Molecular basis for the adaptive evolution of environment-sensing by H-NS proteins
- Department of Chemistry, The University of Vermont, United States
- King Abdullah University of Science and Technology (KAUST), Computational Bioscience Research Center (CBRC), Biological and Environmental Science and Engineering (BESE), Saudi Arabia
- King Abdullah University of Science and Technology (KAUST), Biological and Environmental Science and Engineering (BESE), Saudi Arabia
- Centre de Biochimie Structurale, CNRS, INSERM, Université de Montpellier, France
Abstract
The DNA-binding protein H-NS is a pleiotropic gene regulator in gram-negative bacteria. Through its capacity to sense temperature and other environmental factors, H-NS allows pathogens like Salmonella to adapt their gene expression to their presence inside or outside warm-blooded hosts. To investigate how this sensing mechanism may have evolved to fit different bacterial lifestyles, we compared H-NS orthologs from bacteria that infect humans, plants, and insects, and from bacteria that live on a deep-sea hypothermal vent. The combination of biophysical characterization, high-resolution proton-less nuclear magnetic resonance spectroscopy, and molecular simulations revealed, at an atomistic level, how the same general mechanism was adapted to specific habitats and lifestyles. In particular, we demonstrate how environment-sensing characteristics arise from specifically positioned intra- or intermolecular electrostatic interactions. Our integrative approach clarified the exact modus operandi for H-NS-mediated environmental sensing and suggested that this sensing mechanism resulted from the exaptation of an ancestral protein feature.
Introduction
The histone-like nucleoid-structuring (H-NS) protein is a central controller of the gene regulatory networks in enterobacteria (Williams and Rimsky, 1997). H-NS inhibits gene transcription by coating and/or condensing DNA; an environment-sensing mechanism allows H-NS to liberate these DNA regions for gene expression in response to physicochemical changes (Fang and Rimsky, 2008; Winardhi et al., 2015; Ali et al., 2012). H-NS preferentially binds to AT-rich sequences, which enables its dual role in (1) the organization of the bacterial chromosome and (2) the silencing of horizontally acquired foreign DNAs (Gordon et al., 2011; Landick et al., 2015; Lang et al., 2007; Navarre et al., 2007). The latter mechanism allows bacteria to assimilate foreign DNAs, which, however, are only expressed as a last resort in case of acute threats or stresses (Navarre et al., 2007). Thus, H-NS plays a crucial role in the adaptation, survivability, and antibiotic resistance of bacteria. Given the growing threat of multidrug resistance, H-NS has attracted increasing research interest, with a particular focus on elucidating the molecular mechanisms of adaptive evolution (Ali et al., 2014; Will et al., 2015; Higashi et al., 2016).
H-NS possesses two dimerization domains (site1, residues 1–44; site2, resides 52–82; the numbering of Salmonella typhimurium is adopted throughout the text) and a C-terminal DNA-binding domain (DNAbd, residues 93–137) that is connected through a flexible region (linker, residues 83–92) to site2 (Figure 1; Gordon et al., 2011; Shindo et al., 1995; Bloch et al., 2003; Arold et al., 2010; Gao et al., 2017). The combination of site1 ‘head-to-head’ dimers with site2 ‘tail-to-tail’ dimers allows H-NS to multimerize (Figure 1B). These H-NS multimers form a superhelix that recapitulates the structure of plectonemic DNA, offering a mechanism for a stable concerted DNA coating by H-NS that results in gene silencing (Arold et al., 2010). However, other modes of DNA association by H-NS were also proposed (Qin et al., 2019).
Figure 1
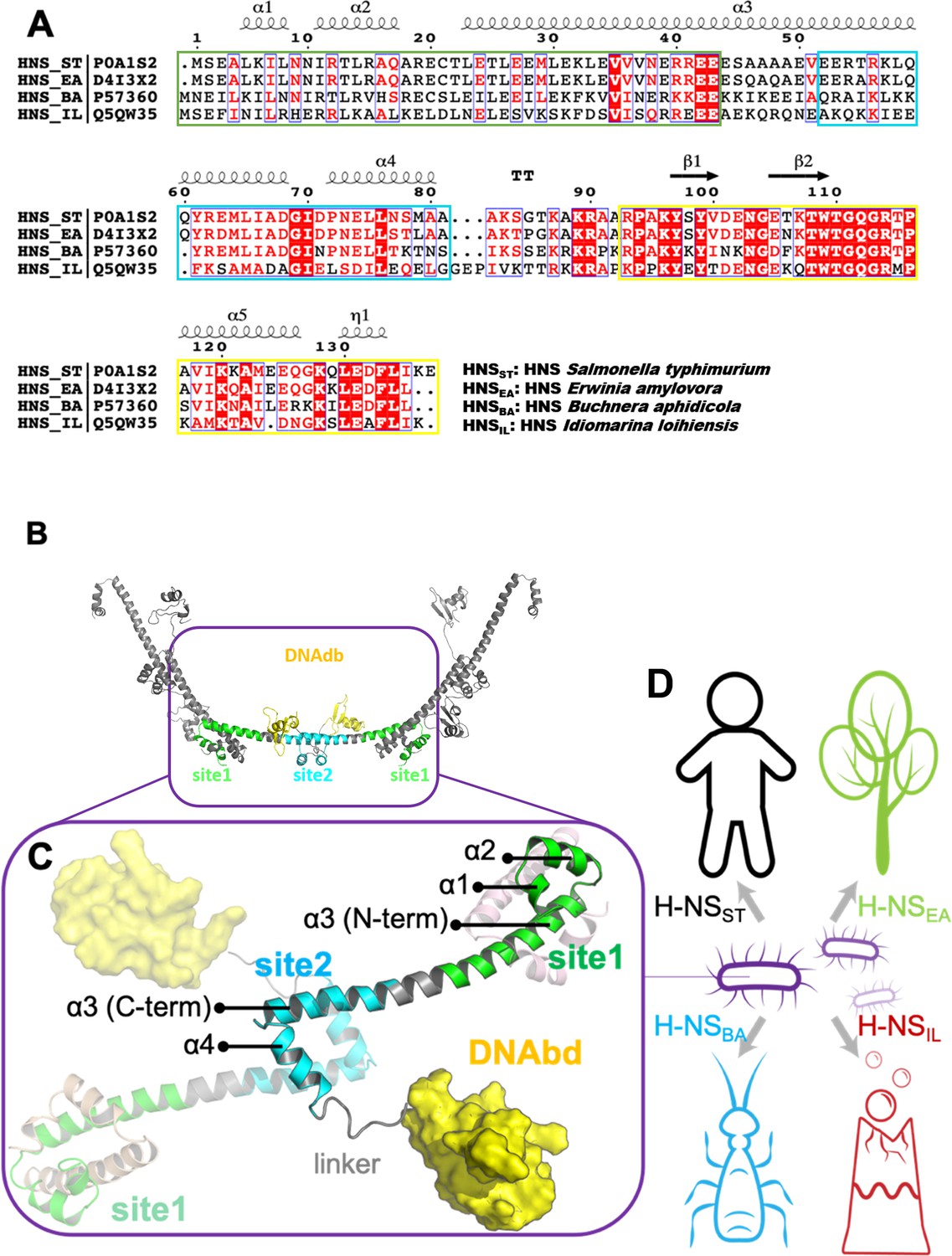
The sequence, structure, and habitat of selected histone-like nucleoid-structuring (H-NS) orthologs.
(A) Sequence alignment of H-NS orthologs (prepared with ESPript 3.0) (Robert and Gouet, 2014): H-NSST (Salmonella typhimurium; UniprotID: P0A1S2), H-NSEA (Erwinia amylovora; UniprotID: D4I3 × 2), H-NSBA (Buchnera aphidicola; UniprotID: P57360), and H-NSIL (Idiomarina loihiensis UniprotID: Q5QW35). The color scheme for sequence similarity is as follows: red background (identical in four orthologs) > red font (similar residues) > black font (totally different). Green, cyan, and yellow frames indicate the site1 (residues 1–44), site2 (resides 52–82), and DNAbd (residues 93–137). (B) Multimer formed by H-NSST. The model was built based on the available experimental structures (PDB accessions 3NR7 and 2L93). One H-NSST dimer shows site1, site2, and DNAbd colored as in (A) (green, cyan, and yellow, respectively). (C) The tetrameric H-NSST model we used for molecular dynamics (MD) is shown. The central dimer has each domain color-coded as in (A) and (B). (D) Illustration of the environment of the selected orthologs.
In a previous study, we showed that site2 of S. typhimurium H-NS is the primary response element to temperature changes (Shahul Hameed et al., 2019). Site2 unfolds at human body temperature, allowing the linker-DNAbd region to associate with site1 to adapt an autoinhibited conformation incapable of binding to DNA. Salinity and pH can also influence the stability of site2 dimers and hence may also affect gene repression by H-NS (Shahul Hameed et al., 2019; van der Valk et al., 2017). Thus, the sensitivity of H-NS to temperature and other physiochemical changes allows human pathogens such as S. typhimurium, Vibrio cholerae, and enterohaemorrhagic Escherichia coli to sense when they enter a homothermic host and adapt their gene expression profiles accordingly.
To date, studies to elucidate environment-sensing of H-NS were almost exclusively conducted with proteins from two model systems, S. typhimurium (e.g. Ali et al., 2014; Navarre et al., 2006; Ali et al., 2013; Hu et al., 2019) and E. coli (e.g. van der Valk et al., 2017; Oshima et al., 2006; White-Ziegler and Davis, 2009; Kahramanoglou et al., 2011; Ueda et al., 2013; Kotlajich et al., 2015), both of which infect humans. Yet, H-NS orthologs are also present in enterobacteria that do not have warm-blooded hosts, raising the question of what biological role H-NS plays in these species. Answering this question requires to determine the structural basis for environment-sensing in H-NS orthologs with drastically different lifestyles. However, the multidomain composition of H-NS hamper conventional structural analysis. Therefore, we combined large-scale molecular simulations and spectroscopic approaches to elucidate how environment-sensing by H-NS may have adapted in different species. This multidisciplinary approach yielded an atomic-level understanding of how H-NS orthologs evolved specific residue substitutions to adapt environment-sensing to their bacterial habitats, and may open new avenues for strategies to combat antibiotic resistance.
