The Drosophila Nab2 RNA binding protein inhibits m6A methylation and male-specific splicing of Sex lethal transcript in female neuronal tissue
- Department of Biology, Emory University, United States
- Department of Cell Biology Emory University School of Medicine, United States
- Graduate Program in Genetics and Molecular Biology, Emory University, United States
- Graduate Program in Biochemistry, Cell and Developmental Biology, Emory University, United States
- Emory Institutional Research and Academic Career Development Award (IRACDA), Fellowships in Research and Science Teaching (FIRST) Postdoctoral Fellowship, United States
- Department of Biochemistry, Duke University School of Medicine, United States
- Department of Neurobiology, Duke University School of Medicine, United States
Abstract
The Drosophila polyadenosine RNA binding protein Nab2, which is orthologous to a human protein lost in a form of inherited intellectual disability, controls adult locomotion, axon projection, dendritic arborization, and memory through a largely undefined set of target RNAs. Here, we show a specific role for Nab2 in regulating splicing of ~150 exons/introns in the head transcriptome and focus on retention of a male-specific exon in the sex determination factor Sex-lethal (Sxl) that is enriched in female neurons. Previous studies have revealed that this splicing event is regulated in females by N6-methyladenosine (m6A) modification by the Mettl3 complex. At a molecular level, Nab2 associates with Sxl pre-mRNA in neurons and limits Sxl m6A methylation at specific sites. In parallel, reducing expression of the Mettl3, Mettl3 complex components, or the m6A reader Ythdc1 rescues mutant phenotypes in Nab2 flies. Overall, these data identify Nab2 as an inhibitor of m6A methylation and imply significant overlap between Nab2 and Mettl3 regulated RNAs in neuronal tissue.
Editor's evaluation
This study provides important new insight into the function of Nab2 protein in regulating Sxl pre-mRNA splicing, a key developmental switch gene in Drosophila. By implicating Nab2 in the modulation RNA methylation on the Sxl transcript, the results illuminate the emerging role of methylation in the regulation of splicing. This proposed role for Nab2 in modulating m6A modification has implications for other systems.
https://doi.org/10.7554/eLife.64904.sa0Introduction
RNA binding proteins (RBPs) play important roles in guiding spatiotemporal patterns of gene expression that distinguish different cell types and tissues within organisms. There are an estimated ~1500 RBPs that distribute between the nucleus and cytoplasm (Gerstberger et al., 2014), and each has the potential to interact with RNAs to modulate post-transcriptional gene expression. Such regulation is particularly critical in highly specialized cells such as neurons (Conlon and Manley, 2017) where regulated alternative splicing of coding regions and 3′UTRs, cleavage/polyadenylation, trafficking, and local translation contribute to precise regulation of gene expression (Brinegar and Cooper, 2016). The critical roles of RBPs in neurons are highlighted by many studies that reveal the importance of this class of proteins in brain development and function (Darnell and Richter, 2012) and by the prevalence of human neurological diseases linked to mutations in genes encoding RBPs (Brinegar and Cooper, 2016). Many of these RBPs are ubiquitously expressed and play multiple roles in post-transcriptional regulation. Thus, defining the key neuronal functions of these proteins is critical to understanding both their fundamental roles and the links to disease.
Among the RBPs linked to human diseases are a group of proteins that bind with high affinity to polyadenosine RNAs, which are termed poly(A) RNA binding proteins or Pabs (Kelly et al., 2010). Functional studies of classical nuclear and cytoplasmic Pabs, which utilize RNA recognition motifs (RRMs) to recognize RNA, have uncovered diverse roles for these proteins in modulating mRNA stability, alternative cleavage, and polyadenylation and translation (Smith et al., 2014). A second, less well-studied, group of Pabs uses zinc-finger (ZnF) domains to bind target RNAs. Among these is the zinc finger Cys-Cys-Cys-His-type containing 14 (ZC3H14) protein, which binds with high affinity to poly(A) RNAs via a set of C-terminal tandem Cys-Cys-Cys-His type ZnF domains (Leung et al., 2009). ZC3H14 is broadly expressed in many tissues and cell types but mutations in the human ZC3H14 gene are associated with a heritable form of intellectual disability (Pak et al., 2011), implying an important requirement for this protein in the central nervous system.
ZC3H14 has well-conserved homologs in eukaryotes, including Saccharomyces cerevisiae Nuclear poly(A)-binding protein 2 (Nab2), Drosophila melanogaster Nab2, Caenorhabditis elegans SUT-2, and murine ZC3H14 (Fasken et al., 2019). Zygotic loss of ZC3H14 in mice and Drosophila impairs neuronal function (Pak et al., 2011; Roxas et al., 2017), while neuron-specific depletion of Drosophila Nab2 is sufficient to replicate these effects (Pak et al., 2011). Reciprocally, expression of human ZC3H14 in Nab2-deficient neurons rescues this defect, demonstrating a high degree of functional conservation between human ZC3H14 and Drosophila Nab2 (Kelly et al., 2014). Collectively, these data focus attention on what are critical, but poorly understood, molecular roles for ZC3H14/Nab2 proteins in neurons.
Neuronal ZC3H14/Nab2 can be divided into two pools: a nuclear pool that accounts for the majority of ZC3H14/Nab2 in the cell, and a small cytoplasmic pool of protein detected in mRNA ribonucleoprotein particles (mRNPs) of axons and dendrites (Leung et al., 2009; Roxas et al., 2017; Bienkowski et al., 2017). Depletion of both pools in Drosophila neurons causes defects in axon projection within the brain mushroom bodies (MBs) (Kelly et al., 2016), a pair of neuropil structures involved in olfactory learning and memory (Armstrong et al., 1998; Heisenberg, 2003), and excess branching of dendrites on peripheral sensory neurons (Corgiat et al., 2022). The Nab2 requirement in MBs is linked to a physical association between Nab2 and the Drosophila Fragile-X mental retardation protein homolog (Wan et al., 2000) in the neuronal cytoplasm and translational repression of shared Nab2-Fmr1 target RNAs (Bienkowski et al., 2017). Genetic data indicate that Nab2 limits dendritic branching through effects on the cytoplasmic planar cell polarity pathway (Corgiat et al., 2022). Despite these insights into a cytoplasmic functions of Nab2, a molecular role of the abundant pool of Nab2 protein in neuronal nuclei remains undefined.
Here, we employ a broad and an unbiased RNA sequencing approach to identify transcriptome-wide changes in the heads of Nab2 loss-of-function mutant flies. While the steady-state levels of most transcripts are not significantly changed, we find a striking effect on splicing of a subset of neuronal RNA transcripts. We focus our analysis on a well-characterized sex-specific alternative splicing event in the Sex-lethal (Sxl) transcript (Salz and Erickson, 2010; Förch and Valcárcel, 2003; Haussmann et al., 2016). Results reveal that Nab2 plays a novel role in regulating the alternative splicing of Sxl in a sex-specific manner. Recent work has revealed a role for m6A RNA methylation by the enzyme Mettl3 in modulating this splicing event (Lence et al., 2016; Kan et al., 2017). Similar to Mettl3, the requirement for Nab2 in alternative splicing of Sxl is only essential for neuronally enriched tissues. Genetic and biochemical experiments support a functional link between m6A methylation and Nab2 in which Nab2 limits m6A on target RNAs. These results demonstrate the role for Drosophila Nab2 in RNA alternative splicing as well as RNA methylation and sex determination in neurons.
Results
Nab2 loss affects levels and processing of a subset of RNAs in the transcriptome of the Drosophila head
To assess the role of Nab2 in regulating the central nervous system transcriptome, a high-throughput RNA sequencing (RNA-Seq) analysis was carried out in triplicate on Nab2 null mutant heads (Nab2ex3 imprecise excision of EP3716) (Pak et al., 2011) and isogenic control heads (Nab2pex41 precise excision of EP3716). To capture any sex-specific differences, heads were collected from both male and female flies of each genotype. Briefly, total RNA from 1-day-old adults was rRNA-depleted and used to generate stranded cDNA libraries that were sequenced (150 cycles) on a NextSeq 500 High Output Flow Cell. This generated a total of approximately 1.1 billion 75 base-pair (bp) paired-end reads (91 million/sample) that were mapped onto the Dmel6.17 release of the Drosophila genome using RNA STAR (Dobin et al., 2013). Read annotation and per-gene tabulation was conducted with featureCounts (Liao et al., 2014), and differential expression analysis was performed with DESeq2 (Love et al., 2014).
RNA sequencing reads across the Nab2 gene are almost completely eliminated in Nab2ex3 mutants, confirming the genetic background and integrity of this analysis pipeline (Figure 1—figure supplement 1). Principal component analysis (PCA) performed with DESeq2 output data confirms that the 12 RNA-seq datasets distribute into four clusters that diverge significantly from one another based on genotype (Nab2ex3 vs. Nab2pex41 control; PC1 58% variance) and sex (male vs. female; PC2 26% variance) (Figure 1A). The DESeq2 analysis detects 3799 and 1545 genes in females and males, respectively, that exhibit statistically significant differences in RNA abundance between Nab2ex3 and control (BH-adjusted p-value/false discovery rate [FDR] < 0.05) (Supplementary file 1). Comparison of fold-changes (Nab2ex3 vs. control) among these significantly different RNAs reveals a high degree of correlation in female vs. male samples (R = 0.79), particularly among RNAs whose levels are most elevated upon Nab2 loss (Figure 1B). Applying a twofold change cutoff (|log2[fold-change]| ≥ 1) trims these sets to 453 significantly changed RNAs in females (294 ‘up,’ 159 ‘down’) and 305 significantly changed RNAs in males (150 ‘up,’ 155 ‘down’) (Figure 1C), which merge into a combined set of 570 significantly affected RNAs that trend similarly in heatmap analysis of mutant vs. control samples (Figure 1D). A majority of the 453 affected ‘female’ RNAs are mRNAs (439) and the remaining are snoRNAs (8), snRNAs (1), pre-rRNAs (1), and tRNAs (4) (Figure 1E). A similar distribution occurs in male heads: a majority of the affected RNAs are mRNAs (297) and the remainder are snoRNAs (4), snRNAs (1), pre-rRNAs (1), and tRNAs (2) (Figure 1E). Overall, the number of significantly changed RNAs (|log2[fold-change]| ≥ 1 and FDR < 0.05) in Nab2ex3 female and male heads represents a small fraction of RNAs detected in heads (2.2 and 3.7% in males and females, respectively), suggesting that Nab2 normally contributes to RNA-specific regulatory mechanisms in Drosophila head tissue.
Figure 1 with 1 supplement see all
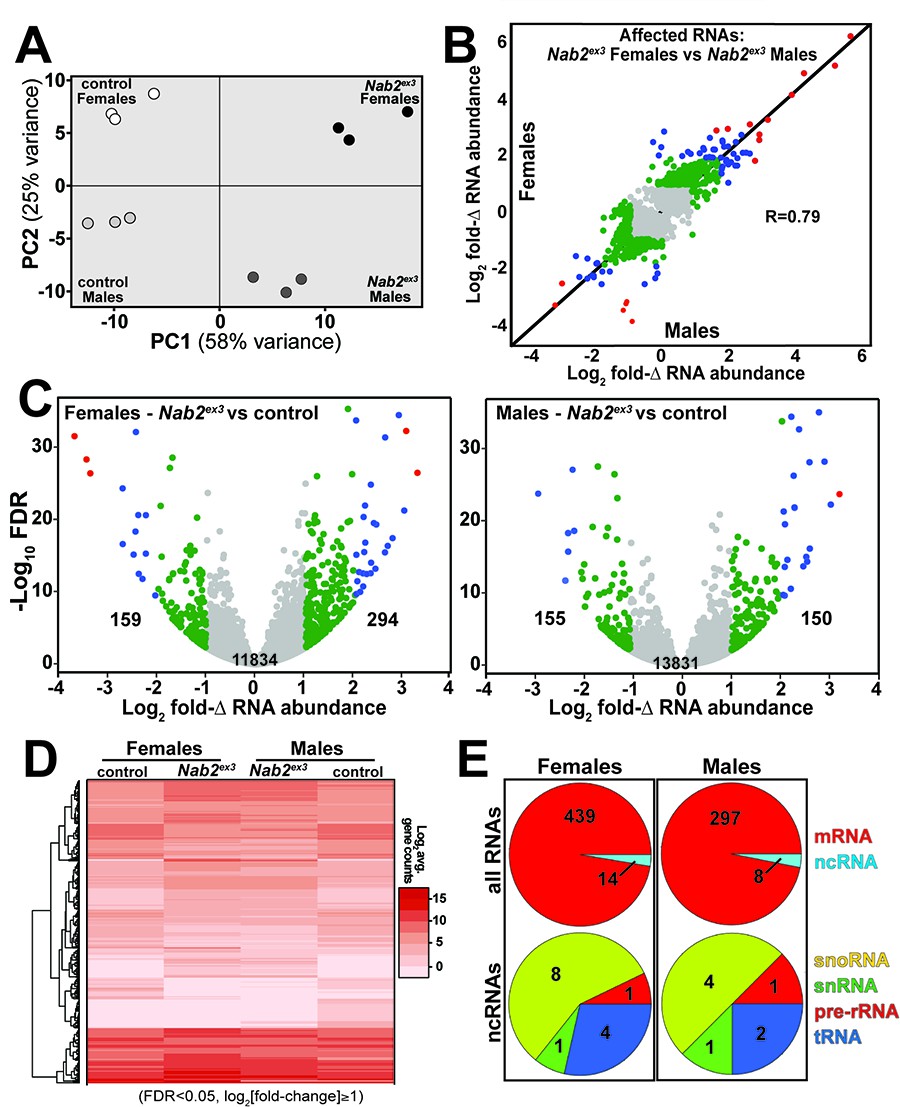
RNA sequencing detects effects of Nab2 loss on the head transcriptome.
