insomniac links the development and function of a sleep-regulatory circuit
- Neuroscience Institute, Department of Neuroscience and Physiology, New York University School of Medicine, United States
- Waksman Institute, Rutgers University, United States
Abstract
Although many genes are known to influence sleep, when and how they impact sleep-regulatory circuits remain ill-defined. Here, we show that insomniac (inc), a conserved adaptor for the autism-associated Cul3 ubiquitin ligase, acts in a restricted period of neuronal development to impact sleep in adult Drosophila. The loss of inc causes structural and functional alterations within the mushroom body (MB), a center for sensory integration, associative learning, and sleep regulation. In inc mutants, MB neurons are produced in excess, develop anatomical defects that impede circuit assembly, and are unable to promote sleep when activated in adulthood. Our findings link neurogenesis and postmitotic development of sleep-regulatory neurons to their adult function and suggest that developmental perturbations of circuits that couple sensory inputs and sleep may underlie sleep dysfunction in neurodevelopmental disorders.
Editor's evaluation
This is an interesting study showing that the short sleep phenotype of inc mutants in Drosophila depends on the loss of the gene at a specific developmental time, and in a specific region, the mushroom bodies (MB). There are very few studies assessing the effects of sleep during development, in any animal species, and thus this paper is a very welcomed addition. The experiments are carefully done, and the conclusions are warranted.
https://doi.org/10.7554/eLife.65437.sa0Introduction
A central goal of sleep research has been elucidating the mechanisms by which genes shape normal sleep patterns and cause sleep disorders. While numerous genes that strongly impact sleep have been identified in humans and in animals ranging from mammals to invertebrates (Chemelli et al., 1999; Chiu et al., 2016; Cirelli et al., 2005; Funato et al., 2016; He et al., 2009; Lin et al., 1999; Raizen et al., 2008), when these genes act to influence sleep is in many cases unresolved. Genes that act in the adult brain to modulate the activity of sleep-regulatory circuits in an ongoing manner have been intensively investigated (e.g. Chemelli et al., 1999; Lin et al., 1999), including with conditional gain-of-function, loss-of-function, and rescue in adult animals (Chiu et al., 2016; Clasadonte et al., 2017; Foltenyi et al., 2007; Guo et al., 2011; Ishimoto and Kitamoto, 2010; Joiner et al., 2006; Van Buskirk and Sternberg, 2007). In contrast, despite great progress in understanding neuronal development (Doe, 2008; Jessell and Sanes, 2000; Sanes and Zipursky, 2020; Tessier-Lavigne and Goodman, 1996; Weinstein and Hemmati-Brivanlou, 1999), developmental mechanisms by which genes influence sleep remain poorly explored, despite the likely relevance of such mechanisms to sleep disturbances in autism and other neurodevelopmental disorders (Angriman et al., 2015; Souders et al., 2017). Notably, the temporal contributions of genes that impact sleep are rarely assessed in a comprehensive manner, and a further challenge has been linking particular genes to developmental processes that control the structure and function of discrete sleep-regulatory circuits.
Here, we assess the temporal contributions of insomniac (inc), a gene whose mutation sharply curtails sleep in Drosophila (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). Pan-neuronal depletion of inc causes short sleep, while restoring inc solely to neurons is largely sufficient to rescue the sleep deficits of inc mutants, indicating that inc impacts sleep chiefly through neurons (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). inc is expressed in the larval, pupal, and adult brain (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011), but when inc acts to influence sleep remains uncertain (Li and Stavropoulos, 2016; Pfeiffenberger and Allada, 2012). inc encodes an adaptor for the Cul3 ubiquitin ligase (Li et al., 2019), which, like inc, is required in neurons for normal sleep (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). Both inc and Cul3 are highly conserved, and mammalian inc orthologs restore sleep to inc mutants (Li et al., 2017), suggesting that functions and substrates of inc are conserved in mammals. Human Cul3 mutations are implicated as a cause of autism and its associated sleep dysfunction (Codina-Solà et al., 2015; Kong et al., 2012; O’Roak et al., 2012), but the underlying mechanisms are unknown. Studies of inc may thus reveal fundamental and conserved mechanisms underlying sleep regulation which are altered in sleep disorders.
Using conditional genetic manipulations of inc, we show that inc acts transiently in developing neurons to impact sleep in adulthood. We furthermore identify developmental defects in inc mutants within the mushroom body (MB), a brain structure that integrates sensory stimuli and regulates sleep. Loss of inc alters MB neurogenesis, causing the overproduction of late-born neurons and changes in postmitotic development that impair the assembly of MB circuits. These developmental alterations persist into adulthood and are associated with specific deficits in the ability of MB neurons to promote sleep in inc adults, in contrast to the anatomy and function of other sleep-regulatory circuits which remain intact. Together, these results elucidate an unexpected mechanism by which inc shapes the development and function of sleep-regulatory neurons to exert a lasting impact on sleep–wake behavior. Our findings additionally suggest that developmental alterations of neurogenesis and within brain centers that integrate sensory inputs may contribute to sleep dysfunction in autism and other neurodevelopmental disorders.
Results
inc acts transiently during a restricted developmental period to impact sleep in adulthood
inc impacts sleep through neurons and is expressed in the developing and adult brain (Figure 1; Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). To assess the temporal mechanisms by which inc impacts sleep, we manipulated inc expression in neurons using the ligand-inducible Q-system (Potter et al., 2010; Riabinina et al., 2015). The Q-system circumvents nonspecific perturbations of sleep caused by other inducible systems and allows constitutive, developmental, and adult manipulations of sleep (Li and Stavropoulos, 2016). We performed a series of conditional rescue experiments in short-sleeping inc1 null mutants bearing a UAS-inc-HA transgene whose expression is induced in neurons by the Q-system upon exposure to quinic acid (Figure 2A). Animals exposed to vehicle throughout development and adulthood slept indistinguishably from inc1 mutants, while animals exposed constitutively to quinic acid exhibited strongly rescued sleep (Figure 2B, C; Figure 2—figure supplement 1), consistent with the rescue conferred by constitutive neuronal expression of inc (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). Anti-HA staining of brains confirmed that the Q-system controlled inc expression as expected: vehicle-fed animals lacked inc-HA signal, while those exposed constitutively to quinic acid expressed inc-HA in the larval, pupal, and adult brain (Figure 2D). We next asked whether inc influences sleep through adult-specific or developmental mechanisms. Animals fed quinic acid in adulthood expressed inc-HA in the adult brain but exhibited no rescue of their sleep deficits (Figure 2B–D; Figure 2—figure supplement 1). In stark contrast, developmental induction of inc-HA from embryonic through pupal stages restored sleep to near wild-type levels (Figure 2B–D; Figure 2—figure supplement 1). These findings indicate that inc is dispensable in adult neurons and acts instead during neuronal development to ultimately impact sleep–wake behavior.
Figure 1

Expression of 3×FLAG-inc driven by inc-Gal4 in the larval, pupal, and adult brain.
Maximal projections are shown for male inc-Gal4; UAS-3×FLAG-inc/+ brains stained with anti-FLAG. For larval brain, projection from a partial z-stack is shown to allow visualization of signal in mushroom body projections (arrowheads). In pupae and adults, signal is prominent in the mushroom body, pars intercerebralis, fan-shaped body, and ellipsoid body. Scale bars, 100 μm.
Figure 2 with 1 supplement see all
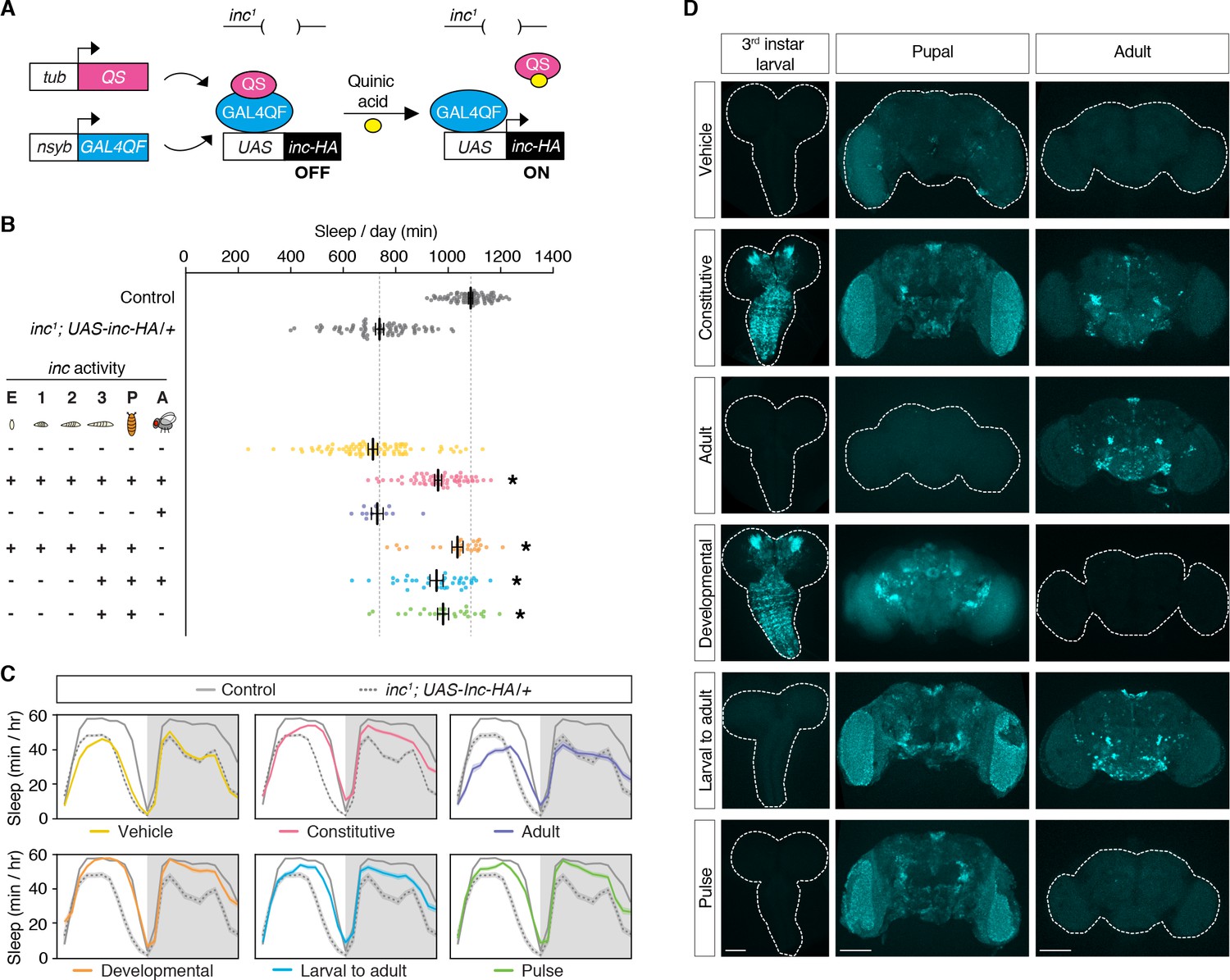
inc acts in a restricted period of neuronal development to impact sleep in adulthood.
(A) Conditional rescue of inc1 mutants using the ligand-inducible Q-system. Quinic acid relieves QS suppression of the pan-neuronally expressed Gal4QF transcriptional activator, inducing UAS-inc-HA in neurons. (B) Total sleep duration of controls (gray) and inc1; UAS-inc-HA/tub-QS; nsyb-GAL4QF/+ animals exposed to quinic acid (+) or vehicle (−) at indicated life stages; embryos (E), larval stages (1–3), pupae (P), and adults (A). Bars represent mean ± standard error of the mean (SEM). n = 11–86. One-way analysis of variance (ANOVA) (F(7,397) = 86.73, p < 0.0001) and Tukey post hoc tests, *p < 0.01 for comparisons to inc1; UAS-inc-HA/+. (C) Average sleep profiles of flies in (B), with induction regimens indicated below. Shading indicates ± SEM. (D) Anti-HA staining of inc1; UAS-inc-HA/tub-QS; nsyb-GAL4QF/+ brains from indicated induction regimens. Scale bars, 100 μm.
