO-CwlOΔcc expression was induced with 0.5 mM IPTG for 3 hr. Cells were collected by centrifugation, resuspended in 50 mL Buffer A (20 mM Tris HCl pH 7.4, 500 mM NaCl, 20 mM Imidazole, and 2X complete protease inhibitor tablets (Roche), and stored at −80 °C. The cell suspension was thawed on ice and lysed by two passes through a cell disruptor (Constant Systems Ltd.) at 25,000 psi. The lysate was clarified by ultracentrifugation at 35,000 rpm for 30 min at 4°C. The supernatant was added to 1 mL Ni2+-NTA beads (Qiagen) and incubated for 1 hr at 4°C. The suspension was loaded into a 10 mL column (BioRad), washed twice with 4 mL Buffer A, and eluted with 2.5 mL Buffer B (20 mM Tris HCl pH 7.4, 500 mM NaCl, 300 mM Imidazole). 10 µL of purified His6-Ulp1 (1.25 mg/ml) was added to the eluate, and the mixture was dialyzed into storage buffer (20 mM Tris HCl pH 8, 100 mM NaCl, 10% glycerol) overnight at 4°C. The next morning 10 µL more His6-Ulp1 was added to the dialysate and incubated for 1 hr at 30°C. The dialysate was mixed with 1 mL of Ni2+-NTA beads for 1 hr at 4°C to remove free His6-Ulp1 and His6-SUMO. The suspension was loaded onto a column and the CwlOΔcc-containing flow-through was collected, aliquoted, and stored at −80°C.
B. subtilis sacculi preparation
Request a detailed protocolWild-type B. subtilis was grown in 500 mL LB at 37°C to an OD600 = 0.5. Cells were pelleted, re-suspended in 10 mL 0.1 M Tris HCl pH 7.5 with 2% SDS (wt/vol), and boiled for 1 hr. The sample was cooled to room temperature and incubated with Proteinase K Solution (Invitrogen) at a final concentration of 0.4 mg/mL at 50°C for 1 hr. Sacculi were pelleted at 20,000 x g and washed five times with 10 mL ddH2O until free of SDS. The sacculi were then subjected to acid hydrolysis by suspension in 10 mL 1 M HCl at 37°C for 4 hr. Sacculi were then pelleted and washed five times with 10 mL ddH2O. Sacculi were distributed into 10 × 1 mL aliquots and stored at −80°C.
Sacculi digestion
Request a detailed protocolB. subtilis sacculi aliquots were resuspended in 1.4 mL cleavage buffer (25 mM MES pH 5.5) and dispersed in a water-bath sonicator for 30 min. Purified CwlOΔcc (final concentration 0.13 mg/mL), LytA (Flores-Kim et al., 2019), final concentration 0.17 mg/mL), and mutanolysin (Sigma Aldrich, 50 units final) were separately added to sonicated sacculi and incubated overnight at 37°C. After the overnight incubation, insoluble material was pelleted at 20,000 g for 15 min and the soluble cleavage products were collected, lyophilized, heat-inactivated (100°C for 20 min), and stored at −80°C. Immediately prior to use, the lyopholized material was resuspended in 50 µL ddH2O. When indicated, CwlOΔcc was inactivated with 20 mM iodoacetamide at room temperature in darkness for 30 min prior to addition to sacculi and overnight incubation.
Assaying soluble cleavage products for inhibition of WalRK signaling
Request a detailed protocolCultures of exponentially growing cells were diluted to an OD600 = 0.01 in LB and grown for 1 hr at 37°C to OD600 ~0.05. 50 µL of soluble sacculi cleavage products (prepared as described above) were added to 450 µL of cells and were incubated at 37°C with aeration. Fluorescent images were acquired before and at indicated times after addition.