(A) Principal component analysis (PCA) of RNA-seq data from three biological replicates of control and Nab2 mutant (Nab2ex3) male and female heads. (B) Correlation scatter plot of log2 fold change (Δ) in abundance of affected RNAs in males and females (log2 average gene counts: gray < 1, 1 ≤ green < 2, 2 ≤ blue < 3, red ≥ 3). (C) Volcano plots of fold-Δ in abundance vs. false discovery rate (FDR -log10) of affected RNAs in Nab2ex3 females and males (dot plot color coding as in B). Elevated (1), reduced (–1), and total RNAs are indicated. (D) Heatmap comparison of significantly changed gene counts (FDR < 0.05;|log2 fold-Δ| 1) in Nab2ex3 females and males vs. sex-matched controls. (E) Pie chart distribution of RNA classes among significantly affected RNAs detected in (C) and (D).
Nab2 loss alters levels of transcripts linked to mRNA processing
To identify functional groups within Nab2-regulated RNAs, Gene Set Enrichment Analysis (GSEA) (Mootha et al., 2003; Subramanian et al., 2005) was performed with the goal of defining enriched Gene Ontology (GO) terms (Ashburner et al., 2000; The Gene Ontology Consortium, 2019) among the significantly changed female and male RNAs identified by DESeq2. This filtering uncovers significant enrichment (p<0.05) for ‘RNA splicing’ GO (GO:0008380) within the upregulated group of RNAs in both sexes (Figure 2A). In Nab2ex3 females, 32 of 155 genes annotated under this term are present among upregulated RNAs; whereas in males, 75 of 159 genes annotated under this term are present among upregulated RNAs (Figure 2A). This enrichment for upregulated splicing-related factors indicates that Nab2 loss could shift splicing patterns in the adult head. Consistent with this hypothesis, mixture of isoforms (MISO) analysis (Katz et al., 2010) of annotated alternative splicing events confirms that Nab2 loss significantly alters splicing patterns within a small number of transcripts in female (48) and male (50) heads (Supplementary file 2) that fall into a variety of GO terms (Figure 2—figure supplement 1). These MISO-called alternative splicing events include 5′ and 3′ alternative-splice site usage, intron retention events, and previously annotated exon skipping events, some of which are detected in the same transcripts (Figure 2B).
Figure 2 with 1 supplement see all
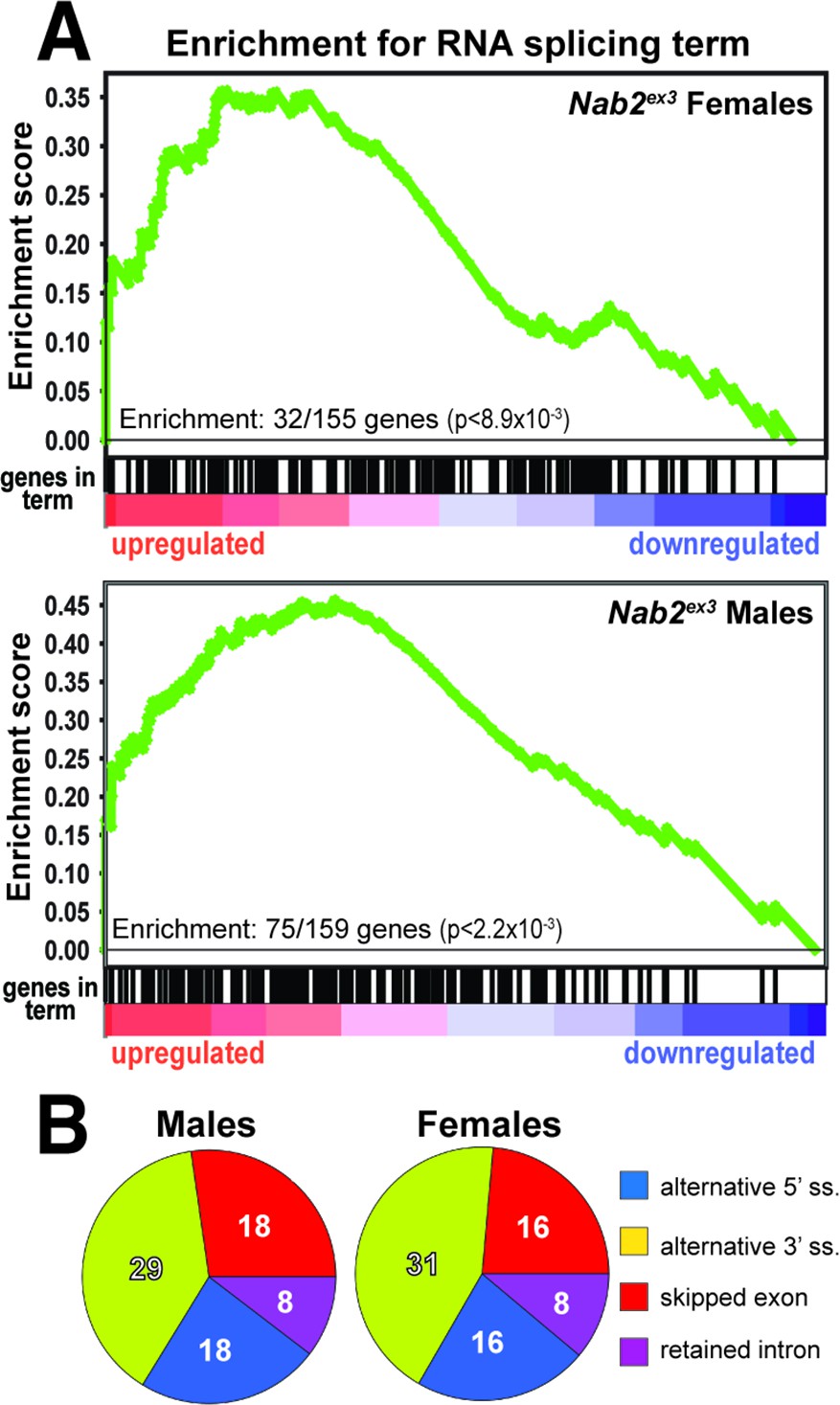
Significantly up-/downregulated RNAs in Nab2ex3 heads are enriched for predicated splicing factors.
(A) Gene Set Enrichment Analysis (GSEA) detects enrichment for the ‘RNA splicing’ Gene Ontology (GO) term in up- and downregulated gene sets in both female (top) and male (bottom) Nab2ex3 datasets. Gene enrichments are indicated with corresponding p-values. (B) Pie chart illustrating the distribution of previously annotated alternative splicing RNA splicing events that are significantly altered in Nab2ex3 mutant female and male heads (ss = splice site).
To test whether Nab2 loss results in unannotated or aberrant splicing events among head RNAs, DEXSeq analysis (Anders et al., 2012) was performed to scan for differential abundance of individual exons relative to other exons within the same transcript. This analysis detects 151 affected RNAs in Nab2ex3 females and 114 in Nab2ex3 males (Table 1), with many top-ranked transcripts encoding factors with roles in behavior, neurodevelopment, and/or neural function (Supplementary file 3). Reanalysis with a lower significance threshold yielded additional transcripts that show evidence of altered post-transcriptional processing in Nab2 mutant heads but did not alter the group of RNAs identified as most significantly affected by Nab2 loss. Among the 151 most affected RNAs, the most statistically significant exon usage change in either sex is female-specific inclusion of exon 3 in the Sex lethal (Sxl) mRNA (p=3.08 × 10–235). This effect on Sxl mRNA in Nab2ex3 females is followed in rank order of statistical significance by enhanced inclusion of exons 1 and 2 of the MIF4GD homolog transcript CG13124, exons 1 and 2 of the voltage-gated ion channel transcript Ih channel (Ih), and exon 1 of the synaptic enzyme transcript Acetylcholine esterase (Ace). In Nab2ex3 males, the top four events are enhanced inclusion of exon 1 of the Ace transcript, exon 1 of the Protein kinase C at 53E (Pkc53E) transcript, exons 1 and 2 of the Rab GTPase pollux (plx) transcript, and exons 1 and 2 of Protein kinase N (Pkn) transcript. In a number of cases, identical exons are affected in both Nab2ex3 sexes and accompanied by retention of the intervening intron (e.g., see CG13124 and Ih traces in Figure 3—figure supplement 1). The robust increase in Sxl exon 3 in Nab2ex3 females is noteworthy both for the central role that differential inclusion of Sxl exon 3 plays in Drosophila sex determination (Harrison, 2007) and because DEXSeq did not detect changes in exon 3 inclusion or abundance in Nab2ex3 males. In light of this sex-specific effect of Nab2 loss on alternative splicing of Sxl exon 3, subsequent analyses focused on the role of Nab2 in Sxl mRNA splicing in female heads.
Table 1
Alternative exon use (DexSeq) in Nab2ex3 head transcriptomes.
| Females | Males | |
|---|---|---|
| No. of alternatively used exons * | 151 | 114 |
| * |(exon usage fold change)| >~1.75 BH Adj. p<0.05 | ||
| Top mis-spliced transcripts | ||
| Females | foldΔ exon usage | BH adj. p-value |
| Sex lethal (Sxl) | 2.86 | 3.08 × 10–235 |
| CG13124 | 2.45 | 1.09 × 10–81 |
| Ih channel | 2.29 | 3.28 × 1063 |
| Ace | 1.81 | 1.02 × 10–59 |
| Males | foldΔ exon usage | BH adj. p-value |
| Ace | 2.02 | 1.88 × 10–169 |
| Pkc53E | 1.74 | 6.12 × 10–102 |
| plx | 2.12 | 9.03 × 10–67 |
| Pkn | 1.84 | 9.04 × 10–67 |
| Bacc | 2.31 | 1.11 × 10–64 |
-
Table 1—source data 1
Files (.bed format, openable as .excel tables) of splicing defects detected by lower threshold analysis than in Table 1.
Abbreviations: A3’SS, alternative 3′ splice site; A5’SS, alternative 5′ splice site; MEE, mutually exclusive exon; RI, retained intron; SE, skipped exon.
- https://cdn.elifesciences.org/articles/64904/elife-64904-table1-data1-v1.zip
Nab2ex3 females exhibit masculinized Sxl splicing in neuron-enriched tissues
The Sxl protein is a female-specific, U-rich RNA binding protein that is best defined for its role acting through the tra-dsx and msl-2 pathways to promote female somatic and germline identity (Penalva and Sánchez, 2003; Gawande et al., 2006). Sxl pre-mRNA is expressed in both males and females, but alternative splicing regulated by m6A RNA methylation and several RBPs leads to female-specific skipping of exon 3 during splicing (Haussmann et al., 2016; Lence et al., 2016; Sakamoto et al., 1992). Because exon 3 includes an in-frame translation ‘stop’ codon, full-length Sxl protein is only made and active in female cells (Bell et al., 1988). The inclusion of Sxl exon 3 in Nab2ex3 mutants would thus implicate Nab2 as a novel component of molecular machinery that controls Sxl pre-mRNA splicing.
Visualizing Sxl RNA-Seq reads with Integrative Genomics Viewer (IGV) (Robinson et al., 2017) confirms a large increase in exon 3 reads in Nab2ex3 females (Nab2ex3 F) relative to control females (control F), and also reveals retention of ~500 bases of intron 3 sequence in Nab2ex3 females (Figure 3A). Normal splicing patterns are detected across all other Sxl intron-exon junctions in both genotypes of males and females, including female-specific exon 9 inclusion. Quantification of reads across the entire Sxl locus detects an ~1.5-fold increase in the overall abundance of the Sxl mRNA in Nab2ex3 females compared to control females. Parallel reverse transcription polymerase chain reaction (RT-PCR) on fly heads using Sxl primers that detect exon 2-exon 4 (control females) and exon 2-exon 3-exon 4 (control males) confirms the presence of the mis-spliced exon 2-exon 3-exon 4 mRNA transcript in Nab2ex3 females (Figure 3B). The exon 2-exon 3-exon 4 mRNA transcript appears to be more abundant in Nab2ex3 female heads than in female heads lacking Mettl3, which encodes the catalytic component of the m6A methyltransferase complex that promotes exon 3 skipping in nervous system tissue (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017). RT-PCR also reveals an ~1 kb band in Nab2ex3 females (arrowhead, Figure 3B) that sequencing identifies as aberrantly spliced transcript that incorporates 503 bases of intron 3 leading up to a cryptic 5′ splice site (i.e., exon 2-exon 3-intron 3503-exon 4), which matches the Sxl intron 3 sequencing reads observed in IGV (see Figure 3A).