Results
To investigate the adaptation of environment-sensing by H-NS, we searched for representatives of H-NS–containing bacteria that have diverse lifestyles. Accordingly, we selected four H-NS orthologs from ~3000 hr-NS-like sequences available in the Uniprot database: (1) H-NSST from S. typhimurium. This bacterium is a pathogen of mammals and uses temperature-sensing to adapt to a presence inside the warm-blooded host. (2) H-NSEA from Erwinia amylovora, which is a plant pathogen that infects apples and pears. Hence, temperature is not a reliable differentiator between free-living and host-based states. (3) H-NSBA from Buchnera aphidicola. This bacterium is an obligate endosymbiont of aphids and has no free-living forms. (4) H-NSIL from Idiomarina loihiensis, which is a free-living bacterium from a deep-sea hydrothermal vent producing large heat gradients. H-NSEA and H-NSBA share more than 60% sequence identity with H-NSST, whereas H-NSIL is only 40% identical to H-NSST (Figure 1A, Supplementary file 1A).
The site1 dimer is markedly more stable than the site2 dimer in the H-NS orthologs
H-NSST site1 and site2 form homodimers to enable H-NS multimerization in a head-to-head/tail-to-tail fashion (Figure 1B; Arold et al., 2010). In concert with the DNA interaction of the individual domains, this homo-oligomerization is required for tight DNA binding and hence gene repression. In our previous study, we showed that only H-NSST site2 dimers unfold and dissociate within a biologically relevant temperature range, whereas site1 dimers remain unaffected (Shahul Hameed et al., 2019). The higher stability of the site1 dimer of H-NSST is explained by a substantially larger contact surface between the two monomers (ca. 3300 Å2 compared to ca. 850 Å2 for site1 and site2, respectively, according to PDBePISA [Krissinel and Henrick, 2007]).
To investigate whether this mechanism is conserved in other H-NS orthologs, we built homology models for H-NSEA, H-NSBA, and H-NSIL using the crystal structure of the H-NSST site1–site2 fragment as a template (PDB ID: 3NR7) (Arold et al., 2010). Next, we constructed a tetrameric model as a minimal representation that conserves all features of the H-NS multimer. This tetramer contained two full-length H-NS monomers (residues 1–137, with templates PDB IDs: 3NR7 and 2L93) and two partial monomers, truncated before site2 (residues 1–52) (Figure 1C). To probe differences in environmental responses of the orthologs, we first used conventional all-atom MD. We simulated all four tetramers (~100,000 atoms in each system; see Materials and methods) for 200 ns at three different conditions (0.15 M NaCl, 293 K; 0.50 M NaCl, 293 K/20°C; or 0.15 M NaCl, 313 K/40 °C) (Supplementary file 1B).
The tetramer simulations at 0.15 M NaCl and 20°C produced a lower residue fluctuation level in site1 (local root-mean-square fluctuation [RMSF] 0.4–1.9 Å) than in site2 (local RMSF 0.5–4.4 Å) for all four orthologs (Supplementary file 1C). The higher stability of the site1 dimer is explained by the generally higher number of nonpolar contacts than in the site2 dimer (Figure 2—figure supplement 1). These contacts involved conserved hydrophobic amino acid residues, notably L5 (or I5) and L8 of α1, L14 of α2, and L23, L26, V36, and V37 (or I37) of α3 (Figure 2). Our in silico mutant stability prediction analysis corroborated qualitatively the importance of hydrophobic residues for stabilizing the site1 dimer, in particular of L5, L8, L23, and L26 (Supplementary file 1D and 1E).
Figure 2 with 2 supplements see all
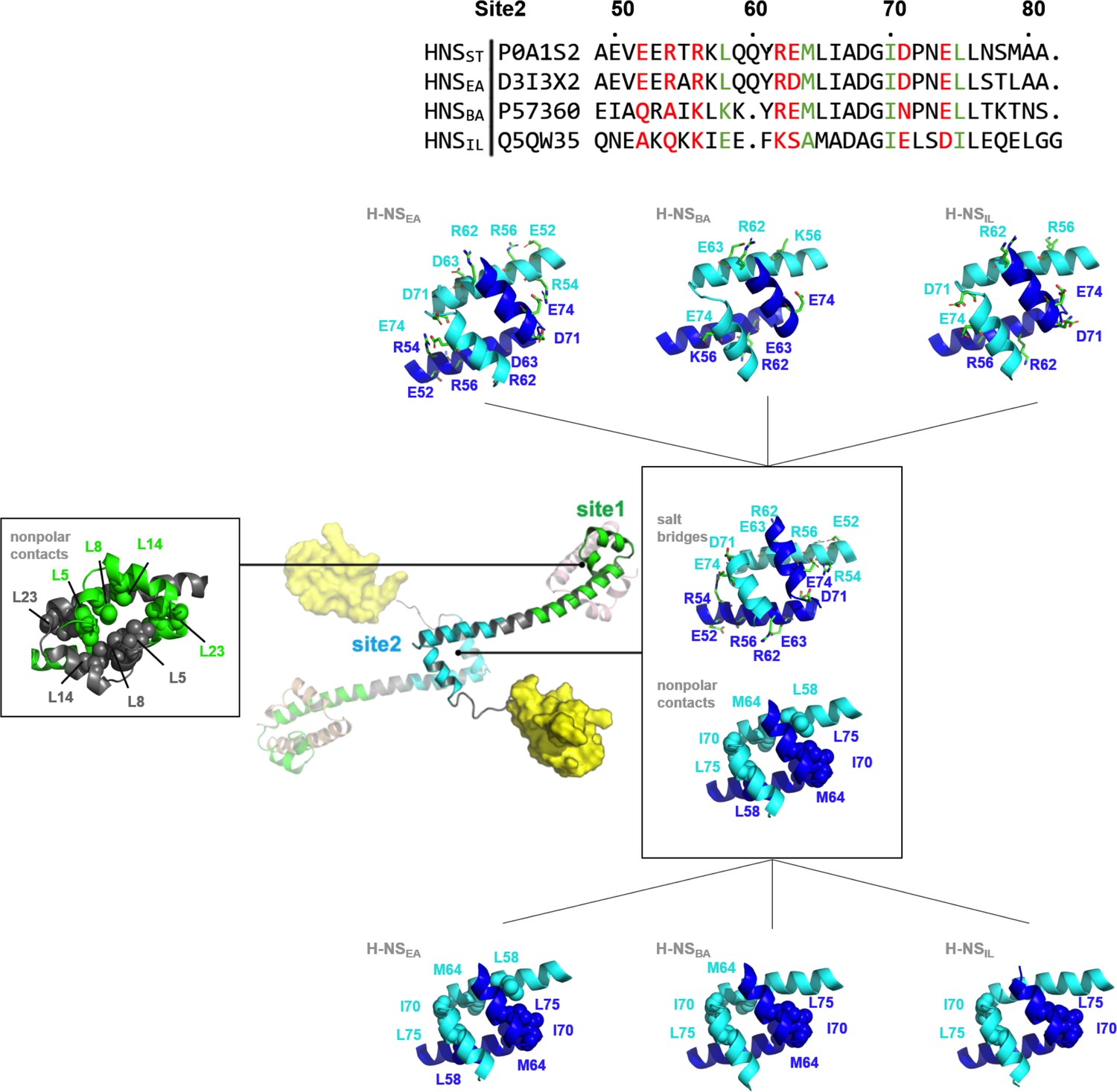
Nonpolar and ionic interactions in histone-like nucleoid-structuring (H-NS) site1 and site2 dimers.
Hydrophobic contact residues are shown as sphere models and polar contact residues as stick models. The two protein chains forming the dimer are color-coded. The site2 sequence alignment indicates in red the residue sites that potentially form salt bridges in H-NSST, and in green the residues that form nonpolar contacts in H-NSST. For additional details, see Figure S1.
These interactions remained formed in all site1 dimers in our tetramer simulations (at 0.15 M NaCl at 20°C) and tetramer simulations at higher salinity (at 0.50 M NaCl at 20°C) or higher temperature (at 0.15 M NaCl at 40°C). Hence, we found that the stability of the site1 dimers resulted mainly from strong and conserved nonpolar packing. Indeed, recombinantly expressed site1 fragments of all four orthologs formed ~15 kDa dimers in size exclusion chromatography–multi-angle light scattering. These dimers were stable within the temperature range relevant for environment-sensing (aggregation temperature, Tagg > 37 °C; Figure 2—figure supplement 2). We concluded that the mechanism observed for H-NSST – where site1 remains stable and the site2 stability is affected by the environment – is conserved in H-NSEA, H-NSBA, and H-NSIL.
