Endoplasmic reticulum-associated SKN-1A/Nrf1 mediates a cytoplasmic unfolded protein response and promotes longevity
- Massachusetts General Hospital, United States
- Harvard Medical School, United States
Abstract
Unfolded protein responses (UPRs) safeguard cellular function during proteotoxic stress and aging. In a previous paper (Lehrbach and Ruvkun, 2016) we showed that the ER-associated SKN-1A/Nrf1 transcription factor activates proteasome subunit expression in response to proteasome dysfunction, but it was not established whether SKN-1A/Nrf1 adjusts proteasome capacity in response to other proteotoxic insults. Here, we reveal that misfolded endogenous proteins and the human amyloid beta peptide trigger activation of proteasome subunit expression by SKN-1A/Nrf1. SKN-1A activation is protective against age-dependent defects caused by accumulation of misfolded and aggregation-prone proteins. In a C. elegans Alzheimer’s disease model, SKN-1A/Nrf1 slows accumulation of the amyloid beta peptide and delays adult-onset cellular dysfunction. Our results indicate that SKN-1A surveys cellular protein folding and adjusts proteasome capacity to meet the demands of protein quality control pathways, revealing a new arm of the cytosolic UPR. This regulatory axis is critical for healthy aging and may be a target for therapeutic modulation of human aging and age-related disease.
https://doi.org/10.7554/eLife.44425.001Introduction
Loss of proteostasis and accumulation of damaged and misfolded proteins is a hallmark of aging (López-Otín et al., 2013). Cells detect protein misfolding and activate unfolded protein responses (UPRs) that adjust protein metabolism to restore proteostasis (Pilla et al., 2017). These changes include inhibition of translation to limit synthesis of new proteins, upregulation of chaperones that mediate protein folding, and enhanced destruction of misfolded proteins via the proteasome or autophagy. Protein damage that accrues over time appears to eventually overcome these homeostatic mechanisms and contributes to the decline in cellular and organismal health during aging. Mutations that persistently increase production of unfolded proteins or that impair their clearance accelerate this process to cause a number of adult-onset neurodegenerative diseases (Hipp et al., 2014). Conversely, increasing the activity of UPR pathways to enhance proteostasis may be a means to combat these diseases or even aging itself (Powers et al., 2009; Taylor and Dillin, 2011).
The proteasome mediates the targeted degradation of misfolded and damaged proteins and is essential for proteostasis and cell viability (Collins and Goldberg, 2017). Impaired proteasome function is associated with aging and age-dependent neurodegenerative diseases (Saez and Vilchez, 2014). The SKN-1A/Nrf1 transcription factor regulates the transcription of proteasome subunit genes to increase proteasome biogenesis when the proteasome is inhibited, for example by proteasome inhibitor drugs (Grimberg et al., 2011; Lehrbach and Ruvkun, 2016; Radhakrishnan et al., 2010; Steffen et al., 2010). This compensatory response is essential for the survival of mammalian cells and C. elegans under conditions of impaired proteasome function (Lehrbach and Ruvkun, 2016; Radhakrishnan et al., 2010). SKN-1A/Nrf1 is an unusual transcription factor that associates with the ER via an N-terminal transmembrane domain (Glover-Cutter et al., 2013; Wang and Chan, 2006). The bulk of SKN-1A/Nrf1 extends into the ER lumen where it undergoes N-linked glycosylation at particular asparagine residues (Radhakrishnan et al., 2014; Zhang et al., 2007). After it is glycosylated, SKN-1A/Nrf1 is translocated from the ER lumen to the cytoplasm by the ER-associated degradation (ERAD) machinery, which also targets this short half-life transcription factor for rapid proteasomal degradation (Lehrbach and Ruvkun, 2016; Steffen et al., 2010). Under conditions of impaired proteasome function, the SKN-1A/Nrf1 half-life is dramatically increased so that some of the protein escapes degradation and enters the nucleus where it can up-regulate target genes (Lehrbach and Ruvkun, 2016; Li et al., 2011; Radhakrishnan et al., 2010; Steffen et al., 2010). All proteasome subunit genes are direct transcriptional targets of SKN-1A/Nrf1 (Niu et al., 2011; Sha and Goldberg, 2014). Activation of SKN-1A/Nrf1 also requires deglycosylation by the peptide N-glycanase PNG-1/NGLY1 and proteolytic cleavage by the aspartic protease DDI-1/DDI2 (Koizumi et al., 2016; Lehrbach and Ruvkun, 2016; Tomlin et al., 2017). It is not yet known whether the SKN-1A/Nrf1 transcription factor regulates proteasome levels in response to other proteotoxic insults.
Here we show that SKN-1A increases proteasome subunit gene expression in response to endogenous misfolded proteins or expression of a foreign aggregation-prone protein, the human amyloid beta peptide. This pathway requires the DDI-1/DDI2 aspartic protease and the PNG-1/NGLY1 peptide N-glycanase, factors that are also required for activation of SKN-1A during proteasome dysfunction. C. elegans mutants that lack SKN-1A show enhanced age-dependent toxicity of misfolding proteins, accelerated tissue degeneration during aging and reduced overall lifespan. Conversely, increasing SKN-1A levels is sufficient to extend C. elegans lifespan. Our data suggests that SKN-1A/Nrf1 mediates an unfolded protein response that adjusts proteasome capacity to ensure protein quality control. This pathway preserves cellular function during aging by limiting accumulation of unfolded and damaged proteins.
Results
Misfolded proteins trigger SKN-1A activation
A transgene expressing GFP from the rpt-3 proteasome subunit gene promoter shows SKN-1A-dependent upregulation in response to proteasome dysfunction (Lehrbach and Ruvkun, 2016). To explore the genetic defects that can activate such proteasome response pathways and the mechanisms that control proteasome subunit gene expression, we performed a large-scale random EMS-mutagenesis screen for mutants that cause increased expression of rpt-3p::gfp. We isolated 21 alleles affecting proteasome subunit genes, including mutations affecting components of the 19S regulatory particle and the 20S catalytic core of the proteasome (Table 1, Figure 1—figure supplement 1). Many of the mutants show temperature sensitive defects in fertility, consistent with previous genetic analysis of proteasome function in C. elegans germline development (Figure 1—figure supplement 2) (Shimada et al., 2006). Some proteasome mutant strains show severe temperature sensitive developmental defects that may reflect temperature-sensitivity of the mutant protein (Table 1). Activation of rpt-3p::gfp in proteasome hypomorphic mutants requires skn-1 and depletion of SKN-1 by RNAi causes larval lethality in all but one of the mutant strains, although skn-1(RNAi) is not larval lethal in wild type (Table 1, Figure 1—figure supplement 1). These data indicate that a wide range of perturbations to proteasome function trigger SKN-1A activation and confirm that compensatory upregulation of proteasome subunit genes by SKN-1A is critical for survival of proteasome dysfunction, either due to mutations or pharmacological inhibition (Keith et al., 2016; Lehrbach and Ruvkun, 2016).
Table 1
Protesome subunit mutants.
https://doi.org/10.7554/eLife.44425.002| genotype | allele effect | Viability at 20ºC | Viability at 25ºC | rpt-3p::gfp induction on skn-1(RNAi) | growth on skn-1(RNAi) |
| wild type | + | Yes | Yes | + | |
| pas-1(mg511) | G82R | Yes | No (Ste) | lost | Lva |
| pbs-2(mg581) | C90Y | Yes | Yes | lost | Lva |
| pbs-2(mg538) | G93E | Yes | Yes | lost | Lva |
| pbs-2(mg530) | D97N | Yes | ND | lost | Lva |
| pbs-3(mg527) | S180L | Yes | Yes | lost | Lva |
| pbs-4(mg539) | M48K | Yes | No (Emb/Lva) | lost | Lva |
| pbs-5(mg509) | 3'UTR | Yes | Yes | lost | Lva |
| pbs-5(mg502) | promoter* | Yes | Yes | lost* | +* |
| rpt-6(mg513) | I302N, P328S | Yes | Yes | lost | Lva |
| rpt-6(mg512) | E278K | Yes | No (Ste) | lost | Lva |
| rpn-1(mg514) | S519F | Yes | No (Ste) | lost | Lva |
| rpn-1(mg537) | G431E | Yes | No (Ste) | lost | Lva |
| rpn-5(mg534) | T76I | Yes | No (Emb/Lva) | lost | Lva |
| rpn-8(mg587) | G73R | Yes | No (Ste) | lost | Lva |
| rpn-8(mg536) | A88V | Yes | No (Ste) | lost | Lva |
| rpn-9(mg533) | G357STOP | Yes | No (Emb/Lva) | lost | Lva |
| rpn-10(mg525) | G114E | Yes | No (Ste) | lost | Lva |
| rpn-10(mg495) | K130STOP | Yes | No (Ste) | lost | Lva |
| rpn-10(mg531) | Frameshift | Yes | ND | lost | Lva |
| rpn-10(mg529) | Q298STOP | Yes | ND | lost | Lva |
| rpn-11(mg494) | E108K | Yes | No (Ste) | lost | Lva |
-
ND: Not determined
Our large genetic screen also identified three alleles of unc-54, which encodes a myosin class II heavy chain (MHC B) expressed in the body wall muscle (Ardizzi and Epstein, 1987; Epstein et al., 1974). UNC-54 is the major MHC B in body wall muscles and unc-54 loss of function mutations cause paralysis. The unc-54 alleles we isolated activate rpt-3p::gfp specifically in body wall muscle cells (Figure 1a,b), unlike the proteasome mutations which activate rpt-3p::gfp in many tissues. To understand how MHC B affects rpt-3p::gfp, we tested other unc-54 alleles. The temperature-sensitive unc-54(e1301) and unc-54(e1157) alleles encode mutant forms of UNC-54/MHC B that are prone to misfold and aggregate (Ben-Zvi et al., 2009; Gidalevitz et al., 2006; Silva et al., 2011). Both unc-54(e1301) and unc-54(e1157) activate expression of rpt-3p::gfp in muscle cells (Figure 1a,b). By contrast, unc-54(e190), a null (deletion) allele that eliminates MHC B expression and causes paralysis regardless of temperature (Dibb et al., 1985), does not activate rpt-3p::gfp (Figure 1a,c,e). Interestingly, all of the unc-54 alleles we isolated in our screen for proteasome subunit activation are missense mutations that cause temperature-sensitive paralysis similarly to unc-54(e1301) (Figure 1c, Figure 1—figure supplement 3). These data suggest that rpt-3p::gfp activation is triggered by the presence of mutant forms of MHC B that are prone to misfold, not simply by loss of MHC B or defective muscle function.