Figure 3 with 2 supplements see all
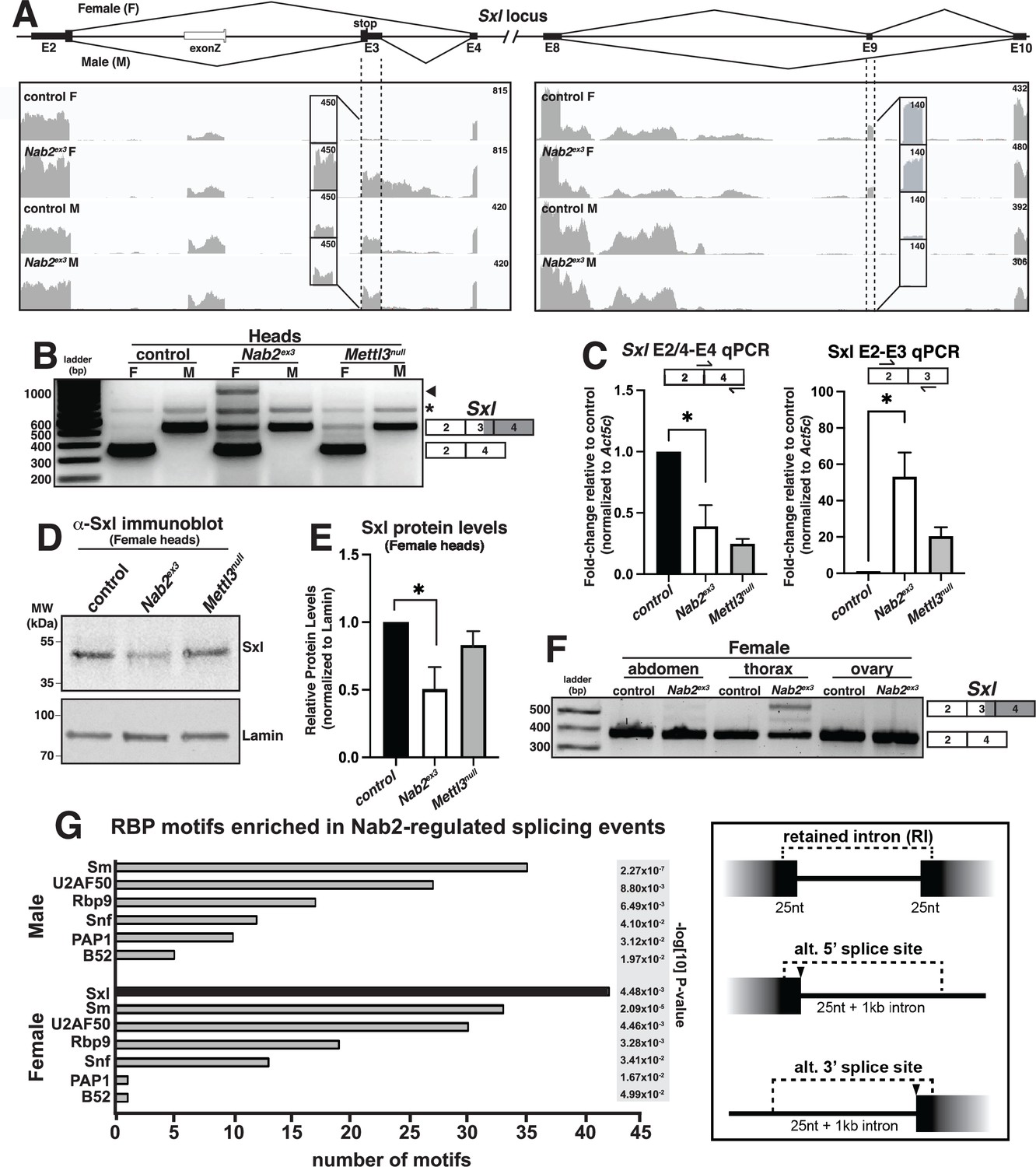
Sxl alternative splicing and protein levels are disrupted in Nab2ex3 female heads.
(A) Top panel: normal Sxl alternative splicing patterns across exon 2-4 and exon 8-10 regions in female (F) and male (M). Bottom panel: corresponding sequencing reads across the Sxl locus in the indicated sexes and genotypes. Dotted lines and boxed insets highlight exon 3 and exon 9 reads. (B) RT-PCR analysis of Sxl mRNA in control, Nab2ex3 and Mettl3null female (F) and male (M) heads. Exon 2-3-4 and exon 2-4 bands indicated. Arrowhead denotes exon 2-3-intron-4 product noted in text. Asterisk is nonspecific product. (C) RT-qPCR analysis of Sxl transcripts in adult female, control Nab2ex3, and Mettl3null heads using the indicated primer sets. Asterisk indicates results that are statistically significant at p-value<0.05. (D) Immunoblot of protein samples from control, Nab2ex3, and Mettl3null female heads. Antibody against female-specific Sxl protein isoform was used to detect Sxl in each sample. Lamin serves as a loading control. Molecular weights are given in kDa and indicated to the left. (E) Quantification of Sxl protein levels in (D) using ImageLab software. Protein levels are normalized to control, with the value for control set to 1.0. Asterisk indicates results that are statistically significant at p-value<0.05. (F) RT-PCR analysis of Sxl mRNA in adult female control and Nab2ex3 tissues with exon 2-3-4 and 2-4 bands indicated. (G) RNA binding protein (RBP) motif enrichment analysis detects predicted Sxl binding sites as the most frequent motif among Nab2-regulated splicing events in female heads. Other enriched motifs are similar between male and female heads. Regions used for motif analysis (retained introns, and alternative 5′ or 3′ splice sites plus flanking sequence) are described in the text and illustrated in the schematic to the right.
-
Figure 3—source data 1
Original agarose gel showing RT-PCR analysis of Sxl mRNA in the relevant genotypes in Figure 3B.
Shown as original (bottom) and inverted (top) grayscale.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig3-data1-v1.zip
-
Figure 3—source data 2
Original western blot of head lysates labeled in Figure 3D using anti-Sxl antibody.
Boxed areas correspond to regions shown in the main figure.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig3-data2-v1.zip
-
Figure 3—source data 3
Original western blot of head lysates labeled in Figure 3D using anti-Lamin antibody.
Boxed areas correspond to regions shown in the main figure.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig3-data3-v1.zip
-
Figure 3—source data 4
Raw agarose gel showing RT-PCR analysis of Sxl mRNA in the relevant genotypes and tissues shown in Figure 3F.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig3-data4-v1.zip
qRT-PCR confirms a statistically significant increase in the inclusion of the male-specific exon 3 in females with a concomitant decrease in the level of correctly spliced (exon 2-exon 4) transcript in both Nab2ex3 and Mettl3null female heads (Figure 3C). Because Sxl exon 3 includes an in-frame translation ‘stop’ codon, we tested whether full-length Sxl protein levels decrease in Nab2ex3 female heads. Indeed, immunoblotting analysis reveals reduced levels of Sxl protein in Nab2ex3 female heads compared to control or Mettl3 null heads (Figure 3D and E). Together, these data implicate Nab2 in post-transcriptional regulation of Sxl splicing and control of Sxl protein levels within female heads.
As all the analysis carried out thus far employed heads as source material, we tested whether Nab2-dependent splicing changes were also detected in other tissues. Significantly, RT-PCR analysis of Sxl mRNA in dissected control and Nab2ex3 females detects exon 3 retention in Nab2ex3 samples prepared from the thorax, but little to no retention in the abdomen and ovary (Figure 3F). This result implies that Nab2 is only necessary to direct Sxl exon 3 exclusion in specific tissues or cell types such as neurons, which are enriched in the head (brain) and thorax (ventral nerve cord). In sum, these data reveal a tissue-specific role for Nab2 in blocking Sxl exon 3 inclusion in females and regulating 5′-splice site utilization at the exon 3-exon 4 junction.
As Sxl is itself an RBP with roles in alternative splicing (Penalva and Sánchez, 2003; Bell et al., 1988), we performed a bioinformatic scan for RBP motifs enriched in proximity to the Nab2-dependent alternative splicing events identified by MISO analysis (see Figure 2B). Input sequences were composed of retained introns plus 25 nt extending into each flanking exon, and alternative splice sites with 25 nt of exon plus 1 kb of adjacent intron (see schematic, Figure 3G). This unbiased scan detected candidate Sxl binding sites as the single most abundant RBP motif within the Nab2-regulated MISO events in females (Figure 3G). Notably, Sxl motifs were not detected as enriched in the male Nab2ex3 MISO dataset, which otherwise strongly resembles the remaining group of female-enriched RBP motifs (e.g., the hnRNPL homolog smooth [sm], RNA binding protein-9 [Rbp9], the U1-SNRNPA homolog sans fille [snf], and the U2-SNRNP component [U2AF50]). The female-specific enrichment for Sxl binding sites raises the possibility that Nab2 regulates a portion of the alternative splicing events indirectly via control of a Sxl-regulated splicing program, or that Sxl and Nab2 proteins target common splicing events. Intriguingly, the Sxl target transformer (tra) and the Tra target double-sex (dsx) (Sánchez et al., 2001; Horabin and Schedl, 1993) were not recovered in the Nab2ex3 MISO or DESeq2 datasets, and IGV reads show little evidence of altered structure of tra and dsx RNAs as compared to Nab2pex41 controls (Figure 3—figure supplement 2). Together, these data suggest that Sxl may not control the tra-dsx pathway in the adult head, or that tra and dsx splicing are only altered in a subset of Nab2ex3 head cells and thus not detectable by bulk RNA-Seq analysis.
The dosage compensation complex contributes to phenotypes in Nab2ex3 mutant females
The decrease in Sxl protein in Nab2ex3 female heads suggests that aberrant inclusion of Sxl exon 3 could contribute to Nab2ex3 phenotypes by reducing Sxl activity. To test this hypothesis, the constitutively female-spliced SxlM8 allele (Barbash and Cline, 1995) was placed as a heterozygote into the background of Nab2ex3 animals. SxlM8 contains a 110 bp deletion spanning the 3′-end of intron 2 and 5′-end of exon 3, and consequently undergoes constitutive splicing to the feminized exon 2-exon 4 variant regardless of sex (Figure 4A, top panel). Consistent with the original report describing SxlM8 (Barbash and Cline, 1995), the allele is male-lethal in both control and Nab2ex3 backgrounds. However, heterozygosity for SxlM8 produces strong rescue of Nab2ex3 mutant female viability from ~4% to 71% (SxlM8/+;;Nab2ex3) (Figure 4A). Female Nab2ex3 siblings that did not inherit the SxlM8 allele also exhibit elevated viability (64%), perhaps due to maternal loading of Sxl mRNA. SxlM8/+;;Nab2ex3 females also show improved locomotion in a negative geotaxis assay (Figure 4B) and lengthened lifespan (Figure 4C) relative to Nab2ex3 females. This female-specific rescue of Nab2ex3 by SxlM8 indicates that partial restoration of Sxl expression can compensate for Nab2 loss.
Figure 4 with 1 supplement see all
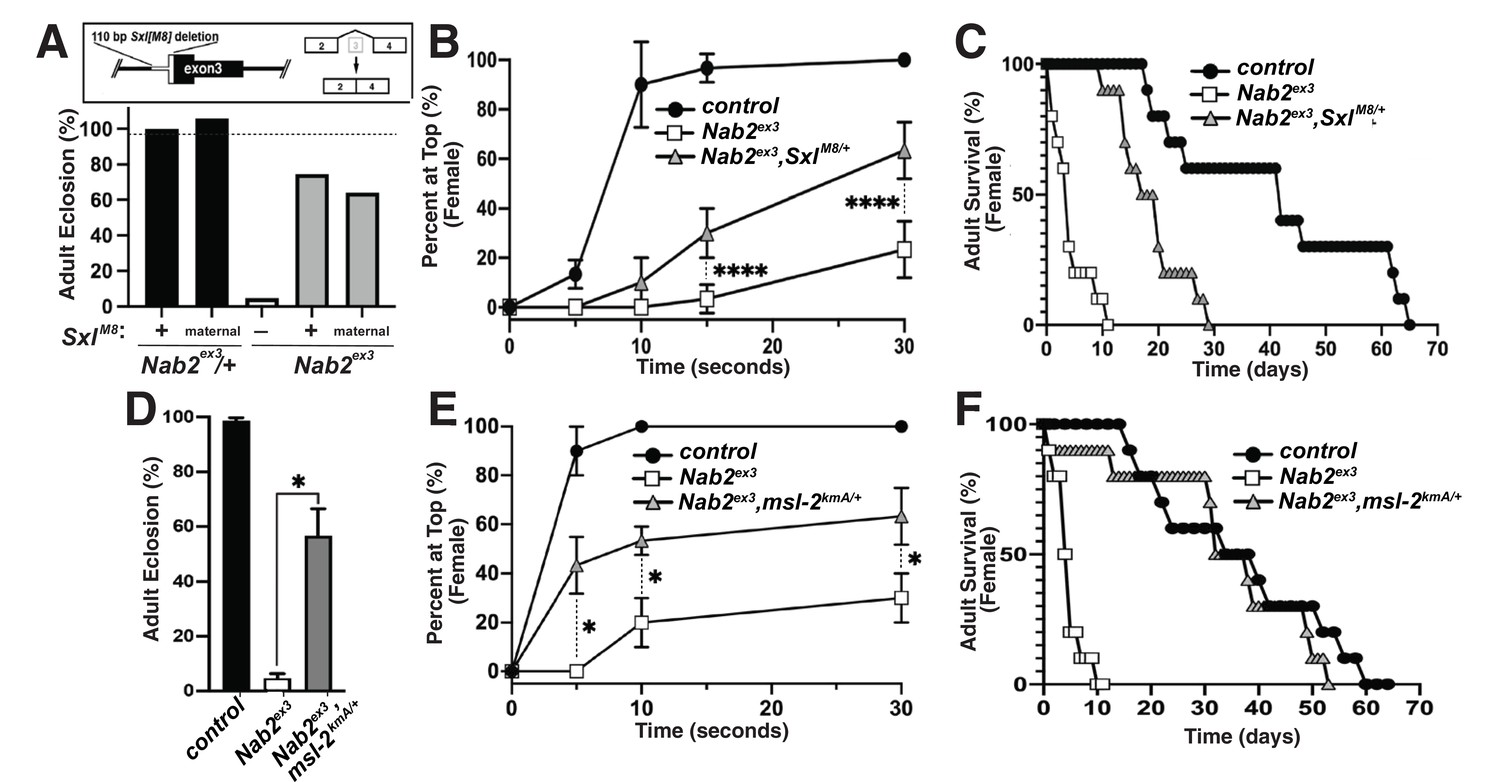
Alleles of SxlM8 or the Dosage Compensation Complex (DCC) component male-specific lethal-2 (msl-2) rescues Nab2 phenotypes.
