The Na+/K+ pump dominates control of glycolysis in hippocampal dentate granule cells
- Department of Neurobiology, Harvard Medical School, United States
Abstract
Cellular ATP that is consumed to perform energetically expensive tasks must be replenished by new ATP through the activation of metabolism. Neuronal stimulation, an energetically demanding process, transiently activates aerobic glycolysis, but the precise mechanism underlying this glycolysis activation has not been determined. We previously showed that neuronal glycolysis is correlated with Ca2+ influx, but is not activated by feedforward Ca2+ signaling (Díaz-García et al., 2021a). Since ATP-powered Na+ and Ca2+ pumping activities are increased following stimulation to restore ion gradients and are estimated to consume most neuronal ATP, we aimed to determine if they are coupled to neuronal glycolysis activation. By using two-photon imaging of fluorescent biosensors and dyes in dentate granule cell somas of acute mouse hippocampal slices, we observed that production of cytoplasmic NADH, a byproduct of glycolysis, is strongly coupled to changes in intracellular Na+, while intracellular Ca2+ could only increase NADH production if both forward Na+/Ca2+ exchange and Na+/K+ pump activity were intact. Additionally, antidromic stimulation-induced intracellular [Na+] increases were reduced >50% by blocking Ca2+ entry. These results indicate that neuronal glycolysis activation is predominantly a response to an increase in activity of the Na+/K+ pump, which is strongly potentiated by Na+ influx through the Na+/Ca2+ exchanger during extrusion of Ca2+ following stimulation.
Editor's evaluation
The authors investigate mechanisms that regulate glycolytic ATP production in neurons. They conclude that the cytosolic Na+, not Ca2+, and the activity of the Na+/K+ pump drive glycolysis. The study is conceptually significant as it seeks to determine how neuronal glycolysis is coupled to electrical activity. The study is thoughtful, uses sophisticated fluorescence lifetime imaging technology and clever experimental designs, and as such provides new insights into how electrical activity regulates glycolysis.
https://doi.org/10.7554/eLife.81645.sa0Introduction
Cellular energy consumption must be balanced by new energy production through the activation of metabolism. In neurons, energy consumption is strongest following excitation (Attwell and Iadecola, 2002; Attwell and Laughlin, 2001; Engl and Attwell, 2015; Howarth et al., 2012; Lennie, 2003; Yu et al., 2018). Accordingly, neuronal excitation triggers a transient activation of glycolysis, which is measured as an increase to neuronal cytoplasmic NADH:NAD+ (NADHCYT) that can last for several minutes after the excitatory event (Díaz-García et al., 2017; Díaz-García et al., 2021a). The specific mechanism that drives neuronal glycolysis activation has not been identified.
It seems highly plausible that glycolysis activation in neurons could be driven primarily by an increase in the activity of ion-ATPases, the ATP-powered ion transporters that move ions against their electrochemical gradients by (through a series of reaction steps; Post et al., 1965; Sen and Post, 1964) converting ATP into ADP and Pi (Biondo et al., 2021; Calì et al., 2018; Chemaly et al., 2018; Kaplan, 2002). Ion-ATPases are estimated to be responsible for most of the energy consumption associated with neuronal signaling and are closely coupled to glycolytic enzymes across many cell types (Ames, 2000; Dhar-Chowdhury et al., 2007; Dzeja and Terzic, 2003; Xu et al., 1995). Moreover, activation of glycolysis by increased ion-ATPase activity could be multimodal: a decrease to cellular ATP would stimulate glycolysis by mass action at the ATP-generating steps, while an increase to cellular ADP and Pi would stimulate glycolysis by allosterically activating phosphofructokinase and by mass action at both the glyceraldehyde-3-phosphate dehydrogenase/phosphoglycerate kinase enzyme complex and pyruvate kinase steps (Fothergill-Gilmore and Michels, 1993; Kemp and Foe, 1983; Nelson et al., 2008; Schöneberg et al., 2013; Tomokuni et al., 2010).
There are several ion-ATPases (also called ion pumps) that regulate neuronal Na+ and Ca2+ changes (Figure 1A). Na+ is handled exclusively by the Na+/K+-ATPase (or Na+/K+ pump), which uses 1 ATP molecule to export 3 Na+ and import 2 K+ across the plasma membrane. Ca2+ is handled by a diverse set of active transporters, including the plasma membrane Ca2+-ATPase (PMCA), which exports 1 Ca2+ across the plasma membrane per ATP (Niggli et al., 1982; Thomas, 2009), and the sarco-/endo-plasmic reticulum Ca2+-ATPase (SERCA), which pumps 2 Ca2+ into the endoplasmic reticulum lumen per ATP (Tran et al., 2009). Aside from ATPases, Ca2+ is also regulated by the Na+/Ca2+-exchanger (NCX) (Lee et al., 2009), a secondary active transporter that uses the electrochemical energy stored in the plasma membrane Na+ gradient (built by the Na+/K+ pump) to actively export 1 Ca2+ by importing 3 Na+. Any increase to neuronal Na+ or Ca2+ will increase the activity of their respective ion transporters.
Figure 1 with 3 supplements see all
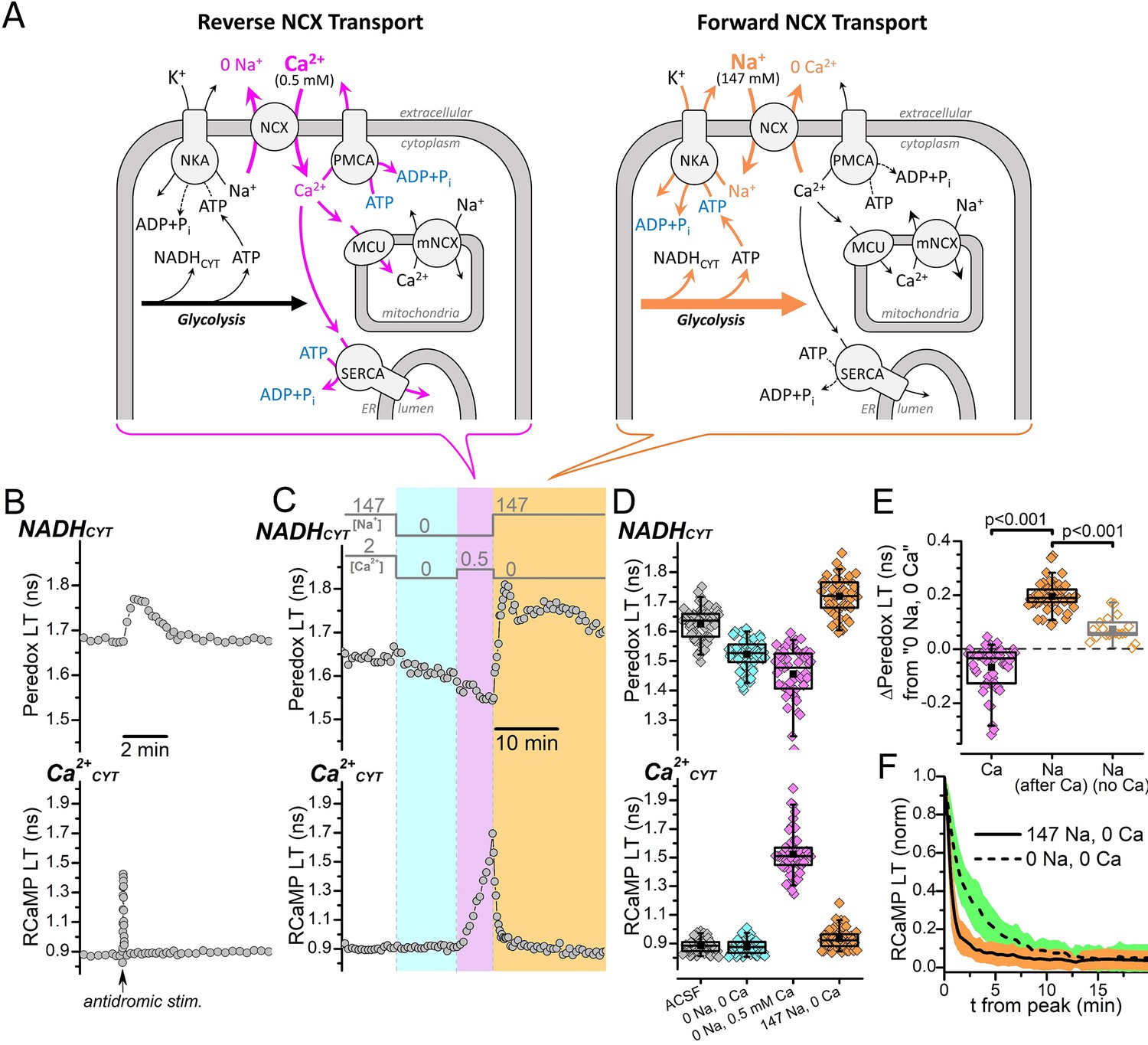
NADHCYT production is strongly influenced by Na+, but not by Ca2+CYT.
(A) Cartoon showing the NCX transport modes activated by different external [Na+] and [Ca2+] conditions and their expected effects on the activities of ion pumps and the production of NADHCYT from glycolysis activation. Reverse NCX transport (left schematic) increases intracellular [Ca2+], which increases the activities Ca2+ pumps and Ca2+ transport into mitochondria (magenta arrows). Forward NCX transport (right schematic) increases intracellular [Na+], which increases the activity of the Na+/K+ pump (orange arrows). The bracket below each schematic indicates the NCX transport mode activated by the external solution changes in (C). Transport stoichiometries are not indicated. Abbreviations: Na+/Ca2+-exchanger (NCX), Na+/K+-ATPase (NKA), plasma membrane Ca2+-ATPase (PMCA), sarco-/endo-plasmic reticulum Ca2+-ATPase (SERCA), mitochondrial Ca2+ uniporter (MCU), mitochondrial Na+/Ca2+-exchanger (mNCX), endoplasmic reticulum (ER). (B) Representative fluorescence lifetime (LT) traces of Peredox (top trace) and RCaMP (bottom trace) from a DGC bathed in ACSF. Antidromic stimulation was delivered at the time point indicated by the arrow along the RCaMP trace, which transiently increases both NADHCYT and Ca2+CYT. (C) Fluorescence LT traces of Peredox (top) and RCaMP (bottom) from a DGC showing how external Na+ and Ca2+ changes affect NADHCYT and Ca2+CYT. The bars above the Peredox trace indicate the external [Na+] and [Ca2+]. NADHCYT was decreased by switching the bath solution from ACSF (147 mM Na+ and 2 mM Ca2+) to a solution with nominally 0 Na+ and 0 Ca2+ (cyan shading). Ca2+CYT was elevated by applying 0.5 mM Ca2+ with 0 Na+ to activate reverse NCX transport (magenta shading), and NADHCYT decreased further. NADHCYT was strongly increased after activating forward NCX transport by the subsequent removal of external Ca2+ and application of 147 mM Na+ (orange shading). (D) Box plots of the fluorescence LTs of Peredox (top) and RCaMP (bottom) showing the effects of the external Na+ and Ca2+ changes performed in panel C across many DGCs (n=53). The external bath conditions for each box plot are listed at the bottom of the RCaMP plot in chronological order from left to right. The colors of each box plot correspond to the colors indicated in (C). The mean Peredox LT values in each condition were: 1.63±0.06 ns in ACSF, 1.52±0.05 ns in 0 Na+ and 0 Ca2+, 1.46±0.09 ns in 0 Na and 0.5 mM Ca2+, and 1.72±0.06 ns in 147 mM Na+ and 0 Ca2+. The mean RCaMP LT values in each condition were: 0.88±0.05 ns in ACSF, 0.88±0.05 ns in 0 Na+ and 0 Ca2+, 1.52±0.15 ns in 0 Na+ and 0.5 mM Ca2+, and 0.93±0.07 ns in 147 mM Na+ and 0 Ca2+. (E) Changes to the Peredox LT relative to the 0 Na+ and 0 Ca2+ condition, after either a Ca2+CYT elevation from reverse NCX transport (Ca, black box, magenta filled diamonds), an influx of Na+ due to forward NCX transport (Na after Ca, black box, orange filled diamonds), or application of Na+ without forward NCX (Na no Ca, gray box, orange open diamonds). The mean Peredox LT changes were: –0.07±0.08 ns (n=53) for Ca2+CYT elevation, 0.20±0.05 ns (n=53) for Na+ influx via forward NCX, and 0.07364±0.04565 ns (n=19) for Na+ application without forward NCX. Statistical significance between ‘Ca’ and ‘Na after Ca’ is indicated by a paired Wilcoxon test and between ‘Na after Ca’ and ‘Na no Ca’ by a Mann-Whitney test. (F) Effect of external Na+ on the return of Ca2+CYT to baseline following a reverse NCX transport-mediated Ca2+ influx. The mean decay of the RCaMP LT following the Ca2+CYT increase (normalized to the peak RCaMP LT value) is shown when the external solution contained either 147 mM Na+ (solid line, orange SD shading, n=53) or 0 Na+ (dashed line, green SD shading, n=49). Decay data in 147 mM Na+ were from the same DGCs as in (D) and (E), while data in 0 Na+ were from the same DGCs as Figure 1—figure supplement 1B; a representative trace of this experiment is shown in Figure 1—figure supplement 1A.