Variations in the site2 sequence alter the sensing sensitivity of H-NS orthologs
Compared to site1 dimers, site2 dimers harbor fewer nonpolar contacts, only involving residues L58 (or I58), Y61 (or F61), M64 (or A64), I70, and L75 (or I75) (Figure 2). Hence, while site1 dimerization was largely maintained by nonpolar packing, site2 dimerization was strongly driven by electrostatic interactions from selective salt bridges. MD simulations revealed that these salt bridges were in a dynamic equilibrium between forming, breaking, and rearranging. These salt bridges were either formed in cis, within the site2 monomer, (e.g. E52-R56 and R62-E63 in H-NSST) or in trans, between two monomers in the site2 dimer (e.g. R54-D71’, R54-E74’, and K57-D68’ in H-NSST, where the apostrophe denotes residues from the second chain; illustrated in Figure 3). In addition to substitutions that delete (E52A in H-NSBA; E63S and D68A in H-NSIL) or weaken (E63Q in H-NSEA; D71N in H-NSBA) these salt bridges, our simulations showed different levels of site2 salt bridge stability among orthologs (Figure 3, Supplementary file 1F): (1) The inter-monomer salt bridge R/K54-E/D74’ was stable in all our simulations at 20°C and 0.15 M NaCl, but less likely to form at an increased temperature (40°C) or salinity (0.50 M NaCl), suggesting that this salt bridge is involved in environmental sensing (Figure 3A). (2) Absent in H-NSIL, the inter-monomer salt bridge K57-D68’ remained formed during all our simulations of H-NSST, H-NSEA, and H-NSBA, indicating a ‘housekeeping’ role for the stability of the site2 dimer in all orthologs except for H-NSIL (Figure 3B).
Figure 3 with 2 supplements see all
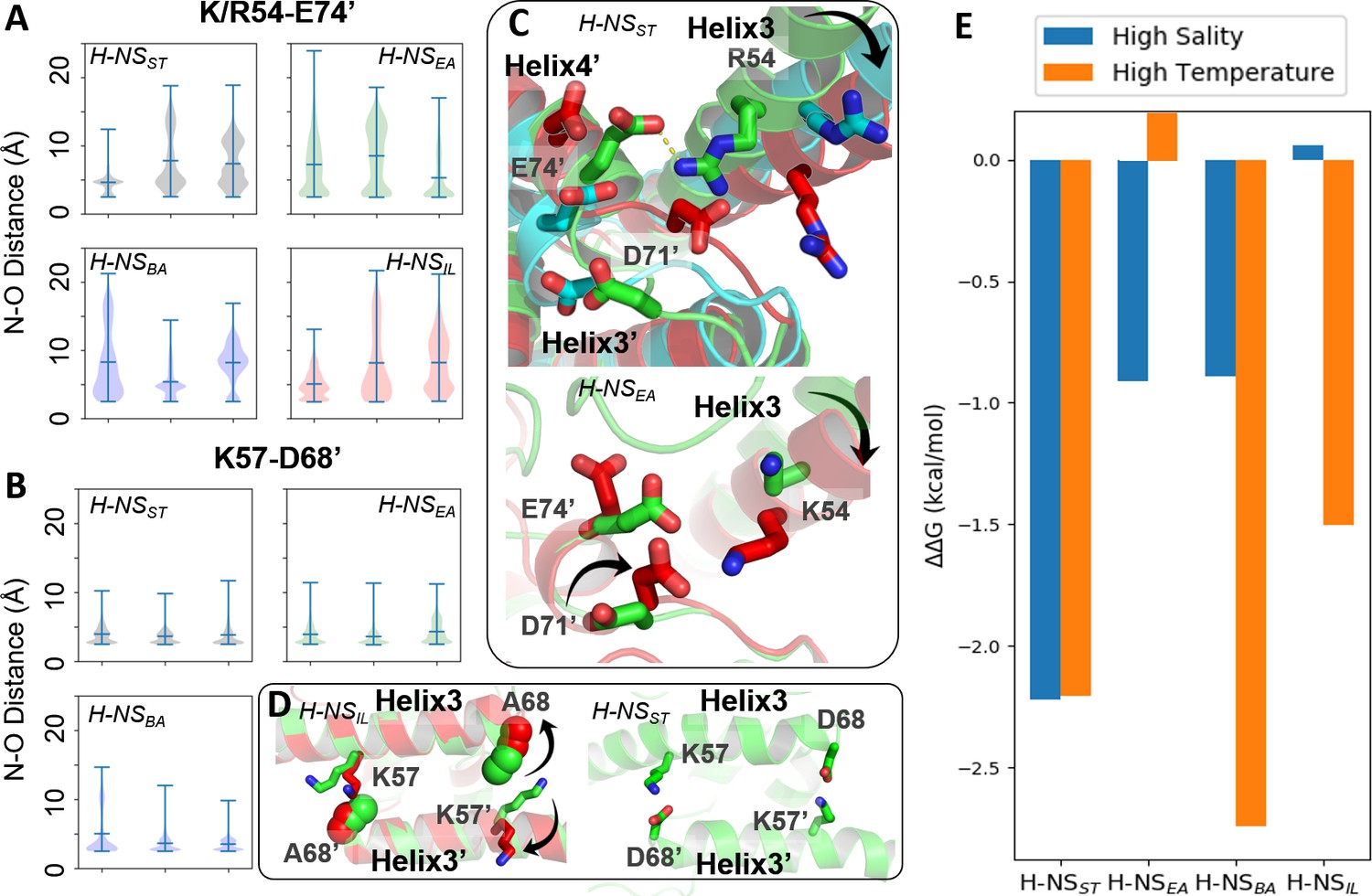
Conserved inter-dimer salt bridges observed in molecular dynamics (MD) simulations.
(A, B) Violin plots of the distance between the nearest side chain nitrogen atom of lysine/arginine and the side chain oxygen atom of aspartic/glutamic acid in the salt bridge. Each subplot shows the results obtained at 20°C, 0.15 M NaCl (left), 40°C, 0.15 M NaCl (middle), and 20°C, 0.50 M NaCl (right). Each ‘violin’ displays the mean value (the bar in the center of the violin), the range (the stretched line), and the distribution of the distance (kernel density on the side). As we use the numbering of H-NSST, there are position shifts in H-NSBA and H-NSIL: R54 to R53, K57 to K56, and D68 to D67 in H-NSBA; R54 to K53 in H-NSIL. (C) Final snapshots of the R54-E74’ salt bridge in H-NSST and K54-D71’ in H-NSEA. Color scheme of the cartoon: 20°C, 0.15 M NaCl (green), 40°C, 0.15 M NaCl (red), and 20°C, 0.50 M NaCl (cyan). (D) Final snapshots of the K57-D/A68 contact in H-NSST and H-NSIL. Same color scheme as (C). (E) Free energy changes as a result of increased salinity or temperature, according to the potential of mean force (PMF) calculated from umbrella sampling.
Our simulations show how specific protein dynamics might modulate the ortholog’s response to salinity or temperature. For example, we observed increased bending of the α3 backbone (annotated by the black arrow in Figure 3C) at high temperature (40°C) or high salinity (0.50 M NaCl) (Figure 3—figure supplement 1). Although α3 bending occurred in all orthologs, it only significantly affected the site2 dimer of H-NSST by separating R54 from E74’ or D71’, suggesting that this mechanism contributed to the salt and temperature sensitivity of H-NSST site2, whereas it was not strong enough to significantly affect site2 stability in other orthologs.
Another example was given by H-NSEA, where an alternative R54-D71’ salt bridge formed whenever the R54-E74’ contact was broken at 40°C. This alternative R54-D71’ salt bridge stabilized the H-NSEA site2 dimer at the higher temperature, suggesting that this compensatory mechanism resulted in a decreased sensitivity to temperature (Figure 3C). H-NSIL provided a final example for a specific response. Compared with the R54-E74’ salt bridge (Figure 3A), the K57-D68’ salt bridge only varied slightly in all our simulations (Figure 3B). However, the substitution D68A in H-NSIL supplanted the electrostatic interaction with a nonpolar interaction, which was broken at 40°C in our simulations (Figure 3D). This effect suggested that H-NSIL had a reduced sensitivity to salinity, while remaining sensitive to temperature.
To complement the dynamics of H-NS orthologs from our conventional MD simulations, we used extensive simulations with umbrella sampling (US) to quantitate the overall site2 stability. We calculated the potential of mean force (PMF) for site2 dimer dissociation (residues 50–82, ~46,000 atoms) of the four H-NS orthologs for three different conditions (low salinity/low temperature, high salinity, or high temperature). The site2 monomers were not constrained and remained structurally flexible during the dissociation process. To ensure convergence in the PMFs, we employed long windows (54 ns) in simulations totaling 52 µs (details provided in the SI; see Figure 3—figure supplement 2 for resulting histograms and PMFs along the dissociation coordinate). According to the free-energy difference between the dimerization and dissociation states (∆G = Gdimer − Gdissociation), we estimated the energetic impact from increased salinity and temperature as follows: ∆∆G = ∆Ghigh salinity or temperature− ∆G293K, 0.15M NaCl (Figure 3E). Notably, high salinity (0.50 M NaCl) or temperature (40°C) decreased the stability of the H-NSST site2 dimer by 2.2 kcal/mol. H-NSBA displayed a similar sensitivity to temperature but a lower sensitivity to salinity, which destabilized the dimer by 1.5 kcal/mol. Interestingly, our data indicated that H-NSEA was only sensitive to salinity, whereas raising the temperature had little impact on the stability of the H-NSEA site2 dimer. Conversely, H-NSIL only responded to temperature, whereas the increased salinity did not affect the stability of its site2 dimer (∆G ~ 0 kcal/mol). Collectively, our conventional MD simulations and PMF calculations suggested how, on the atomic level, changes in the site2 sequence may alter the sensitivity of the H-NS orthologs to different environmental changes.
