CRISPRi is not strand-specific at all loci and redefines the transcriptional landscape
- University of Oxford, United Kingdom
- Suez University, Egypt
Abstract
CRISPRi, an adapted CRISPR-Cas9 system, is proposed to act as a strand-specific roadblock to repress transcription in eukaryotic cells using guide RNAs (sgRNAs) to target catalytically inactive Cas9 (dCas9) and offers an alternative to genetic interventions for studying pervasive antisense transcription. Here, we successfully use click chemistry to construct DNA templates for sgRNA expression and show, rather than acting simply as a roadblock, sgRNA/dCas9 binding creates an environment that is permissive for transcription initiation/termination, thus generating novel sense and antisense transcripts. At HMS2 in Saccharomyces cerevisiae, sgRNA/dCas9 targeting to the non-template strand for antisense transcription results in antisense transcription termination, premature termination of a proportion of sense transcripts and initiation of a novel antisense transcript downstream of the sgRNA/dCas9-binding site. This redefinition of the transcriptional landscape by CRISPRi demonstrates that it is not strand-specific and highlights the controls and locus understanding required to properly interpret results from CRISPRi interventions.
https://doi.org/10.7554/eLife.29878.001Introduction
Eukaryotic genomes are pervasively transcribed but the effect of this inter- and intragenic transcription is not fully understood (Mellor et al., 2016). Of particular interest is the function of antisense transcription, which alters the chromatin in the vicinity of sense promoters (Lavender et al., 2016; Murray et al., 2015; Pelechano and Steinmetz, 2013), and is associated with repression, activation or no change in levels of the corresponding sense transcript (Murray et al., 2015). Discerning the mechanism by which antisense transcription functions in gene regulation is confounded by the difficulty in ablating antisense transcription without direct genetic intervention (Bassett et al., 2014). A new approach, CRISPR interference (CRISPRi) (Qi et al., 2013), can circumvent these issues by avoiding the need to manipulate endogenous DNA sequences. An endonucleolytically dead version of Cas9 (dCas9) is recruited by single base-pairing guide RNAs (sgRNAs) targeting the non-template (NT) DNA strand, where it blocks transcription strand-specifically at the loci tested (Lenstra et al., 2015; Qi et al., 2013). In a further adaptation, dCas9 fusion with transcriptional repressors or activators allows both negative and positive regulations of transcription, respectively (Gilbert et al., 2013; Gilbert et al., 2014), but the strand-specificity is lost and thus is not suitable for strand-specific repression of antisense transcription. The sgRNA compenent of the CRISPRi system consists of two regions: a constant region (82 nt) that binds to dCas9 and a variable region (20 nt) that is responsible for targeting. The modular nature of the sgRNA lends itself to production from DNA templates constructed using synthetic copper(I)-catalysed alkyne-azide cycloaddition (CuAAC) click chemistry (El-Sagheer and Brown, 2010). The stable artificial triazole DNA linker generated is biocompatibile, being read and accurately copied by DNA and RNA polymerases (Birts et al., 2014; El-Sagheer and Brown, 2011; El-Sagheer et al., 2011). We show that click chemistry is an efficient method for sgRNA template construction and that when combined with dCas9, these sgRNAs are as effective as sgRNAs produced from chemically synthesised full-length DNA templates at reducing levels of transcripts in vivo. However, it is still not completely understood how the CRISPRi system functions strand-specifically or why sgRNAs must target the non-template strand.
Here, we use the CRISPRi system to study the effect of blocking antisense transcription at loci with well-characterised sense:antisense transcript pairs. We have previously used a promoter deletion of the antisense transcript SUT650 at the HMS2 locus to show that SUT650 represses HMS2 sense transcription (Nguyen et al., 2014). Now we use CRISPRi to examine the effects of blocking SUT650 antisense transcription to ask (i) whether SUT650 represses HMS2 sense transcription without a genetic intervention and (ii) whether CRISPRi is strand-specific using, in addition to HMS2, an engineered GAL1 gene with a well-characterised antisense transcript (Murray et al., 2012, 2015).
The main conclusion from this study is that CRISPRi at the HMS2 locus is not fully strand-specific and results in (i) premature termination of the sense transcript and (ii) initiation of a new unstable antisense transcript in the vicinity of the sgRNA binding site. As transcription from this new antisense initiation site extends in to the HMS2 promoter, there is no net change in HMS2 sense transcript levels. Thus, CRISPRi redefines the transcriptional landscape at HMS2. This suggests that routine use of CRISPRi for gene expression analysis will require rigorous analysis of transcript integrity and function before conclusions can be drawn.
Results and discussion
DNA templates for sgRNA production can be made using click chemistry
CRISPRi-mediated transcriptional repression requires co-expression of a mature sgRNA and dCas9 (Figure 1A). A small library of single-stranded DNAs, comprised of templates for sgRNA variable regions, were joined to the constant region using click chemistry (Figure 1B) and used successfully as templates for PCR, with no significantly different efficiencies when compared to control full-length synthesised oligonucleotides (Figure 1C). The PCR products were inserted in place of a URA3 selection cassette in the endogenous snR52 locus for expression of a transcript that is then processed to form the mature nuclear-retained sgRNA (Figure 1A). Levels of dCas9 protein were uniform between strains (Figure 1—figure supplement 1). Neither insertion of URA3 into snR52 nor dCas9 expression in the control strains affected growth rate, although strains expressing some sgRNAs grew more slowly indicating a physiological effect (Figure 1D).
