The tRNA thiolation-mediated translational control is essential for plant immunity
- Hubei Hongshan Laboratory, China
- Zhengzhou Tobacco Research Institute of CNTC, China
- College of Life Science and Technology, Huazhong Agricultural University, China
- Shenzhen Branch, Guangdong Laboratory for Lingnan Modern Agriculture, China
- Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, China
- Shenzhen Institute of Nutrition and Health, Huazhong Agricultural University, China
- State Key Laboratory for Managing Biotic and Chemical Threats to the Quality and Safety of Agro-products, Institute of Virology and Biotechnology, Zhejiang Academy of Agricultural Sciences, China
- Key Laboratory of Horticultural Plant Biology, Ministry of Education, College of Horticulture and Forestry Sciences, Huazhong Agricultural University, China
Abstract
Plants have evolved sophisticated mechanisms to regulate gene expression to activate immune responses against pathogen infections. However, how the translation system contributes to plant immunity is largely unknown. The evolutionarily conserved thiolation modification of transfer RNA (tRNA) ensures efficient decoding during translation. Here, we show that tRNA thiolation is required for plant immunity in Arabidopsis. We identify a cgb mutant that is hyper-susceptible to the pathogen Pseudomonas syringae. CGB encodes ROL5, a homolog of yeast NCS6 required for tRNA thiolation. ROL5 physically interacts with CTU2, a homolog of yeast NCS2. Mutations in either ROL5 or CTU2 result in loss of tRNA thiolation. Further analyses reveal that both transcriptome and proteome reprogramming during immune responses are compromised in cgb. Notably, the translation of salicylic acid receptor NPR1 is reduced in cgb, resulting in compromised salicylic acid signaling. Our study not only reveals a regulatory mechanism for plant immunity but also uncovers an additional biological function of tRNA thiolation.
Editor's evaluation
This valuable study provides solid evidence for a role of tRNA thiolation in Arabidopsis immunity through genetic, transcriptomic, and proteomic approaches, specifically that the tRNA mcm5s2U modification affects SA signaling through NPR1 translation.
https://doi.org/10.7554/eLife.93517.sa0Introduction
As sessile organisms, plants are frequently infected by different pathogens, which greatly affect plant growth and development, and cause a tremendous loss in agriculture (Jones and Dangl, 2006; Spoel and Dong, 2012; Yan et al., 2013). To defend against pathogens, plants have evolved sophisticated immune mechanisms. One essential immune regulator is the phytohormone salicylic acid (SA), which plays a central role in immune responses (Vlot et al., 2009; Peng et al., 2021; Yan and Dong, 2014; Zhou and Zhang, 2020). Upon pathogen infection, the biosynthesis of SA is dramatically induced. Plants defective in SA biosynthesis or SA signaling are hyper-susceptible to pathogens (Cao et al., 1997; Rekhter et al., 2019). Several independent forward genetic screens revealed that NONEXPRESSER OF PR GENES 1 (NPR1) is a master regulator of SA signaling (Canet et al., 2010; Cao et al., 1997; Ryals et al., 1997; Shah et al., 1997). In the Arabidopsis npr1 mutant, the SA-mediated immune responses are dramatically reduced. Biochemical and structural studies suggested that NPR1 and its homologs NPR3 and NPR4 are SA receptors (Ding et al., 2018; Fu et al., 2012; Kumar et al., 2022; Wang et al., 2020; Wu et al., 2012; Zhou et al., 2023).
Immune responses involve massive changes in gene expression at transcription, post-transcription, translation, and post-translation levels. Compared with other regulatory mechanisms, the translation regulation mechanism is less well studied. Notably, it is reported that both the pattern-triggered immunity (PTI) and effector-triggered immunity (ETI) involve translational reprogramming (Xu et al., 2017; Yoo et al., 2020). And PABP/purine-rich motif was described as an initiation module for PTI-associated translation (Wang et al., 2022) and CDC123, an ATP-grasp protein, is a key activator of ETI-associated translation (Chen et al., 2023b).
During translation, the code information of mRNA is decoded by transfer RNA (tRNA) molecules, which carry different amino acids. In this sense, the tRNA molecules function as deliverers of the building blocks for translation. The decoding efficiency of tRNAs is affected by their abundance and modifications as well as aminoacyl-tRNA synthetases, amino acid abundance, and elongation factors. Interestingly, an emerging regulatory role for tRNA modifications during elongation has been reported (Delaunay et al., 2016; Schaffrath and Leidel, 2017; Torres et al., 2014).
Currently, more than 150 different tRNA modifications have been identified (Agris et al., 2018). Among them, the 5-methoxycarbonylmethyl-2-thiouridine of uridine at wobble nucleotide (mcm5s2U) is highly conserved in all eukaryotes. The mcm5s2U modification is present in the wobble position of tRNA-Lys(UUU), tRNA-Gln(UUG), and tRNA-Glu(UUC) (Huang et al., 2005; Lu et al., 2005; Sen and Ghosh, 1976). In budding yeast (Saccharomyces cerevisiae), the 5-methoxycarbonylmethyl of uridine (mcm5U) is catalyzed by the Elongator protein (ELP) complex and the Trm9/112 complex, whereas thiolation (s2U) is mediated by the ubiquitin-related modifier 1 (URM1) pathway involving URM1, UBA4, NCS2, and NCS6 (Leidel et al., 2009; Nakai et al., 2004; Noma et al., 2009; Zabel et al., 2008). Loss of the mcm5s2U modification causes ribosome pausing at AAA and CAA codons, which results in defective co-translational folding of nascent peptides and protein aggregation, thereby disrupting proteome homeostasis (Nedialkova and Leidel, 2015; Ranjan and Rodnina, 2017; Rezgui et al., 2013). In yeasts, the mcm5s2U modification was reported to regulate cell cycle, DNA damage repair, and abiotic stress responses (Dewez et al., 2008; Jablonowski et al., 2006; Klassen et al., 2017; Leidel et al., 2009; Nedialkova and Leidel, 2015; Zinshteyn and Gilbert, 2013). In humans, loss of the mcm5s2U modification causes numerous disorders including severe developmental defects, neurological diseases, tumorigenesis, and cancer metastasis (Pan, 2018; Shaheen et al., 2019; Simpson et al., 2009; Torres et al., 2014; Waszak et al., 2020). In plants, loss of the mcm5s2U modification was associated with developmental defects and hypersensitivity to heat stress (Leiber et al., 2010; Nakai et al., 2019; Xu et al., 2020). However, it remains unknown whether the mcm5s2U modification is involved in plant immune responses.
In this study, we found that the mcm5s2U modification is required for plant immunity. Transcriptome and proteome analyses revealed that the mcm5s2U modification is essential for the reprogramming of immune-related genes. Especially, the translation of the master immune regulator NPR1 is compromised in the mcm5s2U mutant. Our study not only expands the biological function of tRNA thiolation but also highlights the importance of translation control in plant immunity.
Results
ROL5 is required for plant immunity
In a study to test the disease phenotypes of some transgenic Arabidopsis, we found that one transgenic line was hyper-susceptible to the bacterial pathogen Pseudomonas syringae pv. Maculicola (Psm) ES4326. The disease symptom resembled that of npr1, in which the master immune regulator NPR1 was mutated (Figure 1A and B). We named this line cgb (for Chao Gan Bing; ‘hyper-susceptible to pathogens’ in Chinese). To identify the causal gene of cgb, we sequenced its genome using the next-generation sequencing technology, which revealed that there was a T-DNA insertion in the fourth exon of ROL5 (AT2G44270; Figure 1C). The insertion was confirmed by genotyping analysis (Figure 1D). In the cgb mutant, the transcript of ROL5 was undetectable (Figure 1E), indicating that cgb was a knock-out mutant. To confirm that ROL5 was the CGB gene, we carried out a complementation experiment by transforming ROL5 into the cgb mutant. As shown in Figure 1A and B, the disease phenotype of the complementation line (COM) was similar to that of wild-type (WT). Moreover, we generated another allele of ROL5 mutant, rol5-c, using the CRISPR-Cas9 gene-editing approach (Wang et al., 2015). In rol5-c, a 2 bp deletion in the first exon of ROL5 causes a frameshift (Figure 1C). As expected, the rol5-c mutant was hyper-susceptible to Psm as cgb (Figure 1A and B). These data strongly suggested that ROL5 is required for plant immunity.