Figure 1 with 5 supplements see all
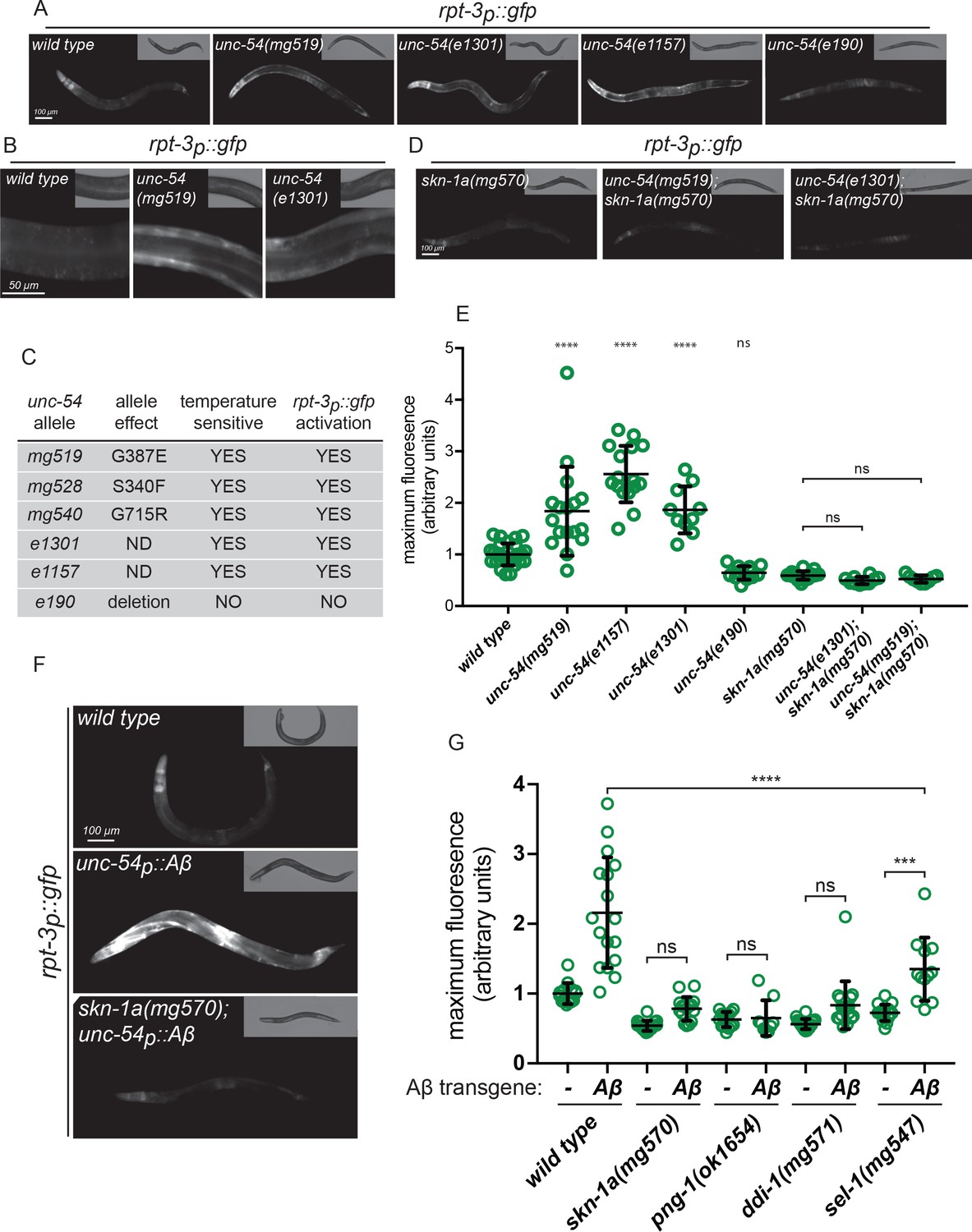
Misfolded proteins activate SKN-1A.
(a, b) Fluorescence images showing rpt-3p::gfp expression in various unc-54 mutants. (c) Temperature dependent paralysis and rpt-3p::gfp effects of unc-54 alleles. (d) Fluorescence images showing rpt-3p::gfp induction in unc-54(mg519) and unc-54(e1301) requires skn-1a. (e) Quantification of rpt-3p::gfp expression in various unc-54 mutants. (f) Fluorescence images showing Aβ expression in muscle increases rpt-3p::gfp fluorescence in wild type but not in skn-1a mutant animals. (g) Quantification of Aβ-induced activation of rpt-3p::gfp in various mutant backgrounds. Panels e and g: ****p<0.0001; ***p<0.001; ns p>0.05. (one-way ANOVA with Tukey’s multiple comparison test), P-value compared to wild type unless otherwise indicated.
Activation of rpt-3p::gfp expression by temperature-sensitive mutant MHC B is completely lost in skn-1a(mg570) mutant animals that lack SKN-1A but retain other SKN-1 isoforms (Figure 1d,e). To test for activation of SKN-1A at the protein level, we used a transgene to ubiquitously express a truncated form of SKN-1A that lacks the DNA binding domain and is fused to GFP at the C-terminus (rpl-28p::skn-1a[∆DBD]::gfp). This protein undergoes the same post-translational regulation as full length SKN-1A (Lehrbach and Ruvkun, 2016). We found increased levels of SKN-1A[∆DBD]::GFP accumulates specifically in the body wall muscle cells of unc-54(e1301) and unc-54(mg519) animals but not in the wild type (Figure 1—figure supplement 4). We conclude that expression of temperature-sensitive mutant UNC-54/MHC B triggers rpt-3p::gfp expression via activation of SKN-1A.
Activation of proteasome subunit expression in animals expressing mutant MHC B might reflect a general response to accumulation of misfolded proteins. To test the model that unfolded proteins engage SKN-1A, we examined the response to another misfolded protein, the human amyloid beta peptide (Aβ). Aβ is derived from the posttranslational processing of the Amyloid precursor protein (APP). Mutations that increase production of Aβ or impair its clearance are associated with Alzheimer’s disease. In Alzheimer’s disease, Aβ forms aggregates that may play an important role in pathogenesis (Selkoe and Hardy, 2016). Transgenic C. elegans that express human Aβ in muscle cells (unc-54p::Aβ) show adult-onset defects in muscle function and serve as a model for the cell biology of Aβ accumulation and toxicity (Link, 2006). We found that Aβ expression in muscle triggers strong muscle-specific activation of rpt-3p::gfp, which is lost in skn-1a(mg570) mutant animals that lack the transmembrane-domain-containing Nrf1 orthologue SKN-1A (Figure 1f).
To test whether SKN-1A activation is broadly associated with protein folding defects, we monitored rpt-3p::gfp activation in hsf-1(sy441) heat shock transcription factor mutants. hsf-1 encodes the C. elegans orthologue of HSF1, which regulates expression of multiple cytoplasmic chaperones under proteotoxic stress conditions such as elevated temperature (Fujimoto and Nakai, 2010). hsf-1(sy441) is a hypomorphic allele that disrupts chaperone regulation (Hajdu-Cronin et al., 2004). hsf-1(sy441) mutant animals develop normally at lower temperatures, but arrest larval development at 25°C, presumably due to the toxic accumulation of misfolded proteins in the cytoplasm. hsf-1(sy441) L4 larvae raised at 20°C show unaltered expression of rpt-3p::gfp compared to the wild type. However, rpt-3p::gfp expression is significantly increased in hsf-1 mutant animals following upshift to 25°C for 24 hr (Figure 1—figure supplement 5). This activation of rpt-3p::gfp in the hsf-1 mutant requires SKN-1A (Figure 1—figure supplement 5). These results indicate that SKN-1A is broadly activated under conditions that increase the cellular burden of unfolded proteins. It is therefore likely that there are many endogenous proteins that, when misfolded, are able to trigger a SKN-1A-dependent response. The effects of the unc-54ts mutants and unc-54p::Aβ indicate that this response is sensitive enough to detect a single - albeit abundant - unfolded protein. Further, at least in muscle, the response is cell autonomously elicited by protein misfolding, but not by mutations - such as the unc-54(e190) deletion - that severely compromise muscle function without misfolded protein expression. This proteasomal response therefore does not depend on cellular or organismal consequences of tissue dysfunction in general. Taken together these data strongly suggest that SKN-1A is activated as part of a cell-autonomous response to cytoplasmic unfolded proteins.
The peptide:N-glycanase PNG-1/NGLY1, the aspartic protease DDI-1/DDI2 and the ERAD component SEL-1/SEL1 are each necessary to activate SKN-1A in response to direct proteasomal insults (Lehrbach and Ruvkun, 2016). To determine if this same genetic pathway is necessary to activate SKN-1A in response to misfolded proteins, we measured activation of rpt-3p::gfp by Aβ in png-1, ddi-1, and sel-1 mutants. The SKN-1A-dependent rpt-3p::gfp transcriptional response to Aβ is lost in png-1(ok1654) and ddi-1(mg571) mutants and is diminished in sel-1(mg457) mutants (Figure 1g). We conclude that related, or possibly identical, mechanisms govern SKN-1A activation by both direct assaults on the proteasome and the presence of misfolded and/or aggregated proteins.
SKN-1A is cell autonomously activated by impaired proteasome function
These data suggest that SKN-1A mediates a cell-autonomous transcriptional response to protein misfolding in muscle cells. SKN-1A also responds to proteasome dysfunction, but whether this response is cell autonomous is not known. We therefore configured a system to induce cell-type specific impairment of proteasome function in body wall muscle cells. Over-expression of an active site mutant of the β5 subunit of the 20S proteasome in otherwise wild-type cells causes proteasome dysfunction in yeast and the mouse (Heinemeyer et al., 1997; Li et al., 2004). We generated a transgene that expresses the corresponding active site mutant of the C. elegans β5 subunit, PBS-5[T65A], under control of the muscle specific myo-3 promoter (myo-3p::pbs-5[T65A]), such that proteasome dysfunction is induced specifically in muscle cells.