Transient increases to neuronal cytoplasmic Ca2+ (Ca2+CYT) induced by excitation are positively correlated with transient increases in subsequent NADHCYT production (Díaz-García et al., 2017; Díaz-García et al., 2021a); in other words, stronger stimulations evoke larger increases to both Ca2+CYT and NADHCYT. At first glance, this positive correlation could be (mis)interpreted as glycolysis being driven primarily by an increase in the activities of Ca2+ pumps (PMCA and SERCA) as Ca2+ is pumped from the cytoplasm. However, this readout of neuronal NADHCYT and Ca2+CYT lacks information about stimulation-induced increases to intracellular Na+, which also likely covary with stimulation strength, so a contribution of increased Na+/K+ pump activity to glycolysis activation cannot be excluded.
The Na+/K+ pump is primed to be the predominant driving force underlying neuronal glycolysis activation. Many reports estimate that the Na+/K+ pump consumes ~50% of total brain energy (Ames, 2000; Astrup et al., 1981; Engl and Attwell, 2015; Milligan and McBride, 1985; Whittam, 1962) and uses substantially more ATP than Ca2+ pumps both during neurotransmission and at rest (Attwell and Laughlin, 2001; Harris et al., 2012; Rolfe and Brown, 1997). Furthermore, since Ca2+CYT regulation in some neurons depends strongly on the NCX (Lee et al., 2009), a transient Ca2+CYT increase could indirectly elevate intracellular [Na+], which would potentiate any increase in Na+/K+ pump activity due to channel-mediated Na+ entry.
To shed light on the mechanism of neuronal glycolysis activation, we investigated the coupling of Na+/K+ pump and Ca2+ pump activities to NADHCYT production in the somas of hippocampal dentate granule cells (DGCs) within acute brain slices by using two-photon fluorescence imaging of genetically encoded fluorescent biosensors and fluorescent dyes. The data provide compelling evidence indicating that increased activity of the Na+/K+ pump is the predominant driver of neuronal glycolysis activation, whereas increased activities of Ca2+ pumps appear to be negligibly coupled to glycolysis.
Results
Cytoplasmic NADH production is strongly influenced by Na+, but not Ca2+
We simultaneously monitored changes in neuronal metabolism and ion fluxes in DGC somas by expressing Peredox and RCaMP, two genetically encoded fluorescent biosensors that report NADH:NAD+ (NADHCYT) and free [Ca2+], respectively (Akerboom et al., 2013; Hung et al., 2011; Mongeon et al., 2016). Analyte binding to Peredox (NADH in competition with NAD+) or RCaMP (Ca2+) alters each sensor’s fluorescence lifetime (LT), the average time that the fluorophore spends in the excited state prior to its return to the ground state (Lakowicz, 2006). The fluorescence LTs of Peredox and RCaMP are increased (i.e., extended in average duration) by NADHCYT or Ca2+CYT increases, respectively.
Our previous observations demonstrated that NADHCYT increases are strongly tied to glycolysis (Díaz-García et al., 2017; Díaz-García et al., 2021a) and occur regardless of changes in mitochondrial metabolism: electrical-stimulation-induced increases to NADHCYT were substantially diminished by iodoacetic acid (an inhibitor of GAPDH) but not by AOA (an inhibitor of MAS) and were not affected by impaired NADH production in mitochondria (i.e., in the presence of inhibitors of both the mitochondrial pyruvate carrier and lactate dehydrogenase; Figure 2D in Díaz-García et al., 2021a). Thus, acute NADHCYT changes arise directly from changes in glycolytic flux and are not strongly influenced by changes in NADH consumption by the mitochondria.
We began our investigations on the coupling of the Na+/K+ pump or Ca2+ pumps to glycolysis activation by recapitulating our previous findings: that electrical stimulation of DGCs transiently increases their NADHCYT production (Díaz-García et al., 2017; Díaz-García et al., 2021a). Representative LT traces of Peredox and RCaMP, when both sensors were expressed in the cytoplasm of a DGC within a hippocampal slice that was bathed in ACSF (Figure 1B), show that an antidromic stimulation event delivered from an electrode placed in the hippocampal hilus causes both a fast transient increase of the RCaMP LT, reflecting a transient increase to Ca2+CYT, and a slower, longer-lasting transient increase of the Peredox LT, reflecting transient overproduction of NADHCYT by glycolysis. But since neuronal stimulation triggers influxes of both Na+ and Ca2+, the glycolysis activation could result from increases to both Na+/K+ pump and Ca2+ pump activities.
To differentiate how changes in the activities of the Na+/K+ pump or Ca2+ pumps affect glycolysis, we measured how NADHCYT production is affected by separate elevations of either intracellular Na+ or Ca2+CYT (Figure 1C). If glycolysis is preferentially coupled to the activity of either the Na+/K+ pump or Ca2+ pumps, then NADHCYT production should be coupled to changes in the levels of the transported ion (Na+ or Ca2+). Simultaneous removal of both Na+ and Ca2+ from the bath solution (by external ion substitution) decreased the Peredox LT from its baseline in ACSF (Figure 1C), which indicates a decrease to NADHCYT; we can attribute this NADHCYT decrease entirely to the removal of Na+, since removing only Ca2+ slightly increases NADHCYT (cf. Figure 6—figure supplement 1b in Díaz-García et al., 2021a). This NADHCYT decrease suggests that the rate of glycolysis can be slowed by depleting the intracellular [Na+], which would decrease the activity of the Na+/K+ pump. The RCaMP LT was not affected by the removal of Na+ and Ca2+ (Figure 1C), indicating that Ca2+CYT was maintained at or below resting levels, near the floor of RCaMP’s dynamic range (Akerboom et al., 2013).
Following the removal of external Na+ and Ca2+, we added each ion back one at a time to separately increase either intracellular Na+ or Ca2+. We increased Ca2+ by applying 0.5 mM external Ca2+ in the nominal absence of external Na+ to facilitate Ca2+ entry via reverse NCX transport, and the expected strong increase in Ca2+CYT was confirmed by an increase in the RCaMP LT (Figure 1C, during the magenta shaded interval). However, this increase to Ca2+CYT was not associated with an increase to NADHCYT. In fact, the opposite occurred: Ca2+CYT elevation decreased the Peredox LT even further from the 0 Na+ 0 Ca2+ bath condition (possibly due to the extrusion of Na+ during reverse NCX; see Discussion). At this point, with Ca2+CYT elevated, we removed 0.5 mM external Ca2+ and re-applied external Na+ to facilitate strong Na+ influx via forward NCX transport (in exchange for Ca2+CYT), which promptly increased NADHCYT. Following the external Na+ application, Ca2+CYT returned to baseline levels (Figure 1C, orange shaded interval).
The decrease to Peredox LT by removing external Na+ and the increase to Peredox LT from re-applying external Na+, across many DGCs (Figure 1D), show that NADHCYT is strongly influenced by Na+. Relative to the Peredox LT in the 0 Na+ 0 Ca2+ bath condition, the Peredox LT was decreased when Ca2+CYT was elevated but increased following external Na+ re-application (Figure 1E), which suggests that glycolysis activation is strongly tied to the activity of the Na+/K+ pump, but not strongly tied to the activity of Ca2+ pumps (nor to the increase in Ca2+CYT per se, e.g., via a signaling mechanism).
Our ion substitution experiments also hinted that both glycolysis activation and Ca2+CYT regulation in DGCs are dependent on forward NCX transport. A Ca2+CYT increase (from reverse NCX transport) returned faster to baseline levels if external Na+ was present (i.e., if forward NCX was activated, Figure 1F; and compare Figure 1C with Figure 1—figure supplement 1A), and NADHCYT did not increase following a Ca2+CYT increase if external Ca2+ was removed without re-applying external Na+ (Figure 1—figure supplement 1A and B). NADHCYT production increased when external Na+ was removed and re-applied without previously elevating Ca2+CYT (i.e., without activating forward NCX, Figure 1—figure supplement 1C and D), but this NADHCYT increase was weaker than when external Na+ was re-applied while Ca2+CYT was elevated (i.e., when forward NCX was activated, Figure 1D and E).
The inability of a Ca2+CYT elevation to measurably stimulate NADHCYT production in the absence of external Na+ suggests that any increase in activity of PMCA and SERCA to pump Ca2+ from the cytoplasm does not substantially activate glycolysis, even during the prolonged Ca2+CYT increases in our ion substitution experiments. But maybe inhibiting one of these Ca2+ pumps, to divert more Ca2+CYT to be extruded by the non-inhibited pump, could boost the activity of the other and consequently amplify its associated effect on glycolysis. We tested this possibility by inhibiting SERCA, which compared to PMCA has more specific pharmacology and uses less ATP per Ca2+ transported. Thapsigargin (1 μM, a specific SERCA inhibitor) increased both the NADHCYT and Ca2+CYT transients evoked by antidromic electrical stimulations in ACSF (Figure 1—figure supplement 2A and B) as well as the ∆Peredox/∆RCaMP ratio (Figure 1—figure supplement 2C). These effects are consistent with SERCA inhibition and a diversion of Ca2+CYT to other clearance pathways that are either more closely coupled to glycolysis or that transport fewer ions per ATP utilized. However, reverse-NCX-mediated Ca2+CYT increases in the presence of 1 μM thapsigargin (and absence of external Na+) still failed to increase NADHCYT (Figure 1—figure supplement 1B), meaning that any putative amplification of PMCA activity after SERCA inhibition was still not enough to drive measurable glycolysis activation.
We have previously shown that electrical stimulation increases DGC mitochondrial Ca2+ (Ca2+MITO) from Ca2+ entry through the mitochondrial Ca2+ uniporter (MCU) (Díaz-García et al., 2021a). Could Ca2+CYT’s failure to activate NADHCYT production during our ion substitution experiments be due to a lack of activation of some unknown Ca2+MITO-dependent process? This seems unlikely, as bath-applied 0.5 mM external Ca2+ in the absence of external Na+ increased the fluorescence LT of mitochondrially targeted RCaMP (Figure 1—figure supplement 3A and B), indicating that Ca2+MITO is increased by reverse-NCX-mediated Ca2+ entry. Ca2+MITO slightly decreased when external Ca2+ was removed, but complete return of Ca2+MITO to baseline levels was not achieved until external Na+ was applied, demonstrating that Ca2+MITO regulation is dependent on Na+, like Ca2+CYT.
In summary, these ion substitution experiments show that neuronal NADHCYT production is closely coupled to the flux of Na+, and that a Ca2+CYT elevation is not sufficient to elevate NADHCYT regardless of whether it is pumped by SERCA, pumped by PMCA, or taken up into mitochondria via MCU. They also show that NADHCYT production is activated more strongly by conditions that favor forward NCX transport that exchanges Ca2+CYT for Na+, and that regulation of both DGC Ca2+CYT and Ca2+MITO depends on Na+. These observations suggest that neuronal glycolysis is both strongly coupled to the activity of the Na+/K+ pump and sensitive to Na+ import via forward NCX transport, which would become activated following an excitatory event where Ca2+CYT must be removed.
Malate-aspartate shuttle activity does not mask a Ca2+-induced increase to NADHCYT
The malate-aspartate shuttle (MAS) is a major pathway that recycles NADHCYT by transferring reducing equivalents from NADHCYT to mitochondria (McKenna et al., 2006; Satrústegui and Bak, 2015). MAS activity is activated by Ca2+CYT elevations but is reduced by Ca2+MITO elevations (Contreras and Satrústegui, 2009; Satrústegui and Bak, 2015), which likely means that neuronal excitation would cause a brief MAS activation followed by a longer inhibition, mirroring the Ca2+CYT and Ca2+MITO transient time courses (Contreras and Satrústegui, 2009). But the state of MAS activity in our ion substitution experiments was uncertain since reverse NCX transport produced prolonged increases to both Ca2+CYT and Ca2+MITO (Figure 1 and Figure 1—figure supplement 3), so it remained a possibility that Ca2+CYT activation of MAS could increase NADHCYT recycling and conceal any small increase to NADHCYT production due to increased activity of Ca2+ pumps. Therefore, we tested if a Ca2+CYT elevation could increase NADHCYT after MAS inhibition (Figure 2).