Figure 1
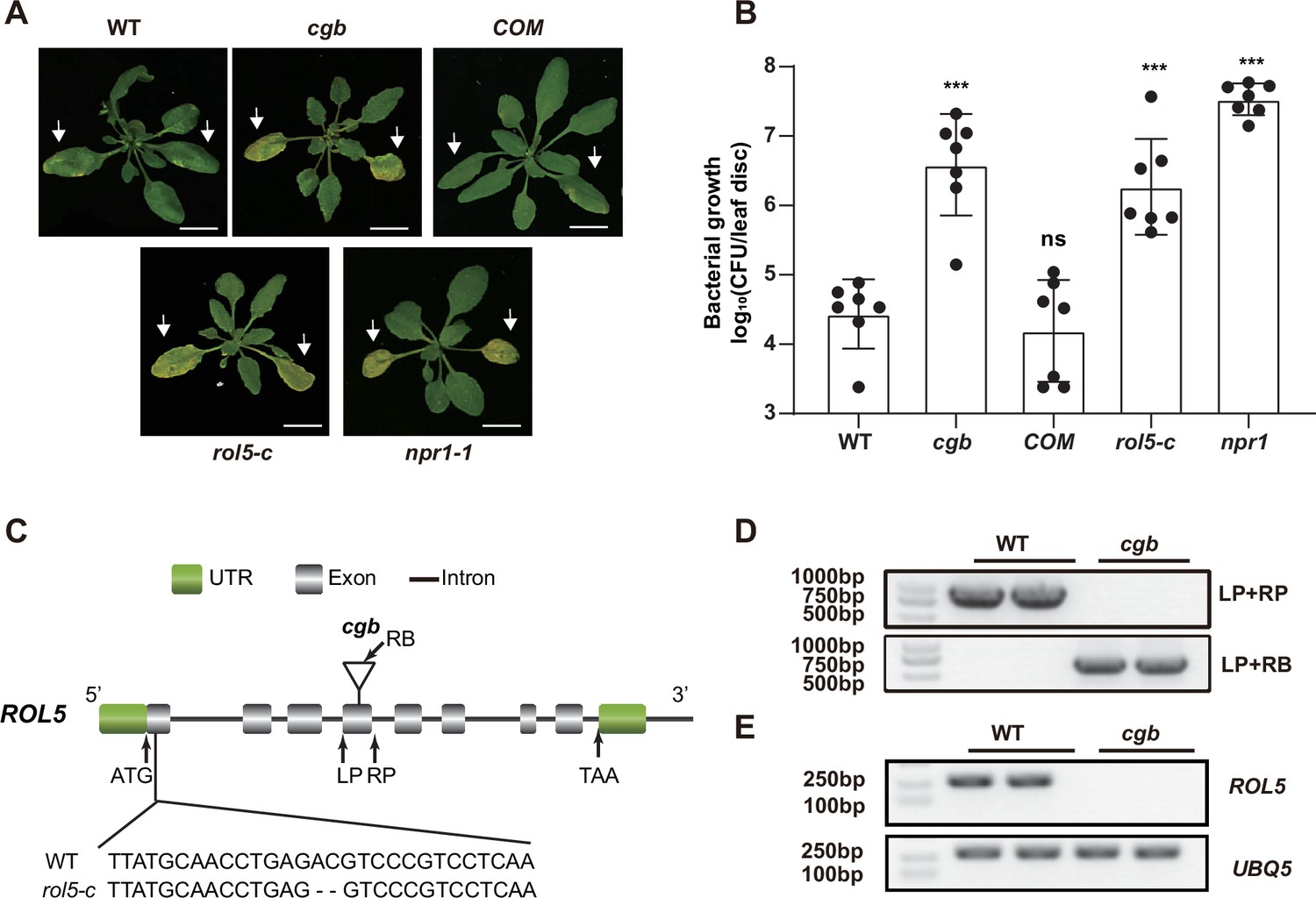
The rol5 mutants are more susceptible to the bacterial pathogen Psm ES4326 than wild-type (WT).
(A) Pictures of Arabidopsis 3 days after infection. The arrows indicate the leaves inoculated with Psm ES4326 (OD600=0.0002). cgb and rol5-c are mutants defective in ROL5. COM, the complementation line of cgb. npr1-1 serves as a positive control. Bar = 1 cm. (B) The growth of Psm ES4326. CFU, colony-forming unit. Error bars represent 95% confidence intervals (n=7). Statistical significance was determined by two-tailed Student’s t-test. ***, p<0.001; ns, not significant. (C) A schematic diagram showing the site of the T-DNA insertion in cgb and the deleted nucleotides in rol5-c. (D) The genotyping results using the primers indicated in C. (E) The transcript of ROL5 is not detectable in cgb. UBQ5 serves as an internal reference gene.
-
Figure 1—source data 1
Source data related to Figure 1B.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig1-data1-v2.xlsx
-
Figure 1—source data 2
Source data related to Figure 1D.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig1-data2-v2.zip
-
Figure 1—source data 3
Source data related to Figure 1E.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig1-data3-v2.zip
ROL5 interacts with CTU2 in Arabidopsis
ROL5 is a homolog of yeast NCS6 (Leiber et al., 2010), which forms a protein complex with NCS2 to catalyze mcm5s2U34 (Figure 2A). The NCS2 homolog in Arabidopsis is CTU2 (Philipp et al., 2014). To test whether ROL5 interacts with CTU2, we first performed yeast two-hybrid assays. Consistent with the previous finding (Philipp et al., 2014), only when ROL5 and CTU2 were co-expressed, the yeasts could grow on the selective medium (Figure 2B), indicating that ROL5 interacts with CTU2 in yeast. To test whether they can interact in vivo, we carried out split luciferase assays in Nicotiana benthamiana. ROL5 was fused with the N-terminal half of luciferase (nLUC) and CTU2 was fused with the C-terminal half of luciferase (cLUC). An interaction between two proteins brings the two halves of luciferase in close proximity, leading to enzymatic activity and production of luminescence that is detectable using a hypersensitive CCD camera. As shown in Figure 2C, the luminescence signal could be detected only when ROL5-nLUC and cLUC-CTU2 were co-expressed. We also performed co-immunoprecipitation (CoIP) assays in N. benthamiana. When ROL5-FLAG was co-expressed with CTU2-GFP, ROL5-FLAG could be immunoprecipitated by the GFP-Trap beads (Figure 2D). To test whether the interaction is direct, we conducted GST pull-down assays. GST-CTU2 and ROL5-His proteins were expressed in Escherichia coli and were purified using affinity resins. As shown in Figure 2E, ROL5-His could be specifically pulled down by GST-CTU2, but not by GST alone, suggesting that ROL5 directly interacts with CTU2.
Figure 2

ROL5 interacts with CTU2.
(A) A schematic diagram showing the function of ROL5 and CTU2. The ROL5 homolog NCS6 and the CTU2 homolog NCS2 form a complex to catalyze the mcm5s2U modification at wobble nucleotide of tRNA-Lys (UUU), tRNA-Gln (UUC), and tRNA-Glu (UUG), which pair with the AAA, GAA, and CAA codons in mRNA, respectively. (B) Yeast two-hybrid assays. The growth of yeast cells on the SD-Trp/Leu/His medium indicates interaction. BD, binding domain. AD, activation domain. (C) Split luciferase assays. The indicated proteins were fused to either the C- or N-terminal half of luciferase (cLUC or nLUC) and were transiently expressed in N. benthamiana. The luminesce detected by a CCD camera reports interaction. (D) Co-immunoprecipitation (CoIP) assays. CTU2-GFP and/or ROL5-FLAG fusion proteins were expressed in N. benthamiana. The protein samples were precipitated by GFP-Trap, followed by western blotting using anti-GFP or anti-FLAG antibodies. (E) GST pull‐down assays. The recombinant GST or GST-CTU2 proteins coupled with glutathione beads were used to pull down His-ROL5, followed by western blotting using anti-His or anti-GST antibodies.