The myo-3p::pbs-5[T65A] transgene causes muscle-specific activation of the rpt-3p::gfp proteasome subunit reporter in a manner closely resembling that caused by mutant MHC B and Aβ (Figure 2a,b). This activation is lost in skn-1a(mg570) mutants, consistent with a SKN-1A-dependent response to proteasome dysfunction (Figure 2a). Wild type animals carrying the myo-3p::pbs-5[T65A] transgene show mildly impaired locomotion compared to non-transgenic controls (Figure 2c). Because impairment of the proteasome may cause age-dependent defects in cellular function, we examined movement of these animals at different ages. The locomotor rate of wild type animals carrying the myo-3p::pbs-5[T65A] transgene is reduced to a similar extent in day 1 and day 7 adults showing that this mild defect is not exacerbated by age (Figure 2c). This suggests that wild-type muscle cells are robust to proteasomal insults and so are able to maintain near-normal function despite the presence of the mutant β5 subunit. By contrast, the myo-3p::pbs-5[T65A] transgene causes complete paralysis in skn-1a(mg570) mutant animals lacking the SKN-1A-mediated proteasomal response pathway (Figure 2d). We conclude that SKN-1A mediates cell-autonomous activation of proteasome subunit genes in response to proteasome impairment, and that this SKN-1A-dependent compensation is essential for maintaining function in muscle cells experiencing proteasome dysfunction.
Figure 2
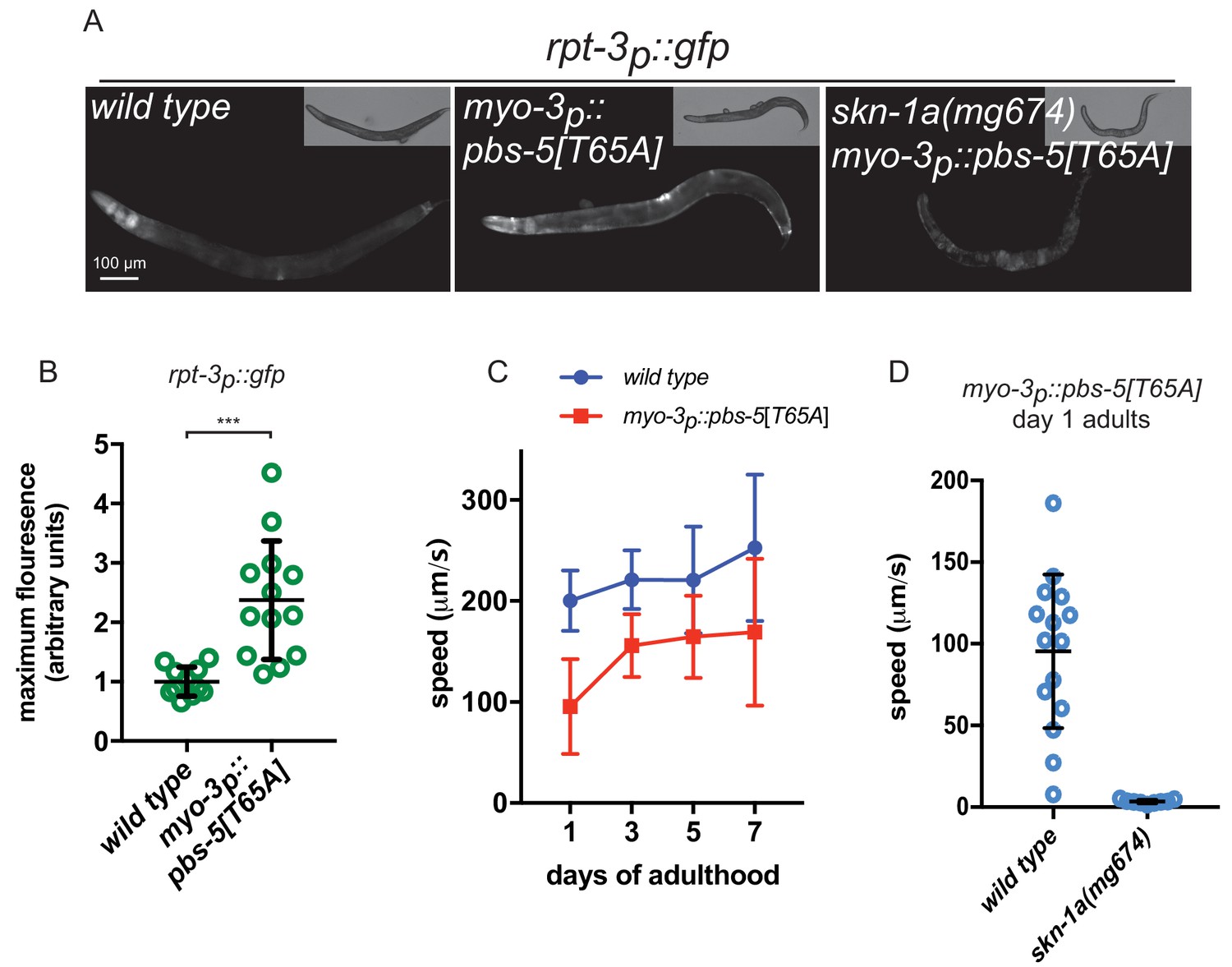
Proteasome impairment in muscle causes cell autonomous activation of SKN-1A.
(a) Fluorescence images showing rpt-3p::gfp expression in animals expressing a dominant negative proteasome subunit in the muscle (myo-3p::pbs-5[T65A]). (b) Quantification of rpt-3p::gfp expression in animals expressing a mutant proteasome subunit in the muscle. ***p<0.001 (Welch’s t-test). (c) Comparison of locomotor rate between wild type and myo-3p::pbs-5[T65A] transgenic animals. (d) Comparison of locomotor rate between wild type and skn-1a mutant animals carrying the myo-3p::pbs-5[T65A] transgene on day 1 of adulthood.
SKN-1A activation by misfolded proteins involves little or no impairment of proteasome function
Aggregation-prone proteins including human Aβ may interact with proteasomes and impair their function (Ayyadevara et al., 2015; Deriziotis et al., 2011; Gregori et al., 1995; Kristiansen et al., 2007; Snyder et al., 2003). To test the possibility that misfolded proteins trigger SKN-1A activation via inhibition of the proteasome, we generated a reporter of proteasome activity, a ubiquitously expressed unstable ubiquitin-GFP fusion protein (rpl-28p::ub(G76V)::gfp). The UB(G76V)::GFP ubiquitin fusion protein is normally degraded by the proteasome, but accumulates to detectable levels if proteasome function is impaired (Johnson et al., 1995; Segref et al., 2014). As expected, this reporter of proteasome activity reveals a muscle-specific proteasomal defect in myo-3p::pbs-5[T65A] transgenic animals (Figure 3a,b). Thus tissue-specific impairment of the proteasome in body wall muscle can be readily detected by monitoring UB(G76V)::GFP levels. Stabilization of UB(G76V)::GFP in PBS-5[T65A]-expressing muscle cells is greatly enhanced in the skn-1a mutant – all mutant animals show accumulation of GFP in all muscle cells and at higher levels than the wild type (Figure 3a,b). These data show that the SKN-1A transcriptional program partially corrects the muscle proteasomal defect caused by the myo-3p::pbs-5[T65A] insult. The severe locomotor defects and paralysis of myo-3p::pbs-5[T65A] animals that lack SKN-1A therefore likely stem from enhanced defects in proteasome function.
Figure 3 with 1 supplement see all
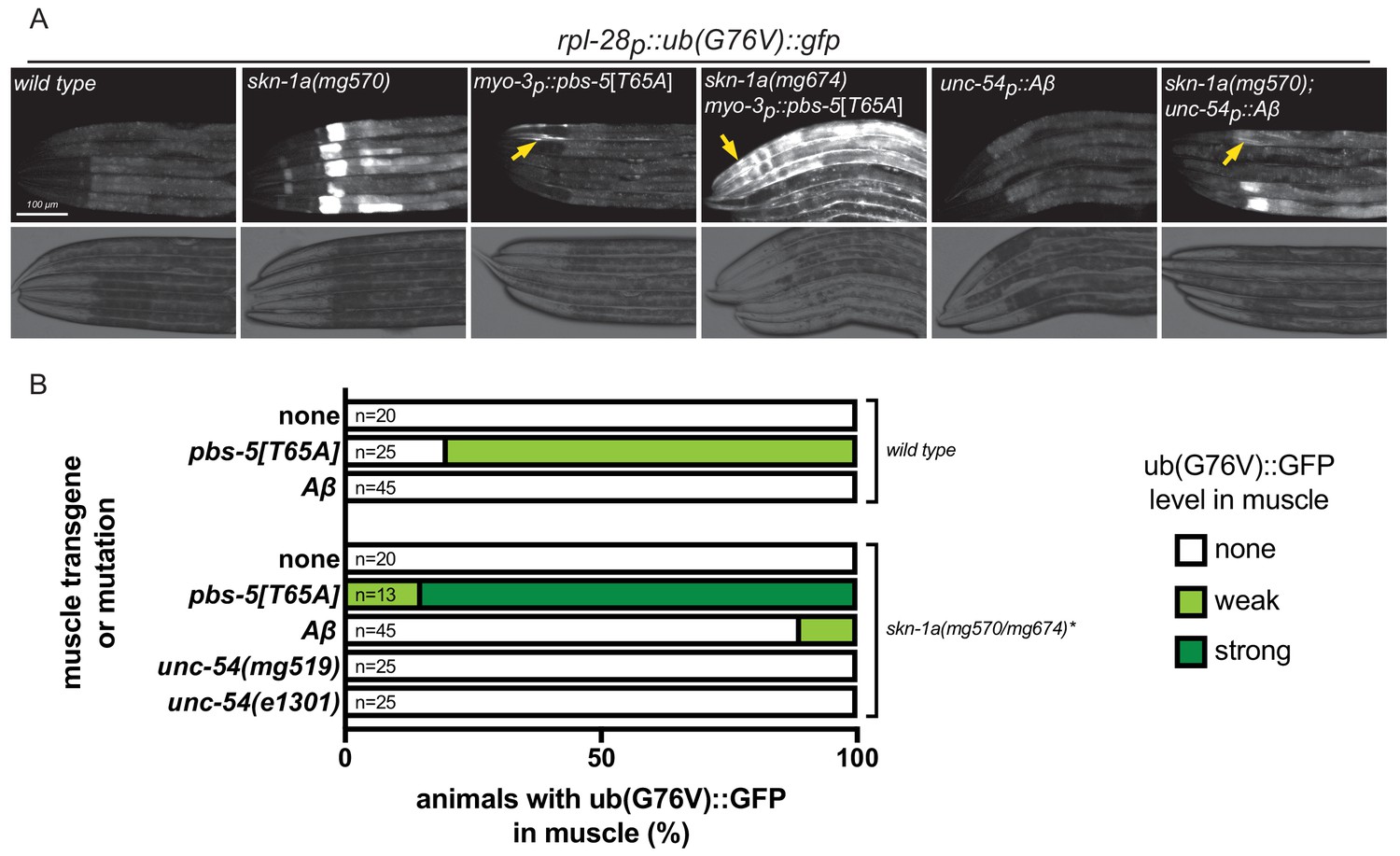
Proteasome function is not impaired in animals expressing misfolded proteins.