We further defined the developmental period in which inc functions, using more precise temporal manipulations. Neuronal induction of inc-HA from the late third instar larval stage through adulthood strongly rescued the inc sleep phenotype (Figure 2B–D; Figure 2—figure supplement 1), indicating that inc is dispensable in embryonic and early larval neurons. Induction of inc activity solely in late third instar larval and pupal neurons, using a pulse of quinic acid exposure (Figure 2D), restored sleep indistinguishably from constitutive neuronal induction (Figure 2B, C; Figure 2—figure supplement 1). The sleep deficits of inc2 animals, which bear an independent inc null allele that can be reverted by Gal4 (Stavropoulos and Young, 2011), were similarly rescued by this pulse of quinic acid (Figure 3A–C; Figure 3—figure supplement 1), confirming that inc activity in this developmental period is sufficient to restore sleep to inc mutants. We next assessed whether inc is required in late third instar larval and pupal neurons for normal sleep in adulthood, by using the Q-system to induce a pulse of inc RNAi. This manipulation markedly decreased sleep (Figure 3D, E; Figure 3—figure supplement 2). Together, these findings indicate that inc acts transiently in neurons of late third instar larvae and pupae to influence adult sleep–wake behavior. During these developmental stages, many neurons of the adult brain are born and assemble into circuits (Truman and Bate, 1988; White and Kankel, 1978).
Figure 3 with 2 supplements see all

Conditional rescue of inc2 mutants and conditional inc RNAi in larval and pupal neurons.
(A) Conditional neuronal rescue of inc2 mutants using the ligand-inducible Q-system. inc2 mutants contain a transposon insertion in the inc 5′UTR immediately upstream of the endogenous start codon. A UAS/TATA element within the transposon terminus permits Gal4-dependent restoration of inc expression (Stavropoulos and Young, 2011). (B) Total sleep duration in inc2; tub-QS/+; nysb-Gal4QF/+ animals exposed to vehicle or quinic acid at the late third instar larval and pupal stages. n = 20–83. One-way analysis of variance (ANOVA) (F(3, 170) = 70.66, p > 0.0001) and Tukey post hoc tests, *p < 0.01 for comparisons to inc2. (C) Average sleep profiles of indicated genotypes from (B). (D) Total sleep duration in tub-QS/UAS-inc-RNAi; nsyb-Gal4QF/UAS-dcr2 animals exposed to vehicle or quinic acid at the late third instar larval and pupal stages. n = 16–24. Student’s t-test, *p < 0.01 for comparison to vehicle-treated control. (E) Average sleep profiles of animals from (D). For (B) and (D), bars represent mean ± SEM. For (C) and (E), shading represents ± SEM.
inc has a critical function in the MB that impacts sleep
To identify neurons that might underlie the developmental impact of inc on sleep, we performed a rescue screen in inc2 mutants. We screened 277 Gal4 lines expressed in sleep-regulatory circuits or randomly selected populations of cells in the brain and identified two drivers, c253-Gal4 and c309-Gal4, that rescued sleep similarly to the pan-neuronal nsyb-Gal4 driver (Figure 4A). After backcrossing to an isogenic background, both drivers retained their ability to rescue most of the sleep phenotypes of inc2 mutants (Figure 4B, C; Figure 4—figure supplement 1). In late third instar larvae and adults, c253-Gal4 and c309-Gal4 are strongly expressed in the MB (Figure 4D), a structure important for sensory integration, associative learning, and sleep regulation (Heisenberg, 2003; Joiner et al., 2006; Pitman et al., 2006). Because c253-Gal4 and c309-Gal4 are also expressed outside of the MB, we used independent genetic manipulations to confirm that inc acts in the MB to influence sleep. inc-Gal4, a driver that bears inc regulatory sequences and fully rescues inc mutants when used to restore inc activity (Li et al., 2017; Stavropoulos and Young, 2011), is expressed in the larval, pupal, and adult MB (Figure 1). We tested whether the rescue conferred by inc-Gal4 was altered by MB-Gal80, a Gal4 suppressor expressed in MB neurons during development and adulthood (Krashes et al., 2007; Pauls et al., 2010). MB-Gal80 partially suppressed the ability of inc-Gal4 to restore sleep to inc1 mutants, indicating that while inc does not influence sleep solely through the MB, inc is required in MB neurons for normal sleep regulation (Figure 4E, F; Figure 4—figure supplement 2).
Figure 4 with 2 supplements see all
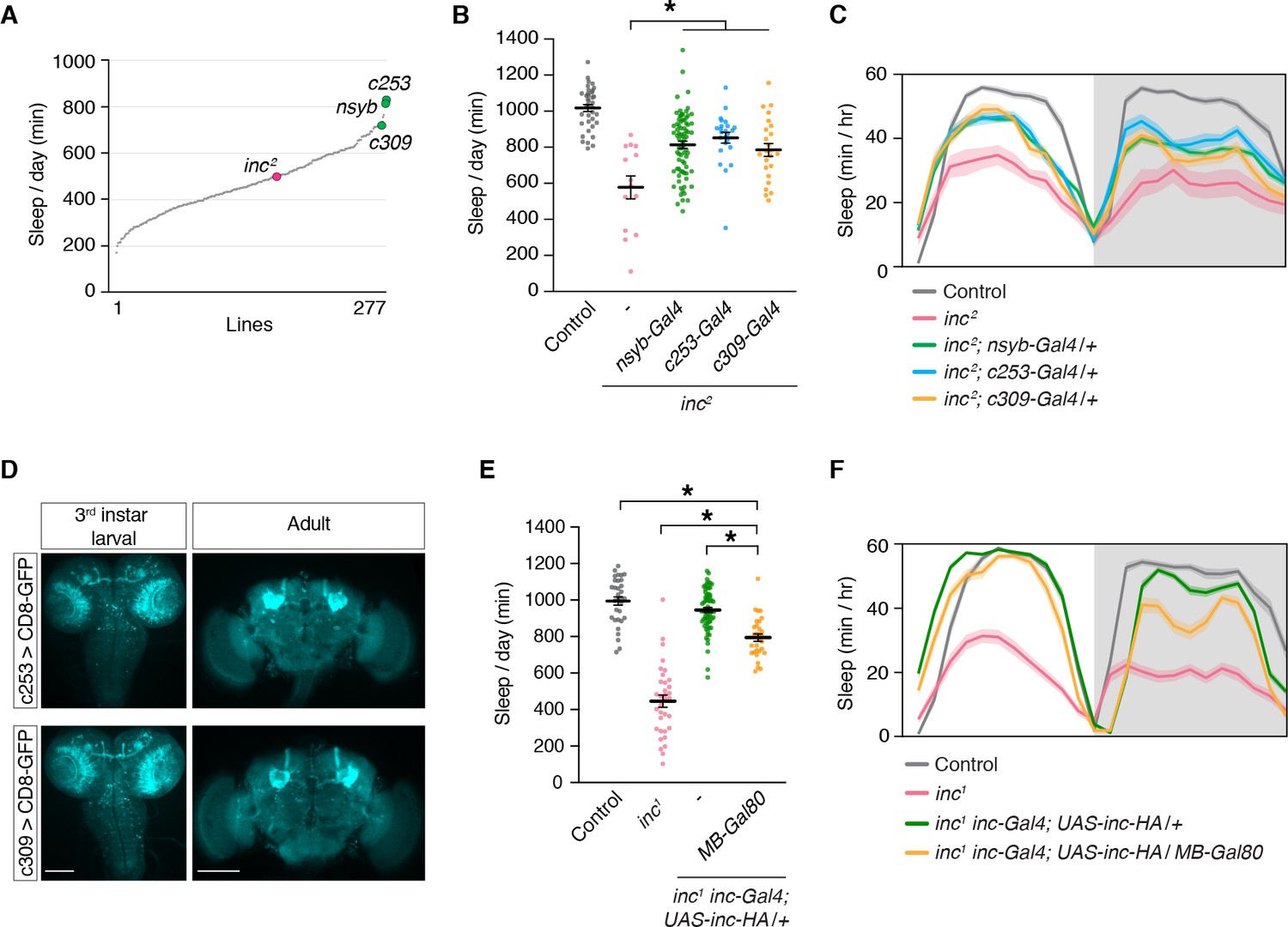
The mushroom body is a critical brain region through which inc impacts sleep.
(A) Mean sleep is plotted for each line in a Gal4 rescue screen of inc2 animals. n ≥ 5 per genotype. (B) c253-Gal4 and c309-Gal4 rescue sleep in inc2 mutants. n = 14–78. One-way analysis of variance (ANOVA) (F(4, 172) = 20.36, p < 0.0001) and Tukey post hoc test, *p < 0.01 for comparisons to inc2. (C) Average sleep profiles of flies in (B). (D) Anti-GFP immunostaining of indicated genotypes. Scale bars, 100 μm. (E) MB-Gal80 suppresses sleep rescue in inc1 inc-Gal4; UAS-inc-HA/+ animals. n = 30–69. One-way ANOVA (F(3, 161) = 121.4, p < 0.0001) and Tukey post hoc tests, *p < 0.01. (F) Average sleep profiles of indicated genotypes from (E). For (B) and (E), bars represent mean ± SEM. For (C) and (F), shading represents ± SEM.
Loss of inc abolishes the sleep-promoting functions of MB neurons but spares the functions of other sleep-regulatory circuits
While different circuits within the MB can promote or inhibit sleep upon activation (Joiner et al., 2006; Pitman et al., 2006; Sitaraman et al., 2015a), ablation of the MB strongly reduces sleep (Joiner et al., 2006; Pitman et al., 2006), suggesting that the integrated activity of the MB is sleep-promoting. To assess whether the sleep-regulatory functions of the MB are altered in inc mutants, we activated MB neurons in adult wild-type and inc1 flies using the dTrpA1 heat-activated cation channel (Hamada et al., 2008). Wild-type control flies lacking Gal4 drivers exhibited no change in total sleep when shifted to 28.5°C for 24 hr, while inc1 flies lacking Gal4 drivers exhibited decreased sleep at this temperature (Figure 5A, B), suggesting that inc mutants are hyperarousable by thermal stimuli, as for mechanical stimuli (Pfeiffenberger and Allada, 2012). Activation of neurons expressing TrpA1 under the control of c253-Gal4 or c309-Gal4 strongly increased sleep in wild-type animals (Figure 5A, B; Figure 5—figure supplement 1), consistent with observations that inactivating synaptic output using the same drivers promotes wakefulness (Pitman et al., 2006). Because c253-Gal4 and c309-Gal4 are expressed in some cells outside of the MB, we also assessed a split-Gal4 driver expressed specifically in MB neurons (Figure 5—figure supplement 2). Using this driver to express TrpA1 and activate MB neurons increased sleep in wild-type animals (Figure 5A, B, ‘pan-MB’). Strikingly, using the same three drivers to activate neurons in inc1 mutants elicited no significant changes in sleep compared to inc1; UAS-TrpA1/+ controls (Figure 5A, B; Figure 5—figure supplement 1), indicating that the sleep-promoting effects of MB activation are abolished in inc mutants.