-
Figure 2—source data 1
Source data related to Figure 2D.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig2-data1-v2.zip
-
Figure 2—source data 2
Source data related to Figure 2E.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig2-data2-v2.zip
The tRNA thiolation is required for plant immunity
Given that CTU2 interacts with ROL5, we reasoned that the ctu2 mutant should have similar phenotypes to rol5 in response to pathogens. To test this, we infected the T-DNA insertion mutant ctu2-1 with Psm ES4326. As expected, the ctu2-1 mutant is hyper-susceptible to the pathogen (Figure 3A and B).
Figure 3
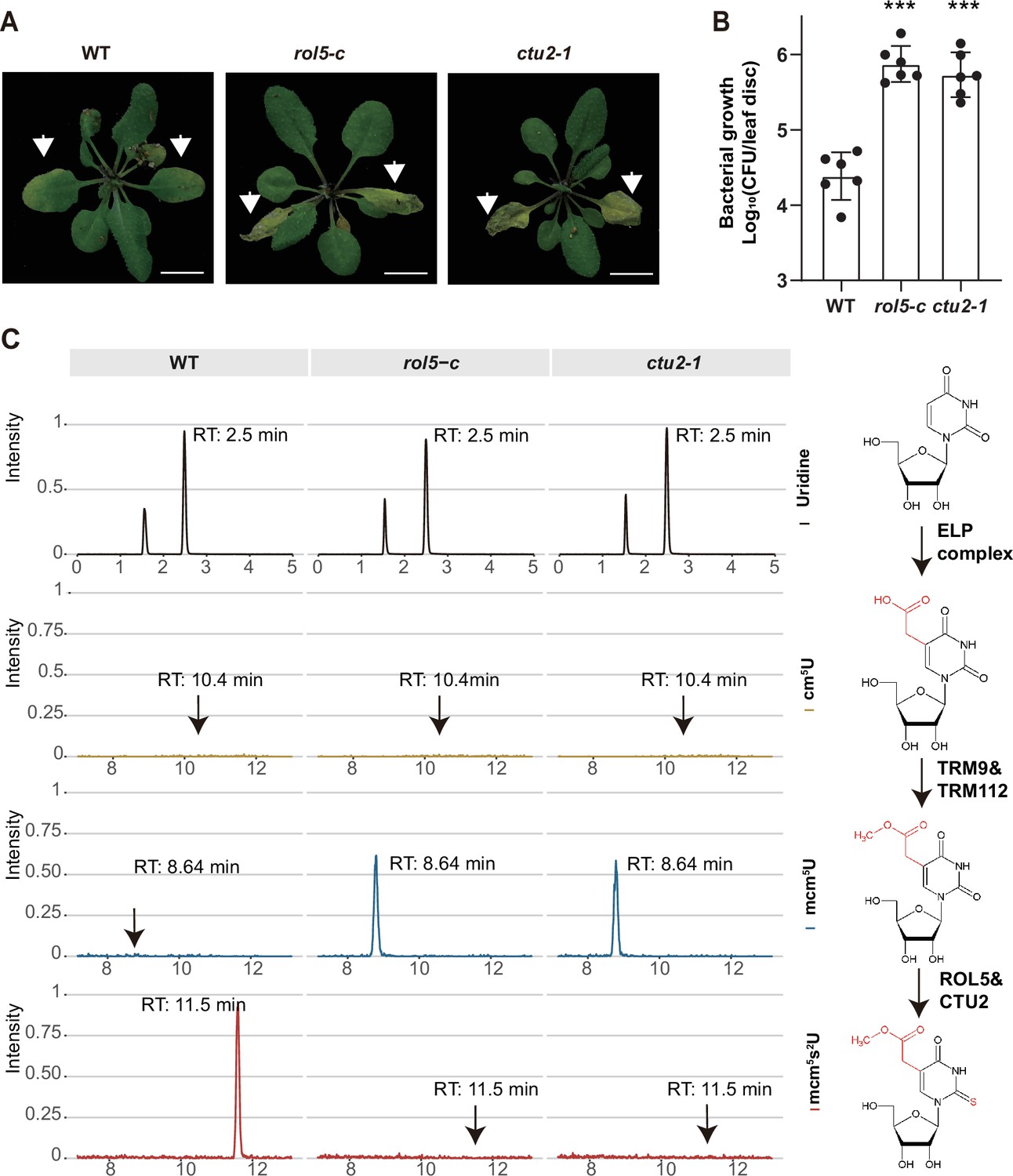
ROL5 and CTU2 are required for mcm5s2U modification and plant immunity.
(A and B) The rol5-c and ctu2-1 mutants are more susceptible to the bacterial pathogen Psm ES4326 than wild-type (WT). (A) Pictures of Arabidopsis plants 3 days after infection. Arrows indicate the leaves inoculated with Psm ES4326. Bar = 1 cm. (B) The growth of Psm ES4326. CFU, colony-forming unit. Error bars represent 95% confidence intervals (n=6). Statistical significance was determined by two-tailed Student’s t-test. ***, p<0.001. (C) The rol5-c and ctu2-1 mutants lack the mcm5s2U modification. The levels of U, cm5U, mcm5U, and mcm5s2U were quantified through high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS) analyses. The intensity and the retention time of each nucleotide are shown. The structure of each nucleotide and the catalyzing enzymes are shown on the right.
-
Figure 3—source data 1
Source data related to Figure 3A.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig3-data1-v2.xlsx
-
Figure 3—source data 2
Source data related to Figure 3C.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig3-data2-v2.xlsx
By using N-acryloylamino phenyl mercuric chloride, which binds thiolated tRNAs, previous studies revealed that tRNA thiolation was defective in the rol5 and ctu2 mutant (Leiber et al., 2010; Philipp et al., 2014). To confirm this result, we measured the mcm5U and mcm5s2U levels in WT, rol5-c, and ctu2-1 using high-performance liquid chromatography coupled with mass spectrometry (HPLC-MS). In WT, mcm5U was almost undetectable (Figure 3C), indicating that it is efficiently transformed into mcm5s2U in Arabidopsis. However, in the rol5-c and ctu2-1 mutants, the mcm5s2U level was undetectable while the mcm5U level was very high, suggesting that both ROL5 and CTU2 are required for mcm5s2U. These data revealed that ROL5 and CTU2 form a complex to catalyze the mcm5s2U modification, which is essential for plant immunity.
Transcriptome and proteome reprogramming are compromised in cgb
To understand why the cgb mutant was hyper-susceptible to pathogens, we performed transcriptome and proteome analyses of the cgb mutant and the COM line. Each sample was divided into two parts, one for transcriptome analysis using RNA sequencing (RNA-seq) approach, and the other for proteome analysis using a tandem mass tag (TMT)-based approach. Principal component analysis showed that the reproducibility between biological replicates was good (Figure 4—figure supplement 1). The differentially expressed genes (DEGs) and the differentially expressed proteins (DEPs) between different samples were identified and quantified through data analysis. Regarding the transcriptome, in COM, 22% (4819) and 27% (5767) of genes were respectively up-regulated or down-regulated after Psm infection (Figure 4A). However, only 18% (3986) and 23% (4913) of genes were respectively up-regulated or down-regulated in cgb. Regarding the proteome, in COM, 16% (1193) and 13% (1021) of proteins were respectively up-regulated or down-regulated after Psm infection (Figure 4B). In contrast, only 12% (909) and 10% (787) of proteins were respectively up-regulated or down-regulated in cgb. Therefore, the numbers of both DEGs and DEPs were reduced in cgb compared to those in COM.