(a) Fluorescence micrographs showing impairment of UB(G76V)::GFP degradation in various genotypes. Arrows indicate UB(G76V)::GFP accumulation in muscle cells. (b) Comparison of UB(G76V)::GFP stabilization in muscles of animals carrying various SKN-1A-activating transgenes or mutations. *The skn-1a mutation used in the pbs-5[T65A] strain is mg674, which is an identical CRISPR-induced lesion to mg570. All animals were examined for UB(G76V)::GFP stabilization in the muscle at the L4 stage. We note that animals lacking SKN-1A show a defect in basal proteasome function, causing accumulation of UB(G76V)::GFP. This basal effect is limited to the intestine, and so we were still able to detect muscle-specific effects.
Mutant UNC-54, Aβ and PBS-5[T65A] all cause SKN-1A activation, as indicated by activation of rpt-3p::gfp. If all three trigger SKN-1A by the same mechanism – that is, by impairing proteasome function – they should also stabilize UB(G76V)::GFP. However, in contrast to myo-3p::pbs-5[T65A], we did not observe stabilization of UB(G76V)::GFP in unc-54p::Aβ transgenics (Figure 3a,b). Because activation of SKN-1A could compensate for an effect of Aβ on proteasome function, we also examined the effect of Aβ in skn-1a(mg570) mutants. unc-54p::Aβ only weakly affected UB(G76V)::GFP levels within the muscle cells of skn-1a mutants: about 10% of skn-1a(mg570) Aβ-expressing animals showed weak accumulation of UB(G76V)::GFP in some muscle cells suggesting a mild impairment of proteasome function (Figure 3a,b). We also tested the effect of unc-54(e1301) and unc-54(mg519) in the skn-1a(mg570) mutant background and found no effect on UB(G76V)::GFP degradation in the muscle (Figure 3b).
In mammalian cells, UbG76V::GFP accumulates only in cells with severely compromised proteasome function, as measured by Suc-LLVY-AMC hydrolysis in cell lysates (Dantuma et al., 2000). It is therefore possible that mutant MHC B and Aβ cause mild defects in proteasome function that are sufficient to activate rpt-3p::gfp without altering steady state levels of UB(G76V)::GFP. To test this possibility, we compared the behavior of the two reporters in animals exposed to very low doses of the proteasome inhibitor bortezomib (Figure 3—figure supplement 1). Because the effect of bortezomib on proteasome function may be masked by SKN-1A-dependent compensation, we monitored UB(G76V)::GFP levels in both wild type and skn-1a mutant animals. We found that very low concentrations of bortezomib (2 ng/ml) cause increased accumulation of UB(G76V)::GFP in skn-1a mutant animals. But wild type animals exposed to bortezomib at the same concentration do not show activation of rpt-3p::gfp. This suggests that monitoring UB(G76V)::GFP accumulation in a skn-1a mutant background serves as a more sensitive indicator of proteasome impairment than rpt-3p::gfp expression in wild type animals. As such, the UB(G76V)::GFP reporter should be sensitive enough to detect impairment of proteasome function, if this were the mechanism through which misfolded MHC B or Aβ cause activation of rpt-3p::gfp. These results therefore suggest that SKN-1A may be activated by misfolded proteins even in the absence of impaired proteasome function.
SKN-1A modulates age-dependent effects of misfolded UNC-54/MHC B
SKN-1A may regulate proteasome capacity to promote clearance of misfolded proteins that may otherwise accumulate and cause cellular dysfunction during aging. If this were the case, we would expect loss of SKN-1A to enhance age-dependent defects in animals expressing misfolded and aggregation-prone proteins. We therefore examined locomotion as a measure of defects in muscle cell function caused by the misfolded proteins that we had identified as activators of SKN-1A. We found no difference in locomotion rate between the wild type and skn-1a(mg570) mutants during the first week of adulthood (Figure 4a). We measured locomotion of unc-54(e1301) and unc-54(mg519) temperature-sensitive myosin heavy chain mutants at 20°C. This condition slows movement but does not cause paralysis of the mutant animals, presumably reflecting partial misfolding of the mutant MHC B. In contrast to wild type, the locomotion of animals harboring unc-54(e1301) or unc-54(mg519) mutations is strongly modulated by age in a SKN-1A-dependent manner (Figure 4b,c). The unc-54ts mutants show a severe locomotion defect on day 1 of adulthood, but remarkably, recover to near-normal rates of locomotion on days 3–7. This suggests that during aging the capacity for correct folding and function of mutant MHC B improves. Although age-dependent changes in proteostasis and physiology are thought to be largely detrimental, this suggests that in some cases they may include activation of protective responses that improve protein folding or function. Strikingly, this beneficial effect of age is entirely dependent on SKN-1A. unc-54(mg519); skn-1a(mg570) double mutants show no age-dependent improvement in locomotion and unc-54(e1301); skn-1a(mg570) double mutants show a slight age-dependent decline in locomotion (Figure 4b,c). Since two independent unc-54ts mutations have the same age-dependent genetic interaction with skn-1a, this is not allele-specific, but rather a general effect of SKN-1A on the function of misfolding MHC B. We measured rpt-3p::gfp expression in day 1 and day 5 unc-54(e1301) and unc-54(mg519) mutant adults. Expression of the rpt-3p::gfp reporter was unchanged, suggesting that SKN-1A activity does not increase as unc-54ts animals age (Figure 4—figure supplement 1). Thus, although SKN-1A is needed for unc-54ts animals to recover locomotion as they age, this is not caused by age-dependent changes in SKN-1A activity.
Figure 4 with 2 supplements see all
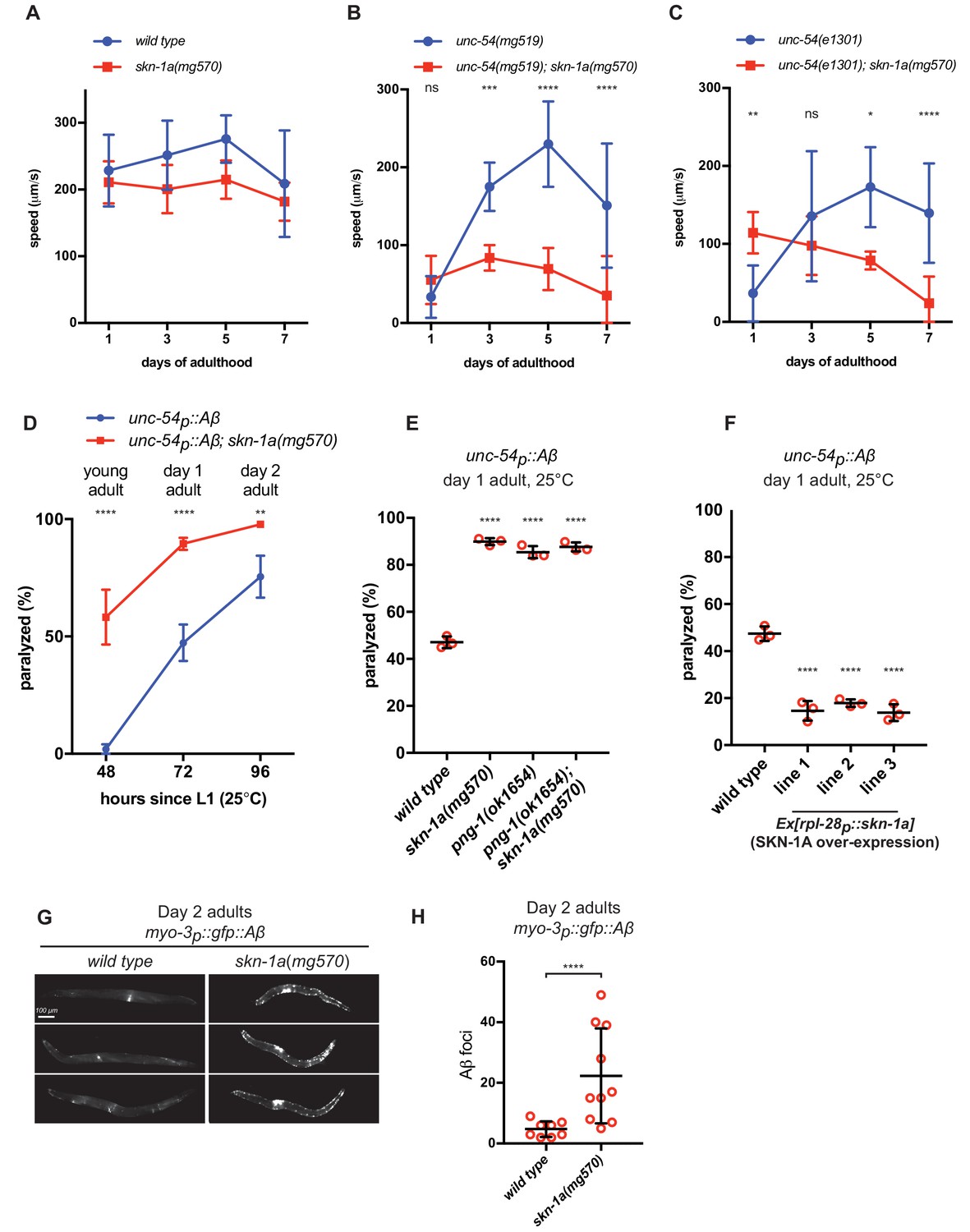
SKN-1A ameliorates age-dependent toxicity of misfolded proteins.
Analysis of locomotion of (a) wild type and skn-1a(mg570) mutant animals, (b) unc-54(mg519) and unc-54(mg519); skn-1a(mg570) double mutant animals and (c) unc-54(e1301) and unc-54(e1301); skn-1a(mg570) double mutant animals during aging. (d) Age-dependent paralysis of wild type and skn-1a(mg570) mutant Aβ expressing animals. Panels b, c, d: ****p<0.0001; ***p<0.001; **p<0.01; *p<0.05; ns p>0.05 indicates P-value compared to the skn-1a(+) control at each time point (two-way ANOVA with Dunnett’s multiple comparisons test). (e) increased paralysis of Aβ expressing with defective SKN-1A activation. (f) reduced paralysis of Aβ expressing animals with increased SKN-1A levels. Panels e and f: ****p<0.0001 compared to wild type (one-way ANOVA with Tukey’s multiple comparisons test). (g) Fluorescence images showing increased accumulation of Aβ::GFP in day two adults in skn-1a(mg570) as compared to wild type. (h) Quantification of Aβ::GFP puncta in wild type and skn-1a(mg570). ****p<0.0001 (Welch’s t-test).
