The deubiquitinase Ubp3/Usp10 constrains glucose-mediated mitochondrial repression via phosphate budgeting
- Institute for Stem Cell Science and Regenerative Medicine (DBT-inStem), India
- Manipal Academy of Higher Education, India
eLife assessment
This study provides valuable insights into the regulation of metabolic flux between glycolysis and respiration in yeast, particularly focusing on the role of inorganic phosphate. The authors propose a novel mechanism involving Ubp3/Ubp10 that potentially mitigates the Crabtree effect, offering substantial, solid evidence through a variety of well-designed assays. This study could reshape our understanding of metabolic regulation with broad biological contexts.
https://doi.org/10.7554/eLife.90293.4.sa0Significance of the findings:
Valuable: Findings that have theoretical or practical implications for a subfield
- Landmark
- Fundamental
- Important
- Valuable
- Useful
Strength of evidence:
Solid: Methods, data and analyses broadly support the claims with only minor weaknesses
- Exceptional
- Compelling
- Convincing
- Solid
- Incomplete
- Inadequate
During the peer-review process the editor and reviewers write an eLife Assessment that summarises the significance of the findings reported in the article (on a scale ranging from landmark to useful) and the strength of the evidence (on a scale ranging from exceptional to inadequate). Learn more about eLife Assessments
Abstract
Many cells in high glucose repress mitochondrial respiration, as observed in the Crabtree and Warburg effects. Our understanding of biochemical constraints for mitochondrial activation is limited. Using a Saccharomyces cerevisiae screen, we identified the conserved deubiquitinase Ubp3 (Usp10), as necessary for mitochondrial repression. Ubp3 mutants have increased mitochondrial activity despite abundant glucose, along with decreased glycolytic enzymes, and a rewired glucose metabolic network with increased trehalose production. Utilizing ∆ubp3 cells, along with orthogonal approaches, we establish that the high glycolytic flux in glucose continuously consumes free Pi. This restricts mitochondrial access to inorganic phosphate (Pi), and prevents mitochondrial activation. Contrastingly, rewired glucose metabolism with enhanced trehalose production and reduced GAPDH (as in ∆ubp3 cells) restores Pi. This collectively results in increased mitochondrial Pi and derepression, while restricting mitochondrial Pi transport prevents activation. We therefore suggest that glycolytic flux-dependent intracellular Pi budgeting is a key constraint for mitochondrial repression.
Introduction
Rapidly proliferating cells have substantial metabolic and energy demands in order to increase biomass (Cai and Tu, 2012; Zhu and Thompson, 2019). This includes a high ATP demand, obtained from cytosolic glycolysis or mitochondrial oxidative phosphorylation (OXPHOS), to fuel multiple reactions (Nelson et al., 2008). Interestingly, many rapidly proliferating cells preferentially rely on ATP from glycolysis/fermentation over mitochondrial respiration even in oxygen-replete conditions, and is the well-known Warburg effect (Vander Heiden et al., 2009; Warburg, 1925). Many such cells repress mitochondrial processes in high glucose, termed glucose-mediated mitochondrial repression or the Crabtree effect (Crabtree, 1929; De Deken, 1966). This is observed in tumors (Vander Heiden et al., 2009), neutrophils (Xia et al., 2021), activated macrophages (Kornberg, 2020), stem cells (Abdel-Haleem et al., 2017; Pacini and Borziani, 2014; Tsogtbaatar et al., 2020), and famously Saccharomyces cerevisiae (De Deken, 1966). Numerous studies have identified signaling programs or regulators of glucose-mediated mitochondrial repression. However, biochemical programs and regulatory processes in biology evolve around key biochemical constraints (Cornish-Bowden, 2016). The biochemical constraints for mitochondrial repression remain unresolved (Diaz-Ruiz et al., 2011; Hammad et al., 2016).
There are two hypotheses on the biochemical principles driving mitochondrial repression. The first proposes direct roles for glycolytic intermediates in driving mitochondrial respiration, by inhibiting specific mitochondrial outputs (Díaz-Ruiz et al., 2008; Rosas Lemus et al., 2018). The second hypothesizes that a competition between glycolytic and mitochondrial processes for mutually required metabolites/co-factors such as pyruvate, ADP, or inorganic phosphate (Pi) could determine the extent of mitochondrial repression (Diaz-Ruiz et al., 2011; Hammad et al., 2016; Koobs, 1972). These are not all mutually exclusive, and a combination of these factors might dictate mitochondrial repression in high glucose. However, any hierarchies of importance are unclear (Rodríguez-Enríquez et al., 2001), and experimental data for the necessary constraints for mitochondrial repression remains incomplete.
One approach to resolve this question has been to identify regulators of metabolic state under high glucose. Post-translational modifications (PTMs) regulated by signaling systems can regulate mitochondrial repression (Broach, 2012; Hitosugi and Chen, 2014; Tripodi et al., 2015). Ubiquitination is a PTM that regulates global proteostasis (Hershko and Ciechanover, 1998; Komander and Rape, 2012), but the roles of ubiquitination-dependent processes in regulating mitochondrial repression are poorly explored. Ubiquitination itself is determined by the balance between ubiquitination and deubiquitinase (DUB) dependent deubiquitination (Pickart and Eddins, 2004). Little is known about the roles of DUBs in regulating metabolic states, making the DUBs interesting candidate regulators of mitochondrial repression.
In this study, using an S. cerevisiae DUB knockout library-based screen, we identified the evolutionarily conserved DUB Ubp3 (mammalian Usp10) as required for mitochondrial repression in high glucose. Loss of Ubp3 resulted in mitochondrial activation, along with a reduction in the glycolytic enzymes - phosphofructokinase 1 (Pfk1) and GAPDH (Tdh2 and Tdh3). This consequently reroutes glucose flux and increases trehalose biosynthesis. This metabolic rewiring increases Pi release from trehalose synthesis, and decreases Pi consumption in glycolysis, to cumulatively increase Pi pools. Using ubp3Δ cells along with independent analysis of wild-type (WT) cells, and isolated mitochondrial fractions, we establish that glycolytic flux-dependent Pi allocations to mitochondria determines mitochondrial activity. Through these data, we propose how intracellular Pi balance as controlled by glycolytic flux is a key biochemical constraint for mitochondrial repression.
Results
A DUB deletion screen identifies Ubp3 as a regulator of glucose-mediated mitochondrial repression
In cells such as S. cerevisiae, high glucose represses mitochondrial activity as well as OXPHOS-dependent ATP synthesis (De Deken, 1966; Postma et al., 1989) (illustrated in Figure 1A). Our initial objective was to identify proteostasis regulators of glucose-mediated mitochondrial repression. We generated and used a DUB deletion strain library of S. cerevisiae (Figure 1—figure supplement 1A), to unbiasedly identify regulators of mitochondrial repression by measuring the fluorescence intensity of a potentiometric dye Mitotracker CMXRos (illustrated in Figure 1B). Using this screen, we identified DUB mutants with altered mitochondrial membrane potential (Figure 1C, Figure 1—figure supplement 1A). Note: WT cells in a respiratory medium (2% ethanol) were used as a control to estimate maximum mitotracker fluorescence intensity (Figure 1—figure supplement 1B).
Figure 1 with 1 supplement see all
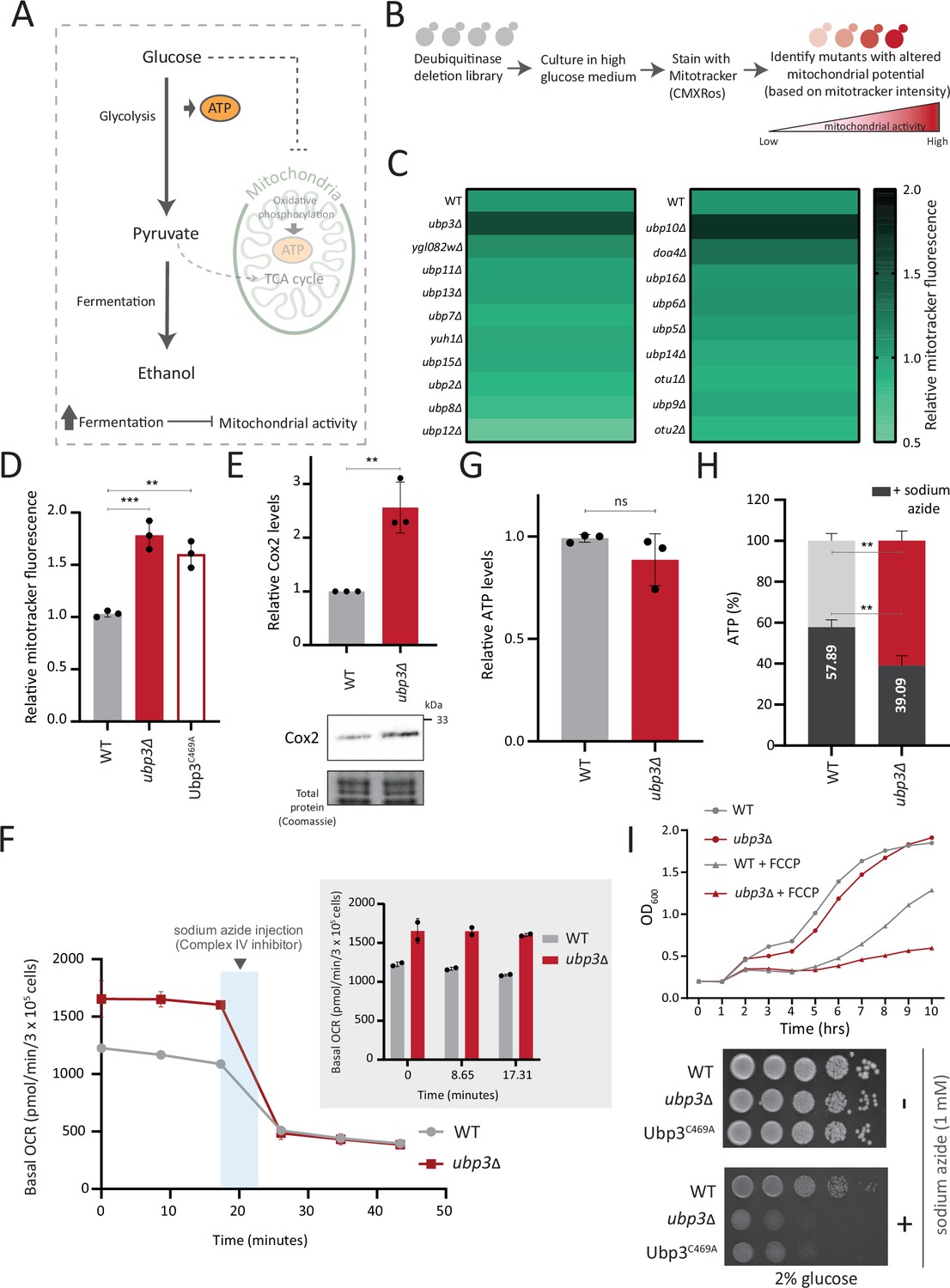
A deubiquitinase (DUB) deletion screen identifies Ubp3 as a regulator of glucose-mediated mitochondrial repression.
(A) Schematic depicting glucose-mediated mitochondrial repression (Crabtree effect). (B) Schematic describing the screen with a yeast DUB knockout (KO) library to identify regulators of Crabtree effect. (C) Identifying DUB knockouts with altered mitochondrial potential. Heat map shows relative mitochondrial membrane potential of 19 DUB deletions in high glucose, from two biological replicates. Also see Figure 1—figure supplement 1A and B. (D) The DUB activity of Ubp3 and repression of mitochondrial membrane potential. Wild-type (WT), ubp3Δ, and Ubp3C469A were grown in high glucose and relative mitochondrial membrane potential was measured. Data represent mean ± SD from three biological replicates (n=3). Also see Figure 1—figure supplement 1D. (E) Effect of loss of Ubp3 on electron transport chain (ETC) complex IV subunit Cox2. WT and ubp3Δ were grown in high glucose, and Cox2 was measured (western blot using an anti-Cox2 antibody). A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. (F) Basal oxygen consumption rate (OCR) in high glucose in ubp3Δ. WT and ubp3Δ were grown in high glucose, and OCR was measured. Basal OCR corresponding to ~3×105 cells, from two independent experiments (n=2), normalized to the OD600 is shown. Bar graph representations are shown in the inset. Data represent mean ± SD. (G) Total ATP levels in ubp3Δ and WT. WT and ubp3Δ were grown in high glucose, and total ATP were measured. Data represent mean ± SD from three biological replicates (n=3). (H) Dependence of ubp3Δ on mitochondrial ATP. WT and ubp3Δ cells were grown in high glucose, and treated with 1 mM sodium azide for 45 min. Total ATP levels in sodium azide treated and untreated cells were measured. Data represent mean ± SD (n=3). (I) Requirement for mitochondrial respiration in high glucose in ubp3Δ. A growth curve of WT and ubp3Δ in high glucose in the presence of oxidative phosphorylation (OXPHOS) uncoupler FCCP (10 µM), and serial dilution growth assay in high glucose in the presence/absence of sodium azide (1 mM) are shown. Data represent mean ± SD (n=2). Also see Figure 1—figure supplement 1H and I. Data information: **p<0.01, ***p<0.001.
-
Figure 1—source data 1
Uncropped and labeled gels and blots for Figure 1.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig1-data1-v1.zip
-
Figure 1—source data 2
Raw unedited gels and blots for Figure 1.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig1-data2-v1.zip
A prominent ‘hit’ was the evolutionarily conserved DUB Ubp3 (Figure 1C), (homologous to mammalian Usp10) (Figure 1—figure supplement 1D). Due to its high degree of conservation across eukaryotes (Figure 1—figure supplement 1D) as well as putative roles in metabolism or mitochondrial function (Isasa et al., 2015; Nostramo et al., 2016; Ossareh-Nazari et al., 2010), we focused our further attention on this DUB. Cells lacking Ubp3 showed an ~1.5-fold increase in mitotracker fluorescence (Figure 1C, Figure 1—figure supplement 1A and C). Cells with catalytically inactive Ubp3 (Ubp3C469A) showed increased mitochondrial potential comparable to ubp3∆ (Figure 1D). This data confirmed that Ubp3 catalytic activity is required to fully repress mitochondrial activity under high glucose. The catalytic site mutation did not affect steady-state Ubp3 levels (Figure 1—figure supplement 1E).
Next, to assess the requirement of Ubp3 for mitochondrial function, we quantified the electron transport chain (ETC) complex IV subunit Cox2 (Fontanesi et al., 2006; Figure 1E). ubp3Δ had higher Cox2 than WT (Figure 1E). As a control, we estimated total mitochondrial content in WT and ubp3Δ cells, using either estimates of the structural protein Tom70, or measuring the fluorescence intensity in strains engineered with mitochondrial targeted mNeonGreen (Dua et al., 2022). There was no increase in the total mitochondrial volume (estimated by measuring the intensity of mitochondria targeted mNeon green) (Figure 1—figure supplement 1F) or mitochondrial outer membrane protein Tom70 (Figure 1—figure supplement 1G). This suggests that the increased Cox2 is not merely because of higher total mitochondrial content. We next measured the basal oxygen consumption rate (OCR) of ubp3Δ, and basal OCR was higher in ubp3Δ, indicating higher respiration (Figure 1F).
Next, we asked if mitochondrial ATP synthesis was higher in ubp3Δ. The total ATP levels in WT and ubp3∆ were comparable (Figure 1G). However, upon treatment with the ETC complex IV inhibitor sodium azide, ATP levels in WT were higher than ubp3∆, contributing to ~60% of the total ATP (Figure 1H). In contrast, the ATP levels in ubp3∆ after sodium azide treatment were ~40% of the total ATP (Figure 1H). These data suggest a higher contribution of mitochondrial ATP synthesis toward the total ATP pool in ubp3∆.