The autoinhibited H-NS conformation is maintained through dynamic electrostatic interactions
In a previous study (Shahul Hameed et al., 2019), we had shown that melting and dissociation of site2 dimers allow H-NSST to adapt a closed conformation in which the linker-DNAbd fragment interacts with a negatively charged region on site1 α3 (Figure 4—figure supplement 1A) and that this autoinhibitory interaction is incompatible with DNA interactions. However, due to extensive signal broadening of mainly linker amides exchanging with water, our conventional proton-detected NMR analysis based on exchangeable amide H/N-observed correlations did not allow confident mapping of the binding site on the C-terminal region (Shahul Hameed et al., 2019; Figure 4—figure supplement 1B). Herein, we overcame this limitation by using proton-less 13C-detected NMR analysis to complete the resonance assignment of the linker-DNAbd fragment (Figure 4 and Figure 4—figure supplement 1C). These complete carbon chemical shifts allowed us to elucidate the structural mechanism of H-NSST autoinhibition fully and, in a second step, to use this understanding to investigate the existence of this closed conformation in the orthologs.
Figure 4 with 1 supplement see all
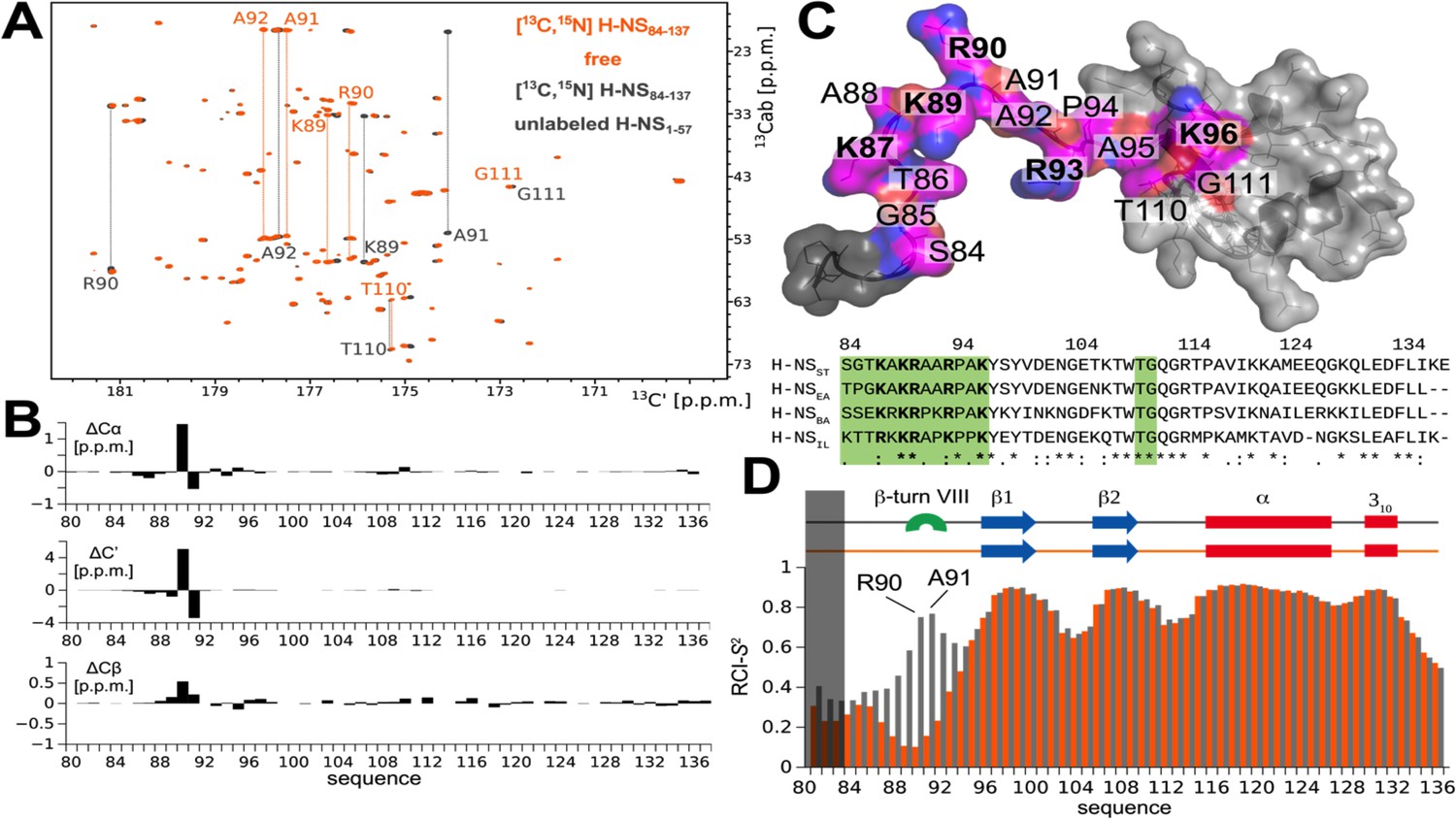
The atomistic details of histone-like nucleoid-structuring (H-NS) autoinhibition revealed by high-resolution proton-less low-γ detected NMR.
(A) The 2D CBCACO correlation 13C-detected spectra of 200 µM 13C,15N H-NSSTCt (orange) and 150 µM 13C,15N H-NSSTCt saturated 1:10 (molar) with unlabeled 1.5 mM H-NS1-57 (dark grey). Given a Kd of ~4 µM (Shahul Hameed et al., 2019) over 99% of H-NSSTCt are expected to be in the complexed form under these conditions. The OX axis holds all of the 13C,15N H-NSSTCt backbone C’ carbonyl chemical shifts correlated with OY (marked 13Cab) where each amino acid stripe crosses with its own Cα and Cβ (linked by thin dotted lines). (B) Top panel: The 13C chemical shift differences as a function of residue number of H-NS1-57 saturated 13C,15N H-NSSTCt and apo form. The most marked changes occur in residues K89, R90, A91, and A92 that predominantly form a β-turn type VIII. (C) Structural model of the H-NSSTCt in transparent surface representation revealing the backbone as ribbon. The structure is based on PDB ID 2L93, but extended N-terminally in random conformation to represent the full sequence of our construct in its apo form. All residues experiencing significant 13C chemical shift changes upon binding to H-NS1-57 are marked in magenta on the structure of the H-NSSTCt; positive residues (R+K) are labeled in bold. Bottom panel: The sequence alignment of the four selected HN-S orthologs highlighting conserved positively charged linker residues (bold) and residues implicated in binding to site1 (green). (D) Secondary structure motifs (red: helix; blue: β-sheet; green: β-turn, present only in the complex) and the RCI-S2 order parameter (describing the backbone dynamics) of ligand-free (orange) and saturated (gray) H-NSSTCt are shown.
We first titrated unlabeled H-NSST site1 (residues 1–57) onto the 13C,15N-labeled H-NSST C-terminal region (CtST, residues 84–137), comprising the linker (residues 84–93) and DNAbd (residues 94–137) (Figure 4A, Figure 4—figure supplement 1A). Motif identification from chemical shifts (MICS) (Shen and Bax, 2013) revealed that the interaction promoted the formation of a short type VIII β-turn in residues 89–92 (MICS confidence coefficient was 0.69). No significantly stable other motif besides the residual random coil was identified (Figure 4B,C). This sharp turn brings the positively charged linker side chains K87, K89, R90, R93 closer to each other than in the free state, presumably as a result of pairing them with opposite charges on site1 (Shahul Hameed et al., 2019; Figure 4B,C).
To further probe the local dynamics of the polypeptide chain, we determined the random-coil-index order parameter RCI-S2 based on the fully assigned 13C-resonances for each residue for the ligand-free and site1-saturated CtST. The dynamics of the well-ordered DNAbd domain remained unchanged with or without site1 present, in agreement with its only minor involvement in the autoassociation (Figure 4D). Conversely, the linker residues 84–95 were disordered without regular secondary motifs in the absence of site1 (RCI-S2 < 0.35). Upon addition of site1, the local dynamics decreased, particularly within the stretch of four amino acids K89-R90-A91-A92 (RCI-S2 > 0.6) that predominantly form the type VIII β-turn according to MICS. Nonetheless, the overall RCI-S2 of the linker remained low, although experimental conditions resulted in >99% of ligand saturation of the labeled CtST, demonstrating that the association with site1 did not substantially restrict the linker’s movements (Figure 4D).
Collectively, our analysis established that the autoinhibitory site1:CtST association was driven by oppositely charged residues located on site1 and the linker, and involved only a small region of the DNAbd. The center of this linker region, residues 89–92, rigidified upon binding and predominantly formed a β-turn conformation. However, the resulting intramolecular interaction was maintained through ‘fuzzy’ charge-pairing that did not fix the partners into a structurally stable complex.
Autoinhibition varies among H-NS orthologs
Having established the detailed autoinhibitory interactions between site1 and the Ct region in H-NSST, we next examined the H-NS orthologs. Based on our structural models (initial homology models and models from conventional MD), the electrostatic surface of the Ct was well conserved across all H-NS orthologs (Figure 5A). This level of conservation was expected, given that this region is also required for DNA association (Gao et al., 2017) – a role that needs to be conserved in all H-NS. Conversely, the site1 surface that binds to Ct was not conserved across all orthologs. While H-NSEA was similar to H-NSST in the overall charge distribution, α3 of H-NSBA showed a distinctly basic surface. H-NSIL displayed an intermediate electrostatic character, with features closer to H-NSST/H-NSEA (Figure 5A). These findings suggested that the stability of the closed conformation varies across orthologs. To test this prediction, we carried out in vitro binding experiments using microscale thermophoresis (MST).