Although the rate of movement of unc-54ts; skn-1a(mg570) double mutant animals is significantly reduced compared to unc-54ts single mutants on later days of adulthood (days 5–7), it is not reduced in day 1 adults. In fact, the locomotor rate of each double mutant is slightly increased compared to the corresponding unc-54ts single mutant on day 1 of adulthood (Figure 4—figure supplement 2). These data show that activation of the proteasome by SKN-1A is required to maintain muscle function in unc-54ts mutant animals as they age, rather than an age-independent requirement for SKN-1A to ensure folding or function of mutant MHC B. SKN-1A is essential for the locomotion of day 1 adults with impaired proteasome function in the muscle (myo-3p::pbs-5[T65A] transgenics; Figure 2e), so these data also confirm that mutant MHC B activates SKN-1A without impairing proteasome function as strongly as myo-3p::pbs-5[T65A]. Taken together, these results indicate that SKN-1A mediates functionally distinct responses to proteasome dysfunction and expression of misfolded proteins in the muscle. SKN-1A is essential for muscle function during proteasome impairment, regardless of age. In contrast, SKN-1A modulates an age-dependent defect in muscle function caused by misfolded MHC B.
SKN-1A mitigates accumulation and toxicity of Aβ
Expression of human Aβ in C. elegans muscle cells causes progressive adult-onset paralysis (Link, 2006). Paralysis is accompanied by aggregation and formation of amyloid fibrils, features also associated with adult-onset neurodegeneration in Alzheimer’s disease (Fay et al., 1998; Link, 1995). Adult-onset paralysis caused by human Aβ in C. elegans muscle is enhanced in skn-1a(mg570) mutants (Figure 4d). The effects of Aβ are also enhanced in png-1(ok1654), consistent with the failure of the png-1 mutant to activate SKN-1A (Figure 4e). The paralysis of png-1(ok1654); skn-1a(mg570) double mutants is not enhanced compared to either single mutant, supporting the model that PNG-1 acts through SKN-1A to mitigate Aβ toxicity. Overexpression of SKN-1A reduces the paralysis caused by muscle-specific Aβ expression in wild type (Figure 4f). These data indicate that proteasome activation by SKN-1A is required and sufficient to mitigate the age-dependent toxic effects of Aβ.
Using animals expressing Aβ fused to GFP (myo-3p::gfp::Aβ), we monitored expression and localization of Aβ in muscles of wild type and skn-1a mutant animals. In day 2 adults, levels of GFP::Aβ were consistently higher in the muscles of skn-1a mutant animals than wild type (Figure 4g), and skn-1a mutant muscles contained many more puncta of localized GFP::Aβ accumulation, suggesting increased formation of Aβ-containing aggregates (Figure 4h). These data suggest that the enhanced adult-onset paralysis in animals that lack SKN-1A is caused by higher levels of Aβ accumulation and aggregation.
ER-associated SKN-1A promotes longevity and healthy aging
Accumulation of misfolded and aggregated proteins is thought to cause decline in cellular function and health during aging (David et al., 2010; López-Otín et al., 2013; Walther et al., 2015). Mutations that affect both SKN-1A and SKN-1C reduce lifespan, but the individual contribution of SKN-1A is not known (Blackwell et al., 2015). We found that skn-1a(mg570), which affects only SKN-1A, causes ~20% reduction in lifespan compared to the wild type (Figure 5a). The lifespan of skn-1a/c(zu67) animals lacking both SKN-1A and SKN-1C is the same as that of skn-1a(mg570) (Figure 5b), showing that the effect of skn-1a/c(zu67) on lifespan can be explained by loss of SKN-1A. The mgTi1[rpl-28p::skn-1a::gfp] single copy transgene expresses a functional SKN-1A::GFP fusion protein under the control of the constitutively active rpl-28 promoter (Lehrbach and Ruvkun, 2016). This transgene rescues the bortezomib sensitivity and maternal effect lethality of skn-1a/c(zu67) mutants. The lifespan of skn-1a/c(zu67); mgTi1[rpl-28p::skn-1a::gfp] animals is not reduced compared to wild type, indicating that SKN-1A is sufficient to confer normal lifespan in the absence of SKN-1C. In fact, the lifespan of the rescued animals was reproducibly longer than the wild type (Figure 5c). This single copy transgene drives expression from the rpl-28 ribosome subunit promoter so that SKN-1A::GFP is likely to be overexpressed compared to endogenous SKN-1A. Other independently isolated single-copy rpl-28p::skn-1a transgenes also extend lifespan (Figure 5d). Thus, SKN-1A is necessary for normal lifespan and sufficient to extend lifespan when over-expressed. Like skn-1a(mg570), the lifespan of png-1(ok1654) mutant animals is reduced compared to wild type (Figure 5e). png-1(ok1654) lifespan is shorter than the skn-1a(mg570) mutant, suggesting that PNG-1 might promote longevity through additional SKN-1A-independent pathways. The lifespan of png-1(ok1654); skn-1a(mg570) double mutants is not further reduced compared to the png-1(ok1654) mutant, indicating that both genes act in the same pathway that controls lifespan (Figure 5f). These data suggest that the PNG-1-dependent processing of SKN-1A following release from the ER is required for this transcription factor to regulate lifespan.
Figure 5

SKN-1A and PNG-1 control lifespan.
(a–f) Experiments showing that SKN-1A and PNG-1 control lifespan, and that SKN-1A accounts for the effect of skn-1a/c mutations on normal lifespan: (a) The lifespan of skn-1a(mg570) mutant animals is reduced compared to the wild type. (b) The lifespan of skn-1a/c(zu67) mutant animals is not further reduced compared to skn-1a(mg570). (c) The reduced lifespan of skn-1a/c(zu67) mutant animals is rescued by a transgene expressing SKN-1A under control of the rpl-28 promoter. (d) Overexpression of SKN-1A increases lifespan. In five independent rpl-28p::skn-1a::gfp lines we found a 10–20% increase in lifespan compared to the wild type. (e) The lifespan of png-1(ok1654) mutant animals is reduced compared to wild type. (f) Removal of SKN-1A does not further reduce the lifespan of png-1(ok1654) mutant animals. For summary of lifespan statistics see Supplementary file 1 (g) Analysis of vulval degeneration in day 7 adults. ***p<0.001; **p<0.01; ns p>0.05; P-value compared to wild type control is shown unless otherwise indicated (one-way ANOVA with Sidak’s multiple comparisons test).
Age-dependent defects in vulval integrity are correlated with reduced C. elegans lifespan and have been proposed as a marker of healthspan. These defects in vulval integrity are increased by skn-1(RNAi), which depletes multiple SKN-1 isoforms (Leiser et al., 2016). skn-1a(mg570) and skn-1a/c(zu67) animals both show dramatically increased age-dependent vulval integrity defects (Figure 5g). This age-dependent vulval degeneration is rescued in skn-1a/c(zu67) animals carrying the mgTi1[rpl-28p::skn-1a::gfp] transgene. Thus, loss of SKN-1A causes the vulval degeneration of skn-1 mutants. png-1(ok1654) mutant animals also show defects in vulval integrity, similar to the skn-1a(mg570) mutant (Figure 5g). Vulval degeneration is not enhanced in the png-1(ok1654); skn-1a(mg570) double mutant, suggesting that both genes act in the same genetic pathway governing vulval integrity during aging. We conclude that regulation of the proteasome by SKN-1A promotes healthy aging and longevity.
Discussion
We have found that unfolded or aggregated proteins elicit a signal transduced by the SKN-1A/Nrf1 transcription factor, which activates proteasome subunit gene expression. This pathway allows cells to respond to protein folding defects by increasing proteasome levels, enabling more efficient destruction of unfolded or aggregated proteins. We show that this pathway mitigates the age-dependent effects of chronic protein misfolding and aggregation, ensures healthy aging and promotes longevity. Collectively, these data reveal a new unfolded protein response pathway that maintains proteostasis and cellular function during aging (Figure 6).
Figure 6
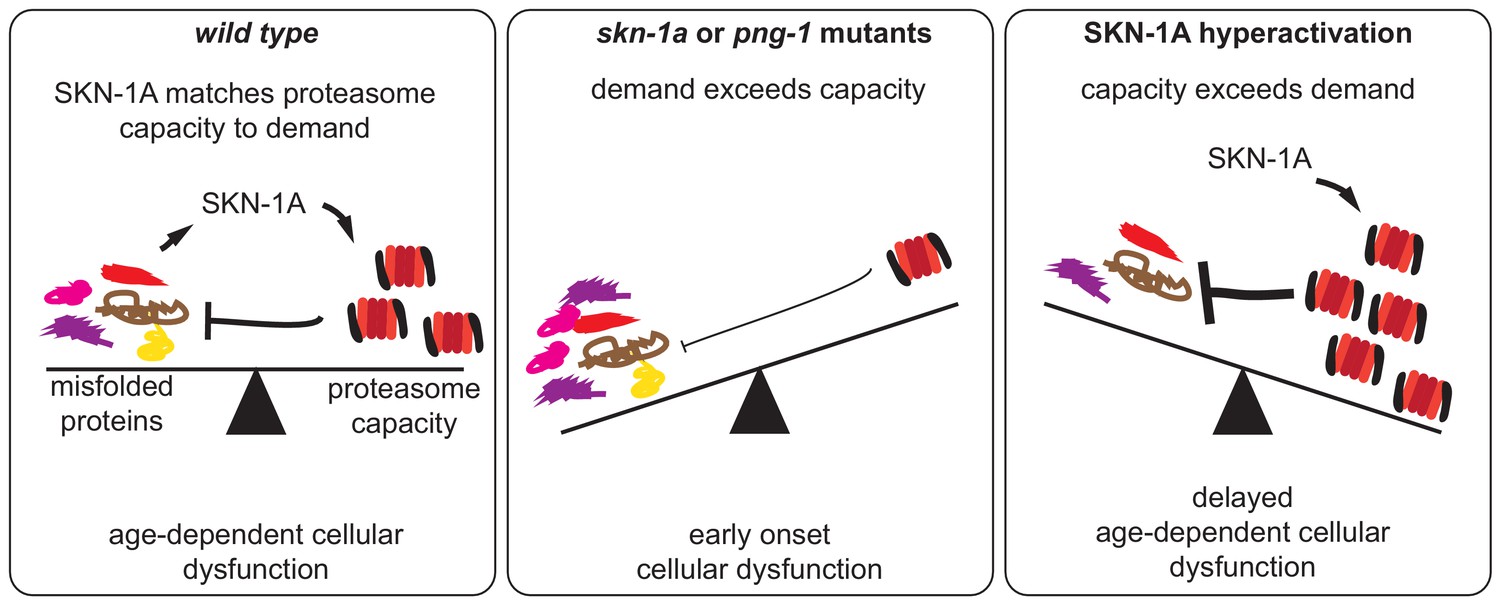
SKN-1A modulates functional decline during aging by adjusting proteasome capacity to meet demand for degradation of misfolded proteins.