Figure 1 with 1 supplement see all
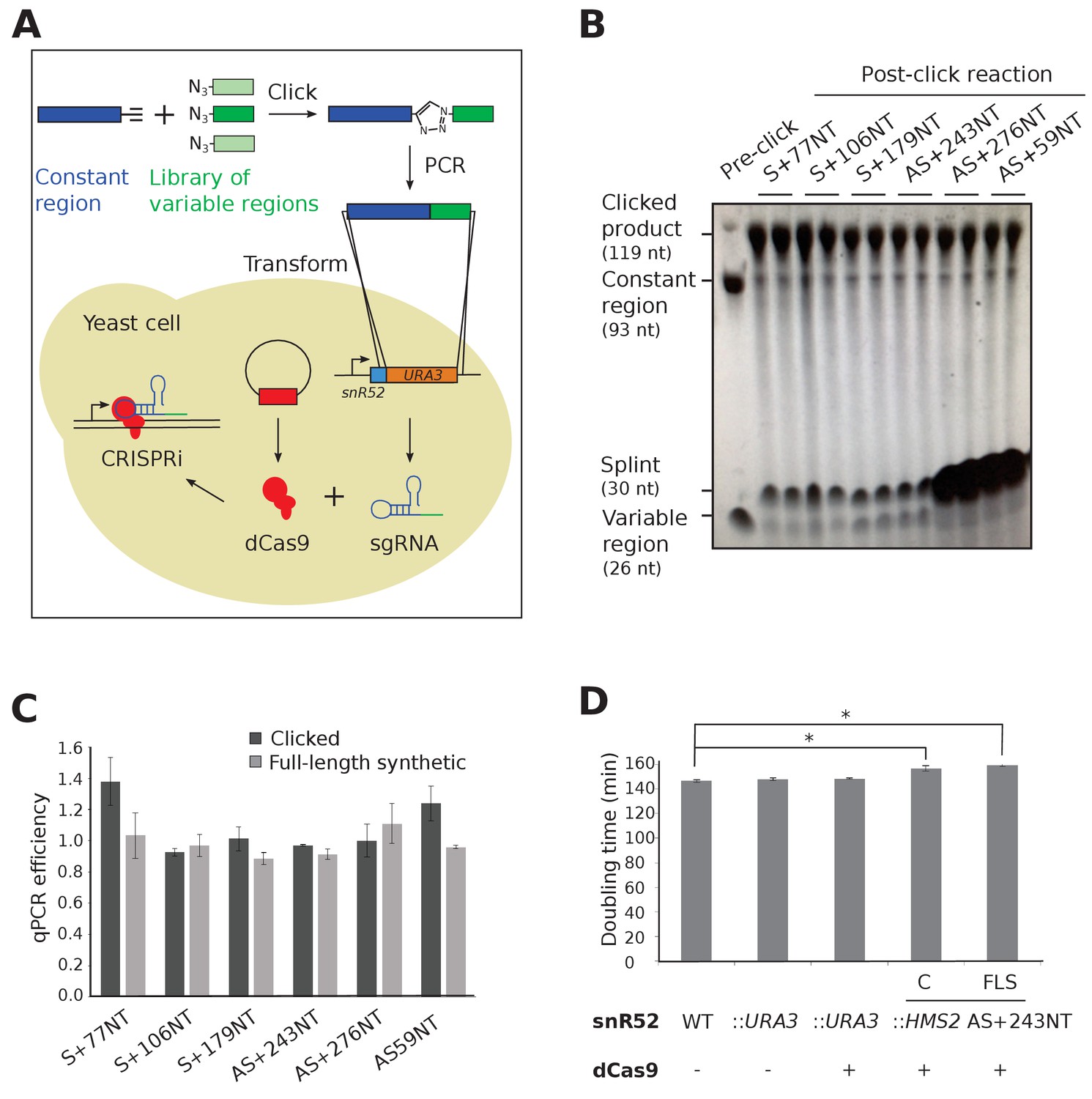
Experimental strategy for sgRNA template synthesis and incorporation into yeast.
(A) An outline of the experiment. DNA templates for sgRNA production were generated using a click chemistry reaction between a single-stranded DNA oligonucleotide with a 3’ alkyne group encoding the constant region (dark blue) and a number of single-stranded DNA oligonucleotides with 5’ azido groups encoding the different variable regions (green). The resulting single-stranded DNA oligonucleotide was purified, amplified by PCR and transformed into yeast to replace the URA3 selection cassette that had previously been introduced into the endogenous snR52 locus. Correct insertion was confirmed by Sanger sequencing. The expression of the sgRNA is driven from the endogenous RNA polymerase III snR52 promoter and processed to produce a mature sgRNA. The sgRNA couples with enzymatically dead Cas9 (dCas9, red), expressed under the control of the TDH3 promoter off a plasmid, to block transcription. See Materials and methods for more detail. (B) A polyacrylamide gel visualised by UV shadowing reveals a high efficiency of the click reaction for all the HMS2 constructs (see Figure 2E and Figure 3—figure supplement 1 for positions of the sgRNA binding sites). The sizes of the variable region, DNA splint, constant region and clicked product are indicated. (C) PCR efficiencies of the clicked and full-length synthetic DNA oligonucleotides as measured by qPCR with a serial dilution series. N = 3, errors are standard error of the mean (SEM), all differences are not significant p>0.05. See Source data 1, tab 2. (D) Doubling times (min) of the indicated yeast strains grown in complete synthetic media minus leucine. C, strains constructed using clicked oligonucleotides; FLS, strains constructed using full-length synthetic oligonucleotides. N = 10, errors are SEM, *p<0.05. See Source data 1, tabs 3–4.
CRISPRi represses the production of antisense transcripts at HMS2 and GAL1
CRISPRi represses transcription when sgRNAs/dCas9 are targeted to the non-template (NT) strand next to a protospacer adjacent motif (PAM) (Qi et al., 2013) (Figure 2A). Firstly, we used an engineered version of GAL1 that has a stable antisense transcript (GAL1 AS) initiating within an ADH1 terminator inserted into the GAL1 coding region (Murray et al., 2012) (Figure 2B). GAL1 AS is present in cells grown in glucose-containing media when the GAL1 gene is repressed and is reduced as cells are switched into galactose-containing media and GAL1 sense is induced (Figure 2C lanes 1–3). We designed sgRNAs adjacent to two PAM sequences on the non-template strand near the antisense transcription start site (TSS) (AS+28NT and AS+112NT) and a third strand-specificity control sgRNA on the template strand in this region (AS + 93T) (Figure 2B). Only sgRNA AS+112NT/dCas9 caused significant (p=0.004) reduction in GAL1 antisense transcript levels, as assessed by Northern blotting (Figure 2C,D).