(A) A single copy of the SxlM8 allele, which harbors a 110 bp deletion that causes constitutive exon 2-4 splicing, partially suppresses lethality of Nab2ex3, both zygotically and maternally (calculated as #observed/#expected). (B, C) SxlM8 dominantly (i.e., M8/+) suppresses previously defined (B) locomotion (as assessed by negative-geotaxis) and (C) lifespan defects in age-matched Nab2ex3 females. (D) Percent of control, Nab2ex3, or msl-2kmA/+;;Nab2ex3 (msl-2 is on the X chromosome) that eclose as viable adults (calculated as #observed/#expected). (E, F) msl-2kmA dominantly (i.e., kmA/+) suppresses previously defined (E) locomotion (as assessed by negative-geotaxis) and (F) lifespan defects in age-matched Nab2ex3 females. Significance values are indicated (*p<0.05, ****p<0.0001).
The absence of any effect on tra or dsx transcripts upon loss of Nab2 (see Figure 3—figure supplement 2) prompted us to analyze the other major role of Sxl, which is to bind to the male-specific lethal-2 (msl-2) mRNA and inhibit its translation in female somatic and germline tissues (Lucchesi and Kuroda, 2015; Keller and Akhtar, 2015). As a result, Msl-2 protein is only expressed in male cells, where it promotes assembly of a chromatin modifying complex termed the Dosage Compensation Complex (DCC; composed of Msl-1, Msl-2, Msl-3, Mof, Mle, and roX1 and roX2 non-coding RNAs), which is recruited to the male X chromosome to equalize X-linked gene expression between males and females (Lucchesi and Kuroda, 2015; Keller and Akhtar, 2015). A number of DCC components are expressed at high levels in the adult nervous system (Amrein and Axel, 1997), which correlates with the tissue-restricted link between Nab2 and Sxl splicing (as in Figure 3F). As a functional test of interactions between Nab2 and the DCC pathway, the msl-2kmA (msl-2killer of males-A) male lethal EMS allele (Bevan et al., 1993) was tested for dominant effects on Nab2ex3 female phenotypes. A single copy of msl-2kmA significantly rescues defects in viability (Figure 4D), locomotion (Figure 4E), and lifespan (Figure 4F) in Nab2ex3 females. Furthermore, a second msl-2 mutant allele over a deficiency that uncovers the msl-2 locus (msl-l227/Exel7016) (Zhou et al., 1995), as well as roX1 and mle loss-of-function alleles, rescue Nab2ex3 mutant phenotypes (Figure 4—figure supplement 1). Given that Msl-2 is not normally active in adult female tissues (Amrein and Axel, 1997; Meller et al., 1997) and its forced expression reduces female viability (Kelley et al., 1995), rescue of Nab2ex3 females by these msl-2, mle, and roX1 alleles provides evidence that the DCC pathway is inappropriately activated upon Nab2 loss. Of note, the msl-2, mle, and roX1 RNAs appear similar in IGV reads from both control and Nab2ex3 adult heads (see Figure 3—figure supplement 2), suggesting that genetic interactions between these loci are not through direct effects of Nab2 loss on post-transcriptional processing of these RNAs in a large fraction of cells.
Loss of the Mettl3 m6A methyltransferase rescues Nab2ex3 phenotypes
Genetic interactions between Nab2, Sxl, and msl-2 alleles are consistent with a role for Nab2 protein in regulating sex-specific splicing of Sxl exon 3. One mechanism that promotes exon 3 exclusion in females is based on N6-methylation of adenosines (m6A) in Sxl pre-mRNA by the Methyltransferase like-3 (Mettl3)-containing methyltransferase complex (Haussmann et al., 2016; Lence et al., 2016). Inactivating mutations in components of this m6A ‘writer’ complex masculinize the pattern of exon 3 splicing in female flies in a manner similar to Nab2ex3 and molecular studies indicate that the Mettl3 complex promotes exon 3 exclusion in females by depositing m6A within Sxl exon 3 and flanking introns (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017; Kan et al., 2021).
To assess Nab2-Mettl3 functional interactions, the Mettl3null allele (formerly known as Ime4null) (Lence et al., 2016) was carefully recombined with Nab2ex3 (the loci are less than 1 cM apart; Figure 5—figure supplement 1). Multiple recombinant Nab2ex3,Mettl3null chromosomes were found to be lethal at pre-larval stages but semi-viable over the Nab2ex3 chromosome; we therefore analyzed phenotypes in Nab2ex3,Mettl3null/+ mutant females. Consistent with prior work (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017), homozygosity for the Mettl3null allele reduces adult viability (Figure 5A), decreases locomotion in a negative geotaxis assay (Figure 5B), and shortens lifespan (Figure 5C). However, Mettl3null heterozygosity has the inverse effect of suppressing each of these defects in Nab2ex3 females: Nab2ex3,Mettl3null/+ mutant females show approximately 3-fold higher viability (Figure 5A), 6-fold higher climbing rates (at the 30 s time point; Figure 5B), and 1.75-fold longer lifespan (Figure 5C) than Nab2ex3 mutant females. As both Nab2 and Mettl3 act within the Drosophila nervous system (Bienkowski et al., 2017; Lence et al., 2016; Kan et al., 2017; Kan et al., 2021; Corgiat et al., 2021; Rounds et al., 2021), we sought to test whether this rescue of Nab2ex3 by reduced Mettl3 stems from cell autonomous roles for both factors within neurons. To address this hypothesis, we expressed a UAS-Mettl3-RNAi transgene in Nab2ex3 neurons using the pan-neuronal driver elav-Gal4 (Lin and Goodman, 1994) (i.e., elav-Gal4;UAS-Mettl3-RNAi;Nab2ex3). Notably, this depletion of Mettl3 only in neurons was sufficient to suppress Nab2ex3-associated defects in both viability (Figure 5D) and locomotion (Figure 5E) in female flies, consistent with a functional interaction between Nab2 and Mettl3 in neurons. Similarly, we reasoned that reducing other components of the m6A ‘writer’ complex could rescue Nab2ex3 defects. Indeed, we observed that heterozygous loss of two other components of the methyltransferase complex required for Sxl exon 3 skipping, female-lethal(2)d (fl(2)d) and virilizer (vir) (Granadino et al., 1990; Hilfiker et al., 1995; Niessen et al., 2001), also suppresses Nab2ex3 mutant phenotypes (Figure 5F and G). Heterozygous loss of fl(2)d in a Nab2ex3 mutant suppresses defects in female locomotion but not viability, while heterozygous loss of virilizer dominantly suppresses defects in both viability and locomotion.
Figure 5 with 1 supplement see all
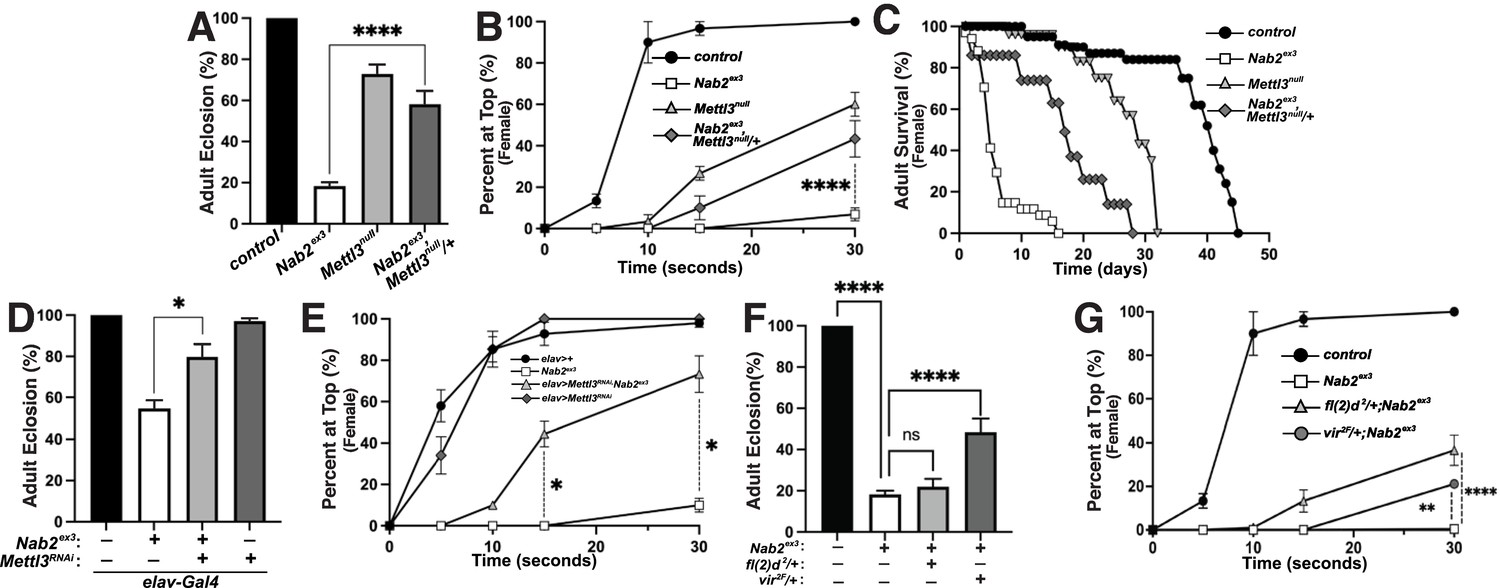
Reduction of the Mettl3 m6A transferase suppresses viability and behavioral defects in Nab2 mutant females.
(A) Percent of control, Nab2ex3, and Nab2ex3,Mettl3null/+ flies that eclose as viable adults (calculated as #observed/#expected). (B) Negative geotaxis of age-matched adult females of the indicated genotypes over time in seconds. (C) Survival of age-matched adult female flies of the indicated genotypes over time in days. (D) Percent of elav >Gal4 alone control, elav-Gal4;;Nab2ex3, elav-Gal4;UAS-Mettl3-RNAi;Nab2ex3, and elav-Gal4;UAS-Mettl3-RNAi flies that eclose as viable adults (calculated as #observed/#expected). Note that baseline Nab2ex3 viability is elevated in the background of the elav-Gal4 transgene, and significantly suppressed by inclusion of UAS-Mettl3 RNAi. (E) Negative geotaxis assay for age-matched adult females of the indicated genotypes over time in seconds. (F) Percent of control, Nab2ex3, Nab2ex3,fl(2)d2/+, or Nab2ex3,vir2F/+ flies that eclose as viable adults (calculated as #observed/#expected). (G) Negative geotaxis of age-matched adult females of the indicated genotypes over time in seconds. Significance values are indicated (*p<0.05, **p<0.01, ****p<0.0001).
Nab2 binds Sxl pre-mRNA and modulates m6A methylation
The finding that reduced Mettl3 expression rescues viability, lifespan, and locomotor defects in Nab2ex3 females indicates that the Mettl3 m6A ‘writer’ complex is required to promote developmental and behavioral defects in Nab2 mutants. However, loss of the same Mettl3 m6A ‘writer’ complex normally causes developmental and behavioral defects that resemble Nab2 mutant phenotypes documented here (Figures 4 and 5), and that are accompanied by Sxl exon 3 inclusion due to hypomethylation of Sxl mRNA (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017; Kan et al., 2021). This paradox could be explained if Sxl exon 3 inclusion in Nab2ex3 females accumulate excess m6A on the Sxl pre-mRNA. To test this hypothesis, a series of primer sets were designed to examine Sxl pre-mRNA and mRNA transcripts by RNA-immunoprecipitation (RIP) and anti-m6A-RIP (MeRIP) (Figure 6A). As illustrated in Figure 6A, the Sxl transcript contains candidate binding sites for both Sxl protein (polyuridine tracts = red ticks) and the polyadenosine RNA binding protein Nab2 protein (AC(A)13 tract = green tick). Approximate mapped sites of m6A methylation (yellow ticks) in the Drosophila head transcriptome are also indicated (Kan et al., 2021; see Figure 6—figure supplement 1 for a more detailed schematic).