We next asked if ubp3Δ required higher mitochondrial activity for growth, using a series of mitochondrial activity inhibitors and comparing relative growth. In high glucose, WT cells show minimal growth inhibition in the presence of sodium azide, indicating lower reliance on mitochondrial function (Figure 1I). Contrastingly, ubp3Δ or Ubp3C469A show a severe growth defect in the presence of the mitochondrial ETC complex inhibitors sodium azide and oligomycin, and the mitochondrial OXPHOS uncoupler FCCP (Figure 1I, Figure 1—figure supplement 1H). Additionally, the loss of Ubp3 in respiration defective cox2-62 cells (Bonnefoy et al., 2001) or Rho0 cells (which lacks mitochondrial DNA) resulted in a severe growth defect (Figure 1—figure supplement 1I). Deletion of ATP synthase subunits Atp1 and Atp10 also results in a severe growth defect in ubp3Δ compared to WT (Figure 1—figure supplement 1I). Together, these results indicate that the loss of Ubp3 makes cells dependent on mitochondrial ATP synthesis in high glucose.
In order to address whether the deletion of Ubp3 might increase ubiquitinated proteins and consequent proteostatic stress, we analyzed the global ubquitination state in ubp3Δ cells. WT and ubp3Δ cells grown under brief (1 hr) heat stress, which increases protein ubiquitination, was used as a control. However, we did not observe a significant increase in the global ubiqutinatin state in ubp3Δ cells (Figure 1—figure supplement 1J). This suggests that the altered mitochondrial metabolism in ubp3Δ cells is unlikely to be due to general proteostatic stress.
Collectively these data show that in 2% glucose, ubp3Δ have high mitochondrial activity, respiration, and rely on this mitochondrial function for ATP production and growth. We therefore decided to use ubp3Δ cells to start delineating requirements for glucose-mediated mitochondrial repression.
Key glycolytic enzymes decrease and glucose flux is rerouted in ubp3Δ cells
Glucose-6 phosphate (G6P) is the central node in glucose metabolism where carbon allocations are made toward distinct metabolic arms, primarily: glycolysis, the pentose phosphate pathway (PPP), and trehalose biosynthesis (Figure 2A). We first compared amounts of two key (‘rate-controlling’) glycolytic enzymes - phosphofructokinase 1 (Pfk1), GAPDH isozymes (Tdh2, Tdh3) (Nelson et al., 2008), along with the enolase isozymes (Eno1, Eno2) in WT and ubp3Δ cells. Pfk1, Tdh2, and Tdh3 substantially decreased in ubp3Δ (but not Eno1 and Eno2) (Figure 2B, Figure 2—figure supplement 1A). Since DUBs can control protein amounts by regulating proteasomal degradation, we asked if the decrease in Pfk1, Tdh2, and Tdh3 in ubp3Δ is due to proteasomal degradation. To test this, we measured the levels of these enzymes in ubp3Δ after treatment with proteasomal inhibitor MG132. We did not observe any rescue in the levels of these enzymes in MG132-treated samples, suggesting that the decreased levels of these enzymes were not due to increased proteasomal degradation (Figure 2—figure supplement 1B). To further understand if these enzyme transcripts are altered in ubp3Δ, we measured the mRNA levels of PFK1, TDH2, and TDH3 in WT, ubp3Δ, and Ubp3C469A cells. The transcripts of all the three genes in ubp3Δ and Ubp3C469A cells decreased (Figure 2—figure supplement 1C), suggesting that Ubp3 regulates the transcripts of these glycolytic enzyme genes. The reduction in the Pfk and GAPDH enzyme amounts was intriguing, because the Pfk and GAPDH steps are critical in determining glycolytic flux (Nelson et al., 2008; Shestov et al., 2014). A reduction in these enzymes could therefore decrease glycolytic flux, and would reroute glucose (G6P) allocations via mass action toward other branches of glucose metabolism, primarily the PPP as well as trehalose biosynthesis (Figure 2A). To assess this, we first measured the steady-state levels of key glycolytic and PPP intermediates, and trehalose in WT or ubp3Δ using targeted LC-MS/MS (Figure 2C, Figure 2—figure supplement 1D). Glucose-6/fructose-6 phosphate (G6P/F6P) increased in ubp3Δ (Figure 2C). Concurrently, trehalose, and the PPP intermediates ribose 5-phosphate (R5P) and sedoheptulose 7-phosphate (S7P), increased in ubp3Δ (Figure 2C).
Figure 2 with 2 supplements see all
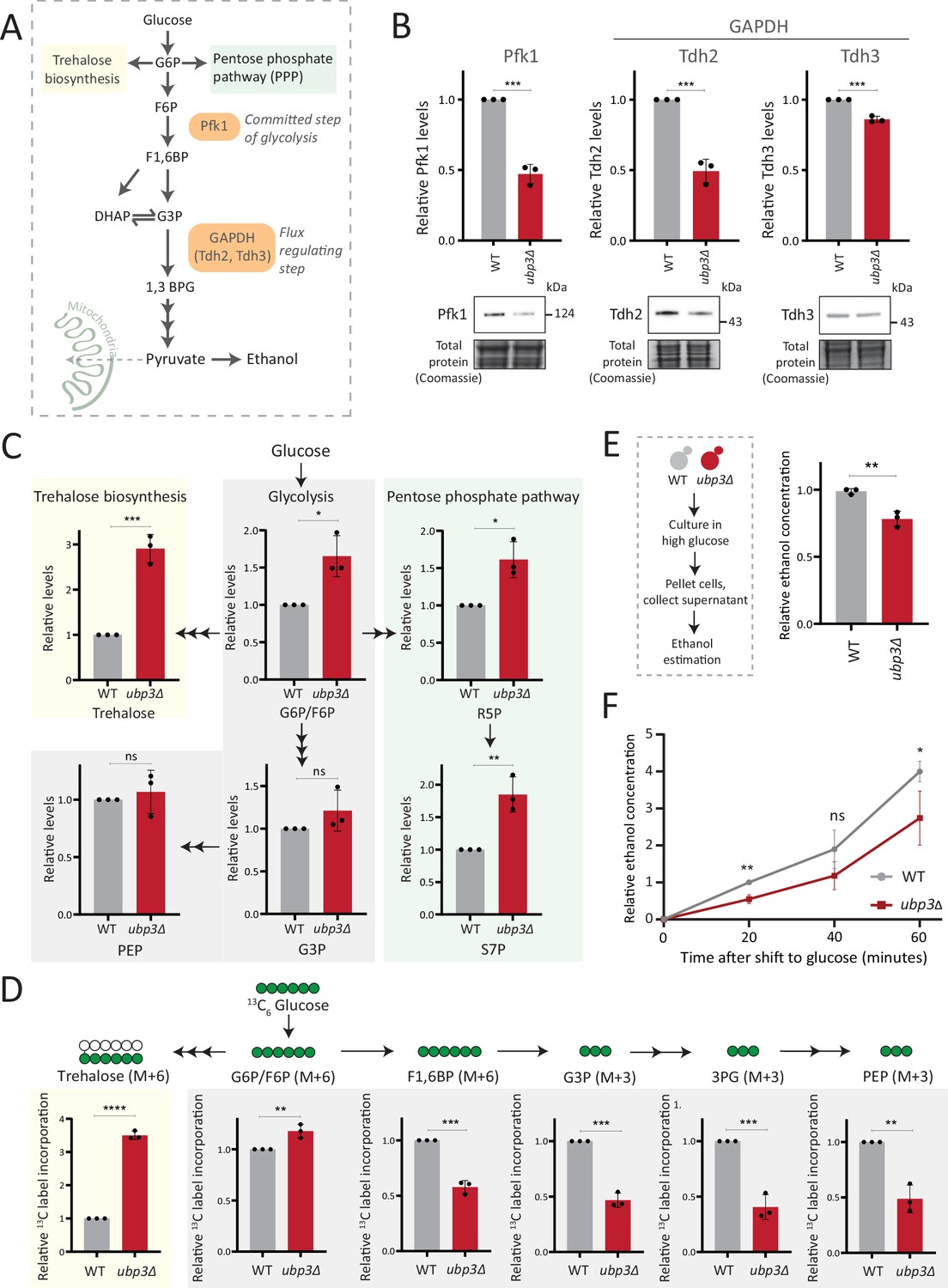
Key glycolytic enzymes decrease and glucose flux is rerouted in ubp3Δ cells.
(A) A schematic illustrating directions of glucose-6 phosphate (G6P) flux in cells. Glucose is converted to G6P, a precursor for trehalose, the pentose phosphate pathway (PPP), and glycolysis. (B) Effect of loss of Ubp3 on key glycolytic enzymes. Wild-type (WT) and ubp3Δ were grown in high glucose and the Pfk1, Tdh2, and Tdh3 levels were measured by western blot using an anti-FLAG antibody. A representative blot (out of three biological replicates, n=3) and their quantification are shown. Data represent mean ± SD. Also see Figure 2—figure supplement 1A. (C) Steady-state metabolite amounts in WT and ubp3Δ in high glucose. Relative steady-state levels of trehalose, major glycolytic, and PPP intermediates were estimated in WT and ubp3Δ. Data represent mean ± SD from three biological replicates (n=3). Also see Appendix 1—table 3. (D) Relative glycolytic and trehalose synthesis flux in WT and ubp3Δ. Relative 13C-label incorporation into trehalose and glycolytic intermediates, after a pulse of 1% 13C6 glucose is shown. Data represent mean ± SD from three biological replicates (n=3). Also see Appendix 1—table 3, Figure 2—figure supplement 1D and E. (E) Ethanol production in ubp3Δ. WT and ubp3Δ were grown in high glucose and ethanol in the media was measured. Data represent mean ± SD from three biological replicates (n=3). (F) Relative rate of ethanol production in WT vs ubp3Δ. WT and ubp3Δ were grown in high glucose (to OD600~0.6), equal numbers of cells were shifted to fresh medium (high glucose) and ethanol concentration in the medium was measured temporally. Data represent mean ± SD from three biological replicates (n=3). Data information: *p<0.05, **p<0.01, ***p<0.001.
-
Figure 2—source data 1
Uncropped and labeled gels and blots for Figure 2.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig2-data1-v1.zip
-
Figure 2—source data 2
Raw unedited gels and blots for Figure 2.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig2-data2-v1.zip
Since steady-state metabolite amounts cannot separate production from utilization, in order to unambiguously assess if glycolytic flux is reduced in ubp3Δ cells, we utilized a pulse labeling of 13C6 glucose, following which the label incorporation into glycolytic and other intermediates was measured. Note that because glycolytic flux is very high in yeast, this experiment would require rapid pulsing and extraction of metabolites in order to stay in a linear range and avoid label saturation. We therefore established a very short time point of label addition, quenching and metabolite extraction post 13C glucose pulse. Since flux saturates/reaches steady state in seconds, we first ensured that the label incorporation into individual metabolites after the 13C glucose pulse was in the linear range, and for early glycolytic intermediates this was seconds after glucose addition. This new methodology is extensively described in Materials and methods, with required controls shown in Figure 2—figure supplement 1F. WT and ubp3Δ cells were grown in high glucose, pulsed with 13C6 glucose, and the relative 13C label incorporation into glycolytic intermediates and trehalose were measured, as shown in the schematic (Figure 2—figure supplement 1E). In ubp3Δ, 13C label incorporation into G6P/F6P as well as trehalose substantially increased (Figure 2D). Contrastingly, 13C label incorporation into glycolytic intermediates F1,6BP, G3P, 3PG, and PEP decreased, indicating decreased glycolytic flux (Figure 2D). We next measured ethanol concentrations and production rates, as an additional output of relative glycolytic rates. We observed decreased steady-state ethanol levels, as well as ethanol production rates in ubp3Δ (Figure 2F).
Glycolysis-derived pyruvate is transported to mitochondria and fuels the trichloroacetic acid (TCA) cycle. Therefore, we asked if the decreased glycolytic rate result in a decrease in the TCA cycle flux as well. To test this, we first measured the steady-state levels of TCA cycle intermediates in WT or ubp3Δ using targeted LC-MS/MS. We did not observe any significant change in the levels of TCA cycle intermediates in ubp3Δ, except malate which showed a significant decrease in ubp3Δ (Figure 2—figure supplement 2A). Next, in order to assess if TCA cycle flux reduces in ubp3Δ cells, WT and ubp3Δ cells were grown in high glucose, pulsed with 13C6 glucose, and the relative 13C label incorporation into TCA cycle intermediates was measured, as shown in the schematic (Figure 2—figure supplement 2B). The kinetics of 13C label incorporation in TCA cycle intermediates are shown in Figure 2—figure supplement 2C. We did not observe any significant change in the relative 13C label incorporation in TCA cycle intermediates in ubp3Δ (Figure 2—figure supplement 2D). Therefore, these data suggest that the decreased glycolytic flux in ubp3Δ does not result in a decrease in TCA cycle flux. The increased respiration and mitochondrial activity in ubp3Δ cells is therefore driven via other factors.
Collectively, these results reveal that that reduced Pfk1 and GAPDH (in ubp3Δ) decrease glucose flux via glycolysis, which results in rewired glucose flux toward trehalose biosynthesis and the PPP.
Rerouted glucose flux results in phosphate (Pi) accumulation
We therefore asked if the proteomic state, as observed in ubp3Δ cells, could provide clues to explain the coupling between mitochondrial derepression and rerouted glucose flux. A recent study by Isasa et al. had systematically quantified the changes in protein levels in ubp3Δ cells (Isasa et al., 2015). We therefore reanalyzed this extensive dataset, looking for changes in proteins that would correlate with these metabolic processes. Notably, we observed increased levels of proteins of the mitochondrial ETC and respiration, and decreased amounts of glucose metabolizing enzymes in ubp3Δ (Figure 3A). Additionally, multiple proteins involved in regulating phosphate (Pi) homeostasis were decreased in ubp3Δ (Figure 3A; Isasa et al., 2015). We had recently uncovered a reciprocal coupling of Pi homeostasis with the different arms of glucose metabolism, particularly trehalose biosynthesis (Gupta et al., 2019; Gupta and Laxman, 2021), and therefore wondered if a glycolytic flux-dependent change in Pi homeostasis had any role in mitochondrial respiration. Our hypothesis was refined based on the reasoning given below.