Figure 5 with 1 supplement see all
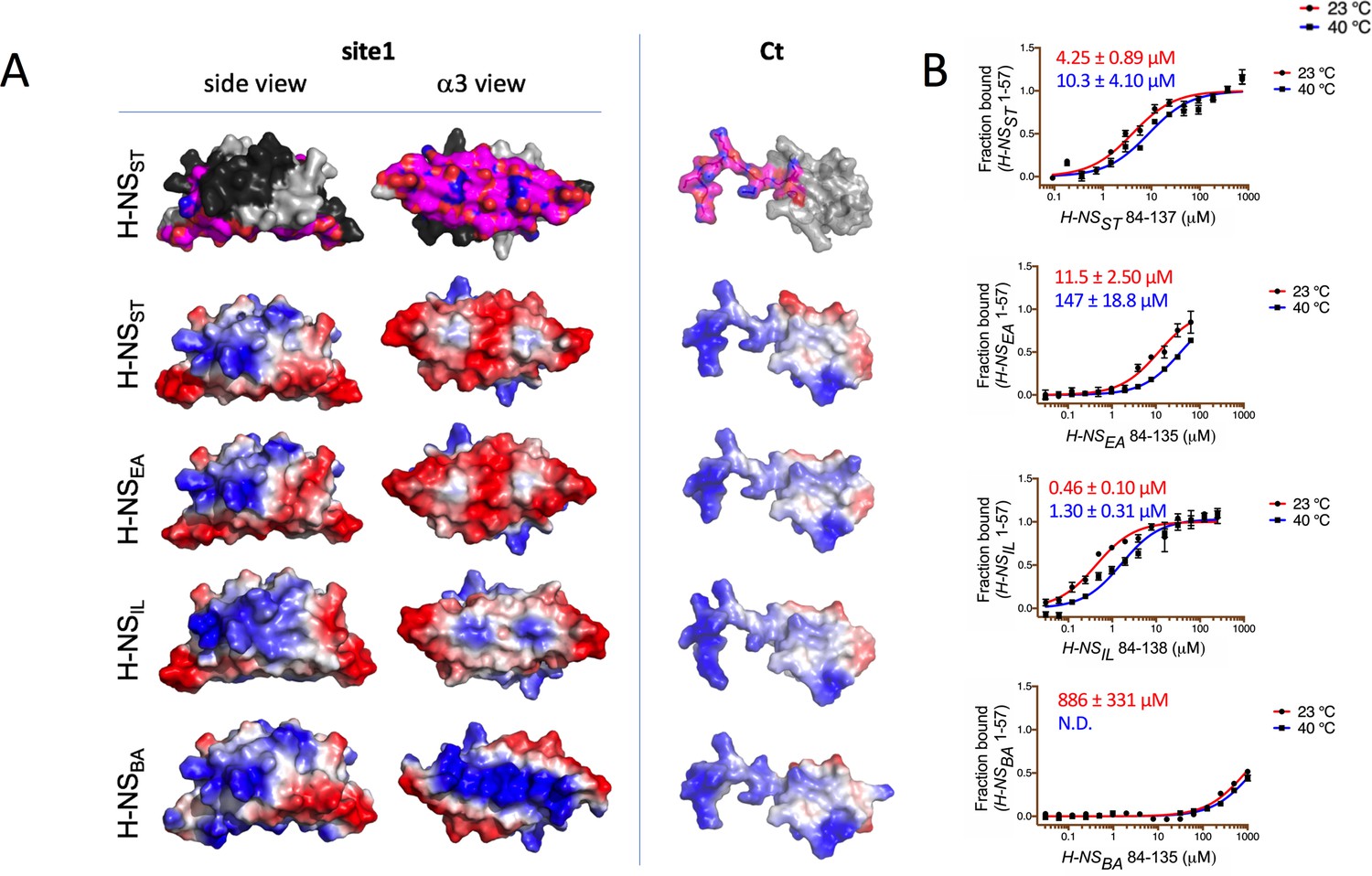
Modeling and in vitro analysis of autoinhibition and multimerization of histone-like nucleoid-structuring (H-NS) orthologs.
(A) Left and middle panels: Surface representation of site1 of H-NS orthologs (modeled on E. coli H-NS, PDB accession 1NI8) shown as side and α3 (bottom) view. Right panel: Ct, comprising the linker-DNAbd fragment, residues 84–137. The top row shows the residues mapped by NMR involved in forming the autoinhibitory closed conformation (Shahul Hameed et al., 2019 and this study). Other rows show the electrostatic surfaces, color-ramped from blue (positive) to red (negative) (calculated and visualized by Pymol). (B) Microscale thermophoresis (MST) titrations of unlabeled linker-DNAbd fragment onto 50 nM of Alexa-488-labeled H-NS 1-57 at 23°C (red) and 40°C (blue). The dissociation constant Kd is color-coded in red (23°C) and blue (40°C). N.D.: not determined.
These binding experiments between site1 and Ct confirmed that the strength of the autoassociation was similar for H-NSST and H-NSEA (Figure 5B). The autoassociation was 10-fold stronger in H-NSIL than in H-NSST, despite a less acidic site1. Hence, autoinhibition in H-NSIL might include additional and/or different interactions. Conversely, H-NSBA did not show a significant capacity for autoassociation, as expected from its markedly more basic site1 surface. In agreement, the double H-NSST site1 mutant E34K/E42K (designed to make the electrostatic site1 surface of H-NSST B. aphidicola like) dramatically lowered its affinity for H-NSSTCt (Figure 5—figure supplement 1). In addition to the reduced electrostatic complementarity, H-NSBACt also has a proline residue (P91) in position 3 of the β-turn region, which is highly unfavorable for this secondary structure element (Creighton, 1990). Indeed, at room temperature, H-NSBACt associated only very weakly with H-NSST site1 (Kd > mM), whereas the Ct of H-NSEA and H-NSIL bound to H-NSST site1 with a similar affinity than H-NSSTCt (Kds were 31.2 ± 3 µM and 18.2 ± 2 µM, respectively Figure 5—figure supplement 1A,B). Given that all four Cts display comparable electrostatic surfaces, the loss of affinity for H-NSBACt supported the importance of the β-turn. Increasing the temperature decreased the self-association strength 2- to 3-fold in H-NSST and H-NSIL and more than 10-fold in H-NSEA (Figure 5B). It also decreased the Kd for H-NSBA to values beyond the measurement range.
We concluded that the strength of the autoinhibitory conformation is mostly modulated by the electrostatic surface characteristics of site1. The Ct is constrained by the requirement to preserve the overlapping DNA binding surface, but can decrease autoinhibition by disrupting the β-turn conformation.
H-NS orthologs show adaptive features in vitro
We next experimentally assessed the response of the H-NS orthologs to physicochemical changes using dynamic light scattering (DLS). DLS provides the average hydration radius RH of the particles in solution, and hence gives a proxy for the tendency of H-NS molecules to form site2-mediated multimers or (still site1-linked) dimers. Thus, the RH is a convoluted signal of both effects, that is the relative strength of site2 multimerization and of the autoinhibitory conformation (if it exists). We measured the RH under different salt concentrations and temperatures. As reported previously, H-NSST showed a clear drop in RH from 10°C to 40 °C (Figure 6A; Shahul Hameed et al., 2019). The marked decrease of RH for curves at 0.15, 0.25, and 0.50 M NaCl indicated a strong inverse correlation between salinity and site2 stability, in agreement with a key role of salt bridges in stabilizing the site2 dimers.
Figure 6
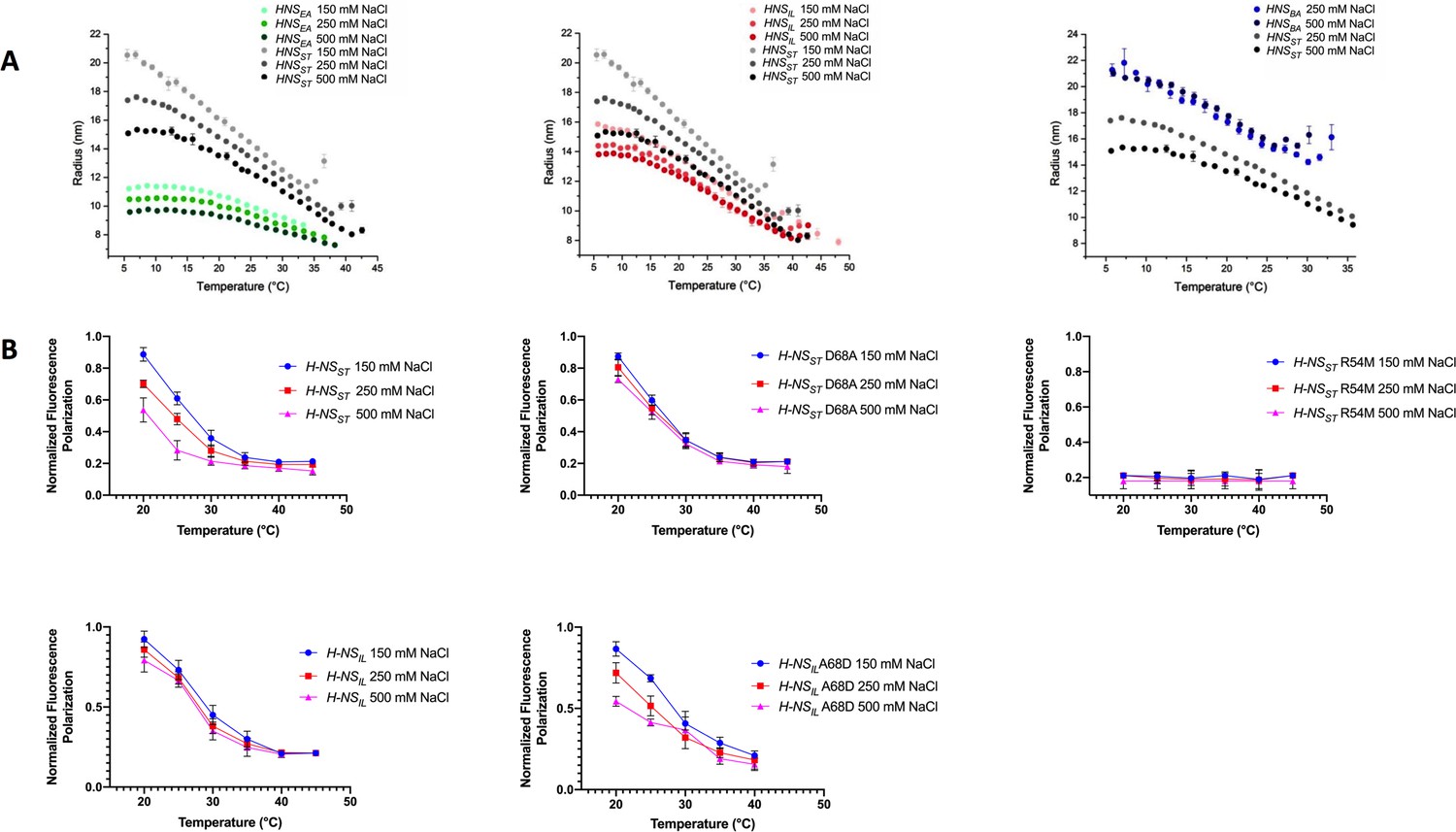
Temperature- and salt-dependent oligomerization of histone-like nucleoid-structuring (H-NS) proteins.