Strain and plasmid construction
Deletion strains
Request a detailed protocolInsertion-deletion mutants were from the Bacillus knock-out (BKE) collection (Koo et al., 2017) or were generated by isothermal assembly (Gibson, 2011) of PCR products followed by direct transformation into B. subtilis. All BKE mutants were back-crossed twice into B. subtilis PY79. All deletions were confirmed by PCR. Antibiotic cassette removal was performed using a temperature-sensitive plasmid that constitutively expresses Cre recombinase (Meeske et al., 2015).
The following oligonucleotide primers were used to make the indicated strains:
∆walRK::erm (oAM475-478); ∆sigI::kan (oYB213/214, oYB215/216); ∆walHI::tet (oGD137/150, oGD151/152); ∆rsgI::spec (oYB178/179, oYB180/181); ∆walH::erm (BKE collection); ∆walI::erm (BKE collection); ∆prkC::erm (BKE collection); Antibiotic cassettes were amplified with (oWX438/439)
Plasmid construction
pYB018 [His-SUMO-lytE(∆lysMx3) (amp)]
Request a detailed protocolpYB018 was generated in a 2-way ligation with an BamHI-XhoI PCR product containing lytEΔlysMx3 (amplified from PY79 genomic DNA using oligonucleotide primers oYB66 and oYB54) and pTD68 (Uehara et al., 2010). The resulting plasmid was sequence-confirmed.
pYB064 [ycgO::Pveg-(optRBS)-lacZ (erm)]
Request a detailed protocolpYB064 was generated in an isothermal assembly reaction (Gibson, 2011) with a PCR product containing Pveg-(optRBS)-lacZ (oligonucleotide primers oYB189 and oYB190 and bMR18 genomic DNA) and pER118 [ycgO::erm] cut with BamHI and EcoRI. pER118 is an ectopic integration vector for insertion at ycgO (E. Riley and D.Z.R. unpublished). The resulting plasmid was sequence-confirmed. pYB066 [ycgO::PlytE-(optRBS)-lacZ (erm)] pYB066 was generated in a 2-way ligation with an EcoRI-HindIII PCR product containing the lytE promoter (oligonucleotide primers oYB195 and oYB196 and PY79 genomic DNA) and pYB064. The resulting plasmid was sequence-confirmed.
pYB069 [amyE::PlytE-(optRBS)-lacZ (kan)]
Request a detailed protocolpYB069 was generated in a 2-way ligation with a BamHI-EcoRI fragment containing PlytE-(optRBS)-lacZ from pYB066 and pER82 [amyE::kan]. pER82 is an ectopic integration vector derived from pDG364 with the kan gene replacing the cat gene.
[yvbJ::Phyperspank-(optRBS)-lysM3-mepM(D,D-endo) (spec)]
Request a detailed protocolwas not able to be propagated in E. coli due to toxicity. Instead, the plasmid was cloned by isothermal assembly (as described below) and directly transformed into bYB952 [ΔlytE::kan yvbJ::cat], to generate strain bYB962 [∆lytE::kan yvbJ::Phyperspank-(optRBS)-lysM3-mepM(D,D-endo) (spec)]. The resulting construct was sequence-confirmed. The isothermal assembly reaction used to produce [yvbJ::Phyperspank-(optRBS)-lysM3-mepM(D,D-endo) (spec)] included (1) a PCR product containing the 5' end of the lytE gene encoding the N-terminal LysM domains (oligonucleotide primers oYB245 and oYB246 and PY79 genomic DNA); (2) a PCR product containing the 3' end of the mepM gene encoding the D,D-endopeptidase domain (oligonucleotide primers oYB247 and oYB248 and E. coli MG1655 genomic DNA) and (3) pMS052 [yvbJ::Phyperspank (spec)] cut by SpeI and SphI. pMS052 is an ectopic integration vector containing the Phyperspank promoter for insertions at the yvbJ locus (M. Stanley and D.Z.R., unpublished).
