Cell Cycle: Micromanaging checkpoint proteins
The kinase Mps1, long known to be the 'boss' in mitotic checkpoint signaling, phosphorylates multiple proteins in the checkpoint signaling cascade.
- The FIRC Institute of Molecular Oncology (IFOM), Italy
- Virginia Tech, United States
- Biocomplexity Institute of Virginia Tech, United States
Micromanagement has a bad reputation. It is frowned upon in the workplace, but it may have benefits in cellular signaling because a single regulator that interferes at multiple steps of a signaling cascade can lead to more reliable signaling. Now, in eLife, Hongtao Yu and colleagues at the University of Texas Southwestern Medical Center – including Zhejian Ji and Haishan Gao as joint first authors – report that a kinase called Mps1 acts as a micromanaging boss of a checkpoint signaling pathway that regulates cell division (Ji et al., 2017).
When cells divide, their chromosomes duplicate and a protein complex called the kinetochore assembles on each chromosome copy. Microtubules then attach to the kinetochores to pull the copies apart and segregate them between the newly forming cells. The mitotic checkpoint is a cellular safeguard that triggers the checkpoint signaling cascade if the microtubules do not attach properly to the kinetochores. In particular, this cascade leads to the formation of the “mitotic checkpoint complex”, which inhibits another multi-protein structure called the anaphase-promoting complex. This inhibition prevents chromosome segregation and the final stages of cell division (Musacchio, 2015).
Hints that Mps1 oversees and controls checkpoint signaling were uncovered decades ago. The overexpression of Mps1 was found to trigger checkpoint signaling even when the microtubules were all properly attached to kinetochores (Hardwick et al., 1996). In order to create the checkpoint signal, Mps1 relied on all other known checkpoint proteins, which suggested that Mps1 is the boss at the top of the signaling cascade.
Over the years, it became clear that Mps1 phosphorylates multiple checkpoint proteins, and also the kinetochore protein KNL1, but the mechanistic details of these events have only recently started to emerge. Phosphorylation of KNL1 leads to the recruitment of the checkpoint protein Bub1 to kinetochores. And work in budding yeast subsequently revealed that Mps1 phosphorylates Bub1 to enable it to bind to another checkpoint protein called Mad1. This interaction was crucial for checkpoint signaling in budding yeast (London and Biggins, 2014), but efforts to detect such an interaction in other organisms were unsuccessful. Now, however, Ji et al. provide strong evidence that a similar interaction occurs in human cells. Similar findings have emerged from research into fission yeast (Mora-Santos et al., 2016; Yuan et al., 2017).
The work of Ji et al. goes further by showing that Mps1 also phosphorylates Mad1 (as opposed to just phosphorylating Bub1 so that it can bind to Mad1). The region of Mad1 that is phosphorylated was known to have an essential role in checkpoint signaling, but its precise function had remained unclear (Heinrich et al., 2014; Kruse et al., 2014). Ji et al. now find that this region binds a protein called Cdc20 that has a central role in cell division as the activator of the anaphase-promoting complex. Checkpoint signaling packs Cdc20 into the mitotic checkpoint complex, thereby blocking its activity: however, this can only happen if Cdc20 first binds to a spindle checkpoint protein called Mad2. This binding occurs in an unusual fashion, with Mad2 changing conformation as it closes around a flexible fragment of Cdc20, just like a car seatbelt wrapping around a passenger. Ji et al. now propose that Mps1-phosphorylated Mad1 positions the flexible Cdc20 segment for capture by Mad2 (Figure 1). This is an intriguing model, and it will be important to corroborate it by structural or biophysical methods.