(A) Dynamic light scattering (DLS) experiments showing changes in hydrodynamic radius (as a proxy of apparent site2 stability) upon changes in salinity and temperature. Data in (B, D) are means ± S.D., n = 3. (B) Fluorescence anisotropy to determine oligomerization of H-NS site1–site2 (residues 1–83) and mutants at various salt concentrations. Unlabeled H-NSST (1–83) was titrated against Alexa-488-labeled H-NSST (1–83), D68A, and R54M mutants. Unlabeled H-NSIL (1–83) was titrated against Alexa-488-labeled H-NSIL (1–83) and A68D mutant.
All three H-NS orthologs displayed a similar behavior overall, further supporting that the general mechanism of site2-mediated multimerization and environment-sensing was preserved. However, we noted important differences in the orthologs’ response characteristics (Figure 6A): (1) Of the four orthologs, H-NSST responded most strongly to salinity and temperature, consistent with the broken R54-E74’ salt bridge and large site2 RMSF in our high-salinity or high-temperature simulations. (2) H-NSEA was less temperature sensitive and showed weaker multimerization than the other orthologs. Indeed, our simulations suggested that H-NSEA site2 can rearrange the inter-dimer salt bridge and form either R54-E74’ or R54-D71’ to maintain site2 stability at higher temperatures. (3) H-NSBA had the highest tendency to multimerize among all the orthologs tested, which might partly be explained by the absence of the autoinhibitory conformation. Compared to H-NSST, our PMF calculations showed a slightly higher sensitivity to temperature and a slightly reduced sensitivity to salinity. Although these tendencies were apparent in our DLS data, these data were also affected by the fact that H-NSBA required more than 150 mM NaCl to stay in solution, but H-NSBA nonetheless aggregated at 30°C. (4) H-NSIL showed a decreased sensitivity to salinity compared to H-NSST, as suggested by our computational analysis (i.e. the lack of the site2 K57-D68’ salt bridge, the lack of salt-promoted free-energy changes, and the attenuated electrostatic site1 surface).
To corroborate these conclusions, we designed several site2 mutants and tested their effect on protein multimerization using fluorescence anisotropy. To eliminate the influence of the autoinhibitory site1:Ct interaction, we used H-NS constructs that lacked the Ct (H-NS 1–83). The normalized fluorescence polarization (NFP) of H-NSST declined between 20°C and ~40°C to a value of 0.2, indicating that the average particle size decreased with temperature, as observed in DLS. And as in DLS, increases in salt lowered the NFP, and hence, the propensity of the particles to form site2-linked multimers (Figure 6B). The H-NSST R54M mutant, disrupting the ‘housekeeping’ salt bridges R54-E74’ and R54-D71’, displayed an NFP of 0.2 at all temperatures and salt concentrations. Thus, this mutant supported the key roles of the R54-mediated salt bridges in H-NS multimerization and environment-sensing. To experimentally assess the role of the K57-D68’ salt bridge in conveying salt sensitivity, we introduced the D68A ‘IL-like’ mutation in H-NSST, and the A68D ‘ST-like’ mutation in H-NSIL. These mutations abrogated salt sensitivity in H-NSST and introduced salt sensitivity in H-NSIL, as predicted by our computational analysis (Figure 6B).
Collectively, our experimental observations revealed significant differences in response to physicochemical parameters, which were in agreement with our predictions based on the molecular features of the H-NS orthologs.
Conclusion
Environment-sensing through the pleiotropic gene regulator H-NS helps S. typhimurium to adapt when it is present inside its host mammal. In a previous study, we had shown that an increase in temperature, and to some extent salinity, dissociates the second dimerization element (site2). Melting of site2 produces two effects: first, it impedes synergistic DNA binding of H-NS multimers, and second, it allows H-NS to adopt an autoinhibitory conformation where DNA binding residues on the C-terminal linker-DNAbd fragment (herein abbreviated as the Ct) associate with the N-terminal site1 dimerization domain (Shahul Hameed et al., 2019). In our current study, we confirmed that site2 is the element that senses changes in physicochemical parameters, and we uncovered additional aspects of this process. In particular, proton-less NMR fully revealed the position and dynamics of the Ct residues involved in the autoinhibitory association with site1. We also showed that the formation of a β-turn in the linker residues 89–91 is associated with the autoinhibited conformation. The Ct residues critical for autoinhibition cannot reach site1 without site2 dissociation (Figure 3—figure supplement 2B), confirming that the closed autoinhibited conformation is mutually exclusive with H-NS multimerization. Our NMR analysis also demonstrated that this autoinhibition is achieved at a low entropic cost, maintaining a high flexibility with respect to the exact distribution of the interacting charges on both site1 and the linker-DNAbd fragment. On the one hand, avoiding the entropic penalty helps the autoinhibitory interaction to prevail against the competing DNA association. (Of note, the covalent link between site1 and the Ct will enhance their local concentration and hence their apparent affinity compared to our measurement based on separate domains in Figure 5B.) On the other hand, the fuzziness of the charge–charge interactions facilitates preserving the capacity for autoinhibition during bacterial evolution and adaptation.
Based on our refined molecular understanding of S. typhimurium H-NS, we then investigated environment-sensing of H-NS orthologs from bacteria that infect plants, bacteria that are endosymbionts of insects, and bacteria that are presumably free-living in or close to a hydrothermal vent. Across all four orthologs, we observed a conceptually similar response to temperature and salt, both overall and on an atomic level, where salt bridges play key roles. This similarity suggests that environment-sensing in H-NS evolved by co-opting an ancestral feature, namely the relative instability of the simple site2 helix-turn-helix dimerization motif. However, marked idiosyncrasies in the response of H-NS orthologs suggest that this ancestral feature was then adapted to fit the current habitat and lifestyle. Thus, our analysis suggests that environment-sensing by H-NS originated from an exaptation followed by adaptation. Our combined computational and experimental structural analysis allowed us to relate the observed in vitro features of this adaptation to events on a residual level: in particular, the salt bridge disposition and stability of site2, and the strength of the autoinhibition governed mostly by the electrostatics of site1 helix α3.
Although other factors inside bacteria can modify the in vitro behavior of the isolated protein, it is interesting to consider these idiosyncrasies with respect to the bacteria’s habitats (Figure 7):
Figure 7
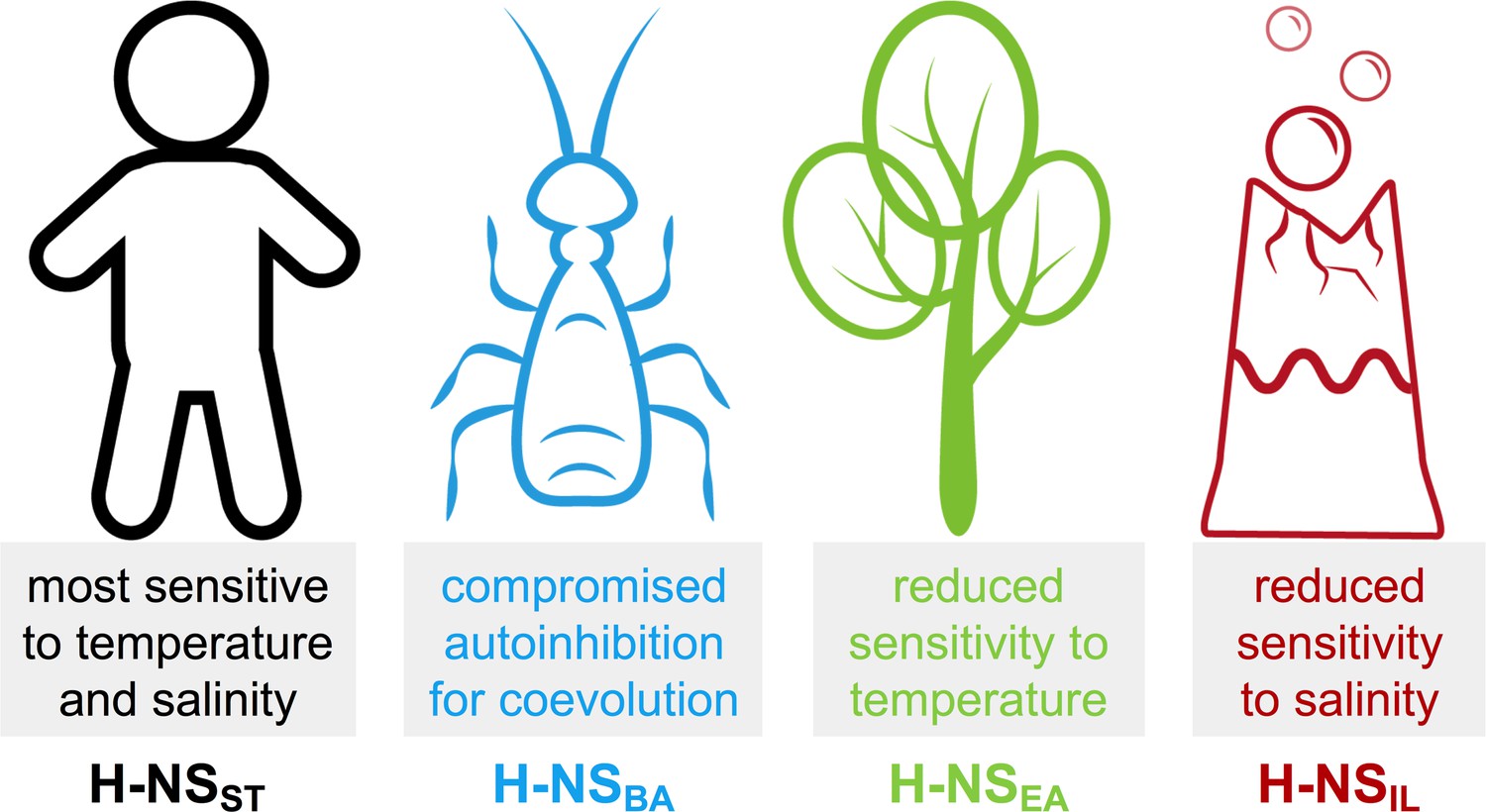
Summary of the most notable adaptations in environment-sensing observed for the histone-like nucleoid-structuring (H-NS) orthologs.