Figure 4 with 1 supplement see all
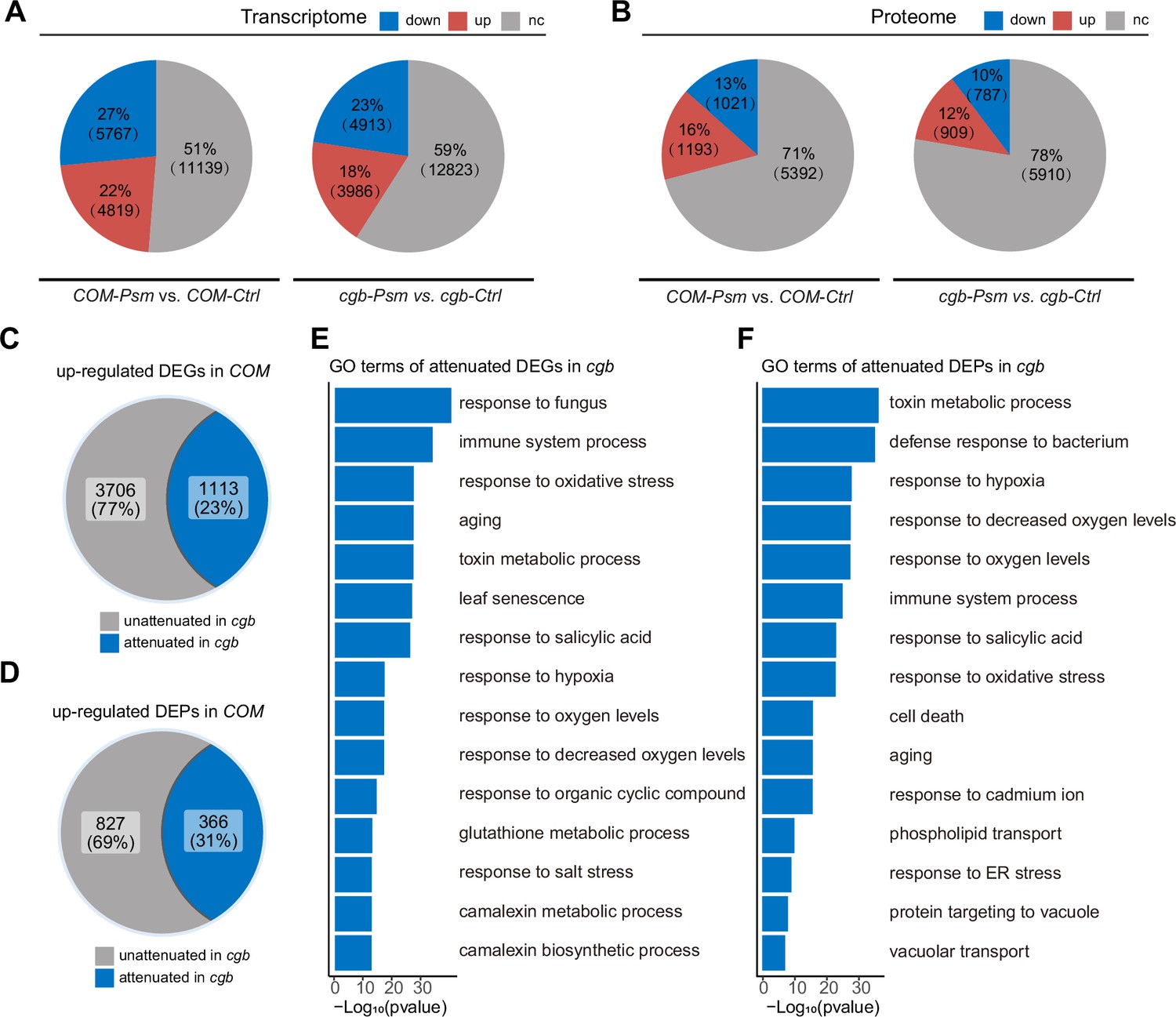
The transcriptome and proteome reprogramming are compromised in cgb.
(A and B) The percentage and the number of the differentially expressed genes (DEGs, p-value <0.05, |Log2Foldchange|>Log21.5, (A)) and the differentially expressed proteins (DEPs, p-value <0.05, |Log2Foldchange|>Log21.2, (B)) after Psm infection in the cgb mutant and the complementation line (COM). Down, down-regulated. Up, up-regulated. Nc, no change. (C and D) The percentage and the number of the attenuated genes (C) and proteins (D) in cgb among the up-regulated DEGs and DEPs in COM. (E and F) Gene Ontology (GO) analysis of the attenuated genes (E) or proteins (E) in cgb. The top 15 significantly enriched GO terms are shown.
-
Figure 4—source data 1
Source data related to Figure 4A.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data1-v2.xlsx
-
Figure 4—source data 2
Source data related to Figure 4B.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data2-v2.xlsx
-
Figure 4—source data 3
Source data related to Figure 4C.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data3-v2.xlsx
-
Figure 4—source data 4
Source data related to Figure 4D.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data4-v2.xlsx
-
Figure 4—source data 5
Source data related to Figure 4E.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data5-v2.xlsx
-
Figure 4—source data 6
Source data related to Figure 4F.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig4-data6-v2.xlsx
To further examine the gene expression defects in cgb, we compared the expression changes after Psm infection between cgb and COM. Among 4819 up-regulated DEGs in COM, the expression changes of 1113 genes were less prominent in cgb than in COM (Figure 4C). These genes were referred to as attenuated genes. Among 1193 up-regulated DEPs in COM, the expression changes of 366 proteins were less prominent in cgb than in COM (Figure 4D). These proteins were named attenuated proteins. Gene Ontology (GO) analysis of the attenuated genes and attenuated proteins revealed that many important biological processes were significantly enriched (Figure 4E and F). These data suggested that both transcriptome and proteome reprogramming were compromised in cgb.
The translation efficiency of immune-related proteins is compromised in cgb
Since the mcm5s2U modification directly regulates translation process (Nedialkova and Leidel, 2015; Schaffrath and Leidel, 2017), we sought to identify the proteins with compromised translation efficiency. The 366 attenuated proteins in cgb may be due to reduced transcription or reduced translation. To distinguish between these two possibilities, we performed Venn diagram analysis between attenuated genes and attenuated proteins, revealing that 261 attenuated proteins were not attenuated at the transcript level, suggesting that the attenuated expression of these proteins is due to reduced translation (Figure 5A). GO analysis of these 261 proteins revealed that some immune-related processes (i.e. response to salicylic acid, defense response to bacterium, and immune system process) were significantly enriched (Figure 5B). Notably, NPR1 is one of these proteins.
Figure 5 with 3 supplements see all
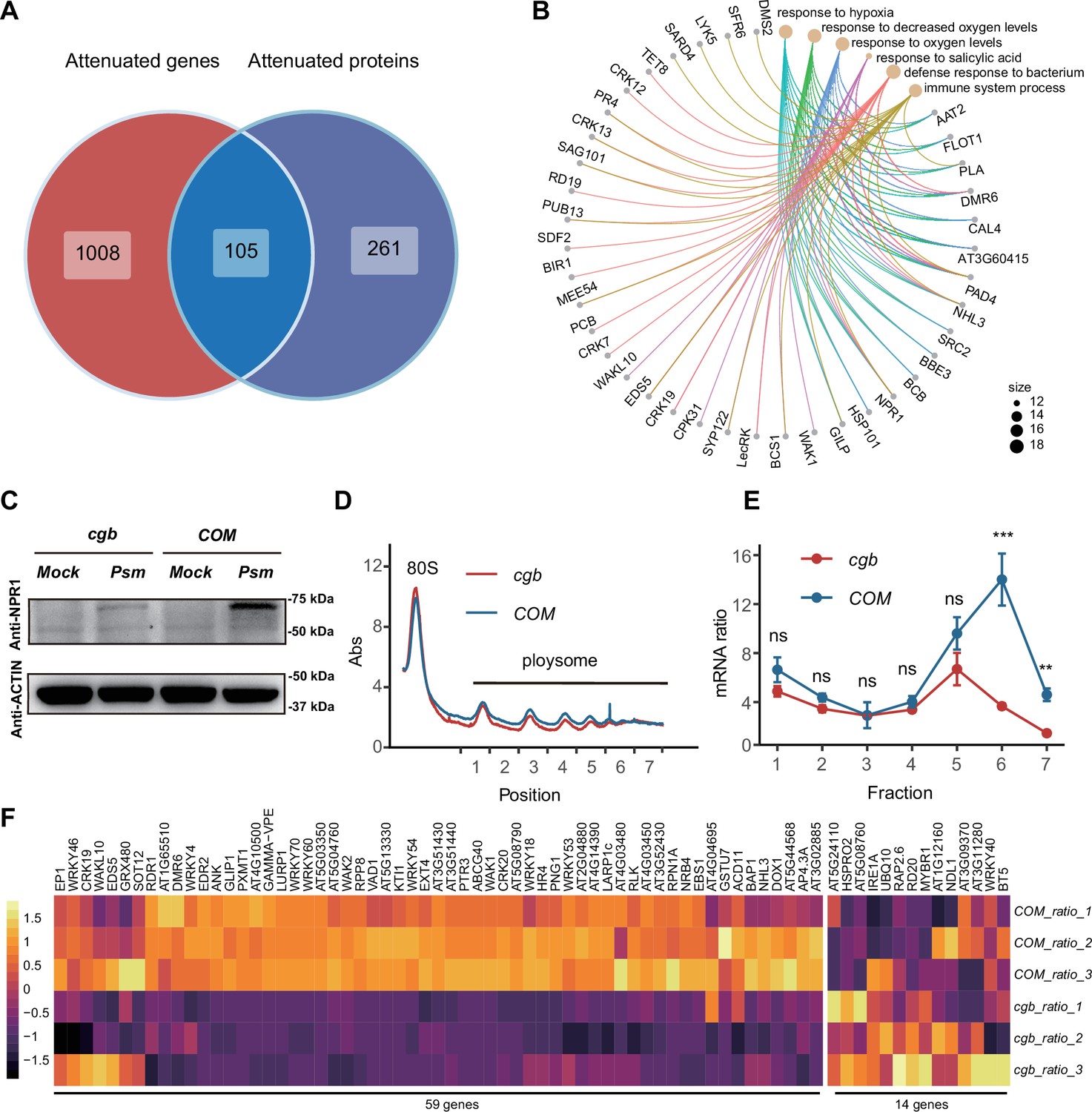
The translation of immune-related proteins is compromised in cgb.