pYB083 [yhdG::Phyperspank-(optRBS)-lytE (cat)]
Request a detailed protocolpYB083 was generated in a 2-way isothermal assembly reaction with a PCR product containing the lytE gene with an optimized RBS (oligonucleotide primers oYB245 and oJM104 and PY79 genomic DNA) and pMS018 [yhdG::Phyperspank (cat)] cut by SpeI and SphI. pMS018 is an ectopic integration vector containing the Phyperspank promoter for insertions at the yvbJ locus (M. Stanley and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pYB087 [yvbJ::Phyperspank-(optRBS)-lytE (spec)]
Request a detailed protocolpYB087 was generated in a 2-way isothermal assembly reaction with a PCR product containing the lytE with an optimized RBS (oligonucleotide primers oYB245 and oJM104 and PY79 genomic DNA) and pMS052 [yvbJ::Phyperspank (spec)] cut by SpeI and SphI. The resulting plasmid was sequence-confirmed.
pYB095 [yvbJ::Phyperspank-(optRBS)-(lysM)3 (cat)]
Request a detailed protocolpYB095 was generated in a 2-way isothermal assembly reaction with a PCR product containing the 5' end of the lytE gene encoding the N-terminal LysM domains (oligonucleotide primers oYB245 and oYB257 and PY79 genomic DNA) and pYB092 [yvbJ::Phyperspank (cat)] cut with NheI and SpeI. pYB092 is an ectopic integration vector containing the Phyperspank promoter for insertions at the yvbJ locus. (Y.B and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pYB097 [yvbJ::Phyperspank-(optRBS)-(lysM)3-yrvJ(amidase) (cat)]
Request a detailed protocolpYB097 was generated in a 2-way isothermal assembly reaction with a PCR product containing the 3' end of the yrvJ gene encoding the amidase domain (oligonucleotide primers oYB260 and oYB261 and PY79 genomic DNA) and pYB095 [yvbJ::Phyperspank-(optRBS)-(lysM)3 (cat)] cut by NheI. The resulting plasmid was sequence-confirmed.
pYB098 [yvbJ::Phyperspank-(optRBS)-(lysM)3-lytH(L,D-endo) (cat)]
Request a detailed protocolpYB098 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the lytH gene encoding the L,D-endopeptidase domain (oligonucleotide primers oYB262 and oYB263 and PY79 genomic DNA) and pYB095 cut with NheI.
pYB099 [yvbJ::Phyperspank-(optRBS)-(lysM)3-cwlK(L,D-endo) (cat)]
Request a detailed protocolpYB099 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the cwlK gene encoding the L,D-endopeptidase domain (oligonucleotide primers oYB264 and oYB265 and PY79 genomic DNA) and pYB095 cut with NheI.
pYB100 [yvbJ::Phyperspank-(optRBS)-(lysM)3-lytF(D,L-endo) (cat)]
Request a detailed protocolpYB100 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the lytF gene encoding the D,L-endopeptidase domain (oligonucleotide primers oYB266 and oYB267 and PY79 genomic DNA) and pYB095 cut with NheI.
pYB101 [yvbJ::Phyperspank-(optRBS)-(lysM)3-cwlS(D,L-endo) (cat)]
Request a detailed protocolpYB101 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the cwlS gene encoding the D,L-endopeptidase domain (oligonucleotide primers oYB268 and oYB269 and PY79 genomic DNA) and pYB095 cut with NheI.
pYB102 [yvbJ::Phyperspank-(optRBS)-(lysM)3-mepS(D,D-endo) (cat)]
Request a detailed protocolpYB102 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the mepS gene encoding the D,D-endopeptidase domain (oligonucleotide primers oYB270 and oYB271 and E. coli MG1655 genomic DNA) and pYB095 cut with NheI.
pYB103 [yvbJ::Phyperspank-(optRBS)-(lysM)3-lytD(glucosaminidase) (cat)]
Request a detailed protocolpYB103 was generated in a 2-way isothermal assembly reaction with a PCR product containing 3' end of the lytD gene encoding the glucosaminidase domain (oligonucleotide primers oYB272 and oYB273 and PY79 genomic DNA) and pYB095 cut with NheI.