Figure 2
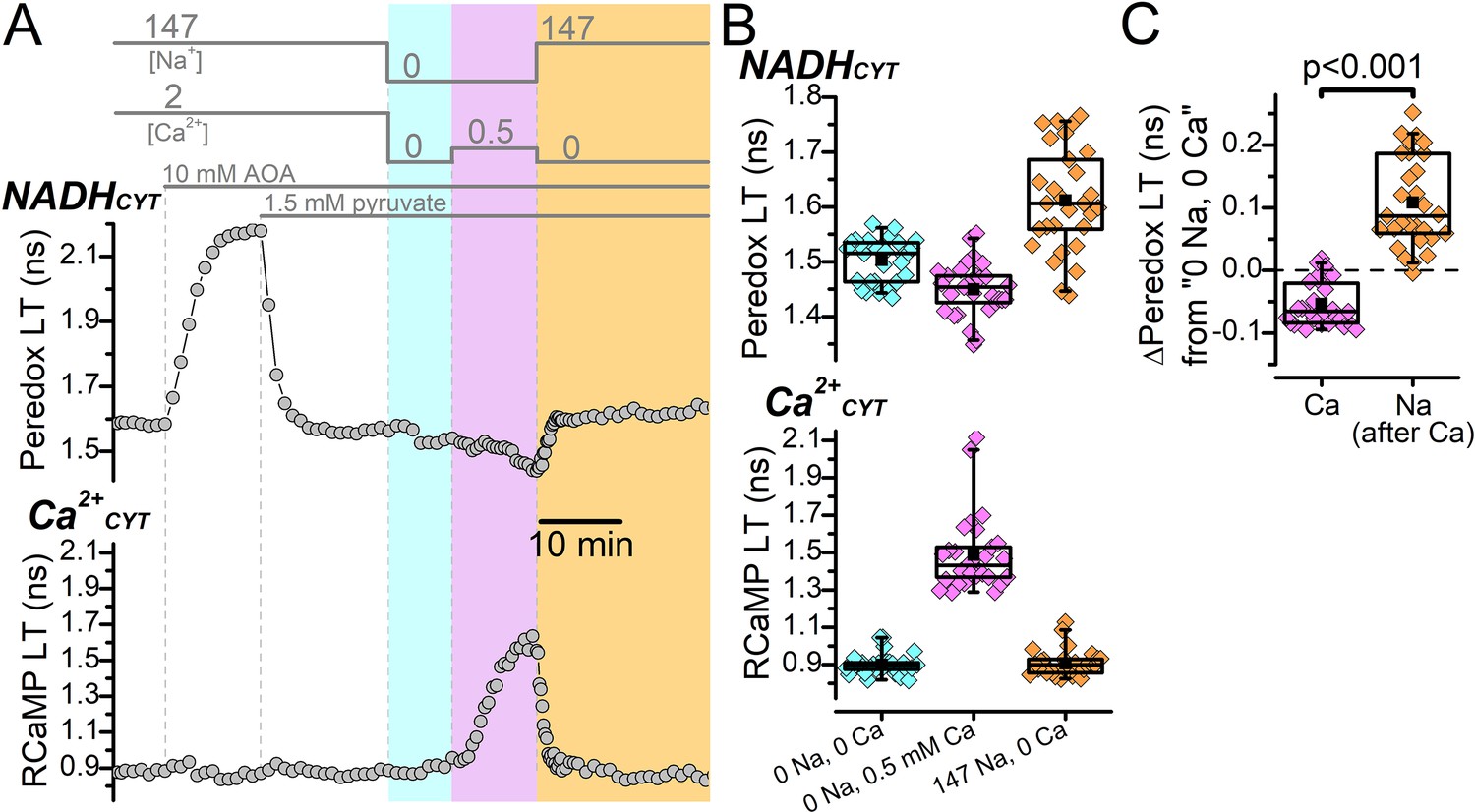
Malate-aspartate shuttle activity does not conceal a possible NADHCYT increase produced by elevated Ca2+CYT.
(A) Representative fluorescence lifetime (LT) traces of Peredox (top) and RCaMP (bottom) from a DGC. External solution changes are indicated by the bars above the Peredox trace. The recording begins in ACSF (147 mM Na+ and 2 mM Ca2+), where the addition of 10 mM AOA strongly increases the Peredox LT. Addition of 1.5 mM pyruvate reverted the Peredox LT approximately back to baseline. Following this, external Na+ and Ca2+ were removed (cyan shading), which slightly decreased the Peredox LT. The addition of 0.5 mM Ca2+ in 0 Na+ to stimulate reverse NCX transport increased the RCaMP LT and further decreased the Peredox LT (magenta shading). Subsequently, the removal of external Ca2+ and re-addition of external Na+ to stimulate forward NCX transport (orange shading) increased the Peredox LT and brought the RCaMP LT back to baseline. (B) Box plots of the Peredox (top) and RCaMP (bottom) fluorescence LTs in the different external solution conditions performed in (A), across many DGCs (n=29). The external [Na+] and [Ca2+] condition after applying AOA and pyruvate is listed across the bottom in chronological order from left to right. The coloring of the data corresponds to the shading in A. The mean Peredox and RCaMP LTs in each external condition were (in ns): 1.50±0.04 in 0 Na+ and 0 Ca2+, 1.45±0.05 in 0 Na+ and 0.5 mM Ca2+, and 1.61±0.09 in 147 Na+ and 0 Ca2+. The mean RCaMP LTs were (in ns): 0.90±0.06 in 0 Na+ and 0 Ca2+, 1.49±0.20 in 0 Na+ and 0.5 mM Ca2+, and 0.91±0.07 in 147 Na+ and 0 Ca2+. (C) Relative change to the Peredox fluorescence LT induced by the addition of 0.5 mM Ca2+ in the absence of external Na+ to stimulate reverse NCX transport (Ca) or 147 mM Na+ in the absence of external Ca2+ to stimulate forward NCX transport (Na). The data are from the same DGCs as in (B). The mean ∆Peredox values were: –0.05±0.04 ns for ‘Ca’ and 0.11±0.07 ns for ‘Na’. Statistical significance from a paired Wilcoxon test is indicated.
Representative fluorescence LT traces from a DGC show that Peredox LT was substantially increased by the application of AOA to block the shuttle’s aspartate aminotransferase enzyme (Figure 2A), as previously described, Díaz-García et al., 2017, which is consistent with an inhibition of NADHCYT recycling and consequent elevation of the cytoplasmic NADH:NAD+. This strong increase to Peredox LT, which approaches the upper end of its dynamic range, precludes the ability to measure further NADHCYT increases, so we applied exogenous pyruvate (1.5 mM) to promote the oxidation of NADH to NAD+ via lactate dehydrogenase and restore the Peredox LT approximately back to its baseline LT in ACSF. We then removed external Na+ and Ca2+, which decreased the Peredox LT from the AOA and pyruvate condition (Figure 2A); and then increased Ca2+CYT by applying 0.5 mM external Ca2+ in zero external Na+ to drive reverse NCX transport. We confirmed the Ca2+CYT increase from the increase to RCaMP LT, but the Ca2+CYT elevation still did not coincide with an increase to NADHCYT. Rather, NADHCYT decreased relative to the 0 Na+ 0 Ca2+ baseline (Figure 2B and C). This demonstrates that an increase to Ca2+CYT still fails to observably stimulate NADHCYT production even when NADHCYT recycling by the MAS is attenuated. Following the Ca2+CYT increase, we removed 0.5 mM external Ca2+ and re-applied external Na+ to activate forward NCX transport (Figure 2A), which increased the Peredox LT (Figure 2B and C). This shows again that NADHCYT production is coupled to the flux of Na+.
Overall, these experiments argue against a concealment of Ca2+CYT-induced NADHCYT production by a Ca2+CYT activation of NADHCYT recycling via the MAS and indicates, once again, that an increase to Na+/K+ pump activity is the major driver of glycolysis.
Activation of glycolysis by Ca2+ is mediated by coupling between the Na+/Ca2+-exchanger and the Na+/K+ pump
The increase to NADHCYT production by an influx of Na+ (Figure 1 and Figure 2) suggests that glycolysis activation in DGCs is strongly tied to an increase in the activity of the Na+/K+ pump. Also, the NADHCYT increase induced by external Na+ application was stronger if Ca2+CYT was elevated compared to when Ca2+CYT was not elevated (Figure 1E), meaning that forward NCX transport, which trades Ca2+CYT for Na+, can potentiate glycolysis activation. By considering these observations together with the apparent insensitivity of glycolysis to increases in Ca2+ pump activity, it seemed logical that a transient Ca2+CYT elevation would activate glycolysis predominantly by stimulating Na+ influx through forward NCX transport, which would then increase the activity of the Na+/K+ pump.
We sought for a way to directly test how NADHCYT is affected by inhibition of the Na+/K+ pump or inhibition of forward NCX transport (Figure 3). Na+/K+ pumps are specifically inhibited by ouabain, although the rodent Na+/K+ pump α1-isozymes have relatively low sensitivity and require millimolar inhibitor concentrations for complete inhibition under physiological conditions (Blanco and Mercer, 1998; Marks and Seeds, 1978). Application of 2 mM ouabain to DGCs bathed in ACSF quickly led to Ca2+ overload and cell rupture (Figure 3—figure supplement 1), which made it impossible to measure how electrical stimulation-induced NADHCYT transients are affected by complete Na+/K+ pump inhibition. Clearly, the activity of the Na+/K+ pump must be substantial, even at rest, but achieving complete Na+/K+ pump inhibition under physiological neuronal conditions is challenging. DGCs bathed in ACSF were more tolerant to application of low-dose (5 μM) ouabain, which inhibits only the rodent Na+/K+ pump α2- and α3-isozymes that have higher inhibitor sensitivities (Blanco and Mercer, 1998), but the stimulation-induced NADHCYT transient was not eliminated (Figure 3—figure supplement 2). This result argues that in these cells, the α1 subunit is adequate to support the activation of glycolysis.
Figure 3 with 2 supplements see all
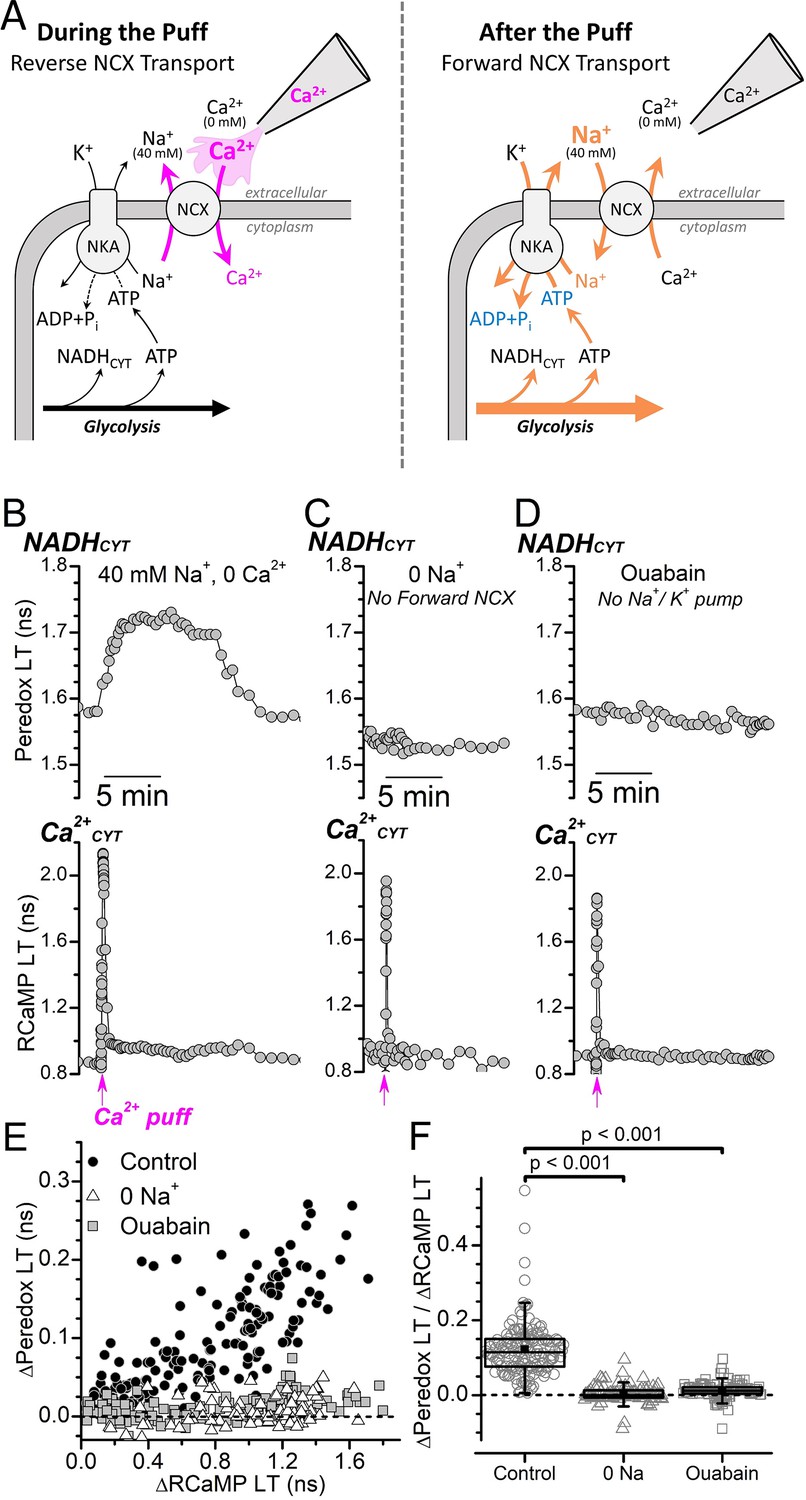
Ca2+CYT transients induced by external Ca2+ puffs only increase NADHCYT production when both forward NCX transport and Na+/K+ pump activity are intact.