Figure 6 with 4 supplements see all
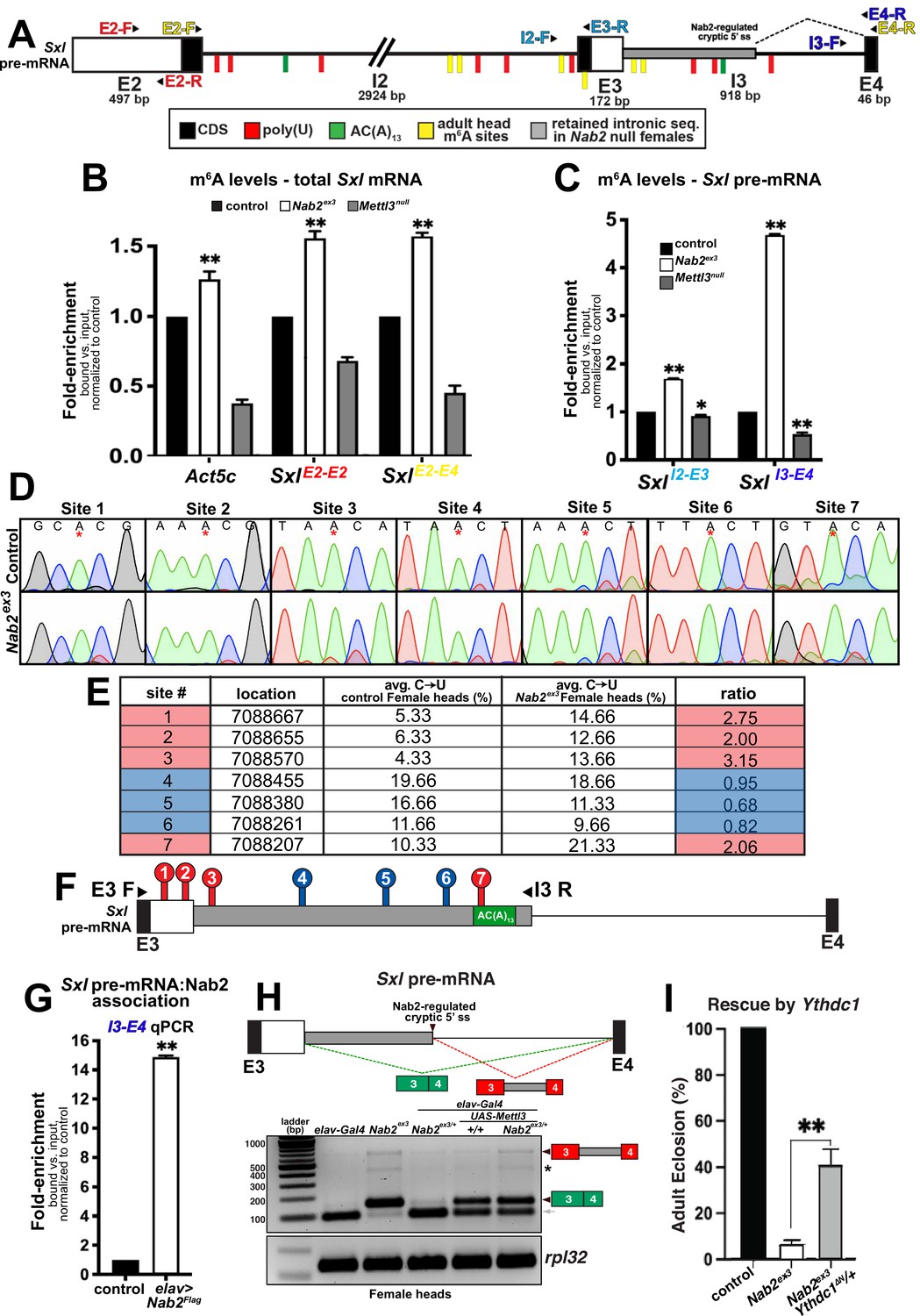
Nab2 associates with the Sxl mRNA and inhibits its m6A methylation.
(A) Diagram of exons (E2, E3, E4) and introns (I2 and I3) of the Sxl pre-mRNA annotated to show coding sequence (CDS; black), the retained intronic region in Nab2ex3 female (gray), and location of color-coded primer pairs (E2-F(orward) and E2-R(everse), E2-F and E4-R, I2-F and E3-R, I3-F, and E4-R), poly(U) sites (red lines), AC(A)13 site (green line), and mapped m6A positions in Drosophila heads (yellow lines) (Kan et al., 2021). (B) RT-qPCR analysis of Act5c and Sxl mRNA present in anti-m6A precipitates of control (control; black), Nab2ex3 (white), or Mettl3null (gray) female heads. The position of Sxl primer pairs is indicated (E2-F+E2-R and E2-F+E4-R). (C) Similar analysis as in (B) using I2-F+E3-R and I3-F+E4-R primer pairs to detect unspliced Sxl transcripts in anti-m6A precipitates of control (black), Nab2ex3 (white), or Mettl3null female heads. (D) Sanger sequencing traces showing C-to-U editing adjacent to m6A sites in control and Nab2ex3 female head RNA samples subjected to DART-sanger sequencing (Meyer, 2019) within the retained intronic region of Sxl pre-mRNA. m6A sites are indicated by red asterisks. (E) Table of the m6A sites (red = hypermethylated in Nab2ex3, blue = no change in Nab2ex3) mapped by DART-sanger sequencing in (D) with the corresponding location (dm6), average C-to-U editing fraction (%), and ratio of C-to-U editing for Nab2ex3 to control female head samples. Data are representative of three biological replicates. (F) Schematic showing the location of the m6A sites mapped by DART within exons (E3 and E4) and intron 3 of Sxl pre-mRNA. Site numbering corresponds to numbering in (E). (G) RT-qPCR analysis with the I3-F+E4-R primer pair in (A) from anti-Flag precipitates of control and elav-Gal4,UAS-Nab2:Flag female heads. (H) Top: schematic of the exon3-exon4 region of Sxl mRNA showing the intron region retained in Nab2ex3 (gray fill) and the normal exon 3-exon 4 splicing product (green fill) and the aberrant exon 3-intron 3503-exon 4 (red-gray fill). Bottom: RT-PCR analysis of Sxl using the E3-E4 primer pair and RNAs harvested from female heads of the indicated genotypes: elav-Gal4 alone, elav-Gal4+Nab2ex3/+, Nab2ex3 mutant, Nab2ex3/+, UAS-Mettl3 alone, or elav>Mettl3+Nab2ex3/+. Arrowheads denote exon 3-exon 4 and exon 3-intron 3503-exon 4 bands are indicated. Small gray arrow indicates Nab2-dependent splice variant. Asterisk marks a nonspecific band. (I) Percent of control, Nab2ex3, or Nab2ex3;Ythdc1ΔN/+ flies that eclose as viable adults (calculated as # observed/# expected).
-
Figure 6—source data 1
Raw agarose gel showing RT-PCR analysis of Sxl mRNA in the relevant genotypes shown in Figure 6H.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig6-data1-v1.zip
-
Figure 6—source data 2
Raw agarose gel showing RT-PCR analysis of RpL32 mRNA in the relevant genotypes shown in Figure 6H.
- https://cdn.elifesciences.org/articles/64904/elife-64904-fig6-data2-v1.zip
To assess the m6A status of total Sxl RNA, MeRIP precipitates from female head lysates (control, Nab2ex3, and Mettl3null) were analyzed by reverse transcription-real time quantitative PCR (RT-qPCR) with the exon 2-exon 2 (E2-E2) primer pair, which amplifies both pre-mRNA and mature mRNA (SxlE2-E2 in Figure 6B). This approach detects reduced Sxl m6A in Mettl3null heads relative to controls, which is consistent with prior studies (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017; Kan et al., 2021), and an increase in Sxl transcript recovered from MeRIP of Nab2ex3 heads, consistent with increased Sxl m6A modification. As additional controls for m6A status, two m6A-methylated Mettl3-target RNAs, Act5c and Usp16 (Lence et al., 2016; Kan et al., 2017; Kan et al., 2021) were analyzed. MeRIP-qPCR indicates that both mRNAs show decreased m6A in Mettl3null and show increased m6A in Nab2ex3 flies (Figure 6—figure supplement 2). Shifting this analysis to qPCR with the Sxl E2-E4 primer set (SxlE2-E4 in Figure 6B), which detects spliced Sxl mRNA, reveals a similar pattern of elevated Sxl m6A in Nab2ex3 heads. Together, these MeRIP-qPCR data argue that Nab2 either inhibits Mettl3-mediated m6A deposition or promotes m6A removal from Sxl mRNA. A prediction of this model is that Nab2 loss should result in increased levels of m6A on Sxl pre-mRNA. MeRIP analysis using the I2-E3 primer pair (SxlI2-E3 in Figure 6C) or the I3-E4 primer pair (SxlI3-E4 in Figure 6C) reveals moderate (1.5-fold) enrichment for intron 2-containing Sxl RNAs in Nab2ex3 heads, and stronger (4.5-fold) enrichment for intron 3-containing RNAs, consistent with elevated m6A on Sxl pre-mRNAs that contain introns 2 and 3.
To more precisely define how loss of Nab2 alters the relative abundance and/or location of m6A deposition along the Sxl transcript in female heads, we utilized in vitro DART-Sanger sequencing (Deamination adjacent to RNA modification Targets followed by Sanger sequencing) (Meyer, 2019; Tegowski et al., 2022; Figure 6D–F). This method overcomes several limitations of traditional antibody-based methods including limited sensitivity and selectivity, and struggle to distinguish m6A from other RNA modifications (i.e., m6Am) (Meyer, 2019). Briefly, in vitro DART-Sanger sequencing involves incubating RNA with a chimeric fusion protein consisting of the deaminating enzyme APOBEC1 fused to the m6A-binding YTH domain of m6A ‘reader’ proteins. As m6A-modified adenosine (A) residues are followed by a cytosine (C) residue in the most common consensus sequence (Meyer, 2019; Linder et al., 2015; Meyer et al., 2012), the APOBEC-YTH fusion recognizes m6A-modified A and deaminates the neighboring C, creating a uracil (U) base, which is read as a thymine (T) during Sanger sequencing. Therefore, C-to-U transitions and the frequency at which they occur permit mapping of m6A location and relative abundance. Thus, this method enables us to define the m6A modification status of the Sxl transcript in control and Nab2ex3 heads. For this experiment, we treated RNA extracted from female control or Nab2ex3 heads with APOBEC1-YTH, and subsequently performed RT-PCR with primers that amplify Sxl exon 3-intron 3503 (E3-I3 as illustrated in Figure 6F). Sanger sequencing and subsequent analysis of C-to-U transitions revealed the presence of seven m6A sites (sites #1–7) within this region (Figure 6D–F, denoted by asterisk in Figure 6D). These sites fall within or adjacent to sites mapped in a previous study of Drosophila head RNAs (Kan et al., 2021). Of the seven m6A modifications mapped within this region, four sites show a statistically significant increases C-to-U transition in Nab2ex3 female heads compared to control (Figure 6D; four sites are denoted by red color in Figure 6E–F). Specifically, sites 1–3 and 7 show 2.00 ratio of m6A modification (calculated as %C-to-U Nab2ex3/%C-to-U control), providing evidence that these sites are methylated to a greater extent in Nab2ex3 female heads compared to control heads. Notably, the m6A modification mapped to site 7 falls within the first adenosine residue of the proposed Nab2 AC(A)13 binding site (see schematic in Figure 6A and F). These results are consistent with a role for Nab2 in inhibiting m6A levels on Sxl pre-mRNA and suggest that modulation of m6A levels may link Nab2 to other RNA targets within the Drosophila head transcriptome. To test whether Nab2 physically associates with Sxl pre-mRNA as a potential mechanism to limit m6A levels, an anti-Flag IP of FLAG-Nab2 was performed from head lysates of adult females expressing N-terminally tagged Nab2 specifically in neurons (elav>Flag:Nab2). RT-qPCR of precipitates analyzed with Sxl I3-E4 primers provides evidence that Nab2 associates with unspliced Sxl pre-mRNA (Figure 6G). In sum, these data provide a molecular framework to interpret Nab2-Mettl3-Sxl genetic interactions in which Nab2 associates with the Sxl pre-mRNA, perhaps via the AC(A)13 site located in I3 (green tick; Figure 6A) and limits levels of m6A on this transcript.