Figure 3 with 1 supplement see all
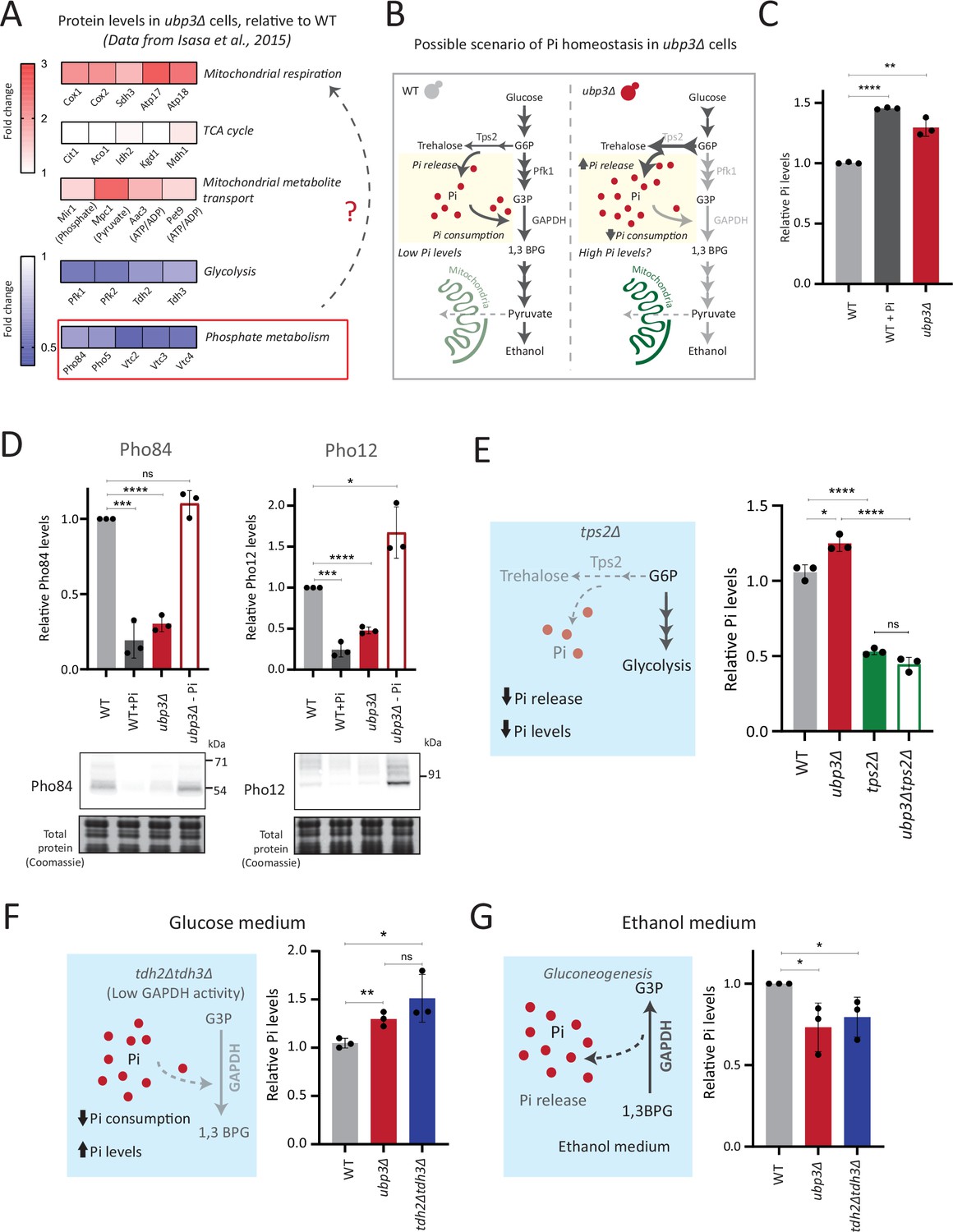
Rerouted glucose flux results in inorganic phosphate (Pi) accumulation.
(A) Changes in protein levels in ubp3Δ (dataset from Isasa et al., 2015). ubp3Δ cells have an increase in proteins involved in mitochondrial respiration and decrease in proteins involved in glucose and phosphate metabolism. (B) Schematic showing maintenance of Pi balance during glycolysis. Trehalose synthesis from glucose-6 phosphate (G6P) releases Pi, and the conversion of G3P to 1,3BPG by GAPDH consumes Pi. In ubp3Δ, trehalose biosynthesis (which releases Pi) increases. ubp3Δ have decreased GAPDH, which will decrease Pi consumption. This increase in Pi release along with decreased Pi consumption could increase cytosolic Pi. (C) Intracellular Pi levels in wild-type (WT) and ubp3Δ. WT and ubp3Δ were grown in high glucose and the total free phosphate (Pi) levels were estimated. WT in high Pi (2% glucose, 10 mM Pi) was a positive control. Data represent mean ± SD from three biological replicates (n=3). Also see Figure 3—figure supplement 1A. (D) Pho regulon responses in WT and ubp3Δ. Protein levels of Pho84-FLAG and Pho12-FLAG were compared between WT grown in high glucose and in high Pi, ubp3Δ in high glucose with or without a shift to a no-Pi medium for 1 hr, by western blot. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. (E) Contribution of trehalose synthesis as a Pi source. WT, tps2Δ, ubp3Δ, and ubp3Δtps2Δ were grown in high glucose and the total Pi levels were estimated. Data represent mean ± SD from three biological replicates (n=3). Also see Figure 3—figure supplement 1B. (F) Loss of GAPDH isozymes Tdh2 and Tdh3 and effect on Pi. WT, ubp3Δ, and tdh2Δtdh3Δ were grown in high glucose and total Pi was estimated. Data represent mean ± SD from three biological replicates (n=3). (G) Pi levels in ubp3Δ and tdh2Δtdh3Δ cells in ethanol medium. WT, ubp3Δ, and tdh2Δtdh3Δ cells were grown in ethanol medium and the total Pi levels were estimated. Data represent mean ± SD from three biological replicates (n=3). Data information: *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001.
-
Figure 3—source data 1
Uncropped and labeled gels and blots for Figure 3.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig3-data1-v1.zip
-
Figure 3—source data 2
Raw unedited gels and blots for Figure 3.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig3-data2-v1.zip
One explanation for mitochondrial repression can be an internal competition for shared metabolites/co-factors between (cytosolic) glycolytic and mitochondrial processes, that mitochondria might not be sufficiently able to access (Diaz-Ruiz et al., 2011; Koobs, 1972; Rodríguez-Enríquez et al., 2001). In this context, a plausible role for inorganic phosphate (Pi) in regulating mitochondrial repression can be hypothesized. Cytosolic glycolysis requires rapid, high consumption of net Pi (Mason et al., 1981; Rodríguez-Enríquez et al., 2001; van Heerden et al., 2014), and this could possibly limit the Pi that is continuously available for mitochondrial use (Brazy et al., 1982; Koobs, 1972), thereby repressing mitochondria. Contextually, the balance between reactions releasing vs consuming Pi could explain changes in global Pi levels (Gupta and Laxman, 2021). Glycolysis is a continuous hub of Pi consumption. In glycolysis, GAPDH catalyzes G3P to 1,3BPG, converting ADP to ATP, while concurrently consuming a molecule of Pi (Hohmann et al., 1996; van Heerden et al., 2014). This Pi that goes into ATP will subsequently be used for nucleotide biosynthesis, polyphosphate biosynthesis, and protein phosphorylation (Gupta and Laxman, 2021; Hunter, 2012). Therefore, we can surmise that in high glycolytic flux, the production of ATP, nucleotides, and polyphosphates is concurrent with Pi consumption (Austin and Mayer, 2020; Hohmann et al., 1996; Ljungdahl and Daignan-Fornier, 2012). Could this reaction therefore limit cytosolic Pi for the mitochondria (as illustrated in Figure 3B)? Notably, ubp3Δ have reduced GAPDH levels and decreased glycolytic flux. Second, trehalose synthesis is a Pi-releasing reaction, and a major source of free Pi that is critical for Pi homeostasis (Gupta et al., 2019; van Heerden et al., 2014). Flux through this reaction is also substantially higher in ubp3Δ. Therefore, these cells might have increased Pi release (via trehalose), coupled with decreased Pi consumption (via decreased GAPDH). We therefore asked if total Pi increases in ubp3Δ (Figure 3B)?
To test this, we directly assessed total Pi levels in ubp3Δ and WT. ubp3Δ cells had higher Pi than WT, and this was comparable to Pi in WT grown in excess Pi (Figure 3C). Similarly, Pi amounts also increased in Ubp3C469A (Figure 3—figure supplement 1A). Therefore, the loss of Ubp3 increases intracellular Pi levels. Next, we asked if ubp3Δ cells exhibit signatures of a ‘high Pi’ state. S. cerevisiae maintains internal Pi balance by controlling the expression of multiple genes collectively known as the Pho regulon (Mouillon and Persson, 2006). The Pho regulon is induced under Pi limitation, and repressed during Pi sufficiency (Gupta et al., 2019; Mouillon and Persson, 2006). We assessed two major Pho proteins (Pho84: a high-affinity membrane Pi transporter, and Pho12: an acid phosphatase) in WT and ubp3Δ in high glucose. ubp3Δ cells have lower amounts of Pho84 and Pho12 (Figure 3D). Further, upon shifting to low Pi for 1 hr, Pho84 and Pho12 increased in ubp3Δ. These data suggest that reduced Pho84 and Pho12 amounts in ubp3Δ are because of increased Pi, and not due to altered Pho regulon function itself (Figure 3D). These data clarify earlier observations from ubp3Δ which noted reduced Pho proteins (Isasa et al., 2015). Therefore, ubp3Δ constitutively have higher Pi, and likely a consequent decrease in Pho proteins.
We next asked if the increased Pi in ubp3Δ is because of altered G6P allocations toward different end-points, particularly trehalose synthesis, which can be a major node of Pi restoration (Gupta et al., 2019; van Heerden et al., 2014). We assessed the contribution of increased trehalose synthesis toward the high Pi in ubp3Δ, by estimating Pi levels in the absence of trehalose 6-phosphate phosphatase (Tps2), which catalyzes the Pi-releasing step in trehalose synthesis. Notably, loss of Tps2 in ubp3Δ decreased Pi (Figure 3E). There was no additive difference in Pi between tps2Δ and ubp3Δtps2Δ (Figure 3E). As an added control, we assessed trehalose in WT and ubp3Δ in the absence of Tps2, and found no difference (Figure 3—figure supplement 1B). Therefore, increasing G6P flux toward trehalose biosynthesis is a major source of the increased Pi in ubp3Δ.
Since the major GAPDH isozymes, Tdh2 and Tdh3, are reduced in ubp3Δ, we directly asked if reducing GAPDH can decrease Pi consumption and increase Pi. To assess this, we generated tdh2Δtdh3Δ cells, which exhibit a growth defect, but are viable, permitting further analysis. tdh2Δtdh3Δ had higher Pi in high glucose (Figure 3F). Expectedly, we observed a decrease in ethanol in tdh2Δtdh3Δ (Figure 3—figure supplement 1C), along with an accumulation of F1,6BP, and G3P, and decreased 3PG and PEP (Figure 3—figure supplement 1D). However, G6P and trehalose levels between WT and tdh2Δtdh3Δ were comparable (Figure 3—figure supplement 1D and E). These data suggest that unlike in ubp3Δ, the increased Pi in tdh2Δtdh3Δ comes mainly from decreased Pi consumption (GAPDH step). To further assess the role of reduced glycolytic flux in increasing Pi (ubp3Δ and tdh2Δtdh3Δ), we measured the Pi in these cells growing in a gluconeogenic medium - 2%, ethanol. In this scenario, the GAPDH catalyzed reaction will be reversed, converting 1,3BPG to G3P, which should release and not consume Pi. Compared to WT, the Pi levels decreased in ubp3Δ and tdh2Δtdh3Δ (Figure 3G), suggesting that the changes in Pi in these mutants are driven by the relative change in Pi release vs consumption.
These results collectively indicate that the combined effect of increased Pi release coming from trehalose synthesis and decreased Pi consumption from reduced GAPDH increase Pi levels in ubp3Δ.
Mitochondrial Pi availability correlates with mitochondrial activity in ubp3Δ
We therefore wondered if this observed Pi increase from the combined rewiring of glucose metabolism resulted in more Pi becoming accessible to the mitochondria. This would effectively result in Pi budgeting between cytosolic glycolysis and mitochondria based on the relative flux in different arms of glucose metabolism, by increasing Pi availability for the mitochondria (Figure 4A). If this were indeed so, a prediction would be that the pool of Pi in the mitochondria of ubp3Δ cells would be higher than WT cells, while the opposite would be expected in the cytosolic fraction of these cells, due to increased Pi in the mitochondria. To test if this were so, we first measured the cytosolic pools of Pi in WT and ubp3Δ. The cytosolic fraction was isolated (extensive experimental details are in the Appendix 1) from WT and ubp3Δ cells, and Pi levels in this fraction were estimated (Figure 4B, Figure 4—figure supplement 1A). We observed significantly reduced Pi in the cytosolic fraction in ubp3Δ cells. Since total cellular Pi amounts is higher in ubp3Δ cells (Figure 3), a decreased cytosolic Pi would be consistent with greater transport of Pi from cytosol to other organelles such as vacuole (where it is stored as polyphosphate) and/or mitochondria. Therefore, we next asked if the mitochondria in ubp3Δ have correspondingly increased Pi. Mitochondria were isolated by immunoprecipitation from WT and ubp3Δ, the isolation efficiency was analyzed (Figure 4—figure supplement 1A), and relative Pi levels compared. Pi levels were normalized to Idh1 (isocitrate dehydrogenase) in isolated mitochondria, since Idh1 protein levels did not decrease in ubp3Δ (Figure 4—figure supplement 1B), and consistent with another study (Isasa et al., 2015). Mitochondrial Pi was higher in ubp3Δ (Figure 4B). We next asked if the increased mitochondrial activity in ubp3Δ is a consequence of high Pi. If this were so, tdh2Δtdh3Δ should partially phenocopy ubp3Δ with respect to mitochondrial activity. Consistently, tdh2Δtdh3Δ have higher mitotracker intensity as well as Cox2 levels compared to WT (Figure 4—figure supplement 1C, Figure 4C). Also consistent with this, a high basal OCR in tdh2Δtdh3Δ was observed (Figure 4D), together indicating high mitochondrial activity in tdh2Δtdh3Δ, which is comparable to ubp3Δ.
Figure 4 with 1 supplement see all
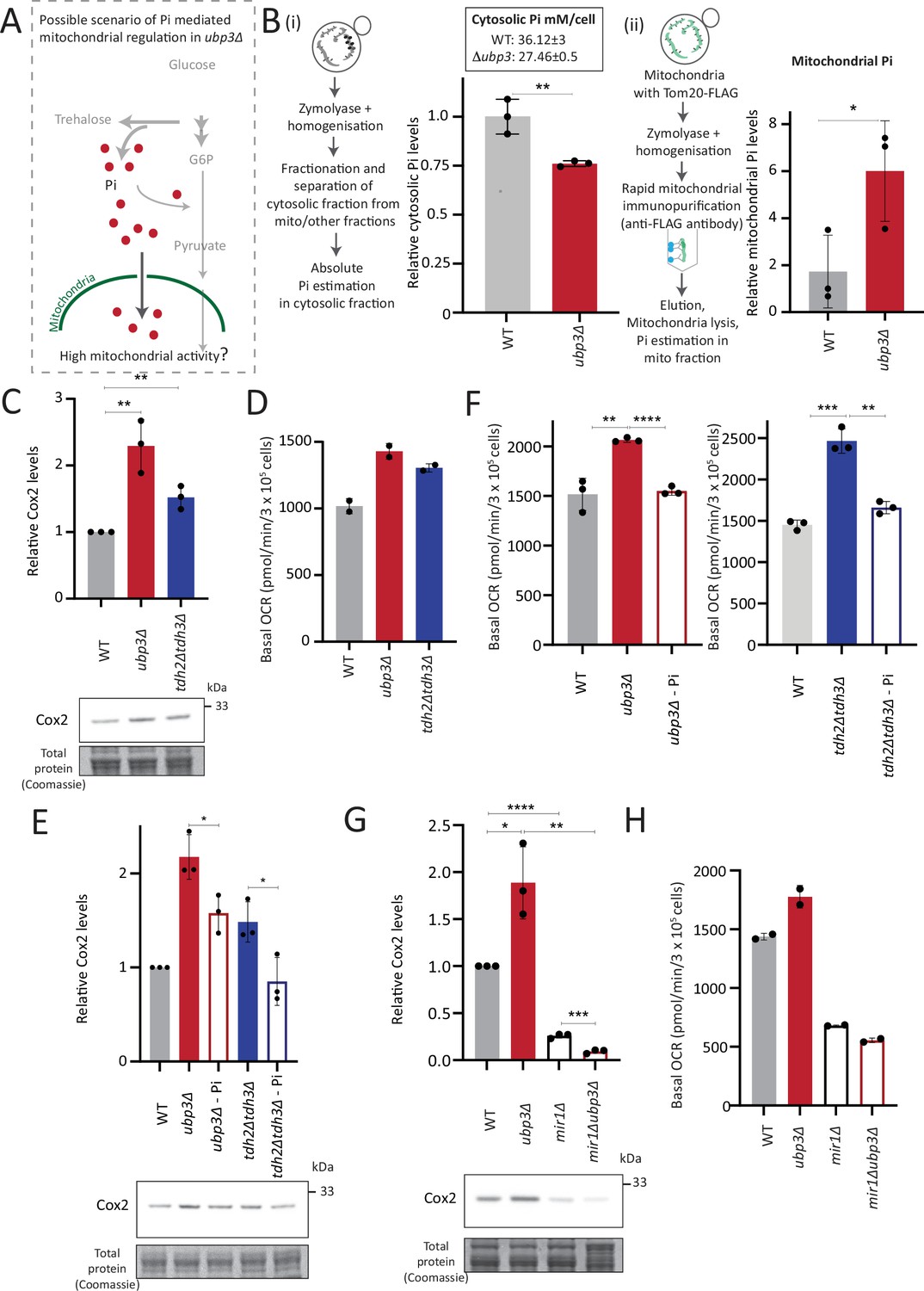
Mitochondrial inorganic phosphate (Pi) availability correlates with mitochondrial activity in ubp3Δ.