Figure 2 with 3 supplements see all

CRISPRi reduces antisense transcripts at GAL1 and HMS2.
(A) A schematic demonstrating the previously reported requirement of non-template strand targeting by the sgRNA/dCas9 complex for strand-specific transcriptional repression. Arrows indicate the direction of transcription. NT, non-template; T, template; red, dCas9; PAM, protospacer adjacent motif; green/blue, sgRNA. (B) A schematic of the engineered GAL1 locus showing the site of insertion of the ADH1 terminator (T, red box), the transcription start site for the stable GAL1 antisense transcript and the positions of the designed sgRNAs (green vertical lines) targeting the non-template (AS+28NT, AS+112NT) and template (AS+93NT) strands with respect to antisense transcription. The position of the Northern blotting probe detecting the GAL1 AS transcript is shown in purple. (C) A Northern blot showing the reduction in GAL1 antisense transcript (black arrowhead) in the strain expressing sgRNA AS+112NT/dCas9 relative to the no sgRNA control (GAL1:ADH1t snR52::URA3 with dCas9). sgRNAs AS+28NT and AS+93T do not alter GAL1 AS levels. Samples were taken from cells grown in glucose (t = 0) and at the indicated times after transfer to galactose-containing media (min). Positions of the rRNA are indicated by the short horizontal lines. Ethidium-bromide-stained rRNA is used as loading control. (D) Quantification of Northern blotting for the GAL1 AS transcript at t = 0 in the control no sgRNA strain and the strain with sgRNA AS+112NT. N = 6, errors are SEM, *p=0.004. See Source data 1, tabs 10–12. (E) A map of the HMS2 locus showing the HMS2 AS transcript, SUT650 (black arrow, transcript B) and positions of the three sgRNAs targeting SUT650 (green vertical lines). The position of the Northern blotting probe to detect SUT650 (H1 AS) is shown by the purple line. (F) A Northern blot probed with HMS2 antisense probe H1 (see schematic in (E)) showing the level of SUT650 (black arrowhead, transcript B) in the no sgRNA control (snR52::URA3 with dCas9) strain and strains expressing the indicated antisense-targeting sgRNAs. Deletion of XRN1 allows detection of a shorter antisense transcript (Bt) in the strain expressing AS+243NT. Positions of the rRNA are indicated by the short horizontal lines. *Represents cross-hybridisation with the 25S rRNA. A blot probed for the 18S rRNA is used as loading control. (G) Quantification of the level of SUT650 (transcript B) reduction in the strains expressing each of the three antisense sgRNAs relative to the control no sgRNA strain. N = 4–8, errors represent SEM, *p<0.05. Click-linked and full-length synthetic sgRNA AS+243NT templates behave similarly. See Source data 1, tabs 5–9. (H) Northern blots with a series of HMS2 antisense-specific probes. A new shorter antisense transcript (Bt) can be detected upon XRN1 deletion in the strains expressing sgRNA AS+243NT/dCas9. Two clones of the CRISPRi strains produced using clicked (C) or full-length synthetic (FLS) DNA oligos for strain construction are shown. Positions of the antisense-specific probes (purple) and the site of sgRNA AS+243NT/dCas9 binding (green vertical line) are shown in the schematic. Positions of the rRNA on the Northern blot are indicated by the short horizontal lines. *Represents cross-hybridisation with the 25S rRNA. Ethidium-bromide-stained rRNA is used as loading control.
Next, we examined the HMS2 locus which has a stable antisense transcript, SUT650, initiating within the 3’ coding region of HMS2 (Nguyen et al., 2014; Pelechano et al., 2013). We designed three SUT650-targeting sgRNAs, located 59, 243 and 276 nucleotides downstream of the major SUT650 TSS on the non-template strand (sgRNAs AS+59NT, AS+243NT, AS+276NT, respectively) (Figure 2E). Note that there is considerable heterogeneity in the SUT650 TSS (Figure 2—figure supplement 1) and thus in some cells, sgRNA AS+59NT may be binding upstream of the SUT650 TSS. AS+243NT/dCas9 significantly (p=0.011) reduced SUT650 (Figure 2F lanes 1 and 3, 2G) and this repression was comparable for the clicked and control full-length synthetic constructs (Figure 2G). However, there was no obvious effect of the other two sgRNAs/dCas9 on SUT650 levels (Figure 2F lanes 1, 4 and 5, 2G). Consistently, we could only detect dCas9 binding to the site of sgRNA AS+243NT binding and not to the other two SUT650-targeting sgRNA binding sites tested (Figure 2—figure supplement 2). CRISPR efficiency has been linked to nucleosome occupancy (Horlbeck et al., 2016b; Isaac et al., 2016), but we found no correlation between the level of repression and the nucleosome occupancy (Knight et al., 2014) at each of the SUT650 sgRNA target regions (Figure 2—figure supplement 3). However, other factors such as sequence determinants can also influence repression (Horlbeck et al., 2016a). These results at GAL1 and HMS2 confirm CRISPRi is suitable for reducing levels of antisense transcripts in yeast but controls are needed for each sgRNA designed to ensure that repression has been achieved.