In light of these m6A data, we revisited the effect of altered Mettl3 gene dosage on Sxl RNA structure. Reducing Mettl3 levels by half (Mettl3null/+) does not significantly alter Sxl splicing patterns in either control females or Nab2ex3 females (Figure 6—figure supplement 3). Because complete removal of Mettl3 is lethal in animals that also lack Nab2, we considered whether overexpressing Mettl3 is sufficient to reproduce Sxl splicing defects we observe in Nab2ex3 female heads. To test this possibility, we compared patterns of Sxl splicing between Nab2ex3 female heads and heads from animals overexpressing Mettl3 in neurons using a UAS-Mettl3 (Lence et al., 2016) transgene driven by elav-Gal4 (Lin and Goodman, 1994; Figure 6—figure supplement 4). As shown in Figure 6H, RT-PCR using primers located in Sxl exon 3 and exon 4 detect mis-spliced exon 3-exon 4 RNA (green boxes) and the aberrant exon 3-intron 3503-exon 4 (red-gray boxes) product in Nab2ex3 homozygous heads, with none or very low levels of these two RNA species in control (elav-Gal4 alone) and Nab2ex3 heterozygous heads. However, overexpression of Mettl3 in neurons is sufficient to produce the exon 3-exon 4 and exon 3-intron 3503-exon 4 RNAs in both control and Nab2ex3 heterozygote heads, thus replicating the effect of Nab2 loss on Sxl splicing. This analysis also identified a Sxl exon 3-exon 4 splicing intermediate in female heads that is approximately 60 nt smaller than the expected exon 3-exon 4 product, which is lost in Nab2ex3 female heads (Figure 6H, gray arrow). Sanger sequencing of this product revealed the presence of a Nab2-regulated cryptic 3′ splice site located within exon 3 that corresponds to the Sxl-RZ, RK and RQ RNAs (see FlyBase).
The increase in m6A levels detected on Sxl pre-mRNA upon loss of Nab2 provides evidence that Nab2 normally limits methylation on some RNAs. Excess m6A on transcripts in Nab2ex3 heads could lead to over-recruitment of the nuclear m6A YTH-domain containing ‘reader’ protein, Ythdc1 (or YT521-B), which regulates nuclear processing of many pre-mRNA targets including the removal of Sxl exon 3 in females (Haussmann et al., 2016). Thus, we tested whether reducing levels of Ythdc1 with the Ythdc1ΔN null allele (Lence et al., 2016) could rescue the lethality of Nab2ex3 mutants. Indeed, heterozygous loss of Ythdc1 increases viability of Nab2ex3 females approximately fivefold (Figure 6I). This finding is consistent with biochemical evidence that Nab2 represses m6A levels on the Sxl RNA and provides additional evidence that Nab2 interacts genetically with multiple elements of the m6A machinery.
Discussion
Through an unbiased high-throughput RNA sequencing approach, we identify a set of head-enriched RNAs in Drosophila whose levels or structure are significantly affected by loss of the Nab2 RBP, with the latter effect on RNA structure traced to splicing defects (including intron retention, alternative 5′ and 3′ splice site usage, and exon skipping) in a small group of approximately 150 transcripts. The top-ranked Nab2-regulated splicing event is skipping of Sxl exon 3 in females, which prior studies Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017 have shown to be guided by m6A methylation of specific sites in the Sxl pre-mRNA. Our biochemical studies reveal that Nab2 inhibits hypermethylation of sites in and around Sxl exon3, and genetic data show that developmental and behavioral phenotypes resulting from Nab2 loss are rescued by decreasing levels of the Mettl3 methyltransferase, other components of the Mettl3 complex, or the nuclear m6A reader protein Ythdc1. Data suggest that Nab2-Mettl3 coregulation of Sxl splicing is most significant in neurons – the effect of Nab2 on Sxl splicing is strongest in tissues that contain CNS components (e.g., brain and ventral nerve cord), while Mettl3 overexpression only in neurons is sufficient to replicate Sxl splicing defects seen in Nab2 mutant heads. This apparent tissue specificity of the link between Nab2 and Mettl3 may help explain neurological defects in mice and humans lacking the Nab2 ortholog ZC13H14, although lethality of animals lacking both Nab2 and Mettl3 is consistent with only partial overlap between RNA targets of these two RBPs.
Because Sxl exon 3 contains a translational termination (stop) codon, inclusion of this exon disrupts female-specific expression of Sxl protein, a U-rich RNA binding protein that controls somatic and germline sexual identity via effects on splicing and translation of target mRNAs (rev. in Penalva and Sánchez, 2003; Moschall et al., 2017). Multiple lines of evidence suggest that Sxl mRNA may be a particularly significant target of Nab2 in neurons: mis-spliced RNAs in Nab2 mutant female heads are enriched for bioinformatically predicted Sxl binding motifs, and the SxlM8 allele that constitutively skips exon 3 (Barbash and Cline, 1995) substantially reverses developmental and behavioral defects in Nab2 null females (Figure 4). Moving downstream of Sxl, alleles of male-specific DCC components, including the direct Sxl target msl-2 (Bashaw and Baker, 1995; Bashaw and Baker, 1997), also rescue phenotypic defects in Nab2 mutant females (Figure 4 and Figure 4—figure supplement 1). Given that these DCC components are not normally expressed or active in females, these data provide evidence that masculinized Sxl splicing and DCC activity contribute to developmental and behavioral defects in Nab2 mutant female flies. Elevated DCC activity could contribute to axon projection defects in female MBs, but this seems unlikely given that Nab2ex3 males develop similar MB axonal defects (Kelly et al., 2016). Overall, these data imply a specific link between Nab2 and the Sxl exon 3 splicing machinery, which is confirmed by strong genetic interactions between Nab2 and the Mettl3 methyltransferase that promotes exon 3 skipping by depositing m6A on Sxl pre-mRNA (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017).
Molecular assays provide key insight into the Nab2-Sxl interaction. A tagged form of Nab2 protein associates with unspliced Sxl pre-mRNA when expressed in brain neurons, and Nab2 loss results in excess m6A on Sxl mRNA as detected by two independent assays used to map m6A sites, meRIP-qPCR and DART. The high resolution of the DART technique allowed us to map m6A sites in the Sxl exon 3-intron 3-exon 4 region that are more highly methylated in Nab2 mutants than in controls, consistent with Nab2 inhibiting m6A accumulation at sites normally modified by the Mettl3 complex. Significantly, these Nab2-regulated methylation sites lie under or adjacent to anti-m6A-CLIP peaks mapped in the Sxl RNA from adult female heads (Kan et al., 2021) and thus complement and extend our knowledge of m6A patterns on Sxl mRNAs expressed in the adult head. Given the known role of m6A in promoting Sxl exon 3 excision (Haussmann et al., 2016; Lence et al., 2016; Kan et al., 2017), these data collectively support a model in which Nab2 interacts with the Sxl pre-mRNA in the nucleus and opposes m6A methylation by the Mettl3 complex, thus ensuring a level of m6A necessary to guide Sxl exon 3 skipping in the female nervous system. We term this a ‘goldilocks’ model in which either too little or too much m6A methylation of the region surrounding Sxl exon 3 can result in its retention in the developing female brain. These data provide the first evidence that the highly conserved Nab2 RBP is a key regulator of splicing in the adult brain, and that Nab2 is required to limit m6A modification of an RNA.
Studies employing the SxlM8 allele indicate that altered Sxl splicing and decreased Sxl protein contribute to Nab2 mutant phenotypes in females. As Sxl is itself an RBP that can control splicing, some fraction of the mis-sliced mRNAs detected by Nab2ex3 high-throughput sequencing may thus be Sxl targets. This hypothesis is supported by the substantial rescue conferred by the SxlM8 allele and the enrichment for candidate Sxl-binding sites among mis-spliced mRNAs in Nab2 mutant female heads. However, splicing of the Sxl target RNA tra is unaffected in the Nab2 mutant RNA-Seq datasets. The lack of effect on tra could be due to lack of read depth in the RNA-seq data, although this does not seem to be the case (see Figure 4—figure supplement 1), or to alternative Sxl target RNAs in adult heads. Unbiased screens for Sxl target RNAs have carried out in ovaries (Primus et al., 2019) and primordial germ cells (Ota et al., 2017), but a similar approach has not been taken in the adult nervous system, where Sxl targets may differ from other tissue types. In this regard, the group of Nab2-regulated RNAs identified here may be enriched for neuronal RNAs that are directly regulated by Sxl.
Although this study focuses on Sxl as a female-specific Nab2 regulated RNA, a large majority of other mis-splicing events in Nab2 mutant head RNAs occur in both males and females. This evidence of a Nab2 role in non-sex-specific splicing events parallels evidence of accumulation of ~100 intron-containing pre-mRNAs in nab2 mutant S. cerevisiae cells (Soucek et al., 2016). Rescue of Nab2 mutant males and females by neuron-restricted expression of human ZC3H14 (Kelly et al., 2014) implies that this specificity may be a conserved in ZC3H14 proteins in higher eukaryotes. Indeed, knockdown of ZC3H14 in cultured vertebrate cells results in pre-mRNA processing defects and intron-specific splicing defects in the few RNAs that have been examined (Morris and Corbett, 2018; Wigington et al., 2016). The basis for Nab2 target specificity in Drosophila heads is not clear but could be due selectivity in binding to nuclear pre-mRNAs (e.g., Sxl) or interactions between Nab2 and partner proteins that define splicing targets.
Site-specific hypermethylation of Sxl resulting from Nab2 loss could arise by several mechanisms, including Nab2 modulating m6A deposition by blocking access of the Mettl3 complex to its target sites, or to Nab2 recruitment of an m6A ‘eraser’. However, recent studies demonstrating that Nab2 and ZC3H14 each co-purify at nearly stoichiometric levels with the exon junction complex (EJC) (Obrdlik et al., 2019; Singh et al., 2012) and that the EJC binding locally excludes Mettl3-mediated m6A deposition on pre-mRNAs (He et al., 2023; Uzonyi et al., 2023; Yang et al., 2022) suggest an alternate model in which Nab2 inhibits m6A deposition in cooperation with the EJC. Notably, the human homolog of the Drosophila protein Virilizer, which is an m6A methyltransferase subunit and splicing factor (Hilfiker et al., 1995; Niessen et al., 2001), was recovered in an IP/mass-spectrometry screen for ZC3H14 nuclear interactors (Morris and Corbett, 2018). This finding raises an additional possibility that ZC3H14/Nab2 modulates m6A methylation via interactions with both the Mettl3 complex and the EJC. Moreover, evidence that m6A modulatory role of Nab2 is not restricted to the Sxl mRNA (see Figure 6B and Figure 6—figure supplement 2) raises the additional hypothesis that changes in abundance or structure of the group of Nab2-regulated RNAs defined in this study (see Figures 1 and 2) are due in part to changes in m6A status.
Prior work has shown that almost all developmental and behavioral defects caused by Nab2 loss can be traced to a Nab2 role within central nervous system neurons (Pak et al., 2011; Kelly et al., 2014; Kelly et al., 2016; Corgiat et al., 2022; Rounds et al., 2021). Suppression of these phenotypes by heterozygosity for SxlM8 or Mettl3null alleles or by neuron-specific Mettl3 RNAi is thus consistent with a mechanism in which Nab2 inhibits steady-state m6A levels on a group of neuronal RNAs, and that Sxl is one of these RNAs in the female brain. However, the lack of statistically significant rescue of Sxl splicing defects by Mettl3 heterozygosity (Figure 6—figure supplement 3) implies that Sxl splicing is only rescued in a small subset of cells or that SxlM8 and Mettl3null heterozygosity rescue Nab2 mutant phenotypes through different mechanisms. While SxlM8 specifically restores a single splicing event in a single mRNA, the Mettl3null allele has the potential to broadly affect m6A levels on multiple RNAs with subsequent effects on multiple m6A-dependent processes in the cytoplasm, including mRNA export to the cytoplasm and translation. One potential candidate mRNA of this type is Wwox, which encodes a conserved WW-domain oxidoreductase that accumulates in brains of Nab2 mutant flies (Corgiat et al., 2021) and is mutated in human spinocerebellar ataxia type 12 (Serin et al., 2018; Mallaret et al., 2014). Significantly, the Wwox RNA has a 3′UTR intron that is retained in Nab2 mutant heads (this study) and contains a candidate m6A site (Kan et al., 2021), suggesting that Wwox RNA may be a target of both Nab2 and Mettl3. Elevated Wwox protein is also detected in the hippocampus of ZC3H14 knockout mice, raising the possibility that Nab2 and ZC3H14 share some common RNA targets across species (Roxas et al., 2017). ZC3H14 has to date not been linked to the m6A mark in mouse or human cells. However, the enrichment for Sxl mis-splicing in neuronal tissue (see Figure 3F) and rescue by ZC3H14 when expressed in neurons of otherwise Nab2-deficient animals (Kelly et al., 2016) supports the hypothesis that the Nab2/ZC3H14 family of RBPs may share an m6A inhibitory role that is specific to neurons, and that excessive m6A methylation of RNAs also contributes to neurological deficits in mice and humans lacking ZC3H14.
Materials and methods
Drosophila stocks and genetics
Request a detailed protocolDrosophila melanogaster stocks and crosses were maintained in humidified incubators at 25°C with 12 hr light-dark cycles. The alleles Nab2ex3 (null), Nab2pex41 (precise excision 41; control), and UAS-Flag-Nab2 have been described previously (Pak et al., 2011; Kelly et al., 2014). Lines from Bloomington Drosophila Stock Center (BDSC): GMR-Gal4 (#1350), elavC155-Gal4 (#458), msl-2227 (#5871), msl-2kmA (#25158), mle9 (#5873), roX1ex6 (#43647), UAS-Mettl3, UAS-Mettl3-RNAi (#80450), fl(2)d2 (#36302), vir2F (#77886). The Mettl3null, UAS-Mettl3, and YthdcΔN alleles were all kind gifts of J-Y. Roignant. The Nab2ex3,Mettl3null and Nab2ex3, YthdcΔN chromosomes were generated by meiotic recombination and confirmed by genomic PCR. A total of 200 recombinant lines were screened to identify Nab2,Mettl3 double mutants.