During aging, misfolded proteins eventually accumulate to levels that disrupt cellular function. SKN-1A adjusts proteasome capacity to meet demand for degradation of damaged and misfolded proteins. This modulates the age-dependent accumulation and toxicity of misfolded proteins, thereby altering the rate of functional decline during aging. In animals lacking this pathway (i.e. skn-1a or png-1 mutants), insufficient proteasome capacity leads to a rapid decline and reduced lifespan. Conversely, enhancement of this pathway (by increasing SKN-1A levels or activity) delays the cellular dysfunction caused by misfolded proteins and extends lifespan.
Diverse proteotoxic insults might be expected to engage SKN-1A, however our genetic analyses suggest that this transcription factor responds selectively to cytosolic unfolded proteins and impaired proteasome activity. Proteasome dysfunction can occur as a consequence of oxidative stress, ER stress, and mitochondrial dysfunction (Bulteau et al., 2001; Menéndez-Benito et al., 2005; Segref et al., 2014). But our unbiased genetic analysis of transcriptional regulation of the proteasome thus far has only pointed to mutations that impair the proteasome itself and the misfolding of a very abundant cytoplasmic protein, UNC-54, as SKN-1A activators. Proteotoxic insults that do not activate SKN-1A/Nrf1 might activate other SKN-1/Nrf isoforms instead; for example oxidative stress activates SKN-1C in C. elegans and Nrf2 in mammalian cells. This suggests that the different SKN-1 isoforms – and mammalian Nrfs – have evolved distinct mechanisms of regulation to allow cells to mount appropriate responses to various types of proteotoxic stress.
Our genetic screen yielded multiple unc-54 alleles rather than a collection of lesions which disrupt the folding of many different proteins. It is possible that only very abundant misfolded proteins will activate rpt-3p::gfp sufficiently to be detected in this screen. Our screen was also designed to isolate viable mutants. Since many of the most highly expressed proteins perform essential functions, this may have prevented us from isolating mutations that disrupt their folding. Muscle cells show higher levels of both basal and induced rpt-3p::gfp expression compared to other tissues (Figure 1—figure supplement 1). Some combination of the abundance of the UNC-54 protein, its function in muscle, and the viability of unc-54 mutants may have conspired to make this gene a major target in our screen. An interesting possibility is that the proteasome is particularly important for the regulated degradation of misassembled sarcomeres of the muscle, a tissue that undergoes rapid protein synthesis and turnover – for example during exercise-mediated muscle growth or atrophy during prolonged inactivity.
A detailed elucidation of the mechanism that links accumulation of unfolded proteins to SKN-1A activation will be of great future interest. Our genetic analysis suggests that activation of SKN-1A by misfolded proteins requires release from the ER by ERAD/SEL-1, deglycosylation by PNG-1 and cleavage by DDI-1. These post-translational processing steps are also required for SKN-1A activation during proteasome dysfunction. This suggests that unfolded proteins impair proteasomal degradation of SKN-1A. This model is compatible with the ability of misfolded proteins to cause proteasome dysfunction (Ayyadevara et al., 2015; Bence et al., 2001; Gregori et al., 1995; Kristiansen et al., 2007; Snyder et al., 2003). However, we detect SKN-1A activation under conditions that have little or no effect on the degradation of a heterologous proteasome substrate. This suggests that proteasome dysfunction is not required for misfolded proteins to trigger SKN-1A activation. One possibility is that SKN-1A is exquisitely sensitive to changes in proteasome substrate load, and is activated by increased delivery of proteins to the proteasome – even if the increased substrate load does not reach a level that exceeds proteasome capacity. It is also possible that SKN-1A interacts directly with sensor(s) of cellular protein folding that regulate its activity or stability. Interestingly, SKN-1A/Nrf1 itself behaves like an unfolded protein: it is a substrate of the ERAD pathway (Lehrbach and Ruvkun, 2016; Steffen et al., 2010), which normally functions to eject misfolded glycoproteins from the ER; SKN-1A/Nrf1 activation requires deglycosylation by PNGase, an enzyme that preferentially acts on denatured glycoproteins (Hirsch et al., 2004); and Nrf1 is prone to form aggregates in the cytoplasm of cells under proteotoxic stress (Sha and Goldberg, 2016). This property could facilitate interactions with cellular sensors of protein folding that may influence SKN-1A/Nrf1 activation. Whatever the mechanism, activation of SKN-1A by misfolded proteins – in the absence of outright proteasome dysfunction – could allow cells to adjust proteasome abundance to meet demand for targeted destruction of damaged or misfolded proteins before they reach levels that compromise cellular function.
Our data are consistent with a model in which SKN-1A boosts various protein quality control pathways that rely on the ubiquitin-proteasome system to eliminate aberrant or damaged proteins. In the case of pathologically misfolding proteins such as Aβ, it is easy to imagine how enhanced elimination of the toxic molecule could limit accumulation over time and so delay the onset of pathology. The explanation for the effects of age and SKN-1A on muscle function in unc-54ts mutants must be more complex. SKN-1A is required for the unusual recovery of locomotor function of unc-54ts animals that occurs as they age. But SKN-1A activity levels do not change as unc-54ts animals get older. This recovery of muscle function is therefore unlikely to be directly mediated by SKN-1A, but likely requires SKN-1A in addition to another unidentified mechanism. It is striking that the function of temperature sensitive mutant proteins, including the mutant MHC B expressed by the e1301 and e1157 alleles, is disrupted by the presence of other misfolded or aggregation-prone proteins (Gidalevitz et al., 2006; Olzscha et al., 2011). By limiting the accumulation of misfolded proteins globally, SKN-1A may create a cellular environment more conducive to the correct folding and function of mutant MHC B.
The accumulation of misfolded and aggregated proteins is a hallmark of aging that has been observed in many species including C. elegans (David et al., 2010; Walther et al., 2015). The effects of SKN-1A and other unfolded protein response pathways on aging and longevity supports the model that protein misfolding and aggregation is a cause rather than a consequence of functional decline during aging (Denzel et al., 2014; Hsu et al., 2003; Walker and Lithgow, 2003). The skn-1 gene is a component in several C. elegans longevity pathways, but the precise mechanism(s) through which skn-1 promotes longevity are not fully understood (Blackwell et al., 2015). The UPR that we have uncovered requires SKN-1A, but not other SKN-1 isoforms, which do not undergo the post-translational modifications necessary for regulation of the proteasome (Lehrbach and Ruvkun, 2016). We show that the lifespan and healthspan effects of skn-1 mutations are largely explained by loss of SKN-1A, and that elevated SKN-1A levels are sufficient to extend lifespan, even in animals that lack SKN-1C. Thus our data suggests that the skn-1 gene primarily promotes longevity by safeguarding proteostasis through SKN-1A/Nrf1-dependent control of proteasome expression and activity.