CRISPRi repression of SUT650 induces a new shorter antisense transcript at HMS2
At HMS2, sgRNA AS+243NT/dCas9 was able to reduce SUT650 transcript levels significantly (Figure 2F,G). Since SUT650 is a substrate for the major cytoplasmic 5’−3’ exonuclease Xrn1 (Figure 2F, lanes 1 and 2), we investigated whether the CRISPRi-induced SUT650 reduction was due to direct repression of SUT650 transcription and/or a reduction in SUT650 stability. Whilst SUT650 was not stabilised upon XRN1 deletion in the strain expressing AS+243NT/dCas9, supporting previous studies that CRISPRi operates via a transcriptional block (Qi et al., 2013) (Figure 2F, lanes 3 and 6, transcript B), we detected a shorter antisense transcript (Figure 2F, lane 6, transcript Bt). To map transcript Bt, we used a series of Northern blotting probes across the locus (Figure 2H). With antisense-specific probes H1 and H2 in the HMS2 gene promoter and 5’ coding region, respectively, we could detect antisense transcript Bt, indicating that it extends into the HMS2 sense promoter. However, probe H4 at the 3’ end of the HMS2 gene was unable to hybridise to Bt, indicating that Bt initiates upstream of this site. So whilst CRISPRi successfully blocked the original SUT650, a new Xrn1-sensitive unstable antisense transcript (Bt) was created. This is potentially due to changes in the chromatin structure caused by AS+243NT/dCas9 binding to this region (Horlbeck et al., 2016b; Isaac et al., 2016) or by blocking SUT650, creating an environment that is permissive for transcription initiation (Murray et al., 2012). We note that using probe H4 we were also unable to detect SUT650 initiating from its WT start site but terminating at the AS+243 NT binding site, due to its small size (~243 nt).
CRISPRi-induced GAL1 AS repression does not affect the GAL1 sense transcript
We tested whether GAL1 AS repression by AS+112NT/dCas9 affected the GAL1 sense transcript. Previously we mutated a TATA-like sequence to ablate antisense transcription but observed no difference in GAL1 sense transcript levels in the population under repressive or activating conditions (Murray et al., 2015). Using CRISPRi, we also observed no significant change in GAL1 sense transcript levels or size (Figure 3A,B). Additionally, we observed no leaky expression of the GAL1 sense transcript under repressive conditions (glucose) when the GAL1 AS transcript was blocked (Figure 3A, t = 0). To support a strand-specific transcriptional block of antisense transcription, GAL1 sense transcript polyA site usage was unaffected by sgRNA AS+112NT/dCas9 (Figure 3C). Furthermore, XRN1 deletion in this strain also did not affect polyA site usage, ruling out a partial double-stranded transcriptional block and subsequent destabilisation of the resulting truncated sense transcript (Figure 3C).
Figure 3 with 1 supplement see all
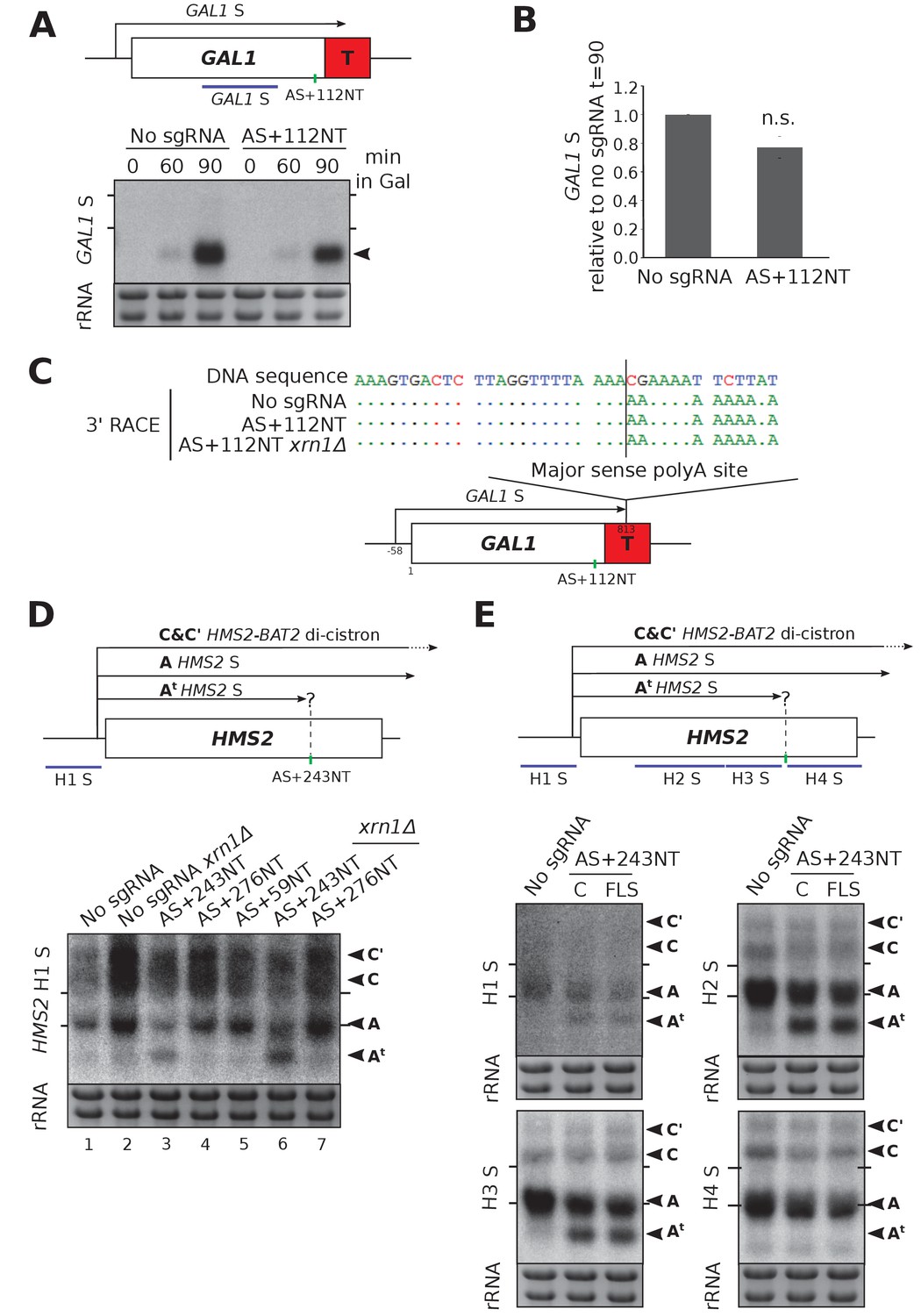
HMS2 and GAL1 sense transcripts are differently affected by blocking of their respective antisense transcripts.