H-NSST had the highest sensitivity to temperature and salt, in agreement with the critical role of H-NSST in helping Salmonella adapt its gene expression profile depending on if it is inside or outside a warm-blooded mammal.
In comparison, we found that the response to temperature was markedly attenuated in H-NSEA. E. amylovora is the causing agent of fire blight, a contagious disease that mostly affects apples and pears (Vrancken et al., 2013). The reduced sensitivity of H-NS to temperature may reflect the minor importance of this factor in an environment of ambient temperature in temperate climate zones.
B. aphidicola is an intracellular symbiont of aphids that is maternally transmitted to the next generation via the ovaries (Douglas, 1998). B. aphidicola co-evolved with aphids for more than 150 million years, and despite having the highest sequence identity (61%) to H-NSST of all orthologs, H-NSBA showed the least conserved features among the orthologs tested, indicating that adaptive evolution was achieved by only minor changes. H-NSBA site2 interactions were stronger than those of other orthologs, and the features promoting the autoinhibitory form were compromised. Hence, H-NSBA may provide a stronger and more robust repression of the genes that it controls. In vitro, H-NSBA was the least stable ortholog tested and had already started to aggregate above 30°C, in agreement with the fact that B. aphidicola cannot survive temperatures of 35°C for extended periods.
Despite having a sequence identity least similar to H-NSST (41%), H-NSIL maintained an overall similar response profile. However, with an aggregation temperature of 45–50°C, H-NSIL was the most heat stable, especially at low pH and high salinity, as expected for a thermophilic and halophilic bacterium. Moreover, autoinhibition was 10-fold stronger than in Salmonella H-NS and relatively little affected by heat. The natural environment of I. loihiensis (hot hydrothermal fluids venting into cold seawater) provides a temperature range from 4°C to 163°C (Donachie et al., 2003), suggesting that temperature-sensing by H-NSIL is biologically relevant. The attenuated response of H-NSIL to salinity might reflect the capacity of I. loihiensis to grow in 20% (wt/vol) NaCl medium.
Our integrative approach provided atomistic insights on how residue-level substitutions on a protein support adaptation of organisms to different lifestyles.
Materials and methods
Using the CHARMM36 all-atom force field, we performed conventional MD simulations of H-NS tetramers and simulations with US enhanced sampling of site2 dimers in GROMACS. For DLS and MST, recombinant protein production and measurements were adapted from Shahul Hameed et al., 2019. However, we fluorescently labeled site1 for DLS, instead of the Ct. For NMR, 13C,15N-labeled S. typhimurium H-NS84-137 was expressed in minimal M9 media with 5 g/L of [U-13C] glucose and 1 g/L of 15NH4Cl salt. Proton and low-γ detected high-resolution NMR spectroscopy was carried out on a 700MHz Bruker Avance NEO spectrometer equipped with a 5 mm TXO cryogenic probe optimised for 15N and 13C direct detection at 25°C. Details are shown below.
Computational methods
Model preparation
Request a detailed protocolWe built our homology model of full-length H-NS (UniprotID: P0A1S2) based on orthologs in SwissModel (Arnold et al., 2006) with the templates for the dimerization domain (PDB ID: 3NR7) and the DNA-binding domain (PDB ID: 2L93). The site2 dimer models were initiated in an anti-parallel configuration, while the tetramer models were constructed according to the crystal packing (PDB ID: 3NR7). Maestro (Schrödinger, Inc) was used to construct the full-length model from different domains.
Simulation setup
Request a detailed protocolOur simulations were carried out by GROMACS (Wassenaar et al., 2013) (MD simulations of tetramers and PMF simulations of site2 dimers). All the models were solvated in a TIP3P water box, with counterions to neutralize the charges and additional NaCl for the desired salinity. Each tetramer system contains ~33,000 TIP3P water molecules, counter ions, and 150 or 500 mM NaCl, totaling ca. 100,000 atoms in a periodic box 13 × 9 × 9 nm3. All simulations were performed following a minimization, 250 ps equilibration in the NVT and NPT ensemble with Berendsen temperature and pressure coupling, and a production stage NPT (20 or 40°C, 1 bar). The CHARMM36 force field (Best et al., 2012) was used with the cmap correction. The particle mesh Ewald (PME) technique (Darden et al., 1993) was used for the electrostatic calculations. The van der Waals and short-range electrostatics were cut-off at 12.0 Å with switch at 10 Å.
The PMF simulations were carried out with the MD program GROMACS (Wassenaar et al., 2013) using the umbrella sampling technique. The CHARMM36 force field was also used. Each site2 monomer of the center of mass (COM) distance was chosen as the dissociation pathway and used for enhanced sampling. After 500 ps equilibrium with the NPT ensemble, initial structures for windows along the reaction coordinates were generated with steered MD. In the steered MD simulation, one chain was pulled away along the direction of increasing the COM distance with a force constant of k = 12 kcal mol−1 Å−2, until the COM distance reached 25 Å. The windows were taken within a range of 0–25 Å. The umbrella windows were optimized at the 0.3 Å interval to ensure sufficient overlap. There are about 80 windows per simulation, and each window was simulated with a force constant of 1.2 kcal mol−1 Å−2. All PMF simulations converged in 54 ns per window (Figure 3—figure supplement 2A). The helicity percentage of initial and final structures was measured for each window. For all windows, the helicity percentage was approximately 87.5% for the initial models and 85% for the final ones (Figure 3—figure supplement 2B).
Experimental methods
Protein production
Request a detailed protocolS. typhimurium H-NS1-57,C21S, H-NS1-57,C21S,E34K,E42K, H-NS1-83, C21S, H-NS1-83,C21S,D68A, H-NS1-83,C21S,R54F, H-NS84-137, and H-NS84-137,K89E,R90E were cloned and produced as described previously (Shahul Hameed et al., 2019). E. amylovora (H-NS1-57, H-NS82-134), B. aphidicola (H-NS1-57, H-NS84-135), and I. loihiensis (H-NS1-57, H-NS1-83, H-NS1-83,A68D, and H-NS85-138) genes were individually cloned into pGEX6P-1, expressed, and purified as described previously (Shahul Hameed et al., 2019). For the high-resolution nuclear magnetic resonance (NMR) studies, the uniformly double 13C,15N-labeled S. typhimurium H-NS84-137 (with additional GPLG residues before S84) was expressed in minimal M9 media with 5 g/L of [U-13C] glucose and 1 g/L of 15NH4Cl salt. The unlabeled S. typhimurium H-NS1-57 N-terminal domain was expressed and purified as described before (Shahul Hameed et al., 2019). The final NMR buffer was 50 mM NaCl, 2% (vol/vol) D2O, 20 mM Bis–Tris at pH 6.5%, and 0.002% NaN3.
Dynamic light scattering
Request a detailed protocolFor DLS measurements, H-NS from S. typhimurium, E. amylovora, B. aphidicola, and I. loihiensis were expressed as N-terminal mCherry fusion proteins with an N-term His tag in E. coli BL21 using the expression vector pET28b. The linker sequence SAGGSASGASG was inserted between mCherry and H-NS proteins to avoid steric clashes in the dimer. Bacteria were grown in LB medium, induced with 1 mM IPTG at 25°C overnight. Cells were harvested and resuspended in lysis buffer (50 mM Tris pH8, 500 mM NaCl, 10 mM Imidazole with addition of lysozyme, DNase I, and 1% triton X-100) and lysed by mild sonication. Proteins and bacterial membranes were separated by centrifugation (30 min, at 15,000 × g), and the supernatant was applied to Ni-NTA beads (Qiagen) for 2 hr. The column was washed thoroughly with 50 mM Tris pH8, 500 mM NaCl, 10 mM Imidazole, and protein was then eluted with 50 mM Tris pH8, 500 mM NaCl, 400 mM imidazole, and 1 mM dithiothreitol. After dialysis in 50 mM HEPES pH7.4, 300 mM NaCl, 0.5 mM TCEP, eluted protein was further purified by ion-exchange chromatography using either MonoQ or MonoS column (GE) in the same buffer. Protein multimerization was observed in the combination of different salt (150, 250, and 500 M NaCl) and pH (6, 7, and 8) conditions. For this, 100 mM MES, MOPS, and HEPES buffers were used, with proteins at concentrations ranging from 125 to 500 μM, in a final volume of 100 μL. Dynamic light scattering measurements were performed in 96-well plates (Greiner) using a DynaPro plate reader-II (Wyatt Technologies). A triplicate of three wells was measured for every sample with five acquisitions of 5 s for every well. The machine was cooled with gaseous nitrogen, with a starting temperature of 5℃, followed by an increase to 60℃ at a ramp rate set so that each well is measured every 1℃. Data were analyzed with DYNAMICS software (Wyatt Technologies) as temperature dependence and exported for further fitting on Origin software using a Logistic Fit. The presented results are mean values with standard error mean determined from the triplicate sample.