(A) Cartoon depicting the expected effect of a local external Ca2+ puff on ion transport by the NCX and subsequent activation of the Na+/K+ pump. (Left) A pipette containing Ca2+ is placed near the soma of a DGC within a slice that is bathed in solution containing 40 mM Na+ and 0 Ca2+ solution. A brief pulse of positive pressure is applied to the pipette to transiently increase the local external [Ca2+] and stimulate reverse NCX transport (magenta arrows), leading to Ca2+ import and an increase to Ca2+CYT. (Right) In the aftermath of the puff, the local external [Ca2+] decreases as the small volume of puffed Ca2+ mixes with large volume of the 40 mM Na+ 0 Ca2+ bath solution, which leads to forward NCX transport that stimulates Na+ extrusion and an increase to Na+/K+ pump activity (orange arrows). Abbreviations: Na+/Ca2+-exchanger (NCX), Na+/K+-ATPase (NKA). (B–D) Representative fluorescence lifetime (LT) traces of Peredox (top traces) and RCaMP (bottom traces) from a DGC bathed in either 40 mM Na+ and 0 Ca2+ solution (B, Control), 0 Na+ and 0 Ca2+ to block forward NCX transport (C, 0 Na+), or in 40 mM Na+ and 0 Ca2+ with 5 mM ouabain to block the Na+/K+ pump (D, Ouabain). Puffs of Ca2+ were delivered at the timepoint indicated along the bottom of the RCaMP LT traces (magenta arrows). (E) Scatterplot of the Ca2+-puff-induced transient changes to the Peredox and RCaMP LTs of DGCs in 40 mM Na+ and 0 Ca2+ (Control, black circles, n=172), in 0 Na+ and 0 Ca2+ (0 Na+, white triangles, n=102), or in 40 mM Na+ and 0 Ca2+ with 5 mM ouabain (Ouabain, gray squares, n=114). The dashed line indicates where ∆Peredox=0. Analysis was restricted to recordings with transient RCaMP LT half-decay times <35 s. (F) Ratio of ∆Peredox to ∆RCaMP for the same Ca2+-puff-induced LT transients as in panel E, except excluding those recordings where the ∆RCaMP LT was <0.2 ns to avoid noisy ratio calculations due to small ∆RCaMP values. The dashed line indicates where ∆Peredox/∆RCaMP=0. The mean ∆Peredox/∆RCaMP in each condition was: 0.12±0.07 in 40 mM Na+ and 0 Ca2+ (Control, n=147), 0.01±0.02 in 0 Na+ and 0 Ca2+ (0 Na+, n=96), and 0.00±0.02 in 40 mM Na+ and 0 Ca2+ with 5 mM ouabain (Ouabain, n=99). Statistical significance from Mann-Whitney tests is indicated.
We successfully established a condition amenable to millimolar ouabain application by reducing the external [Na+] to 40 mM and removing external Ca2+ to prevent unintended reverse NCX transport. Stimulation-induced Ca2+CYT transients using an electrode were not possible for DGCs bathed in this condition, but we evoked similar Ca2+CYT transients by applying puffs of extracellular Ca2+ to transiently produce reverse-NCX transport (Figure 3A). A brief puff of Ca2+ to a DGC within a slice bathed in 40 mM Na+ and 0 Ca2+ (Figure 3B) induced a fast transient increase to the RCaMP LT, reflecting a fast increase to Ca2+CYT, followed by a transient increase to the Peredox LT, reflecting an increase to NADHCYT. This shows that a transient Ca2+CYT elevation can, indeed, activate glycolysis under these ionic conditions. But for a DGC within a slice that was instead bathed in zero external Na+ to prevent the NCX-mediated influx of Na+ in exchange for the elevated Ca2+CYT, a Ca2+ puff still evoked a fast Ca2+CYT transient but did not increase NADHCYT (Figure 3C). Similarly, the NADHCYT increase that would normally follow the Ca2+ puff was prevented for a DGC within a slice bathed with 5 mM ouabain to block Na+/K+ pumps (Figure 3D).
A scatter plot of the transient Peredox LT and RCaMP LT changes that were evoked by Ca2+ puffs (Figure 3E) illustrates the positive correlation between NADHCYT production and Ca2+CYT increases for DGCs bathed in 40 mM Na+ and 0 Ca2+, where both forward NCX and Na+/K+ pump activities are intact. But NADHCYT production was deeply attenuated when DGCs were bathed without external Na+ or with 5 mM ouabain, even with large elevations of Ca2+CYT (∆RCaMP>1.2 ns). The mean ∆Peredox/∆RCaMP ratio (Figure 3F) for Ca2+-puff-evoked transients in 40 mM external Na+ (0.122±0.005, n=147) was reduced to nearly 0 after blocking forward NCX transport by removing external Na+ (0.003±0.002, n=96) or after blocking the Na+/K+ pump with ouabain (0.013±0.002, n=99).
These data demonstrate that Ca2+CYT can activate glycolysis in the DGC soma if it is traded for Na+ through forward NCX transport, which increases the activity of the Na+/K+ pump. The lack of Ca2+-puff-induced NADHCYT responses when either forward NCX transport or the Na+/K+ pump was blocked further substantiates that any increase in the activity of Ca2+ pumps has a negligible effect on glycolysis activation.
Antidromic stimulation-induced Na+ transients depend on Ca2+
The stimulation of NADHCYT production by Ca2+CYT through coupling between forward NCX transport and the Na+/K+ pump (Figure 3) indicates that Na+ influx during forward NCX transport can activate glycolysis. But how does the amount of Na+ imported by the NCX to extrude Ca2+CYT compare to the total Na+ influx induced by neuronal excitation? We investigated this by measuring intracellular [Na+] changes during electrical stimulation with and without Ca2+ entry (Figure 4A).
Figure 4 with 1 supplement see all

Antidromic stimulation-induced [Na+] transients depend strongly on Ca2+ entry.
(A) Cartoon depicting antidromic stimulation-induced Na+ and Ca2+ fluxes. Left, Control: Stimulation in ACSF triggers Na+ and Ca2+ influx through their respective voltage-gated Na+ and Ca2+ channels. Following the Ca2+CYT elevation, the NCX will import Na+ to drive Ca2+ extrusion. Both the Na+ influx through NaV and through the NCX contribute to the total intracellular [Na+] increase (blue arrows). Right, After CaV Block: Inhibiting Ca2+ influx through CaV (indicated by the red X) prevents Ca2+CYT elevation, which strongly reduces Na+ influx through the NCX. Abbreviations: voltage-gated Na+ channel (NaV), voltage-gated Ca2+ channel (CaV), Na+/Ca2+-exchanger (NCX). (B) Representative fluorescence traces showing antidromic stimulation-induced transient changes to SBFI ∆F/F (top) and RCaMP lifetime (LT) (bottom) from a DGC bathed in ACSF with synaptic blockers. The stimulation was delivered at the timepoint indicated by the arrow at the bottom of the RCaMP trace. The SBFI ∆F/F axis is inverted to illustrate an increase to [Na+] as an upward deflection. (C) Antidromic stimulation-induced transient changes to SBFI ∆F/F and RCaMP LT in the presence of 3 μM isradipine and 20 μM CdCl2 for the same DGC as in (B). The stimulation was delivered at the timepoint indicated by the arrow below the RCaMP trace. (D) Average, interpolated, antidromic stimulation-induced SBFI ∆F/F and RCaMP LT transients (n=13) before (ACSF, black trace and shading) and after CaV block (Israd+Cd2+, magenta trace and shading). The means are indicated by the solid line, SEM is indicated by the darker shading, and SD is indicated by the lighter shading. (E) Paired box plots showing the antidromic stimulation-induced peak transient SBFI ∆F/F amplitudes before (ACSF, black box) and after CaV block (Israd+Cd2+, magenta box) for the same DGCs as in (D). The mean peak SBFI ∆F/F values were: –0.29±0.07 in ACSF and –0.15±0.04 with Israd+Cd2+. Statistical significance from a paired sample t-test is indicated. (F) Paired box plots showing the ∆RCaMP LT before (ACSF, black box) after CaV block (Israd+Cd2+, magenta box) for the same DGCs as in (D) and (E). The mean ∆RCaMP LTs were: 0.30±0.13 ns in ACSF and 0.04±0.03 ns with Israd+Cd2+. Statistical significance from a paired sample t-test is indicated. (G) Scatterplot of antidromic stimulation-induced transient changes to SBFI ∆F/F against ∆RCaMP LT before (ACSF, gray circles) and after CaV block (Israd +Cd2+, magenta squares). Data are from the same DGCs as in (D–F).
We recorded stimulation-induced changes in intracellular [Na+] and Ca2+CYT simultaneously by loading SBFI (Harootunian et al., 1989), a Na+-sensitive fluorescence dye, into RCaMP-expressing DGCs via single-cell electroporation. Representative traces of the SBFI relative fluorescence intensity (∆F/F) and RCaMP LT from a DGC bathed in ACSF show that antidromic stimulation evoked a transient decrease to SBFI ∆F/F (note the inverted y-axis), reflecting an increase to [Na+], and a transient increase to RCaMP LT, reflecting an increase to Ca2+CYT (Figure 4B). SBFI ∆F/F transients were longer in duration than the RCaMP LT transient, regardless of whether they were evoked by antidromic stimulation or synaptic stimulation in the absence of synaptic blockers (Figure 4 and Figure 4—figure supplement 1), indicating that stimulation-induced intracellular [Na+] elevation outlasts Ca2+CYT elevation. A scatterplot of the transient changes to SBFI ∆F/F and RCaMP LT shows that intracellular [Na+] and Ca2+CYT changes are positively correlated when evoked by either stimulation paradigm (Figure 4—figure supplement 1D); while this is consistent with a positive dependence of intracellular [Na+] on Ca2+CYT, a positive correlation between [Na+] and Ca2+CYT increases would also be observed if neuronal stimulation evokes proportional yet independent changes to both ions.
To directly test if somatic intracellular [Na+] changes depend on Ca2+ entry, we measured how antidromic stimulation-induced [Na+] transients were affected by blocking Ca2+ channels (Figure 4A). Blocking voltage-gated Ca2+ channels with isradipine and Cd2+ (Berjukow et al., 2000; Lansman et al., 1986) attenuated the antidromic stimulation-induced RCaMP LT transient (Figure 4C and D), indicating a strong reduction to Ca2+ entry and Ca2+CYT elevation. The SBFI ∆F/F transient became smaller after blocking Ca2+ entry (Figure 4C and D), demonstrating that stimulation-induced intracellular [Na+] changes depend on Ca2+CYT elevation; the peak of the SBFI Na+ transient (as ∆F/F) was reduced to 54.6±19.3% of the original response and the Ca2+ transient (as ∆RCaMP LT) was reduced to 14.5±12.5% (n=13) (Figure 4E and F).
A scatterplot of the antidromic stimulation-induced transient changes to SBFI ∆F/F and ∆RCaMP LT in ACSF or in the presence of isradipine and Cd2+ (Figure 4G) shows that blocking Ca2+ entry shifts the peak SBFI ∆F/F down and to the left, which indicates that a major component of somatic [Na+] changes is dependent on Ca2+CYT. This means that Na+ imported by forward NCX transport in response to Ca2+CYT extrusion comprises a substantial proportion of the Na+ that enters the soma following stimulation, which provides clarity to the apparent dependence of antidromic stimulation-induced glycolysis activation on Ca2+CYT elevation (Díaz-García et al., 2021a): an elevation to Ca2+CYT potentiates Na+ influx, which ultimately leads to a stronger activation of the Na+/K+ pump than when Ca2+ entry is blocked.
Discussion
Glycolysis in neurons is transiently increased following stimulation (Díaz-García et al., 2017; Fox et al., 1988; Fox and Raichle, 1986). To understand the mechanism that drives this increase to glycolysis following neuronal electrical activity, we tested the responsiveness of NADHCYT to changes in intracellular Na+ and Ca2+CYT, which affect energy consumption by ion-ATPases. We have shown in hippocampal DGC somas that NADHCYT is strongly influenced by changes in intracellular Na+ and that activation of NADHCYT increases by Ca2+CYT is nearly completely dependent on ion transport coupling between the NCX and the Na+/K+ pump. This means that nearly all of the transient glycolysis increase following a stimulation is a response to an activation of the Na+/K+ pump to extrude the Na+ that enters either through Na+ channels or through the NCX to power active Ca2+ transport.
