Tbx5 drives Aldh1a2 expression to regulate a RA-Hedgehog-Wnt gene regulatory network coordinating cardiopulmonary development
- Center for Stem Cell and Organoid Medicine (CuSTOM), Division of Developmental Biology, Perinatal Institute, Cincinnati Children’s Hospital Medical Center, United States
- Department of Pediatrics, University of Chicago, United States
- Department of Pathology, University of Chicago, United States
- Department of Human Genetics, University of Chicago, United States
- University of Cincinnati, College of Medicine, Department of Pediatrics, United States
Abstract
The gene regulatory networks that coordinate the development of the cardiac and pulmonary systems are essential for terrestrial life but poorly understood. The T-box transcription factor Tbx5 is critical for both pulmonary specification and heart development, but how these activities are mechanistically integrated remains unclear. Here using Xenopus and mouse embryos, we establish molecular links between Tbx5 and retinoic acid (RA) signaling in the mesoderm and between RA signaling and sonic hedgehog expression in the endoderm to unveil a conserved RA-Hedgehog-Wnt signaling cascade coordinating cardiopulmonary (CP) development. We demonstrate that Tbx5 directly maintains expression of aldh1a2, the RA-synthesizing enzyme, in the foregut lateral plate mesoderm via an evolutionarily conserved intronic enhancer. Tbx5 promotes posterior second heart field identity in a positive feedback loop with RA, antagonizing a Fgf8-Cyp regulatory module to restrict FGF activity to the anterior. We find that Tbx5/Aldh1a2-dependent RA signaling directly activates shh transcription in the adjacent foregut endoderm through a conserved MACS1 enhancer. Hedgehog signaling coordinates with Tbx5 in the mesoderm to activate expression of wnt2/2b, which induces pulmonary fate in the foregut endoderm. These results provide mechanistic insight into the interrelationship between heart and lung development informing CP evolution and birth defects.
Introduction
Proper integration of the cardiac and pulmonary systems begins during early embryogenesis and is essential for terrestrial life. A key feature of cardiopulmonary (CP) development is evolutionarily conserved bi-directional paracrine signaling between the foregut endoderm, which gives rise to pulmonary epithelium, and the cardiogenic mesoderm (Xie et al., 2012; Rankin et al., 2016; Steimle et al., 2018). The interplay between these signals and lineage-specific transcription factors (TFs) to control lineage-specific gene regulatory networks (GRNs) for heart and lung morphogenesis is poorly understood. A better understanding of these CP GRNs will provide insight into the orchestration of heart and lung development and inform the molecular basis of life-threatening CP birth defects.
The vertebrate heart forms from two distinct populations of cardiac progenitor cells in the anterior lateral plate mesoderm (lpm), termed the first and second heart fields, respectively (FHF and SHF; Kelly et al., 2014). The FHF differentiates first and forms the early heart tube, including portions of the two atria and left ventricle. The SHF contributes to the anterior and posterior poles of the developing heart and differentiates later. The anterior SHF (aSHF) is characterized by the expression of Fgf8, Fgf10, and Tbx1 and generates the right ventricle, portions of the outflow tract, and pharyngeal mesoderm (Rochais et al., 2009; Kelly et al., 2014). The posterior SHF (pSHF) is characterized by the expression of Tbx5, Osr1, and Foxf1, (Xie et al., 2012; Hoffmann et al., 2014; Steimle et al., 2018) and generates the atrial septum and sinus venosus. A subset of the pSHF marked by Isl1, Gli1, and Wnt2 expression contains multipotent cardiopulmonary progenitors (CPPs) that give rise to lung mesenchyme, pulmonary vasculature, and myocardium of the inflow tract (Peng et al., 2013). CPPs are both the recipient and source of reciprocal signaling with the adjacent pulmonary endoderm essential for heart and lung development.
The T-box TF Tbx5 is a key player coordinating CP organogenesis. Numerous studies in vertebrate animal models over the past 20 years have documented conserved Tbx5 expression initially in the FHF and then later in the pSHF. Heterozygous mutations in humanTBX5 cause Holt–Oram syndrome with congenital heart anomalies including atrial septal defects and hypoplastic left heart (Li et al., 1997; Ryan and Chin, 2003). Tbx5−/− null mutant mice die between E9.5 and E10.5 with severe cardiac deficiencies and a failure of pulmonary development (Bruneau et al., 2001; Xie et al., 2012; Hoffmann et al., 2014; Steimle et al., 2018; De Bono et al., 2018). While significant advances have been made in understanding the TBX5-regulated GRNs controlling cardiomyocyte development (Kathiriya et al., 2021), how TBX5 coordinates heart and lung organogenesis is less clear. We recently showed that Tbx5 is non-cell-autonomously required to activate expression of Hedgehog (Hh) ligands in the adjacent foregut endoderm, which are essential for both heart and lung development (Steimle et al., 2018). Endodermal Hh signals back to the lpm stimulating Gli TFs, which cooperate with Tbx5 to directly activate expression of mesodermal Wnt2/2b signals that are essential to induce pulmonary fate in the adjacent foregut endoderm (Steimle et al., 2018; Goss et al., 2009; Harris-Johnson et al., 2009). Tbx5 is thereby required for establishing the reciprocal mesoderm—endoderm—mesoderm signaling loop that coordinates CP development. A major unanswered question is how Tbx5 non-cell-autonomously activates sonic hedgehog (Shh) ligand expression in the foregut endoderm.
Retinoic acid (RA) signaling is a strong candidate for the Tbx5-dependent signal that activates endodermal Shh expression. Like Tbx5 mutants, RA deficient embryos have reduced Shh expression in the foregut (Wang et al., 2006; Rankin et al., 2016) and manifest multiple cardiac and pulmonary defects, like Tbx5 mutants (Zaffran et al., 2014; Xavier-Neto et al., 2015; Perl and Waxman, 2019; Sirbu et al., 2020). RA, a derivative of vitamin A, is produced in the lpm by the aldehyde dehydrogenase enzyme Aldh1a2, which converts cellular retinaldehyde into RA (Niederreither et al., 1999; Metzler and Sandell, 2016). Aldh1a2 and Tbx5 are co-expressed in a subset of the pSHF and previous studies have shown that RA patterns the SHF by promoting Tbx5+ pSHF identity whilst repressing Tbx1+ aSHF fate (Niederreither et al., 2001; Sirbu et al., 2008; Ryckebusch et al., 2008; Deimling and Drysdale, 2009; Ryckebüsch et al., 2010; Rydeen and Waxman, 2016; Rankin et al., 2016; De Bono et al., 2018). How the regional production of RA is controlled to pattern the SHF and regulate shh expression remains unknown.
In this study, we demonstrate that RA signaling is the link between mesodermal Tbx5 activity and endodermal Shh expression. We further define the molecular basis by which Tbx5 drives RA signaling and by which RA signaling drives Shh expression. Specifically, Tbx5 directly maintains expression of Aldh1a2 in pSHF via an evolutionarily conserved intronic enhancer, and Tbx5/Aldh1a2-dependent RA signaling directly activates Shh transcription in the foregut endoderm via an evolutionarily conserved MACS1 endoderm enhancer. We conclude that Tbx5 coordinates CP development by controlling expression of the RA-producing enzyme Aldh1a2, and that this RA signal initiates a mesenchyme-epithelial signaling cascade that controls both Hh/Wnt-dependent lung induction and SHF patterning. Hh/Gli and Tbx5 then cooperate to promote Wnt2/2b expression and lung induction. This work unifies previously unconnected observations to resolve the molecular basis of a mesoderm-endoderm-mesoderm signaling network that coordinates pulmonary induction and SHF cardiac patterning.
Results
Tbx5 regulates cardiopulmonary development and Aldh1a2 expression in mouse
To investigate the Tbx5-regulated GRN that coordinates heart and lung development, we re-examined our published RNA-seq data of CP tissue (containing both foregut mesoderm and endoderm) micro-dissected from wild-type (WT) and Tbx5−/− mouse embryos at E9.5 (Steimle et al., 2018). Differential expression analysis revealed 1588 upregulated genes and 1480 downregulated genes and in the absence of Tbx5 (≥1.5 fold change and 5% FDR) (Figure 1A; Figure 1—source data 1; Steimle et al., 2018). Reduced expression of Hh signaling components (Shh, Ihh, and Patch2), Hh-targets (Hhip and Gli1), the lung-inducing Wnt2/2b ligands, and pulmonary progenitor marker Nkx2-1, indicated a loss of pulmonary fate in Tbx5−/− mutant CPP tissue (Figure 1B). We examined the relationship between pSHF/lung and aSHF/pharyngeal transcripts in Tbx5−/− embryos by intersecting the Tbx5-regulated transcriptome with gene sets from recent single-cell RNA-seq studies of the developing E7.75–E9.5 mouse heart and foregut that define aSHF, pSHF, pharynx, and lung progenitor cells (de Soysa et al., 2019; Han et al., 2020, Supplementary files 2 and 3). We found that 25% of genes (91/366) specifically enriched in aSHF or pharyngeal cells, but only 5% of genes (10/213) specifically enriched in pSHF or lung progenitors, overlapped with transcripts upregulated in the Tbx5−/− mutants (*p<0.0001, hypergeometric probability test, HGT) (Figure 1A). On the other hand, 34% of the pSHF or lung marker genes (72/213) but only 6% of the pSHF+ pharynx enriched genes (21/366) were downregulated in Tbx5−/− mutants (*p<0.001, HGT) (Figure 1A). The aSHF-enriched genes upregulated in Tbx5−/− CP tissue included well-known patterning genes Hand1, Irx3, Irx5, Mef2c, Meg3, and Tlx1 as well as FGF signaling components and targets including Fgf8, Fgf10, Spry1, Spry2, and Dusp6 (Figure 1B). Gene set enrichment analysis (GSEA) confirmed a statistically significant overrepresentation of aSHF/pharynx genes among upregulated genes (normalized enrichment score [NES]=1.58; p<0.0001) and overrepresentation of pSHF/lung transcripts among the downregulated genes (NES=–1.99; p<0.0001) in the Tbx5−/− CP tissue (Figure 1—figure supplement 1A,B). Thus, Tbx5 mutant mouse embryos exhibit a reduction of the pSHF transcriptional program and gain of aSHF gene expression in the pSHF domain, consistent with recent reports (De Bono et al., 2018).
Figure 1 with 1 supplement see all
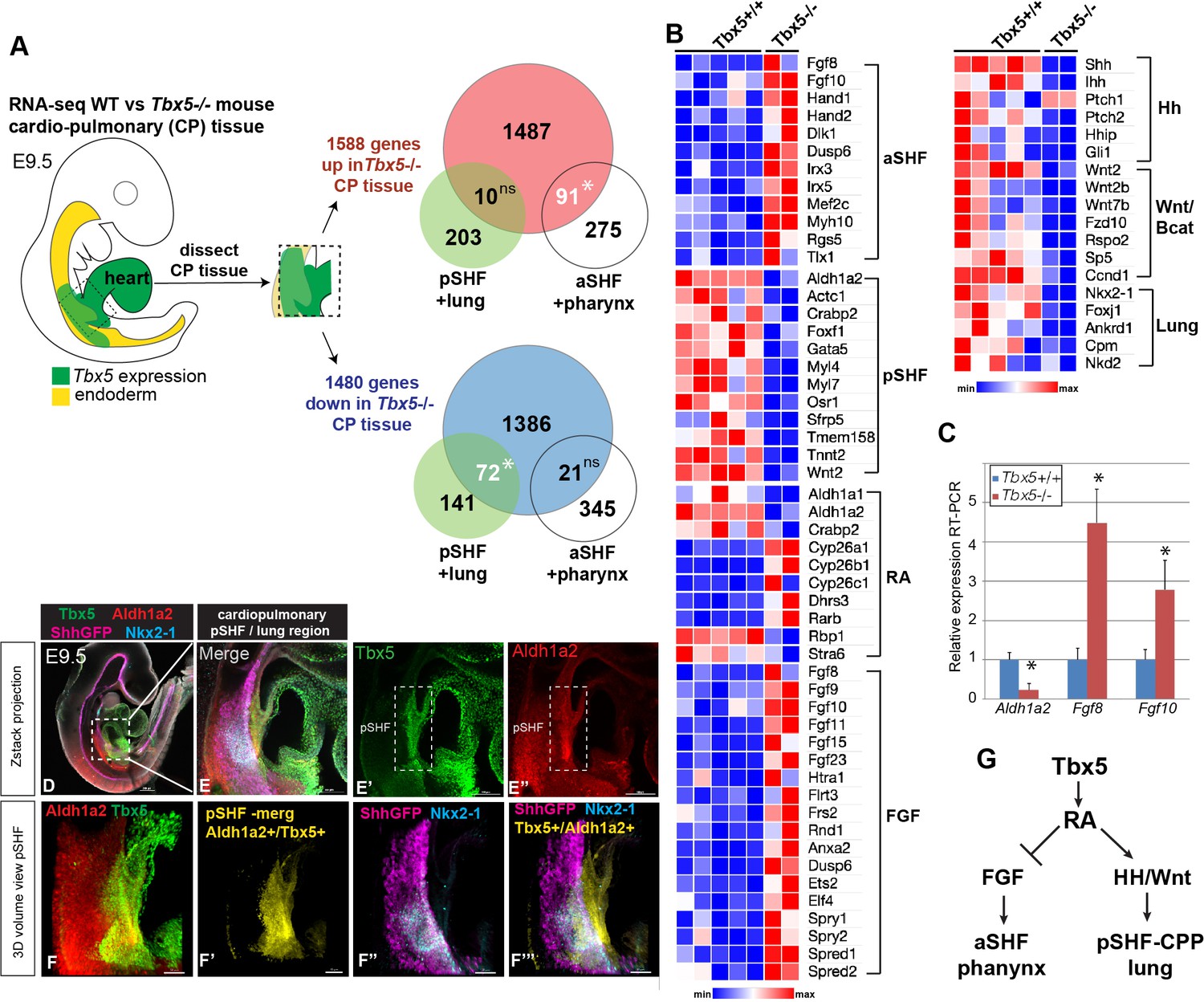
Tbx5 is required for posterior cardiopulmonary (CP) development.