(A) Venn diagram analysis of attenuated genes and proteins. (B) Gene Ontology (GO) analysis of the 261 attenuated proteins. The top 6 significantly enriched GO terms are shown. (C) Western blot analysis of NPR1 protein levels. The 7-day-old seedlings grown on 1/2 MS medium were treated with buffer (10 mM MgCl2, pH 7.5, Mock) or Psm ES4326 (OD600=0.2) for 48 hr. (D) Polysome profiling results. Abs, the absorbance of sucrose gradient at 254 nm. The numbers on the X-axis indicate the polysomal fractions subjected to qPCR analyses. (E) The qPCR analyses. The relative mRNA level of NPR1 in different fractions or in total mRNA was normalized against UBQ5. The ratio between the relative mRNA levels in each fraction and in total mRNA was shown (n=3). Statistical significance was determined by two-tailed Student’s t-test. **, p<0.01; ***, p<0.001; ns, not significant. (F) The heatmap showing the expression changes of salicylic acid (SA)-responsive genes after pathogen infection.
-
Figure 5—source data 1
Source data related to Figure 5A.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data1-v2.xlsx
-
Figure 5—source data 2
Source data related to Figure 5B.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data2-v2.xlsx
-
Figure 5—source data 3
Source data related to Figure 5C.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data3-v2.zip
-
Figure 5—source data 4
Source data related to Figure 5D.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data4-v2.xlsx
-
Figure 5—source data 5
Source data related to Figure 5E.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data5-v2.xlsx
-
Figure 5—source data 6
Source data related to Figure 5F.
- https://cdn.elifesciences.org/articles/93517/elife-93517-fig5-data6-v2.xlsx
To verify the expression of NPR1, we performed RT-qPCR and western blot analysis. Consistent with transcriptome and proteome data, the transcription levels of NPR1 were similar between COM and cgb both before and after Psm ES4326 infection (Figure 5—figure supplement 1), whereas the NPR1 protein level was much higher in COM than that in cgb after Psm ES4326 infection (Figure 5C). To further confirm that the translation of NPR1 was reduced in cgb, we carried out ribosome profiling experiment. Compared with COM, the polysome fractions in cgb were reduced (Figure 5D), suggesting that the overall translation efficiency is lower in cgb. As expected, the relative mRNA levels of NPR1 in multiple polysome fractions were significantly lower in cgb than in COM (Figure 5E).
The reduced NPR1 protein level in cgb suggested that SA signaling is compromised. To test this possibility, we examined the expression of all the genes (118) belonging to the GO term ‘response to salicylic acid’. In our transcriptome data, we could detect the expression of 73 genes, among which 59 genes (80.8%) were reduced in cgb compared with COM (Figure 5F). To further examine the defects of SA signaling in cgb, we performed SA-mediated protection assay. The Arabidopsis plants were treated with benzothiadiazole (BTH), a functional analog of SA, for 24 hr before infection. As expected, the growth of Psm ES4326 was reduced in BTH-treated COM, but not cgb and npr1 (Figure 5—figure supplement 2). These results suggested that SA signaling is indeed compromised in the cgb mutant.
To investigate the genetic relationship between CGB and NPR1, we generated the cgb npr1 double mutant and examined its disease phenotypes. We found that cgb npr1 was significantly more susceptible than either npr1 or cgb (Figure 5—figure supplement 3). There are two possible reasons for the observed additive effects of cgb and npr1. First, the translation of NPR1 was reduced rather than completely blocked in cgb (Figure 5C). In other words, NPR1 still has some function in cgb. But in the cgb npr1 double mutant, the function of NPR1 is completely abolished, which explains why cgb npr1 was more susceptible than cgb. Second, in addition to NPR1, some other immune regulators (such as PAD4, EDS5, and SAG101) were also compromised in cgb (Figure 5B), which explains why cgb npr1 was more susceptible than npr1.
Discussion
Upon pathogen infections, plants need to efficiently reprogram their gene expression, allowing the transition from growth to defense. However, how translation contributes to the immune response is not well studied. It is known that tRNA thiolation is required for efficient protein expression (Nedialkova and Leidel, 2015; Schaffrath and Leidel, 2017). Here, we show that tRNA thiolation is abolished in the cgb mutant (Figure 3), leading to disease hyper-susceptibility (Figure 1). We found that the translation of many immune-related proteins was reduced in cgb (Figure 5). Therefore, our study strongly suggested that tRNA thiolation is required for plant immunity, revealing an additional mechanism underlying plant immune responses. It is possible that tRNA thiolation is a regulatory step during immune responses. However, since many defense-related proteins are up-regulated after pathogen infection (Figure 4B), we cannot rule out the possibility that tRNA thiolation just becomes a limiting factor due to the high demand of translation resource during immune responses. More studies are required to distinguish these two possibilities.
The SA receptor NPR1 is the master regulator of SA signaling. NPR1 can function as a transcription coactivator to regulate gene expression and an adaptor of ubiquitin E3 ligase to mediate protein degradation (Yu et al., 2022; Yu et al., 2021; Zavaliev et al., 2020). It has been shown that the activity of NPR1 is regulated at multiple levels including post-translational modifications such as phosphorylation, ubiquitination, S-nitrosylation, and sumoylation (Saleh et al., 2015; Spoel et al., 2009; Tada et al., 2008). However, how NPR1 is regulated at the translational level is unknown. Here, we show that the tRNA thiolation-mediated translation control is required for the optimal expression of NPR1 (Figure 5B and D), revealing an additional layer of regulation for NPR1.
The tRNA thiolation modification is highly conserved in eukaryotes. However, its biological functions in plants are less well understood. Previously, it was reported that tRNA thiolation regulates the development of root hairs, chloroplasts, and leaf cells (Leiber et al., 2010; Philipp et al., 2014). Recently, it was found that tRNA thiolation is required for heat stress tolerance (Xu et al., 2020). Our study revealed an additional biological function of tRNA thiolation in plant immunity. It will also be interesting to test whether tRNA thiolation is required for responses to other stresses such as drought, salinity, and cold.