Neuronal glycolysis is strongly influenced by the Na+/K+ pump, but not by Ca2+ pumps
Coupling between glycolysis and Na+/K+ pump activity has been reported across many tissues and cell types (Andersen and Marmarou, 1992; Balaban and Bader, 1984; Hasin and Barry, 1984; Hellstrand et al., 1984; James et al., 1996; James et al., 1999; Knull, 1978; Lipton and Robacker, 1983; Lynch and Balaban, 1987; Mercer and Dunham, 1981; Minakami et al., 1964; Paul et al., 1979; Paul, 1983; Proverbio and Hoffman, 1977; Weiss and Hiltbrand, 1985; Whittam et al., 1964; Whittam and Ager, 1965). Our lab previously reported a link between the activity of the Na+/K+ pump α3-isozyme and glycolysis activation in DGCs of acute mouse hippocampal slices: the potentiation to antidromic stimulation-induced NADHCYT production after increasing channel-mediated influx of Na+ with α-pompilidotoxin in the absence of external Ca2+ (i.e., when both Na+ influx through the NCX and Ca2+ influx through channels are blocked) could be reversed by strophanthidin at low micromolar concentrations (Díaz-García et al., 2021a). We have shown here in DGCs that NADHCYT production decreases with depletion of Na+ and increases with repletion of Na+ (Figure 1), which strongly suggests that the rate of neuronal glycolysis mirrors changes in intracellular [Na+] and in the activity of the Na+/K+ pump. Moreover, DGCs could not withstand complete Na+/K+ pump inhibition when bathed in ACSF (Figure 3—figure supplement 1), meaning that Na+/K+ pump activity is essential to counteract Na+ leak and/or influx through Na+-coupled transporters even when neurons are at rest.
The small NADH:NAD+ increase produced by removing only external Ca2+ that we observed during our previous studies (Díaz-García et al., 2021a) is also consistent with a predominant influence of Na+ on glycolysis: since Na+ conductance through the NALCN (Na+-leak channel, non-selective), which regulates DGC resting membrane potential and excitability (S.-Y. Lee et al., 2019), is higher in the absence of external Ca2+ (Kschonsak et al., 2020; Lu et al., 2010), removing external Ca2+ likely elevates Na+ influx through the NALCN and increases Na+/K+ pump activity.
The sensitivity of glycolysis to Ca2+ pump activity is less clear. The small increases to the antidromic stimulation-induced ∆Peredox/∆RCaMP ratio after applying inhibitors that target SERCA (thapsigargin; Figure 1—figure supplement 2) or PMCA (E6-berbamine or calmidazolium; Brini et al., 2013; Díaz-García et al., 2021a) do suggest some involvement of Ca2+ pumps in regulating stimulation-induced Ca2+CYT increases, but our observations that NADHCYT was not increased by the elevation of Ca2+CYT alone, either by reverse NCX transport during ion substitution or Ca2+ puffs in either zero external Na+ or 5 mM ouabain, indicate that ATP consumption associated with the direct pumping of Ca2+ does not have a measurable effect on glycolysis (Figures 1—3, and Figure 1—figure supplement 1). This may indicate that Ca2+ pumps are either not closely coupled to glycolytic enzymes in DGCs or that Ca2+ pump density is small with respect to Na+/K+ pumps. It appears that the density of Na+/K+ pumps outweighs the density of Ca2+ pumps in hippocampus since hippocampal homogenates have ouabain-sensitive ATPase activity ~2- to 3-fold larger than Ca2+-sensitive ATPase activity (Gutierres et al., 2014; Spohr et al., 2022; Teixeira et al., 2020). Even if we assume that Ca2+ pumps and Na+/K+ pumps are coupled to glycolysis equally, the lower density of Ca2+ pumps would make their glycolytic (and total cellular) ATP consumption less than the ATP consumed by Na+/K+ pumps.
Ca2+ pumps have an apparent K0.5 for Ca2+ activation of ~0.1 μM and relatively slow maximal turnover rates of ~100 s–1 (characteristic of ion pumps; Glitsch, 2001; Meyer et al., 2017), meaning that they are well suited for tuning the resting Ca2+CYT but not for regulating large dynamic Ca2+CYT increases (Caroni and Carafoli, 1981). It is likely that Ca2+ pump transport capacity would saturate quickly in response to the 0.3–1 μM increases in Ca2+ during some stimulations. If an increase to Ca2+ pump activity was the predominant driver of NADHCYT production, the saturation of their Ca2+ transport during larger Ca2+CYT increases would likely cause a ‘rounding off’ of the Peredox LT change for stimulations where RCaMP LT changes are >0.5 ns; but the near linearity of the ∆Peredox-∆RCaMP relationship for RCaMP LT changes of 0.05–1 ns (Díaz-García et al., 2017) argues against such a scenario.
The Ca2+CYT-dependence of aerobic glycolysis activation arises from ion transport coupling between the Na+/Ca2+-exchanger and the Na+/K+ pump
Our observations here that glycolysis activation in DGCs is strongly coupled to the activity of the Na+/K+ pump but not Ca2+ pumps was seemingly at odds with the ~70% reduction to antidromic stimulation-induced NADHCYT production by blocking Ca2+CYT elevation (Díaz-García et al., 2021a). However, the reduction to NADHCYT production is fully explained by the involvement of forward NCX transport in DGC Ca2+CYT regulation (Figure 1F; Lee et al., 2012) and by the close coupling between the ion transport of the NCX and the Na+/K+ pump.
The NCX has an apparent K0.5 for internal Ca2+ of ~1 μM (Blaustein and Santiago, 1977; Collins et al., 1992; Miura and Kimura, 1989) and a transport rate 10–30 times faster than the slow turnover rates of ATPases (Blaustein and Lederer, 1999; Caroni et al., 1980). This means that NCX transport activity would be near the lower end of its total capacity when neurons are at rest (when Ca2+CYT is ~100 nM) but could rapidly respond to stimulation-induced Ca2+CYT increases and exchange it for Na+. Unfortunately, most NCX inhibitors are unspecific or do not block the forward transport mode (Iwamoto et al., 2004; Iwamoto and Kita, 2006; Secondo et al., 2015; Sharikabad et al., 1997), so the most effective way to test the involvement of forward NCX transport in glycolysis activation is to replace external Na+ with an inert ion that cannot be transported by the NCX (i.e., choline).
The transient NADHCYT production evoked by Ca2+ puffs in 40 mM Na+ and 0 Ca2+ was eliminated when external Na+ was completely removed (i.e., replaced with choline) or when ouabain was added (Figure 3), meaning that both forward NCX transport and Na+/K+ pump activity are required for a Ca2+CYT elevation to measurably activate glycolysis. This unequivocally demonstrates that the exchange of Ca2+CYT for Na+ by the NCX is closely coupled to Na+ extrusion by the Na+/K+ pump, and that this coupling is tightly associated with the activation of glycolysis. Coupling between the NCX and Na+/K+ pump within discrete microdomains has also been reported in cardiac tissue (Mohler et al., 2005; Zhang et al., 2009; Zhang et al., 2011).
Strong coupling between the NCX, the Na+/K+ pump, and glycolysis activation can also explain two of our other observations: First, it can explain why the stimulation-induced ∆Peredox/∆RCaMP ratio is increased by inhibition of SERCA or PMCA. Blocking either of these Ca2+ pumps likely increases the Ca2+ that is exported by the NCX, which would amplify the associated Na+ influx through the NCX and accelerate the ATP consumption by the Na+/K+ pump to extrude Na+. Second, it can explain why NADHCYT production was decreased when Ca2+CYT was elevated by reverse NCX transport (Figure 1 and Figure 1—figure supplement 1). For reverse NCX transport to import 1 Ca2+ from the extracellular environment, the NCX must export 3 Na+ (Figure 1A). This means that an increase to Ca2+CYT would occur at the same time as a decrease to intracellular [Na+], which would decrease the activity of the Na+/K+ pump and, consequently, decrease the rate of glycolysis and its associated NADHCYT production. The decrease to cytosolic NADH:NAD+ from reverse NCX transport further supports the strong association between the energy consumed to extrude Na+ and glycolysis and implies that even decreasing the Na+/K+ pump activity from an already reduced steady-state level of activity (due to the depletion of intracellular [Na+] from prior application of the 0 Na 0 Ca solution) is more influential on the rate of glycolysis than any putative strong activation of Ca2+ pumps by a Ca2+CYT increase.
The ∆Peredox/∆RCaMP ratio evoked by Ca2+ puffs in 40 mM Na+ and 0 Ca2+ (~0.1, Figure 3E) is ~40–50% of the ∆Peredox/∆RCaMP ratio evoked by electrical stimulation in ACSF (~0.2–0.25) (Díaz-García et al., 2017; Díaz-García et al., 2021a), meaning that the peak activation of glycolysis relative to the Ca2+CYT change is smaller when evoked by Ca2+ puffs compared to electrical stimulation. This could be due to the absence of channel-mediated Na+ entry during Ca2+ puffs compared to electrical stimulation or to the reduced external [Na+] in Ca2+ puff experiments (40 mM) compared to the external [Na+] during electrical stimulation in ACSF (147 mM), which would slow down the kinetics of Ca2+CYT-dependent Na+ influx through forward NCX transport, consequently leading to a less potent activation of the Na+/K+ pump; both the quantity and the rate of Na+ influx are likely to be important determinants of the NADHCYT transient amplitude and time course.
Glycolysis does not appear to be influenced by intracellular [Na+] per se, but rather by the downstream effect of [Na+] on Na+/K+ pump activity. If glycolytic enzymes could be directly stimulated by an increase to [Na+], then complete inhibition of the Na+/K+ pump should have facilitated stronger NADHCYT responses. But this was not the case, as NADHCYT responses evoked by Ca2+ puffs were strongly attenuated in the presence of 5 mM ouabain (Figure 3), a condition that allows for Na+ influx through the NCX but prevents Na+ extrusion through the Na+/K+ pump. In contrast to the strong attenuation by 5 mM ouabain, the increase to the NADHCYT transients evoked by synaptic electrical stimulation after applying 5 μM ouabain in ACSF (Figure 3—figure supplement 2) is not as straightforward and cannot be attributed solely to an inhibition of the Na+/K+ pump. First, 5 μM ouabain does not substantially inhibit rodent α1-pumps (Marks and Seeds, 1978; Sweadner, 1979); we know that 5 μM ouabain only causes partial Na+/K+ pump inhibition in our acute slice preparation from the mouse because it does not cause Ca2+CYT overload (unlike 5 mM ouabain in ACSF), which means that the plasma membrane Na+ gradient in the presence of 5 μM ouabain remains sufficient to prevent a reversal of NCX transport. Second, the stimulation-induced Ca2+CYT transients were increased, which is, perhaps, reflective of an effect on synaptic release.
We think that the absence of strong NADHCYT responses to Ca2+CYT increases when Na+ is unavailable for exchange via NCX (Figure 1 and Figure 3) is quite informative on how both ions influence glycolysis activation, even though it is true that the rapid ionic changes that occur through channels during neuronal activity following electrical stimulation might obey somewhat different rules than the slower ionic changes we are able to make by driving ion fluxes through a secondary ion transporter. Since we have determined that NADHCYT production is strongly increased by an influx of Na+ but not increased by large increases in Ca2+CYT (Figures 1—3) it is logical to infer that a transient increase to NADHCYT production following electrical stimulation is primarily coupled to the energy consumed to directly pump Na+ rather than to directly pump Ca2+. The amplitudes of RCaMP LT increases that we achieve with both the ion substitution experiments (Figure 1) and the Ca2+ puff experiments (Figure 3) are also similar to the typical amplitude of RCaMP LT increase when evoked by electrical stimulation (Figure 4 and Díaz-García et al., 2017; Díaz-García et al., 2021a), meaning that Ca2+CYT is increased to similar levels by all experimental paradigms regardless of their site of entry (i.e., channels or NCX).
Na+ entry via the Na+/Ca2+-exchanger contributes substantially to total Na+ entry in the soma
It seemed that stimulation-induced Na+ influx via the NCX could be substantial given that a transient Ca2+CYT increase could only activate glycolysis if both forward NCX transport and the Na+/K+ pump were active (Figure 3) and that blocking Ca2+CYT strongly reduces the NADHCYT response (Díaz-García et al., 2021a). The ~50% reduction to the stimulation-induced SBFI ∆F/F peak amplitudes after blocking Ca2+ entry confirmed that Na+ influx via forward NCX transport contributes greatly to the total Na+ influx in the soma (Figure 4). We think it is unlikely that the reduction to the [Na+] transient after blocking Ca2+ entry resulted from less Na+ entering through voltage-gated Ca2+ channels, since Na+ permeation through these channels is blocked by external Ca2+ with a Kd of ~1 µM (Almers and McCleskey, 1984; Tsien et al., 1987).
It is interesting to note that the positive relationship between the amplitudes of stimulation-induced transient increases to NADHCYT and Ca2+CYT appears to intercept the origin (cf. Figure 2; Díaz-García et al., 2017) while the relationship between the amplitudes of transient intracellular [Na+] and Ca2+CYT increases does not (Figure 4—figure supplement 1). Perhaps weaker stimulations that evoke small increases to intracellular [Na+] without a measurable increase to Ca2+CYT have only very weak effects on glycolysis: it could be that smaller [Na+] increases have proportionally more clearance by diffusion through the cytoplasm (Hodgkin and Keynes, 1956; Pusch and Neher, 1988) or that the energy needed to regulate smaller [Na+] increases (without sustained Na+ influx through forward NCX transport) is mostly derived from ATP buffering systems known to compartmentalize near sites of high ATP turnover (Kültz and Somero, 1995; Lange et al., 2002; Ovádi and Saks, 2004; Wallimann et al., 1992), which could subsequently replenish their energy storage pools without large steady-state changes to glycolytic flux. Directly evaluating the role of these ATP buffering enzymes, that is, creatine kinase and adenylate kinase, in shaping the stimulation-induced transient glycolysis activation in neurons will be a key area for future exploration.