RNA sequencing (RNA-Seq) on Drosophila heads
Request a detailed protocolRNA-Seq was performed on three biological replicates of 60 newly eclosed adult female and male Drosophila heads genotype (control and Nab2ex3 mutants). Heads were collected on dry ice, lysed in TRIzol (Thermo Fisher), phase-separated with chloroform, and ran through a RNeasy Mini Kit purification column (QIAGEN). Samples were treated with DNase I (QIAGEN) to remove DNA contamination and transported to the University of Georgia’s Genomics and Bioinformatics Core for sequencing. rRNA was depleted using a Ribo-Zero Gold Kit (Illumina) and cDNA libraries were prepared using a KAPA Stranded RNA-Seq Kit (Roche). Quality control steps included initial Qubit quantification along with RNA fragment size assessment on an Agilent 2100 Bioanalzyer before and after rRNA depletion. The cDNA libraries were then sequenced for 150 cycles on a NextSeq 500 High Output Flow Cell (Illumina) set to generate paired-end, 75 base-pair (bp) reads. Total sequencing yield across all samples was 81.48 Gbp, equivalent to about 1.1 billion reads in total and 91 million reads per sample. Sequencing accuracy was high; 93.52% of reported bases have a sequencing quality (Q) score ≥ 30.
Read mapping, differential expression, and visualization
Request a detailed protocolRaw read FASTA files were analyzed on the Galaxy web platform (usegalaxy.org; Afgan et al., 2018). The BDGP6 release Drosophila melanogaster genome (dos Santos et al., 2015) from release 92 of the Ensembl database (Yates et al., 2020) was used as input for subsequent read mapping, annotation, and visualization. Briefly, reads from all four NextSeq500 flow cell lanes were concatenated using the Galaxy Concatenate datasets tail-to-head (cat) tool and mapped using RNA STAR (Dobin et al., 2013) with default parameters with some modifications. For each Galaxy tool, version numbers and exact parameters used are detailed in the following table:
| Galaxy software and parameters | ||
|---|---|---|
| Tool | Concatenate datasets tail-to-head (cat) | Default parameters |
| Galaxy version 0.1.0 | ||
| Tool | RNA STAR | Default parameters with the following exceptions: |
| Galaxy version 2.5.2b-0 | Read type: paired | |
| Reference genome: from history (using Ensembl FASTA and GTF referenced in text) | ||
| Tool | featureCounts | Default parameters with the following exceptions: |
| Galaxy version 1.6.0.3 | Gene annotation file: history (Ensembl GTF referenced in text) | |
| Count fragments instead of reads: enabled | ||
| GFF gene identifier: gene_name | ||
| Strand specificity: stranded-reverse | ||
| Tool | DESeq2 | Default parameters with the following exceptions: |
| Galaxy version 2.11.40.1 | Factors: four levels, each a group of three biological replicates | |
| Output normalized counts table – true | ||
| Output all levels vs. all levels – true | ||
| Tool | DEXSeq-Count | Default parameters with the following exceptions: |
| Galaxy version 1.20.1 | In 'read count' mode: strand-specific library – yes, reverse | |
| Tool | DEXSeq | Default parameters with the following exception: |
| Galaxy version 1.20.1 | Visualize results? – no | |
| Gene Ontology (GO) software and parameters | ||
| Tool | GO2MSIG | Parameters: |
| web interface | Data source: NCBI gene2go | |
| Taxon ID – 7227 | ||
| Evidence codes: include EXP, IDA, IEP, IGI, IMP, IPI, ISS, TAS | ||
| Propagate associations – true | ||
| Use gene – symbol | ||
| Repress IDs – no | ||
| Create genesets for – [1 top-level domain only] | ||
| Max. geneset size – 700 | ||
| Min. geneset size – 15 | ||
| Output format – gmt | ||
| Database release – April 2015 | ||
| Tool | GSEA Desktop for Windows | Default parameters with the following exceptions: |
| v4.0.3 | For up- and downregulated transcripts in Nab2ex3 vs. control: | |
| GSEA-Preranked | zip-report – true | |
| plot_top_x – 100 | ||
| create_svgs – true | ||
| Collapse – No collapse | ||
| For alternatively spliced transcripts in Nab2ex3 vs. control: | ||
| zip-report – true | ||
| Minimum gene set size – 5 | ||
| create_svgs – true | ||
| Collapse – No collapse | ||
| Tool | AmiGO 2 | Default parameters |
| web interface | ||
Mapped reads were assigned to exons and tallied using featureCounts (Liao et al., 2014) default parameters with some modifications noted above. Differential expression analysis was conducted for all 12 samples using DESeq2 (Love et al., 2014; Galaxy version 2.11.40.1) and default parameters with some modifications noted above. Differential exon usage was analyzed using Galaxy version 1.20.1 of DEXSeq (Anders et al., 2012) and the associated Galaxy tool DEXSeq-Count in both ‘prepare annotation’ and ‘count reads’ modes. Both tools were run with the Ensembl GTF with default parameters with some modifications noted above. Unlike with DESeq2, female samples and male samples were compared in independent DEX-Seq analyses. Outputs from all tools were downloaded from Galaxy for local analysis, computation, and visualization.
Custom R scripts were written to generate volcano plots and heatmaps. Additional R packages used include ggplot2 (Wickham, 2016) and ggrepel (Slowikowski, 2019). R scripts were written and compiled in RStudio (RStudio Team, 2018). Principal component analysis was conducted on Galaxy. Mapped reads were visualized in the IGV (Robinson et al., 2017) and annotated based on data available on FlyBase (Thurmond et al., 2019). Significant fold change values in either male or female from DESeq2 (adj. p-val<0.05 and |log2FC| > 1) were plotted, with the color indicating the fold change threshold reached in either males or females. Significantly DE genes (adj. p-val<0.05 and |log2FC| > 1) were classified by type, as indicated by their gene ID.
MISO analysis
Request a detailed protocolMISO (Katz et al., 2010) version 0.5.4 was used to determine percent spliced in (PSI) values for annotated alternative 3′ splice sites, alternative 5′ splice sites, and retained introns for each sample separately as follows. Alternative splicing annotations were generated using the rnaseqlib (a direct link to script is provided at https://rnaseqlib.readthedocs.io/en/clip/) script, gff_make_annotation.py, with flags--flanking-rule commonshortest --genome-label dm6. Replicates for each sample were pooled, and only full-length, mapped reads (76 bp) were used for the MISO analysis as MISO requires all reads input to be of the same length. MISO was run with the flag-prefilter, and the output was then input into the script, summarize_miso, with the flag --summarize-samples. Next, differential, alternative 5′ and 3′ splice sites, and differential retained introns, were determined comparing Nab2ex3 and control for males and females, separately, using the script, compare_miso, with flag --compare-samples. The output of compare miso was then input into the script, filter_events, with the flags --filter --num-inc 10 --num-exc 10 --num-sum-inc-exc 50 --delta-psi 0.3 --bayes-factor 10, to obtain the final differential PSI values.
GO analysis
Request a detailed protocolGSEA software (Subramanian et al., 2005) was employed for GO analysis (The Gene Ontology Consortium, 2019). For clarity, analyses were conducted separately for each of the three top-level GO domains: molecular function, biological process, and cellular component. GSEA-compatible GO term gene sets for D. melanogaster were acquired using the GO2MSIG web interface (Powell, 2014). GSEA Desktop for Windows, v4.0.3 (Broad Institute) was then used to identify two distinct classes of GO terms, independently for females and males: (1) terms enriched among up- and downregulated transcripts in Nab2ex3 compared to controls, and (2) terms enriched among transcripts alternatively spliced in Nab2ex3 compared to controls. For the first class, inputs consisted of all genes whose expression could be compared by DESeq2 (i.e., adjusted p-value ≠ NA). For the second class, inputs consisted of all genes with previously annotated alternative splicing events according to MISO. To identify the first class of GO terms, genes were ranked by log2 (fold change) calculated by DESeq2 and analyzed by the GSEA-Pre-ranked tool. To identify the second class of GO terms, genes with were ranked by the absolute value of the difference in PSI comparing Nab2ex3 and control calculated by MISO. This second ranking was analyzed by the GSEA-Preranked tool. Enriched GO terms (nominal p-value<0.05) identified for the first class were evaluated manually, surfacing multiple terms directly related to splicing. Enriched GO terms (nominal p-value<0.05) for the second class were ordered by normalized enrichment score (NES) and evaluated to identify the top ‘independent’ GO terms. Terms were defined as ‘independent’ by reference to their position in the GO hierarchy as reported on each term’s ‘Inferred Tree View’ page of the AmiGO2 GO database web tool (Carbon et al., 2009). ‘Independent’ terms had no parent, child, or sibling terms in the GO hierarchy associated with a higher NES than their own.
RBPs motif enrichment analysis using MISO analysis
Request a detailed protocolRNA sequences were analyzed at differentially retained introns and alternative 3′ and 5′ splice sites obtained from the MISO analysis on males and females separately (Nab2ex3 mutants vs. control). The sequence for each of these went 25 bp into the exon(s) of interest and 1 kb into the intron of interest. In the case of alternative 3′ and 5′ splice sites, the sequences went 25 bp into the exon starting from the alternative spice site that is closest to the center of the exon (i.e., the inner-most splice site), and 1 kb into the intron starting from that inner-most spice site. To convert these to RNA sequences, DNA sequences were first obtained using fastaFromBed (Quinlan and Hall, 2010), and then all T’s were converted to U’s with a custom script. To obtain putative binding sites for RBPs at these sequences, the sequences were then input into fimo using the flags --text --max-strand and the ‘Ray2013_rbp_Drosophila_melanogaster.meme’ file (Grant et al., 2011).
RNA isolation for RT-PCR and real-time qPCR
Request a detailed protocolTotal RNA was isolated from adult tissues with TRIzol (Invitrogen) and treated with DNase I (QIAGEN). For RT-PCR, cDNA was generated using SuperScript III First Strand cDNA Synthesis (Invitrogen) from 2 μg of total RNA, and PCR products were resolved and imaged on 2% agarose gels (Bio-Rad image). Quantitative real-time PCR (qPCR) reactions were carried out in biological triplicate with QuantiTect SYBR Green Master Mix using an Applied Biosystems StepOne Plus real-time machine (ABI). Results were analyzed using the ΔΔCT method, normalized as indicated (e.g., to Act5C), and plotted as fold-change relative to control.
Primers used for RT and qPCR analysis
Request a detailed protocol| Name | Sequence | Detects |
|---|---|---|
| Sxl pre-mRNA | Fwd: AGAACCAAAACTCCCTTACAGC Rev: GTGAGTGTCTTTCGCTTTTCG | Intron 2-exon 3 |
| Sxl pre-mRNA | Fwd: ACCAATAACCGACAACACAATC Rev: ACATCCCAAATCCACGCCCACC | Intron 3-exon 4 |
| Sxl mRNA | Fwd: GCTGAGCGCCAAAACAATTG Rev: AGGTGAGTTTCGGTTTTACAGG | Exon 2-exon 2 |
| Sxl RT-PCR | Fwd: ACACAAGAAAGTTGAACAGAGG Rev: CATTCCGGATGGCAGAGAATGG | Exon 2-3-4 |
| Sxl RT-PCR | Fwd: CTCTCAGGATATGTACGGCAAC Rev: CATTCCGGATGGCAGAGAATGG | Exon 2-3-4 |
| Sxl RT-PCR | Fwd: AGTATGTAGTTTTTATTTGCACGGG Rev: CATTCCGGATGGCAGAGAATGG | Exon 3-4 |
| Sxl mRNA exon 2-4 transcript | Fwd: GATTGAATCTCGATCATCGTTC Rev: CATTCCGGATGGCAGAGAATGG | Exon 2-exon 4 |
| Sxl mRNA exon 3-4 transcript | Fwd: CGAAAAGCGAAAGACACTCACTG Rev: CATTCCGGATGGCAGAGAATGG | Exon 3-exon 4 |
| Act5C | Fwd: GAGCGCGGTTACTCTTTCAC Rev: ACTTCTCCAACGAGGAGCTG | Actin5C |
| USP-16-45-RF | Fwd: ACACTTGGTCACGTCGTTCA Rev: GGGCGCGCTCTTGAATTTAC | USP-16 |
Immunoblotting
Request a detailed protocolFor analysis of Sxl protein levels, Drosophila were reared at 25°C. Heads of newly eclosed flies were collected on dry ice. Protein lysates were prepared by homogenizing heads in 0.5 mL of RIPA-2 Buffer (50 mM Tris-HCl, pH 8; 150 mM NaCl; 0.5% sodium deoxtcholate; 1% NP40; 0.1% SDS) supplemented with protease inhibitors (1 mM PMSF; Pierce Protease Inhibitors; Thermo Fisher Scientific). Samples were sonicated 3 × 10 s with 1 min on ice between repetitions, and then centrifuged at 13,000 × g for 15 min at 4°C. Protein lysate concentration was determined by Pierce BCA Protein Assay Kit (Life Technologies). Head lysate protein samples (40–60 µg) in reducing sample buffer (50 mM Tris HCl, pH 6.8; 100 mM DTT; 2% SDS; 0.1% Bromophenol Blue; 10% glycerol) were resolved on 4–20% Criterion TGX Stain-Free Precast Polyacrylamide Gels (Bio-Rad), transferred to nitrocellulose membranes (Bio-Rad), and incubated for 1 hr in blocking buffer (5% non-fat dry milk in 0.1% TBS-Tween) followed by overnight incubation with anti-Sxl monoclonal antibody (1:1000; DHSB #M18) diluted in blocking buffer. Primary antibody was detected using species-specific horse-radish peroxidase (HRP) conjugated secondary antibody (Jackson ImmunoResearch) with enhanced chemiluminescence (ECL, Sigma).