(A) A hypothetical mechanism of cytosolic free Pi controlling mitochondrial activity by regulating mitochondrial Pi availability. (B) Cytosolic and mitochondrial Pi amounts in wild-type (WT) vs ubp3Δ. The cytosolic fraction was isolated by centrifugation (see Appendix 1), and in separate experiments, mitochondria were isolated by immunoprecipitation from WT and ubp3Δ and mitochondrial Pi estimated. (i) Cytosolic Pi levels (relative as well as absolute) and (ii) mitochondrial Pi levels (normalized to Idh1) are shown. Data represent mean ± SD from three biological replicates (n=3) respectively for the cytosolic and mitochondrial measurements. Also see Figure 4—figure supplement 1A and B. (C) Cox2 protein in tdh2Δtdh3Δ. WT, ubp3Δ, and tdh2Δtdh3Δ were grown in high glucose and Cox2 protein was estimated. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. (D) Basal oxygen consumption rate (OCR) levels in tdh2Δtdh3Δ. WT, ubp3Δ, and tdh2Δtdh3Δ were grown in high glucose and basal OCR was measured from two independent experiments (n=2). Data represent mean ± SD. Also see Figure 4—figure supplement 1C. (E) Comparative Pi amounts and Cox2 levels in ubp3Δ, tdh2Δtdh3Δ, WT cells. WT cells were grown in high glucose, ubp3Δ and tdh2Δtdh3Δ were grown in high glucose and low Pi, and Cox2 protein was estimated. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. Also see Figure 4—figure supplement 1D and F. (F) Pi amounts and basal OCR in ubp3Δ and tdh2Δtdh3Δ vs WT cells. WT cells were grown in high glucose, ubp3Δ and tdh2Δtdh3Δ were grown in high glucose and low Pi, and basal OCR was measured from three independent experiments (n=3). Data represent mean ± SD. (G) Effect of loss of mitochondrial Pi transporter Mir1 on Cox2 protein. WT, ubp3Δ, mir1Δ, and mir1Δubp3Δ were grown in high glucose and Cox2 amounts compared. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. (H) Relationship of mitochondrial Pi transport and basal OCR in WT vs ubp3Δ. WT, ubp3Δ, mir1Δ, and mir1Δubp3Δ cells were grown in high glucose and basal OCR was measured from two independent experiments (n=2). Data represent mean ± SD. Data information: *p<0.05, **p<0.01, ****p<0.0001.
-
Figure 4—source data 1
Uncropped and labeled gels and blots for Figure 4.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig4-data1-v1.zip
-
Figure 4—source data 2
Raw unedited gels and blots for Figure 4.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig4-data2-v1.zip
We next asked if higher Pi is necessary to increase mitochondrial activity in ubp3Δ and tdh2Δtdh3Δ. Since glycolysis is defective in ubp3Δ and tdh2Δtdh3Δ, it is necessary to distinguish the effect of high Pi vs. only the effect of low glycolysis in activating mitochondria. Logically, if decreased glycolysis (independent of Pi) is sufficient to activate mitochondria, bringing down the Pi levels in ubp3Δ to that of WT should not affect mitochondrial activity. Notably, ubp3Δ grown in low (1 mM) Pi have Pi levels similar to WT in standard (normal Pi) medium (Figure 4—figure supplement 1D). ubp3Δ grown in low Pi also had decreased ethanol, suggesting reduced glycolysis (Figure 4—figure supplement 1E). Therefore, we used this condition to further understand the role of Pi in inducing mitochondrial activity. Mitotracker fluorescence decreased in both ubp3Δ and tdh2Δtdh3Δ in low Pi (Figure 4—figure supplement 1F). Note: Basal mitotracker fluorescence in WT also decreases in low Pi, which is consistent with a required role of Pi for mitochondrial activity (Figure 4—figure supplement 1F). Similarly, Cox2 levels were reduced in both ubp3Δ and tdh2Δtdh3Δ (Figure 4E). Consistent with both reduced mitotracker intensity and Cox2 levels, basal OCR also decreased in both ubp3Δ and tdh2Δtdh3Δ in low Pi (Figure 4F). As an additional control, we used Rho0 strains (which have no mitochondrial DNA and therefore lack functional ETC) to compare basal OCR. The expectation in these strains is that basal OCR will not change if Pi changes. Consistently, we did not observe any significant difference in basal OCR in WT, ubp3Δ and ubp3Δ in low Pi in a Rho0 strain background (which lacks mitochondrial DNA) (Figure 4—figure supplement 1G). These data collectively suggest that high intracellular Pi is necessary to increase mitochondrial activity in ubp3Δ and tdh2Δtdh3Δ.
Next, we asked how mitochondrial Pi transport regulates mitochondrial activity. Mir1 and Pic2 are mitochondrial Pi transporters, with Mir1 being the major Pi transporter (Murakami et al., 1990; Zara et al., 1996). We first limited mitochondrial Pi availability in ubp3Δ by knocking out MIR1. In mir1Δ, the increased Cox2 observed in ubp3Δ was no longer observed (Figure 4G). Consistent with this, we observed no further increase in the basal OCR in mir1Δubp3Δ compared to mir1Δ (Figure 4H). Furthermore, mir1Δ showed decreased Cox2 as well as basal OCR even in WT cells (Figure 4G and H). These data together suggest that mitochondrial Pi transport is critical for increasing mitochondrial activity in ubp3Δ, and in maintaining basal mitochondrial activity even in high glucose.
As a control, no significant increase in Mir1 and Pic2 was observed in ubp3Δ (Figure 4—figure supplement 1H), suggesting that ubp3Δ do not increase mitochondrial Pi by merely increasing the Pi transporters, but rather by increasing available Pi pools.
Taken together, these data suggest the possibility that the altered Pi homeostasis in ubp3Δ cells increases the mitochondrial Pi pool. This increased mitochondrial Pi pool correlates with increased mitochondrial activity. Decreasing mitochondrial Pi by either reducing total Pi or by reducing mitochondrial Pi transport decreases mitochondrial activity.
Mitochondrial Pi availability constrains mitochondrial activity under high glucose
So far, these data suggest that the cytosolic Pi available for the mitochondria can determine the extent of mitochondrial activity. Therefore, we further investigated if mitochondrial Pi allocation was a necessary constraint for glucose-mediated mitochondrial repression.
To test this, we first asked how important mitochondrial Pi transport was to switch to increased respiration. In WT yeast, low glucose or glycolytic inhibition will result in increased respiration (Broach, 2012). What happens therefore if we restrict mitochondrial Pi in this context? For this, we measured the basal OCR in WT and mir1Δ after switching from high (2%) to low (0.1%) glucose. We observed a significant increase in the basal OCR in WT but not in mir1Δ (Figure 5A). The alternate scenario is after glycolytic inhibition. We assessed the role of mitochondrial Pi in this context, by inhibiting glycolysis using 2-deoxyglucose (2DG). WT, but not mir1Δ, increased their OCR (respiration) upon a 1 hr treatment with 2DG (Figure 5B). Consistent with this, mitotracker fluorescence increased with an increase in 2DG in WT, but not in mir1Δ (Figure 5—figure supplement 1A). We further asked if the mitochondrial Pi transporter itself glucose repressed, and therefore assessed Mir1 amounts in high and low glucose. Mir1 levels are higher upon a shift to low glucose, and in cells grown in 2% ethanol, suggesting that Mir1 is glucose repressed (Figure 5C, Figure 5—figure supplement 1B). These data suggest that mitochondrial Pi transport is necessary for increasing mitochondrial activity after glucose derepression.
Figure 5 with 1 supplement see all
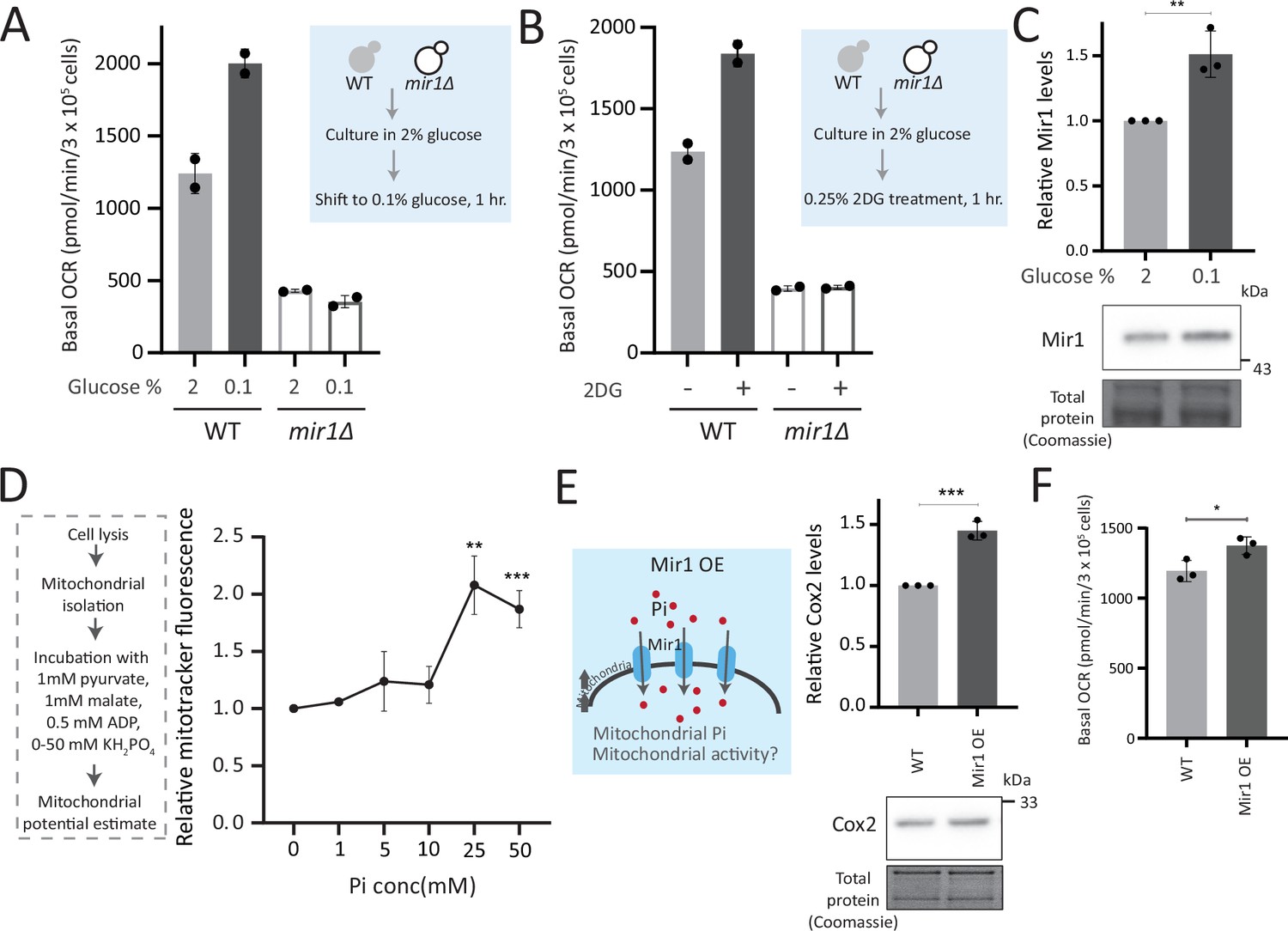
Mitochondrial inorganic phosphate (Pi) availability constrains mitochondrial activity under high glucose.
(A) Relationship of mitochondrial Pi transport and respiration after glucose removal. Wild-type (WT) and mir1Δ cells were cultured in high (2%) glucose and shifted to low (0.1%) glucose for 1 hr. The normalized basal oxygen consumption rate (OCR) from two independent experiments (n=2) are shown. Data represent mean ± SD. (B) Requirement of mitochondrial Pi transport for switch to respiration upon glycolytic inhibition by 2-deoxyglucose (2DG). WT and mir1Δ cells were cultured in high glucose and treated with or without 0.25% 2DG for 1 hr. Basal OCR was measured from two independent experiments (n=2). Data represent mean ± SD. Also see Figure 5—figure supplement 1A. (C) Glucose-dependent regulation of Mir1. Cells (with Mir1-HA) were grown in high glucose and shifted to low glucose (0.1% glucose) for 1 hr, and Mir1 levels compared. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. Also see Figure 5—figure supplement 1B. (D) Increasing Pi concentrations and mitochondrial activity in isolated mitochondria. Mitochondria were isolated from WT cells grown in high glucose, incubated with 1 mM pyruvate, 1 mM malate, 0.5 mM ADP, and 0–50 mM KH2PO4. The mitochondrial activity was estimated by mitotracker fluorescence intensity, and intensities relative to the sample with 0 mM KH2PO4 is shown. Data represent mean ± SD from three biological replicates (n=3). (E) Effect of overexpressing Mir1 on Cox2 protein. WT (containing empty vector) and Mir1 overexpressing (Mir1OE) cells were grown in high glucose and Cox2 levels were estimated. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. Also see Figure 5—figure supplement 1G. (F) Effect of overexpressing Mir1 on basal OCR. The basal OCR in WT (containing empty vector) and Mir1OE in high glucose was measured from three independent experiments (n=3). Data represent mean ± SD. Data information: *p<0.05, **p<0.01, ***p<0.001.
-
Figure 5—source data 1
Uncropped and labeled gels and blots for Figure 5.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig5-data1-v1.zip
-
Figure 5—source data 2
Raw unedited gels and blots for Figure 5.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig5-data2-v1.zip
We next asked whether just adding external Pi was sufficient to increase mitochondrial activity, when cells are in high glucose. In medium supplemented with excess Pi, the internal Pi increases as seen earlier (Figure 3C). Therefore, a simplistic assumption would be that the addition of external Pi to cells in high glucose would also increase mitochondrial Pi. However, an alternate possibility presents itself wherein since glycolytic flux is already high in glucose, supplementing Pi will continue to fuel glycolysis. Indeed, this was originally observed by Harden and Young in 1908, where adding Pi increased fermentation (Harden and Young, 1906). In such a scenario, there could be an increase in the cytosolic Pi but not the mitochondrial Pi. We estimated the cytosolic and mitochondrial Pi in this condition where excess Pi was externally supplemented. Notably, cells grown in high Pi had increased cytosolic Pi, but decreased mitochondrial Pi (Figure 5—figure supplement 1C), without any changes in total mitochondria volume or amounts (Figure 5—figure supplement 1D). Furthermore, directly adding Pi to cells growing in high glucose also decreased basal OCR (Figure 5—figure supplement 1E), consistent with decreased mitochondrial Pi. These data indicate that in high glucose, simply supplementing Pi will not increase Pi access to the mitochondria, and instead results in an accumulation of Pi in the cytosol (Figure 5—figure supplement 1C). We therefore now asked, if we inhibit glycolytic flux and then supplement Pi, what would happen to mitochondrial activity. For this, we treated cells with 2DG, and subsequently added Pi and measured the OCR (Figure 5—figure supplement 1F). In this case, supplementing Pi increased the basal OCR (Figure 5—figure supplement 1F). Collectively, these data suggest that a combination of decreasing glycolysis and increasing Pi can together increase respiration. Next, in order to directly test mitochondrial activation based on external Pi availability, we isolated mitochondria, and estimated activity in vitro upon adding increasing Pi. Mitochondrial activity increased with increased Pi, with maximum activity observed with 25 mM Pi supplemented (Figure 5D). In a complementary experiment, we overexpressed the Mir1 transporter in WT cells, to increase Pi within mitochondria (Figure 5—figure supplement 1G). Mir1-OE cells have higher Cox2 levels and basal OCR (Figure 5E and F). Therefore, increasing Pi transport to mitochondria is sufficient to increase mitochondrial activity in high glucose.