(A) A Northern blot probed for the GAL1 sense transcript (black arrow) in the control strain with no sgRNA (GAL1 ADH1t snR52::URA3 with dCas9) and in the strain expressing sgRNA AS+112NT/dCas9. Samples were taken from cells grown in glucose (t = 0) and at the indicated times after transfer to galactose-containing media (min). Ethidium-bromide-stained rRNA is used as a loading control. (B) Quantification of the Northern blot shown in (A) at t = 90. N = 4, error bars represent SEM, change is statistically non-significant (n.s.) p=0.062. See Source data 1, tabs 10–12. (C) A schematic showing the results of GAL1 sense transcript 3’ end mapping by RACE in the strains indicated. All distances are shown relative to the sense TSS. Transcript cleavage and polyadenylation occurs beyond the position of the GAL1 AS-blocking AS+112NT sgRNA shown (green vertical line). (D) A Northern blot probed with HMS2 sense probe H1 (purple) showing the level of HMS2 sense (black arrowhead, transcript A) in the no sgRNA control (snR52::URA3 with dCas9) strain and strains expressing the indicated antisense-targeting sgRNAs. A truncated sense transcript (At) was also detected in the strain expressing AS+243NT. C and C’ are read-through HMS2-BAT2 transcripts (see [Nguyen et al., 2014]). Positions of the rRNA are indicated by the short horizontal lines. Ethidium-bromide-stained rRNA is used as loading control. (E) Northern blots with a series of HMS2 sense-specific probes detecting the regions indicated in the schematic. The position of the sgRNA AS+243NT/dCas9 binding site is shown (green vertical line). Positions of the rRNA are indicated by the short horizontal lines. Ethidium-bromide-stained rRNA is used as loading control.
CRISPRi-induced SUT650 repression truncates the HMS2 sense transcript
Previous work shows that reducing SUT650 transcription increases HMS2 sense transcript levels (Nguyen et al., 2014). However, blocking SUT650 by AS+243NT/dCas9 did not similarly increase HMS2 sense levels (Figure 3D, lanes 1 and 3). To our surprise, in addition to the full-length HMS2 sense transcript (A), we detected a considerably shortened HMS2 sense transcript (At). Since the combined levels of the truncated and full-length transcripts are similar to those in the strain not expressing an sgRNA, independent of the presence of XRN1 (Figure 3D, lanes 1 and 3 or 2 and 6), we hypothesised that the AS+243NT/dCas9 complex bound within the HMS2 coding region may be causing partial premature sense transcription termination, leading to transcript At. Thus, we mapped the 3’ end of HMS2 S by Northern blotting using a series of strand-specific probes across the locus (Figure 3E). Both transcripts A and At could clearly be detected using the probes upstream of the AS+243NT/dCas9 binding site (probes H1, H2, H3), but the truncated transcript was undetectable using probe H4 downstream of this site. Thus, the transcriptional block caused by AS+243NT/dCas9 is not strand-specific and re-defines the transcriptional landscape at the HMS2 locus by creating a chromatin environment that is suitable for both transcription initiation and termination. To see if we could more directly target and block HMS2 sense transcripts, we designed three sgRNAs to bind the non-template strand adjacent to PAM sites near the HMS2 sense TSS (S+77NT, S+106NT, S+179NT) and monitored their effects on both HMS2 sense and antisense transcripts (Figure 3—figure supplement 1). However, these sgRNAs gave no CRISPRi repression as indicated by the unchanged transcript levels and integrity.
CRISPRi-induced antisense repression at HMS2 and GAL1 is not as efficient as previous methods
We compared the repression efficiency of CRISPRi with our previously used genetic interventions. At GAL1, mutation of a TATA-like sequence within the ADH1 terminator greatly reduced the level of the GAL1 antisense transcript (Figure 4A,B) (Murray et al., 2015). Lowered GAL1 AS transcript levels could result from either reduced transcription or transcript destabilisation (or a combination of the two). To differentiate between these possibilities, we deleted the major cytoplasmic 5’−3’ exonuclease XRN1. Since XRN1 deletion in the TATA mutant strain only slightly increased GAL1 AS levels, this suggests that most repression was at the level of transcription rather than altering transcript stability (Figure 4B,C). By contrast, XRN1 deletion in the strain expressing AS+112NT/dCas9 did result in some upregulation of GAL1 AS (Figure 4B,C), indicating that antisense transcript repression was not as great in this strain. Unlike at HMS2 (Figure 2F,G), this stabilised GAL1 AS transcript was the same size as in the control and so likely represents stabilisation of the transcripts escaping repression rather than novel transcripts.