Figure 5 with 2 supplements see all
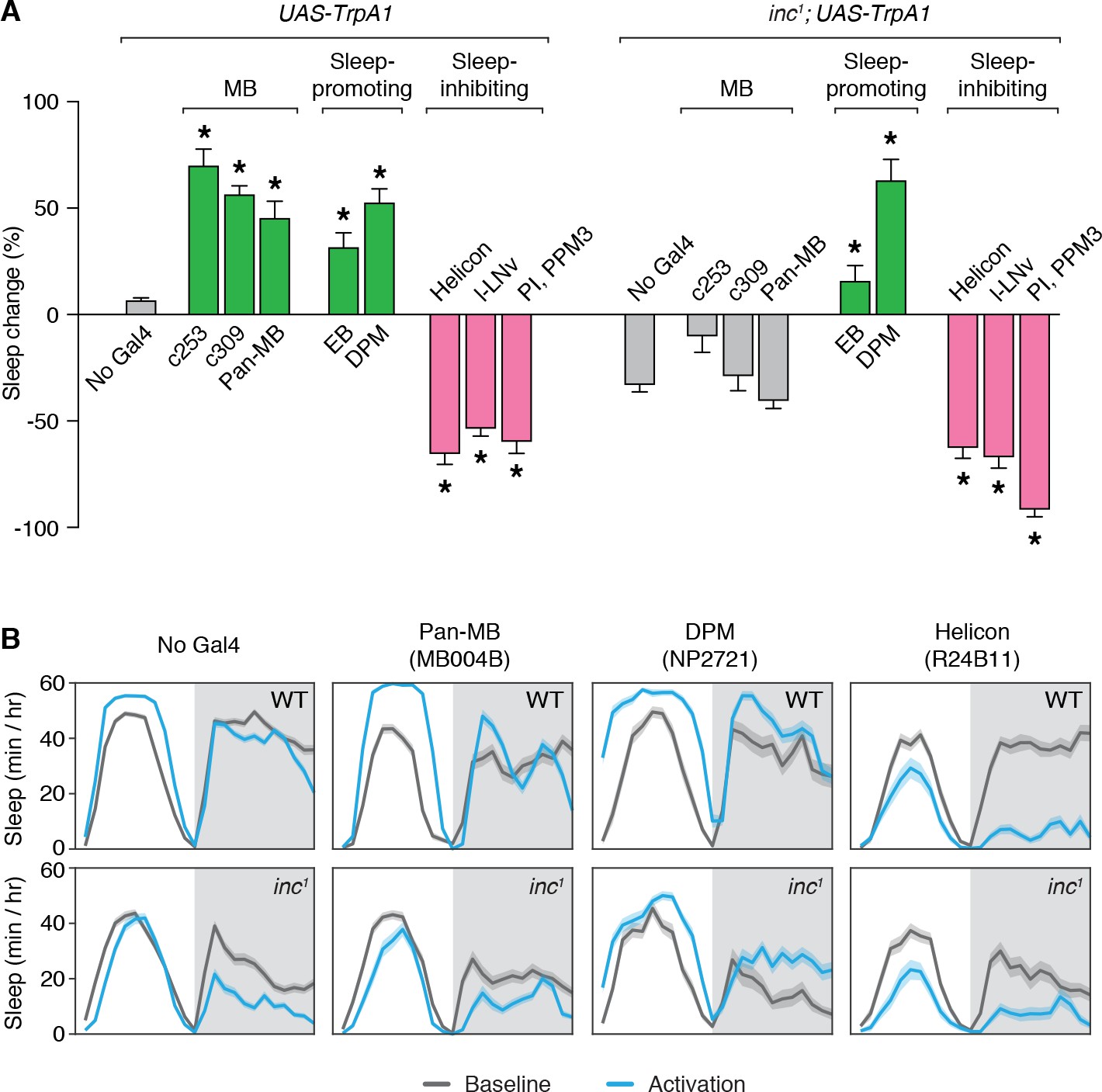
Sleep-promoting functions of the mushroom body are impaired in inc mutants.
(A) Thermogenetic activation of neuronal populations expressing TrpA1 in control and inc1 animals. Percent change in sleep (mean ± SEM) elicited by activation is shown. n = 31–144. Control and inc1 animals expressing dTrpA1 are compared to no-Gal4 controls (UAS-dTrpA1/+ and inc1; UAS-dTrpA1/+, respectively). *p < 0.01 for Dunnet’s post hoc comparisons after one-way analysis of variance (ANOVA) for control animals (F(8, 528) = 92.12, p < 0.0001) or inc1 mutants (F(8, 452) = 50.01, p < 0.0001). Green and pink bars indicate drivers that significantly promote or inhibit sleep, respectively; gray bars indicate no significant change with respect to controls. (B) Average sleep profiles of animals from (A) on the baseline day and during thermogenetic activation. Shading represents ± SEM.
To test whether the loss of inc specifically impairs the sleep-regulatory functions of MB neurons or causes more general deficits in sleep regulation, we assessed other neuronal populations that influence sleep. Activation of sleep-promoting populations that include ellipsoid body R5 (EB) (Liu et al., 2016) or Dorsal Paired Medial (DPM) neurons (Haynes et al., 2015) increased sleep similarly in wild-type and inc1 animals (Figure 5A, B; Figure 5—figure supplement 1). Conversely, activation of sleep-inhibiting populations that include Helicon (Donlea et al., 2018), l-LNv (Sheeba et al., 2008), or pars intercerebralis and dopaminergic PPM3 neurons (PI, PPM3) (Dubowy et al., 2016) strongly decreased sleep in wild-type and inc1 animals (Figure 5A, B; Figure 5—figure supplement 1). The functions of these populations thus appear to be intact in inc mutants, suggesting that the loss of inc specifically impairs the sleep-regulatory functions of MB neurons. These findings, together with the developmental time-of-action of inc and its requirement within the MB for normal sleep, suggest that inc acts developmentally in MB neurons to have a lasting impact on their sleep-regulatory functions in adulthood.
inc regulates the production and anatomy of late-born MB neurons
During the critical developmental period through which inc impacts sleep, MB neurons are born and assemble into adult circuits (Ito and Hotta, 1992; Lee et al., 1999). In each brain hemisphere, four MB neuroblasts proliferate to yield ~2000 neurons comprising seven sequentially born subtypes (γd, γm, α´/β´ap, α´/β´m, α/βp, α/βs, and α/βc) that project axons into distinct lobes (γ, α´/β´, and α/β) (Aso et al., 2014a; Ito et al., 1997; Ito and Hotta, 1992; Kurusu et al., 2002; Lee and Luo, 1999; Tanaka et al., 2008; Truman and Bate, 1988; Zhu et al., 2003). Chemical ablation of the MB by exposing first instar larvae to hydroxyurea, an inhibitor of DNA replication, causes sleep deficits in adulthood (Joiner et al., 2006; Pitman et al., 2006). The sleep deficits caused by MB ablation are similar to but less severe than those of inc mutants, including reductions in sleep across the day and decreased sleep consolidation (Figure 6A–F). These findings and the partial suppression of inc rescue by MB-Gal80 (Figure 4E, F; Figure 4—figure supplement 2) support the notion that reduced sleep in inc mutants results from impairments in the MB, alongside effects in additional neuronal populations.
Figure 6
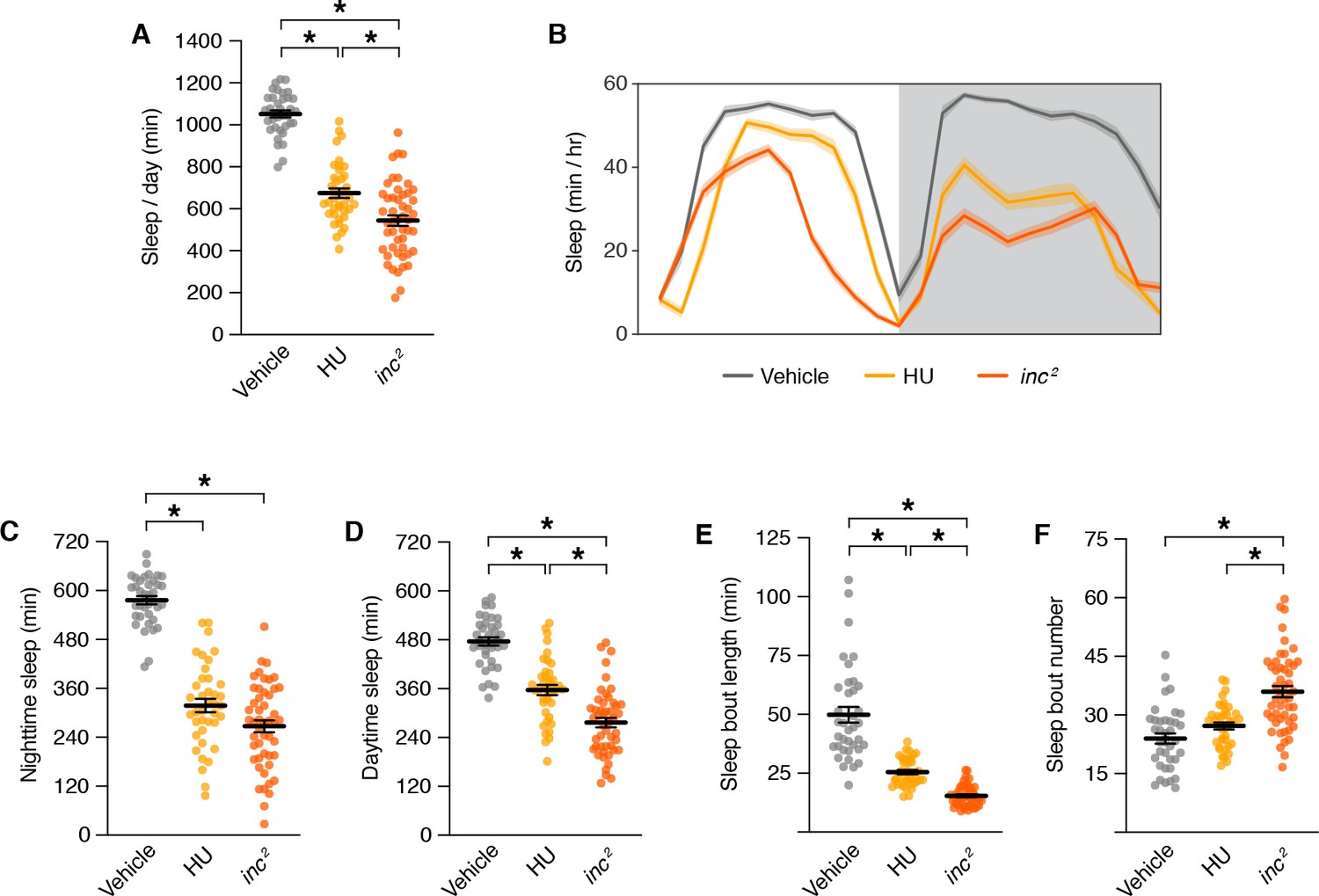
Sleep phenotypes for mushroom body ablation and inc mutants.
Sleep parameters for inc2 mutants and animals exposed to vehicle or hydroxyurea (HU). For all panels, n = 37–49; *p < 0.01 for post hoc tests. (A) Total sleep. One-way analysis of variance (ANOVA) (F(2, 122) = 132.9, p < 0.0001) and Tukey post hoc tests. (B) Average daily sleep profiles. Shading represents ± SEM. (C) Nighttime sleep. One-way ANOVA (F(2, 122) = 126.6, p < 0.0001) and Tukey post hoc tests. (D) Daytime sleep. One-way ANOVA (F(2, 122) = 74.32, p < 0.0001) and Tukey post hoc tests. (E) Sleep bout length. Kruskal–Wallis (p < 0.0001) and Dunn’s post hoc tests. (F) Sleep bout number. One-way ANOVA (F(2, 122) = 24.89, p < 0.0001) and Tukey post hoc tests. For (A) and (C–F), bars represent mean ± SEM.