Mitochondrial pyruvate transport is also required for mitochondrial respiration (Timón-Gómez et al., 2013). We asked where Pi availability stands in a hierarchy of constraints for mitochondrial derepression, as compared to mitochondrial pyruvate transport. We measured the amounts of the Mpc3 subunit of the mitochondrial pyruvate carrier (MPC) complex (Bender et al., 2015; Timón-Gómez et al., 2013). Mpc3 protein increases in ubp3Δ (Figure 5—figure supplement 1H). This also correlated with the unimpaired TCA cycle flux (Figure 2—figure supplement 2D) and the increased mitochondrial activity. Interestingly, in ubp3Δ grown in low Pi, Mpc3 further increased (Figure 5—figure supplement 1H), but as shown earlier this condition cannot increase OCR or mitochondrial activity (Figure 4F, Figure 4—figure supplement 1F). Basal Mpc3 levels decrease in mir1Δ, but upon shifting to 0.1% glucose, Mpc3 increases in both WT and mir1Δ, with higher levels in mir1Δ (Figure 5—figure supplement 1I). Therefore, even where Mpc3 is high (ubp3Δ in low Pi, and mir1Δ in low glucose), mitochondrial activity remains low if Pi is restricted (Figure 4F and H). There was also no decrease in basal OCR in mpc3Δ in high glucose, and the basal OCR increased to the same level as of WT after shifting to 0.1% glucose (Figure 5—figure supplement 1J). Since Mpc3 changes with mitochondrial Pi availability (Figure 5—figure supplement 1H), we also measured Mpc3 in the Mir1OE. No further changes in Mpc3 were observed in Mir1OE (Figure 5—figure supplement 1K), indicating that increasing mitochondrial Pi alone need not increase Mpc3. Overall, although Mpc3 levels correlate with decreased glycolysis (ubp3Δ - Figure 5—figure supplement 1H, ubp3Δ in low Pi - Figure 5—figure supplement 1H, low glucose - Figure 5—figure supplement 1I), increased Mpc3 alone cannot increase mitochondrial activity and respiration in the absence of adequate mitochondrial Pi.
Collectively, mitochondrial Pi availability constrains glucose-mediated mitochondrial repression. Increasing available pools of Pi to enter the mitochondria is sufficient to induce mitochondrial activity.
Repression of mitochondrial respiration via Pi budgeting is conserved in Ubp3 mutants across diverse yeast genetic backgrounds
So far, we have identified a role for intracellular Pi budgeting as a constraint for mitochondrial activity under high glucose. These were all carried out using a robust, prototrophic yeast strain from a CEN.PK background. S. cerevisiae however, while Crabtree positive, have tremendous genetic diversity (Peter et al., 2018). We therefore asked if this mitochondrial repression through Pi budgeting (mediated by Ubp3 function) is conserved across other strains of S. cerevsiae as well. To test this, we generated Ubp3 deletion mutants in different genetic backgrounds of S. cerevsiae, including BY4742, W303, and Σ1278. In all these strain backgrounds, we observed a significant increase in mitotracker fluorescence intensity and Cox2 protein levels in ubp3Δ (Figure 6A and B). This suggests the role of Ubp3 as a regulator of mitochondrial repression, independent of the genetic background of the yeast (S. cerevsiae) strain. To further assess if loss of Ubp3 shows a concurrent increase in Pi levels, we measured the total Pi levels in WT and ubp3Δ in a W303 strain background. We observed a significant increase in total Pi levels in ubp3Δ cells in this strain (Figure 6C), similar to what we observed in the CEN.PK strain (Figure 3C). Finally, to test if the altered Pi budgeting regulates the mitochondrial activity in these cells, we measured the basal OCR in WT, ubp3Δ, and ubp3Δ in low (1 mM) Pi medium, in the W303 strain background. Consistent with the increase in mitotracker fluorescence intensity and Cox2 protein levels, we observed a significant increase in the basal OCR in ubp3Δ cells (Figure 6D). This increase was not observed in ubp3Δ in a low Pi medium (Figure 6D). This suggests that the role of altered Pi budgeting in regulating mitochondrial respiration is conserved in other genetic backgrounds of S. cerevisiae.
Figure 6
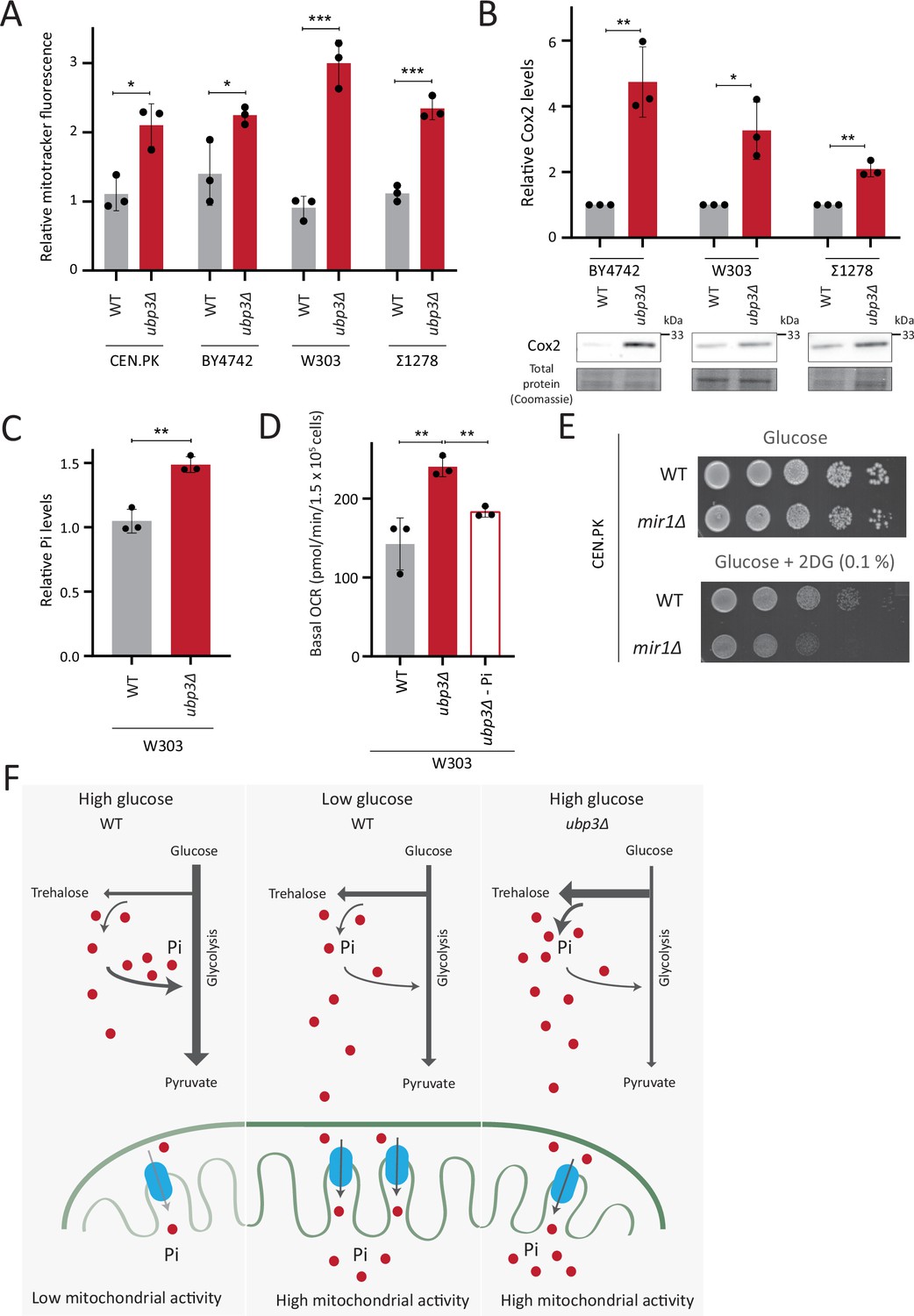
Repression of mitochondrial respiration via inorganic phosphate (Pi) budgeting is conserved in Ubp3 mutants across diverse yeast genetic backgrounds.
(A) Effect of loss of Ubp3 on mitochondrial membrane potential in different yeast strains. Wild-type (WT) and ubp3Δ cells (in CEN.PK as also shown earlier in the manuscript, BY4742, W303, and Σ1278 strains of S. cerevisiae) were grown in high glucose and relative mitochondrial membrane potential was measured. Data represent mean ± SD from three biological replicates (n=3). (B) Effect of loss of Ubp3 on electron transport chain (ETC) complex IV subunit Cox2. WT and ubp3Δ (in BY4742, W303, and Σ1278 strains of S. cerevisiae) were grown in high glucose, and Cox2 was measured. A representative blot (out of three biological replicates, n=3) and their quantifications are shown. Data represent mean ± SD. (C) Intracellular Pi levels in WT and ubp3Δ in W303 strain background. WT and ubp3Δ (in W303 strain background) were grown in high glucose and the total free phosphate (Pi) levels were estimated. Data represent mean ± SD from three biological replicates (n=3). (D) Effect of low Pi on the basal oxygen consumption rate (OCR) in WT and ubp3Δ cells in W303 strain background. WT cells were grown in high glucose and ubp3Δ were grown in high glucose and low Pi, and basal OCR was measured. Data represent mean ± SD (n=3). (E) Requirement of mitochondrial Pi transport for growth after 2-deoxyglucose (2DG) treatment. Shown are serial dilution growth assays in high glucose in the presence and absence of 0.1% 2DG, using WT and mir1Δ cells. The results after 40 hr incubation/30°C are shown. (F) A model illustrating how mitochondrial Pi availability controls mitochondrial activity. In high glucose, the decreased Pi due to high Pi consumption in glycolysis, along with the glucose-mediated repression of mitochondrial Pi transporters, decreases mitochondrial Pi availability. This reduces mitochondrial activity. In low glucose, increased mitochondrial Pi transporters and lower glycolytic flux increases mitochondrial Pi, leading to enhanced mitochondrial activity. In ubp3Δ cells in high glucose, high trehalose synthesis and lower glycolytic flux results in an increase in Pi. This increases mitochondrial Pi availability and thereby the mitochondrial activity. Data information: *p<0.05, **p<0.01, ***p<0.001.
-
Figure 6—source data 1
Uncropped and labeled gels and blots for Figure 6.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig6-data1-v1.zip
-
Figure 6—source data 2
Raw unedited gels and blots for Figure 6.
- https://cdn.elifesciences.org/articles/90293/elife-90293-fig6-data2-v1.zip
Finally, we asked how important mitochondrial Pi transport was for growth, under glycolytic inhibition. Consistent with the requirement for mitochondrial Pi transport to increase mitochondrial respiration upon glycolytic inhibition (Figure 5B), WT cells only exhibit a slightly decreased growth in the presence of 2DG (Figure 6E). In contrast, mir1Δ show a severe growth defect upon 2DG treatment (Figure 6E), revealing a synergetic effect of combining 2DG with inhibiting mitochondrial Pi transport. Therefore, the combined inhibition of glycolysis and mitochondrial Pi transport restricts the growth of glycolytic cells.
Collectively, our data suggests a conserved role for intracellular Pi budgeting in regulating mitochondrial repression in high glucose and the role of mitochondrial Pi transport in regulating adaptation for growth under glycolytic inhibition.
Discussion
In this study, we highlight a role for Pi budgeting between cytosolic glycolysis and mitochondrial processes (which compete for Pi) in constraining mitochondrial repression (Figure 6F). Ubp3 controls this process (Figure 1), by maintaining the amounts of the glycolytic enzymes Pfk1 and GAPDH (Tdh2 and Tdh3) and thereby allows high glycolytic flux. At high fermentation rates, glycolytic enzymes levels at maximal activity maintain high glycolytic rates (effectively following zero-order kinetics), and therefore, changes in the enzyme levels will have a direct, proportionate effect on flux (Grigaitis and Teusink, 2022). The loss of Ubp3 decreases glycolytic flux, resulting in a systems-level, mass-action-based rewiring of glucose metabolism where more G6P is routed toward trehalose synthesis and PPP (Figure 2, Figure 6F). Indeed, inhibiting just phosphofructokinase can reroute glucose flux from glycolysis to PPP (Hollinshead et al., 2016; Miyazawa et al., 2017; Yi et al., 2012), and our study now permits contextualized interpretations of these results. Such reallocations of glucose flux will collectively increase overall Pi, coming from the combined effect of increased Pi release from trehalose synthesis, and decreased Pi consumption via reduced GAPDH. This altered intracellular Pi economy increases Pi pools available to mitochondria, and increasing mitochondrial Pi is necessary and sufficient to increase respiration in high glucose (Figure 4, Figure 5, Figure 6). Mitochondrial Pi transport maintains mitochondrial activity in high glucose, and increases mitochondrial activity in low glucose (Figure 4, Figure 5). Finally, the mitochondrial Pi transporter Mir1 itself decreases in high glucose (Figure 5). Therefore, this glucose-dependent repression of Mir1 also restricts mitochondrial Pi availability (and thereby activity) in high glucose.
Traditionally, loss-of-function mutants of metabolic enzymes are used to understand metabolic state regulation. This approach negates nuanced investigations, since metabolic enzymes are often essential for viability. Furthermore, metabolic pathways have multiple, contextually regulated nodes, through which cells maintain their metabolic state. Therefore, alternate approaches to identify global regulators of metabolic states (as opposed to single enzymes) might uncover ways via which multiple nodes are simultaneously tuned, and can reveal unanticipated systems-level principles of metabolic state rewiring. In this study of glucose-mediated mitochondrial repression, the loss of the DUB Ubp3 decreases glycolytic flux by reducing the enzymes at two critical nodes in the pathway - Pfk1 and GAPDH. Unlike loss-of-function mutants, a reduction in amounts will only rewire metabolic flux. By ‘hitting’ multiple steps in glycolysis simultaneously, ubp3Δ have decreased Pi consumption, as well as increased Pi release. Such a cumulative phenomenon reveals more than inhibiting only GAPDH, where increased Pi comes only from reduced Pi consumption, and not from increased trehalose biosynthesis. Our serendipitous identification of a regulator which regulates multiple steps in glucose metabolism to change the metabolic environment now suggests a general basis of mitochondrial regulation that would have otherwise remained hidden. Separately, finding the substrates of Ubp3 and whether Ubp3 directly regulates glycolytic enzymes are exciting future research questions requiring concurrent innovations in accessible chemical-biological approaches to study DUBs.