Figure 4
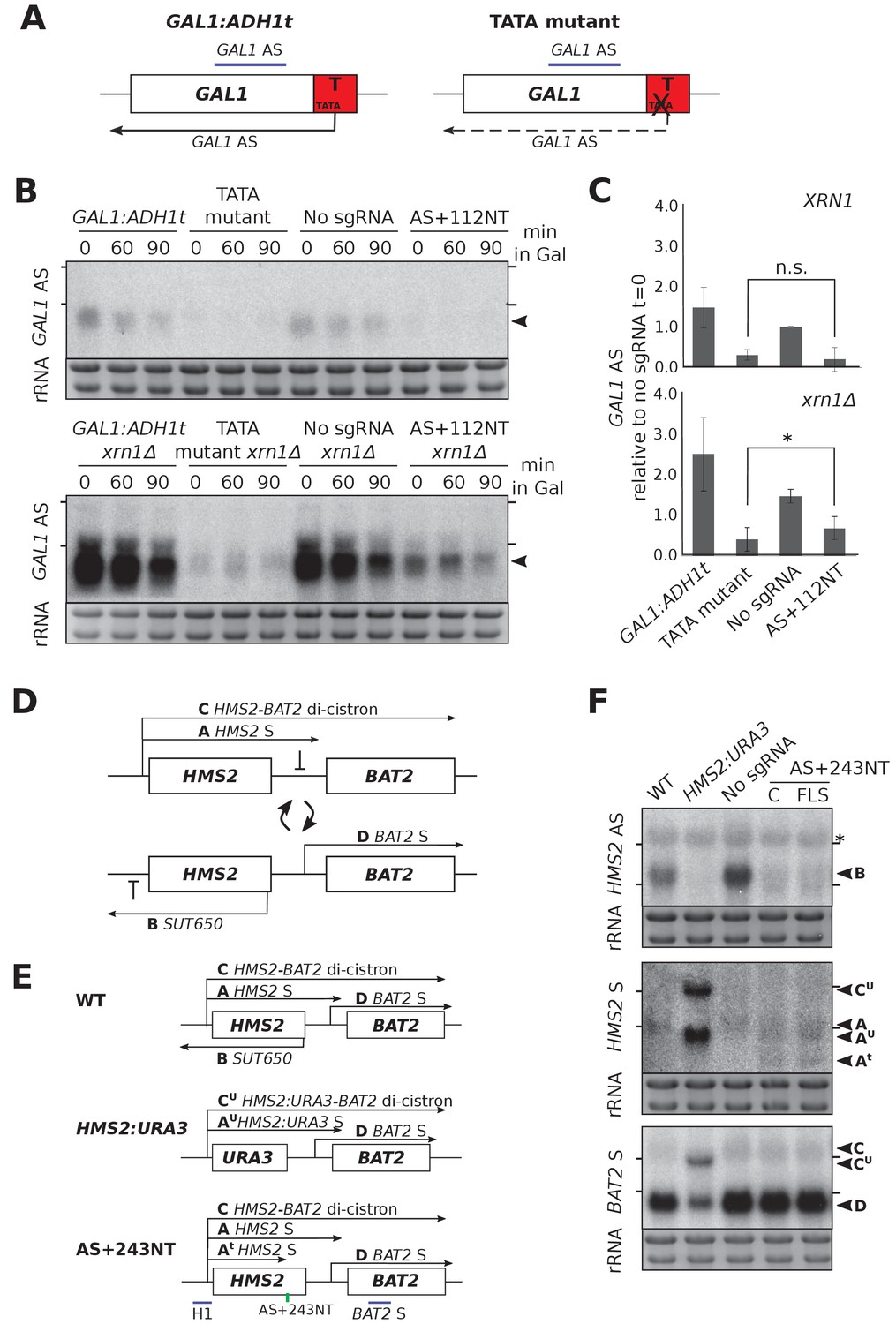
CRISPRi block of SUT650 and GAL1 AS is not as efficient as previous genetic interventions.
(A) A schematic showing the previously used genetic intervention to repress the GAL1 AS transcript (Murray et al., 2015). In the TATA mutant construct, a TATA box within the engineered ADH1 terminator (T) is scrambled and the level of GAL1 antisense transcript is reduced. (B) A Northern blot probed for the GAL1 antisense transcript (black arrowhead) comparing the two methods of reducing antisense at this locus in the presence (top blot) or absence (bottom blot) of XRN1. All samples were run on the same gel and the blots were exposed to film for the same time. Samples were taken from cells grown in glucose (t = 0) and at the indicated times after transfer to galactose-containing media (min). Positions of the rRNA are indicated by the short horizontal lines. Ethidium-bromide-stained rRNA is used as loading control. (C) Quantification of the Northern blot in (B) at t = 0. N = 2, errors represent the SEM, *p=0.026, n.s. non-significant. See Source data 1, tab 12. (D) A schematic of the HMS2-BAT2 locus, showing the HMS2 sense and antisense transcripts (A and B), the HMS2-BAT2 di-cistronic transcripts (C and C’) and the BAT2 sense transcript (D). Cells switch between sense- and antisense-dominant states in which the indicated transcripts are produced (Nguyen et al., 2014). (E) The HMS2-BAT2 locus and associated transcripts in the wild-type (WT) strain and strains that repress the SUT650 transcript. In the HMS2:URA3 strain, the entire coding region of HMS2 is replaced by that of URA3. (F) Northern blots comparing the efficiency of SUT650 reduction by replacement of the HMS2 coding region with that of URA3 versus CRISPRi with sgRNA AS+243NT and the relative effects of these two strategies on the HMS2 sense and BAT2 transcripts shown in (E). The WT (BY4741) and HMS2.URA3 strains have been transformed with plasmid pRS315 to allow for growth on complete synthetic media lacking leucine and thus comparisons with the CRISPRi strains. Positions of the rRNA are indicated by the short horizontal lines. *Represents cross-hybridisation with the 25S rRNA. Ethidium-bromide-stained rRNA is used as loading control.
Next we compared the efficacy of AS+243NT/dCas9 with previous experiments to ablate SUT650, where we replaced the entire HMS2 coding region, including the antisense TSS with the URA3 coding region (HMS2:URA3) (Nguyen et al., 2014). This successful block of SUT650 transcription increased levels of the HMS2 sense transcript, the di-cistronic HMS2-BAT2 transcript and consequently decreased levels of the downstream gene, BAT2, due to transcriptional interference (Figure 4D–F). Whilst CRISPRi resulted in a similar decrease in SUT650 as HMS2:URA3, neither the HMS2, BAT2 nor HMS2-BAT2 di-cistronic transcripts were altered (Figure 4F). The discovery that whilst AS+243NT/dCas9 represses SUT650, a new unstable HMS2 antisense transcript is induced and the sense transcript is prematurely terminated could explain why blocking SUT650 using CRISPRi and the URA3 gene body replacement strategies did not give the same results (Figure 4F). Thus, CRISPRi is not as effective as a genetic mutation in reducing levels of either the GAL1 or HMS2 AS transcripts.
Concluding remarks
Although CRISPRi has been used to strand-specifically repress antisense transcription at GAL10 (Lenstra et al., 2015) and GAL1 (this work), here we demonstrate that this is not true at HMS2. This may reflect the vastly different transcription levels for the galactose-inducible genes (high) compared to HMS2 (low). Furthermore, a study in human K562 cells found that CRISPRi-induced repression did not correlate with which strand was targeted at 49 genes, although the mechanism behind this observation was not investigated (Gilbert et al., 2014).