To determine whether inc mutants have anatomical changes in the adult MB that might disrupt its sleep-regulatory functions, we examined MB neurons expressing UAS-Myr-GFP-2A-RedStinger, a bicistronic reporter that marks projections and nuclei (Daniels et al., 2014). Specifically, we used split-Gal4 drivers that label MB neuron subtypes born in embryos (γd), late larval stages (α´/β´), and in pupae (α/βc) (Aso et al., 2014a), to assess whether the loss of inc might preferentially alter subtypes whose birth and development coincides with the critical period through which inc impacts sleep. Consistent with this notion, we observed prominent changes in the number and anatomy of larval- and pupal-born MB neurons in inc mutants. While embryonic-born γd neurons were present in similar numbers in adult brains of controls and inc1 mutants (control, 102 ± 4; inc1, 94 ± 2) (Figure 7A, B), the number of larval-born α´/β´ neurons was increased 58% in inc1 animals (control, 141 ± 13; inc1, 223 ± 24), and the number of pupal-born α/βc neurons was doubled (control, 223 ± 11; inc1, 458 ± 45). The surplus of α´/β´ and α/βc neurons varied between left and right hemispheres in individual inc1 brains and this variation was greatest for α/βc neurons, the last-born in the MB (Figure 7A, C; Figure 7—figure supplement 1), indicating that inc mutants have a stochastic and cumulative defect in MB neurogenesis. Four clusters of α/βc neurons were present in control animals, reflecting their birth from four MB neuroblasts (Ito et al., 1997; Ito and Hotta, 1992; Truman and Bate, 1988), whereas inc1 mutants exhibited an average of nearly seven clusters (control, 3.7 ± 0.2; inc1, 6.8 ± 0.6) (Figure 7A, D; Figure 7—figure supplement 1), suggesting an origin from aberrant or excess neuroblasts. The numbers of other sleep-regulatory neurons, including those of the dorsal fan-shaped body (dFB) and DH44+ neurons, were unchanged in inc mutants (Figure 7A, I), indicating that neuronal overproduction in inc mutants is specific to the MB or manifests preferentially within this neuronal lineage. These findings indicate that inc regulates neurogenesis, a fundamental process regulated by proteins conserved from flies to mammals (Doe, 2008; Knoblich, 2008), and suggest that alterations in early nervous system development can exert a lasting impact on sleep.
Figure 7 with 2 supplements see all
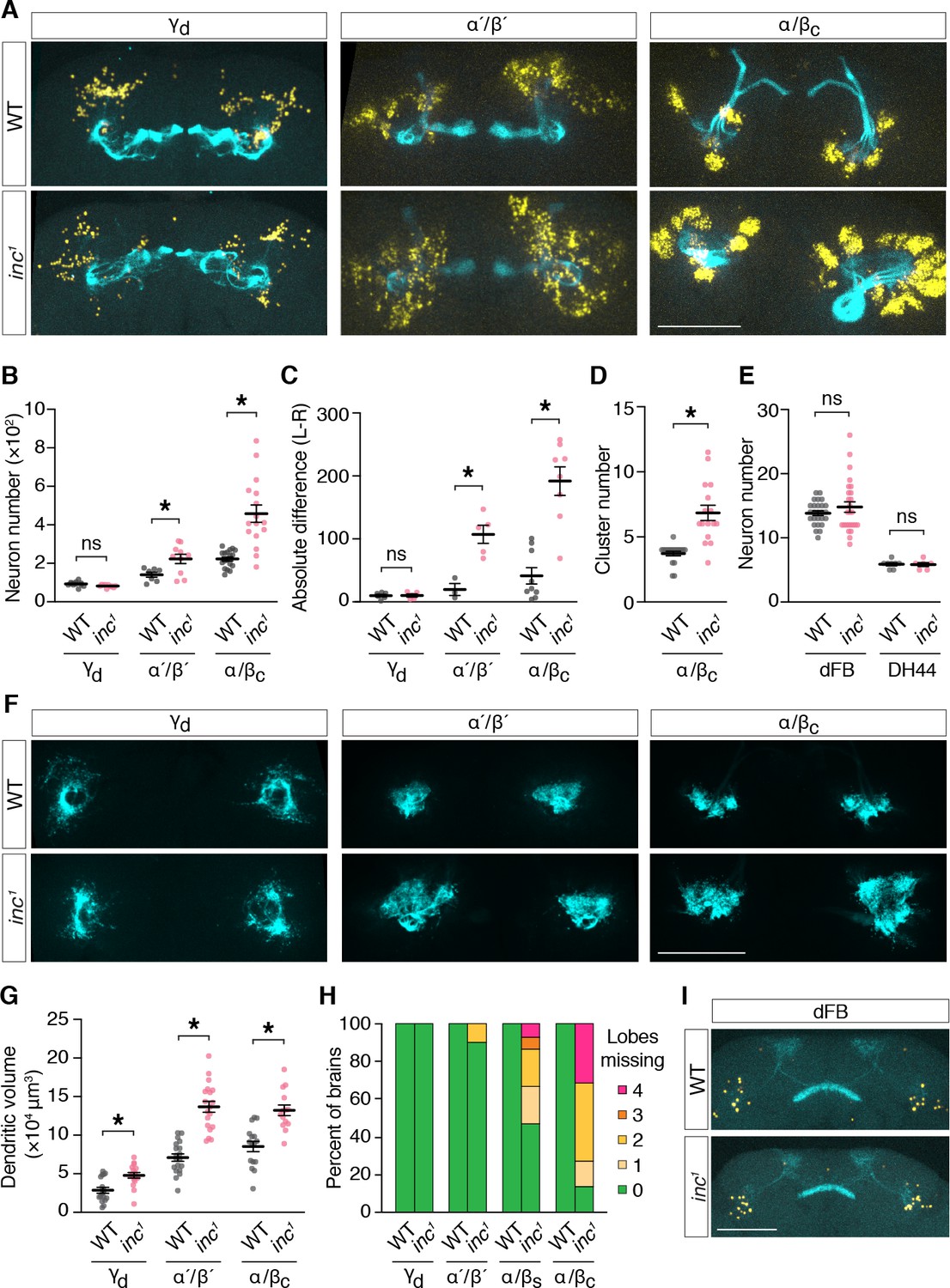
inc regulates neurogenesis and anatomy of late-born mushroom body (MB) neurons.
(A) Adult control and inc1 brains expressing UAS-MyrGFP-2A-RedStinger in indicated MB neuron subtypes, stained with anti-GFP (cyan) and anti-dsRed (yellow). (B) MB neuron number per hemisphere. γd, n = 10–11; α ́/β ́, n = 7–10; α/βc, n = 16–18. *p < 0.01, Welch’s t-test. (C) Absolute difference in MB neuron number between left and right brain hemispheres; γd, n = 5–6; α ́/β ́, n = 3–5; α/βc, n = 8–9. *p < 0.01, Welch’s t-test. (D) Number of α/βc neuron clusters per hemisphere. n = 16–18. *p < 0.01, Welch’s t-test. (E) Numbers of dorsal fan-shaped body (dFB) and DH44+ neurons. dFB, n = 26; DH44+, n = 6–8. ns, p > 0.01, Welch’s t-test. (F) Adult control and inc1 brains expressing UAS-DenMark-smGdP-V5 in indicated MB neuron subtypes, stained with anti-GFP. (G) Dendrite volume per hemisphere. γd, n = 16–17; α ́/β ́, n = 19; α/βc, n = 14–16. *p < 0.01, Welch’s t-test. (H) Quantification of axonal projection defects for MB neuron subtypes. Colored bars represent the number of MB lobes in each brain entirely lacking axonal myr-GFP signal. See also panel (A). n = 10–25. (I) Adult control and inc1 brains expressing UAS-MyrGFP-2A-RedStinger in dFB neurons. All scale bars represent 100 μm. For (B–E) and (G), bars represent mean ± SEM.
To further assess MB anatomy in inc mutants, we examined axons marked with myr-GFP and separately examined dendrites by expressing DenMark (Nicolaï et al., 2010). Axons of embryonic-born γd neurons exhibited no obvious changes in inc1 mutants (Figure 7A, H). In contrast, axons of larval- and pupal-born MB neurons exhibited morphological defects whose severity correlated with neuronal overproduction and birth order (Figure 7A, H). While α´/β´ axons were absent from MB lobes in a minority (10%) of inc1 brains, axons of α/βs neurons, the penultimate to be born, were missing from MB lobes in 53% of inc1 brains (1.07 ± 0.33 missing lobes per brain) (Figure 7H). Axons of last-born α/βc neurons showed the most severe defects; they failed to project into lobes in 86% of inc brains (2.23 ± 0.3 missing lobes per brain), fasciculated from ectopic neuronal clusters, and often aggregated near the peduncle (Figure 7A, H; Figure 7—figure supplement 1). The dendrites of γd, α´/β´, and α/βc neurons occupied enlarged territories in inc mutants but otherwise appeared normal (Figure 7F, G). Expansions in dendritic volume for α´/β´ and α/βc subtypes paralleled increases in the numbers of these neurons (Figure 7A, B), while increases for γd dendrites occurred independently of neuron number, consistent with functions of inc in postmitotic γd neurons or non-cell autonomous mechanisms. Axons and dendrites of other sleep-regulatory circuits, including those of the dFB, CRZ+ neurons, and PDF+ circadian pacemaker neurons, exhibited no obvious changes in inc mutants (Figure 7I; Figure 7—figure supplement 2), suggesting that alterations of neuronal anatomy in inc mutants are specific to the MB. These findings indicate that increases in the numbers of late-born MB neurons in inc mutants are associated with changes in postmitotic development expected to perturb circuit assembly and function. In particular, the altered axons of multiple MB neuron subtypes are unlikely to form normal circuits with their targets that influence sleep, including dopaminergic neurons, MB output neurons, and recurrent connections to the MB (Aso et al., 2014b; Sitaraman et al., 2015a; Sitaraman et al., 2015b).
Discussion
Here, we have used temporally restricted genetic manipulations to show that inc acts during neuronal development to ultimately impact sleep in adulthood. While many genes are known to act in adults to impact sleep, developmental mechanisms underlying sleep regulation have only recently gained attention (Chakravarti Dilley et al., 2020; Gong et al., 2021; Iwasaki et al., 2021; Xie et al., 2019). Our results underscore the importance of unbiased temporal genetic manipulations to define critical periods through which genes impact sleep, and suggest that genes may influence sleep through unappreciated developmental mechanisms. A clear implication of these findings is that variations in human sleep patterns, including pathological disruptions of sleep, may have a developmental origin.
Reciprocal conditional manipulations have been critical in revealing surprising developmental and adult contributions of genes to neuronal function and behavior. In one notable example, anxiety-like behaviors in mice caused by mutations of the 5-HT1A serotonin receptor were found to be rescued by developmental expression of the receptor (Gross et al., 2002). Withdrawal of receptor expression in adulthood had no measurable consequences on anxiety-like behavior, and adult-specific receptor expression failed to provide rescue, indicating the necessity and sufficiency of the receptor during development (Gross et al., 2002). A second noteworthy example is provided by a mouse model of Rett syndrome, a neurodevelopmental disorder caused by mutation of MECP2, a transcriptional regulator. Conditional MeCP2 expression solely in adulthood was found to be sufficient to rescue mutant phenotypes, indicating a critical period for MeCP2 function in adults rather than during brain development (Guy et al., 2007; Guy et al., 2012). Inactivation of MECP2 specifically in adulthood causes MECP2 mutant phenotypes (McGraw et al., 2011), confirming its adult requirement. By analogy, various genes that influence sleep might act developmentally or in adulthood in a manner that cannot be anticipated in the absence of conditional manipulations.
inc activity is required in neurons for normal sleep, and conversely, restoring inc solely to neurons is largely sufficient to rescue the short sleep of inc mutants (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). Our conditional neuronal manipulations of inc span embryonic development through adulthood and indicate that inc expression in neurons of late third instar larvae and pupae is sufficient to rescue sleep in inc mutants to near wild-type levels, indistinguishable from the rescue provided by constitutive neuronal inc expression (Pfeiffenberger and Allada, 2012; Stavropoulos and Young, 2011). Extending this developmental pulse of neuronal inc expression into adulthood does not augment the rescue of inc sleep phenotypes, nor does expressing inc only in adult neurons restore sleep to inc animals. inc expression in embryonic, early larval, and adult neurons thus appears dispensable for normal sleep. Instead, inc is required at a time coincident with the birth and development of many adult neurons, including those of the MB (Ito and Hotta, 1992; Lee et al., 1999; White and Kankel, 1978). While our findings suggest that the MB is not the sole brain structure through which inc impacts sleep, they establish a vital role for inc in regulating MB development and its sleep-regulatory functions.