pYB114 [yvbJ::Pspank-(optRBS)-lytE (spec)]
Request a detailed protocolpYB114 was generated in a 2-way isothermal assembly reaction with a PCR product containing the lytE gene and optimized RBS (oligonucleotide primers oYB245 and oJM104 from PY79 genomic DNA) and pMS050 [yvbJ::Pspank (spec)] cut by SpeI and SphI. pMS050 is an ectopic integration vector containing the Pspank promoter for insertion at the yvbJ locus (M. Stanley and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pYB139 [ycgO::PyocH-(optRBS)-lacZ (erm)]
Request a detailed protocolpYB139 was generated in a 2-way ligation with an EcoRI-HindIII fragment containing the yocH promoter from pGD055 [amyE::PyocH-(optRBS)-venus (cat)] and pYB064 [ycgO::Pveg-(optRBS)-lacZ (erm)] cut with the same enzymes.
pYB140 [ycgO::PiseA-(optRBS)-lacZ (erm)]
Request a detailed protocolpYB140 was generated in a 2-way ligation with an EcoRI-HindIII fragment containing the iseA promoter from pGD056 [amyE::PiseA-(optRBS)-venus (cat)] and pYB064 cut with the same enzymes.
pYB142 [yhdG::PxylA-(nativeRBS)-lytE (kan])
Request a detailed protocolpYB142 was generated in a 2-way ligation with an EcoRI-BamHI fragment containing PxylA-(nativeRBS)-lytE from pYB061 [yvbJ:: PxylA-(nativeRBS)-lytE (cat)] (Brunet et al., 2019) and pCB059 [yhdG::Pspank (kan)] cut with the same enzymes. pCB059 is an ectopic integration vector for insertions at the yhdG locus (R. Barajas and D.Z.R., unpublished).
pYB159 [yvbJ::Phyperspank-(optRBS)-lytE(C247S) (spec)]
Request a detailed protocolpYB159 was generated in a 3-way isothermal assembly reaction. Two of the fragments contained the 5' and 3' regions of the lytE gene PCR amplified from B. subtilis PY79 genomic DNA using oYB245 and oGD165 and oGD166 and oJM104. Assembly of these two products generated the C247S mutation. The third fragment was pMS052 [yvbJ::Phyperspank (spec)] cut by SpeI and SphI. The resulting plasmid was sequence-confirmed.
pYB161 [yvbJ::lytE(WalR box2 mut) (spec)]
Request a detailed protocolpYB161 was generated in a 3-way isothermal assembly reaction. Two of the fragments contained the 5' and 3' regions of the lytE gene PCR amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oYB352 and oYB336, and oYB337 and oYB353. Assembly of these two products generated the WalR binding site box 2 mutations (WalR box2 mut). The third fragment was pCB043 [yvbJ::spec] cut with EcoRI and BamHI. pCB043 is an ectopic integration vector for insertions at the yvbJ locus (R. Barajas and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pYB162 [yvbJ::lytE (spec)]
Request a detailed protocolpYB162 was generated in a 2-way isothermal assembly reaction with a PCR product containing the lytE gene (amplified from PY79 genomic DNA with oligonucleotide primers oYB352 and oYB353) and pCB043 [yvbJ::spec] cut with EcoRI and BamHI. The resulting plasmid was sequence-confirmed.