The ELP complex is composed of six proteins, with ELP1, ELP2, and ELP3 forming a core sub-complex, and ELP4, ELP5, and ELP6 forming an accessory sub-complex. The ELP complex catalyzes the cm5U modification, which is the precursor of mcm5s2U catalyzed by ROL5 and CTU2. As expected, the mcm5s2U modification was undetectable in the elp mutants such as elp3 and elp6 mutants (Leitner et al., 2015; Mehlgarten et al., 2010). Interestingly, similar to the rol5 and ctu2 mutants, the elp2 and elp3 mutants were hyper-susceptible to pathogens (DeFraia et al., 2010; Defraia et al., 2013; Wang et al., 2013). In addition to tRNA modification, the ELP complex has several other distinct activities including histone acetylation, α-tubulin acetylation, and DNA demethylation (Wang et al., 2013). Therefore, it is difficult to dissect which activity of the ELP complex contributes to plant immunity. However, the only known activity of ROL5 and CTU2 is to catalyze tRNA thiolation. Considering that the elp, rol5, and ctu2 mutants are all defective in tRNA thiolation, it is likely the tRNA modification activity of the ELP complex underlies its function in plant immunity.
Previous studies have identified numerous pathogen-responsive genes through transcriptome analysis (Zhang et al., 2020). However, the correlation between mRNAs and proteins is not always that strong (Lahtvee et al., 2017; Schwanhäusser et al., 2011). Given that proteins are major players in cellular functions, it is necessary to study immune responses at the protein level. Through high-throughput proteome analysis, we found 2215 proteins differentially accumulated after Psm infection in Arabidopsis (Figure 4). To our knowledge, this is the largest dataset of pathogen-responsive proteins in Arabidopsis. We believe that this dataset will provide a good research resource for future studies on plant immunity.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Arabidopsis thaliana) | ROL5 | TAIR | AT2G44270 | |
| Gene (Arabidopsis thaliana) | CTU2 | TAIR | AT4G35910 | |
| Genetic reagent (Arabidopsis thaliana) | cgb | This paper | It contains a T-DNA insertion in the fourth exon of ROL5 and is hypersusceptible to pathogen. | |
| Genetic reagent (Arabidopsis thaliana) | COM | This paper | It contains the coding sequence of ROL5 driven by 35S promoter in cgb. | |
| Genetic reagent (Arabidopsis thaliana) | rol5-c | This paper | The mutant was generated using CRISPR-Cas9 system. It contains a 2-bp deletion in the first exon of ROL5. | |
| Genetic reagent (Arabidopsis thaliana) | ctu2-1 | ABRC | SALK_032692 | |
| Genetic reagent (Arabidopsis thaliana) | npr1-1 | Cao et al., 1997 | ||
| Strain, strain Background (Escherichia coli) | BL21 | TransGen | Cat # CD901-02 | Electrocompetent cells |
| Strain, strain background (Escherichia coli) | DH5α | TransGen | Cat # CD201-01 | Electrocompetent cells |
| Strain, strain background (Agrobacterium tumefaciens) | GV3101 | Sangon | Cat # B528430 | Electrocompetent cells |
| Strain, strain background (Saccharomyces cerevisiae) | AH109 | Clontech | Cat # 630489 | Electrocompetent cells |
| Strain, strain background (Pseudomonas syringae pv. Maculicola) | Psm 4326 | Durrant et al., 2007 | ES4326 | |
| Antibody | Anti-NPR1 (Rabbit polyclonal) | From Dr. Li Yang | WB(1:3000) | |
| Antibody | Anti-His (Mouse monoclonal) | Abclonal | Cat # AE003 | WB(1:5000) |
| Antibody | Anti-GST (Mouse monoclonal) | Abclonal | Cat # AE001 | WB(1:5000) |
| Antibody | Anti-FLAG (Mouse monoclonal) | Promoter | WB(1:5000) | |
| Antibody | Anti-GFP (Mouse monoclonal) | Promoter | WB(1:5000) | |
| Other | GFP-Trap | chromotek | Cat # gtma | |
| Other | Hypersil GOLD | Thermo Fisher | Cat # 25005-254630 |
Plant material and growth conditions
Request a detailed protocolAll Arabidopsis seeds used in this study are in Columbia-0 background. The npr1-1 mutant was described previously (Cao et al., 1997). The cgb mutant and the complementation line were generated in this study. The mutant of ctu2-1 (SALK_032692) was purchased from ABRC. The rol5-c mutant was generated using EC1-based CRISPR-Cas9 system (Wang et al., 2015). All seeds were sterilized with 2% Plant Preservative Mixture-100 (Plant Cell Technology) at 4°C in the dark for 2 days and then were plated on Murashige and Skoog (MS) medium with 1% sucrose and 0.3% phytagel. The plants were grown under long-day conditions at 22°C (16 hr of light/8 hr of dark; supplied by white-light tubes).
Strains and growth conditions
Request a detailed protocolE. coli strain DH5α for molecular cloning was cultured in LB medium at 37°C. E. coli strain BL21 (DE3) for recombinant protein expression was cultured in LB medium at 16°C. Agrobacterium tumefaciens strain GV3101 for transformation was cultured in Yeast Extract Beef (YEB) medium at 28°C. Psm ES4326 for infection assay was cultured in King’s B (KB) medium at 28°C. Yeast strain AH109 for yeast two-hybrid assay was cultured in Yeast Peptone Dextrose (YPD) medium or SD medium at 28°C.
Vector constructions
Request a detailed protocolThe vectors were constructed using the digestion-ligation method or a lighting cloning system (Biodragon Immunotechnology). For complementation experiment, ROL5 was inserted into Nco I/Xba I-digested pFGC5941. For pull-down assays, CTU2 was inserted into BamH I/Xho I-digested pGEX-6P-1; ROL5 was inserted into Nco I/Hind III-digested pET28a. For split luciferase assays, ROL5 and CTU2 were cloned into the Kpn I/Sal I-digested pJW771 and pJW772, respectively. For yeast two-hybrid assays, ROL5 and CTU2 were cloned into EcoR I/BamH I-digested pGBKT7 and pGADT7. For CoIP assays, ROL5-FLAG and CTU2-GFP were cloned into Nco I/Xba I-digested pFGC5941. To generate rol5-c, the target sequence was designed and cloned into pHEE401 as described previously (Wang et al., 2015). The primer sequences used for cloning are listed in Appendix 1—table 1.
Reverse transcription and qPCR
Request a detailed protocolThe total RNA or the RNA in ribosome fractions was extracted using TRIzol Reagent (Invitrogen). The cDNA was synthesized using HiScript II Q RT SuperMix (Vazyme). The qPCR analyses were performed using the AceQ qPCR SYBR Green Master Mix (Vazyme). UBQ5 was used as the internal reference gene. Primers used for qPCR are listed in Appendix 1—table 1.
Pathogen infection
Request a detailed protocolThe third and fourth leaves of 3-week-old Arabidopsis plants were infiltrated with Psm ES4326 (OD600=0.0002) using a needleless syringe. Three days after infection, the leaves were sampled to measure the growth of Psm ES4326 as described previously (Durrant et al., 2007). For SA-mediated protection assay, the 3-week-old Arabidopsis plants were treated with 600 μM BTH (Syngenta) for 24 hr before infection.
Yeast two-hybrid assays
Request a detailed protocolMatchmaker GAL4 Two-Hybrid System (Clontech) was used and the assays were performed according to the user manual. Briefly, the bait (in pGBKT7) and prey (in pGADT7) vectors were co-transformed into the yeast strain AH109. The protein interactions were determined by yeast growth on SD/-Leu/-Trp/-His/ medium. The empty vectors were used as negative controls.
In vitro pull-down assays
Request a detailed protocolThe GST pull-down assays were performed as previously described (Chen et al., 2023a , Chen et al., 2023b). Briefly, ROL5-His, GST, and GST-CTU2 proteins were expressed in E. coli BL21 (DE3). GST (5 μg) and GST-CTU2 (5 μg) were coupled to glutathione beads (GE Healthcare Life Sciences) and then were incubated with ROL5-His (10 μg) in 0.5 mL binding buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, and 2 mM DTT) at 4°C for 2 hr. The beads were washed three times with washing buffer (binding buffer plus 2% NP-40), boiled in 1× SDS loading buffer, and analyzed by western blot using anti-GST (Abclonal) or anti-His (Abclonal) antibodies.