Our findings reveal that inc governs neurogenesis, a fundamental process regulated by genes and pathways conserved from flies to mammals (Doe, 2008; Knoblich, 2008), and suggest that alterations of neurogenesis can cause lasting changes in sleep–wake behavior. The cellular and molecular mechanisms underlying altered neurogenesis in inc mutants, including the stochastic nature of these phenotypes and their apparent restriction to the MB, are of particular interest. inc null mutations are viable (Stavropoulos and Young, 2011), in contrast to the lethality of mutations that globally alter neurogenesis (Betschinger et al., 2006; Lee et al., 2006a; Lee et al., 2006b; Rolls et al., 2003; Vaessin et al., 1991), consistent with the notion that altered neurogenesis in inc mutants manifests preferentially or specifically within the MB. The stochastic nature of neurogenic defects in inc mutants and the overproduction of neurons with projection defects are reminiscent of phenotypes of mushroom body defect (mud) mutants (Guan et al., 2000; Hovhanyan and Raabe, 2009; Prokop and Technau, 1994). In mud mutants, infrequent errors in asymmetric neuroblast division give rise to excess neuroblasts and MB neurons (Bowman et al., 2006; Siller et al., 2006). Similar alterations in neuroblast proliferation in inc mutants may account for the stochastic and cumulative defects in the production of late-born MB neurons; a subtle defect in neuroblast proliferation would be expected to manifest particularly in the MB lineage, the longest in the fly brain. Our results do not yet distinguish the cellular populations through which inc regulates neurogenesis. One possibility is that inc acts in neurons to promote their differentiation, analogous to lola and midlife crisis, genes whose absence causes neurons to dedifferentiate and acquire the proliferative character of neuroblasts (Carney et al., 2013; Southall et al., 2014). Another possibility is that inc functions in neuroblasts, like mud, to govern their asymmetric division.
Our studies and recent findings (Gong et al., 2021) suggest that proper regulation of neurogenesis is essential for normal sleep and that altered neurogenesis in discrete circuits can cause lifelong sleep dysfunction. Intriguing but fragmentary evidence suggests that other genes whose mutation impacts sleep might similarly alter neurogenesis. wide awake (wake), whose mutation causes short sleep in Drosophila (Liu et al., 2014; Zhang et al., 2015), was characterized in an independent study as banderuola (bnd) and shown to regulate the asymmetric division of neuroblasts (Mauri et al., 2014). An interesting possibility yet to be assessed is whether sleep phenotypes of wake/bnd mutants might arise developmentally or through neuroblasts. Similarly, while short sleep phenotypes caused by mutations in the potassium channel subunits encoded by Shaker and Hyperkinetic (Bushey et al., 2007; Cirelli et al., 2005) are thought to reflect their role in regulating excitability in specific adult neurons (Kempf et al., 2019; Pimentel et al., 2016), developmental functions that could contribute to their impact on sleep remain unexplored. Notably, mutations in the Shaker ortholog Kv1.1 analogous to those that strongly reduce sleep in Drosophila (Cirelli et al., 2005; Gisselmann et al., 1989) cause megencephaly and neuronal overproduction in mammals, implicating Kv1.1 in regulating neurogenesis (Chou et al., 2021; Donahue et al., 1996; Petersson et al., 2003; Yang et al., 2012). Explicit tests of whether wake/bnd and Shaker impact sleep through adult or developmental mechanisms, or through a combination of the two, await conditional temporal analysis.
While further manipulations of inc are required to elucidate the precise developmental mechanisms by which it impacts sleep, Cul3 is known to regulate various aspects of neuronal development. Clonal analysis of Cul3 mutations in Drosophila indicates that Cul3 is required for normal axonal arborization and dendritic elaboration within the MB, as well as axonal fasciculation (Zhu et al., 2005). These phenotypes overlap those of inc mutants, although direct comparisons are complicated by the pleiotropic nature of Cul3 mutations, which dysregulate multiple adaptor and substrate pathways. Mosaic analysis of inc is required to discern its developmental functions in postmitotic neurons, to compare its phenotypes with Cul3, and to distinguish cell autonomous and non-cell autonomous mechanisms. In mammals, Cul3 mutations alter neurogenesis, cortical lamination, neuronal migration, synaptic development, and cause behavioral deficits (Amar et al., 2021; Dong et al., 2020; Fischer et al., 2020; Rapanelli et al., 2021). inc and Cul3 are present at synapses in flies and mammals (Kikuma et al., 2019; Li et al., 2017) and are required at the Drosophila larval neuromuscular junction for synaptic homeostasis (Kikuma et al., 2019), a process proposed to be a core function of sleep (Tononi and Cirelli, 2003). The impact of inc on the development and function of central synapses has yet to be assessed, and whether such functions contribute to inc sleep phenotypes remains unknown. As a Cul3 adaptor, inc may engage multiple molecular targets and cellular pathways. Identifying and manipulating inc substrates are thus important goals in elucidating the mechanisms through which inc impacts neuronal development and sleep–wake behavior.
The loss of inc causes enduring developmental and functional impairments in the MB, a structure important for sensory integration, learning, and sleep regulation. The MB integrates olfactory (de Belle and Heisenberg, 1994; Heisenberg et al., 1985), gustatory (Keene and Masek, 2012; Masek and Scott, 2010), visual (Li et al., 2020; Vogt et al., 2016), and thermal inputs (Frank et al., 2015; Hong et al., 2008; Shih et al., 2015), and its activity is altered by sleep pressure (Bushey et al., 2015; Sitaraman et al., 2015a). The MB may thus integrate and filter sensory stimuli to promote sleep in appropriate environmental conditions, in a manner modulated by learning and sleep history. The anatomical defects in inc mutants may render the MB hypersensitive to sensory stimuli, alter functions of the MB that link learning and sleep (Berry et al., 2015; Cervantes-Sandoval et al., 2017; Haynes et al., 2015; Seugnet et al., 2011; Seugnet et al., 2008), or impair the relay of sensory input from MB neurons to downstream sleep-promoting circuits (Aso et al., 2014b; Sitaraman et al., 2015a). While MB circuits and genetic pathways that act in the MB to influence sleep have been manipulated with increasing precision (Aso et al., 2014b; Cavanaugh et al., 2016; Guo et al., 2011; Joiner et al., 2006; Pitman et al., 2006; Sitaraman et al., 2015a; Sitaraman et al., 2015b; Yi et al., 2013), much remains unknown about the function of the MB in sleep regulation, and additional analysis is required to elucidate how inc lesions might alter discrete circuits within the MB and signaling to their targets.
While sensory hypersensitivity and sleep dysfunction are hallmarks of autism and other neurodevelopmental disorders, the underlying mechanisms remain obscure. Given the conserved functions of Cul3–inc complexes and the associations of Cul3 lesions with autism (Kong et al., 2012; Li et al., 2017; O’Roak et al., 2012), elucidating inc substrates and their contributions to neurogenesis and neuronal anatomy may provide insights into brain development, tumorigenesis, and sleep disorders.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Antibody | α-HA (rat monoclonal) | Roche | Cat# 11867431001, RRID:AB_390919 | (1:100) |
| Antibody | α-Brp (mouse monoclonal) | DSHB | Cat# nc82, RRID:AB_2314866 | (1:20 and 1:50) |
| Antibody | α-FLAG (mouse monoclonal) | Sigma-Aldrich | Cat# F1804, RRID:AB_262044 | (1:100) |
| Antibody | α-GFP (mouse monoclonal) | DSHB | Cat# GFP-G1, RRID:AB_2619561 | (1:1000) |
| Antibody | α-GFP (rabbit polyclonal) | Thermo Fisher Scientific | Cat# A11122, RRID:AB_221569 | (1:2000) |
| Antibody | α-dsRed (rabbit polyclonal) | Takara Bio | Cat# 632496, RRID:AB_10013483 | (1:1000) |
| Antibody | α-FasII (mouse monoclonal) | DSHB | Cat# 8 C6, RRID:AB_2314391 | (1:50) |
| Antibody | α-mouse Alexa Fluor 488 (donkey polyclonal) | Thermo Fisher Scientific | Cat# A21202, RRID:AB_141607 | (1:1000) |
| Antibody | α-rabbit Alexa Fluor 488 (donkey polyclonal) | Thermo Fisher Scientific | Cat# A21206, RRID:AB_2535792 | (1:1000) |
| Antibody | α-rat Alexa Fluor 488 (donkey polyclonal) | Thermo Fisher Scientific | Cat# A21208, RRID:AB_2535794 | (1:1000) |
| Antibody | α-rabbit Alexa Fluor 568 (donkey polyclonal) | Thermo Fisher Scientific | Cat# A10042, RRID:AB_2534017 | (1:1000) |
| Antibody | α-mouse Alexa Fluor 647 (donkey polyclonal) | Thermo Fisher Scientific | Cat# A31571, RRID:AB_162542 | (1:1000) |
| Chemical compound, drug | Hydroxyurea | Sigma-Aldrich | H8627 | |
| Genetic reagent (D. melanogaster) | w1118 | Bloomington Drosophila Stock Center | RRID:BDSC_5905 | Ryder et al., 2004 |
| Genetic reagent (D. melanogaster) | inc1 | Stavropoulos lab | FLYB:FBal0266013 | Stavropoulos and Young, 2011; BDSC #5,905 background |
| Genetic reagent (D. melanogaster) | inc2 | Stavropoulos lab | FLYB:FBal0162225 | Stavropoulos and Young, 2011; BDSC #5,905 background |
| Genetic reagent (D. melanogaster) | tub-QS; nsyb-Gal4QF | Christopher Potter | Riabinina et al., 2015; Li and Stavropoulos, 2016; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | inc-Gal4 | Stavropoulos lab | Stavropoulos and Young, 2011; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | inc1inc-Gal4 | Stavropoulos lab | Li et al., 2017; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | nsyb-Gal4 | Julie Simpson | Simpson, 2016; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | c253-Gal4 (MB) | Bloomington Drosophila Stock Center | RRID:BDSC_6980 | Pitman et al., 2006; BDSC #5,905 background; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | c309-Gal4 (MB) | Bloomington Drosophila Stock Center | RRID:BDSC_6906 | Connolly et al., 1996; Pitman et al., 2006 Joiner et al., 2006; Aso et al., 2009; BDSC #5,905 background; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | c929-Gal4 (l-LNv) | Amita Sehgal | Hewes et al., 2000; Hewes et al., 2003; Sheeba et al., 2008; Parisky et al., 2008; Shang et al., 2008; iso31 background; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | c584-Gal4 (PI, PPM3) | Amita Sehgal | Martin et al., 1999; Dubowy et al., 2016; iso31 background; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | R69F08-Gal4 (EB) | Mark Wu | Liu et al., 2016; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | R24B11-Gal4 (Helicon) | Bloomington Drosophila Stock Center | RRID:BDSC_49070 | Donlea et al., 2018 |
| Genetic reagent (D. melanogaster) | R23E10-Gal4 (dFB) | Bloomington Drosophila Stock Center | RRID:BDSC_49032 | Donlea et al., 2014 |
| Genetic reagent (D. melanogaster) | NP2721-Gal4 (DPM) | Leslie Griffith | Wu et al., 2011; Haynes et al., 2015; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | DH44-Gal4 | Bloomington Drosophila Stock Center | RRID:BDSC_39347 | Cavanaugh et al., 2014 |
| Genetic reagent (D. melanogaster) | pdf-Gal4 | Stavropoulos lab | Renn et al., 1999 | |
| Genetic reagent (D. melanogaster) | crz-Gal4 | Stavropoulos lab | Tayler et al., 2012 | |
| Genetic reagent (D. melanogaster) | MB004B (pan-MB) | Yoshinori Aso | Sitaraman et al., 2015a | |
| Genetic reagent (D. melanogaster) | MB607B (ɣd) | Yoshinori Aso | Sitaraman et al., 2015a | |
| Genetic reagent (D. melanogaster) | MB370B (α'β'm + α'β'ap) | Yoshinori Aso | Sitaraman et al., 2015a | |
| Genetic reagent (D. melanogaster) | MB185B (αβs) | Yoshinori Aso | Sitaraman et al., 2015a | |
| Genetic reagent (D. melanogaster) | MB594B (αβc) | Yoshinori Aso | Sitaraman et al., 2015a | |
| Genetic reagent (D. melanogaster) | MB-Gal80 | Michael Young | Krashes et al., 2007 | |
| Genetic reagent (D. melanogaster) | UAS-3xFLAG-Inc | Stavropoulos lab | Li et al., 2017; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | UAS-inc-HA | Stavropoulos lab | Li et al., 2017; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | UAS-inc-RNAi | Vienna Drosophila Resource Center | FLYB:FBst0453067 | Dietzl et al., 2007; Stavropoulos and Young, 2011 |
| Genetic reagent (D. melanogaster) | UAS-dcr2 | Bloomington Drosophila Stock Center | RRID:BDSC_24651 | Dietzl et al., 2007; BDSC #5,905 background |
| Genetic reagent (D. melanogaster) | UAS-TrpA1 | Stavropoulos lab | Hamada et al., 2008; BDSC #5,905 background | |
| Genetic reagent (D. melanogaster) | UAS-MyrGFP-2A-RedStinger | Barry Ganetzky | Daniels et al., 2014 | |
| Genetic reagent (D. melanogaster) | 5xUAS-DenMark::smGdP-V5 | Bloomington Drosophila Stock Center | RRID:BDSC_62138 | Nern et al., 2015 |
| Genetic reagent (D. melanogaster) | 5xUAS-IVS-Syt1::smGdP-HA | Bloomington Drosophila Stock Center | RRID:BDSC_62142 | Nern et al., 2015 |
| Genetic reagent (D. melanogaster) | 20xUAS-IVS-CD8-GFP | Bloomington Drosophila Stock Center | RRID:BDSC_32194 | Pfeiffer et al., 2010 |
| Genetic reagent (D. melanogaster) | NP1227-Gal4 | Kathy Nagel | Okada et al., 2009; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | R2-Split Gal4 | Greg Suh | Liu et al., 2016; used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | R72G06-Gal4 | Mark Wu | used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | VT64246-Gal4 | Leslie Griffith | used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | c305a-Gal4 | Leslie Griffith | used in inc[2] rescue screen | |
| Genetic reagent (D. melanogaster) | P{GMR49E09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38692 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR49F01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38694 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR49F02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38695 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR49G06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38707 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR51G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38797 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR53B06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38863 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR53C04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_38871 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR54F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39081 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55A03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39095 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55B12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39103 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55D01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39110 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55D05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39112 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55F07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39128 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55G11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39132 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR56H02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39164 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR56H09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39166 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR58E10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39184 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR58H05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39198 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39209 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59E09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39220 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59H05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39229 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60C01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39240 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60D05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39247 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60H12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39268 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR64A11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39289 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR64F03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39309 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR64G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39316 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65B04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39336 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65D06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39352 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39353 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR67A04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39396 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR69C02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39483 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR71D01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39579 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR72H03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39799 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR74H01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39872 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR76F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39937 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR77H03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39976 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR78A01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_39985 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR78G06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40013 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR79A01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40021 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR79B08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40029 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR83H01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40368 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR85C07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40422 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR87A08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40473 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR92G09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40629 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93C06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40647 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40662 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93H07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40669 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR94D04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40681 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR94E07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40688 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR94F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40694 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR95E08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40710 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR95F11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_40714 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR40B09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41235 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR40E08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41238 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR41G11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41244 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR42F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41253 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60D10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41284 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65C03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41290 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR74B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41301 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR87B02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41316 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65B09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_41353 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR34C12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45219 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR45D10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45323 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60G12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45360 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR23G07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45493 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26C01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45518 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48D06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45774 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR20E01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45837 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25G01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_45851 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR53G07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46041 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR55G02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46070 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR35H03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46205 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR46H09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46275 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR58G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46410 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59H01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46423 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR64D08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46539 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65C05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46554 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65H08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46566 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR69H02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46620 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR70G11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46641 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR71E04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46658 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR72A04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46665 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR73D06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46692 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR56F05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46714 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR77A04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_46976 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR80C12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47059 