pYB164 [ycgO::PlytE(walR box2 mut)-(optRBS)-lacZ (erm)]
Request a detailed protocolpYB164 was generated in a 2-way ligation with an EcoRI-HindIII PCR product containing PlytE(WalR box2-mut) (amplified from pYB161 [yvbJ::lytE(WalR box2 mut) (spec)] with oligonucleotide primers oYB195 and oYB196) and pYB064 [ycgO::Pveg-(optRBS)-lacZ (erm)] cut with the same enzymes. The resulting plasmid was sequence-confirmed. pYB169 [yhdG::PxylA-(optRBS)-walI (erm)] pYB169 was generated in a 2-way isothermal assembly reaction with a PCR product containing the walI gene with optimized RBS (amplified from PY79 genomic DNA with oligonucleotide primers oYB351 and oYB361) and pCB106 [yhdG::PxylA (erm)] cut with HindIII and XhoI. pCB106 is an ectopic integration vector containing the PxylA promoter for insertions at the yhdG locus (R. Barajas and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pYB170 [yhdG::PxylA-(optRBS)-walI∆ECD(36-280) (erm)]
Request a detailed protocolpYB170 was generated in a 2-way isothermal assembly reaction with a PCR product containing walI∆ECD(36-280) (amplified from PY79 genomic DNA with oligonucleotide primers oYB359 and oYB361) and pCB106 [yhdG::PxylA (erm)] cut with HindIII and XhoI. The resulting plasmid was sequence-confirmed.
pYB177 [amyE::PlytE-(optRBS)-venus (cat)]
Request a detailed protocolpYB177 was generated in 2-way isothermal assembly reaction with a PCR product containing the lytE promoter (amplified from PY79 genomic DNA with oligonucleotide primers oYB376 and oYB377) and pGD015 [amyE::(optRBS)-venus (cat)] cut with EcoRI. The resulting plasmid was sequence-confirmed.
pYB190 [ycgO::Phyperspank-(optRBS)-iseA (erm)]
Request a detailed protocolpYB190 was constructed in a 2-way ligation with a PCR product containing iseA (amplified from PY79 genomic DNA with oligonucleotide primers oYB398 and oYB399) cut with SpeI and HindIII, and pCB089 [ycgO::Phyperspank (erm)] cut with the same enzymes. pCB089 is an ectopic integration vector containing the Phyperspank promoter for insertions at the ycgO locus (R. Barajas and D.Z.R., unpublished). The resulting plasmid was sequence-confirmed.
pAM187 [yvbJ::Pspank-(nativeRBS)-walRK (spec)]
Request a detailed protocolpAM187 was constructed in a 2-way ligation with a PCR product containing the walR and walK genes (amplified from B. subtilis PY79 genomic DNA with oligonucleotide primers oAM473 and oAM474) cut with XmaI and SpeI and pMS050 [yvbJ::Pspank (spec)] cut with the same enzymes. The resulting plasmid was sequence-confirmed. pGD015 [amyE::(optRBS)-venus (cat)] pGD015 was constructed in a 2-way isothermal assembly reaction with a PCR product containing the venus gene and an optimized RBS (amplified from plasmid pLPT10 (kindly provided by Johan Paulsson) using oligonucleotide primers oGD59 and oGD60) and pDG364 [amyE::cat] cut with EcoRI and BamHI. The resulting plasmid was sequence-confirmed.
pGD018 [amyE::PiseA-(optRBS)-venus (cat)]
Request a detailed protocolpGD018 was created in a 2-way isothermal assembly reaction with a PCR product containing the iseA promoter (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD68 and oGD69) and cloned pGD015 [amyE::(optRBS)-venus (cat)] cut with EcoRI. The resulting plasmid was sequence-confirmed.
pGD021 [yhdG::PxylA-spoIVFB(E44Q)-gfp (kan)]
Request a detailed protocolpGD021 was constructed in a 2-way ligation with a HindIII-BamHI fragment containing spoIVFB(E44Q)-gfp from pKM260 [(ycgO::PIVF-spoIVF(E44Q)-gfp (erm)] and pMS033 [yhdG::PxylA (kan)] cut with the same enzymes. pMS033 is an ectopic integration vector with PxylA for insertions at the yhdG locus (M. Stanley and D.Z.R., unpublished). The resulting plasmid was sequence confirmed.
