Functional reconstitution of a bacterial CO2 concentrating mechanism in Escherichia coli
- Department of Molecular and Cell Biology, University of California, Berkeley, United States
- Department of Plant and Environmental Sciences, Weizmann Institute of Science, Israel
- Max Planck Institute of Molecular Plant Physiology, Germany
Abstract
Many photosynthetic organisms employ a CO2 concentrating mechanism (CCM) to increase the rate of CO2 fixation via the Calvin cycle. CCMs catalyze ≈50% of global photosynthesis, yet it remains unclear which genes and proteins are required to produce this complex adaptation. We describe the construction of a functional CCM in a non-native host, achieved by expressing genes from an autotrophic bacterium in an Escherichia coli strain engineered to depend on rubisco carboxylation for growth. Expression of 20 CCM genes enabled E. coli to grow by fixing CO2 from ambient air into biomass, with growth in ambient air depending on the components of the CCM. Bacterial CCMs are therefore genetically compact and readily transplanted, rationalizing their presence in diverse bacteria. Reconstitution enabled genetic experiments refining our understanding of the CCM, thereby laying the groundwork for deeper study and engineering of the cell biology supporting CO2 assimilation in diverse organisms.
Introduction
Nearly all carbon in the biosphere enters by CO2 fixation in the Calvin-Benson-Bassham cycle (Bassham, 2003; Bassham et al., 1954; Benson, 2002; Field et al., 1998; Raven, 2009). Ribulose Bisphosphate Carboxylase/Oxygenase - commonly known as rubisco - is the CO2 fixing enzyme in this cycle (Kawashima and Wildman, 1971; Weissbach et al., 1956; Wildman, 2002) and likely the most abundant enzyme on Earth (Bar-On and Milo, 2019).
As rubisco is abundant and central to biology, one might expect it to be an exceptional catalyst, but it is not. Photosynthetic rubiscos are modest enzymes, with carboxylation turnover numbers (kcat) ranging from 1 to 10 s−1 (Badger et al., 1998; Flamholz et al., 2019; Iñiguez et al., 2020; Jordan and Ogren, 1983; Savir et al., 2010; Tcherkez et al., 2006). Moreover, all known rubiscos catalyze a competing oxygenation of the five-carbon organic substrate, ribulose 1, 5-bisphosphate (Bathellier et al., 2018; Bowes and Ogren, 1972; Cleland et al., 1998). Rubisco oxygenation represents a ‘waste’ of cellular resources on two fronts: it fails to generate any new organic carbon and also produces a molecule (2-phosphoglycolate) that is not part of the Calvin cycle and therefore must be recycled through a salvage pathway to keep the cycle going (Busch, 2020).
Rubisco arose >2.5 billion years ago, when Earth’s atmosphere contained little O2 and abundant CO2 (Fischer et al., 2016; Shih et al., 2016). In this environment, rubisco’s eponymous oxygenase activity could not have hindered carbon fixation or the growth of CO2-fixing organisms. Present-day atmosphere, however, poses a problem for plants and other autotrophs: their primary carbon source, CO2, is relatively scarce (≈0.04%) while a potent competing substrate, O2, is abundant (≈21%).
CO2 concentrating mechanisms (CCMs) arose multiple times over the last 2 billion years (Flamholz and Shih, 2020; Raven et al., 2017) and overcame rubisco’s limitations by concentrating CO2 near the enzyme (Figure 1A). In an elevated CO2 environment, most rubisco active sites will be occupied with CO2 and not O2. As such, high CO2 is expected to increase the rate of carboxylation and competitively inhibit oxygenation (Bowes and Ogren, 1972) thereby improving overall carbon assimilation (Figure 1B). Today, at least four varieties of CCMs are found in plants, algae, and bacteria (Flamholz and Shih, 2020; Raven et al., 2017), organisms with CCMs are collectively responsible for ≈50% of global net photosynthesis (Raven et al., 2017), and some of the most productive human crops (e.g. maize and sugarcane) rely on CCMs.