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR81C04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47087 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR81D04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47094 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR91A08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47148 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR91G01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47175 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR92H11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47211 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93B04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47215 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93D01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47221 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93D06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47224 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93G11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47238 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR94H10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47268 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR16D12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47325 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR16H05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47327 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10E03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47447 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR42E09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47589 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR52A01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47634 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR70A09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47720 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR72F10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47731 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR74G04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47742 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10A11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47839 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10A12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47840 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13C06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47860 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR19G10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47887 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR21C11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47898 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR30F07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47911 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR44G12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47933 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR52F09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_47943 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR28F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48083 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR33H11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48119 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR50A07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48179 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR51B08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48183 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR52C05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48190 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR54H12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48205 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR58F01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48213 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48215 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59C12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48219 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59E04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48221 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10D10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48261 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10H09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48277 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR67B06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48294 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR73H09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48318 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR87C01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48389 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR89C02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48404 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR92A08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48414 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93C08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48417 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR93F02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48422 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR95F03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48433 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10E07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48440 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR11C07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48448 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR12B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48490 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR12D12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48506 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR12G09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48525 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48548 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13D09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48561 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13E04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48565 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13E06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48566 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR13F04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48573 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14C08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48606 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR20F01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48610 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14E05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48642 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14E06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48643 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14E09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48645 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14E12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48647 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14F11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48653 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14G08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48661 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14H02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48664 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR15B07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48678 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR15D11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48690 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR15E09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48696 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR16E03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48727 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR17B12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48759 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR17D02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48764 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR17G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48782 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR18D04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48811 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR18D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48813 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR18F04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48820 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR18G06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48826 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR19F05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48855 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR20F04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48904 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR21C09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48936 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR21D02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48939 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR21D06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48942 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR22C12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48978 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR22E06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_48986 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR22H10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49005 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR23B04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49016 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR23C06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49023 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR23E10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49032 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR23F05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49035 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24A08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49058 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49070 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24C06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49073 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24C07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49074 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24C10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49075 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24E05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49081 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24F03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49086 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR24H03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49098 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25A01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49102 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25A06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49105 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25C01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49115 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25C03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49117 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25E04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49125 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR25H06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49144 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26B04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49158 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49164 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26B12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49165 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26C11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49171 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26E02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49179 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26E07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49182 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR26F09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49194 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR27A02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49207 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10E06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49236 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR14B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49255 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR15B03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49261 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR18G02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49278 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR32D08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49357 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR35F09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49371 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR60F05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49405 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR28E01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49457 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR29A12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49478 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR30B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49522 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR43D09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49553 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR47E07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49568 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49572 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR52F11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49579 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR59A05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49593 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR65H10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49614 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR66A03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49615 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR30G03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49646 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR31F06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49684 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR31G04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49686 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR31H05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49692 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR32E04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49717 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR33H07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49760 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR34B11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49774 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR34C08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49780 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR35B08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49818 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR10G02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49825 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR11E05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49827 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR19C10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49831 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR19E12-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49835 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR20D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49848 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR20E08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49851 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR21H06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49866 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR22F03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49875 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR35D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49908 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR37E08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49958 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR37F05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49961 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR38A11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49980 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR38B06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_49986 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR38E08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50008 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR39C07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50039 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR39E10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50053 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR39G09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50064 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR40C07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50080 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR42D11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50156 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR44B03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50200 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR44B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50202 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR44D02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50205 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR45D05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50227 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR45G01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50241 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR45G05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50243 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR45H11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50248 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR46B05-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50253 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR47D07-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50304 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR47F04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50319 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR47G08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50328 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR47H01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50330 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48A03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50339 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48A08-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50341 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48B10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50352 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48C06-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50357 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48E02-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50367 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48G01-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50381 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48G04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50383 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48H04-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50392 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48H10-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50395 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR48H11-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50396 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR49A09-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50403 | Jenett et al., 2012; used in inc[2] rescue screen |
| Genetic reagent (D. melanogaster) | P{GMR49C03-GAL4}attP2 | Bloomington Drosophila Stock Center | RRID:BDSC_50414 | Jenett et al., 2012; used in inc[2] rescue screen |
Fly food and culture
Request a detailed protocolFly food was prepared in batches containing the following ingredients: 1800 g cornmeal (Labscientific, FLY-8010-20), 1800 ml molasses (Labscientific, FLY-8008-16), 744 g yeast (Labscientific, FLY-8040-20F), 266 g agar (Mooragar, 41084), 56 g methyl paraben (Sigma, H3647), 560 ml alcohol (Fisher, A962P4), 190 ml propionic acid (Fisher, A258500), and 47 l of water. Unless indicated otherwise, crosses were performed with five females and three males in vials (28.5 mm diameter × 95 mm height) containing standard fly food supplemented with dry yeast (Fleischmann, B000LRFVHE). Crosses were cultured at 25°C in 12 hr light–dark (LD) cycles.
To prepare food for conditional induction of the Q-system, solid fly food was melted in a microwave oven and allowed to cool before addition of quinic acid or vehicle. Quinic acid solution was freshly prepared essentially as described (Riabinina et al., 2015). 10 g of quinic acid (Sigma, 138622) was dissolved in 30 ml of water and the pH was adjusted to 6.5 with 10 mM NaOH. A volume of quinic acid solution containing the equivalent of 0.66 g of quinic acid (~2.4 ml) was added for each 10 ml of melted fly food and mixed well; ~12.4 ml was distributed to each empty vial. Food was allowed to cool and subsequently stored at 4°C prior to use. Vehicle food was prepared similarly, substituting an equal volume of water.
Conditional Q-system induction
Request a detailed protocolThree sets of conditional induction experiments were performed. The first set contained vehicle treatment and constitutive, developmental-specific, and adult-specific induction regimens. The second set included vehicle, constitutive induction, and induction from the late third instar larval stage through adulthood. The third set included vehicle, constitutive induction, and a pulse of induction from the late third instar larval stage through pupal stages. Initiation, maintenance, or termination of induction at desired developmental stages was achieved by transferring larvae, pupae, and/or adults to food containing quinic acid or vehicle as described below. Within each set of experiments, w1118 and inc1 controls were exposed to vehicle and quinic acid induction regimens, and all animals underwent the same physical transfers in parallel. Sleep of w1118 and inc1 animals was not altered by exposure to vehicle or quinic acid, as described previously (Li and Stavropoulos, 2016), nor by physical transfer at larval, pupal, or adult stages. Vehicle-treated w1118 and inc1 animals, pooled across all three sets of experiments, are shown in Figure 2B. Two to three independent biological replications were performed for all induction experiments.