pGD022 [yhdG::PxylA-(optRBS)-mciZ (kan)]
Request a detailed protocolpGD022 was constructed in a 2-way ligation with a HindIII-BamHI fragment containing the mciZ gene from pRB099 [(yvbJ::PxylA-mciZ (erm)] and pMS033 [yhdG::PxylA (kan)] cut with the same enzymes. The resulting plasmid was sequence confirmed.
pGD055 [amyE::PyocH-(optRBS)-venus (cat)]
Request a detailed protocolpGD055 was constructed in a 2-way isothermal assembly reaction with a PCR product containing the yocH promoter (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD148 and oGD149) and pGD015 cut with EcoRI. The resulting plasmid was sequenced-confirmed. pGD061 [yhdG::PxylA-(optRBS)-mciZ (phleo)] pGD061 was constructed in a 2-way ligation with a HindIII-BamHI fragment containing the mciZ gene from pRB099 [(yvbJ::PxylA-mciZ (erm)] and pCB109 [yhdG::phleo] cut with the same enzymes. pCB109 is an ectopic integration vector for insertions in the yhdG locus (R. Barajas and D.Z.R., unpublished).
pGD062 [yhdG::walRK (kan)]
Request a detailed protocolpGD062 was constructed in a 2-way isothermal assembly reaction with a PCR product containing walRK (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD172 and oGD173) and pCB037 [yhdG::kan] cut with EcoRI and SpeI. pCB037 is an ectopic integration vector for insertions in the yhdG locus (R. Barajas and D.Z.R., unpublished). The plasmid was sequenced and used for the construction of pGD073.
pGD073 [yhdG::walRKHI (kan)]
Request a detailed protocolpGD073 was constructed in a 2-way isothermal assembly reaction using a PCR product containing walKHI (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD188 and oGD189) and pGD062 cut with XhoI. XhoI cuts in the middle of walK; this feature was used to insert walKHI into pGD062. The resulting plasmid was sequenced-confirmed.
pGD090 [yhdG::walRK(Δ44–167)-walHI (tet)]
Request a detailed protocolpGD090 was constructed in a 3-way isothermal assembly reaction with a PCR product containing walR-walK(1-43) (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD172 and oGD215); a PCR product containing walK(168-611)-walHI (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD214 and oGD189) and pCB036 [yhdG::tet] cut with EcoRI and XhoI. pCB036 is an ectopic integration vector for insertions in the yhdG locus (R. Barajas and D.Z.R., unpublished). The resulting plasmid was sequenced-confirmed.
pGD101 [ycgO::PxylA-(optRBS)-walH (kan)]
Request a detailed protocolpGD101 was constructed in a 2-way isothermal assembly reaction with a PCR product containing walH with an optimized RBS (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD227 and oGD228) and pCB136 [yhdG::PxylA (kan)] cut with XhoI and BamHI. pCB136 is an ectopic integration vector with a PxylA promoter for insertions into the yhdG locus (R. Barajas and D.Z.R., unpublished). The resulting plasmid was sequenced-confirmed.
pGD102 [ycgO::PxylA-(optRBS)-walHΔECD(61-455) (kan)]
Request a detailed protocolpGD102 was constructed in a 2-way isothermal assembly reaction with a PCR product containing walHΔECD(61-455) and an optimized RBS (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD227 and oGD229) and pCB136 cut with XhoI and BamHI. The resulting plasmid was sequenced-confirmed.
Bacterial two-hybrid plasmids
pGD115 [T25-walK]
Request a detailed protocolpGD115 was constructed in a 2-way ligation with a PCR product containing the walK gene (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD278 and oGD279) cut with XbaI and KpnI and pKT25 cut with the same enzymes. The resulting plasmid was sequenced-confirmed.
pGD118 [T18-walH]
Request a detailed protocolpGD118 was constructed in a 2-way ligation with a PCR product containing the walH gene (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD282 and oGD283) cut with XbaI and KpnI and pCH364 cut with the same enzymes. The resulting plasmid was sequenced-confirmed.