Because phosphates are ubiquitous, it is challenging to identify hierarchies of Pi-dependent processes in metabolic state regulation (Gupta and Laxman, 2021). Phosphate transfer reactions are the foundation of metabolism, driving multiple, thermodynamically unfavorable reactions (Kamerlin et al., 2013; Westheimer, 1987). Contextually, the laws of mass action predict that the relative rates of these reactions will regulate overall Pi balance, and contrarily the Pi allocation to Pi-dependent reactions will determine reaction rates (Gupta and Laxman, 2021; van Heerden et al., 2014). Additionally, cells might control Pi allocations for different reactions via compartmentalizing Pi in organelles, to spatially restrict Pi availability (Booth and Guidotti, 1997; Solesio et al., 2021; Vila et al., 2022). Our data collectively suggest a paradigm where the combination of factors regulates mitochondrial Pi and thereby activity. In glycolytic yeast cells growing in high glucose, mitochondrial Pi availability becomes restricted due to higher utilization of Pi in glycolysis compared to mitochondria. Consistent with this, a rapid decrease in Pi upon glucose addition has been observed (Hohmann et al., 1996; Koobs, 1972; Rodríguez-Enríquez et al., 2001). Interesting, in vitro studies with isolated mitochondria from tumor cells also find that decreasing Pi levels decreases respiration (Rodríguez-Enríquez et al., 2001), which would be consistent with this scenario. Further, supplementing Pi correlates with decreased mitochondrial repression in tumors (Brin and Mckee, 1956; Koobs, 1972). By increasing Pi through a systems-level rewiring of glucose metabolism (such as in ubp3Δ cells), cells can collectively increase mitochondrial access to Pi. This Pi budgeting determines mitochondrial activity. Supplementing Pi under conditions of low glycolysis (where mitochondrial Pi transport is enhanced), as well as directly supplementing Pi to isolated mitochondria, increases respiration (Figure 5, Figure 5—figure supplement 1). Notably, this increased respiration does not happen upon directly supplementing Pi to highly glycolytic WT cells, where the Pi increases in cytosol, without increasing mitochondrial Pi (Figure 5—figure supplement 1C). Therefore, in order to derepress mitochondria, a combination of increased Pi along with decreased glycolysis is required. An additional systems-level phenomenon that might regulate Pi transport to the mitochondria is the decrease in cytosolic pH upon decreased glycolysis (Dechant et al., 2010; Orij et al., 2011). The cytosolic pH in highly glycolytic cells is ~7, and decreasing glycolysis results in cytosolic acidification (Dechant et al., 2010; Orij et al., 2011). Therefore, under conditions of decreased glycolysis (2DG treatment, deletion of Ubp3, and decreased GAPDH activity), cytosolic pH becomes acidic. Since mitochondrial Pi transport itself is dependent on the proton gradient, a low cytosolic pH would favor mitochondrial Pi transport (Hamel et al., 2004). Therefore, under conditions of decreased glycolysis (2DG treatment, or loss of Ubp3, or decreased GAPDH activity), where cytosolic pH would be acidic, increasing cytosolic Pi might indirectly increase mitochondria Pi transport, thereby leading to increased respiration. Alternately, increasing mitochondrial Pi transporter amounts can achieve the same result, as seen by overexpressing Mir1 (Figure 5). A similar observation has been reported in Arabidopsis, reiterating an evolutionarily conserved role for mitochondrial Pi in controlling respiration (Jia et al., 2015). Relatedly, glycolytic inhibition can suppress cell proliferation in Warburg-positive tumors (O’Neill et al., 2019; Pelicano et al., 2006), or inflammatory responses (Soto-Heredero et al., 2020). However, these cells survive by switching to mitochondrial respiration (Lu et al., 2015; Shiratori et al., 2019), requiring alternate approaches to prevent their proliferation (Cheng et al., 2012). Inhibiting mitochondrial Pi transport in combination with glycolytic inhibition could restrict the proliferation of Warburg/Crabtree-positive cells. It is important to highlight that our experiments, whether involving Pi supplementation or Pi limitations, maintain the cellular Pi concentration within the millimolar range, and are conducted within a short timeframe (~1 hr). This differs significantly from Pi starvation studies, where cells are subjected to prolonged and complete Pi deprivation. In those contexts, cells trigger extensive metabolic adaptations in order to sustain available Pi pools including an increase in mitochondrial membrane potential which can be independent of respiration (Ouyang et al., 2024).
Since its discovery in the 1920s, the phenomenon of accelerated glycolysis with concurrent mitochondrial repression has been intensely researched. Yet, the biochemical constraints for glucose-mediated mitochondrial repression remains unresolved. One hypothesis suggests that the availability of glycolytic intermediates might determine the extent of mitochondrial repression. F1,6BP inhibits complex III and IV of the ETC in Crabtree-positive yeast (Diaz-Ruiz et al., 2011; Díaz-Ruiz et al., 2008; Hammad et al., 2016; Rosas Lemus et al., 2018). Similarly, the ratio between G6P and F1,6BP regulates the extent of mitochondrial repression (Díaz-Ruiz et al., 2008; Rosas Lemus et al., 2018). Although G6P/F6P accumulates in ubp3Δ (Figure 2C), this is not the case in tdh2Δtdh3Δ (GAPDH mutant) (Figure 3—figure supplement 1D), suggesting that G6P/F6P accumulation in itself is not the criterion to increase mitochondrial activity. Separately, the competition for common metabolites/co-factors between glycolysis and respiration (such as ADP, Pi, or pyruvate) could drive this phenomenon (Diaz-Ruiz et al., 2011; Koobs, 1972). Here, we observe that Mpc3-mediated mitochondrial pyruvate transport alone cannot increase respiration. An additional consideration is the possible contribution of changes in ADP in regulating mitochondrial activity, where the use of ADP in glycolysis might limit mitochondrial ADP. Therefore, when Pi changes as a consequence of glycolysis, it could be imagined that a change in ADP balance can coincidentally occur. However, prior studies show that even though cytosolic ADP decreases in the presence of glucose, this does not limit mitochondrial ADP uptake, or decrease respiration, due to the very high affinity of the mitochondrial ADP transporter (Diaz-Ruiz et al., 2011; Rodríguez-Enríquez et al., 2001). These collectively reiterate the importance of Pi access and transport to mitochondria in constraining mitochondrial respiration. Indeed, this interpretation can also contextually explain observations from other model systems where mitochondrial Pi transport seems to regulate respiration (Scheibye-Knudsen and Quistorff, 2009; Seifert et al., 2015).
We parsimoniously suggest that Pi access to the mitochondria as a key constraint for mitochondrial repression under high glucose. In a hypothetical scenario, a single-step event in evolution, reducing mitochondrial Pi transporter amounts, and/or increasing glycolytic flux (to deplete cytosolic Pi), will result in whole-scale metabolic rewiring to repress mitochondria. More elaborate regulatory events can easily be imagined as subsequent adaptations to enforce mitochondrial repression. Given the central role played by Pi, something as fundamental as access to Pi will constrain mitochondrial repression. Concurrently, the rapid incorporation of Pi into faster glycolysis can give cells a competitive advantage, while also sequestering Pi in the form of usable ATP. Over the course of evolution, this could conceivably drive other regulatory mechanisms to enforce mitochondrial repression, leading to the currently observed complex regulatory networks and signaling programs observed in the Crabtree effect, and other examples of glucose-dependent mitochondrial repression.
Materials and methods
Statistics and graphing
Request a detailed protocolUnless otherwise indicated, statistical significance for all indicated experiments were calculated using unpaired Student’s t-tests (GraphPad Prism 9.0.1). Graphs were plotted using GraphPad Prism 9.0.1.
Yeast strains, media, and growth conditions
Request a detailed protocolA prototrophic CEN.PK strain of S. cerevisiae (WT) (van Dijken et al., 2000) was used unless mentioned otherwise. Strains are listed in Appendix 1—table 1. Gene deletions, chromosomal C terminal-tagged strains were generated by PCR-mediated gene deletion/tagging (Longtine et al., 1998). Mitochondria-targeted mNeon strain (Mito-mNeon green) is described in Dua et al., 2022. The cox2-62 strain is described in Bonnefoy et al., 2001. Media compositions, growth conditions, and CRISPR-Cas9-based mutagenesis are described in Extended methods (Appendix 1).
Mitotracker fluorescence
Request a detailed protocolMitotracker fluorescence was measured using Thermo Varioscan LUX multimode plate reader (579/599 excitation/emission). Detailed protocol is described in Extended methods (Appendix 1). Mitotracker fluorescence were normalized using OD600 of each sample and relative fluorescence intensity calculated.
Protein extraction and western blotting
Request a detailed protocolTotal protein was precipitated, extracted using TCA as described earlier (Vengayil et al., 2019). Blots were quantified using ImageJ software. Detailed protocol is described in Extended methods (Appendix 1).
Basal OCR measurement
Request a detailed protocolThe basal OCR was measured using Agilent Seahorse XFe24 analyzer. Basal OCR readings were normalized for cell number (using OD600 of samples) in each well. The detailed methods are described in Extended methods (Appendix 1).
Mitochondrial volume estimation
Request a detailed protocolHigh-resolution 3D fluorescence experiments were performed on an inverted confocal laser scanning microscope (Carl Zeiss LSM 780 or Olympus FV3000). For each imaging field of view, sequential z-stacks were acquired for each excitation channel. 488 nm laser excitation for mNeonGreen and 561 nm laser excitation for Mitotracker CMXros dye were used respectively. Images taken were deconvolved and analyzed further in ImageJ software with custom-written routines. Mitochondria segmentation and quantification was done using the Mitochondria Analyzer plugin (Chaudhry et al., 2020) in ImageJ. For visualization, maximum intensity projection of 3D images was used.
RNA extraction and RT-qPCR
Request a detailed protocolThe RNA extraction was done using the hot phenol extraction method as described in Vengayil et al., 2019. The isolated RNA was DNase treated, and used for cDNA synthesis. Superscript III reverse transcriptase enzyme (Invitrogen) was used for cDNA synthesis and RT-qPCR was performed using KAPA SYBR FAST qRT PCR kit (KK4602, KAPA Biosystems). Taf10 was used as a control for normalization and the fold change in mRNA levels were calculated by 2-ΔΔct method.
ATP, ethanol, and Pi measurements
Request a detailed protocolATP levels were measured by ATP estimation kit (Thermo Fisher A22066). Ethanol concentration in the medium was estimated using potassium dichromate-based assay described in Sriariyanun et al., 2019, with modifications. Pi was estimated using a malachite green phosphate assay kit (Cayman Chemicals, 10009325). Detailed sample collection and assay protocols are described in Extended methods (Appendix 1).
Metabolite extraction and analysis by LC-MS/MS
Request a detailed protocolThe steady-state levels and relative 13C label incorporation into metabolites were estimated by quantitative LC-MS/MS methods as described in Walvekar et al., 2018. Detailed methodology is extensively described in Extended methods (Appendix 1). Peak area measurements are listed in Supplementary file 1.
Mitochondrial isolation
Request a detailed protocolMitochondria was isolated by immunoprecipitation as described in Chen et al., 2017; Liao et al., 2018, with modifications. The detailed protocol is described in Extended methods (Appendix 1). The eluted mitochondria were used in malachite green assay for Pi estimation, boiled with SDS-glycerol buffer for western blots or incubated with mitotracker CMXROS with mitochondrial activation buffer for mitotracker assays.
Cytosolic fraction isolation
Request a detailed protocolCytosolic fraction was isolated from spheroplasts by centrifugation as described in detail in Appendix 1. The total protein amounts in the cytosolic fraction was estimated by BCA protein estimation assay and the Pi levels were estimated by malachite green assay.
Appendix 1
Extended methods
Media and growth conditions
Media used in this study are high glucose (1% yeast extract, 2% peptone, and 2% glucose), low glucose (1% yeast extract, 2% peptone, and 0.1% glucose), and ethanol (1% yeast extract, 2% peptone, and 2% ethanol). The YPD-low Pi medium was prepared as previously described (Kaneko et al., 1982). Briefly, 1 L YP medium was prepared with 10g yeast extract and 20g peptone in 800ml water. To this, 10 ml of 1M MgS04 and 10ml aqueous ammonia were added and incubated for 30 mins at room temperature (RT), to precipitate inorganic phosphate. The precipitate was filtered and the pH of the clear solution adjusted to 5.8 using HCl, and the volume made up to 1 L. This medium was autoclaved and sterile dextrose was added to prepare the YPD-no Pi medium. The low Pi medium was prepared by adding an indicated concentration of filter-sterilized KH2PO4 solution. For experiments involving shift to low glucose, cells were grown in high glucose (2%) and at OD600 ~0.6, cells were pelleted and shifted to low glucose (0.1%), for one hour. For experiments involving a shift to a no-Pi or low glucose medium, cells were subcultured in high glucose till OD600~0.6, pelleted by centrifugation (1000 x g, 2 min at RT), washed, and shifted to no-Pi or low glucose medium for 1 hour. For experiments involving measurement of basal OCR at different Pi concentrations (Figure 5—figure supplement 1), cells were grown in 5 mM Pi medium (prepared by removing Pi from YPD medium and adding KH2PO4 at the required concentration) and at OD600 ~0.6, the cells were supplemented with additional Pi at required final concentrations. MG132 was used at a final concentration of 100 µM and the experiments involving MG132 addition were performed in pdr5Δ background to prevent MG132 efflux by Pdr5 transporter. Experiments involving heat stress were performed by incubating cells at 42°C for one hour.
CRISPR-Cas9 based mutagenesis
To generate the Ubp3C469A strain, cells were transformed a constitutive Cas9 expressing plasmid (Addgene plasmid 43802, DiCarlo et al., 2013). Guide RNA (gRNA) sequences (forward and reverse) were annealed and cloned into a pMEL13 plasmid. Strains were then transformed with gRNA plasmid and homology repair (HR) fragments with the base pair mutation. The HR fragment was generated by site-directed mutagenesis PCR. Positive clones were selected using drug, and sequenced to confirm the mutation. Oligonucleotides used are listed in Appendix 1—table 2.
Sample preparation for mitotracker fluorescence assay
Cells were grown to OD600~0.6, 1 OD600 cells were collected by centrifugation (1000 x g, 1 min at RT), and resuspended in 1 ml fresh media. Mitotracker CMXRos (M7512 ThermoFisher) was added to a final concentration of 200 nM and incubated at 30°C in a shaking incubator. Cells were washed and resuspended in fresh media and fixed with 2% formaldehyde for 20 minutes. Fixed cells were washed in 1xPBS, pH 7.4, and resuspended in 1 ml 1xPBS. 300 µl of this was aliquoted onto 96 well plates in replicates.
Mitotracker based screen to identify DUBs that regulate mitochondrial repression
The deubiquitinase deletions were made by PCR mediated gene deletion, DUB deletion mutants generated were grown in high glucose and the mitochondrial membrane potential measured by the mitotracker assay described earlier. For performing the screen, the 19 DUB knockouts were divided into two groups- each containing 10 and 9 DUB KOs respectively and the screen was performed separately for each group in replicates. WT cells grown in high glucose indicated the basal mitochondrial potential and WT cells grown in ethanol was used as a positive control for each group. The mitotracker fluorescence intensity were normalised to OD600 of each mutant and the fluorescence intensity relative to the WT cells were calculated. The relative fluorescence intensity for the replicate samples are shown in Figure 1—figure supplement 1A, mean fluorescence intensities of the replicates (relative to WT) were calculated and plotted as heat maps (Figure 1C).
Serial dilution growth assay
For serial dilution-based growth assays, cells were grown in standard high glucose to OD600~0.8, collected by centrifugation (1000 x g, 1 min at RT), washed with water, serial dilutions made (OD600=1, 0.1, 0.01, 0.001, 0.0001), 5 µl of each dilution were spotted onto agar plates, incubated at 30°C and growth monitored.
Protein extraction and western blotting
The cells were grown in the indicated medium to OD600~0.8 and pelleted by centrifugation(1000 x g, 2 min at RT). Total protein was precipitated and extracted using trichloroacetic acid (TCA), and resuspended in SDS/glycerol buffer. The supernatant was collected after centrifugation, and total proteins were estimated by BCA assay (BCA assay kit, G-Biosciences). Protein samples were normalized to ensure the same protein amounts in all the samples, in SDS/glycerol buffer. Samples were resolved on 4–12% bis-tris gels (Invitrogen, NP0336BOX), using MOPS running buffer (50 mM tris, 50 mM MOPS, 0.1% SDS, and 1 mM EDTA), the gels cut so that the relevant portion of the gel is transferred to a nitrocellulose membrane (GE Healthcare, 10600003), while a lower or higher region was stained with coomassie blue for protein normalization, and blots were developed using the following antibodies: anti-HA mouse (Sigma-Aldrich 11583816001), anti-FLAG mouse (Sigma-Aldrich F1804), anti-Cox2 mouse (Invitrogen MTCO2 459150), anti-ubiquitin (P4D1 mouse mAb, CST), anti-Idh1 goat (Sigma-Aldrich SAB2501682). Horseradish peroxidase-conjugated secondary antibody was from Sigma-Aldrich (mouse and rabbit) and Thomas scientific (goat). Chemiluminescence was detected by using Western Bright ECL HRP substrate, Advansta, K12045.