In the first set of experiments, developmental-specific induction was achieved by setting crosses on food containing quinic acid, allowing animals to develop and pupate in the same vials, and transferring adult males within 2–3 hr of eclosion to fresh vials with vehicle-containing food to terminate Q-system induction. Adult animals were maintained in these vials for 3–4 days, anesthetized with CO2, and transferred to DAM tubes with vehicle-containing food for measurement of sleep. For adult-specific induction, crosses were set on vehicle food and animals developed in the same vials. Adult males eclosing from these cultures were transferred within 2–3 hr of eclosion to fresh vials with food containing quinic acid, maintained on this food for 3–4 days, and transferred to DAM tubes containing food with quinic acid for measurement of sleep. For constitutive induction and vehicle treatment, food containing quinic acid or vehicle, respectively, was used throughout, along with the same transfer procedure.
In the second set of experiments, induction from the late third instar larval stage through adulthood was achieved as follows: crosses were set on vehicle-containing food and wandering third instar larvae from these cultures were gently collected with blunt forceps and examined under brief phosphate-buffered saline (PBS) immersion to select males by visual identification of gonads as described (Kerkis, 1931). Larvae were transferred to recipient vials containing isogenic w1118 larvae and pre-churned quinic acid food; these recipient cultures were initiated in parallel with experimental crosses to allow food consistency to be maintained during Q-system induction. Adult animals bearing mini-white-marked transgenes were transferred within 2–3 hr of eclosion to fresh vials containing quinic acid food to maintain Q-system induction. Three- to four-day-old adults were subsequently transferred to DAM tubes with food containing quinic acid for measurement of sleep. Constitutive induction and vehicle treatment were performed similarly, using appropriate food and the same transfer procedure.
In the third set of experiments, a pulse of Q-system induction specific to late third instar larval and pupal stages was achieved as follows: crosses were set on vehicle food and male wandering third instar larval progeny were selected and transferred to w1118 recipient vials containing pre-churned quinic acid food as described above. To prevent adult exposure to quinic acid, pupae bearing mini-white-marked transgenes were identified at approximately the P13–P14 stage by pigmented eyes and black wings (Ashburner et al., 2005; Bainbridge and Bownes, 1981) and gently dislodged from vial walls with a paintbrush and transferred to the walls of fresh vials containing vehicle food. Three- to four-day-old adults eclosing from these vials were transferred to DAM tubes containing vehicle food for measurements of sleep. Constitutive induction and vehicle treatment were performed similarly, using appropriate food and the same transfer procedure.
inc2 rescue screen
Request a detailed protocolinc2 virgins were crossed to male flies carrying Gal4 transgenes and a minimum of five male progeny were screened for each genotype. A total of 277 Gal4 lines were screened, including 266 randomly selected drivers and 11 drivers previously characterized for expression in sleep-regulatory circuits. To select random lines, 4088 lines from the FlyLight collection available from the Bloomington Drosophila Stock Center were assigned sample numbers. Using the randperm command in Matlab, 300 lines were randomly selected. Expression patterns for these lines in the Janelia Flylight database were examined; 84 lines were excluded due to very low levels of expression, very broad expression patterns unlikely to be useful for functional mapping, or because expression data were unavailable. Expression patterns for the remaining 216 lines ranged from broad to sparse. This procedure for random selection was applied iteratively to yield 266 lines. Top-ranking hits from the initial screen were rescreened in independent crosses. Rescreening of c253-Gal4 and c309-Gal4 was performed after backcrossing each line six generations to an isogenic w1118 stock (BDSC #5905) (Ryder et al., 2004).
MB ablation
Request a detailed protocolMB ablation was performed essentially as described previously (de Belle and Heisenberg, 1994). Egg collection was performed on grape juice agar plates containing a spot of rehydrated dry yeast. w1118 larvae at the first instar stage were transferred to a well of a 24-well plate bearing a spot of rehydrated dry yeast paste, containing water vehicle or 50 mg/ml hydroxyurea (Sigma, H8627). After 4–5 hr, larvae were collected and washed briefly with distilled water on a Nitex mesh filter (Genesee Scientific, 57–102) to remove yeast and subsequently transferred to vials containing standard food. Vials were cultured at 25°C in LD cycles and adult animals eclosing from these cultures were assayed for sleep as described below. MB ablation was verified in adult brains in a separate cohort of animals by staining with anti-FasII primary antibody (1:50, DSHB) and Alexa 488-conjugated donkey anti-mouse secondary as described below. Vehicle-treated animals exhibited MB lobes demarcated with FasII signal (100%, n = 9), while hydroxyurea-treated animals exhibited complete MB ablation as indicated by the lack of residual FasII staining (100%, n = 12); FasII signal within the EB was observed in all brains, providing a control for staining of the MB.
Immunohistochemistry
Request a detailed protocolAll fixing, washing, and incubation steps for immunohistochemistry were performed on a nutator. To assess conditional induction of inc-HA using the Q-system, larval, pupal, and adult brains were dissected from inc1; UAS-inc-HA/tub-QS; nsyb-Gal4QF/+ males. Wandering third instar male larvae were selected by visual identification of gonads as described above. Larval brains were dissected in ice-cold PBS, fixed with 4% paraformaldehyde in PBS for 30 min at room temperature, and washed 3× 15 min in PBS containing 0.2% Triton X-100 (PBST). Male pupae at stage P13–P14 were identified by the staging criteria described above and the presence of sex combs. Pupal brains were dissected in ice-cold PBST, fixed with 4% paraformaldehyde in PBST for 30 min at room temperature, and washed 3× 15 min in PBST. To prepare adult brains, 2- or 4-day-old whole male adults were fixed with 4% paraformaldehyde in PBST for 3 hr at 4°C and washed 3× 15 min in PBST at room temperature prior to brain dissection in PBST. After dissection, all brains were blocked with 5% normal donkey serum (NDS) (Lampire Biological, 7332500) in PBST at room temperature for 30–60 min. Samples were incubated overnight at 4°C in rat anti-HA (1:100; Sigma, 11867431001) and mouse anti-Brp (1:20, DSHB, nc82) antibodies prepared in 5% NDS in PBST. Brains were subsequently washed 3× 15 min in PBST at room temperature, incubated overnight at 4°C in Alexa 488 donkey anti-rat (1:1000; Life Technologies, A21208) and Alexa 647 donkey anti-mouse (1:1000, Life Technologies A31571) antibodies prepared in 5% NDS in PBST, washed 3× 15 min at room temperature in PBST, and mounted on microscope slides (Fisher, 1255015) in Vectashield (Vector Labs, H-1000).
For all other immunohistochemistry, adult brains of 4-day-old males were dissected in PBST, fixed with 4% paraformaldehyde in PBST for 30 min at room temperature, and washed 3× 20 min in PBST at room temperature. Brains of male wandering third instar larvae and stage P13–P14 pupae were dissected, fixed, and stained as described above for Q-system experiments. Primary antibodies were mouse anti-FLAG (1:100; Sigma, F1804), rabbit anti-GFP (1:2000; Fisher, A11122), mouse anti-GFP (1:1000; DSHB, GFP-G1), rabbit anti-dsRed (1:1000; Takara, 632496), and mouse anti-Brp (1:50, DSHB, nc82). Secondary antibodies were Alexa 488 donkey anti-rabbit (1:1000; Life Technologies, A21206), Alexa 488 donkey anti-mouse (1:1000; Life Technologies, A21202), and Alexa 568 donkey anti-rabbit (1:1000; Life Technologies, A10042).
Imaging and quantitation of neuron number and cluster number
Request a detailed protocolAll imaging was performed on a Zeiss LSM800 confocal microscope, using a 10X air objective to capture z-stacks at 512 × 512 pixel resolution with 1 μM z-slices, unless indicated otherwise. All imaging settings were identical for each experiment comprising control and experimental brains stained in parallel.
To quantify MB neuron numbers, wild-type and inc1 brains expressing UAS-MyrGFP-2A-RedStinger under the control of split-Gal4 drivers were imaged as described above. For each neuron subtype, wild-type and inc1 brains were assigned sample numbers and a subset, randomly selected using the randperm command in Matlab, was imaged at higher resolution with a 63X oil objective. Both hemispheres of brains were imaged, capturing dsRed and myr-GFP channels separately. Only a single hemisphere could be imaged for two wild-type brains, one each in the γd and α´/β´ groups, due to sample compression by the objective. z-stacks encompassing nuclei were captured at 512 × 512 resolution for γd neurons and at 1024 × 1024 resolution for α´/β´ and α/βc neurons; 2 μM z-slices were used to ensure that all nuclei (diameter ~3 μM) were segmented in at least one optical section.
High resolution z-stacks were assigned a random letter code and neurons were counted in a single-blind manner by two independent experimenters. Nuclei of γd and α´/β´ neurons exhibited minimal overlap along the z-axis, allowing nuclei to be counted in maximum intensity z-projections using the Cell Counter plug-in in ImageJ; visual inspection of z-stacks in parallel allowed overlapping nuclei to be differentiated. Dense distribution of α/βc neurons prohibited accurate counting in single maximum intensity z-projections; maximum intensity z-projections were generated for every 10 z-slices, yielding three to four maximum intensity z-projections representing 20 μM each. To improve visualization of densely clustered α/βc nuclei, background was subtracted using a rolling ball/sliding paraboloid algorithm (radius set to the size of the largest nucleus: 50 pixels) and image intensity display range was adjusted (minimum: 5; maximum: 175). Processed maximum intensity z-projections representing 20 μM each were then merged into a single z-stack for manual counting using the Cell Counter plug-in in ImageJ; to avoid double-counting of nuclei segmented in adjacent z-projections, the original unprocessed z-stack was examined in parallel. The variation in MB neuron counts between experimenters, calculated as the absolute difference between the two counts divided by their mean, was (mean ± SEM) 2.0% ± 0.2% for γd; 1.5% ± 0.3% for α´/β´; and 2.6% ± 0.3% for α/βc. Where neuron counts were different for a given hemisphere, the average was plotted. Numbers of γd neurons in wild-type animals were intermediate between those reported in prior studies (Aso et al., 2014a; Shih et al., 2019), while numbers of α´/β´ and α/βc neurons were lower, likely reflecting conservative assignment of nuclei in our study and the use of different antibodies and reporters (Aso et al., 2014a; Shih et al., 2019). α´/β´ counts obtained using the MB370B driver were similar to previously reported numbers of α´/β´m neurons; because MB370B labels α´/β´m neurons strongly and α´/β´ap neurons weakly, the lower absolute numbers of α´/β´ neurons in our studies may reflect detection sensitivity and correspond chiefly to α´/β´m neurons.
To count the number of α/βc neuron clusters in wild-type and inc1 brains, the same randomly selected samples used to quantify neuron numbers were assessed in a single-blind manner by two independent experimenters. Each z-stack was analyzed using a combination of visual inspection of z-sections and rotating the image stack in three dimensions using the 3D Viewer plug-in in ImageJ (threshold: 0; resampling factor: 2). A group of nuclei distributed continuously along all axes was classified as a cluster; a continuous gap at least one nuclear diameter in width across all axes was used to define cluster edges and discrete clusters. Cluster counts were identical for wild-type brains; total cluster counts for inc1 brains differed by 8.2% ± 0.2% (mean ± SEM) between experimenters. Where cluster counts were different for a given hemisphere, the average was plotted.
DH44 and dFB somata were counted in a single-blind manner by two independent experimenters as described for γd and α´/β´ neurons, using DH44-Gal4 to drive UAS-MyrGFP-2A-RedStinger and 23E10-Gal4 to drive 5× UAS-IVS-Syt1::smGdP-HA. Numbers of dFB and DH44 neurons were identical between two independent experimenters.