Viability and lifespan analysis
Request a detailed protocolViability at 25°C was measured by assessing eclosion rates of among 100 wandering L3 larvae collected for each genotype, and then reared in a single vial. Hatching was recorded for 5–6 d. At least three independent biological replicates per sex/genotype were tested and significance was calculated using grouped analysis on GraphPad (Prism). Lifespan was assessed at 25°C as described previously (Morton et al., 2020). In brief, newly eclosed animals were collected, separated by sex, placed in vials (10 per vial), and transferred to fresh vials weekly. Survivorship was scored daily. At least three independent biological replicates per vial of each genotype were tested, and significance was calculated using grouped analysis on GraphPad (Prism).
Locomotion assays
Request a detailed protocolNegative geotaxis was tested as previously described (Morton et al., 2020). Briefly, newly eclosed flies (day 0) were collected, divided into groups of 10 male or females, and kept in separate vials for 2–5 d. Cohorts of age-matched flies were then transferred to a 25 mL graduated cylinder for analysis. At least three biological replicates per sex were analyzed per genotype using GraphPad (Prism).
Flag and m6A RNA immunoprecipitation (Flag-RIP and MeRIP)
Request a detailed protocolThe FLAG-RIP and MeRIP protocols were performed using previously described protocols (Bienkowski et al., 2017) and (Lence et al., 2016) with some modification. Briefly, three replicates of 30 newly eclosed female flies were collected in 1.5 mL Eppendorf tubes and frozen in dry ice. Heads were removed with a 5.5 Dumont tweezer and homogenized with a mortar/pestle in Isolation buffer (50 mM Tris-HCl pH 8.1, 10 mM EDTA, 150 mM NaCl, and 1% SDS, 50 mM NaCl). This preparation was diluted into IP buffer (50 mM HEPES, 150 mM NaCl, 5 mM EDTA, 0.5 mM DTT, 0.1% NP-40) supplemented with protease inhibitors (Roche) and RNasin Plus Inhibitor (Promega). Lysates were incubated with anti-Flag (M2 clone; Sigma) or anti-m6A (Synaptic Systems) antibody and recovered on magnetic Protein G Dynabeads (Invitrogen). After overnight incubation at 4°C with rocking, beads were washed 5× in IP buffer and RNA was isolated from antibody-bead precipitates, or controls (input samples) using TRIzol (Thermo Fisher). Samples were treated with DNase I and RNA was purified using RNeasy Kit (QIAGEN).
Deamination adjacent to RNA modification targets (DART)
Request a detailed protocolAPOBEC-YTH and APOBEC-YTHmut were purified and in vitro DART-Sanger sequencing assays were performed as previously described (Tegowski et al., 2022) with minor modifications. Briefly, total RNA was isolated from adult heads with TRIzol (Invitrogen) and treated with DNase I (NEB). RNA was isolated once more with TRIzol (Invitrogen) to remove DNase I and DNase I Buffer (NEB). Next, 200 ng of purified RNA from Drosophila heads was incubated with 1000 ng of purified DART protein in DART buffer (10 mM Tris-HCl, pH 7.4, 50 mM KCl, 0.1 M ZnCl2) and 1 µL of RNaseOUT (Invitrogen) in a total volume of 200 µL for 4 hr at 37°C. RNA was isolated with the QIAGEN Plus Micro Kit (QIAGEN) and stored at –80°C before being thawed for downstream Sanger sequencing analysis. cDNA was made using iScript Reverse Transcription Supermix (Bio-Rad). PCR amplification of Sxl pre-mRNA was carried out with Phusion High Fidelity PCR Kit (NEB). The resulting PCR product was PCR-purified using the QIAGEN PCR Purification Kit (QIAGEN). Samples were submitted for Sanger sequencing (McLabs) and %C-to-U editing was quantified using EditR software (Kluesner et al., 2018).
Primers used for DART PCR and Sanger sequencing
Request a detailed protocol| Name | Sequence | Detects |
|---|---|---|
| Sxl DART-PCR | Fwd: ACATATTTTTTTTCACAGCCCAG Rev: TCAAAACGATCCCCCAGTTAT | Exon 3-intron 3 |
| Sxl DART Sanger Seq | Fwd: TTTTCACAGCCCAGAAAGAAGC | Exon 3-intron 3 |
Statistical analysis
Request a detailed protocolGroup analysis on biological triplicate experiments was performed using two-way ANOVA (Turkey’s multiple-comparison test) on GraphPad (Prism) version 8.4.2 (464). Sample sizes (n) and p-values are denoted in the text, figures, and/or figure legends and indicated by asterisks (e.g., *p<0.05).
Data availability
Sequencing data have been deposited in GEO under accession code GSE162531.
-
NCBI Gene Expression OmnibusID GSE162531. The Nab2 RNA binding protein promotes sex-specific splicing of Sex lethal in Drosophila neuronal tissue.
References
-
The Galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 updateNucleic Acids Research 46:W537–W544.https://doi.org/10.1093/nar/gky379
-
Detecting differential usage of exons from RNA-seq dataGenome Research 22:2008–2017.https://doi.org/10.1101/gr.133744.111
-
Metamorphosis of the mushroom bodies; large-scale rearrangements of the neural substrates for associative learning and memory in DrosophilaLearning & Memory 5:102–114.
-
Gene Ontology: tool for the unification of biologyNature Genetics 25:25–29.https://doi.org/10.1038/75556
-
On the Allelism of killer-of-male and male-specific lethal mutationsDrosophila Information Service 72:125.https://doi.org/10.1093/genetics/96.1.165
-
AmiGO: online access to ontology and annotation dataBioinformatics 25:288–289.https://doi.org/10.1093/bioinformatics/btn615
-
RNA-binding proteins in neurodegeneration: mechanisms in aggregateGenes & Development 31:1509–1528.https://doi.org/10.1101/gad.304055.117
-
The RNA-binding protein Nab2 regulates the proteome of the developing Drosophila brainThe Journal of Biological Chemistry 297:100877.https://doi.org/10.1016/j.jbc.2021.100877
-
Cytoplasmic RNA-binding proteins and the control of complex brain functionCold Spring Harbor Perspectives in Biology 4:a012344.https://doi.org/10.1101/cshperspect.a012344
-
STAR: ultrafast universal RNA-seq alignerBioinformatics 29:15–21.https://doi.org/10.1093/bioinformatics/bts635
-
Splicing regulation in Drosophila sex determinationProgress in Molecular and Subcellular Biology 31:127–151.https://doi.org/10.1007/978-3-662-09728-1_5
-
A census of human RNA-binding proteinsNature Reviews Genetics 15:829–845.https://doi.org/10.1038/nrg3813
-
FIMO: scanning for occurrences of a given motifBioinformatics 27:1017–1018.https://doi.org/10.1093/bioinformatics/btr064
-
Sex determination: controlling the masterCurrent Biology 17:R328–R330.https://doi.org/10.1016/j.cub.2007.03.012
-
Mushroom body Memoir: from maps to modelsNature Reviews Neuroscience 4:266–275.https://doi.org/10.1038/nrn1074
-
Regulated splicing of the Drosophila sex-lethal male Exon involves a blockage mechanismMolecular and Cellular Biology 13:1408–1414.https://doi.org/10.1128/mcb.13.3.1408-1414.1993
-
The M(6)A pathway facilitates sex determination in DrosophilaNature Communications 8:15737.https://doi.org/10.1038/ncomms15737
-
The MSL complex: juggling RNA-protein interactions for dosage compensation and beyondCurrent Opinion in Genetics & Development 31:1–11.https://doi.org/10.1016/j.gde.2015.03.007
-
Recognition of polyadenosine RNA by the zinc finger domain of nuclear poly(A) RNA-binding protein 2 (Nab2) is required for correct mRNA 3’-end formationThe Journal of Biological Chemistry 285:26022–26032.https://doi.org/10.1074/jbc.M110.141127
-
EditR: a method to quantify base editing from sanger sequencingThe CRISPR Journal 1:239–250.https://doi.org/10.1089/crispr.2018.0014
-
Dosage compensation in DrosophilaCold Spring Harbor Perspectives in Biology 7:a019398.https://doi.org/10.1101/cshperspect.a019398
-
DART-Seq: an antibody-free method for global M(6)A detectionNature Methods 16:1275–1280.https://doi.org/10.1038/s41592-019-0570-0
-
Transcripts Immunoprecipitated with Sxl protein in primordial germ cells of Drosophila embryosDevelopment, Growth & Differentiation 59:713–723.https://doi.org/10.1111/dgd.12408
-
RNA binding protein sex-lethal (Sxl) and control of Drosophila sex determination and dosage compensationMicrobiology and Molecular Biology Reviews 67:343–359.https://doi.org/10.1128/MMBR.67.3.343-359.2003
-
Variant review with the integrative Genomics viewerCancer Research 77:e31–e34.https://doi.org/10.1158/0008-5472.CAN-17-0337
-
Control of Drosophila sex-lethal pre-mRNA splicing by its own female-specific productNucleic Acids Research 20:5533–5540.https://doi.org/10.1093/nar/20.21.5533
-
Poly(A)-Binding proteins are required for diverse biological processes in MetazoansBiochemical Society Transactions 42:1229–1237.https://doi.org/10.1042/BST20140111
-
Detecting M(6)A with in vitro DART-SeqMethods in Molecular Biology 2404:363–374.https://doi.org/10.1007/978-1-0716-1851-6_20
-
The gene Ontology resource: 20 years and still going strongNucleic Acids Research 47:D330–D338.https://doi.org/10.1093/nar/gky1055
-
Flybase 2.0: the next generationNucleic Acids Research 47:D759–D765.https://doi.org/10.1093/nar/gky1003
-
Characterization of Dfmr1, a Drosophila melanogaster Homolog of the fragile X mental retardation proteinMolecular and Cellular Biology 20:8536–8547.https://doi.org/10.1128/MCB.20.22.8536-8547.2000
-
BookGgplot2: Elegant Graphics for Data AnalysisSpringer International Publishing.https://doi.org/10.1007/978-3-319-24277-4
-
The Polyadenosine RNA-binding protein, zinc finger Cys3His protein 14 (Zc3H14), regulates the pre-mRNA processing of a key ATP synthase subunit mRNAThe Journal of Biological Chemistry 291:22442–22459.https://doi.org/10.1074/jbc.M116.754069
-
Exon junction complex shapes the M(6)A EpitranscriptomeNature Communications 13:7904.https://doi.org/10.1038/s41467-022-35643-1
Article and author information
Author details
J Christopher Rounds

Funding
National Institutes of Health (R01 MH107305)
- Ken Moberg
- Anita Corbett
National Institutes of Health (F31 NS103595)
- Binta Jalloh
National Institutes of Health (F31 HD088043)
- J Christopher Rounds
National Institutes of Health (R25 GM125598)
- Anita Corbett
National Institutes of Health (F31 HD079226)
- Rick S Bienkowski
National Institutes of Health (F32 GM125350)
- Derrick J Morton
National Institutes of Health (RM1 HG011563)
- Kate Meyer
National Institutes of Health (R01 MH118366)
- Kate Meyer
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
Stocks obtained from the Bloomington Drosophila Stock Center/BDSC (NIH P40OD018537) were used in this study. We thank members of the Moberg and Corbett Labs for helpful discussions and advice, and B Bixler, J Tanquary, and C Bowen for their contributions. We also thank M Alabady (Georgia Genomics and Bioinfomatics Core) for technical support, and T Lence and J-Y Roignant (Lausanne) for the gift of the Mettl3 allele, and T Cline (Berkeley) for discussions and insights on the Sxl[M8] allele. This work was funded by grants from the National Institute of Health to KHM and AHC (R01 MH10730501), KDM (NIGMS RM1 HG011563; R01 MH118366), JCR (F31 HD088043), BJ (F31 NS103595), and CLL (F31 NS127545). BJ and BEB were also supported during portions of the study by the Emory Initiative to Maximize Student Development (NIH R25 GM125598).
Copyright
© 2023, Jalloh, Lancaster et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,267
- views
-
- 205
- downloads
-
- 14
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 14
- citations for umbrella DOI https://doi.org/10.7554/eLife.64904
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The Drosophila Nab2 RNA binding protein inhibits m6A methylation and male-specific splicing of Sex lethal transcript in female neuronal tissue
eLife 12:e64904.
https://doi.org/10.7554/eLife.64904




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}