(A) Schematic of an E9.5 mouse embryo highlighting the dissected CP tissue (containing foregut mesoderm and endoderm) profiled by bulk RNA-seq. Venn diagrams show genes differentially expressed in wild-type (WT) versus Tbx5−/− CP tissue (>1.5 fold change, 5% FDR; Steimle et al., 2018, GSE75077) intersected with gene sets from single-cell RNA-seq studies defining aSHF+ pharynx cells versus pSHF/CPP+ lung progenitor cells (de Soysa et al., 2019, GSE126128; Han et al., 2020, GSE136689) (Supplementary files 2 and 3). Statistically significant intersection based on hypergeometric tests. *p<0.0001. (B) Transcriptome analysis of Tbx5−/− CP tissue suggests disrupted SHF pattering and failed pulmonary development with reduced RA and increased FGF signaling. Heat map of selected differentially expressed genes in WT Tbx5+/+ (n=5) and Tbx5−/− mutant (n=2) CP tissue grouped by domain of expression or pathway. (C) RT-qPCR validation of decreased Aldh1a2 and increased Fgf8, Fgf10 expression in E9.5 WT and Tbx5−/− CP tissue. Relative mean expression+S.D. *p<0.05 Student’s t-test relative to WT littermates. (D–F) Whole-mount immunostaining of E9.5 Shh:GFP mouse embryos show that Tbx5 (green) and Ald1a2 (red) expression overlaps in a subset of the pSHF (yellow in (F’) and (F”’)) adjacent to the Nkx2-1+/Shh:GFP+ pulmonary domain (F”, F”’). Scale bar in (D) = 200 μM, (E–E”) = 100 μM, and (F–F”’) = 50 μM. (G) Model of the proposed Tbx5-RA signaling networks in the cardiopulmonary tissue. Also see Figure 1—figure supplement 1, Figure 1—figure supplement 1– tables 1-3. aSHF, anterior second heart field; fg endo, foregut endoderm; ns, not significant; oft, outflow tract; pSHF, posterior SHF; RA, retinoic acid; ventr, ventricle.
-
Figure 1—source data 1
Differentially expressed genes in mouse E9.5 micro-dissected cardiopulmonary progenitor (CPP) tissue based on bulk RNA-seq (Steimle et al., 2018, GSE GSE75077).
1588 upregulated genes and 1480 downregulated genes were observed in Tbx5 knockout CPP tissue (≥1.5 fold change and 5% FDR).
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig1-data1-v2.xlsx
These changes in pSHF gene expression suggested a possible loss of RA signaling. Indeed, genes that promote RA signaling were downregulated in Tbx5−/− CP tissue, including Aldh1a2, Crabp2, which promotes nuclear shuttling of RA, and Rbp1, a cytosolic chaperone of the RA precursor retinol (Figure 1B). On the other hand, enzymes that attenuate RA signaling, including Cyp26a1, Cyp26b1, Cyp26c1, and Dhrs3, were increased in Tbx5−/− pSHF/CPP tissue. Reduced RA-signaling in Tbx5−/− CP tissue was also consistent with increased expression of multiple TGFβ pathway components and targets (Figure 1—figure supplement 1C), which are known to be suppressed by RA during foregut and heart development (Chen et al., 2007; Li et al., 2010; Ma et al., 2016).
The observation that Aldh1a2 expression was reduced in the Tbx5−/− pSHF whereas FGF signaling components and targets were increased is consistent with the known role of RA in negatively regulating Fgf8/Fgf10+ aSHF fate (Ryckebusch et al., 2008; Sirbu et al., 2008; Rydeen and Waxman, 2016). RT-qPCR of dissected E9.5 CP tissue validated the RNA-seq analysis with Aldh1a2 being dramatically downregulated in Tbx5−/− mutants while Fgf8 and Fgf10 were upregulated (Figure 1C). Immunostaining of transgenic Shh:GFP embryos confirmed previous reports that Aldh1a2 is co-expressed with Tbx5 in a subset of pSHF cells adjacent to the foregut Shh/Nkx2-1-expressing pulmonary endoderm (Figure 1D–F” and Figure 1—figure supplement 1D-E'' Hochgreb et al., 2003; Ryckebusch et al., 2008; De Bono et al., 2018; de Soysa et al., 2019). Co-expression of Tbx5 and Aldh1a2 transcripts in the pSHF adjacent to Tbx1+ aSHF and Shh + endoderm were also evident in an online spatial atlas of single-cell gene expression (Figure 1—figure supplement 1F; Lohoff et al., 2021). To further define the spatial distinct aSHF-FGF and pSHF-RA signaling domains, we generated 3D reconstructions using serial sections of Aldh1a2, Fgf8, Fgf10, and Shh in-situ hybridizations from the WT mouse E9.5 (Figure 1—figure supplement 1G-H). Together these observations were consistent with the hypothesis that Tbx5 regulates CP development by controlling expression of Aldh1a2, which in turn establishes a local domain of RA activity in the pSHF that suppresses aSHF fate and promotes pulmonary development.
Tbx5 regulates cardiopulmonary development and maintains Aldh1a2 expression in Xenopus
Since Tbx5−/− mutant mouse embryos die shortly after E9.5 from cardiac insufficiency (Bruneau et al., 2001; Xie et al., 2012), we turned to Xenopus to elucidate the molecular mechanisms by which Tbx5 coordinates CP development. Xenopus larva can live for many days without a functional heart, by absorbing oxygen from the water, and their experimental advantages facilitate epistatic analysis of signaling pathways.
Previous studies have shown that Tbx5-regulated CP development is conserved between Xenopus and mouse: Tbx5 loss-of-function (LOF) in Xenopus, either by CRISPR/CAS9-mediated mutation or morpholino (MO) knockdown, phenocopies the mouse Tbx5−/− phenotype with severe cardiac hypoplasia, a failure to induce Nkx2-1+ lung progenitors and the foregut tube fails to separate into distinct trachea and esophagus (Steimle et al., 2018; Brown et al., 2005; Figure 2—figure supplement 1A).
Analysis of control and Tbx5 depleted Xenopus embryos showed that, like in mice, Tbx5 and Aldh1a2 were co-expressed in the foregut lpm/pSHF (Figure 2A and B) and that Xenopus Tbx5 is required for aldh1a2 expression (Figure 2C–F). Both X. laevis Tbx5-MO morphant and X. trop tbx5 CRISPR/CAS9 mutant embryos exhibited a loss or strong reduction of aldh1a2 transcripts and Aldh1a2 protein in the foregut lpm at NF34 (a timepoint similar to mouse E9.5) (Figure 2C–F; Figure 2—figure supplement 1B). Quantification of the Aldh1a2 immunostaining in 3D volume renderings of the fg lpm/pSHF domain of Tbx5 morphants and mutants revealed that Aldh1a2 protein was only expressed on average to approximately 28% (p=0.0009) and 33% (p≤0.0001) of WT levels, respectively (Figure 2—figure supplement 1B and Figure 3K). Analysis of transgenic Wnt/β-catenin reporter embryos Tg(WntRE:dGFP) (Tran et al., 2010), confirmed the failure of Wnt-dependent pulmonary induction in the ventral foregut of Tbx5-deficient embryos (Figure 2C and D). Importantly, co-injection of human TBX5 RNA rescued aldh1a2 expression and pulmonary development (Figure 2D–F, Figure 2—figure supplement 1B, and Figure 3). A time course analysis revealed that loss of Tbx5 resulted in a downregulation of aldh1a2 expression in the foregut lpm starting at NF25, but not at early somitogenesis stages (NF15) (Figure 2E and F). These results demonstrate that Tbx5 is required to maintain aldh1a2 expression in the foregut lpm, and that it regulates a conserved transcriptional program in Xenopus and mouse to coordinate SHF patterning and lung induction.
Figure 2 with 1 supplement see all

Tbx5 maintains aldh1a2 expression in Xenopus foregut lpm.
-
Figure 2—source data 1
Summary of in-situ hybridization results from Xenopus CAS9/CRISPR injection experiments in F0 embryos.
The tbx5 exon5 sgRNA (targeting the DNA binding domain; Steimle et al., 2018) causes approximately 40% of injected embryos to have a molecular phenotype as revealed by in-situ hybridization. 2 nl of a mixture containing 50 pg/nl sgRNA with 0.5 ng/nl Cas9 protein (PNA Bio CP01-20) was injected on either side of the sperm entry point at the 1 cell stage (total of 200 pg sgRNA and 2 ng Cas9 protein per embryo), embryos were cultured to NF34 and assayed by in-situ hybridization for the indicated genes.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig2-data1-v2.xlsx
Figure 3 with 1 supplement see all
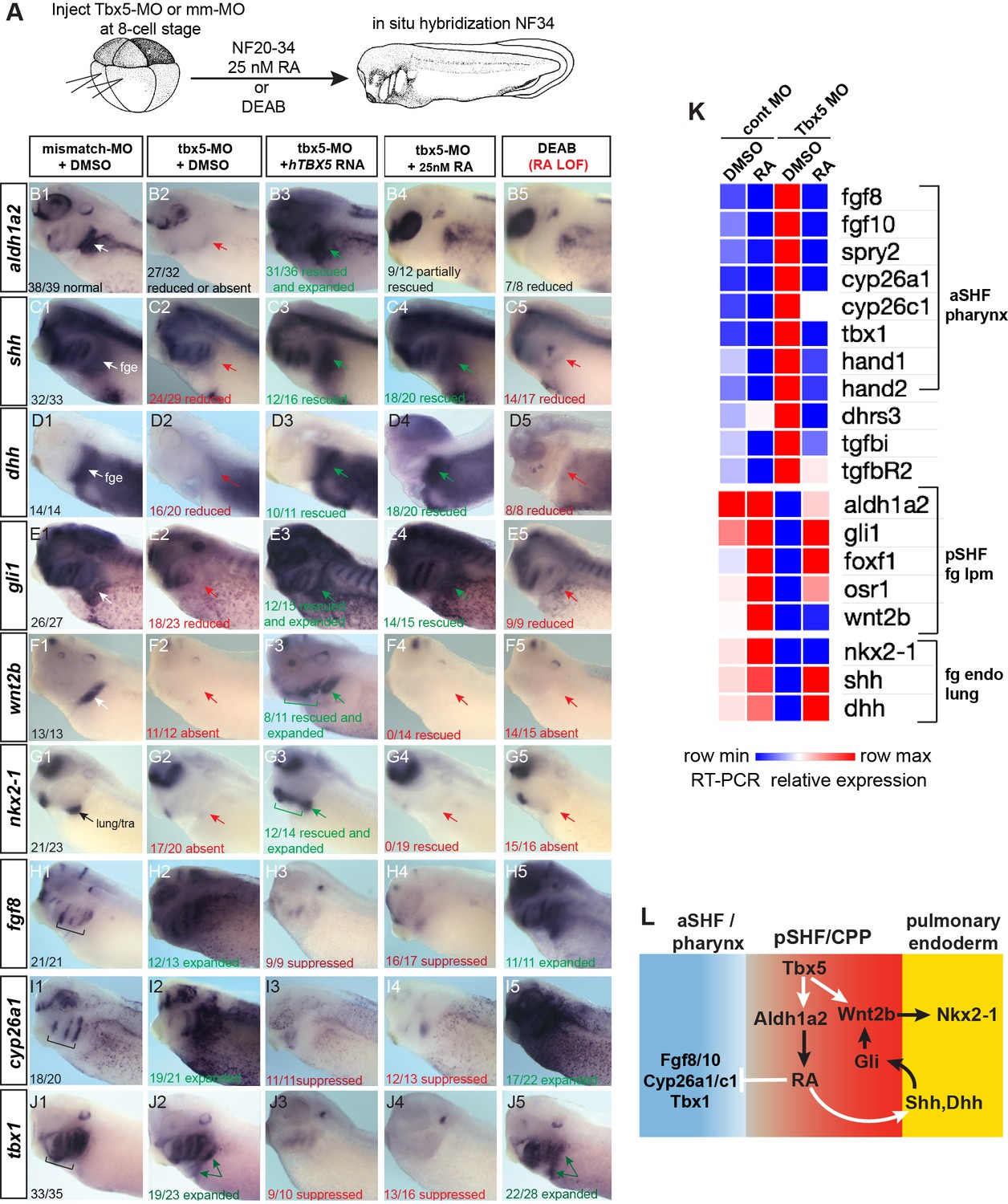
Tbx5 regulates Xenopus cardiopulmonary development in part via RA.