Seahorse assay
1 ml of the XF Calibrant solution was aliquoted into each well of the utility plate and the sensor cartridge plate was hydrated overnight as per the manufacturer’s instructions. The sensor cartridge was loaded with complex IV inhibitor sodium azide and equilibrated in the Seahorse XFe24 analyzer one hour prior to the start of the assay. The culture plate was coated with 50 µl poly-L-lysine and incubated for 1 hour at RT. Excess poly-L-lysine was removed and the plate dried at 30°C for 30 minutes. The cells were grown in standard high glucose to an OD600~0.6. The samples were aliquoted to poly-L-lysine coated wells at a final cell number in each well of ~3x10^5. The plate was centrifuged for 2 min at 100xg (acceleration 2, brake 2) and incubated at 30°C for 30 minutes. The plate was loaded to the Seahorse XFe24 analyzer and basal OCR measured over time. A minimum of three measurements were taken with intermittent 2 min mixing and waiting steps. This was followed by sodium azide injection to the sample wells, and three measurements were taken with intermittent 2 min mixing and waiting steps.
Sample preparation for ATP estimation assay
Cells were grown in high glucose and at OD600 ~0.8, 10 OD600 were pelleted at 4°C. The pellet was treated with 300 µl ice-cold TCA (5%), resuspended and incubated in ice for 15 mins. The suspension was diluted such that the final TCA concentration is 0.1% using 20 mM Tris-HCl, Ph 7.0. For calculating the contribution of mitochondria to total ATP, cells were grown in high glucose and at OD600 ~0.6, sodium azide was added at a final concentration of 1 mM and incubated for 45 minutes at 30°C in a shaking incubator. The reaction mixture for ATP measurements were prepared as per manufacturer’s instructions and luminescence was measured using Sirius luminometer (Tiertek Berthold). The relative ATP concentrations were calculated, graphs were plotted and using GraphPad prism 9.0.1.
Sample preparation for ethanol estimation
Cells were grown in appropriate media conditions to an OD600~1, 10 ml of cells collected, and centrifuged at 3000 x g at 4°C. 5 ml of the supernatant was collected in a fresh tube and 1ml of Tri-n-butyl phosphate (TBP) was added. The mixture was vortexed for 5 minutes and centrifuged at 3420xg for 5 minutes to separate the phases. 500 µl of the top, clear phase was transferred to a fresh tube and 500 µl of potassium dichromate reagent was added (10% w/v of K2Cr2O7 in 5 M of H2SO4), followed by vortexing for 1 minute. The mixture was incubated at RT for 10 minutes and 200 µl of the bottom layer was transferred to a 96 well plate. The absorbance in each well was measured at 595 nm. A standard curve was plotted using medium containing different concentrations of added ethanol, and was used to calculate the ethanol concentration in samples. For calculating the rate of ethanol production, WT and ubp3Δ cells were grown in high glucose and at OD600~0.6, equal number of cells were shifted to fresh high glucose medium. The supernatant from 10ml cells were collected, and ethanol concentration in the medium was estimated at different time points after the shift (normalised to OD600 at each time point, to consider the growth difference between WT and ubp3Δ cells).
Sample preparation for Pi estimation by malachite green assay
The cells were grown to OD600~0.8, pelleted by centrifugation (1000 x g, 2 min at RT), pelleted cells were washed and resuspended in 1 ml ice-cold HPLC grade water (to avoid Pi contamination) and lysed by bead beating (3 x 20s, 1 min intermittent cooling). Lysates were centrifuged (14000 rpm, 1 min at 4°C) and 800 µl of the clear supernatant collected. Protein concentrations of the supernatant were estimated using BCA assay (BCA assay kit, GBiosciences). Lysates corresponding to 1µg total protein was used for the Pi estimation assay.
Metabolite extraction and analysis by LC-MS/MS
Steady state measurements of glycolytic intermediates
For measuring steady-state levels of glycolytic intermediates, trehalose, and PPP intermediates, cells were grown in standard high glucose medium to an OD600~0.8, 5 OD600 cells were collected, quenched in 60% methanol at -40°C, and metabolites were extracted as described in Walvekar et al., 2018. The extracted metabolites were separated by Synergi 4µm Fusion-RP 80 Å LC column (150 x 4.6 mm, Phenomenex) on Waters Acquity UPLC system, and measured as described earlier (Walvekar et al., 2018), in negative polarity mode. Solvents used: 5 mM ammonium acetate in water (solvent A) and 100% acetonitrile (solvent B). ABSciex QTRAP 6500 mass spectrometer was used and data acquisition was done usingAnalyst 1.6.2 (Sciex). Detailed flow parameters are described elsewhere (Walvekar et al., 2018). MultiQuant version 3.0.1 was used for data analysis. The parent and product ion masses used for the analysis are listed in Appendix 1—table 3. The peak areas for individual metabolites were calculated and values were plotted relative to WT. The retention time for different metabolites and peak area obtained after analysis are listed in Supplementary file 1. The statistical significance was calculated using unpaired Student’s t-test (GraphPad prism9.0.1).
Measurement of 13C carbon flux into glycolysis and trehalose
The carbon flux into glycolysis and trehalose was estimated by measuring the relative incorporation of 13C label into glycolytic intermediates and trehalose after a pulse of 13C6 glucose. Cells were grown in high glucose medium with 1% glucose and at OD600~0.6, the cells were shifted to fresh medium with 1% glucose. The OD600 was measured 30 minutes after the shift and cells corresponding to ~5 OD600 were collected, and 13C6 glucose (Cambridge Isotope Laboratories, CLM-1396) was pulsed at a final concentration of 1%, making the total glucose concentration in the medium 2% (1% labelled + 1% unlabelled). Note: Since label saturation into glycolytic intermediates happens instantaneously (within seconds) after 13C glucose pulse, the time points for sample collection should be determined prior to the experiment by plotting total label percentage over time for individual metabolites to calculate the time point at which label saturation takes place. For measuring 13C label incorporation, the cells were collected at 3s (for F1,6BP, G3P, 3PG and PEP), 10s (for G6P/F6P) and 4 mins (for trehalose) after 13C6 glucose pulse, based on the percentage label incorporation plotted in figure S2C. The cells were quenched in 60% methanol at -40°C, and metabolites were extracted as described in Walvekar et al., 2018. The extracted metabolites were detected as described earlier. The relative 13C label incorporation in ubp3Δ cells were measured by calculating the area under the peak for individual 13C labelled metabolites (using MultiQuant version 3.0.1) and dividing it by the peak area of that specific metabolite in the WT sample. The change in 13C labelled metabolites relative to WT samples were plotted. For calculating the total 13C label percentage of a metabolite, the peak area of 13C labelled metabolites were divided by the sum of peak areas of that metabolite (labelled plus unlabelled) and expressed as percentage. The total label percentage was plotted over time for individual metabolites to calculate the time point at which label saturation takes place (Figure S2C). The retention time for different labelled and unlabelled metabolites and peak area obtained after analysis are listed in Supplementary file 1. The statistical significance was calculated using unpaired Student’s t-test (GraphPad prism 9.0.1).
Sample preparation for cytosolic fraction separation and mitochondria isolation by immunoprecipitation
Cells with Tom20 (for mitochondria isolation) and Eno1 (for cytosolic fraction isolation) endogenously tagged at the C terminus with a 3X FLAG epitope tag were grown in 300 ml high glucose medium and at OD600~ 0.8, the cells were pelleted at 1500 x g for 5 minutes at 4°C. The cells were washed in water, pelleted and weighed. The cells were then incubated in Tris-DTT buffer (0.1 M Tris-SO4, 10 mM DTT, pH 9.4) (5 ml/g pellet weight) for 15 mins at 30°C in a shaking incubator. The cells were washed in SEH buffer (0.6 M sorbitol, 20 mM HEPES-KOH, 2mM MgCl2, pH 7.4) (5 ml/g pellet weight) and incubated with Zymolyase 20T (0832092, MP biomedicals) dissolved in SEH buffer for 60 minutes, at 30°C in a shaking incubator. The spheroplasts produced by Zymolyase treatment were collected by centrifugation at 4500 x g for 5 mins at 4°C, washed with ice-cold SEH buffer (5 ml/g pellet weight) and resuspended in ice-cold SEH buffer with protease inhibitors (1 mM PMSF, 0.5 mg/ml pepstatin A and 0.5 µg/ml leupeptin) (5 ml/g pellet weight). The resuspended spheroplasts were homogenised using a Dounce homogeniser, centrifuged at 1500 x g for 5 mins and the supernatant was collected. The supernatant was centrifuged at 12000 x g for 10 mins at 4°C. The supernatant cytosolic fraction was collected and the pellets were resuspended in 700 µl ice-cold SEH buffer to obtain the mitochondria-enriched fraction. To purify mitochondria by immunoprecipitation, the mitochondria-enriched fraction was incubated with 1.5 mg Dynabeads protein G (Invitrogen) conjugated with 10 µg of anti-FLAG antibody (mouse, Sigma-Aldrich F1804), for 60 minutes at 4°C. The beads were separated using a magnet, washed 3 times in ice-cold SEH buffer and the mitochondria were eluted in 200 ul SEH buffer with FLAG peptide (1.5 mg/ml).
Mitotracker assay using isolated mitochondria
For mitotracker assays with isolated mitochondria (Figure 5D), the mitochondria enriched fraction resuspended in SEH buffer was centrifuged twice- 700 x g for 5mins and 1500 x g for 5 mins at 4°C. The supernatant was collected and centrifuged at 12000 x g for 10 mins at 4°C and the pellet was resuspended in ice-cold SEH buffer. The protein concentration of this crude mitochondria was estimated by BCA protein estimation assay and mitochondria corresponding to 5 µg protein was incubated in 200 µl SEH buffer containing 1 mM pyruvate, 1 mM malate, 0.5 mM ADP and 0 – 50 mM KH2PO4 for 30 minutes at 30°C. Mitotracker CMXROS was added at a final concentration of 200 nM, followed by incubation at 30°C for 30 minutes. The fluorescence intensities were measured using Thermo Varioscan LUX multimode plate reader at 579/599 excitation/emission, and fluorescence intensities relative to samples containing 0 mM KH2PO4 was calculated. Statistical significance was calculated using unpaired Student’s t-test (GraphPad prism 9.0.1).
Appendix 1—table 1
List of strains used in this study.
| SL no. | Strain name | Genotype | Description | Source |
|---|---|---|---|---|
| 1 | CEN.PK a (WT) | Mat a | haploid strain of CEN.PK MAT ‘a’ mating type | van Dijken et al., 2000 |
| 2 | DUBs KO strains | CEN.PK a::hphMX6 | Deletion of the individual deubiquitinases (see Figure 1—figure supplement 1A) | This study |
| 3 | Ubp3C469A | CEN.PK a UBP3C469A | Catalytically inactive Ubp3 mutant | This study |
| 4 | Δatp1 | CEN.PK a Δatp1::natMX6 | Deletion of ATP1 gene | This study |
| 5 | Δatp1 Δubp3 | CEN.PK a Δubp3::hphMX6 Δatp1:: natMX6 | Deletion of ATP1 gene in a UBP3 deletion strain | This study |
| 6 | Δatp10 | CEN.PK a Δatp10::natMX6 | Deletion of ATP10 gene | This study |
| 7 | Δatp10 Δubp3 | CEN.PK a Δubp3::hphMX6 Δatp10:: natMX6 | Deletion of ATP10 gene in a UBP3 deletion strain | This study |
| 8 | Pfk1-FLAG | CEN.PK a PFK1 3x FLAG::natMX6 | C terminal tagged Pfk1 | This study |
| 9 | Pfk1-FLAG Δubp3 | CEN.PK a PFK1-3x FLAG::natMX6 Δubp3::hphMX6 | C terminal tagged Pfk1 in a UBP3 deletion strain | This study |
| 10 | Tdh2-FLAG | CEN.PK a TDH2-3x FLAG::natMX6 | C terminal tagged Tdh2 | This study |
| 11 | Tdh2-FLAG Δubp3 | CEN.PK a TDH2-3x FLAG::natMX6 Δubp3::hphMX6 | C terminal tagged Tdh2 in a UBP3 deletion strain | This study |
| 12 | Tdh3-FLAG | CEN.PK a TDH3-3x FLAG::natMX6 | C terminal tagged Tdh3 | This study |
| 13 | Tdh3-FLAG Δubp3 | CEN.PK a TDH3-3x FLAG::natMX6 Δubp3::hphMX6 | C terminal tagged Tdh3 in a UBP3 deletion strain | This study |
| 14 | Eno1-FLAG | CEN.PK a ENO1-3x FLAG::natMX6 | C terminal tagged Eno1 | This study |
| 15 | Eno1-FLAG Δubp3 | CEN.PK a ENO1-3x FLAG::natMX6 Δubp3::hphMX6 | C terminal tagged Eno1 in a UBP3 deletion strain | This study |
| 16 | Eno2-FLAG | CEN.PK a ENO2-3x FLAG::natMX6 | C terminal tagged Eno2 | This study |
| 17 | Eno2-FLAG Δubp3 | CEN.PK a ENO2-3x FLAG::natMX6 Δubp3::hphMX6 | C terminal tagged Eno2 in a UBP3 deletion strain | This study |
| 18 | Δtdh2 Δtdh3 | CEN.PK a Δtdh2::natMX6 Δtdh3:: kanMX6 | Deletion of TDH2 gene in a TDH3 deletion strain | This study |
| 19 | Δtps2 | CEN.PK a Δtps2::hphMX6 | Deletion of TPS2 gene | This study |
| 20 | Δtps2 Δubp3 | CEN.PK a Δtps2::hphMX6 Δubp3::natMX6 | Deletion of UBP3 gene in a TPS2 deletion strain | This study |
| 21 | Mir1-HA | CEN.PK a MIR1-6xHA::natMX6 | C terminal tagged Mir1 | This study |
| 22 | Mir1-HA Δubp3 | CEN.PK a MIR1-6xHA::natMX6 Δubp3::hphMX6 | C terminal tagged Mir1 in a UBP3 deletion strain | This study |
| 23 | Pic2-HA | CEN.PK a PIC2-6xHA::natMX6 | C terminal tagged Pic2 | This study |
| 24 | Pic2-HA Δubp3 | CEN.PK a PIC2-6xHA:: natMX6 Δubp3::hphMX6 | C terminal tagged Pic2 in a UBP3 deletion strain | This study |
| 25 | Δmir1 | CEN.PK a Δmir1::natMX6 | Deletion of Mir1 | This study |
| 26 | Δmir1 Δubp3 | CEN.PK a Δmir1::natMX6 Δubp3::hphMX6 | Deletion of Mir1 in a UBP3 deletion strain | This study |
| 27 | WT+ Empty vector | CEN.PK a pG6PD:: kanMX6 | haploid strain of CEN.PK MAT ‘a’ with an empty vector with kanamycin resistance | This study |
| 28 | Mir1-HA OE | CEN.PK a pG6PD-Mir1-6xHA:: kanMX6 | Mir1-HA overexpression under the constitutive G6PD promoter | This study |
| 29 | Mpc3-FLAG | CEN.PK a MPC3- 3xFLAG::natMX6 | C terminal tagged Mpc3 | This study |
| 30 | Mpc3-FLAG Δubp3 | CEN.PK a MPC3-3xFLAG:: natMX6 Δubp3::hphMX6 | C terminal tagged Mpc3 in a UBP3 deletion strain | This study |
| 31 | Mpc3-FLAG Mir1-HA OE | CEN.PK a MPC3-3xFLAG:: natMX6 pG6PD-Mir1-6xHA:: kanMX6 | C terminal tagged Mpc3 in Mir1-HA overexpression | This study |
| 32 | Δmpc3 | CEN.PK a Δmpc3::natMX6 | Deletion of Mpc3 | This study |
| 33 | Tom20-FLAG | CEN.PK a Tom20-3xFLAG::natMX6 | C terminal tagged Tom20 | This study |
| 34 | Tom20-FLAG Vph1-HA | CEN.PK a Tom20-3xFLAG:: natMX6 Vph1-6xHA:: hphMX6 | C terminal tagged Vph1 in c terminal tagged Tom20 | This study |
| 35 | Mito-Mneon green | CEN.PK a HO::PCYC1-SU9m Neongreen-TCYC1-KanMX6 | Mneon gene with a mitochondria targeted sequence at the N terminus | Dua et al., 2022 |
| 36 | cox2-62 | leu2 Δarg8 ΔURA3 ura3-52 kar1-1 ade2-101 | cox2-62 ρ+, Cox2 with deletion of -295 to +363 relative to AUG | Bonnefoy et al., 2001 |
| 37 | W303 | MAT a leu2-3,-112;his3-11,-15;trp11;ura3-1; ade2-1;can1-100 | WT W303 strain | Ralser et al., 2012 |
| 38 | W303 Δubp3 | MAT a leu2-3,-112;his3-11,-15;trp11;ura3-1; ade2-1;can1-100Δubp3::hphMX6 | Deletion of Ubp3 in W303 | This study |
| 39 | BY4742 | MAT α his3Δ1:leu2Δ0:lys2Δ0: MET15:ura3Δ0 | WT BY4742 strain | Winston et al., 1995 |
| 40 | BY4742 Δubp3 | MAT α his3Δ1:leu2Δ0:lys2Δ0: MET15:ura3Δ0 Δubp3::hphMX6 | Deletion of Ubp3 in BY4742 | This study |
| 41 | Σ1278 | MAT a | WT Σ1278 strain | Isolate via Fink lab |
| 42 | Σ1278 Δubp3 | MAT a ura3-52 Δubp3::hphMX6 | Deletion of Ubp3 in BY4742 | This study |
| 43 | S288C | MAT a | WT S288C strain | This study |
| 44 | Rho0 | CEN.PK Mat a | Lacks mitochondrial DNA, generated using EtBr treatment | This study |
| 45 | Rho0 Δubp3 | CEN.PK Mat a | Deletion of Ubp3 in Rho0 strain | This study |
Appendix 1—table 2
Oligonucleotides used for CRISPR-Cas9 based mutagenesis.