pGD120 [T18-walI]
Request a detailed protocolpGD120 was constructed in a 2-way ligation with a PCR product containing the walI gene (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD284 and oGD285) cut with XbaI and KpnI and pCH364 cut with the same enzymes. The resulting plasmid was sequenced-confirmed.
pGD122 [T18-walHI]
Request a detailed protocolpGD122 was constructed in a 2-way ligation with a PCR product containing the walHI genes (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oGD282 and oGD285) cut with XbaI and KpnI and pCH364 cut with the same enzymes. The resulting plasmid was sequenced-confirmed.
pGD123 [T25-walK(∆44–167)]
Request a detailed protocolpGD123 was constructed in a 2-way ligation with a PCR product containing the walK(∆44–167) gene (amplified with genomic DNA from bGD500 using oligonucleotide primers oGD278 and oGD279) cut with XbaI and KpnI and pKT25 cut with the same enzymes. The resulting plasmid was sequenced-confirmed.
pJM063 [PT7-SUMO-6xHis-cwlOΔcc]
Request a detailed protocolpJM063 was constructed in a 2-way isothermal assembly reaction with a PCR product containing the C-terminal domain of CwlO (cwlOΔcc, amino acids 334–473) (amplified from B. subtilis PY79 genomic DNA using oligonucleotide primers oJM117 and oJM320) and pTB146 (Bendezú et al., 2009) cut with SacI and BamHI. The resulting plasmid was sequenced-confirmed.
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files.
References
-
Effects of oxygen on biofilm formation and the AtlA autolysin of Streptococcus mutansJournal of Bacteriology 189:6293–6302.https://doi.org/10.1128/JB.00546-07
-
Two-Component signal transduction as a target for microbial Anti-Infective therapyAntimicrobial Agents and Chemotherapy 42:1529–1536.https://doi.org/10.1128/AAC.42.7.1529
-
LysM, a widely distributed protein motif for binding to (peptido)glycansMolecular Microbiology 68:838–847.https://doi.org/10.1111/j.1365-2958.2008.06211.x
-
Extracytoplasmic PAS-like domains are common in signal transduction proteinsJournal of Bacteriology 192:1156–1159.https://doi.org/10.1128/JB.01508-09
-
Tearing down the wall: peptidoglycan metabolism and the WalK/WalR (YycG/YycF) essential two-component systemAdvances in Experimental Medicine and Biology 631:214–228.https://doi.org/10.1007/978-0-387-78885-2_15
-
A two-component signal transduction system essential for growth of Bacillus subtilis: implications for anti-infective therapyJournal of Bacteriology 180:6375–6383.
-
Promoter selectivity of the Bacillus subtilis RNA polymerase A and H HoloenzymesJournal of Biochemistry 124:89–97.https://doi.org/10.1093/oxfordjournals.jbchem.a022102
-
Characterization of new L,D-endopeptidase gene product CwlK (previous YcdD) that hydrolyzes peptidoglycan in Bacillus subtilisMolecular Genetics and Genomics 278:371–383.https://doi.org/10.1007/s00438-007-0255-8
-
A role for the essential YycG sensor histidine kinase in sensing cell divisionMolecular Microbiology 79:503–522.https://doi.org/10.1111/j.1365-2958.2010.07464.x
-
Enzymatic assembly of overlapping DNA fragmentsMethods in Enzymology 498:349–361.https://doi.org/10.1016/B978-0-12-385120-8.00015-2
-
Novel antibacterial compounds specifically targeting the essential WalR response regulatorThe Journal of Antibiotics 63:127–134.https://doi.org/10.1038/ja.2010.4
-
Peptide inhibitor of cytokinesis during sporulation in Bacillus subtilisMolecular Microbiology 68:588–599.https://doi.org/10.1111/j.1365-2958.2008.06173.x
-
Ligand-binding PAS domains in a genomic, cellular, and structural contextAnnual Review of Microbiology 65:261–286.https://doi.org/10.1146/annurev-micro-121809-151631
-
Regulation of a new cell wall hydrolase gene, cwlF, which affects cell separation in Bacillus subtilisJournal of Bacteriology 180:2549–2555.