(A) Schematic of the experimental design. (B–J) Exogenous RA rescues Tbx5 LOF, while inhibition of RA phenocopies Tbx5 LOF. Whole-mount in-situ hybridization of NF34 X. laevis embryos after the indicated experimental treatments: injection of negative control 3 bp mismatch-MO (10 ng), Tbx5-MO (10 ng), Human TBX5 RNA (hTBX5; 100 pg), and/or 25 nM RA, 10 µM DEAB, DMSO vehicle control from NF20-34. The numbers of embryos with the observed expression pattern are indicated. Arrows indicate the relevant expression domain in the cardiopulmonar (CP) tissue. Brackets indicate the aSHF/pharyngeal domain. (K) Heat map showing relative expression from RT-PCR analysis of NF34 CP-foregut (fg) tissue dissected from control or Tbx5-MO injected embryos and treated with or without RA from NF20 to NF34. Each row is the average from the three biological replicates (n=4 explants per replicate). (L) Diagram of the proposed GRN model at NF25–35 showing the key role of Aldh1a2-dependent RA signaling downstream of Tbx5. White arrows indicate relationships tested in the above experiments and black arrows are demonstrated from the previous publications. Also see Figure 3—figure supplement 1, and related source data files. GRN, gene regulatory network; LOF, loss-of-function; MO, morpholino; RA, retinoic acid.
-
Figure 3—source data 1
Xenopus explant RT-qPCR source data. Explants were dissected at NF20, cultured 48 hr±DMSO or 25 nM all-trans retinoic acid (RA); harvested at NF34; three biological replicates for each condition; n=4 pooled explants in each replicate; pooled explants came from 2 to 3 separate fertilization/injection experiments.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig3-data1-v2.xlsx
Tbx5 regulates cardiopulmonary development via RA signaling
A detailed analysis of Tbx5-MO embryos by in-situ hybridization and RT-PCR showed that many of the CP genes that were misregulated in mouse Tbx5−/− CP tissue were also misregulated in Xenopus (Figure 3A–K). In addition to a loss of aldh1a2, pSHF markers osr1, foxf1, gli1, and wnt2b and pulmonary endoderm markers shh, dhh, and nkx2-1 were reduced, while pharyngeal/aSHF markers fgf8, fgf10, tbx1, cyp26a1, cyp26c1, spry2, hand1, hand2, dhrs3, tgfbR2, and tgfbi were all upregulated. In total, all 19 transcripts tested exhibited changes in gene expression similar to Tbx5−/− mice. Interestingly in the Tbx5-MO embryos, we observed changes in gene expression beyond just the CP region, including the kidney, pharynx, and head all of which are known to be regulated by RA, FGF, and/or Hh signaling. This suggests that non-cell-autonomous effects in Tbx5 depleted embryos are likely due to changes in secreted factors.
We hypothesized that the disrupted CP development in Tbx5-deficient Xenopus embryos was primarily caused by reduced Aldh1a2-dependent RA signaling. To address this, we tested whether blocking endogenous RA could phenocopy loss of Tbx5 or if addition of exogenous RA could rescue the Tbx5 LOF phenotype (Figure 3A). We suppressed endogenous RA synthesis by addition of the Aldh enzyme inhibitor DEAB between NF20 and NF34, the time when aldh1a2 expression was Tbx5-dependent. This phenocopied the Tbx5 LOF with loss of pSHF and pulmonary markers and an expansion of aSHF gene expression (Figure 3B–J). While DEAB allowed temporal-specific inhibition, in some instances pharmacological reagents can have off-target effects. Therefore, we also depleted Aldh1a2 protein by targeted microinjection of MOs into the foregut region, which phenocopied the DEAB treatment (Figure 3—figure supplement 1A-E).
Importantly, we could partially rescue the Tbx5-MO, Aldh1a2-MO, and DEAB phenotypes with exogenous RA between NF20 and NF34, using a physiological concentration of 25 nM (Horton and Maden, 1995; Mic et al., 2003; Sheikh et al., 2014; Figure 3B–J; Figure 3—figure supplement 1E). Addition of RA suppressed the expanded expression domains of aSHF markers fgf8, fgf10, spry2, cyp26a1, cyp26c1, and tbx1 in Tbx5/RA-deficient embryos (Figure 3B–J; Figure 3—figure supplement 1E). RA was also sufficient to rescue endodermal expression of shh and dhh as well as expression of known Hh-target genes gli1, foxf1, and osr1 in the foregut lpm of Tbx5-MO embryos and explants (Figure 3B–K). However, exogenous RA did not rescue expression of the pulmonary-inducing wnt2/2b ligands nor the lung marker nkx2-1. In contrast, addition of recombinant WNT2B protein to Tbx5-MO foregut explants was sufficient to rescue nkx2-1+ lung fate but not shh nor dhh expression (Figure 3—figure supplement 1F,G), consistent with previous reports that Tbx5 directly promotes wnt2/2b transcription (Steimle et al., 2018).
These results combined with our previous data suggest that Tbx5 promotes CP development by multiple mechanisms, which are experimentally separable (Figure 3L). First, by maintaining aldh1a2 expression, Tbx5 ensures robust RA signaling required for SHF pattern and induction of endodermal shh/dhh expression, and second, by cooperating with Hh to activate mesodermal expression of Wnt2/2b which promotes pulmonary induction.
Tbx5 directly activates Aldh1a2 transcription and indirectly represses Fgf8 via RA
In preliminary experiments, we found that expression of a doxycycline (Dox) inducible Tbx5 transgene during the directed differentiation of mouse embryonic stem cells (mESCs) into cardiac fate (Kattman et al., 2011; Steimle et al., 2018) was sufficient to increase Aldh1a2 expression and suppress Fgf8 and Fgf10 levels (Figure 4A). However, in these experiments, it was unclear whether Tbx5 regulated Aldh1a2 or Fgf expression directly or indirectly.
Figure 4 with 2 supplements see all
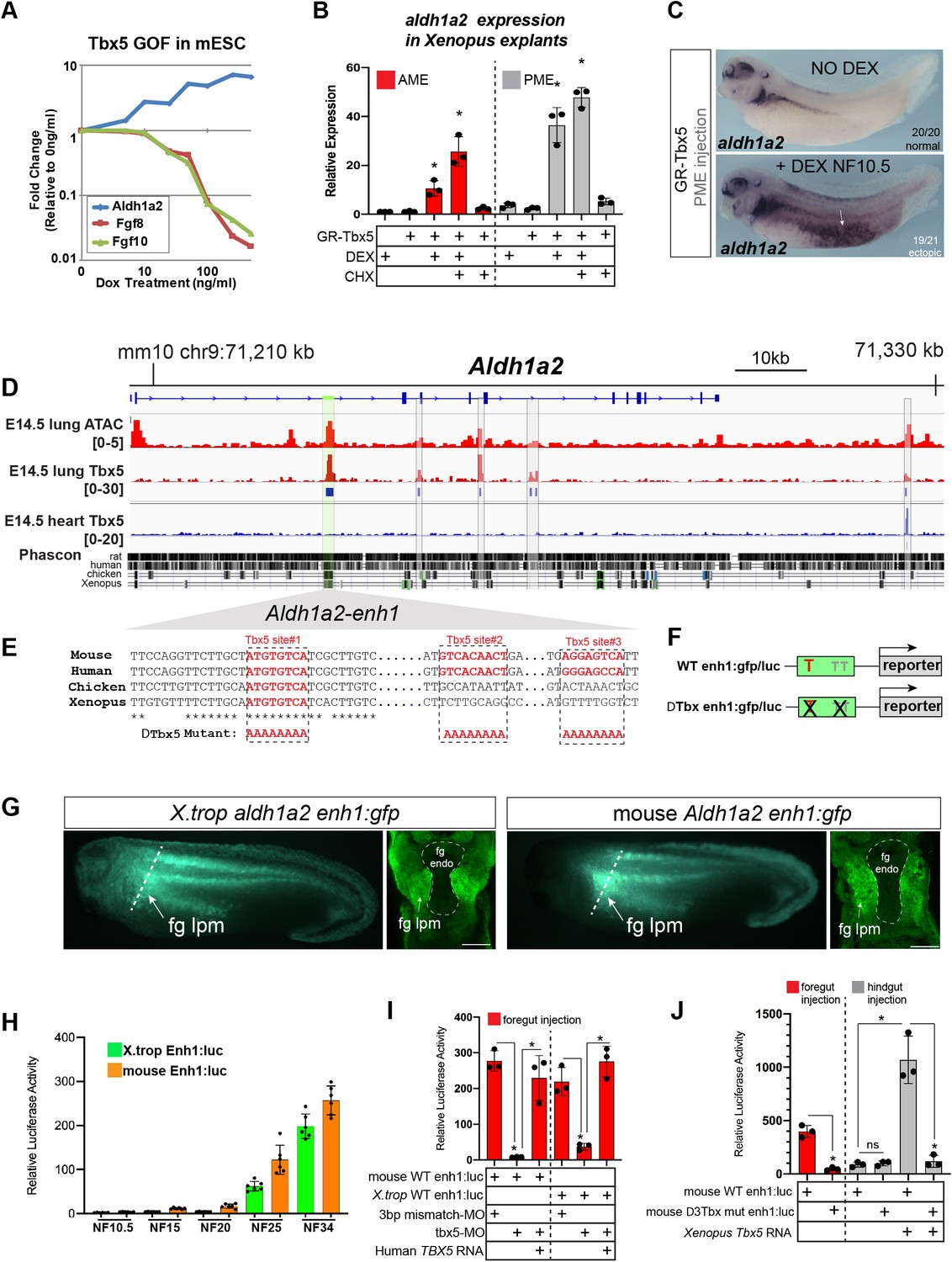
Tbx5 directly activates aldh1a2 transcription via an evolutionarily conserved first intron enhancer.
(A) Dox-inducible Tbx5 activated Aldh1a2 expression and repressed Fgf8 and Fgf10 during the directed differentiation of mouse embryonic stem cells (mESCs) into cardiac progenitors in a dose-dependent manner. (B) Tbx5 directly activated aldh1a2 expression in Xenopus anterior or posterior mesendoderm (AME, PME) explants. RT-qPCR shows that aldh1a2 transcription was induced by DEX activated GR-Tbx5 in the presence of translation inhibitor CHX. Graphs show mean relative expression ± standard deviation from N=3 biological replicates, four explants/replicate. *p<0.05, pair-wise Student’s t-test relative to uninjected, untreated explants. (C) Whole-mount in-situ hybridization of aldh1a2 expression of X. laevis NF34 embryos injected with GR-Tbx5 (100 pg) into the PME with or without DEX. (D) Genome browser of the mouse Aldh1a2 locus showing Tbx5 ChIP-seq tracks from E14.5 mouse lung (GSE167207) and E14.5 mouse heart (Burnicka-Turek et al., 2020, GSE139803) as well as ATAC-seq track from the ENCODE project (Castillo et al., 2010; Davis et al., 2018; ENCSR335VJW). Tbx5 ChIP-seq peaks in the E14.5 lung are indicated in blue. Phascon multiple species conservation track shows that the prominent Tbx5-bound first intron enhancer (enh1) is evolutionarily conserved from mammals to Xenopus. (E) Multiple species sequence alignment of enh1 reveals one Tbx5 DNA-binding site conserved from mammals to Xenopus and two additional mammalian-specific Tbx5 sites, which were mutated in reporter constructs. (F) Schematic of the Wild-type (WT) and mutant (ΔTbx) enh1:gfp and enh1:luciferase reporter constructs. (G) Both the Xenopus and mouse intronic enh1 enhancer are sufficient to drive GFP expression in the foregut lpm in Xenopus transgenic assays. (H) Time course of Xenopus and mouse enh1:luc reporter activity injected into X. laevis CP-foregut tissue, reflects endogenous Tbx5-dependent aldh1a2 expression between NF25 and NF34. Graphs show mean relative luciferase activity ± standard deviation. N=5 biological replicates/time point with five embryos/replicate. *p<0.05, parametric two-tailed paired t-test. (I) The Xenopus and mouse aldh1a2 enh1 reporter constructs are regulated by Tbx5. Graphs show relative mean luciferase activity ± standard deviation of reporters injected into CP-foregut tissue with control mm-MO, Tbx5-MO, and/or human TBX5 RNA. N=3 biological replicates/time point with five embryos/replicate. *p<0.05, parametric two-tailed paired t-test. (J) The three putative Tbx5 motifs in the mouse aldh1a2-enh1 enhancer are required for reporter activity in the CP-foregut tissue and Tbx5-dependent activation in the hindgut. Graphs show mean relative luciferase activity ± standard deviation. N=5 biological replicates/time point with five embryos/replicate. *p<0.05, parametric two-tailed paired t-test. Also see Figure 4—figure supplement 1, Figure 4—figure supplement 2 and related source data files. CP, cardiopulmonary.
-
Figure 4—source data 1
Xenopus explant RT-qPCR source data. GR-Tbx5 directly activated aldh1a2 in Xenopus anterior or posterior mesendoderm explants. NF10.5 anterior or posterior mesendoderm explants were treated for 2 hr in CHX prior to 6 hr in CHX+DEX.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Figure 4H Luciferase source data.