Figure 1
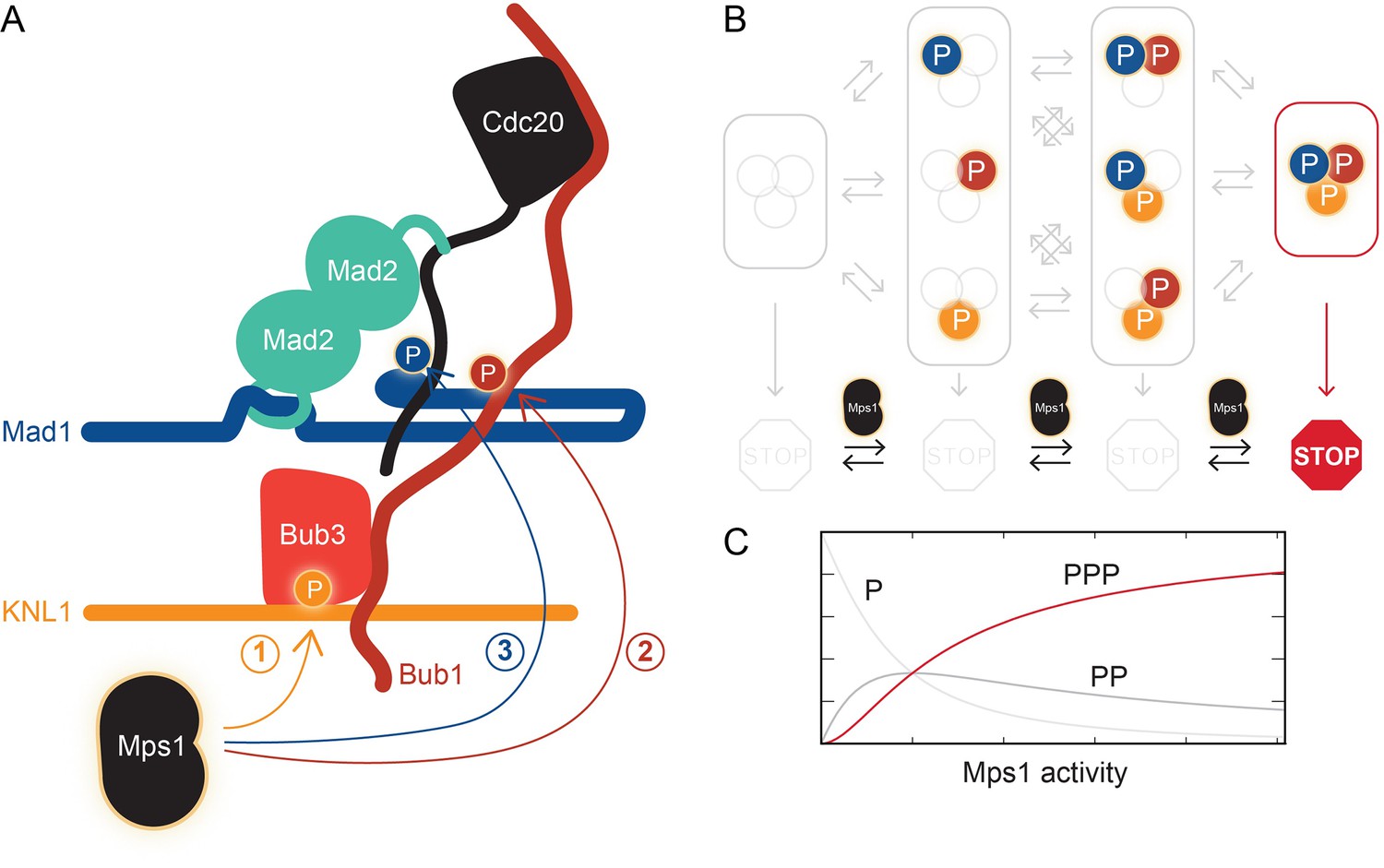
The kinase Mps1 and its role in mitotic checkpoint signaling.
(A) Mps1 phosphorylates (P) three different proteins to promote the assembly of the mitotic checkpoint complex. It phosphorylates the kinetochore protein KNL1 to recruit the checkpoint protein complex Bub1-Bub3 to KNL1 (1). It phosphorylates Bub1, which allows this protein to interact with another checkpoint protein, Mad1 (2). It also phosphorylates Mad1, which promotes the binding of Mad2 to the regulatory protein Cdc20 (3). Ji et al. propose that phosphorylated Mad1 binds to Cdc20, thereby positioning the latter for capture by Mad2. (B) The checkpoint (represented by the STOP sign) is only active when Mps1 has phosphorylated all three proteins, KNL1, Bub1, and Mad1. (C) Checkpoint activity (y-axis) plotted as a function of Mps1 kinase activity (x-axis) for the phosphorylation of one (P), two (PP) or all three sites (PPP).
Further support for this model comes from a recent study that used a technique called FRET (which probes the distance between fluorescently labeled molecules) to follow the assembly of the mitotic checkpoint complex over time (Faesen et al., 2017). This work demonstrated that the binding of Mad2 to Cdc20 is the rate-limiting step in the assembly process, and that the phosphorylation of Mad1 by Mps1 is crucial for the process to occur efficiently. The data from both reports contain many other gems for mitotic checkpoint aficionados, and we encourage all checkpoint enthusiasts to take a read.
But back to micromanagement. Is it significant that Mps1 influences multiple interactions throughout the checkpoint signaling pathway? The phosphorylation of multiple substrates in a single pathway conceptually resembles the phosphorylation of a single substrate at multiple sites. As opposed to a single phosphorylation event, multi-site phosphorylation can lead to more interesting behaviors (Ferrell and Ha, 2014). In particular, the output of the signaling pathway can be negligible when the activity of the Mps1 kinase is low, but it can increase abruptly if kinase activity rises above a certain threshold (Figure 1C). This could prevent spurious Mps1 kinase activity from activating the checkpoint at the wrong time. Ji et al. speculate that Mps1 phosphorylates one substrate after the other in a “cascade”, though this remains to be tested.
Lastly, if you have a micromanaging boss, you want to keep her or him in check. Close contact between Mps1 and the kinetochore protein KNL1 is sufficient to trigger checkpoint signaling, even if there are no unattached kinetochores in the cell (Aravamudhan et al., 2015; Yuan et al., 2017). Hence, for the checkpoint to work properly, Mps1’s access to KNL1 needs to be under tight control and should only happen if there is a problem with microtubule attachment to kinetochores. Despite some initial work, this crucial part of the regulation is still only partly understood (Aravamudhan et al., 2015; Hiruma et al., 2015; Ji et al., 2015).
References
-
Ultrasensitivity part II: multisite phosphorylation, stoichiometric inhibitors, and positive feedbackTrends in Biochemical Sciences 39:556–569.https://doi.org/10.1016/j.tibs.2014.09.003
-
The molecular biology of spindle assembly checkpoint signaling dynamicsCurrent Biology 25:R1002–R1018.https://doi.org/10.1016/j.cub.2015.08.051
Article and author information
Author details
Publication history
Copyright
© 2017, Ciliberto et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,481
- views
-
- 264
- downloads
-
- 7
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 7
- citations for umbrella DOI https://doi.org/10.7554/eLife.25001
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Cell Cycle: Micromanaging checkpoint proteins
eLife 6:e25001.
https://doi.org/10.7554/eLife.25001




{kind=link}