This work extends our previous hypothesis (Nguyen et al., 2014), allowing us to propose that, rather than acting as a roadblock, the binding of the sgRNA/dCas9 complex at HMS2 creates a new chromatin environment, either directly or indirectly via the blocking of antisense transcription, which is permissible for transcription initiation and termination. Thus, new transcription units are generated that result in novel sense and antisense transcripts of varying stabilities that are therefore not always detectable. This redefinition of the transcriptional landscape highlights the levels of controls and locus understanding needed before results from using CRISPRi can be interpreted.
Materials and methods
Key resource table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Gene (Sacccharomyces cerevisiae) | HMS2 | YJR147W; SGD ID S000003908; RRID:SCR_003093; SGD, RRID:SCR_004694 | ||
| Gene (S. cerevisiae) | GAL1 | YBR020W; SGD ID S000000224; RRID:SCR_003093; SGD, RRID:SCR_004694 | ||
| Gene (S. cerevisiae) | BAT2 | YJR148W; SGD ID S000003909 RRID:SCR_003093; SGD, RRID:SCR_004694 | ||
| Gene (S. cerevisiae) | snR52 | SGD ID S000006443; RRID:SCR_003093; SGD, RRID:SCR_004694 | ||
| Strain, strain background (S. cerevisiae) | see Supplementary file 1C | see Supplementary file 1C | Saccharomyces cerevisiae, NCBITaxon:1247190; SGD, RRID:SCR_004694 | see Supplementary file 1C |
| Genetic reagent (S. cerevisiae) | see Supplementary file 1C | see Supplementary file 1C | see Supplementary file 1C | see Supplementary file 1C |
| Antibody | Tubulin (rat monoclonal YL1/2) | Abcam | Abcam ab6160 RRID:AB_305328 | (1:3333) |
| Antibody | Cas9 (rabbit polyclonal) | Diagenode | C15310258 RRID:AB_2715516 | (1:5000 Western blot); 5 µl/ChIP |
| Recombinant DNA reagent | see Supplementary file 1A | Eurofins Genomics | see Supplementary file 1A | see Supplementary file 1A |
| Recombinant DNA reagent | Plasmid with pTDH-dCas9 | (Qi et al., 2013) Addgene | Addgene #46920 RRID:SCR:002037 | PMID:23849981 |
| Sequence-based reagent | single-stranded DNA templates for variable and constant sgRNA regions | This study | see Supplementary file 1A | synthesised by phosphoramidite oligonucleotide synthesis with 3’-alkyne or 5’-azido modifications (El-Sagheer et al., 2011) |
| Sequence-based reagent | see Supplementary files 1A,B,D,E,F | Eurofins Genomics | see Supplementary files 1A,B,D,E,F | see Supplementary files 1A,B,D,E,F |
| Commercial assay or kit | QIAquick PCR Purification Kit | Qiagen; RRID:SCR_008539 | Cat# 28106 | |
| Chemical compound, drug | SYBR green qPCR master mix | Bioline | Cat# QT605-20 | |
| Software, algorithm | Off-spotter sgRNA algorithm | https://cm.jefferson.edu/Off-Spotter// | ||
| Software, algorithm | Rotor-Gene Q Series Software | https://www.qiagen.com/us/resources/ | ||
| Software, algorithm | Fiji/Image J | https://imagej.net/Fiji | ImageJ, RRID:SCR_003070 |
sgRNA design
Request a detailed protocolPAM sequences within 300 bp of the TSS in question were identified and the 20 bp immediately adjacent to these were used to design the variable regions of the sgRNAs. These sequences were run through an off-spotter algorithm (https://cm.jefferson.edu/Off-Spotter//) to minimise any off-target effects (Pliatsika and Rigoutsos, 2015). The sequence for the engineered constant region and SUP4 terminator were taken from (DiCarlo et al., 2013; Mali et al., 2013). The HMS2 sgRNA templates were synthesised as two DNA oligonucleotides to be joined by click chemistry, with the majority of the constant region on one oligonucleotide that could be used for all reactions. To increase the efficiency of the click reaction, the click linkage was designed between a dCpT dinucleotide within the Cas9 handle region. The sequences for the HMS2 and GAL1 sgRNA templates are shown in Supplementary file 1A. Full-length sgRNA templates were also synthesised for the GAL1 sgRNAs and as a control for the HMS2 click chemistry constructs (Eurofins Genomics, Germany).
Click chemistry
Request a detailed protocolSingle-stranded DNA oligonucleotides complementary to the constant and variable sgRNA regions were synthesised by standard phosphoramidite oligonucleotide synthesis with 3’-alkyne or 5’-azido modifications respectively (El-Sagheer et al., 2011) (Supplementary file 1A). The click chemistry reaction was performed with a Cu1 catalyst and 24–30 nt splint DNAs (El-Sagheer et al., 2011). PCR was performed for second strand synthesis, amplification and incorporation of sequences homologous to the site of insertion in S. cerevisiae. PCR efficiencies were obtained using qPCR performed three times on a Corbett 6000 Rotorgene with four serial 10-fold dilutions in triplicate. Analysis was performed using Rotor-Gene Q Series Software (Qiagen, Hilden, Germany).
Strain construction
Request a detailed protocoldCas9, without fusion to a transcriptional repressor domain, was expressed from a plasmid (# 46920 Addgene [Qi et al., 2013]) under the control of the TDH3 promoter. The DNA templates for sgRNA production were integrated in place of an exogenous URA3 cassette immediately downstream of the splice site in the endogenous snR52 locus using homologous recombination followed by 5-FOA selection. Correct insertion was confirmed by genomic DNA Sanger sequencing. SNR52 is a Pol III-transcribed C/D box small nucleolar RNA (snoRNA) gene and so does not undergo extensive post-transcriptional processing such as capping and polyadenylation, and transcripts from this locus are retained in the nucleus. Additionally, the locus contains a self-splicing site that produces a mature transcript without additional machinery, allowing precise production of a mature sgRNA without any unwanted extensions. Primer sequences for strain construction are listed in Supplementary file 1B.