Temporal analyses of mouse and Xenopus tropicalis enh1-driven luciferase activity in the foregut.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig4-data2-v2.xlsx
-
Figure 4—source data 3
Luciferase source data. NF34 analysis of mouse and Xenopus tropicalis enh1-driven luciferase activity in the foregut±mismatch MO, tbx5-MO,±Human TBX5 RNA.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig4-data3-v2.xlsx
-
Figure 4—source data 4
Luciferase source data. NF34 analysis of mouse WT Enh1 or Tbx motif mutant Enh1-driven luciferase activity in fg or in hg±Tbx5 RNA.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig4-data4-v2.xlsx
We therefore examined whether Tbx5 was sufficient to directly activate aldh1a2 transcription in Xenopus. We injected RNA encoding a dexamethasone (DEX) inducible Glucocorticoid receptor (GR)-Tbx5 fusion protein (Horb and Thomsen, 1999) into either the anterior or posterior mesoderm. We then induced GR-Tbx5 nuclear translocation at gastrula stage before endogenous tbx5 is normally expressed by addition of DEX, with or without the translation inhibitor cycloheximide (CHX) to block secondary protein synthesis (Figure 4—figure supplement 1A). GR-Tbx5 activated precocious aldh1a2 transcription in both the anterior and posterior tissue even in the presence of CHX, demonstrating direct activation (Figure 4B). In-situ hybridization of NF34 embryos confirmed robust, ectopic activation of aldh1a2 by GR-Tbx5 (Figure 4C). In contrast, suppression of fgf8 transcription by GR-Tbx5 was sensitive to CHX, demonstrating indirect repression (Figure 4—figure supplement 1A,B). We hypothesized that Tbx5 indirectly represses fgf8 via Aldh1a2-dependent RA production since RA is known to directly repress Fgf8 transcription in the mouse SHF (Kumar et al., 2016). We tested this by inhibiting Aldh activity with DEAB which prevented the suppression of fgf8 by GR-Tbx5 (Figure 4—figure supplement 1A-C). These data demonstrate that Tbx5 directly activates aldh1a2 transcription and indirectly suppresses fgf8 expression via RA.
Tbx5 maintains Aldh1a2 transcription via an evolutionarily conserved intronic enhancer
We next sought to identify Aldh1a2 enhancers that are directly regulated by Tbx5, predicting that these would be evolutionarily conserved across terrestrial vertebrates. Since a number of putative enhancers have been documented for the mouse Aldh1a2 locus (Castillo et al., 2010; Vitobello et al., 2011; Huang et al., 2012), we focused on the murine genome. To identify Tbx5-bound enhancers in the CP lineage, we performed Tbx5 chromatin immunoprecipitation followed by high-throughput sequencing (ChIP-seq) of E14.5 fetal mouse lungs as lung mesenchyme is derived from the E9.5 CPPs (Peng et al., 2013). ChIP-seq uncovered five Tbx5-bound regions at the Aldh1a2 locus. Comparing the lung ChIP-seq data to our previously published Tbx5 ChIP-seq from E14.5 heart (Steimle et al., 2018), we found that four of the five Tbx5-bound sites were lung-specific and not bound by Tbx5 in the fetal heart (Figure 4D). Among the four Tbx5-bound sites, only one peak in the Aldh1a2 first intron, which we refer to as enh1 (for ‘enhancer 1;’ Figure 4D, Figure 4—figure supplement 2), showed strong evolutionarily conservation from mammals to Xenopus. The enh1 region also had a strong ATAC-seq peak in E14.5 lungs consistent with open enhancer chromatin (Figure 4D). Sequence analysis of enh1 revealed multiple predicted Tbx5 DNA-binding motifs, one of which was perfectly conserved amongst human, mouse, chicken, and Xenopus (Figure 4E; Figure 4—figure supplement 2).
We tested the ability of both the mouse and X. trop enh1 intronic enhancers to drive reporter expression in Xenopus transgenics and luciferase reporter assays (Figure 4F–J). In transgenics, both the mouse and X. trop enh1 enhancers drove GFP expression in the foregut lpm/pSHF at NF34 overlapping endogenous aldh1a2 (Figure 4G; Figure 4—figure supplement 1D-E''). We noted however that the transgene expression domain in the lpm was broader than endogenous aldh1a2 (Figure 4G), moreover, enh1 did not drive expression in the somites (Figure 4—figure supplement 1D-E), suggesting other enhancers must refine the lpm expression and promote somitic expression in vivo. To quantitatively assess temporal and spatial enhancer activity, we micro-injected enh1 reporters into blastomeres targeting the future CP-foregut or hindgut regions and assayed luciferase activity at a range of developmental stages, from gastrula to tailbud (Figure 4H; Figure 4—figure supplement 1F,G). Neither the mouse nor X. trop enh1 enhancers drove significant reporter activity during early development at NF10.5, NF15, or NF20; however, at NF25 and N34 both the mouse and X. trop enh1 enhancers were active in the foregut but not hindgut (Figure 4H; Figure 4—figure supplement 1F,G). This coincides with the timing at which endogenous aldh1a2 expression is Tbx5-dependent (Figure 2). Taken together, these data demonstrate that the evolutionarily conserved enh1 regulates the temporal and spatial transcription of aldh1a2 in the foregut lpm/pSHF.
We next tested Tbx5 regulation of the enh1 enhancer by combining reporter assays with LOF or gain-of-function (GOF) experiments. Tbx5-MO knockdown resulted in a dramatic reduction of the mouse and X. trop enh1 reporter activity in CP-foregut tissue at NF34, which was rescued by injection of human TBX5 RNA (Figure 4I). Moreover, injection of Xenopus or human TBX5 RNAs were sufficient to ectopically induce robust enh1 reporter activity in hindgut tissue, which does not express endogenous tbx5 (Figure 4J). Mutation of the single Tbx5-binding site that was perfectly conserved amongst human, mouse, chicken, and Xenopus enh1 resulted in a 48% (p=0.0046) and 60% reduction (p=0.0031) of the mouse and frog reporter activity in the foregut respectively, and also significantly blunted their response to ectopic Tbx5 in the hindgut (Figure 4—figure supplement 1H,I). Mutation of all three putative Tbx5 motifs conserved amongst mammals (Figure 4J) largely abolished reporter activity in both the foregut and hindgut (Figure 4J). We conclude Tbx5 directly maintains Aldh1a2 expression via multiple T-box motifs found in an evolutionarily conserved first intron enhancer.
FGF gain-of-function phenocopies Tbx5-loss-of-function in Xenopus
In light of the finding that Tbx5-dependent RA signaling suppresses fgf8 and fgf10, we tested if a temporal FGF GOF would phenocopy Tbx5 LOF (Figure 5A). We treated WT CP-foregut explants with recombinant FGF8 protein from NF20 to NF34, the period when exogenous RA was sufficient to rescue Tbx5 LOF. As predicted FGF8 treatment largely phenocopied Tbx5 depletion with increased expression of aSHF/pharyngeal markers tbx1, fgf10, spry2, and cyp26a1, as well as reduced expression of pSHF and pulmonary endoderm genes wnt2b, shh, gli1, and nkx2-1 (Figure 5B). We also observed reduced expression of tbx5 and aldh1a2 consistent with a feedback loop where FGF restricts Tbx5/Aldh1a2-mediate RA signaling (Figure 5C).
Figure 5 with 1 supplement see all
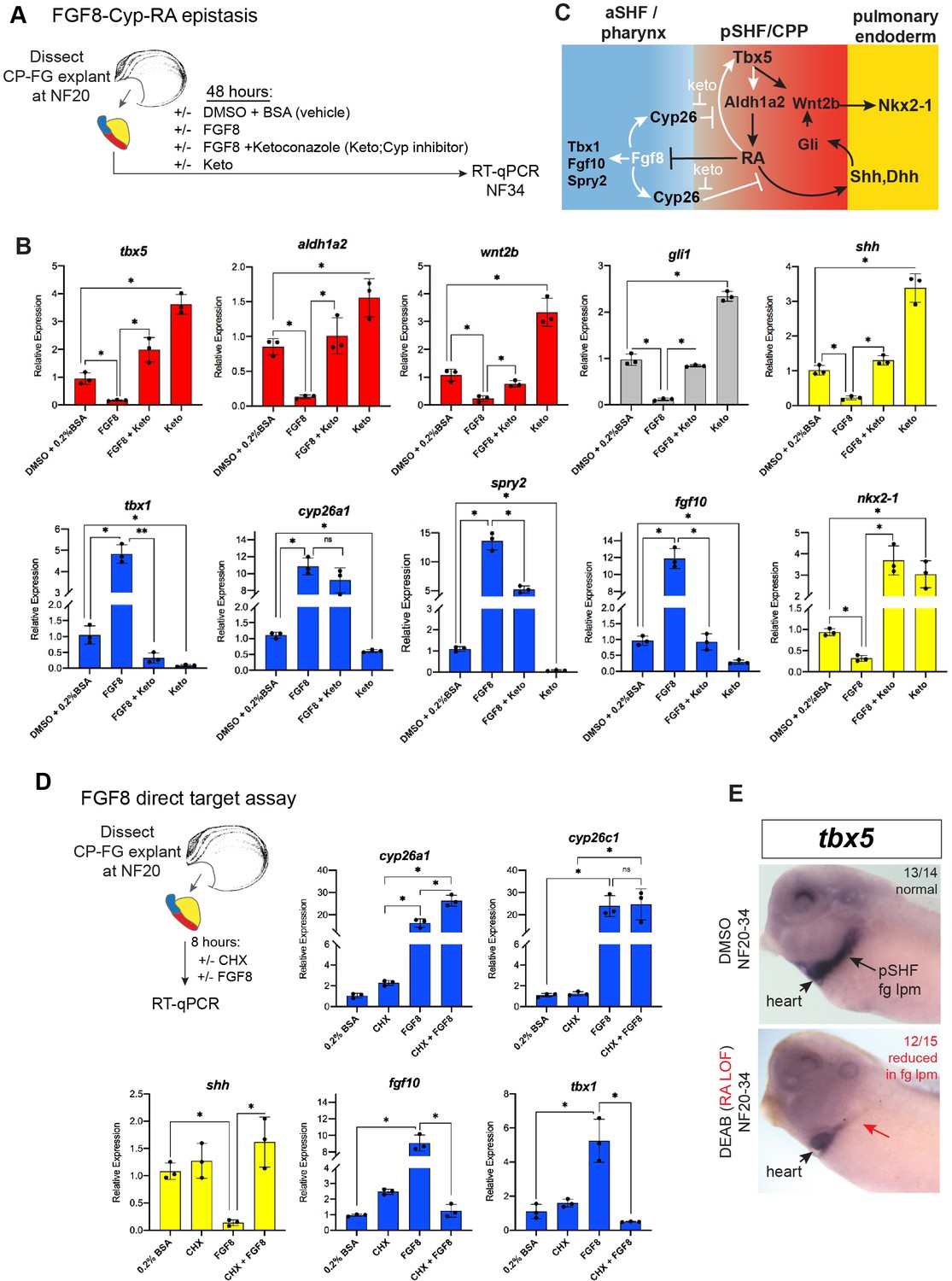
FGF8 gain-of-function (GOF) phenocopies Tbx5 loss-of-function in Xenopus.
(A) Schematic of FGF8 GOF assay in Xenopus cardiopulmonary foregut (CP-FG) explants dissected at NF20, and treated with vehicle controls (DMSO+0.2% BSA) or the indicated combinations of 100 ng/ml FGF8b and/or 0.5 µM ketoconazole (Cyp-inhibitor), harvested at NF34, and analyzed via RT-qPCR. (B) RT-qPCR showing mean relative expression of genes for pSHF (red), aSHF (blue), and pulmonary endoderm (yellow), ± standard deviation from N=3 biological replicates (four explants/replicate). *p<0.05, parametric two-tailed paired t-test. (C) Model depicting the observed FGF8 GOF results. White arrows indicate relationships tested in these experiments. (D) FGF8 direct target gene assay in Xenopus CP foregut explants, demonstrating that FGF8 directly activates cyp26a1, cyp26c1 and indirectly suppresses shh. Explants dissected at NF20 were pre-treated with 1 µM cycloheximide (CHX) for 2 hr prior to culture in 100 ng/ml FGF8b+CHX for 6 hr followed by RT-qPCR analysis. Graphs display mean relative expression ± standard deviation from N=3 biological replicates that contained four explants/replicate. *p<0.05, parametric two-tailed paired t-test. (E) RA signaling is required for the tbx5 expression in the fg lpm/pSHF domain, but not the heart. Embryos were cultured in 10 µM DEAB from NF20 to NF34 and assayed by in-situ hybridization. Number of embryos assayed and with the observed expression pattern is indicated. Also see Figure 5—figure supplement 1 and related source data files. aSHF, anterior second heart field; pSHF, posterior second heart field.
-
Figure 5—source data 1
RT-qPCR analysis of Xenopus foregut CP explants treated with FGF8±ketoconazole (Cyp26 inhibitor). Explants were dissected at NF20, cultured 48 hr±DMSO or recombinant Human FGF8a (200 ng/ml)±20 µM ketoconazole and assayed at NF34. RT-qPCR source data.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig5-data1-v2.xlsx
-
Figure 5—source data 2
RT-qPCR source data of Xenopus foregut cardiopulmonary explants treated with FGF8±CHX (cycloheximide).