Proton and low-γ detected high-resolution NMR spectroscopy
Request a detailed protocolAll NMR measurements were done on 700 MHz Bruker Avance NEO spectrometer equipped with a 5 mm cryogenic TXO direct detection probe optimized for 15N and 13C direct detection at 25°C. The sequence-specific backbone resonance assignments of visible H/N correlations on 1H-detected spectra of S. typhimurium H-NS84-137 protein at 200 μM concentration in Apo and H-NS1-57-saturated (1.5 mM) forms were achieved with classical set of triple-resonance experiments, that is HNCA, HncoCA, HNCO, HNcaCO, HNCACB, CBCAcoNH (Sattler, 1999) and previously published assignments (Shindo et al., 1995). The 100% complete sets of Cα, Cβ, and C’ resonances for Apo and H-NS1-57-saturated (1.5 mM) forms covering the entire protein sequence, together with the residues not visible on H/N correlation 1H-detected experiments (due to amide exchange with water), were achieved with intra-residue 2D (H)CACO (c_hcaco_ia3d, 16 scans) and (H)CACBCO (c_hcbcaco_ia3d, 32 scans) supported with sequential (H)CANCO (c_hcanco_ia3d, 96 scans) 13C-detected experiments (Gray et al., 2012). The low-γ, 13C-detected experiments mentioned above were started with 1H-excitation in order to enhance the sensitivity and recorded in in-phase and anti-phase mode for the virtual decoupling. All spectra were processed in NMRpipe and analyzed in CARA and Sparky software. The random-coil-index order parameters RCI-S2 and secondary motifs, like β-turn, for Apo and H-NS1-57-saturated (1.5 mM) forms were determined from complete lists of Cα, Cβ (except glycines), N, C’ chemical shifts with the TalosN and MICS programs, respectively. The significance of chemical shift perturbations (CSPs) was established as follows: We calculated the combined CSP of backbone 13C atoms, that is CSP = sqrt(ΔσCα^2 + ΔσCO^2), and selected the residues that are above the median+1.5*IQR (interquartile range) as the cut-off (1.5*IQR corresponds to ~2.7*S.D.)
MST for protein–protein interactions
Request a detailed protocolH-NS (residues 1–57) from S. typhimurium and its double mutant (E34K, E42K), E. amylovora, B. aphidicola, and I. loihiensis were individually labeled N-terminally with fluorescent Alexa-488-TFP (Thermo Scientific) and then unlabeled C-term of those proteins and S. typhimurium double mutant K89E, R90E were titrated against Alexa-488-labeled N-term correspondingly and the final results were plotted as described previously (Shahul Hameed et al., 2019).
Fluorescence anisotropy to determine protein oligomerization
Request a detailed protocolH-NSST 1-83,C21S and H-NSIL 1-83 were N-terminally labeled with Alexa-488-TFP (Thermo Scientific) (Shahul Hameed et al., 2019). 200 µM of unlabeled H-NSST 1-83,C21S, H-NSST 1-83,C21S,D68A, H-NSST 1-83,C21S,R54F, H-NSIL 1-83, and H-NSIL 1-83, A68D were added to 1 µM of the corresponding labeled H-NS proteins with the final volume of 25 µL. The proteins were incubated for 30 min, and then measurements were recorded at a temperature ranging from 20°C to 45°C with intervals of 5°C. Measurements were using black/clear 384 well plate (Corning) with PHERAstar FS microplate reader (BMG Labtech) installed with a fluorescence polarization filter. The excitation wavelength was 480 nm, and emission was 520 nm. Polarization with a gain of 40% was used to measure the initial fluorescence polarization of 1 µM protein. Polarization was calculated using MARS data analysis software (Pastor-Flores et al., 2020).
Data availability
NMR chemical shift assignments were deposited at the BMBR https://betadeposit.bmrb.wisc.edu/ with IDs 50239 and 50240.
References
-
Silencing of foreign DNA in BacteriaCurrent Opinion in Microbiology 15:175–181.https://doi.org/10.1016/j.mib.2011.12.014
-
Structural Insights into the Regulation of Foreign Genes in Salmonella by the Hha/H-NS ComplexJournal of Biological Chemistry 288:13356–13369.https://doi.org/10.1074/jbc.M113.455378
-
Silencing by H-NS potentiated the evolution of SalmonellaPLOS Pathogens 10:e1004500.https://doi.org/10.1371/journal.ppat.1004500
-
Optimization of the additive CHARMM all-atom protein force field targeting improved sampling of the backbone φ, ψ and side-chain χ(1) and χ(2) dihedral anglesJournal of Chemical Theory and Computation 8:3257–3273.https://doi.org/10.1021/ct300400x
-
The H-NS dimerization domain defines a new fold contributing to DNA recognitionNature Structural & Molecular Biology 10:212–218.https://doi.org/10.1038/nsb904
-
Particle mesh Ewald: An N ⋅log( N ) method for Ewald sums in large systemsThe Journal of Chemical Physics 98:10089–10092.https://doi.org/10.1063/1.464397
-
Idiomarina loihiensis sp. nov., a halophilic gamma-Proteobacterium from the Lō'ihi submarine volcano, Hawai'iInternational Journal of Systematic and Evolutionary Microbiology 53:1873–1879.https://doi.org/10.1099/ijs.0.02701-0
-
Nutritional interactions in insect-microbial symbioses: aphids and their symbiotic Bacteria buchneraAnnual Review of Entomology 43:17–37.https://doi.org/10.1146/annurev.ento.43.1.17
-
New insights into transcriptional regulation by H-NSCurrent Opinion in Microbiology 11:113–120.https://doi.org/10.1016/j.mib.2008.02.011
-
Detection of disordered regions in globular proteins using ¹³C-detected NMRProtein Science : A Publication of the Protein Society 21:1954–1960.https://doi.org/10.1002/pro.2174
-
Direct and indirect effects of H-NS and fis on global gene expression control in Escherichia coliNucleic Acids Research 39:2073–2091.https://doi.org/10.1093/nar/gkq934
-
Inference of macromolecular assemblies from crystalline stateJournal of Molecular Biology 372:774–797.https://doi.org/10.1016/j.jmb.2007.05.022
-
H-NS and RNA polymerase: a love-hate relationship?Current Opinion in Microbiology 24:53–59.https://doi.org/10.1016/j.mib.2015.01.009
-
Deciphering key features in protein structures with the new ENDscript serverNucleic Acids Research 42:W320–W324.https://doi.org/10.1093/nar/gku316
-
Heteronuclear multidimensional NMR experiments for the structure determination of proteins in solution employing pulsed field gradientsProgress in Nuclear Magnetic Resonance Spectroscopy 34:93–158.https://doi.org/10.1016/S0079-6565(98)00025-9
-
H-NS uses an autoinhibitory conformational switch for environment-controlled gene silencingNucleic Acids Research 47:2666–2680.https://doi.org/10.1093/nar/gky1299
-
Mixing MARTINI: electrostatic coupling in hybrid atomistic-coarse-grained biomolecular simulationsThe Journal of Physical Chemistry B 117:3516–3530.https://doi.org/10.1021/jp311533p
-
Genome-wide identification of H-NS-controlled, temperature-regulated genes in Escherichia coli K-12Journal of Bacteriology 191:1106–1110.https://doi.org/10.1128/JB.00599-08
-
Integrated circuits: how transcriptional silencing and counter-silencing facilitate bacterial evolutionCurrent Opinion in Microbiology 23:8–13.https://doi.org/10.1016/j.mib.2014.10.005
Article and author information
Author details
Umar F Shahul Hameed

Funding
King Abdullah University of Science and Technology (FCC/1/1976-25)
- Stefan T Arold
National Institutes of Health (R01GM129431)
- Jianing Li
King Abdullah University of Science and Technology (FCC/1/1976-21)
- Stefan T Arold
National Science Foundation (CAREER CHE-1945394)
- Jianing Li
- Xiaochuan Zhao
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Research by UH, VK, FH, AK, ML, LJ, and SA reported in this work was supported by the King Abdullah University of Science and Technology (KAUST) through the baseline fund and the Award No FCC/1/1976–25 from the Office of Sponsored Research (OSR). CL and JMR were partially supported by the National Institutes of Health award (R01GM129431 to JL). XZ and JL were partially supported by the National Science Foundation (CAREER CHE-1945394 to JL). We acknowledge support from the KAUST Bioscience and Imaging core laboratories and the computational resources from the Vermont Advanced Compute Core (VACC) and the Anton supercomputer in Pittsburgh Supercomputing Center (PSC), and thank M Cusack (KAUST Research Support Services) for editorial help.
Copyright
© 2021, Zhao et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,709
- views
-
- 261
- downloads
-
- 23
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 23
- citations for umbrella DOI https://doi.org/10.7554/eLife.57467
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Molecular basis for the adaptive evolution of environment-sensing by H-NS proteins
eLife 10:e57467.
https://doi.org/10.7554/eLife.57467




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}