Yeast growth
Request a detailed protocolThe strains used in this study are listed in Supplementary file 1C. Strains were grown to mid-log at 30°C in complete synthetic media lacking leucine (for dCas9 plasmid selection). For experiments studying the GAL1 locus, yeast were grown to mid-log in rich media (YP 2% D/YP 2% Gal) so that the GAL1 induction kinetics were similar to what we had observed previously (Murray et al., 2012; 2015). dCas9 expression was unaffected by the temporary absence of plasmid selection (Figure 1—figure supplement 1).
Yeast growth controls
Request a detailed protocolAssessment of growth in liquid culture (CSM-leucine) was achieved using a Bioscreen spectrophotometer that automatically measures the optical density of cultures at 30°C every 20 min for 24 hr. Doubling times were extracted from the gradient of the curves during logarithmic growth.
Northern blotting
Request a detailed protocolNorthern blotting was performed as before (Murray et al., 2012; Nguyen et al., 2014) using asymmetric PCR or in vitro transcription with T7 RNA polymerase to generate the radioactive strand-specific probes for GAL1 and HMS2, respectively. Primers for these reactions are listed in Supplementary file 1D. Northern blots were quantified using Fiji/ImageJ (Schindelin et al., 2012), and images were acquired using a FLA 7000 phosphorimager (GE Healthcare). Images in the figures are scans of exposures to X-ray film.
Western blotting
Request a detailed protocoldCas9 expression was confirmed after each experiment using an anti-Cas9 antibody (Diagenode C15310258) at 1:5000 dilution and anti-Tubulin (Abcam ab6160) at 1:3333.
Chromatin immunoprecipitation
Request a detailed protocolYeast cells were fixed with 1% formaldehyde (30 min, 22°C) followed by the addition of glycine to 125 mM for 5 min. Cell pellets were collected by centrifugation (3000 rpm, 5 min) before washing twice with 10 ml cold PBS. Cells were re-suspended in cold FA-150 buffer (10 mM HEPES pH 7.9, 150 mM NaCl, 0.1% SDS, 0.1% sodium deoxycholate, 1% Triton X-100) and broken using 1-ml glass beads on a MagnaLyser (Roche) at (2 × 1 min, 6000 rpm, 4°C). Fixed chromatin was sheared by sonication using a biorupter (Diagenode, 30 min, 30 s on, 30 s off, high setting), cleared by centrifugation (10,000 rpm, 15 min, 4°C) and incubated with 5 µl anti-Cas9 antibody (Diagenode C15310258) and 30 µl protein A-dynabeads pre-blocked with 200 µg/ml yeast tRNA, 200 µg/ml BSA and 200 µg/ml glycogen (15–20 hr, 4°C). Beads and attached chromatin were collected using a magnetic rack and washed with TSE-150 buffer (20 mM Tris-Cl pH 8.0, 150 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100) for 3 min, TSE-500 buffer (20 mM Tris-Cl pH 8.0, 500 mM NaCl, 2 mM EDTA, 0.1% SDS, 1% Triton X-100) for 3 min, LiCl buffer (0.25 M LiCl, 10 mM Tris-Cl pH 8.0, 1 mM EDTA, 1% dioxycholate, 1 % NP-40) for 15 min and twice with TE, all at 22°C. After washing, chromatin was eluted from the beads for 30 min at 65°C with elution buffer (0.1 M NaHCO3, 1% SDS). Addition of 350 mM NaCl and incubation for 3 hr at 65°C reversed the cross-links before treatment of samples with RNase A for 1 hr at 37°C and proteinase K overnight at 65°C. DNA was purified using a PCR-purification kit (Qiagen) and eluted in 400 µl 1 mM Tris-Cl pH 8.0. Input DNA was diluted accordingly. Real-time quantitative PCR (qPCR) was carried out using a Corbett 6000 Rotorgene and Sybr green qPCR master mix (Bioline). Data ([IP - no antibody control]/input) were expressed as a percentage of the input (relative to sgRNA +276 NT). Real-time qPCR primers are listed in Supplementary file 1E.
3’ RACE mapping
Request a detailed protocolMapping of the 3’ end of the GAL1 sense transcript was performed as (Nguyen et al., 2014). Primers are listed in Supplementary file 1F.
References
-
Transcription of click-linked DNA in human cellsAngewandte Chemie International Edition 53:2362–2365.https://doi.org/10.1002/anie.201308691
-
Genome engineering in Saccharomyces cerevisiae using CRISPR-Cas systemsNucleic Acids Research 41:4336–4343.https://doi.org/10.1093/nar/gkt135
-
Efficient RNA synthesis by in vitro transcription of a triazole-modified DNA templateChemical Communications 47:12057–12058.https://doi.org/10.1039/c1cc14316f
-
Sense and antisense transcription are associated with distinct chromatin architectures across genesNucleic Acids Research 43:7823–7837.https://doi.org/10.1093/nar/gkv666
-
Gene regulation by antisense transcriptionNature Reviews Genetics 14:880–893.https://doi.org/10.1038/nrg3594
-
Fiji: an open-source platform for biological-image analysisNature Methods 9:676–682.https://doi.org/10.1038/nmeth.2019
Article and author information
Author details
Funding
Biotechnology and Biological Sciences Research Council (BB/J001694/2)
- Tom Brown
- Jane Mellor
Wellcome (209897/Z/17/Z)
- Anna R Lamstaes
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
The authors thank Jack Feltham for assistance with sgRNA template construction.
Copyright
© 2017, Howe et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,826
- views
-
- 711
- downloads
-
- 32
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 32
- citations for umbrella DOI https://doi.org/10.7554/eLife.29878
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
CRISPRi is not strand-specific at all loci and redefines the transcriptional landscape
eLife 6:e29878.
https://doi.org/10.7554/eLife.29878




{kind=link}
{kind=link}
{kind=link}
{kind=link}