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig5-data2-v2.xlsx
FGF signaling is known to promote the expression of RA-degrading Cyp26 enzymes (Shiotsugu et al., 2004; Deimling and Drysdale, 2011; Rydeen and Waxman, 2016), but it is unclear whether this is by direct transcriptional regulation. Therefore, we repeated the FGF8 experiments in the presence of CHX and found that indeed cyp26a1 and cyp26c1 were still upregulated by FGF8, demonstrating direct activation (Figure 5D). In contrast, the ability of FGF8 to suppress shh was CHX sensitive, demonstrating indirect repression (Figure 5D). We hypothesized that FGF8 indirectly suppresses expression of shh and other RA-dependent pSHF genes by promoting Cyp26-mediated RA degradation (Figure 5C). To test this, we treated CP-foregut explants with both FGF8 and the CYP enzyme inhibitor ketoconazole (keto). Keto blocked the ability of FGF8 to suppress shh, dhh, tbx5, aldh1a2, wnt2b, and nkx2-1 (Figure 5B), indicating that FGF indeed acts via Cyp-dependent RA degradation. Consistent with Cyp-mediated RA degradation being a major factor in endogenous CP patterning, keto treatment alone elevated expression of pSHF (tbx5, aldh1a2, and wnt2b) and pulmonary endoderm genes (shh and nkx2-1), whilst decreasing aSHF markers (fgf8, fgf10, and tbx1) (Figure 5B), similar to exogenous RA treatment (Figure 3). Moreover, knockdown of Cyp26a1 and Cyp26c1 by targeted MO injection phenocopied the ketoconazole treatment (Figure 6—figure supplement 1A,B). Interestingly, inhibition or knockdown of Cyp26 resulted in increased tbx5 levels suggesting that RA promotes its expression. Indeed, in-situ hybridization showed tbx5 expression in the pSHF/foregut lpm, but not in the FHF/heart tube, reduced by DEAB indicating that it requires RA (Figure 5E). Combined with our finding that Tbx5 directly maintains aldh1a2 expression, these data identify a RA-Tbx5 positive feedback loop in the pSHF.
RA directly promotes shh transcription through the evolutionarily conserved MACS1 endoderm enhancer
Our data suggest that RA from the Aldh1a2-expressing lpm is a likely candidate to activate Hh ligand expression in the endoderm. We tested whether exogenous RA could directly activate shh and dhh transcription in Xenopus foregut endoderm explants where the tbx5/aldh1a2+ lpm, the source of endogenous RA, had been removed (Figure 6A and B). Without the RA-producing lpm, the foregut endoderm did not express shh nor dhh; however, addition of exogenous RA rescued their expression, even in the presence of CHX, demonstrating direct activation (Figure 6B). As controls, RA also rescued expression of the known direct RA-target hnf1b, whereas the known indirect target ptf1a was not rescued in the presence of CHX (Figure 6B).
Figure 6 with 2 supplements see all

RA-RAR directly activates shh transcription in the Xenopus foregut endoderm via an evolutionarily conserved MACS1 enhancer.
(A) Schematic of direct RA target gene assay. Foregut endoderm (fg endo; yellow) was dissected from foregut mesoderm (fg meso; red) at NF25, pre-treated with 1 µM cycloheximide (CHX) for 2 hr prior to culture in 25 nM RA + CHX (or DMSO vehicle control) for 6 hr followed by RT-qPCR analysis. (B) RA directly activates shh and dhh expression in the presence of CHX. Graphs show mean relative expression ± standard deviation from N=3 biological replicates (four explants/replicate). Endoderm genes are shown in yellow, mesoderm makers in red confirm dissections. *p<0.05, parametric two-tailed paired t-test. (C) Genome browser of the human SHH locus showing the evolutionarily conserved MACS1 distal enhancer (green shading) embedded in an intron of the RNF32. Published ChIP-seq tracks of RXR, H3K4me1, and H3K27ac1 from hPSC-derived foregut endoderm (Vinckier et al., 2020, GSE104840; Wang et al., 2015, GSE54471). (D) MACS1 enhancer contains multiple RAR/RXR DNA-binding half sites, two of which are highly conserved. Schematics of the wild-type and mutant MACS1:luciferase reporter constructs. (E) Luciferase reporter assay in Xenopus show that the Human and X. tropicalis MACS1 enhancers are activated by RA via the RAR/RXR DNA-binding sites. 50 pg of MACS1:luciferase reporter +5 pg pRL-TK reporter were microinjected±250 pg of dominant-negative RARa RNA into either the C1 foregut (fg; red bars) or C4 hindgut (hg; gray bars) blastomeres and luciferase activity was assayed at NF34. 10 μM DEAB treatment was from NF20 to NF34. Mean relative luciferase activity ± standard deviation, from N=6 biological replicates/time point with five embryos/replicate. *p<0.05, parametric two-tailed paired t-test relative to WT MACS1:luc in the foregut (fg). Also see Figure 6—figure supplement 1, Figure 6—figure supplement 2 and related source data files. ns, not significant.
-
Figure 6—source data 1
RT-qPCR source data of Xenopus foregut endoderm explants treated for 2 hr in cycloheximide (CHX) prior to 6 hr in CHX +25 nM RA.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig6-data1-v2.xlsx
-
Figure 6—source data 2
Luciferase source data of Xenopus tropicalis and human Shh MACS1-enhancer-reporter activity at NF34.
- https://cdn.elifesciences.org/articles/69288/elife-69288-fig6-data2-v2.xlsx
Previous work has identified an evolutionarily conserved distal Shh enhancer called MACS1 (for mammalian-amphibian-conserved sequence 1), which is located more than 800 kb from Shh, within an intron of the Rnf32 gene (Sagai et al., 2009; Tsukiji et al., 2014; Sagai et al., 2017). The MACS1 enhancer is able to dive transcription in mouse foregut endoderm but the signals and TFs that control Shh expression via the MACS1 enhancer are unknown. An analysis of publicly available ChIP-seq data from human foregut endoderm, differentiated from pluripotent stem cells (hPSCs) in part by RA treatment (Vinckier et al., 2020; Wang et al., 2015), revealed binding of the RA nuclear receptor RXR at the human SHH MACS1 enhancer as well as H3K4me1 and H3K27ac1, epigenetic marks indicative of enhancer activation (Figure 6C). Sequence analysis of the MACS1 enhancer predicted multiple RXR/RAR nuclear RA receptor half sites (Penvose et al., 2019), two of which were evolutionarily conserved between human, mouse, chicken, and Xenopus (Figure 6D; Figure 6—figure supplement 1), suggesting that RA directly activates SHH transcription.
We functionally interrogated human and X. tropicalis SHH MACS1 enhancer activity in Xenopus luciferase assays (Figure 6E) and found both could drive robust reporter activity in foregut but not hindgut endoderm, demonstrating spatial specificity (Figure 6E). Disruption of endogenous RA signaling via DEAB treatment (NF20–34) or injection of dominant-negative RAR alpha RNA (dN-RARa) abolished human and X. trop MACS1 enhancer activity (Figure 6E). Moreover, exogenous RA could activate the enhancer in foregut explants lacking the RA-producing lpm. Mutation of the two highly conserved RAR/RXR half sites in the MACS1 enhancers dramatically reduced reporter activity in the foregut (Figure 6E) as well as a significantly blunted activation by exogenous RA in isolated endoderm explants (Figure 6—figure supplement 2). These data demonstrate that RA signaling directly stimulated shh transcription in foregut endoderm, via conserved RAR/RXR motifs in the shh MACS1 enhancer.
Discussion
Tbx5 regulates a RA-HH-Wnt GRN that coordinates SHF patterning and pulmonary specification
Our findings reveal complex and evolutionarily conserved interconnected signaling networks downstream of Tbx5 that coordinate early development of the cardiac and pulmonary systems (modeled in Figure 7). We identify the following aspects of this SHF mesoderm—pulmonary endoderm signaling network: (1) Direct Tbx5 activation of an aldh1a2 enhancer, which maintains aldh1a2 transcription in pSHF mesoderm; RA is in turn required to maintain tbx5 expression in the pSHF, establishing a positive feedback loop between Tbx5 and RA; (2) Tbx5-RA and FGF-Cyp form mutually antagonistic modules, with the Tbx5-RA loop promoting pSHF/CPP identity and suppressing aSHF fate, and Cyp-mediated RA degradation refining the spatial domain of RA activity; and (3) Direct RXR/RAR activation of the MACS1 enhancer at the shh locus, which provides a mechanism underlying the cell-non-autonomous activation of endodermal Hh ligand expression by Tbx5/Aldh1a2-dependent RA signaling in the pSHF. Reception of Hh signaling in the pSHF mesoderm activates Gli TFs, which cooperate with Tbx5 to directly activate wnt2/2b transcription; Wnt2/2b then induce pulmonary fate in the foregut endoderm (Hoffmann et al., 2014; Rankin et al., 2016; Steimle et al., 2018; Goddeeris et al., 2008). Thus, during CP development, Tbx5 regulates the production of three key paracrine signals, RA and Wnt directly via Tbx5-dependent enhancers and Hh indirectly via a RA-dependent enhancer. These interdependent signaling loops ensure that the lung primordia and pSHF-derived atria and pulmonary vessels from adjacent to one another, in preparation for the coordinated morphogenesis and functional integration of these two organ systems during development.
Figure 7
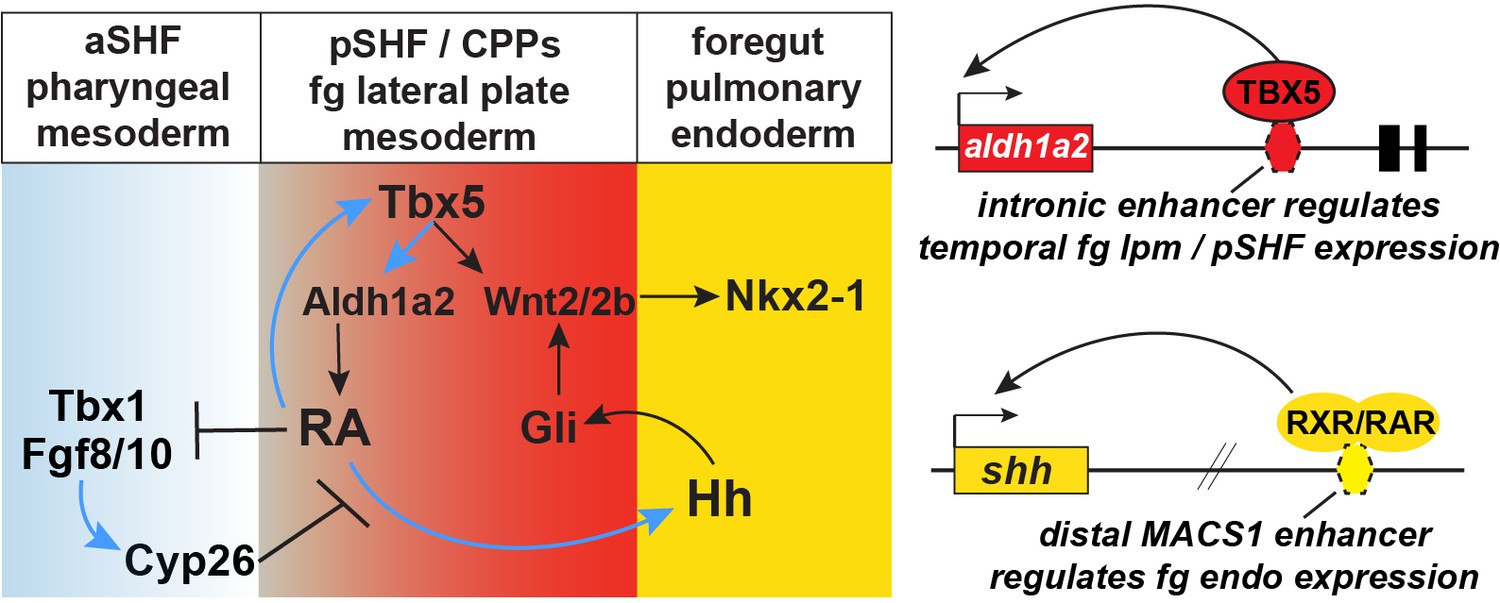
Model of the Tbx5 regulated GRN coordinating SHF pattering and pulmonary induction.
Our data indicate that between NF25and NF35 in Xenopus and around E9.5 in mice, Tbx5 directly maintains Aldh1a2 expression and a RA-Tbx5 positive feedback loop in the pSHF, which is necessary for Hh ligand expression, Wnt2/2b-dependent pulmonary fate induction, and SHF patterning. Blue arrows in the model indicate relationships demonstrated in this study. Tbx5/Aldh1a2-dependent RA signaling restricts FGF/Cyp activity in the aSHF, promotes pSHF identity, and drives expression of shh in pulmonary foregut endoderm. The aldh1a2 enh1 enhancer is directly regulated by Tbx5 and the shh MACS1 enhancer is regulated by RA/RXR/RAR. aSHF, anterior second heart field; GRN, gene regulatory network; pSHF, posterior second heart field.
T-box TFs and RA: a conserved regulatory node disrupted in cardiopulmonary and limb birth defects
Integrated regulatory loops between Tbx5, RA, and FGF regulate limb development and lung branching morphogenesis in addition to SHF cardiac development (Nishimoto et al., 2015; Arora et al., 2012; Feneck and Logan, 2020). We show that Tbx5 and RA form a positive feedforward loop in the pSHF; in this domain, Tbx5 directly maintains Aldh1a2-dependent RA production while RA maintains tbx5 expression. This is consistent with reports that RA is required for the expression of Tbx5 in SHF but not the FHF during early in mouse heart development (De Bono et al., 2018; Stefanovic et al., 2020 ). We predict that this is equivalent to the RA-dependent maintenance of tbx5 that we observed in Xenopus. In the developing limb bud, RA response elements in a regulatory element at Tbx5 are required for enhancer activity and other enhancers at the Tbx5 locus have been identified that can activate transcription in the heart and limb (Minguillon et al., 2012; Smemo et al., 2012; Cunningham et al., 2018), it remains to be determined whether these enhancers are also directly regulated by RA/RAR/RXR or control expression in the pSHF. Regardless, Tbx5 and RA are from a shared module in both SHF and limb development (Nishimoto et al., 2015). Limb defects and atrioventricular septal defects, caused by altered pSHF development, are both a facet of the phenotypic spectrum observed in Holt–Oram syndrome in human patients with TBX5 mutations (Steimle and Moskowitz, 2017). This raises the intriguing possibility that Tbx5-RA interactions were an evolutionary innovation in both limb and CP mesoderm in the adaptation to terrestrial life and that disrupting the Tbx5-RA feedforward loop is a component of TBX5-associated birth defects. Overall, this work provides a framework for understanding the developmental basis of the human birth defects observed in Holt–Oram Syndrome.