| Ubp3C469A gRNA forward | AGAACTCATAAAACAAATGTgtttt |
| Ubp3C469A gRNAreverse | ACATTTGTTTTATGAGTTCTgatca |
| HR fragment Ubp3C469A forward | CAAAATACCAGTCCATTCCATTATTCCAAGAGGCATAATTAACAGAGCCAACATTGCTTTTATGAGTTCT |
| HR fragment Ubp3C469A reverse | ACGTTAATTACATCAATAAATGGCTTACAGTAGAGTAACACTTGTAACACAGAACTCATAAAAGCAATGT |
Appendix 1—table 3
List of parent ion mass and product ion mass (Q1/Q3) used for detection of metabolites.
| Metabolite | Parent Ion (Q1) mass | Product Ion (Q3) mass | Collision energy (V) | Retention time |
|---|---|---|---|---|
| Glucose 6-phosphate (G6P) / Fructose 6-phosphate (F6P) | 259 | 97 | -20 | 2.68 |
| 13C_G6P/F6P_6 | 265 | 97 | -20 | 2.68 |
| Fructose 1,6-bisphosphate (F16BP) | 339 | 97 | -20 | 2.42 |
| 13C_F16BP_6 | 345 | 97 | -20 | 2.42 |
| Trehalose | 341.3 | 179.3 | -17 | 3.85 |
| 13C_Trehalose_6 | 347.3 | 185.3 | -17 | 3.85 |
| 13C_Trehalose_12 | 353.3 | 185.3 | -17 | 3.85 |
| Ribose 5-phosphate (R5P) | 229 | 97 | -20 | 2.69 |
| Sedoheptulose-7-phosphate (S7P) | 289 | 97 | -20 | 2.68 |
| Glyceraldehyde 3-phosphate (G3P) | 169 | 97 | -20 | 2.65 |
| 13C_G3P_3 | 172 | 97 | -20 | 2.65 |
| Phosphoenol pyruvate (PEP) | 167 | 79 | -12 | 2.45 |
| 13C_PEP_3 | 170 | 79 | -12 | 2.45 |
| 3-phosphoglycerate (3PG) | 185 | 97 | -20 | 2.53 |
| 13C_3PG_3 | 188 | 97 | -20 | 2.53 |
| Pyruvate | 299.1 | 91.1 | 28 | 8.89 |
| Citrate | 508 | 385 | 7 | 8.04 |
| 13C_ Citrate _2 | 510 | 387 | 7 | 8.03 |
| 13C_ Citrate _3 | 511 | 388 | 7 | 8.03 |
| 13C_ Citrate _4 | 512 | 389 | 7 | 7.69 |
| 13C_ Citrate _5 | 513 | 390 | 7 | 7.91 |
| 13C_ Citrate _6 | 514 | 391 | 7 | 8.42 |
| 2-Ketoglutarate (2-KG) | 462 | 339 | 11 | 8.68 |
| 13 C_2-KG_1 | 463 | 340 | 11 | 8.67 |
| 13 C_2-KG_2 | 464 | 341 | 11 | 8.67 |
| 13 C_2-KG_3 | 465 | 342 | 11 | 8.63 |
| 13 C_2-KG_4 | 466 | 343 | 11 | 8.6 |
| 13 C_2-KG_5 | 467 | 344 | 11 | 8.69 |
| Succinate | 329 | 206 | 15 | 8.21 |
| 13C_Succinate_1 | 330 | 207 | 15 | 7.32 |
| 13C_Succinate_2 | 331 | 208 | 15 | 7.32 |
| 13C_Succinate_3 | 332 | 209 | 15 | 6.97 |
| 13C_Succinate_4 | 333 | 210 | 15 | 6.96 |
| Fumarate | 327 | 91.2 | 34 | 7.55 |
| 13C_Fumarate_1 | 328 | 91.2 | 34 | 7.74 |
| 13C_Fumarate_2 | 329 | 91.2 | 34 | 7.32 |
| 13C_Fumarate_3 | 330 | 91.2 | 34 | 7.32 |
| 13C_Fumarate_4 | 331 | 91.2 | 34 | 7.32 |
| Malate | 345 | 91.2 | 33 | 7.09 |
| 13C_Malate_1 | 346 | 91.2 | 33 | 7.08 |
| 13C_Malate_2 | 347 | 91.2 | 33 | 7.08 |
| 13C_Malate_3 | 348 | 91.2 | 33 | 7.2 |
| 13C_Malate_4 | 349 | 91.2 | 33 | 6.61 |
| Oxaloacetate | 448 | 325 | 10 | 9.92 |
| 13C_Oxaloacetate_1 | 449 | 326 | 10 | 8.67 |
| 13C_Oxaloacetate_2 | 450 | 327 | 10 | 8.67 |
| 13C_Oxaloacetate_3 | 451 | 328 | 10 | 8.63 |
| 13C_Oxaloacetate_4 | 452 | 329 | 10 | 8.6 |
Data availability
All data generated or analysed during this study are included in the manuscript and supporting files; all raw data are provided as a supplementary file.
References
-
The emerging facets of non-cancerous warburg effectFrontiers in Endocrinology 8:279.https://doi.org/10.3389/fendo.2017.00279
-
Phosphate transport in yeast vacuolesThe Journal of Biological Chemistry 272:20408–20413.https://doi.org/10.1074/jbc.272.33.20408
-
Metabolic requirement for inorganic phosphate by the rabbit proximal tubule: evidence for a crabtree effectThe Journal of Clinical Investigation 70:53.https://doi.org/10.1172/JCI110603
-
The inhibition of respiration by glucose, fructose, and mannose in the Ehrlich mouse ascites tumorCancer Research 16:364–368.
-
Driving the cell cycle through metabolismAnnual Review of Cell and Developmental Biology 28:59–87.https://doi.org/10.1146/annurev-cellbio-092910-154010
-
A pipeline for multidimensional confocal analysis of mitochondrial morphology, function, and dynamics in pancreatic β-cellsAmerican Journal of Physiology. Endocrinology and Metabolism 318:E87–E101.https://doi.org/10.1152/ajpendo.00457.2019
-
Observations on the carbohydrate metabolism of tumoursThe Biochemical Journal 23:536–545.https://doi.org/10.1042/bj0230536
-
The Crabtree effect: a regulatory system in yeastJournal of General Microbiology 44:149–156.https://doi.org/10.1099/00221287-44-2-149
-
Mitochondrial oxidative phosphorylation is regulated by fructose 1,6-bisphosphate. A possible role in crabtree effect induction?The Journal of Biological Chemistry 283:26948–26955.https://doi.org/10.1074/jbc.M800408200
-
The Warburg and Crabtree effects: On the origin of cancer cell energy metabolism and of yeast glucose repressionBiochimica et Biophysica Acta (BBA) - Bioenergetics 1807:568–576.https://doi.org/10.1016/j.bbabio.2010.08.010
-
Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systemsNucleic Acids Research 41:4336–4343.https://doi.org/10.1093/nar/gkt135
-
DarT-mediated mtDNA damage induces dynamic reorganization and selective segregation of mitochondriaThe Journal of Cell Biology 221:e202205104.https://doi.org/10.1083/jcb.202205104
-
The Crabtree and Warburg effects: do metabolite-induced regulations participate in their induction?Biochimica et Biophysica Acta 1857:1139–1146.https://doi.org/10.1016/j.bbabio.2016.03.034
-
The ubiquitin systemAnnual Review of Biochemistry 67:425–479.https://doi.org/10.1146/annurev.biochem.67.1.425
-
Why nature chose phosphate to modify proteinsPhilosophical Transactions of the Royal Society B 367:2513–2516.https://doi.org/10.1098/rstb.2012.0013
-
Multiplexed, proteome-wide protein expression profiling: yeast deubiquitylating enzyme knockout strainsJournal of Proteome Research 14:5306–5317.https://doi.org/10.1021/acs.jproteome.5b00802
-
Why nature really chose phosphateQuarterly Reviews of Biophysics 46:1–132.https://doi.org/10.1017/S0033583512000157
-
The ubiquitin codeAnnual Review of Biochemistry 81:203–229.https://doi.org/10.1146/annurev-biochem-060310-170328
-
The immunologic Warburg effect: Evidence and therapeutic opportunities in autoimmunityWiley Interdisciplinary Reviews. Systems Biology and Medicine 12:e1486.https://doi.org/10.1002/wsbm.1486
-
The importance of inorganic phosphate in regulation of energy metabolism of Streptococcus lactisThe Journal of Biological Chemistry 256:1861–1866.https://doi.org/10.1016/S0021-9258(19)69886-8
-
Intracellular pH is a tightly controlled signal in yeastBiochimica et Biophysica Acta (BBA) - General Subjects 1810:933–944.https://doi.org/10.1016/j.bbagen.2011.03.011
-
Cancer stem cell theory and the warburg effect, two sides of the same coin?International Journal of Molecular Sciences 15:8893–8930.https://doi.org/10.3390/ijms15058893
-
Glycolysis inhibition for anticancer treatmentOncogene 25:4633–4646.https://doi.org/10.1038/sj.onc.1209597
-
Ubiquitin: structures, functions, mechanismsBiochimica et Biophysica Acta 1695:55–72.https://doi.org/10.1016/j.bbamcr.2004.09.019
-
Enzymic analysis of the crabtree effect in glucose-limited chemostat cultures of Saccharomyces cerevisiaeApplied and Environmental Microbiology 55:468–477.https://doi.org/10.1128/aem.55.2.468-477.1989
-
Multisite control of the crabtree effect in ascites hepatoma cellsEuropean Journal of Biochemistry 268:2512–2519.https://doi.org/10.1046/j.1432-1327.2001.02140.x
-
The role of glycolysis-derived hexose phosphates in the induction of the Crabtree effectJournal of Biological Chemistry 293:12843–12854.https://doi.org/10.1074/jbc.RA118.003672
-
The mitochondrial phosphate carrier: role in oxidative metabolism, calcium handling and mitochondrial diseaseBiochemical and Biophysical Research Communications 464:369–375.https://doi.org/10.1016/j.bbrc.2015.06.031
-
Glycolysis - a key player in the inflammatory responseThe FEBS Journal 287:3350–3369.https://doi.org/10.1111/febs.15327
-
A rapid spectrophotometric method for quantitative determination of ethanol in fermentation productsOriental Journal of Chemistry 35:744–750.https://doi.org/10.13005/ojc/350234
-
Post-translational modifications on yeast carbon metabolism: Regulatory mechanisms beyond transcriptional controlBiochimica et Biophysica Acta (BBA) - General Subjects 1850:620–627.https://doi.org/10.1016/j.bbagen.2014.12.010
-
Energy metabolism regulates stem cell pluripotencyFrontiers in Cell and Developmental Biology 8:87.https://doi.org/10.3389/fcell.2020.00087
-
An interlaboratory comparison of physiological and genetic properties of four Saccharomyces cerevisiae strainsEnzyme and Microbial Technology 26:706–714.https://doi.org/10.1016/S0141-0229(00)00162-9
-
The E3 ubiquitin ligase Pib1 regulates effective gluconeogenic shutdown upon glucose availabilityThe Journal of Biological Chemistry 294:17209–17223.https://doi.org/10.1074/jbc.RA119.009822
-
The metabolism of carcinoma cellsThe Journal of Cancer Research 9:148–163.https://doi.org/10.1158/jcr.1925.148
-
Metabolic regulation of cell growth and proliferationNature Reviews. Molecular Cell Biology 20:436–450.https://doi.org/10.1038/s41580-019-0123-5
Article and author information
Author details
Funding
Wellcome Trust/DBT India Alliance (IA/S/21/2/505922)
- Sunil Laxman
Department of Biotechnology, Ministry of Science and Technology, India (DBT SRNBIOS)
- Sunil Laxman
Department of Science and Technology, Ministry of Science and Technology, India (IF170236)
- Vineeth Vengayil
Department of Science and Technology, Ministry of Science and Technology, India (CRG/2019/004772)
- Sunil Laxman
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication. For the purpose of Open Access, the authors have applied a CC BY public copyright license to any Author Accepted Manuscript version arising from this submission.
Acknowledgements
We thank all SL lab members, Ben Tu, Anand Bachhawat, Vijay Jayaraman for comments and suggestions. We acknowledge the NCBS/inStem/CCAMP mass spectrometry facility for LC-MS/MS support. We thank Aayushee Khanna for help with experiments and Gaurav Singh for help with microscopy. VV acknowledges funding support from DST-INSPIRE fellowship (IF170236) from the Department of Science and Technology (DST), Govt. of India, SN and SA acknowledges intramural funding from the inStem PhD program. SL acknowledges support from the DST SERB CRG grant CRG/2019/004772, a DBT-Wellcome India Alliance Senior Fellowship (IA/S/21/2/505922), and institutional support from inStem.
Version history
- Sent for peer review:
- Preprint posted:
- Reviewed Preprint version 1:
- Reviewed Preprint version 2:
- Reviewed Preprint version 3:
- Version of Record published:
Cite all versions
You can cite all versions using the DOI https://doi.org/10.7554/eLife.90293. This DOI represents all versions, and will always resolve to the latest one.
Copyright
© 2023, Vengayil et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 3,136
- views
-
- 276
- downloads
-
- 14
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 5
- citations for umbrella DOI https://doi.org/10.7554/eLife.90293
-
- 9
- citations for Version of Record https://doi.org/10.7554/eLife.90293.4
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The deubiquitinase Ubp3/Usp10 constrains glucose-mediated mitochondrial repression via phosphate budgeting
eLife 12:RP90293.
https://doi.org/10.7554/eLife.90293.4




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}