Estimation of neuronal stimulation-induced changes to intracellular [Na+] and Na+/K+ pump activity
SBFI calibration curves in rodent hippocampal neurons (Baeza-Lehnert et al., 2019; Diarra et al., 2001; Gerkau et al., 2019; Meier et al., 2006; Rose et al., 1999) reported apparent Na+ Kd values of 18–42 mM Na+. However, most were performed with ouabain concentrations of only 50–100 μM, well below the millimolar concentrations needed to fully inhibit the rodent Na+/K+ pump α1 isozyme under physiological conditions; the relatively high ouabain-resistance of rodent α1 pumps would also be strongly increased in calibration solutions that substitute external Na+ with high concentrations of K+ since external K+ competes with binding of the inhibitor to the pump’s externally accessible E2 conformation (Hansen and Skou, 1973; Kanai et al., 2021).
Despite the limitations of these in situ SBFI calibrations in rodent neurons, we use Rose et al.’s calibration of SBFI ∆F/F excited at 790 nm in brain slices with a Na+ Kd of 26 mM (Rose et al., 1999) to estimate the stimulation-induced [Na+] changes in our experiments (which reflect the net movement of Na+ through influx and extrusion pathways). Our estimations assume a baseline intracellular [Na+] of 13 mM, which is the reported value for hippocampal slice CA1 neurons (Mondragão et al., 2016) and nearly identical to the K0.5 for activation of internal Na+-dependent, ATP-induced current produced by α1β1 Na+/K+ pumps (which likely set baseline neuronal internal [Na+]; Azarias et al., 2013; Blanco, 2005; Blanco and Mercer, 1998) in excised patches when internal Na+ is substituted with K+ (Meyer et al., 2017; Meyer et al., 2019; Meyer et al., 2020).
The mean peak amplitude of antidromic stimulation-induced SBFI ∆F/F transients evoked in ACSF was –25.6±4.6% (Figure 4), reflecting an average transient [Na+] increase of ~25 mM from baseline. How would this intracellular [Na+] change affect neuronal Na+/K+ pump activity? Neurons express the α1 and α3 isoforms of the Na+/K+ pump (Blanco and Mercer, 1998). Under physiological conditions, both α1 and α3 pumps are rate limited by the binding of internal Na+ since their K0.5,Na+ is similar to, or above, physiological intracellular [Na+] while their K0.5,K+ is below physiological extracellular [K+]. Activation of Na+/K+ pumps by internal Na+ is also highly cooperative (Hill coefficient of ~3, corresponding to three transported Na+ Crambert et al., 2000; Hasler et al., 1998; Meyer et al., 2017; Meyer et al., 2019; Meyer et al., 2020). Based on the reported curves of Na+/K+ pump activation by internal Na+ (Crambert et al., 2000; Meyer et al., 2019), a 25 mM increase to neuronal [Na+] from a baseline of 13 mM would likely increase peak α1β1 activity ~2-fold (to ~95% of total activity) and α3 activity ~9-fold (to ~60% of total activity).
Blocking Ca2+ entry reduced the antidromic stimulation-induced peak SBFI ∆F/F to –11.5±5.5% (Figure 4), a ~55% decrease from the transient in ACSF that corresponds nonlinearly to a reduction of the average [Na+] increase from ~25 to ~8 mM. The estimated 8 mM stimulation-induced increase to internal [Na+] when Ca2+ entry is inhibited (i.e., when Na+ influx via forward NCX transport is reduced) would increase α1β1 activity ~1.5-fold (to ~80% of total activity) and α3 activity ~3-fold (to ~25% of total activity).
It must be noted that the proportional influence of NCX-mediated Na+ entry on glycolysis activation could be dependent on the stimulation paradigm. Antidromic stimulation in the presence of synaptic blockers does not involve entry of Na+ through NMDA or AMPA receptors. This means that most channel-mediated Na+ influx that occurs from this stimulation method is through voltage-gated channels, which, in DGCs, are highly localized to the axon initial segment with weak, if any, staining in the soma (Kole et al., 2008; Kress et al., 2008; Kress et al., 2010). Thus, it is not unexpected that Na+ influx via forward NCX transport has such a prominent effect on the antidromic stimulation-induced glycolysis activation in the soma. Soma SBFI transients evoked by either antidromic or synaptic stimulation could reach similar peak amplitudes (Figure 4—figure supplement 1), but the contribution of Na+ influx through forward NCX transport to synaptically evoked [Na+] changes could not be tested because blocking voltage-gated Ca2+ entry would prevent synaptic vesicle release.
NCX involvement in glycolysis activation could also vary in different neuronal compartments. Reported SBFI transient measurements indicate that antidromic stimulation-induced [Na+] changes in the soma are typically smaller than transients in the dendrites (Fleidervish et al., 2010; Kole et al., 2008). Measuring the ion and metabolite dynamics in dendrites will be an important step for understanding whether the mechanism of glycolysis activation in dendrites is similar to or different than the mechanism in somas.
On the mechanism of neuronal stimulation-induced aerobic glycolysis activation in somas
Based on our observations here and previously (Díaz-García et al., 2017; Díaz-García et al., 2021a) regarding neuronal stimulation-induced [Na+], Ca2+CYT, and NADHCYT dynamics in the soma, we can begin to compile a more comprehensive understanding of the mechanism underlying transient aerobic glycolysis activation in response to neuronal excitation.
Neuronal stimulation immediately increases both intracellular [Na+] and Ca2+CYT. The transient Ca2+CYT elevation lasts at most a few seconds (slightly longer than the duration of the stimulus; RCaMP1h has a t1/2 decay of 410 ms; Akerboom et al., 2013) before returning to baseline due to buffering by cytosolic proteins and removal from the cytosol. This means that any transient activation of Ca2+ pumps by Ca2+CYT would likely only be a few seconds in duration before returning to steady state. In any case, we know that this produces no measurable activation of glycolysis since similar Ca2+CYT transients evoked by Ca2+ puffs in the absence of external Na+ or presence of ouabain were not associated with NADHCYT increases. It is also clear that Ca2+CYT is rapidly exported across the plasma membrane by forward NCX transport, which loads the neuron with Na+. Na+ is not buffered in the cytoplasm and is actively extruded back across the plasma membrane exclusively by the Na+/K+ pump. The transient intracellular [Na+] increase lasts for a minute or more (longer than the Ca2+CYT transient), during which an increase to the activity of Na+/K+ pumps would be sustained. Both the sensitivity of NADHCYT production to Na+ and the ablation of Ca2+CYT-evoked NADHCYT transients by inhibition of either forward NCX transport or the Na+/K+ pump demonstrate that pumping Na+ is the predominant activity that drives the transient activation of aerobic glycolysis.
Stimulation also triggers a rapid increase to Ca2+MITO via MCU that can last several minutes (Díaz-García et al., 2021a) and requires Na+ in order to return to the baseline (Figure 1—figure supplement 3). The requirement of Na+ for Ca2+MITO extrusion likely indicates a Na+-coupled Ca2+ efflux mechanism, as reported in other cell types (Palty and Sekler, 2012; Parpura et al., 2016; Saotome et al., 2005). Interestingly, the attenuation of Ca2+MITO elevation from MCU knockdown reduces the stimulation-induced cytosolic ∆Peredox/∆RCaMP by ~50% (Díaz-García et al., 2021a), which is consistent with Ca2+MITO extrusion stimulating glycolysis. The extent to which the energetic burden of Ca2+MITO extrusion is placed onto glycolysis, and, more specifically, the Na+/K+ pump, or elsewhere will be an important area for future exploration.
Given the close coupling of the Na+/K+ pump to neuronal glycolysis, it will be exciting to measure how changes in Na+/K+ pump activity affect the flux of other glycolytic metabolites using biosensors that report lactate (Koveal et al., 2022; San Martín et al., 2013), pyruvate (San Martín et al., 2014), and glucose (Díaz-García et al., 2019), or how changes in Na+/K+ pump activity affect cellular energy status (i.e., [ATP] [Imamura et al., 2009] and ATP:ADP [Tantama et al., 2013]). These investigations will ultimately provide a more complete picture of the relationship between ion homeostasis and metabolism.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (Mus musculus, M and F) | C57BL/6NCrl | Charles River | RRID:IMSR_CRL:27 | |
| Recombinant DNA reagent | AAV.CAG.Peredox.WPRE.SV40 | Mongeon et al., 2016 | Addgene #73807 | |
| Recombinant DNA reagent | AAV.hSyn.RCaMP1h.WPRE.SV40 | Akerboom et al., 2013 | ||
| Recombinant DNA reagent | AAV.hSyn.mito-RCaMP1h.WPRE.SV40 | Díaz-García et al., 2021a | ||
| Chemical compound, drug | Isradipine | Abcam | Cat: ab120142, CAS: 75695-93-1 | |
| Chemical compound, drug | NBQX (6-Nitro-7- sulfamoylbenzo [f]quinoxaline-2,3-dione, Disodium Salt) | Toronto Research Chemicals | Cat: N550005, CAS: 479347-86-9 | |
| Chemical compound, drug | D-AP5 (D-(-)–2-Amino- 5-phosphonopentanoic acid) | Abcam | Cat: ab120003, CAS: 79055-68-8 | |
| Chemical compound, drug | Picrotoxin | Sigma-Aldrich | Cat: P1675 CAS: 124-87-8 | |
| Chemical compound, drug | CdCl2 | Sigma-Aldrich | Cat: 202908, CAS: 10108-64-2 | |
| Chemical compound, drug | Poly-L-lysine | Sigma-Aldrich | Cat: P4832 | |
| Chemical compound, drug | Aminooxyacetate (O-(carboxymethyl)hydroxylamine hemihydrate) | Sigma-Aldrich | Cat: C13408 CAS: 2921-14-4 | |
| Chemical compound, drug | Pyruvic acid | Sigma-Aldrich | Cat: 107360, CAS: 127-17-3 | |
| Chemical compound, drug | Thapsigargin | Santa Cruz Biotechnology | Cat: sc-24017A, CAS: 67526-95-8 | |
| Chemical compound, drug | Ouabain | Sigma-Aldrich | Cat: O3125, CAS: 11018-89-6 | |
| Chemical compound, drug | SBFI K+ salt (fluorescent dye) | Ion Biosciences | Cat: 2022B | |
| Other | Glass capillaries, Borosilicate, standard wall, no filament, 4 in., O.D. 1.5 mm | WPI | Cat: 1B150-4 | For microelectrodes and pipettes |
| Other | Glass coverslips, 12 mm circle No.1 | VWR | Cat: 48366–251 | For brain slice handling |
Animals
Experiments were performed using male and female wild-type mice (C57BL/6NCrl, Charles River Laboratories), which were housed in a barrier facility in individually ventilated cages with ad libitum access to standard chow (PicoLab 5053). All experiments followed approved IACUC protocols and the NIH Guide for the Care and Use of Laboratory Animals and Animal Welfare Act. All procedures were approved by the Harvard Medical Area Standing Committee on Animals.
Viral vectors
Request a detailed protocolDNA constructs encoding Peredox, RCaMP, or mito-RCaMP biosensors were packaged into adeno-associated virus (AAV) vectors using either the Penn Vector Core at University of Pennsylvania or the Viral Core Facility at Children’s Hospital in Boston, MA. AAV vectors encoding RCaMP were also produced in our laboratory, as previously described (Kimura et al., 2019). The AAV8 serotype was used for Peredox and AAV9 for RCaMP and mito-RCaMP. AAVs were aliquoted and stored at –80°C.
Intracranial injections
Request a detailed protocolPostnatal day 1 or 2, mice were injected intracranially with AAV to express biosensors in the hippocampus (Díaz-García et al., 2021b). Mice were cryoanesthetized and injected with 150 nL AAV using an UltraMicroPump III (WPI, Sarasota, FL) microinjector in two locations of each hemisphere at the following coordinates relative to lambda: (1) 0 mm anterior-posterior; ±1.9 mm medial-lateral; –2.0 mm dorsal-ventral; and (2) 0 mm anterior-posterior; ±2.0 mm medial-lateral; –2.3 mm dorsal-ventral. Injected pups were placed on a heating pad and allowed to recover before being returned to their cages and were administered daily ketoprofen (10 mg/kg) subcutaneously for up to 3 days.
Hippocampal slice preparation
Request a detailed protocolInjected mice between 14 and 24 days old were anesthetized with isoflurane, decapitated, and the brain was removed into ice-cold slicing solution containing (in mM) 87 NaCl, 2.5 KCl, 1.25 NaH2PO4, 25 NaHCO3, 75 sucrose, 25 D-glucose, 0.5 CaCl2, and 7 MgCl2 (~335 mOsm/kg) and bubbled with 95% O2 and 5% CO2. The brain was glued by the dorsal side, embedded into 2% agarose in phosphate-buffered saline, and submerged in a chamber with ice-cold slicing solution. Horizontal 275 μm brain slices were cut using a compresstome (VF-310-0Z, Precisionary) and immediately transferred to a chamber with ACSF containing (in mM) 120 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 10 D-glucose, 2 CaCl2, and 1 MgCl2 (~290 mOsm/kg) that was warmed to 36°C and bubbled with 95% O2 and 5% CO2. The slices rested on a mesh bottom to adequately perfuse both sides of the tissue. After 35 min, the chamber was cooled to room temperature and the brain slices therein were used for the next 4 hr.