Enhancers controlling the reciprocal RA-HH mesoderm and endoderm signaling
We found that Tbx5-dependent RA signaling directly promotes Shh expression in foregut endoderm via RAR/RXR half-site motifs in the evolutionarily conserved Shh MACS1 enhancer (Sagai et al., 2009; Tsukiji et al., 2014; Sagai et al., 2017; Penvose et al., 2019). Other endodermal TFs that contribute to Shh expression in the foregut have been described, including FoxA2, Meis3, Islet1, Tbx2, and Tbx3 (Lin et al., 2006; diIorio et al., 2007; Tamplin et al., 2008; Mesbah et al., 2012; Lee et al., 2019; Genga et al., 2019), but whether they directly function on the Shh MACS1 enhancer, potentially in cooperation with RA/RAR/RXRs, remains unknown.
Previous studies have defined multiple enhancers that regulate different temporal and spatial expression domains of Aldh1a2 during development, with input from T-box TFs as a reoccurring theme. For example, the T-box TFs VegT, Eomesodermin, and Brachyury, regulate aldh1a2 in the Xenopus gastrula mesoderm via cis-regulatory elements near the promoter (Gentsch et al., 2013; Faial et al., 2015; Tosic et al., 2019). Subsequent expression of Aldh1a2 in the paraxial mesoderm and early lpm is promoted by Hox/Pbx/Meis TF complexes acting on a first intron enhancer (Vitobello et al., 2011) that is distinct from the enh1 enhancer identified here. Indeed, the Tbx5-responsive enh1 transgene did not drive expression in the paraxial mesoderm-derived somites.
The fidelity of these T-box/RA modules is essential for avoiding common cardiovascular birth defects affecting both the aSHF and pSHF. While Tbx5 and RA act in a positive feedback loop during pSHF patterning, Tbx1 and RA have an antagonistic relationship in the aSHF. Interestingly Tbx1, which can act as a transcriptional repressor, is known to spatially restrict Aldh1a2 expression (Guris et al., 2006; Aggarwal et al., 2006; Ryckebüsch et al., 2010), although it is currently unknown whether this activity is direct. On the other hand, RA suppresses tbx1 expression in both Xenopus pSHF (our study) and in mice (Ryckebüsch et al., 2010). In mouse, both loss of Tbx1 and aberrant RA synthesis can result in cardiovascular defects similar to human DiGeorge syndrome patients. Moreover, genetically removing one copy of Aldh1a2, thereby reducing the level of RA, ameliorates the cardiovascular malformations in Tbx1 heterozygous embryos (Ryckebüsch et al., 2010; Vermot et al., 2003). Taken together, these observations suggest that the opposing actions of Tbx5 and Tbx1 act as a mechanistic toggle, wherein RA is activated by Tbx5 to promote the CP program in the pSHF and restrict aSHF identity. In the aSHF Tbx1 suppresses RA production and the CP program. We speculate that Tbx1 and Tbx5 may engage T-box elements in the same enh1 enhancer, with Tbx5 promoting Aldh1a2 in the pSHF and Tbx1 inhibiting Aldh1a2 transcription in the aSHF as a transcriptional mechanism contributing to posterior/anterior patterning of the SHF.
Identification of the specific transcriptional enhancers that mediate the reinforcing signaling loops that pattern the SHF is essential for the genotype-phenotype interpretation of animal model and patient CP defects. The enhancers we identified by which Tbx5 directly activates aldh1a2 transcription and RA directly activates shh are both highly conserved amongst air breathing terrestrial vertebrate species, suggesting a potential role in CP evolution. Previous work has identified a single nucleotide variant in a TBX5 enhancer that contributes to human CHD (Smemo et al., 2012). Identification of the enhancers modulating the essential signaling pathways for heart development will contribute to the curation of whole-genome sequencing, refining the search space for functional non-coding variants and allowing the nomination of non-coding SNPs that may alter the function of known enhancers and thereby contribute to CHD risk.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background(Xenopus tropicalis, females) | Wild-typeadult females | Nasco | LM00823 | |
| Strain, strain background(X. tropicalis, males) | Wild-typeadult males | Nasco | LM00822 | |
| Strain, strain background(X. laevis, females) | Wild-typeadult females | Nasco | LM00531 | |
| Strain, strain background(X. laevis, males) | Wilt-typeadult males | Nasco | LM00715 | |
| Genetic reagent(X. tropicalis) | Xtr.Tg(WntREs: dEGFP)Vlemx | National Xenopus Resource (NXR) Center,Woods Hole, MA | RRID:NXR_1094 | X. tropicalis Wnt/Bcat reporter line |
| Genetic reagent(X. laevis) | Xla.Tg(WntREs: dEGFP)Vlemx, | NXR | RRID:NXR_0064 | X. laevis Wnt/Bcat reporter line |
| Genetic reagent(X. laevis) | Xla.Tg.(nkx2-5:GFP)Mohun | NXR | RRID:NXR_0030 | X. laevis Nkx2-5:GFP reporter line |
| Strain, strain background(Mus musculus) | CD-1 | Charles River Labs | Strain Code022RRID:IMSR_CRL:022 | WT mice |
| Genetic reagent(M. musculus) | Shhtm1(EGFP/cre)Cjt | Jax Labs | JAX: 005622RRID:IMSR_JAX:005622 | Shh:GFP mice |
| Cell line(M. musculus) | Tbx5OE-mESC line | Steimle et al., 2018 | Steimle et al., 2018 | |
| Chemical compound, drug | Doxycycline | Sigma-Aldrich | D9891 | |
| Antibody | (Rabbit polyclonal) anti-Aldh1a2 | Abcam | ab96060, RRID:AB_10679336 | IF (1:500) |
| Antibody | (MouseMonoclonal)Anti-Aldh1/2 | Santa Cruz Biotechnology | sc-166362, RRID:AB_2009458 | IF (1:500) |
| Antibody | (Mouse monoclonal) anti-Sox2 | Abcam | ab79351; RRID:AB_10710406 | IF (1:1000) |
| Antibody | (Rabbit polyclonal) anti-Nkx2-1 (H-190) | Santa CruzBiotechnology | sc-13040X; RRID:AB_793532 | IF (1:500) |
| Antibody | (Mouse monoclonal) anti-Fibronectin (4H2) | Developmental Studies Hybridoma Bank | DSHB #4H2; RRID:AB_2721949 | IF (1:2000) |
| Antibody | (Chicken polyclonal) anti-GFP | Aves Labs | GFP-1020; RRID:AB_10000240 | IF (1:1000) |
| Antibody | (Goat polyclonal)anti-Tbx5 | Santa Cruz Biotechnology | sc-17866, RRID:AB_2200827 | IF (1:300)ChIP: 5 µg |
| Recombinant DNA reagent | pI-SceI-d2EGFP plasmid | Addgene | Addgene_32674 | For meganuclease transgenics |
| Recombinant DNA reagent | pRL-TK(plasmid) | Promega | E2241 | |
| Recombinant DNA reagent | pGL4.23 luc2/miniP(plasmid) | Promega | E8411 | |
| Recombinant DNA reagent | pCS2+ GR-xTbx5 | Addgene | Addgene 117248 | |
| Recombinant DNA reagent | pCS2+ xTbx5 | Addgene | Addgene 117247 | |
| Recombinant DNA reagent | pCSf107mT-Gateway-3′myc | Addgene | Addgene 67617 | |
| Recombinant DNA reagent | pENTR223Human TBX5 | Horizon Discovery | OHS5894-202500411 | |
| Commercial assay or kit | Gateway LR Clonase II enzyme mix | Thermo Fisher Scientific | 11791020 | |
| Commercial assay or kit | mMessage mMachine SP6 RNA synthesis kit | Thermo Fisher Scientific | AM1340 | |
| Peptide, recombinant protein | FGF8b | R&D Systems | 423-F8-025 | |
| Peptide, recombinant protein | WNT2B | R&D Systems | 3900-WN-025 | |
| Commercial assay or kit | TRIzol | Thermo Fisher Scientific | 15596018 | |
| Commercial assay or kit | Direct-zolMiniprep plus kit | Thermo Fisher Scientific | R2070 | |
| Commercial assay or kit | SuperscriptVILO mastermix | Thermo Fisher Scientific | 11755050 | |
| Commercial assay or kit | PowerUP2× SYBR Green MasterMix | Thermo Fisher Scientific | A25742 | |
| Commercial assay or kit | Firefly Luciferase 2.0 kit | Biotium | 30085-1 | |
| Commercial assay or kit | Renilla Luciferase2.0 kit | Biotium | 30082-1 | |
| Chemical compound, drug | DEAB | Sigma-Aldrich | D86256 | |
| Chemical compound, drug | All-trans retinoic acid(RA) | Sigma-Aldrich | R2625 | |
| Chemical compound, drug | Ketoconazole | Tocris | Tocris#1103 | |
| Chemical compound, drug | Cycloheximide | Sigma-Aldrich | C4859 | |
| Chemical compound, drug | Dexamethasone | Sigma-Aldrich | Sigma D4902 | |
| Peptide, recombinant protein | CAS9 | PNA Bio | CP01-20 | |
| Sequence-based reagent | X. tropicalis tbx5 exon5 sgRNA | IDT DNA | GGGGTTCTGATATGAAGTGA | Steimle et al., 2018 |
| Sequence-based reagent | X. laevis Tbx5 MO1 | GeneTools | 5′-TTA GGA AAG TGT CTC TGG TGT TGC C -3′; | Brown et al., 2005 |
| Sequence-based reagent | X. laevis Tbx5 3 bp mismistach MO1 | GeneTools | 5′-TCA GTA AAG TAT CTC TGG TGT TGC C-3′ | This paper |
| Sequence-based reagent | X. laevis Tbx5 MO2 | GeneTools | 5′-CAT AAG CCT CCT CTG TGT CCG CCA T-3 | Brown et al., 2005 |
| Sequence-based reagent | X. laevis Tbx5 3 bp mismatch MO2 | GeneTools | 5′-TAT CAG ACT CCT CTG TGT CCG CCA T-3′ | This paper |
| Sequence-based reagent | X. laevis Aldh1a2-MO | GeneTools | 5′-GCA TCT CTA TTT TAC TGG AAG TCAT-3′ | Strate et al., 2009 |
| Sequence-based reagent | X. laevis Cyp26a1-MO | GeneTools | 5′-TAG TGA GCA GAG TAT ACA GAT CCA T-3′ | Janesick et al., 2013 |
| Sequence-based reagent | X. laevis Cyp26c1-MO | GeneTools | 5′-TAC AAG ATG TTC CTC CTT GAG ATC A-3′ | Yu et al., 2016 |
| Commercial assay, kit | Protein G-conjugated magnetic beads | Life Technologies | 1,003D | |
| Commercial assay, kit | NEBNext Ultra DNA Library Prep Kit | New England Biolabs | E7370S | |
| Commercial assay, kit | Sera-Mag magnetic beads | GE | 6515-2105-050-250 | |
| Software, algorithm | Morpheus | Broad Institute | https://software.broadinstitute.org/morpheusRRID:SCR_017386 | |
| Commercial assay, kit | SceI mega-nuclease enzyme | New England Biolabs | R0694S | Use within 1 month of purchase, store at –80°C |
| Commercial assay, kit | Dispase | Corning Life Sciences | 354235 | Use at 10 U/ml on Xenopus explants |
Xenopus methods
Xenopus embryo injections
Request a detailed protocolWT adult X. laevis and X. tropicalis frogs were purchased from Nasco (Fort Atkinson, WI). Adult transgenic X. laevis and X. tropicalis Wnt/B-catenin reporter (Xla.Tg(WntREs:dEGFP)Vlemx, NXR_0064; and Xtr.Tg(WntREs:dEGFP)Vlemx, NXR_1094), and adult transgenic X. laevis nkx2-5:GFP (Xla.Tg.(nkx2-5:GFP)Mohu, NXR_0030) frogs were purchased from the National Xenopus Resource (RRID:SCR_013713). Ovulation, in-vitro fertilization and natural mating, embryo de-jellying, and microinjection were performed as described (Sive et al., 2000). Plasmids for Xenopus GR-Tbx5 (Addgene 117248), Xenopus Tbx5 (Addgene 117247) (Horb and Thomsen, 1999), and Xenopus dominant-negative RARa (Sharpe and Goldstone, 1997) were previously described. Human TBX5 (Horizon Discovery OHS5894-202500411) was gateway sub-cloned from its entry vector pENTR223 into the expression vector pCSf107mT-Gateway-3′myc (Addgene 67617) using clonase (ThermoFisher 11791020) according to manufacturer’s instructions. Linearized plasmid templates were used to make mRNA for injection using the Ambion mMessage mMachine SP6 RNA Synthesis Kit (ThermoFisher AM1340). Total amounts of injected mRNA were as follows: GR-Tbx5 RNA, 125 pg; dN-RARa, 200 pg; and human TBX5-myc, 100 pg. Previously validated translation-blocking MOs against Tbx5 (Brown et al., 2005; Steimle et al., 2018), Aldh1a2 (Strate et al., 2009), Cyp26a1 (Janesick et al., 2013), and Cyp26c1 (Yu et al., 2016) were injected at the 8-cell stage (for Tbx5-MO: a mixture of 2.5 ng each MO1 +2 per dorsal marginal zone (dmz) in X. laevis; mixture of 0.5 ng each MO1 +2 per dmz in X. tropicalis). MOs were purchased from GeneTools (Philmath, OR) and were as follows: Tbx5-MO1: 5′-TTA GGA AAG TGT CTC TGG TGT TGC C-3′; a negative control Tbx5 mismatch MO1 with three nucleotides mutated: 5′-TCA GTA AAG TAT CTC TGG TGT TGC C-3′; Tbx5-MO2: 5′-CAT AAG CCT CCT CTG TGT CCG CCA T-3′; Tbx5 3 bp mismatch MO2: 5′-TAT CAG ACT CCT CTG TGT CCG CCA T-3′ (mismatch bases are indicated in bold, underlined); Aldh1a2-MO: 5′-GCA TCT CTA TTT TAC TGG AAG TCAT-3′; Cyp26a1 MO: 5′-TAG TGA GCA GAG TAT ACA GAT CCA T-3′; and Cyp26c1 MO: 5′-TAC AAG ATG TTC CTC CTT GAG ATC A-3′.