-
Bordatella pertussis adenylate cyclase: a toxin with multiple talentsTrends in Microbiology 7:172–176.https://doi.org/10.1016/S0966-842X(99)01468-7
-
The lytE gene of Bacillus subtilis 168 encodes a cell wall hydrolaseJournal of Bacteriology 180:749–752.
-
Role in cell permeability of an essential two-component system in Staphylococcus aureusJournal of Bacteriology 181:3666–3673.
-
BookExperiments in Molecular GeneticsCold Spring Harbor: Cold Spring Harbor Laboratory.
-
Peptidoglycan hydrolase LytF plays a role in cell separation with CwlF during vegetative growth of Bacillus subtilisJournal of Bacteriology 181:3178–3184.
-
An antibiotic-inducible cell wall-associated protein that protects Bacillus subtilis from autolysisJournal of Bacteriology 189:4671–4680.https://doi.org/10.1128/JB.00403-07
-
YycH regulates the activity of the essential YycFG two-component system in Bacillus subtilisJournal of Bacteriology 187:5419–5426.https://doi.org/10.1128/JB.187.15.5419-5426.2005
-
YycH and YycI interact to regulate the essential YycFG two-component system in Bacillus subtilisJournal of Bacteriology 189:3280–3289.https://doi.org/10.1128/JB.01936-06
-
Robust growth of Escherichia coliCurrent Biology 20:1099–1103.https://doi.org/10.1016/j.cub.2010.04.045
-
Condensin promotes the juxtaposition of DNA flanking its loading site in Bacillus subtilisGenes & Development 29:1661–1675.https://doi.org/10.1101/gad.265876.115
-
An intermolecular binding mechanism involving multiple LysM domains mediates carbohydrate recognition by an endopeptidaseActa Crystallographica Section D Biological Crystallography 71:592–605.https://doi.org/10.1107/S139900471402793X
-
Characterization of a new Bacillus subtilis peptidoglycan hydrolase gene, yvcE (named cwlO), and the enzymatic properties of its encoded proteinJournal of Bioscience and Bioengineering 98:174–181.https://doi.org/10.1016/S1389-1723(04)00262-2
Article and author information
Author details
Genevieve S Dobihal

Funding
National Institute of General Medical Sciences (GM086466)
- David Z Rudner
National Institute of General Medical Sciences (GM127399)
- David Z Rudner
National Institute of Allergy and Infectious Diseases (U19 AI109764)
- David Z Rudner
National Institute of Allergy and Infectious Diseases (F32AI36431)
- Josué Flores-Kim
European Molecular Biology Organization (Long-Term Fellowship)
- Yannick R Brunet
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank all members of the Bernhardt-Rudner super-group past and present for helpful advice, discussions, and encouragement; Paula Montero Llopis and the HMS Microscopy Resources on the North Quad (MicRoN) core for advice on microscopy; Jeff Meisner for the CwlO expression plasmid and Chris Sham for enzymatic characterization; Alex Meeske for plasmid construction; Johan Paulsson for plasmids; Hoong Chuin Lim for help with fluorescence quantification; James Hoch and Hendrik Szurmant for advice and reagents; Andrew Kruse and Michael Springer for helpful discussions. Support for this work comes from the National Institute of Health Grants GM086466, GM127399, U19 AI109764 (DZR). YRB was funded in part by an EMBO Long-Term Fellowship. JFK was funded in part National Institutes of Health Grant F32AI36431.
Copyright
© 2019, Dobihal et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,437
- views
-
- 820
- downloads
-
- 72
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 72
- citations for umbrella DOI https://doi.org/10.7554/eLife.52088
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Homeostatic control of cell wall hydrolysis by the WalRK two-component signaling pathway in Bacillus subtilis
eLife 8:e52088.
https://doi.org/10.7554/eLife.52088




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}