Recording solutions and pharmacology
Request a detailed protocolThe brain slices were adhered to glass coverslips coated with poly-L-lysine (P4832, Sigma-Aldrich), placed in a bath chamber mounted to the microscope, and superfused with solutions maintained at 33–34°C and bubbled with 95% O2 and 5% CO2 at a rate of 5 mL/min. All solutions were ~290 mOsm/kg and contained 25 μM D-AP5, 5 μM NBQX, and 100 μM picrotoxin (unless otherwise specified) to block synaptic transmission. Ca2+-free solution contained (in mM) 120 NaCl, 2.5 KCl, 1 NaH2PO4, 26 NaHCO3, 10 D-glucose, and 4.1 MgCl2. Na+- and Ca2+-free solution contained (in mM) 120 Choline-Cl, 1.5 KCl, 1 KH2PO4, 26 Choline-HCO3, 10 D-glucose, and 4.1 MgCl2. 1 mM EGTA was added to the Ca2+-free solutions from a 0.5 M stock.
For Ca2+-puff experiments, the 40 mM Na+ and Ca2+-free solution was obtained by mixing Ca2+-free solution with Na+- and Ca2+-free solution; all solutions for those experiments also contained 0.1 mM EGTA.
Thapsigargin (1 μM) was added from a 10 mM stock in DMSO. Aminooxyacetate (AOA, 10 mM) was added from a 2 M stock in water. Pyruvate (1.5 mM) was added from a 1 M aqueous stock, pH 7.3 with N-methyl-D-glucamine. Ouabain was either directly dissolved (for 2–5 mM) prior to recording or added from a 5 mM stock in water (for 5 μM). Isradipine (3 μM) was added from a 50 mM DMSO stock. CdCl2 (20 μM) was added from a 100 mM stock in water.
Electrical stimulation
Request a detailed protocolStimulation trains (of 100 μs pulse width) were delivered as previously described (Díaz-García et al., 2017; Díaz-García et al., 2021a) using a concentric bipolar electrode (FHC, Bowdoin, ME) mounted on a motorized micromanipulator (Burleigh PCS-6000, ThorLabs, Sterling, VA) and connected to an A360 stimulus isolation unit (WPI, Sarasota, FL). DGCs were stimulated antidromically (100 pulses, 50 Hz, 750–1500 μA) by placing the electrode in the hippocampal hilus, or synaptically (60 pulses, 20 Hz, 100–500 μA) by placing the electrode in the molecular layer.
Ca2+ puffs
Request a detailed protocolBorosilicate glass pipettes (with a tip diameter of 5 μm and resistance of 1 MΩ when filled with ACSF) were fabricated on a micropipette puller (P-97 Flaming/Brown, Sutter Instruments), backfilled with 100–250 mM CaCl2 and placed into a pipette holder mounted on a motorized micromanipulator with a closed pressure system connected to a 1 mL syringe. The pipette tip was placed adjacent to groups of DGCs expressing Peredox and RCaMP biosensors. Pressure delivery was controlled by a solenoid valve (NResearch) in series with the syringe line that was triggered by a digital/analog actuator (NTE Electronics). Positive pressure was applied using the syringe, and Ca2+ puffs were delivered by opening the solenoid for 0.5–5 s beginning 300 ms after the start of image acquisition.
Single-cell electroporation of SBFI
Request a detailed protocolSodium-binding benzofuran isophthalate (SBFI) is a fluorescent Na+-indicator (Harootunian et al., 1989). We loaded the SBFI K+ salt (Ion Biosciences, San Marcos, TX) version into DGCs using single-cell electroporation methods derived from Nevian and Helmchen, 2007; we chose this method rather than loading by patch pipette to avoid diluting cytoplasmic metabolites and soluble expressed biosensor, and also to avoid potentially toxic effects on the slice from applying Pluronic-F127 and DMSO to load the membrane-permeant SBFI-acetoxymethyl ester version.
Borosilicate glass pipettes were fabricated (with a tip diameter of 1 μm and resistance of 3 MΩ when filled with ACSF), loaded with 4 mM SBFI-K+ in distilled H2O, and placed onto a pipette holder with a closed pressure system mounted on a motorized micromanipulator. The SBFI solution was contacted by a chlorided silver wire connected to an A360 stimulus isolation unit, and an Ag-AgCl reference electrode was placed in the bath chamber and connected to the same stimulation device. The pipette tip was placed directly next to the soma of an RCaMP-expressing DGC. No positive pressure was applied. A single 10 ms pulse of negative 1.0–1.2 μA loaded the DGC with SBFI. Recovery of the membrane potential from these electroporation pulses takes ~1–2 min (Nevian and Helmchen, 2007). We waited ≥15 min after the electroporation to start the image acquisition and electrically stimulate the SBFI-loaded, RCaMP-expressing DGCs.
The intracellular [dye] from electroporation is estimated to be ~20% of the pipette concentration (Nevian and Helmchen, 2007). Thus, the intracellular [Na+] will not be substantially buffered by the ~0.5–1 mM [SBFI] in our recordings (Fleidervish et al., 2010; Mondragão et al., 2016), meaning that intracellular [Na+] is set by cellular mechanisms and SBFI merely responds to changes in [Na+]. Also, the complexation of Na+ with crown ether molecules (the Na+-sensitive moiety of SBFI) is nearly diffusion-controlled, with very rapid rates of formation and dissociation (Adamic et al., 1986; Liesegang et al., 1977). This means that the response time of SBFI fluorescence changes should be fast compared to almost all neuronal processes.
Two-photon fluorescence imaging
Fluorescence imaging data were acquired using a Thorlabs Bergamo II microscope (Thorlabs Imaging Systems, Sterling, VA) equipped with an Olympus LUMPLFLN 60×/W (NA 1.0) objective lens, hybrid photodetectors R11322U-40 (Hamamatsu Photonics, Shizuoka, Japan), and a Chameleon Vision-S tunable Ti-Sapphire mode-locked laser (80 MHz,~75 fs pulses; Coherent, Santa Clara, CA). The excitation wavelength was 790 nm. Fluorescence emission light from the Peredox and RCaMP biosensors was split with an FF562-Di03 dichroic mirror and bandpass filtered for green (FF01-525/50) and red (FF01-641/75) light; a red 670/50 bandpass filter was used for experiments where SBFI and RCaMP were multiplexed. The photodetector and laser sync signals were preamplified and then digitized at 1.25 GHz using a field-programmable gate array board (PC720 with FMC125 and FMC122 modules, 4DSP, Austin, TX). A modified version of the ScanImage software written in Matlab (Pologruto et al., 2003) (provided by B. Sabatini and modified by G.Y.) controlled the laser, microscope, and image acquisition (128×128 pixels, scanning rate of 2 ms per line).
Time-correlated single-photon counting was performed using laboratory-built firmware and software to determine the arrival time of each photon relative to the laser pulse. The fluorescence LT was determined from a nonlinear least-squares fit to the photon arrival histograms in Matlab (Mathworks, Natick, MA) convolved with a Gaussian for the impulse response function (Yasuda et al., 2006). Fluorescence intensity was determined from the total photon counts.
Data analysis
Request a detailed protocolFluorescence images were analyzed offline using Matlab R2014b software. Regions of interest (ROIs) were defined around individual DGC somas and photon statistics were calculated for all pixels within the ROI. LT values were calculated by fitting the photon arrival histograms with a biexponential decay function (convolved with a Gaussian for the impulse response function [Yasuda et al., 2006] up to 8 ns after the peak photon arrival time). The time constant of this fit is denoted as the ‘tau8‘ value (Díaz-García et al., 2019); using tau8 values minimizes both the fit variability (by restricting the averaging to the approximate time window of the actual data) and the variability between the LTs recorded on different experimental setups with different data acquisition windows.
RCaMP signals were unmixed from Peredox as previously described (Díaz-García et al., 2017; Díaz-García et al., 2021a). RCaMP signals were unmixed from SBFI bleedthrough into the red 670/50 bandpass filter following previous methods (Díaz-García et al., 2017) but using an unmixing ratio of 0.053, since the red channel photon counts from SBFI-loaded DGCs (not expressing RCaMP) were (mean ± SD) 5.3±1.2% (n=42) of the green channel photon counts. Stimulation-induced SBFI intensity recordings were analyzed using Origin 8.1 (OriginLab, Northampton, MA) and Python 3 (https://www.anaconda.com/, RRID – SRC:008394, Numpy and Pandas libraries). Individual transient fluorescence intensity traces were baseline subtracted and the peak ∆F/F was determined as the minimum intensity value.
Average time traces of transient fluorescence LT or intensity changes were created using Python. Data acquisition times for individual fluorescence traces were binned to the nearest second for data acquired before and after the stimulation event (which were acquired every 10–60 s) or the nearest 10 ms for data acquired during the stimulation (which were acquired every ~250 ms). Traces from all stimulation-induced transients in the data set were interpolated and merged onto a single time axis, from which the means, standard deviations, and standard error of the means were determined.
Statistical analysis was performed using Origin 8.1 software. Data sets were tested for normality (α=0.05) using a Shapiro-Wilk test. Data sets that met normality criteria were compared using a paired t-test, or two-sample t-test. Data sets that did not meet normality criteria were compared using a paired sample Wilcoxon test or Mann-Whitney test for unpaired samples. Data are reported as mean ± standard deviation (unless otherwise indicated). For box plots, the means are indicated by the filled square, medians are indicated by the horizontal bar, and 5–95% ranges are indicated by whiskers.
Figures were created using Origin 8.1 and PowerPoint (Microsoft, Redmond, WA).
Reagents
All chemical reagents used to make the brain slicing and artificial cerebrospinal fluid (ACSF) solutions were obtained from Sigma-Aldrich (St. Louis, MO).
Data availability
All data generated or analysed during this study are included in the manuscript.
References
-
Sodium ion complexation by ionizable crown ethers in methanol-water solvents. A thermodynamic and kinetic evaluation of sidearm interactionThe Journal of Physical Chemistry 90:6571–6576.https://doi.org/10.1021/j100282a030
-
Genetically encoded calcium indicators for multi-color neural activity imaging and combination with optogeneticsFrontiers in Molecular Neuroscience 6:2.https://doi.org/10.3389/fnmol.2013.00002
-
CNS energy metabolism as related to functionBrain Research. Brain Research Reviews 34:42–68.https://doi.org/10.1016/s0165-0173(00)00038-2
-
An energy budget for signaling in the grey matter of the brainJournal of Cerebral Blood Flow and Metabolism 21:1133–1145.https://doi.org/10.1097/00004647-200110000-00001
-
The neural basis of functional brain imaging signalsTrends in Neurosciences 25:621–625.https://doi.org/10.1016/s0166-2236(02)02264-6
-
A specific and essential role for Na, K-ATPase α3 in neurons co-expressing α1 and α3The Journal of Biological Chemistry 288:2734–2743.https://doi.org/10.1074/jbc.M112.425785
-
Studies on the relationship between glycolysis and (Na+ + K+) -ATPase in cultured cellsBiochimica et Biophysica Acta 804:419–426.https://doi.org/10.1016/0167-4889(84)90069-7
-
Molecular mechanism of calcium channel block by isradipineJournal of Biological Chemistry 275:22114–22120.https://doi.org/10.1074/jbc.M908836199
-
Diseases caused by mutations in the Na+/K+ pump α1 gene ATP1A1American Journal of Physiology. Cell Physiology 321:C394–C408.https://doi.org/10.1152/ajpcell.00059.2021
-
Isozymes of the Na-K-ATPase: heterogeneity in structure, diversity in functionAmerican Journal of Physiology-Renal Physiology 275:F633–F650.https://doi.org/10.1152/ajprenal.1998.275.5.F633
-
Na, K-ATPase subunit heterogeneity as a mechanism for tissue-specific ion regulationSeminars in Nephrology 25:292–303.https://doi.org/10.1016/j.semnephrol.2005.03.004
-
Sodium/calcium exchange: its physiological implicationsPhysiological Reviews 79:763–854.https://doi.org/10.1152/physrev.1999.79.3.763
-
The plasma membrane calcium pump in health and diseaseThe FEBS Journal 280:5385–5397.https://doi.org/10.1111/febs.12193
-
The PMCA pumps in genetically determined neuronal pathologiesNeuroscience Letters 663:2–11.https://doi.org/10.1016/j.neulet.2017.11.005
-
The Ca2+-pumping ATPase of heart sarcolemma. Characterization, calmodulin dependence, and partial purificationThe Journal of Biological Chemistry 256:3263–3270.