The failure of proteasome-dependent protein quality control systems is intimately linked to neurodegeneration. Intracellular inclusions that contain ubiquitinated proteins are a central feature of essentially all neurodegenerative diseases (Alves-Rodrigues et al., 1998). Depletion of Nrf1 in the mouse brain causes neurodegeneration accompanied by formation of ubiquitin-containing inclusions in young animals (Lee et al., 2011). A recent study has suggested that pharmacological activation of Nrf1 is protective in a mouse model of one age-dependent neurodegenerative condition – spinal and bulbar muscular atrophy (Bott et al., 2016), and our data indicates SKN-1A/Nrf1 is similarly protective in a C. elegans model of Alzheimer’s disease. We therefore suggest that increasing the activity of Nrf1 may be beneficial for human aging and treatment of various adult-onset neurodegenerative diseases.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (E. coli) | E. coli OP50 | CGC | OP50 | |
| Strain, strain background (E. coli) | E. coli HT115 | CGC | HT115 | |
| Strain, strain background (C. elegans) | unc-54(e1301) I. | CGC | CB1301 | |
| Strain, strain background (C. elegans) | dvIs2 | CGC | CL2006 | unc-54::Aβ |
| Strain, strain background (C. elegans) | dvIs37 | CGC | CL2331 | myo-3::gfp::Aβ |
| Strain, strain background (C. elegans) | mgIs72 II | Lehrbach and Ruvkun, 2016 | GR2183 | rpt-3::gfp integrated array |
| Strain, strain background (C. elegans) | pbs-5(mg502) I; mgIs72 II | Lehrbach and Ruvkun, 2016 | GR2184 | proteasome mutant |
| Strain, strain background (C. elegans) | mgIs72 II; skn-1(mg570) IV | Lehrbach and Ruvkun, 2016 | GR2197 | |
| Strain, strain background (C. elegans) | mgIs72 II; ddi-1(mg571) IV | Lehrbach and Ruvkun, 2016 | GR2211 | |
| Strain, strain background (C. elegans) | unc-119(ed3) III; mgTi4 | Lehrbach and Ruvkun, 2016 | GR2212 | rpl-28::ha::skn-1a::gfp::tbb-2 |
| Strain, strain background (C. elegans) | unc-119(ed3) III; mgTi5 | Lehrbach and Ruvkun, 2016 | GR2213 | rpl-28::ha::skn-1a::gfp::tbb-2 |
| Strain, strain background (C. elegans) | mgIs72 II; sel-1(mg547) V | Lehrbach and Ruvkun, 2016 | GR2215 | Strain, strain |
| background (C. elegans) | unc-119(ed3) III; skn-1(zu67) IV; mgTi1 | Lehrbach and Ruvkun, 2016 | GR2221 | rpl-28::skn-1a::GFP::tbb-2 rescues skn-1(zu67) |
| Strain, strain background (C. elegans) | png-1(ok1654) I; mgIs72 II | Lehrbach and Ruvkun, 2016 | GR2236 | |
| Strain, strain background (C. elegans) | skn-1(mg570) IV | Lehrbach and Ruvkun, 2016 | GR2245 | |
| Strain, strain background (C. elegans) | png-1(ok1654) I | CGC | GR2246 | |
| Strain, strain background (C. elegans) | png-1(ok1654) I; skn-1(mg570) IV | this study | GR3089 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs77 V | this study | GR3090 | rpl-28::ub(G76V)::gfp::tbb-2, myo-3::mcherry marker. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-119(ed3) III; mgTi15 | this study | GR3091 | rpl-28::skn-1a::GFP::tbb-2. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-119(ed3) III; mgTi17 | this study | GR3092 | rpl-28::HA::skn-1a::GFP::tbb-2. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | skn-1(mg570) IV; mgIs77 V | this study | GR3094 | rpl-28::ub(G76V)::gfp::tbb-2. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; pas-1(mg511) V | this study | GR3141 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-10(mg525) I; mgIs72 II | this study | GR3142 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; rpn-1(mg514) IV | this study | GR3143 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-5(mg509) I; mgIs72 II | this study | GR3144 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; rpt-6(mg513) III | this study | GR3145 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-10(mg495) I; mgIs72 II | this study | GR3146 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; rpt-6(mg512) III | this study | GR3147 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs78 IV | this study | GR3148 | myo-3::H2B::mcherry::SL2::pbs-5[T65A] (pNL47). Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; mgIs78 IV | this study | GR3149 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-10(mg529) I; mgIs72 II | this study | GR3150 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-2(mg530) I; mgIs72 II | this study | GR3151 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-10(mg531) I; mgIs72 II | this study | GR3152 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e190) I; mgIs72 II | this study | GR3153 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs78 IV; mgIs77 V | this study | GR3154 | myo-3::H2B::mcherry::SL2::pbs-5[T65A] and Ub(G76V)::gfp. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-11(mg494) mgIs72 II | this study | GR3155 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I; mgIs72 II | this study | GR3156 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I | this study | GR3157 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; skn-1 (mg674) mgIs78 IV | this study | GR3158 | mg674 causes G2STOP in SKN-1A. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e1157) I; mgIs72 II | this study | GR3159 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e1301) I; mgIs72 II | this study | GR3160 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg528) I; mgIs72 II | this study | GR3161 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg540) I; mgIs72 II | this study | GR3162 | unc-54ts. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | skn-1(mg674) mgIs78 IV | this study | GR3163 | mg674 causes G2STOP in SKN-1A. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e1301) I; mgIs72 II; skn-1(mg570) IV | this study | GR3164 | Reagent requests: see Materials and methods. |
| Strain, strain background (C. elegans) | unc-54(e1301) I; skn-1(mg570) IV | this study | GR3165 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I; mgIs72 II; skn-1(mg570) IV | this study | GR3166 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I; skn-1(mg570) IV | this study | GR3167 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | skn-1(mg674) mgIs78/nT1[qIs51] IV; mgIs77/nT1[qIs51] V | this study | GR3168 | skn-1(mg674) mgIs78; mgIs77 animals are very sick, use balancer to maintain. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e1301) I; skn-1 (mg570) IV; mgIs77 V | this study | GR3169 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I; skn-1 (mg570) IV; mgIs77 V | this study | GR3170 | Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-3(mg527) mgIs72 II | this study | GR3171 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-2(mg581) I; mgIs72 II | this study | GR3172 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-9(mg533) mgIs72 II | this study | GR3173 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-8(mg587) I; mgIs72 II | this study | GR3174 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-5(mg534) mgIs72 II | this study | GR3175 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | rpn-8(mg536) I; mgIs72 II | this study | GR3176 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 Il; rpn-1(mg537) IV | this study | GR3177 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-2(mg538) I; mgIs72 II | this study | GR3178 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | pbs-4(mg539) I; mgIs72 II | this study | GR3179 | proteasome mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; dvIs2 | this study | GR3180 | Amyloid beta + rpt-3::gfp. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; dvIs2; skn-1(mg570) IV | this study | GR3181 | Amyloid beta + rpt-3::gfp in skn-1a mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | skn-1(mg570) IV; mgIs77 V; dvIs2 | this study | GR3182 | unc-54::Aβ+Ub(G76V):: gfp in skn-1a mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs77 V; dvIs2 | this study | GR3183 | unc-54::Aβ+Ub(G76V)::gfp. Reagent requests: s ee Materials and methods |
| Strain, strain background (C. elegans) | skn-1(mg570) IV; dvIs2 | this study | GR3184 | unc-54::Aβ in skn-1a mutant. Reagent requests: see Materials and methods |
| strain, strain background (C. elegans) | skn-1(mg570) IV; dvIs37 | this study | GR3185 | myo-3::gfp::Aβ in skn-1a mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | png-1(ok1654) I; dvIs2 | this study | GR3186 | unc-54::Aβ in a png-1 mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | png-1(ok1654) I; skn-1(mg570) IV; dvIs2 | this study | GR3187 | unc-54::Aβ in png-1 skn-1a double mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; ddi-1(mg571) IV; dvIs2 | this study | GR3188 | unc-54::Aβ in ddi-1 mutant + rpt-3::gfp. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | png-1(ok1645) I; mgIs72 II; dvIs2 | this study | GR3189 | unc-54::Aβ in png-1 mutant + rpt-3::gfp. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | dvIs2; mgEx813 | this study | GR3190 | skn-1a overexpression (pNL214), array marked by myo-2::mcherry. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | dvIs2; mgEx814 | this study | GR3191 | skn-1a overexpression (pNL214), array marked by myo-2::mcherry. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | dvIs2; mgEx815 | this study | GR3192 | skn-1a overexpression (pNL214), array marked by myo-2::mcherry. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | mgIs72 II; sel-1(mg547) V; dvIs2 | this study | GR3193 | unc-54::Aβ in sel-1 mutant + rpt-3::gfp. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | hsf-1(sy441) I; mgIs72 | this study | GR3291 | rpt-3::gfp, hif-1 mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-119(ed3) III; mgEx831 | this study | GR3292 | rpl-28p::skn-1a[∆DBD]:: gfp marked by myo-2::mcherry and unc-119(+). Reagent requests: s ee Materials and methods |
| Strain, strain background (C. elegans) | unc-54(e1301) I; mgEx831 | this study | GR3293 | rpl-28p::skn-1a[∆DBD]::gfp, unc-54ts mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | unc-54(mg519) I; mgEx831 | this study | GR3294 | rpl-28p::skn-1a[∆DBD]::gfp, unc-54ts mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | hsf-1(sy441) I; mgIs72; skn-1a(mg570) | this study | GR3295 | rpt-3::gfp, hif-1, skn-1a double mutant. Reagent requests: see Materials and methods |
| Strain, strain background (C. elegans) | skn-1(zu67) IV/nT1 [unc-?(n754) let-?](IV;V) | CGC | EU1 | |
| Strain, strain background (C. elegans) | wild type | CGC | N2 | |
| Recombinant DNA reagent (plasmid) | rpl-28::skn-1a::tbb-2 | Lehrbach and Ruvkun, 2016. | pNL214 | Reagent requests: see Materials and methods |
| Recombinant DNA reagent (plasmid) | myo-3::mcherry::his-58:: SL2::pbs-5[T65A] | this study | pNL47 | Reagent requests: see Materials and methods |
| Recombinant DNA reagent (plasmid) | rpl-28::ub(G76V)::gfp::tbb-2 | this study | pNL121 | Reagent requests: see Materials and methods |
| Chemical compound, drug | Bortezomib | L C Laboratories | Cat#B1408 | |
| Software, algorithm | ImageJ | NIH | https://imagej.nih.gov/ij/ | |
| Software, algorithm | Zen | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen.html | |
| Software, algorithm | Ape (A plasmid editor) | M Wayne Davis | http://jorgensen.biology.utah.edu/wayned/ape/ | |
| Software, algorithm | Graphpad Prism | Graphpad | https://www.graphpad.com/scientific-software/prism/ |
C. elegans maintenance and genetics
Request a detailed protocolC. elegans were maintained on standard media at 20°C (unless otherwise indicated) and fed E. coli OP50. A list of strains used in this study is provided in the Key Resources Table. RNAi was performed as described in Kamath and Ahringer (2003). Mutagenesis was performed by treatment of L4 animals in 47 mM EMS for 4 hr at 20°C. Some strains were provided by the CGC, which is funded by NIH Office of Research Infrastructure Programs (P40 OD010440). png-1(ok1654) was generated by the C. elegans Gene Knockout Project at the Oklahoma Medical Research Foundation, part of the International C. elegans Gene Knockout Consortium.
Identification of EMS induced mutations by whole genome sequencing
Request a detailed protocolGenomic DNA was prepared using the Gentra Puregene Tissue kit (Qiagen, #158689) according to the manufacturer’s instructions. Genomic DNA libraries were prepared using the NEBNext genomic DNA library construction kit (New England Biololabs, #E6040), and sequenced on a Illumina Hiseq instrument. Deep sequencing reads were analyzed using Cloudmap (Minevich et al., 2012).
Transgenesis
Request a detailed protocolCloning was performed by isothermal/Gibson assembly (Gibson et al., 2009). All plasmids used for transgenesis are listed in the Key Resources Table. All constructs were assembled in pNL43 (Lehrbach and Ruvkun, 2016) or in pBluescript. The SKN-1 constructs used in this study are described in Lehrbach and Ruvkun (2016). Extra-chromosomal arrays were generated using myo-2::mcherry as a co-injection marker. EMS mutagenesis was used to induce integration of extrachromosomal arrays. The myo-3p::pbs-5[T65A] construct was generated to expresses mcherry:: histone(H2B) and mutant PBS-5 from an artificial operon under control of the myo-3 promoter, which drives expression specifically in the body wall muscle (myo-3p::mcherry::H2B::SL2::PBS-5[T65A]). The mcherry::H2B serves to confirm the tissue specific expression of the transgene. A DNA fragment containing the 5’UTR, coding sequence and 3’UTR of pbs-5 was cloned and site-directed mutagenesis was used to introduce the T65A mutation. The altered pbs-5 DNA fragment was then cloned into pBluescript with the myo-3 promoter (a 2169 bp fragment immediately upstream of the myo-3 start codon) and mcherry fused in-frame to the his-58 (H2B) coding sequence (a 373 bp fragment containing the his-58 open reading frame). The ub(G76V)::gfp construct was generated to drive ubiquitous expression of UB(G76V)::GFP under control of the rpl-28 promoter. A synthesized DNA fragment encoding ubiquitin was cloned in frame with GFP to generate the UB(G76V)::GFP coding sequence. The G76V mutation was introduced by the oligos used for Gibson assembly. This was inserted into pNL43 with the rpl-28 promoter (605 bp immediately upstream of the rpl-28 start codon) and tbb-2 3’UTR (376 bp immediately downstream of the tbb-2 stop codon).