CoIP assays
Request a detailed protocolThe CoIP assays were performed as previously described (Chen et al., 2021). 35S:ROL5-FLAG and 35S:CTU2-GFP were transformed into A. tumefaciens GV3101. 35S:ROL5-FLAG strain (OD600=1) was mixed with the same volume of buffer or 35S:CTU2-GFP strain (OD600=1) and was infiltrated into N. benthamiana leaves. After 48 hr, the infiltrated leaves were ground in liquid nitrogen and were resuspended in IP buffer (20 mM Tris-HCl pH 7.5, 50 mM NaCl, 1 mM EDTA, 0.1% SDS, 1% Triton X-100, 1 mM PMSF, 100 μM MG132, 1× protease inhibitor cocktail) for total protein extraction. The lysates were incubated with GFP-Trap magnetic beads (Chromotek) at 4°C for 2 hr. The beads were washed using washing buffer (20 mM Tris-HCl pH 7.5, 150–500 mM NaCl, 1 mM EDTA, 1 mM PMSF, 1× Protease Inhibitor Cocktail) and then boiled in 1× SDS loading buffer. The western blotting was performed using anti-FLAG (Promoter) and anti-GFP (Promoter) antibodies.
Split luciferase assays
Request a detailed protocolSplit luciferase assay was performed as described previously (Chen et al., 2008). The constructs were transformed into A. tumefaciens strain GV3101 (OD600=1). The resultant strains were then infiltrated into leaves of N. benthamiana. After 48 hr, 1 mM luciferin (GOLDBIO) was applied onto leaves and the images were captured using Lumazone imaging system equipped with 2048B CCD camera (Roper).
Quantification of tRNA modifications
Request a detailed protocolQuantification of tRNA modifications was performed using liquid chromatography coupled with mass spectrometry according to a previous study (Su et al., 2014). Total tRNA was extracted using a microRNA kit (Omega Bio-Tek). Five micrograms of tRNA were hydrolyzed in 10 μL enzymic buffer (1 U benzonase, 0.02 U phosphodiesterase I, and 0.02 U alkaline phosphatase) at 37°C for 3 hr. The UHPLC system (Thermo Fisher Scientific) coupled with TSQ Altis Triple Quadrupole Mass Spectrometer (Thermo Fisher Scientific) was used for quantification of tRNA modification. For the liquid chromatography, the Hypersil GOLD HPLC column (3 µm, 150×2.1 mm2; Thermo Fisher Scientific) was used. The solvent gradient was set as the protocol (Su et al., 2014). The Tracefinder software (Thermo Fisher Scientific) was further used for peak assignment, area calculation, and normalization. Corresponding structures and molecular masses were obtained from the Modomics database (https://iimcb.genesilico.pl/modomics/modifications).
RNA and protein extraction for transcriptome and proteome analysis
Request a detailed protocolThe samples were ground in liquid nitrogen and divided into two parts, one for transcriptome analysis and the other for proteome analysis. Total RNA was extracted using TRIzol Reagent (Invitrogen). Library preparation and RNA-sequencing were performed by Novogene Cooperation. Total proteins were extracted using phenol-methanol method (Deng et al., 2007). The protein concentration was determined with 2D Quant Kit (GE Healthcare Life Sciences) using bovine serum albumin as a standard.
Proteome analysis
Request a detailed protocolFor trypsin digestion, 60 μg proteins of each sample were reduced with 20 mM Tris-phosphine for 60 min at 30°C. Cysteines were alkylated with 30 mM iodoacetamide for 30 min at room temperature in the dark. Proteins were precipitated with 6 volumes of cold acetone overnight and then dissolved in 50 mM triethylammonium bicarbonate (TEAB). Proteins were digested with trypsin (protease/protein = 1/25, wt/wt) overnight at 37°C.
For TMT labeling, each sample containing 25 μg of peptide in 50 mM TEAB buffer was combined with its respective 10-plex TMT reagent (Thermo Fisher Scientific) and incubated for 1 hr at room temperature. Three biological replicates were labeled respectively for each sample, in which COM was labeled with 126-, 127N-, and 128C- of the 10-plex TMT reagent, while cgb was labeled with 129N-, 130C-, and 131- of the 10-plex TMT reagents. The labeling reactions were stopped by the addition of 2 μL of 5% hydroxylamine.
For LC-MS/MS analysis, multiplexed TMT-labeled samples were combined, vacuum dried, reconstituted in 2% acetonitrile and 5 mM ammonium hydroxide (pH 9.5), and separated with the Waters Acquity BEH column (C18, 1.7 μm, 100 mm, Waters) using UPLC system (Waters) at a flow rate of 300 μL/min. Total of 24 fractions were collected, combined into 12 fractions, and vacuum dried for LC-MS/MS analysis. The solvent gradient was set as previously described (Deng et al., 2007). Samples were then analyzed on an Ultimate 3000 nano UHPLC system (Thermo Fisher Scientific) coupled online to a Q Exactive HF mass spectrometer (Thermo Fisher Scientific). The trapping column (PepMap C18, 100 Å, 100 μm×2 cm, 5 μm) and an analytical column (PepMap C18, 100 Å, 75 μm i.d.×50 cm long, 2 μm) were used for separation of the samples. The solvent gradient and MASS parameters were set as previously described (Deng et al., 2007).
Transcriptome data analysis
Request a detailed protocolRaw reads were processed and aligned to the Arabidopsis genome (https://www.arabidopsis.org) using STAR (v.2.6.1a). Genes with over 10 reads were filtered and processed using DESeq2 (v.1.22.2) to identify the DEGs (p-value <0.05, |Log2FoldChange|>Log21.5) (Love et al., 2014).
Proteome data analysis
Request a detailed protocolRaw data were processed using Proteome Discoverer (v.2.2.0.388) and aligned to Arabidopsis genome (https://www.arabidopsis.org) with the SEQUEST HT search engine. Searches were configured with static modifications for the TMT reagents (+229.163 Da). The precursor mass tolerance was set as 10 ppm; the fragment mass tolerance was set as 0.02 Da; the trypsin missed cleavage was set as 2. The reversed sequence decoy strategy was used to control peptide false discovery. The peptides with q scores <0.01 were accepted, and at least one unique peptide was required for matching a protein entry for its identification. PSMs (peptide spectrum matches) results were processed with DESeq2 (v.1.22.2) to identify the DEPs (p-value <0.05, |Log2FoldChange|>Log21.2).
GO and heatmap analysis
Request a detailed protocolThe DEGs or proteins were analyzed by using Clusterprofile (v.3.18.1) (Yu et al., 2012). The heatmap analysis was processed by using pheatmap2 (v.1.0.12).
Analysis of NPR1 protein level
Request a detailed protocolThe seedlings were ground in liquid nitrogen and were resuspended in lysis buffer (50 mM Tris-HCl, pH 7.4, 150 mM NaCl, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS, 200 mM DTT, 1 mM PSMF, 50 µM MG132, 1× protease inhibitor cocktail). After centrifuging, the supernatants were mixed with the same volume of 2× SDS loading buffer and were incubated at 75°C for 15 min. The western blotting was performed using an anti-NPR1 antibody (provided by Li Yang from China Agricultural University).
Ribosome profiling
Request a detailed protocolThe ribosome profiling was performed as previously described with some modifications (Hsu et al., 2016; Xu et al., 2017). The plant sample (0.05–0.1 g) was ground in liquid nitrogen and extracted with 1 mL ribosome lysis buffer (200 mM Tris-HCl pH 8.0, 200 mM KCl, 35 mM MgCl2, 1% Triton X-100, 100 μM MG132, 1 mM DTT, and 100 μg/mL cycloheximide), followed by ultracentrifugation at 4°C for 2 hr (38,000 rpm, Beckman, SW41 rotor) through a 20–60% sucrose gradient (40 mM Tris-HCl, pH 8.4, 20 mM KCl, 10 mM MgCl2, and 50 μg/mL cycloheximide) prepared by Gradient Master (Biocomp Instruments). The profiling signals were recorded by Piston Gradient Fractionator (Biocomp Instruments).