Figure 1 with 2 supplements see all
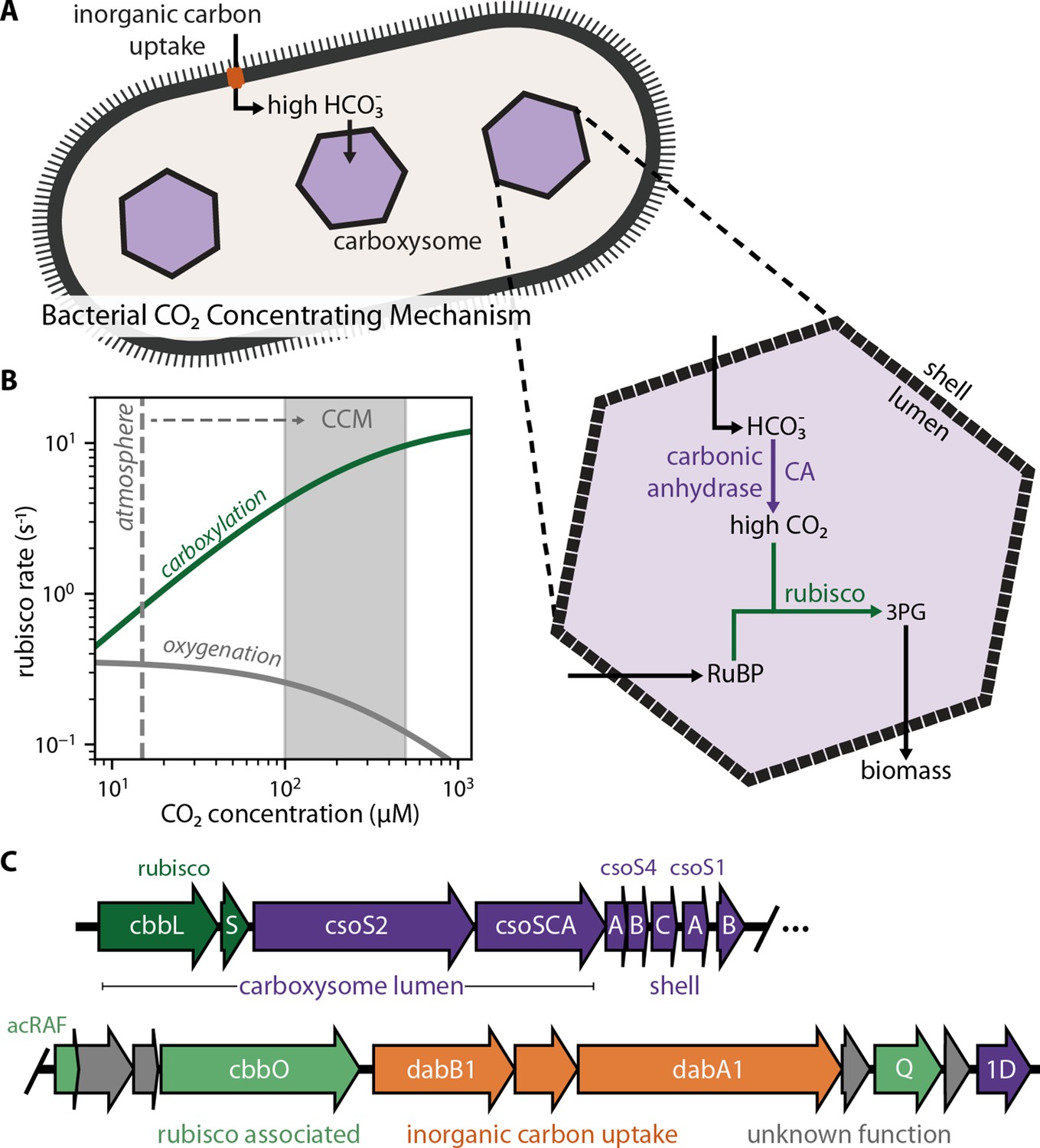
Twenty genes form the basis of a bacterial CCM.