-
SERCA control of cell death and survivalCell Calcium 69:46–61.https://doi.org/10.1016/j.ceca.2017.07.001
-
Calcium signaling in brain mitochondriaJournal of Biological Chemistry 284:7091–7099.https://doi.org/10.1074/jbc.M808066200
-
Transport and pharmacological properties of nine different human Na, K-ATPase isozymesThe Journal of Biological Chemistry 275:1976–1986.https://doi.org/10.1074/jbc.275.3.1976
-
The regulation of ion channels and transporters by glycolytically derived ATPCellular and Molecular Life Sciences 64:3069–3083.https://doi.org/10.1007/s00018-007-7332-3
-
In situ calibration and [H+] sensitivity of the fluorescent Na+ indicator SBFIAmerican Journal of Physiology. Cell Physiology 280:C1623–C1633.https://doi.org/10.1152/ajpcell.2001.280.6.C1623
-
Quantitative in vivo imaging of neuronal glucose concentrations with a genetically encoded fluorescence lifetime sensorJournal of Neuroscience Research 97:946–960.https://doi.org/10.1002/jnr.24433
-
Phosphotransfer networks and cellular energeticsThe Journal of Experimental Biology 206:2039–2047.https://doi.org/10.1242/jeb.00426
-
Non-signalling energy use in the brainThe Journal of Physiology 593:3417–3429.https://doi.org/10.1113/jphysiol.2014.282517
-
Evolution of glycolysisProgress in Biophysics and Molecular Biology 59:105–235.https://doi.org/10.1016/0079-6107(93)90001-z
-
Relation between activity-induced intracellular sodium transients and ATP dynamics in mouse hippocampal neuronsThe Journal of Physiology 597:5687–5705.https://doi.org/10.1113/JP278658
-
Electrophysiology of the sodium-potassium-ATPase in cardiac cellsPhysiological Reviews 81:1791–1826.https://doi.org/10.1152/physrev.2001.81.4.1791
-
Fluorescence ratio imaging of cytosolic free Na+ in individual fibroblasts and lymphocytesJournal of Biological Chemistry 264:19458–19467.https://doi.org/10.1016/S0021-9258(19)47322-5
-
Myocardial metabolic inhibition and membrane potential, contraction, and potassium uptakeThe American Journal of Physiology 247:H322–H329.https://doi.org/10.1152/ajpheart.1984.247.2.H322
-
Role of beta-subunit domains in the assembly, stable expression, intracellular routing, and functional properties of Na, K-ATPaseThe Journal of Biological Chemistry 273:30826–30835.https://doi.org/10.1074/jbc.273.46.30826
-
Experiments on the injection of substances into squid giant axons by means of a microsyringeThe Journal of Physiology 131:592–616.https://doi.org/10.1113/jphysiol.1956.sp005485
-
Updated energy budgets for neural computation in the neocortex and cerebellumJournal of Cerebral Blood Flow and Metabolism 32:1222–1232.https://doi.org/10.1038/jcbfm.2012.35
-
Molecular determinants of Na+/Ca2+ exchange (NCX1) inhibition by SEA0400The Journal of Biological Chemistry 279:7544–7553.https://doi.org/10.1074/jbc.M310491200
-
Linkage of aerobic glycolysis to sodium-potassium transport in rat skeletal muscle. Implications for increased muscle lactate production in sepsisThe Journal of Clinical Investigation 98:2388–2397.https://doi.org/10.1172/JCI119052
-
Stimulation of both aerobic glycolysis and Na+-K+-ATPase activity in skeletal muscle by epinephrine or amylinThe American Journal of Physiology 277:E176–E186.https://doi.org/10.1152/ajpendo.1999.277.1.E176
-
Biochemistry of Na, K-ATPaseAnnual Review of Biochemistry 71:511–535.https://doi.org/10.1146/annurev.biochem.71.102201.141218
-
Allosteric regulatory properties of muscle phosphofructokinaseMolecular and Cellular Biochemistry 57:147–154.https://doi.org/10.1007/BF00849191
-
Association of glycolytic enzymes with particulate fractions from nerve endingsBiochimica et Biophysica Acta 522:1–9.https://doi.org/10.1016/0005-2744(78)90316-9
-
Ion transport in gills of the euryhaline fish Gillichthys mirabilis is facilitated by a phosphocreatine circuitThe American Journal of Physiology 268:R1003–R1012.https://doi.org/10.1152/ajpregu.1995.268.4.R1003
-
Subcellular targeting of metabolic enzymes to titin in heart muscle may be mediated by DRAL/FHL-2Journal of Cell Science 115:4925–4936.https://doi.org/10.1242/jcs.00181
-
Methylation determines the extracellular calcium sensitivity of the leak channel NALCN in hippocampal dentate granule cellsExperimental & Molecular Medicine 51:1–14.https://doi.org/10.1038/s12276-019-0325-0
-
The cost of cortical computationCurrent Biology 13:493–497.https://doi.org/10.1016/s0960-9822(03)00135-0
-
Ultrasonic absorption kinetic studies of the complexation of aqueous Li+, Na+, Rb+, Tl+, Ag+, NH4+, and Ca2+ by 18-crown-6Journal of the American Chemical Society 99:3240–3243.https://doi.org/10.1021/ja00452a006
-
Glycolysis and brain function: [K+] o stimulation of protein synthesis and K+ uptake require glycolysisFederation Proceedings 42:2875–2880.
-
Coupling of aerobic glycolysis and Na+-K+-ATPase in renal cell line MDCKThe American Journal of Physiology 253:C269–C276.https://doi.org/10.1152/ajpcell.1987.253.2.C269
-
A heterogeneous ouabain-ATPase interaction in mouse brainLife Sciences 23:2735–2744.https://doi.org/10.1016/0024-3205(78)90654-9
-
Properties of the new fluorescent Na+ indicator corona green: comparison with SBFI and confocal Na+ imagingJournal of Neuroscience Methods 155:251–259.https://doi.org/10.1016/j.jneumeth.2006.01.009
-
Membrane-bound ATP fuels the Na/K pump. Studies on membrane-bound glycolytic enzymes on inside-out vesicles from human red cell membranesThe Journal of General Physiology 78:547–568.https://doi.org/10.1085/jgp.78.5.547
-
On the effect of hyperaldosteronism-inducing mutations in Na/K pumpsJournal of General Physiology 149:1009–1028.https://doi.org/10.1085/jgp.201711827
-
FXYD protein isoforms differentially modulate human Na/K pump functionJournal of General Physiology 152:e202012660.https://doi.org/10.1085/jgp.202012660
-
Energy costs of ion pumping by animal tissuesThe Journal of Nutrition 115:1374–1382.https://doi.org/10.1093/jn/115.10.1374
-
The control of erythrocyte glycolysis by active cation transportBiochimica et Biophysica Acta (BBA) - General Subjects 90:434–436.https://doi.org/10.1016/0304-4165(64)90219-3
-
Sodium-calcium exchange current. Dependence on internal Ca and Na and competitive binding of external Na and CaThe Journal of General Physiology 93:1129–1145.https://doi.org/10.1085/jgp.93.6.1129
-
Extrusion versus diffusion: mechanisms for recovery from sodium loads in mouse CA1 pyramidal neuronsThe Journal of Physiology 594:5507–5527.https://doi.org/10.1113/JP272431
-
Cytosolic NADH-NAD+ redox visualized in brain slices by two-photon fluorescence lifetime biosensor imagingAntioxidants & Redox Signaling 25:553–563.https://doi.org/10.1089/ars.2015.6593
-
The purified Ca2+ pump of human erythrocyte membranes catalyzes an electroneutral Ca2+-H+ exchange in reconstituted liposomal systemsThe Journal of Biological Chemistry 257:2350–2356.https://doi.org/10.1016/S0021-9258(18)34929-9
-
On the origin of intracellular compartmentation and organized metabolic systemsMolecular and Cellular Biochemistry 256–257:5–12.https://doi.org/10.1023/b:mcbi.0000009855.14648.2c
-
The mitochondrial Na+/Ca2+ exchangerCell Calcium 52:9–15.https://doi.org/10.1016/j.ceca.2012.02.010
-
Functional compartmentalization of oxidative and glycolytic metabolism in vascular smooth muscleAmerican Journal of Physiology-Cell Physiology 244:C399–C409.https://doi.org/10.1152/ajpcell.1983.244.5.C399
-
ScanImage: flexible software for operating laser scanning microscopesBioMedical Engineering OnLine 2:13.https://doi.org/10.1186/1475-925X-2-13
-
A phosphorylated intermediate in adenosine triphosphate-dependent sodium and potassium transport across kidney membranesThe Journal of Biological Chemistry 240:1437–1445.
-
Membrane compartmentalized ATP and its preferential use by the Na,K-ATPase of human red cell ghostsThe Journal of General Physiology 69:605–632.https://doi.org/10.1085/jgp.69.5.605
-
Two-photon Na+ imaging in spines and fine dendrites of central neuronsPflugers Archiv 439:201–207.https://doi.org/10.1007/s004249900123
-
Mitochondrial membrane potential modulates regulation of mitochondrial Ca2+ in rat ventricular myocytesAmerican Journal of Physiology. Heart and Circulatory Physiology 288:H1820–H1828.https://doi.org/10.1152/ajpheart.00589.2004
-
Structure and allosteric regulation of eukaryotic 6-phosphofructokinasesBiological Chemistry 394:977–993.https://doi.org/10.1515/hsz-2013-0130
-
Pharmacological characterization of the newly synthesized 5-amino-N-butyl-2-(4-ethoxyphenoxy)-benzamide hydrochloride (BED) as a potent NCX3 inhibitor that worsens anoxic injury in cortical neurons, organotypic hippocampal cultures, and ischemic brainACS Chemical Neuroscience 6:1361–1370.https://doi.org/10.1021/acschemneuro.5b00043
-
Stoichiometry and localization of adenosine triphosphate-dependent sodium and potassium transport in the erythrocyteJournal of Biological Chemistry 239:345–352.https://doi.org/10.1016/S0021-9258(18)51788-9
-
Two molecular forms of (Na+ + K+) -stimulated ATPase in brain. Separation, and difference in affinity for strophanthidinThe Journal of Biological Chemistry 254:6060–6067.
-
A thermodynamic model of the cardiac sarcoplasmic/endoplasmic Ca2+ (SERCA) pumpBiophysical Journal 96:2029–2042.https://doi.org/10.1016/j.bpj.2008.11.045
-
Calcium channels: mechanisms of selectivity, permeation, and blockAnnual Review of Biophysics and Biophysical Chemistry 16:265–290.https://doi.org/10.1146/annurev.bb.16.060187.001405
-
Functional compartmentation of glycolytic versus oxidative metabolism in isolated rabbit heartThe Journal of Clinical Investigation 75:436–447.https://doi.org/10.1172/JCI111718
-
The dependence of the respiration of brain cortex on active cation transportThe Biochemical Journal 82:205–212.https://doi.org/10.1042/bj0820205
-
The connexion between active cation transport and metabolism in erythrocytesThe Biochemical Journal 97:214–227.https://doi.org/10.1042/bj0970214
-
Evaluating the gray and white matter energy budgets of human brain functionJournal of Cerebral Blood Flow and Metabolism 38:1339–1353.https://doi.org/10.1177/0271678X17708691
-
Phospholemman regulates cardiac Na+/Ca2+ exchanger by interacting with the exchanger’s proximal linker domainAmerican Journal of Physiology. Cell Physiology 296:C911–C921.https://doi.org/10.1152/ajpcell.00196.2008
-
Residues 248-252 and 300-304 of the cardiac Na+/Ca2+ exchanger are involved in its regulation by phospholemmanAmerican Journal of Physiology. Cell Physiology 301:C833–C840.https://doi.org/10.1152/ajpcell.00069.2011
Article and author information
Author details
Carlos Manlio Díaz-García

Funding
National Institute of Neurological Disorders and Stroke (R01 NS102586)
- Gary Yellen
National Institute of General Medical Sciences (R01 GM124038)
- Gary Yellen
National Institute of Neurological Disorders and Stroke (F32 NS116105)
- Dylan J Meyer
National Institute of Neurological Disorders and Stroke (F32 NS100331)
- Carlos Manlio Díaz-García
Harvard Mahoney Neuroscience Institute (Postdoctoral Fellowship)
- Dylan J Meyer
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank the members of the Yellen lab for their valuable discussions. The authors thank Dr Juan Ramón Martínez François and Dr Pablo Artigas for their critical review of the manuscript. This work was supported by research grants from the National Institutes of Health (R01 NS102586 and R01 GM124038 to GY) and Postdoctoral Fellowships from the National Institutes of Health (F32 NS116105 to DJM and F32 NS100331 to CMDG) and the Harvard Mahoney Neuroscience Institute Fund (to DJM).
Ethics
All experiments were performed in compliance with the NIH Guide for the Care and Use of Laboratory Animals and the Animal Welfare Act. The Harvard Medical Area Standing Committee on Animals approved all procedures involving animals. (Animal Welfare Assurance Number A3431-01, Protocol IS00001113-3).
Copyright
© 2022, Meyer et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,721
- views
-
- 357
- downloads
-
- 42
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 42
- citations for umbrella DOI https://doi.org/10.7554/eLife.81645
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The Na+/K+ pump dominates control of glycolysis in hippocampal dentate granule cells
eLife 11:e81645.
https://doi.org/10.7554/eLife.81645




{kind=link}
{kind=link}
{kind=link}
{kind=link}