Analysis of axonal projections and dendritic volume
Request a detailed protocolTo analyze axonal projections and dendritic volume, image stacks were captured using a 10X objective at 512 × 512 resolution with 1 μM z-slices. Axonal projection defects were assessed in maximum intensity z-projections. The number of horizontal and/or vertical lobes missing myr-GFP signal entirely was counted for each brain. To quantify dendritic volume, the Threshold command in ImageJ was applied to z-stacks to select dendrites based on DenMark immunofluorescence; high signal to noise allowed unambiguous demarcation of dendrites and clear separation from background. The same minimum and maximum threshold values were applied to all wild-type and inc1 brains stained in parallel in an experiment and captured the entirety of dendritic signal for all samples. A single rectangular region of interest of minimal area encompassing dendritic signals from both brain hemispheres across all z-slices was drawn for each z-stack. Dendritic volume was quantified using the Voxel Counter plug-in in ImageJ.
Sleep analysis
Request a detailed protocolThree- to four-day-old male flies eclosing from LD-entrained cultures raised at 25°C were loaded in glass tubes (5 mm diameter × 65 mm length) containing standard food or appropriate food for Q-system experiments as described above. Animals were monitored for 5–7 days at 25°C in LD cycles using DAM2 monitors (Trikinetics). Locomotor activity data were collected in 1 min bins. Inactive periods of 5 min or longer were classified as sleep. The first 36–48 hr of data were discarded to allow acclimation of animals to tubes, and 3–5 integral days of data were analyzed beginning with ZT0. Dead animals were excluded from analysis by a combination of automated filtering and visual inspection of locomotor traces. Matlab code used to analyze sleep is available in Source code 1.
Thermogenetic activation
Request a detailed protocolCrosses were set on standard fly food as described above and cultured at 21.5°C. One- to four-day-old male flies eclosing from these cultures were assayed for 5 days in LD cycles. Animals were maintained at 21.5°C for the first 60–72 hr of the assay, including 36–48 hr of acclimation and the subsequent baseline day beginning at ZT0. Temperature was increased to 28.5°C for 24 hr to activate dTrpA1, followed by 24 hr of recovery at 21.5°C. The percent change in sleep was calculated for each animal by subtracting the amount of sleep on the baseline day from the amount of sleep on the activation day and dividing this difference by the amount of sleep on the baseline day. The percent change in sleep for individual animals was averaged for each genotype.
Gal4 drivers used to express TrpA1 were as follows: pan-MB, MB004B split-Gal4; MB, c253-Gal4 and c309-Gal4; EB, R69F08-Gal4; DPM, NP2721-Gal4; Helicon, R24B11-Gal4; l-LNV, c929-Gal4; PI, PPM3, c584-Gal4.
Statistics
One-way analysis of variance (ANOVA) and Tukey post hoc tests were used for comparisons between more than two groups of animals for total sleep, daytime sleep, nighttime sleep, and sleep bout number; for comparisons of these sleep parameters between two groups, unpaired two-sided Student’s t-tests were used. Kruskal–Wallis tests and Dunn’s post hoc tests were used for comparisons of sleep bout length between more than two groups of animals; for comparison between two groups, Mann–Whitney tests were used. One-way ANOVA and Dunnett’s post hoc tests were used for comparisons of percent change in sleep. Unpaired two-sided Welch’s t-tests were used for pairwise comparisons of neuron number, cluster number, and dendrite volume.
Data availability
Data for all figures and code used to analyze sleep are included in the supporting files.
References
-
Sleep in children with neurodevelopmental disabilitiesNeuropediatrics 46:199–210.https://doi.org/10.1055/s-0035-1550151
-
BookDrosophila: A Laboratory Handbook (2nd ed)New York: Cold Spring Harbor Laboratory Press.
-
The mushroom body of adult Drosophila characterized by GAL4 driversJournal of Neurogenetics 23:156–172.https://doi.org/10.1080/01677060802471718
-
Staging the metamorphosis of Drosophila melanogasterJournal of Embryology and Experimental Morphology 66:57–80.https://doi.org/10.1242/dev.66.1.57
-
Drosophila Hyperkinetic mutants have reduced sleep and impaired memoryThe Journal of Neuroscience 27:5384–5393.https://doi.org/10.1523/JNEUROSCI.0108-07.2007
-
Reduced sleep in Drosophila Shaker mutantsNature 434:1087–1092.https://doi.org/10.1038/nature03486
-
Neural stem cells: balancing self-renewal with differentiationDevelopment 135:1575–1587.https://doi.org/10.1242/dev.014977
-
Morphological and functional reversal of phenotypes in a mouse model of Rett syndromeBrain: A Journal of Neurology 135:2699–2710.https://doi.org/10.1093/brain/aws096
-
Drosophila mushroom body mutants are deficient in olfactory learningJournal of Neurogenetics 2:1–30.https://doi.org/10.3109/01677068509100140
-
Mushroom body memoir: from maps to modelsNature Reviews. Neuroscience 4:266–275.https://doi.org/10.1038/nrn1074
-
Structural brain mutants: mushroom body defect (mud): a case studyJournal of Neurogenetics 23:42–47.https://doi.org/10.1080/01677060802471700
-
Proliferation pattern of postembryonic neuroblasts in the brain Drosophila melanogasterDevelopmental Biology 149:134–148.https://doi.org/10.1016/0012-1606(92)90270-q
-
Induction of Mutant Sik3Sleepy Allele in Neurons in Late Infancy Increases Sleep NeedThe Journal of Neuroscience 41:2733–2746.https://doi.org/10.1523/JNEUROSCI.1004-20.2020
-
Development: The decade of the developing brainCurrent Opinion in Neurobiology 10:599–611.https://doi.org/10.1016/s0959-4388(00)00136-7
-
Optogenetic induction of aversive taste memoryNeuroscience 222:173–180.https://doi.org/10.1016/j.neuroscience.2012.07.028
-
Evaluation of Ligand-Inducible Expression Systems for Conditional Neuronal Manipulations of Sleep in DrosophilaG3: Genes, Genomes, Genetics 6:3351–3359.https://doi.org/10.1534/g3.116.034132
-
Central complex substructures are required for the maintenance of locomotor activity in Drosophila melanogasterJournal of Comparative Physiology. A, Sensory, Neural, and Behavioral Physiology 185:277–288.https://doi.org/10.1007/s003590050387
-
Limited taste discrimination in DrosophilaPNAS 107:14833–14838.https://doi.org/10.1073/pnas.1009318107
-
Gamma-aminobutyric acid (GABA)-mediated neural connections in the Drosophila antennal lobeThe Journal of Comparative Neurology 514:74–91.https://doi.org/10.1002/cne.21971
-
Drosophila larvae establish appetitive olfactory memories via mushroom body neurons of embryonic originThe Journal of Neuroscience 30:10655–10666.https://doi.org/10.1523/JNEUROSCI.1281-10.2010
-
Truncation of the Shaker-like voltage-gated potassium channel, Kv1.1, causes megencephalyThe European Journal of Neuroscience 18:3231–3240.https://doi.org/10.1111/j.1460-9568.2003.03044.x
-
Improved and expanded Q-system reagents for genetic manipulationsNature Methods 12:219–222.https://doi.org/10.1038/nmeth.3250
-
Drosophila aPKC regulates cell polarity and cell proliferation in neuroblasts and epitheliaThe Journal of Cell Biology 163:1089–1098.https://doi.org/10.1083/jcb.200306079
-
Large ventral lateral neurons modulate arousal and sleep in DrosophilaCurrent Biology 18:1537–1545.https://doi.org/10.1016/j.cub.2008.08.033
-
Parallel circuits control temperature preference in Drosophila during ageingNature Communications 6:7775.https://doi.org/10.1038/ncomms8775
-
Nuclear Transcriptomes of the Seven Neuronal Cell Types That Constitute the Drosophila Mushroom BodiesG3: Genes, Genomes, Genetics 9:81–94.https://doi.org/10.1534/g3.118.200726
-
Rationally subdividing the fly nervous system with versatile expression reagentsJournal of Neurogenetics 30:185–194.https://doi.org/10.1080/01677063.2016.1248761
-
Control of Sleep by Dopaminergic Inputs to the Drosophila Mushroom BodyFrontiers in Neural Circuits 9:73.https://doi.org/10.3389/fncir.2015.00073
-
Sleep in Children with Autism Spectrum DisorderCurrent Psychiatry Reports 19:34.https://doi.org/10.1007/s11920-017-0782-x
-
Dedifferentiation of neurons precedes tumor formation in Lola mutantsDevelopmental Cell 28:685–696.https://doi.org/10.1016/j.devcel.2014.01.030
-
Neuronal assemblies of the Drosophila mushroom bodyThe Journal of Comparative Neurology 508:711–755.https://doi.org/10.1002/cne.21692
-
The molecular biology of axon guidanceScience 274:1123–1133.https://doi.org/10.1126/science.274.5290.1123
-
Sleep and synaptic homeostasis: a hypothesisBrain Research Bulletin 62:143–150.https://doi.org/10.1016/j.brainresbull.2003.09.004
-
Epidermal growth factor signaling induces behavioral quiescence in Caenorhabditis elegansNature Neuroscience 10:1300–1307.https://doi.org/10.1038/nn1981
-
Neural inductionAnnual Review of Cell and Developmental Biology 15:411–433.https://doi.org/10.1146/annurev.cellbio.15.1.411
-
Kv1.1-dependent control of hippocampal neuron number as revealed by mosaic analysis with double markersThe Journal of Physiology 590:2645–2658.https://doi.org/10.1113/jphysiol.2012.228486
-
Requirement of Cul3 for axonal arborization and dendritic elaboration in Drosophila mushroom body neuronsThe Journal of Neuroscience 25:4189–4197.https://doi.org/10.1523/JNEUROSCI.0149-05.2005
Article and author information
Author details
Nicholas Stavropoulos

Funding
Howard Hughes Medical Institute (International Student Research Fellowship)
- Qiuling Li
National Institute of Neurological Disorders and Stroke (R01NS112844)
- Nicholas Stavropoulos
National Institute of Neurological Disorders and Stroke (R21NS111304)
- Nicholas Stavropoulos
G. Harold and Leila Y. Mathers Foundation
- Nicholas Stavropoulos
Whitehall Foundation (2013-05-78)
- Nicholas Stavropoulos
Alfred P. Sloan Foundation
- Nicholas Stavropoulos
Leon Levy Foundation
- Nicholas Stavropoulos
Brain and Behavior Research Foundation (NARSAD Young Investigator)
- Nicholas Stavropoulos
Sleep Research Society Foundation (J. Christian Gillin, M.D. Research Grant)
- Nicholas Stavropoulos
Irma T. Hirschl Trust (Career Scientist Award)
- Nicholas Stavropoulos
The funders had no role in study design, data collection, and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank C Desplan, N Ringstad, M Shirasu-Hiza, and members of the Stavropoulos lab for comments on the manuscript; Y Aso, B Ganetzky, L Griffith, W Joiner, W Li, K Nagel, C Potter, A Sehgal, J Simpson, G Suh, M Wu, M Young, the Bloomington Drosophila Stock Center, the Vienna Drosophila Resource Center, and the Janelia Flylight collection for fly stocks; and DSHB for antibodies. This work was supported by an International Student Research Fellowship from the Howard Hughes Medical Institute (HHMI) to QL and by grants from the National Institutes of Health (R01NS112844 and R21NS111304), the Mathers Foundation, Whitehall Foundation grant 2013-05-78, fellowships from the Alfred P Sloan and Leon Levy Foundations, a NARSAD Young Investigator Award from the Brain and Behavior Foundation, the J Christian Gillin, M.D. Research Award from the Sleep Research Society Foundation, and a Career Scientist Award from the Irma T Hirschl/Weill-Caulier Trust to NS.
Copyright
© 2021, Li et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,040
- views
-
- 265
- downloads
-
- 15
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 15
- citations for umbrella DOI https://doi.org/10.7554/eLife.65437
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
insomniac links the development and function of a sleep-regulatory circuit
eLife 10:e65437.
https://doi.org/10.7554/eLife.65437




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}