For F0 CISPR-mediated indel mutations, a sgRNA targeting X. trop tbx5 exon 5 (DNA-binding domain) that causes frameshift mutations was synthesized in-vitro as previously described (Steimle et al., 2018). This exon5 sgRNA causes approximately 40% of injected embryos to have a phenotype (Figure 2—source data 1). Briefly, 2 nl of a mixture containing 50 pg/nl sgRNA with 0.5 ng/nl Cas9 protein (PNA Bio CP01-20) was injected on either side of the sperm entry point at the 1-cell stage (total of 200 pg sgRNA and 2 ng Cas9 protein per embryo).
For Xenopus whole embryo small-molecule treatments, embryos were cultured in 0.1× MBS +50 µg/ml gent with concentrations of 1 µM dexamethasone, 25 nM RA, or 10 µM DEAB (Sigma D86256). In all experiments, corresponding amounts of vehicle controls (DMSO or 0.2% fatty-acid free BSA) were used.
Gastrula or CP-foregut explants (containing both endoderm and lpm) were micro-dissected in 1× MBS +50 μg/ml gentamycin sulfate (gent; MP Biochemicals 1676045)±10 U/ml dispase (Corning Life Sciences 354235; to help remove the lpm) and were cultured in 0.5× MBS +0.2% fatty acid free BSA (Fisher BP9704)+50 μg/ml gent with the following concentrations of factors: 1 μM dexamethasone (DEX; Sigma D4902); 1 μM cycloheximide (CHX; Sigma C4859); 25 nM all-trans RA (Sigma R2625); 100 ng/ml WNT2B (R&D Systems 3900-WN-025); 1 μM DEAB (Sigma D86256); 100 ng/ml FGF8b (R&D Systems 423-F8-025); and 0.5 μM ketoconazole (Tocris 1103). In CHX experiments, explants were treated for 2 hr in CHX prior to DEX+CHX treatment for 6 hr.
Xenopus RT-qPCR
Request a detailed protocolXenopus explants were dissected from embryos of 2–3 separate fertilization/injection experiments, frozen on dry ice in 200 μl of TRIzol (ThermoFisher 15596018), and stored at –80°C. RNA was extracted using TRIzol and purified using the Direct-zol RNA miniprep plus kit (ZymoResearch R2070); 500 ng RNA was used in cDNA synthesis reactions using Superscript Vilo Mastermix (ThermoFisher 11755050), all according to the manufacturer’s instructions. qPCR reactions were carried out using PowerUp Mastermix (ThermoFisher A25742) on ABI StepOnePlus or QuantStudio3 machines. Xenopus RT-qPCR primer sequences are listed in Supplementary file 1. Relative expression, normalized to ubiquitously expressed odc, was determined using the 2−ΔΔCt method. Graphs display the average 2−ΔΔCt value ± standard deviation. Statistical significance (p<0.05) was determined using parametric two-tailed paired t-test, relative to uninjected, untreated explants. Each black dot in the RT-qPCR graphs represents an independent biological replicate containing four explants. Heat map of Xenopus RT-qPCR gene expression was generated using Morpheus software (https://software.broadinstitute.org/morpheus/) and shows the average 2−ΔΔCt value from three biological replicates for each condition.
Xenopus in-situ hybridization
Request a detailed protocolIn-situ hybridization of Xenopus embryos was performed as described (Sive et al., 2000) with minor modifications. Briefly, embryos were fixed overnight at 4°C in MEMFA (0.1 M MOPS, 2 mM EGTA, 1 mM MgSO4, and 3.7% formaldehyde), washed 3× 5 min in MEMFA without formaldehyde, dehydrated directly into 100% ethanol, washed 5–6 times in 100% ethanol, and stored at −20°C for at least 24 hr. Proteinase K (ThermoFisher AM2548) on day 1 was used at 2 µg/ml for 10 min on stage NF15, NF25 embryos and 5 µg/ml on NF34 embryos; hybridization buffer included 0.1% SDS; RNAse A (ThermoFisher 12091021) used at 0.5 µg/ml; and anti-DIG-alkaline phosphatase antibody (Sigma 11093274910) used at 1:5000 in MAB buffer (100 mM Maleic acid, 150 mM NaCl, and pH 7.5) + 10% heat-inactivated lamb serum (Gibco 16070096) + 2% blocking reagent (Sigma 11096176001). Anti-sense DIG-labeled in-situ probes were generated using linearized plasmid cDNA templates with 10× DIG RNA labeling mix (Sigma 11277073910) according to the manufacturer’s instructions.
Xenopus immunofluorescence
Embryos were fixed in 100 mM HEPES (pH 7.5), 100 mM NaCl, 2.7% methanol-free formaldehyde for 2 hr at room temperature, dehydrated directly into Dent’s post-fixative (80% Methanol/20% DMSO), washed five times in Dent’s, and stored in Dent’s at −20°C for at least 48 hr. Embryos were serially rehydrated (75%, 40%, 25% methanol) into PBS +0.1% TritonX-100 (PBSTr). Embryos were then cut in a transverse plane through the pharynx and posterior to the liver to create a foregut sample using a fine razor blade on a 2% agarose-coated dish in PBSTr. Foreguts were subjected to antigen retrieval in 1× R-universal epitope recovery buffer (Electron Microscopy Sciences 62719-10) for 1 hr at 60–65°C, washed 2× 10 min in PBSTr, blocked for 1–2 hr in PBSTr +10% normal donkey serum (Jackson ImmunoResearch 017-000-001) + 11% DMSO at room temperature, and incubated overnight at 4°C in this blocking solution+primary antibodies: chicken anti-GFP (Aves GPF-1020; diluted 1:1000), mouse anti-Sox2 (Abcam ab79351; 1:1000), rabbit anti-Aldh1a2 (Abcam ab96060; 1:500), and goat anti-Tbx5 (Santa Cruz Biotechnology sc-17866, 1:350). Secondary antibodies were donkey anti-chicken 488, donkey anti-rabbit Cy3, and donkey anti-mouse Cy5, donkey anti-goat 405 (Jackson ImmunoResearch 703-546-155, 711-166-152, 715-175-151, and 705-476-147, respectively; all used at 1:1000 dilution). After extensive washing in PBSTr, samples were incubated overnight at 4°C in PBSTr +0.2% DMSO+secondary antibodies. Samples were again extensively washed in PBSTr, dehydrated into 100% methanol, washed five times in 100% methanol, cleared, and imaged in Murray’s Clear (two parts benzyl benzoate, one part benzyl alcohol) on a metal slide with glass coverslip bottom using a Nikon A1R confocal microscope to obtain optical sections.
Xenopus luciferase assays
Request a detailed protocolThe Xenopus tropicalis v9.1 genome on Xenbase.org; RRID:SCR_003280 (Karimi et al., 2018) was used to define Xenopus enhancers. For Xenopus luciferase assays, the sequences of the mouse Aldh1a2 Enh1 (chr9:71241739–71242765; mm10 genome), X. trop aldh1a2 enh1 (chr3:89631924–89632943; v9.1/xenTro9 genome), X.trop shh MACS1(chr6:9535614–9536245; v9.1/xenTro9 genome), and human SHH MACS1 (chr7:156459384–156460049; hg19 genome) enhancers, as well as their respective mutant forms, were commercially synthesized (GenScript USA, Piscataway NJ; or IDT DNA, Coralville, IA) and cloned into the pGL4.23 firefly luc2/miniP vector (Promega E8411). For enh1 enhancer assays, embryos were co-injected with 5 pg of pRL-TK:renilla luciferase plasmid (Promega E2241) + 50 pg of the pGL4.23 luc2/miniP enhancer:luciferase plasmid and the following amounts of MOs or mRNAs into each dorsal marginal zone (dmz) region of 4–8 cell embryos: 3.5 ng of tbx5-MO or 3 bp mismatch-MO; 62.5 pg GR-Tbx5 RNA; 50 pg Xenopus Tbx5 RNA; 50 pg human TBX5-myc RNA. For hindgut mesendoderm injections, the luciferase reporters were injected into the ventral-posterior marginal zone at the 4–8 cell stage±Tbx5 RNA. For analysis of Shh MACS1 enhancer activity in endoderm, C1 (foregut) or C4 (hindgut) blastomeres were injected at the 16- or 32-cell stage with 5 pg pRL-TK+50 pg MACS1:luc±100 pg dnRARa RNA.
Each biological replicate contained a pool of five embryos, obtained from 2 to 3 separate fertilization/injection experiments which were frozen on dry ice in a minimal volume of 0.1× MBS and stored at –80°C. To assay luciferase activity samples were lysed in 100 µl of 100 mM TRIS-Cl pH 7.5, centrifuged for 10 min at ~13,000×g and then 25 µl of the clear supernatant lysate was used separately in firefly (Biotium #30085-1) and renilla (Biotium 300821) luciferase assays according to the manufacturer’s instructions. Relative luciferase activity was determined by normalizing firefly to renilla levels for each sample. Graph show the average relative luciferase activity ± standard deviation with dots showing values of biological replicates. Statistical significance was determined by parametric two-tailed paired t-test, *p<0.05.
Xenopus transgenesis
Request a detailed protocolTransgenesis was carried out using the I-SceI meganuclease procedure (Ogino et al., 2006; Pan et al., 2006; Rankin et al., 2009 ). Xenopus transgenic plasmids were constructed using the pI-SceI-d2EGFP plasmid backbone (Addgene 32674). First, a fragment containing the mouse or X. trop enh1 enhancers upstream of a minimal TATA box promoter (Tran et al., 2010) flanked by duplicated copies of the 250 bp chick B-globin HS4 insulator (Allen and Weeks, 2009; Rankin et al., 2011) was commercially synthesized (GenScript USA) and cloned into the ApaI/XhoI sites of pBluescript II KS+ (Agilent 212207). ApaI/XhoI digestion released this fragment, and it was ligated into ApaI/XhoI digested pI-SceI-d2EGFP plasmid. The meganuclease reaction contained 200 ng DNA, 2.5 μl I-SceI enzyme (New England Biolabs R0694S; kept at –80°C and used within 1 month of purchase) in 20 μl total volume and was incubated at 37°C for 30 min. 5 nl was then injected two times into 1 cell embryo on either side of the sperm entry point (10 nl total of meganuclease reaction injected per embryo). We observed 14/102 (~13%) and 21/183 (11%) GFP+full transgenic embryos using the mouse and X. trop enh1 constructs, respectively, from two independent injection experiments. As a negative control, 0/87 embryos were GFP positive when injected using reactions that omitted the I-SceI enzyme.
Mouse methods
RNA-seq
Request a detailed protocolRNA-seq of the micro-dissected E9.5 WT and Tbx5−/− pSHF/CPP was previously published and is available on GEO (Steimle et al., 2018, GSE75077). Heat maps were generated using Morpheus software (https://software.broadinstitute.org/morpheus/). Columns in the Figure 1 heat map represent biological replicates (Tbx5+/+ WT N=5, Tbx5−/− N=2), and each column replicate contained n=4 pooled CP dissected regions.
RT-qPCR, in-situ hybridization, and immunofluorescence
Request a detailed protocolRT-qPCR of dissected, pooled (n=4) mouse E9.5 pSHF/CPP regions was performed as described (Steimle et al., 2018), cDNA generated using SuperScript III First-Strand Synthesis SuperMix (ThermoFisher 18080051), and qPCR was performed using PowerUp Mastermix (ThermoFisher A25742). Gene-expression levels were normalized by Gapdh and RT-qPCR primers are listed in Supplementary file 1. In-situ hybridization on mouse embryos was performed as described (Hoffmann et al., 2009). Shh probe was provided by Elizabeth Grove (University of Chicago). Immunofluorescence of WT CD-1 (Charles River) or Shh:GFP (B6.129 × 1(Cg)-Shhtm6Amc/J; Jax Labs Stock Number #008466) mouse embryos was performed as described (Rankin et al., 2016; Rankin et al., 2018) using mouse anti-Aldh1a2 (Santa Cruz Biotechnology sc-166362; 1:1000), goat anti-Tbx5 (Santa Cruz Biotechnology sc-17866, 1:300), rabbit anti-Nkx2-1 (Santa Cruz Biotechnology sc-13040 H-190, 1:500), and chicken anti-GFP (Aves Labs GFP-1020, 1:1000).