Appendix 1
Appendix 1—table 1
The primers used in this study.
| Name | Sequence(5'–3') | Application |
|---|---|---|
| ROL5-F1 | ACATTACAATTACATTTACAATTACATGGAGGCCAAGAACAAGAA | For complementation |
| ROL5-R1 | GGGTCTTAATTAACTCTCTAGATTAGAAATCCAGAGATCCACAT | |
| ROL5-F2 | CGGAATTC ATGGAGGCCAAGAACAAGA | For Y2H |
| ROL5-R2 | CGGGATCC TTAGAAATCCAGAGATCCAC | |
| CTU2-F1 | CGGAATTC ATGGCTTGTAATTCCTCAG | |
| CTU2-R1 | CGGGATCC TTAGACAACCTCTTCATCGT | |
| ROL5-F3 | GGGGTACCATGGAGGCCAAGAACAAGA | For split luc |
| ROL5-R3 | GCGTCGACGAAATCCAGAGATCCAC | |
| CTU2-F2 | GGGGTACCATGGCTTGTAATTCCTCAG | |
| CTU2-R2 | GCGTCGACTTAGACAACCTCTTCATCGT | |
| GUS-F | acgcgtcccggggcggtaccATGGTAGATCTGAGGGTAAA | |
| GUS-R | cgaaagctctgcaggtcgacCTATTGTTTGCCTCCCTGCTG | |
| ROL5-F0 | TGACTGCTCCCTACCTGTCGAGTTTTAGAGCTAGAAATAGC | For CRISPR mutant of ROL5 |
| ROL5-R0 | AACGAGACGTCCCGTCCTCAAACAATCTCTTAGTCGACTCTAC | |
| ROL5-BsF | ATATATGGTCTCGATTGACTGCTCCCTACCTGTCGAGTT | |
| ROL5-BsR | ATTATTGGTCTCGAAACGAGACGTCCCGTCCTCAAACAA | |
| ROL5-F4 | TTGAAAGGTTTACATCTTGGAAT | For sequencing of target sites |
| ROL5-R4 | AAAGGTGATTGCTTAGATTCTGATT | |
| ROL5-F5 | CTCAAAAACCTCATAAAAGCACTCT | |
| ROL5-R5 | AACTGCGTCACTGTCTTTACTCT | |
| ROL5-F6 | TTAAGAAGGAGATATACCATGGGCATGGAGGCCAAGAACAAGA | For protein expression |
| ROL5-R6 | GAGTGCGGCCGCAAGCTTTTAGAAATCCAGAGATCCAC | |
| CTU2-F3 | TTCCAGGGGCCCCTGGGATCCATGGCTTGTAATTCCTCAG | |
| CTU2-R3 | AGTCACGATGCGGCCGCTCGAGTTAGACAACCTCTTCATCGT | |
| ROL5-F7 | CAATTACATTTACAATTACATGGAGGCCAAGAACAAGA | For co-immunoprecipitation |
| ROL5-R7 | GGGTCTTAATTAACTCTCTAGATTTGTCATCATCGTCTTTG | |
| CTU2-F4 | CAATTACATTTACAATTACATGGCTTGTAATTCCTCAGG | |
| CTU2-R4 | GGGTCTTAATTAACTCTCTAGATTACTTGTACAGCTCGTCCA | |
| cgb-LP | GTATGAGAAGTGATTGAGTATGTG | For genotyping |
| cgb-RP | TCGATGTGCACCTACTTAATCTAC | |
| cgb-RB | CTAATGAGTGAGCTAACTCAC | |
| ctu2-LP | TCACATTGCATTGAATCATCC | For genotyping |
| ctu2-RP | TCAAATTTAGCACATGGGACC | |
| ROL5-F1 | GGAGCTGCGTTATTGAAAGTAG | For qPCR |
| ROL5-R1 | CCACGATATGCATTAGGAGAGT | |
| UBQ5-F1 | GAAGATCCAAGACAAGGAAGGA | |
| UBQ5-R1 | CTTCTTCCTCTTCTTAGCACCA | |
| NPR1-P1 | ATGATTTCTACAGCGACGCTAA | |
| NPR1-P2 | GACTTCGTAATCCTTGGCAATC |
Data availability
RNA sequencing datasets have been deposited to GSE database with an accession number GSE183087. The mass spectrometry proteomics data have been deposited to the ProteomeXchange Consortium via the iProX partner repository with the dataset identifier PXD028189. Data analysis scripts are available on GitHub (copy archived at Zheng, 2022). Source data files have been provided for Figures 1B, 1D, 1E, 2D, 2E, 3B, 3C, Figure 4, and Figure 5.
-
NCBI Gene Expression OmnibusID GSE183087. The thiolation modification of tRNA is essential for plant immunity.
-
ProteomeXchangeID PXD028189. The thiolation modification of tRNA is essential for plant immunity.
References
-
RAD51 supports DMC1 by inhibiting the SMC5/6 complex during meiosisThe Plant Cell 33:2869–2882.https://doi.org/10.1093/plcell/koab136
-
Elp3 links tRNA modification to IRES-dependent translation of LEF1 to sustain metastasis in breast cancerThe Journal of Experimental Medicine 213:2503–2523.https://doi.org/10.1084/jem.20160397
-
A proteomics study of brassinosteroid response in arabidopsisMolecular & Cellular Proteomics 6:2058–2071.https://doi.org/10.1074/mcp.M700123-MCP200
-
The TOR pathway modulates the structure of cell walls in arabidopsisThe Plant Cell 22:1898–1908.https://doi.org/10.1105/tpc.109.073007
-
Elongator function in tRNA wobble uridine modification is conserved between yeast and plantsMolecular Microbiology 76:1082–1094.https://doi.org/10.1111/j.1365-2958.2010.07163.x
-
Yeast Nfs1p is involved in thio-modification of both mitochondrial and cytoplasmic tRNAsThe Journal of Biological Chemistry 279:12363–12368.https://doi.org/10.1074/jbc.M312448200
-
Trna wobble modification affects leaf cell development in Arabidopsis thalianaPlant & Cell Physiology 60:2026–2039.https://doi.org/10.1093/pcp/pcz064
-
Salicylic acid: Biosynthesis and signalingAnnual Review of Plant Biology 72:761–791.https://doi.org/10.1146/annurev-arplant-081320-092855
-
Thio-modification of trna at the wobble position as regulator of the kinetics of decoding and translocation on the ribosomeJournal of the American Chemical Society 139:5857–5864.https://doi.org/10.1021/jacs.7b00727
-
Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degenerationHuman Molecular Genetics 18:472–481.https://doi.org/10.1093/hmg/ddn375
-
How do plants achieve immunity? defence without specialized immune cellsNature Reviews. Immunology 12:89–100.https://doi.org/10.1038/nri3141
-
Role of tRNA modifications in human diseasesTrends in Molecular Medicine 20:306–314.https://doi.org/10.1016/j.molmed.2014.01.008
-
Salicylic acid, a multifaceted hormone to combat diseaseAnnual Review of Phytopathology 47:177–206.https://doi.org/10.1146/annurev.phyto.050908.135202
-
Perception of the plant immune signal salicylic acidCurrent Opinion in Plant Biology 20:64–68.https://doi.org/10.1016/j.pbi.2014.04.006
-
Salicylic acid and ethylene coordinately promote leaf senescenceJournal of Integrative Plant Biology 63:823–827.https://doi.org/10.1111/jipb.13074
Article and author information
Author details
Funding
National Natural Science Foundation of China (31970311)
- Shunping Yan
HZAU-AGIS Cooperation Fund (SZYJY2022004)
- Shunping Yan
National Natural Science Foundation of China (32000373)
- Xiaodan Yu
National Natural Science Foundation of China (32270306)
- Shunping Yan
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We are grateful to Dr. Pascal Genschik for critical revision, Dr. Zhipeng Zhou for helpful discussion, Dr. Peng Chen for technical support, and Dr. Li Yang for providing the anti-NPR1 antibody. This work is supported by the National Natural Science Foundation of China (31970311, 32000373, and 32270306), HZAU-AGIS Cooperation Fund (SZYJY2022004), and BaiChuan Program.
Copyright
© 2024, Zheng, Chen et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,472
- views
-
- 272
- downloads
-
- 18
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 18
- citations for umbrella DOI https://doi.org/10.7554/eLife.93517
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
The tRNA thiolation-mediated translational control is essential for plant immunity
eLife 13:e93517.
https://doi.org/10.7554/eLife.93517




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}