Genome modification by CRISPR/Cas9
Request a detailed protocolThe mgIs78[myo-3p::mcherry::H2B::SL2::PBS-5[T65A]] transgene is integrated within chromosome IV and appears to be tightly linked to skn-1. The skn-1a(mg674) allele is identical to mg570 and was generated as described in Lehrbach and Ruvkun (2016) using dpy-10(cn64) as a co-CRISPR marker by injection of mgIs78 transgenic animals.
Microscopy
Request a detailed protocolFor rpt-3p::gfp, rpl-28p::Ub(G76V)::gfp and myo-3p::gfp::Aβ transgenics, bright field and GFP fluorescence images were collected using a Zeiss AxioZoom V16, equipped with a Hammamatsu Orca flash 4.0 digital camera camera, and using Axiovision software. For rpl-28p::skn-1a[∆DBD]::gfp, DIC and GFP fluorescence images were collected using a Zeiss Axio Image Z1 microscope, equipped with a Zeiss AxioCam HRc digital camera, using Axiovision software. Images were processed using ImageJ software. For all fluorescence images, images shown within the same figure panel were collected using the same exposure time and then processed identically in ImageJ. To quantify rpt-3p::gfp expression, the maximum pixel intensity within a transverse section approximately 25 μm posterior to the pharynx of adult animals was measured using imageJ. To quantify UB(G76V)::GFP stabilization in muscle, images of transgenic animals were manually inspected in imageJ. Weak stabilization was recorded if animals contained low but detectable levels of UB(G76V)::GFP in any part of the body wall muscle (16-bit pixel intensity greater than 500). Strong stabilization was recorded if animals contained higher levels of UB(G76V)::GFP in any part of the body wall muscle (16-bit pixel intensity greater than 2000). Aβ foci were counted using the find maxima tool in imageJ.
Bortezomib treatment for imaging
Request a detailed protocolPlates were supplemented with bortezomib (LC Laboratories #B1408) by spotting a bortezomib solution on top of NGM plates seeded with OP50. The bortezomib solution was allowed to dry into the plate before adding L4 stage animals. These animals were allowed to reproduce, and reporter expression was imaged in the next generation. All treatment conditions contained less than 0.001% DMSO and bortezomib treated worms were compared to DMSO-treated control animals.
Sterility assay
Request a detailed protocolL4 animals were selected from mixed stage cultures that had been maintained without starvation for at least two generations and shifted to 20°C or 25°C. In the next generation, L4 animals were picked individually to fresh plates and returned to the same temperature. The production of progeny was monitored over the following 5 days. Animals that produced no progeny were recorded as sterile, all other animals (regardless of brood size or viability of progeny) were recorded as fertile. Fertility of at least 10 animals was assessed for each strain at each temperature. All strains used in fertility assays contained the mgIs72 transgene.
Aβ-induced paralysis assay
Request a detailed protocolFor each assay at least 100 starvation-synchroized L1 stage animals were raised at 25°C. Animals grown under this condition reach adulthood after ~48 hr. Starting at 48 hr, animals were scored for paralysis every 24 hr. Animals were scored as paralyzed if they showed no sign of movement after tapping the plate or gently prodding the animal.
unc-54ts paralysis assay
Request a detailed protocolL4 animals were selected from mixed stage cultures that had been maintained without starvation for at least two generations and shifted to 20°C or 25°C. When the majority of the progeny had reached adulthood, adult animals were scored for paralysis. Animals were scored as paralyzed if they showed no sign of movement after tapping the plate or gently prodding the animal. At least 100 animals for each strain under each condition were scored.
Measurement of locomotor rate (speed)
Request a detailed protocolLocomotor assays were initiated by selecting L4 animals from mixed stage cultures that had been maintained without starvation for at least two generations. L4 animals were maintained for a further 24 hr to assay day one adults, or for correspondingly longer periods to assay day 3, 5 and 7 adults. For assays in which locomotion was measured on multiple days, a single population of animals was maintained and repeatedly tested. Animals that had bagged or ruptured were removed from analysis since these defects impair locomotion but do not reflect changes in body wall muscle function. To assay locomotor rate, each animal was transferred to a fresh plate seeded with OP50 and then removed after 1 min. An image of the tracks left in the lawn by each animal was collected. The distanced travelled was then measured using imageJ and used to calculate average speed.
Lifespan analysis
Request a detailed protocolLifespan assays were initiated by selecting L4 animals from mixed stage cultures that had been maintained without starvation for at least two generations. Animals were transferred to fresh plates on day three and then every 2 days until reproduction ceased and every 3–5 days thereafter. Animals were checked for survival at least every other day. Animals that died by bagging or crawling off the plates were censored. Animals that died due to age-related vulval integrity defects (after ceasing reproduction, when such defects can be distinguished from bagging) were not censored from analysis, as this is a major mode of age-dependent lethality of some of the mutants analyzed. Survival curves, calculation of mean lifespan and statistical analysis was performed in R using the ‘survival’ package. The log-rank (Mantel-Haenszel) test was used to compare survival curves. Statistics for all assays (including replicate assays not shown in main figures) are shown in Supplementary file 1.
Scoring of age-related vulval integrity defects
Request a detailed protocolAssays to measure age-related vulval integrity defects were initiated by selecting L4 animals from mixed-stage cultures that had been maintained without starvation for at least two generations. Animals were transferred to fresh plates on days 3 and 5 of the assay. On days 5 and 7, animals were checked for rupture, and the cumulative total number of animals ruptured during the first week of adulthood recorded. 30–80 animals were analyzed in each assay. At least three replicate assays were performed for each genotype.
Statistical analysis
Request a detailed protocolStatistical analyses of lifespan data are described in the lifespan analysis section. All other statistical analyses were performed using Graphpad Prism. All biological replicates were performed with independent populations of animals.
Data availability
All data analyzed or generated in this study are included in the figures and supporting files.
References
-
Ubiquitin, cellular inclusions and their role in neurodegenerationTrends in Neurosciences 21:516–520.https://doi.org/10.1016/S0166-2236(98)01276-4
-
SKN-1/Nrf, stress responses, and aging in Caenorhabditis elegansFree Radical Biology and Medicine 88:290–301.https://doi.org/10.1016/j.freeradbiomed.2015.06.008
-
A small-molecule Nrf1 and Nrf2 activator mitigates polyglutamine toxicity in spinal and bulbar muscular atrophyHuman Molecular Genetics 25:1979–1989.https://doi.org/10.1093/hmg/ddw073
-
Oxidative modification and inactivation of the proteasome during coronary occlusion/reperfusionJournal of Biological Chemistry 276:30057–30063.https://doi.org/10.1074/jbc.M100142200
-
A mutant affecting the heavy chain of myosin in Caenorhabditis elegansJournal of Molecular Biology 90:291–300.https://doi.org/10.1016/0022-2836(74)90374-X
-
In vivo aggregation of beta-amyloid peptide variantsJournal of Neurochemistry 71:1616–1625.https://doi.org/10.1046/j.1471-4159.1998.71041616.x
-
Amyloid beta-protein inhibits ubiquitin-dependent protein degradation in vitroThe Journal of Biological Chemistry 270:19702–19708.
-
Basic leucine zipper protein Cnc-C is a substrate and transcriptional regulator of the Drosophila 26S proteasomeMolecular and Cellular Biology 31:897–909.https://doi.org/10.1128/MCB.00799-10
-
The active sites of the eukaryotic 20 S proteasome and their involvement in subunit precursor processingJournal of Biological Chemistry 272:25200–25209.https://doi.org/10.1074/jbc.272.40.25200
-
Proteostasis impairment in protein-misfolding and -aggregation diseasesTrends in Cell Biology 24:506–514.https://doi.org/10.1016/j.tcb.2014.05.003
-
A proteolytic pathway that recognizes ubiquitin as a degradation signalJournal of Biological Chemistry 270:17442–17456.https://doi.org/10.1074/jbc.270.29.17442
-
Endoplasmic reticulum stress compromises the ubiquitin-proteasome systemHuman Molecular Genetics 14:2787–2799.https://doi.org/10.1093/hmg/ddi312
-
Coping with protein quality control failureAnnual Review of Cell and Developmental Biology 33:439–465.https://doi.org/10.1146/annurev-cellbio-111315-125334
-
Biological and chemical approaches to diseases of proteostasis deficiencyAnnual Review of Biochemistry 78:959–991.https://doi.org/10.1146/annurev.biochem.052308.114844
-
The amyloid hypothesis of Alzheimer's disease at 25 yearsEMBO Molecular Medicine 8:595–608.https://doi.org/10.15252/emmm.201606210
-
Proteasomal ubiquitin receptor RPN-10 controls sex determination in Caenorhabditis elegansMolecular Biology of the Cell 17:5356–5371.https://doi.org/10.1091/mbc.e06-05-0437
-
Aggregated and monomeric alpha-synuclein bind to the S6' proteasomal protein and inhibit proteasomal functionJournal of Biological Chemistry 278:11753–11759.https://doi.org/10.1074/jbc.M208641200
-
Aging as an event of proteostasis collapseCold Spring Harbor Perspectives in Biology 3:a004440.https://doi.org/10.1101/cshperspect.a004440
-
Nrf1 is targeted to the endoplasmic reticulum membrane by an N-terminal transmembrane domain. inhibition of nuclear translocation and transacting functionThe Journal of Biological Chemistry 281:19676–19687.https://doi.org/10.1074/jbc.M602802200
Article and author information
Author details
Nicolas J Lehrbach

Funding
Grace Science Foundation
- Nicolas J Lehrbach
- Gary Ruvkun
National Institutes of Health (R01 AG016636)
- Gary Ruvkun
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Copyright
© 2019, Lehrbach and Ruvkun
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 4,265
- views
-
- 682
- downloads
-
- 81
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 81
- citations for umbrella DOI https://doi.org/10.7554/eLife.44425
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Endoplasmic reticulum-associated SKN-1A/Nrf1 mediates a cytoplasmic unfolded protein response and promotes longevity
eLife 8:e44425.
https://doi.org/10.7554/eLife.44425




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}