Reconstructions of whole-mount in-situ hybridizations were generated using previously published methods (Steimle et al., 2018). In brief, images were obtained and pre-processed using Adobe Photoshop CS3 Extended (version 10.0.1, http://www.adobe.com) and reconstructed with AMIRA (version 5.3.2, http://www.amira.com). Manual review of each image in the stack was performed and corrections were made when necessary. LabelFields for gene expression and tissue were generated from the same series of sections using separate CastField and LabelVoxel modules. The SurfaceGen module was used to generate surfaces from these LabelFields. Gene expression models for two different genes were initially aligned using the Landmark (two sets) module, and a minimum of three landmarks were used to align the separate models. These landmarks were located using the pharyngeal endoderm and ventral edge of the SHF. Final alignments were fine-tuned manually using the Transform editor.
Digital in-situ hybridization
Request a detailed protocolDigital in-situ hybridization for Aldh1a2, Tbx1, Tbx5, and Shh was generated using the single cells Spatial Mouse Atlas (Lohoff et al., 2021; https://crukci.shinyapps.io/SpatialMouseAtlas/). Images are centered around the cardiothoracic region with cardiomyocytes highlighted.
Mouse ESCs
Request a detailed protocolThe inducible Tbx5OE-mESC line was previously generated (Steimle et al., 2018) and differentiated to the cardiac lineage as described (Kattman et al., 2011). Doxycycline (Sigma D9891; concentrations of 0, 5, 10, 25, 50, 100, 250, and 500 ng/ml) was applied at the cardiac progenitor-like stage (day 6) and cells were harvested for RNA 24 hr later.
ChIP-seq
Request a detailed protocolChIP-seq was performed using dissected whole lungs from E14.5 CD-1 mouse embryos obtained from Charles River. Chromatin was prepared as previously described (Steimle et al., 2018). For immunoprecipitation, the chromatin extract was incubated with 5 µg of the anti-TBX5 antibody (Santa Cruz Biotechnology sc-17866; Lot #G1516) at 4°C for >12 hr in a total volume of 200 μl. The immune complexes were captured by Protein G-conjugated magnetic beads (Life Technologies, 1003D) and washed as previously described (Steimle et al., 2018). The captured chromatin was eluted in ChIP Elution Buffer (10 mM Tris-HCl, pH 8.0, 1 mM EDTA, 1% SDS, and 250 mM NaCl) at 65°C. After RNase and proteinase K treatment and reverse cross-linking, DNA was purified. High-throughput sequencing libraries from ChIP and input DNA were prepared using NEBNext Ultra DNA Library Prep Kit (New England Biolabs, E7370S). During library preparation, adaptor-ligated DNA fragments of 200–650 bp in size were selected before PCR amplification using Sera-Mag magnetic beads (GE, 6515-2105-050-250). DNA libraries were sequenced using Illumina Hi-seq instruments (single-end 50 base) by the Genomics Core Facility at the University of Chicago.
Bioinformatics
ChIP-seq analysis
Request a detailed protocolRaw sequencing reads were aligned to the mm10 genome using Bowtie2 (Langmead and Salzberg, 2012) and SAMtools (Li et al., 2009) requiring a minimum mapping quality of 10 (−q 10). Pooled peak calling was performed using default settings of MACS2 callpeak (Zhang et al., 2008) with a q-value set to 0.05 and tag size set to 6 (−q 0.05 s 6). A fold-enrichment track was generated using MACS2 with the bdgcmp function (−m FE) for visualization on the IVG genome browser (Thorvaldsdottir et al., 2012). Public data reanalyzed in this study was downloaded from GEO either as Bigwig files or raw reads which were processed as described above.
RNA-seq analysis
Request a detailed protocolRNA-seq of the micro-dissected E9.5 WT and Tbx5−/− pSHF/CPP was previously published and is available on GEO (Steimle et al., 2018, GSE75077). This RNA-seq data was re-analyzed using Computational Suite for Bioinformaticians and Biologists (CSBB – v3.0.0) using ProcessPublicData module (https://github.com/praneet1988/Computational-Suite-For-Bioinformaticians-and-Biologists), (Chaturvedi, 2019). Differentially expressed genes (DEGs) between Tbx5−/− and WT were identified using RUVSeq, with a threshold of 1.5 fold change and 5% FDR. Expression heat maps were generated using Morpheus (https://software.broadinstitute.org/morpheus/).
DEGs were compared with gene sets from single-cell RNA-seq defining aSHF versus pSHF (de Soysa et al., 2019, GSE126128) and pharynx versus CPP+ lung progenitor cells (Han et al., 2020, GSE136689) from the early mouse embryo. We created a cardio-pharyngeal enriched gene set by combining marker genes of aSHF and pharynx mesendoderm, and a CP gene set by combining markers pSHF, pulmonary mesoderm, and lung endoderm. Overlaps in gene sets were visualized by Venn diagrams and significant overlaps were defined by HGTs. In addition, we assessed the enrichment of upregulated and downregulated DEGs from the Tbx5−/− embryos compared to the single-cell data sets by GSEA (Subramanian et al., 2005).
Data availability
ChIP-seq data generated in this study is available from the Gene Expression Omnibus (GEO) accession number GSE167207.
-
NCBI Gene Expression OmnibusID GSE167207. TBX5 ChIP from the Fetal Lung.
-
NCBI Gene Expression OmnibusID GSE75077. Whole genome transcriptomic experiments in mice heart development system.
-
NCBI Gene Expression OmnibusID GSE126128. Single-cell analysis of cardiogenesis reveals basis for organ level developmental defects.
-
NCBI Gene Expression OmnibusID GSE136689. Single cell sequencing of dissected mouse foreguts at embryonic day 8.5 to 9.5.
-
NCBI Gene Expression OmnibusID GSE139803. TBX5 ChIP from the Fetal Heart.
-
NCBI Gene Expression OmnibusID GSE104840. LSD1-mediated enhancer silencing enables endocrine cell development through attenuation of retinoic acid signaling.
-
NCBI Gene Expression OmnibusID GSE54471. Developmental Competence Encoded at the Level of Enhancers.
References
-
Dissection of Tbx1 and Fgf interactions in mouse models of 22q11DS suggests functional redundancyHuman Molecular Genetics 15:3219–3228.https://doi.org/10.1093/hmg/ddl399
-
Bacteriophage phiC31 integrase mediated transgenesis in Xenopus laevis for protein expression at endogenous levelsMethods in Molecular Biology 518:113–122.https://doi.org/10.1007/978-1-59745-202-1_9
-
Transcriptional Patterning of the Ventricular Cardiac Conduction SystemCirculation Research 127:e94–e106.https://doi.org/10.1161/CIRCRESAHA.118.314460
-
The Encyclopedia of DNA elements (ENCODE): data portal updateNucleic Acids Research 46:D794–D801.https://doi.org/10.1093/nar/gkx1081
-
Retinoic acid regulates anterior-posterior patterning within the lateral plate mesoderm of XenopusMechanisms of Development 126:913–923.https://doi.org/10.1016/j.mod.2009.07.001
-
Fgf is required to regulate anterior-posterior patterning in the Xenopus lateral plate mesodermMechanisms of Development 128:327–341.https://doi.org/10.1016/j.mod.2011.06.002
-
Xenbase: a genomic, epigenomic and transcriptomic model organism databaseNucleic Acids Research 46:D861–D868.https://doi.org/10.1093/nar/gkx936
-
Heart fields and cardiac morphogenesisCold Spring Harbor Perspectives in Medicine 4:a015750.https://doi.org/10.1101/cshperspect.a015750
-
Fast gapped-read alignment with Bowtie 2Nature Methods 9:357–359.https://doi.org/10.1038/nmeth.1923
-
The Sequence Alignment/Map format and SAMtoolsBioinformatics 25:2078–2079.https://doi.org/10.1093/bioinformatics/btp352
-
Isl1 is upstream of sonic hedgehog in a pathway required for cardiac morphogenesisDevelopmental Biology 295:756–763.https://doi.org/10.1016/j.ydbio.2006.03.053
-
Identification of a Tbx1/Tbx2/Tbx3 genetic pathway governing pharyngeal and arterial pole morphogenesisHuman Molecular Genetics 21:1217–1229.https://doi.org/10.1093/hmg/ddr553
-
Hox genes regulate the onset of Tbx5 expression in the forelimbDevelopment 139:3180–3188.https://doi.org/10.1242/dev.084814
-
Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouseDevelopment 128:1019–1031.
-
High-throughput transgenesis in Xenopus using I-SceI meganucleaseNature Protocols 1:1703–1710.https://doi.org/10.1038/nprot.2006.208
-
I-SceI meganuclease-mediated transgenesis in XenopusDevelopmental Dynamics 235:247–252.https://doi.org/10.1002/dvdy.20608
-
Reiterative Mechanisms of Retinoic Acid Signaling during Vertebrate Heart DevelopmentJournal of Developmental Biology 7:11.https://doi.org/10.3390/jdb7020011
-
Improved cre reporter transgenic XenopusDevelopmental Dynamics 238:2401–2408.https://doi.org/10.1002/dvdy.22043
-
New doxycycline-inducible transgenic lines in XenopusDevelopmental Dynamics 240:1467–1474.https://doi.org/10.1002/dvdy.22642
-
Signaling pathways controlling second heart field developmentCirculation Research 104:933–942.https://doi.org/10.1161/CIRCRESAHA.109.194464
-
T-box genes and cardiac developmentBirth Defects Research Part C 69:25–37.https://doi.org/10.1002/bdrc.10001
-
Retinoid receptors promote primary neurogenesis in XenopusDevelopment 124:515–523.
-
Retinoic acid controls heart anteroposterior patterning by down-regulating Isl1 through the Fgf8 pathwayDevelopmental Dynamics 237:1627–1635.https://doi.org/10.1002/dvdy.21570
-
Role of carotenoids and retinoids during heart developmentBiochimica et Biophysica Acta. Molecular and Cell Biology of Lipids 1865:158636.https://doi.org/10.1016/j.bbalip.2020.158636
-
BookEarly Development of Xenopus Laevis: A Laboratory ManualCold Spring Harbor Lab Press.
-
Regulatory variation in a TBX5 enhancer leads to isolated congenital heart diseaseHuman Molecular Genetics 21:3255–3263.https://doi.org/10.1093/hmg/dds165
-
TBX5: A Key Regulator of Heart DevelopmentCurrent Topics in Developmental Biology 122:195–221.https://doi.org/10.1016/bs.ctdb.2016.08.008
-
tegrative Genomics Viewer (IGV): high-performance genomics data visualization and explorationBriefings in Bioinformatics 14:178–192.https://doi.org/10.1093/bib/bbs017
-
A novel regulatory element for Shh expression in the lung and gut of mouse embryosMechanisms of Development 131:127–136.https://doi.org/10.1016/j.mod.2013.09.003
-
Retinoic acid regulates morphogenesis and patterning of posterior foregut derivativesDevelopmental Biology 297:433–445.https://doi.org/10.1016/j.ydbio.2006.05.019
-
Signaling through retinoic acid receptors in cardiac development: Doing the right things at the right timesBiochimica et Biophysica Acta 1849:94–111.https://doi.org/10.1016/j.bbagrm.2014.08.003
-
Retinoids and cardiac developmentJournal of Developmental Biology 2:50–71.https://doi.org/10.3390/jdb2010050
-
Model-based analysis of ChIP-Seq (MACSGenome Biology 9:R137.https://doi.org/10.1186/gb-2008-9-9-r137
Article and author information
Author details
Michael J Herriges

Funding
Eunice Kennedy Shriver National Institute of Child Health and Human Development (P01HD093363)
- Aaron M Zorn
National Heart, Lung, and Blood Institute (R01HL092153)
- Ivan P Moskowitz
National Heart, Lung, and Blood Institute (R01HL124836)
- Ivan P Moskowitz
National Institute of General Medical Sciences (T32GM007183)
- Jeffrey D Steimle
National Heart, Lung, and Blood Institute (T32HL007381)
- Jeffrey D Steimle
- Ariel B Rydeen
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank members of the Zorn and Wells labs for suggestions and Lisa Sandell (University of Louisville) for discussions. The authors thank Nicole Edwards and Leslie Brown for help generating and genotyping Shh:GFP mouse embryos.
Ethics
Mouse and Xenopus experiments were performed according to Institutional Animal Care and Use Committee (IACUC) protocols (University of Chicago protocol 71737; Cincinnati Children's Hospital protocol 2019-0053).
Copyright
© 2021, Rankin et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 2,085
- views
-
- 369
- downloads
-
- 27
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 27
- citations for umbrella DOI https://doi.org/10.7554/eLife.69288
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Tbx5 drives Aldh1a2 expression to regulate a RA-Hedgehog-Wnt gene regulatory network coordinating cardiopulmonary development
eLife 10:e69288.
https://doi.org/10.7554/eLife.69288




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}