(A) The bacterial CCM consists of at least two essential components - energy-coupled inorganic carbon uptake and carboxysome structures that encapsulate rubisco with a carbonic anhydrase (CA) enzyme (Desmarais et al., 2019; Kaplan et al., 1980; Price and Badger, 1989a, Price and Badger, 1989b; Rae et al., 2013; Shively et al., 1973). Transport generates a large cytosolic HCO3- pool, which is rapidly converted to high carboxysomal CO2 concentration by the carboxysomal CA (Mangan et al., 2016; McGrath and Long, 2014). (B) Elevated CO2 increases the rubisco carboxylation rate (green) and suppresses oxygenation by competitive inhibition (grey). [O2] was set to 270 μM for rate calculations. A more detailed version of this calculation is described in Figure 1—figure supplement 1. (C) H. neapolitanus CCM genes are mostly contained in a 20 gene cluster (Desmarais et al., 2019) expressing rubisco and its associated chaperones (green), carboxysome structural proteins (purple), and an inorganic carbon transporter (orange). Supplementary file 1 gives fuller description of the functions of these 20 genes along with a per-gene bibliography. Figure 1—figure supplement 2 demonstrates that the operon beginning with acRAF indeed encodes a functional inorganic carbon transporter.
CCMs are particularly common among autotrophic bacteria: all Cyanobacteria and many Proteobacteria have CCM genes (Kerfeld and Melnicki, 2016; Rae et al., 2013). Bacterial CCMs rely on two crucial features: (i) energy-coupled inorganic carbon uptake at the cell membrane and (ii) a 200+ MDa protein organelle called the carboxysome that encapsulates rubisco with a carbonic anhydrase enzyme (Desmarais et al., 2019; Kaplan et al., 1980; Price and Badger, 1989a, Price and Badger, 1989b; Rae et al., 2013; Shively et al., 1973). In the prevailing model of the carboxysome CCM (Fridlyand et al., 1996; Mangan et al., 2016; McGrath and Long, 2014), inorganic carbon uptake produces a high, above-equilibrium cytosolic HCO3- concentration (≈30 mM) that diffuses into the carboxysome, where carbonic anhydrase activity produces a high carboxysomal CO2 concentration that promotes efficient carboxylation by rubisco (Figure 1A–B).
As CCMs accelerate CO2 fixation by rubisco, there is great interest in transplanting them into crops (Ermakova et al., 2020; McGrath and Long, 2014). Carboxysome-based CCMs are especially attractive because they natively function in single cells and appear to rely on a tractable number of genes (Lin et al., 2014; Long et al., 2018; Occhialini et al., 2016; Orr et al., 2020). Modeling suggests that introducing bacterial CCM components could improve plant photosynthesis (McGrath and Long, 2014), especially if aspects of plant physiology can be modulated via genetic engineering (Wu et al., 2019). However, expressing bacterial rubiscos and carboxysome components has, so far, uniformly resulted in transgenic plants displaying impaired growth (Lin et al., 2014; Long et al., 2018; Occhialini et al., 2016; Orr et al., 2020). More generally, as our understanding of the genes and proteins participating in the carboxysome CCM rests mostly on loss-of-function genetic experiments in native hosts (Baker et al., 1998; Cai et al., 2009; Cannon et al., 2001; Desmarais et al., 2019; Marcus et al., 1986; Ogawa et al., 1987; Price and Badger, 1989a), it is possible that some genetic, biochemical, and physiological aspects of CCM function remain unappreciated. We therefore sought to test whether current understanding is sufficient to reconstitute the bacterial CCM in a non-native bacterial host, namely Escherichia coli.
Using a genome-wide screen in the CO2-fixing proteobacterium Halothiobacillus neapolitanus, we recently demonstrated that a 20-gene cluster encodes all activities required for the CCM, at least in principle (Desmarais et al., 2019). These genes are detailed in Supplementary file 1 and include rubisco large and small subunits, the carboxysomal carbonic anhydrase, seven structural proteins of the ɑ-carboxysome (Bonacci et al., 2012), an energy-coupled inorganic carbon transporter (Desmarais et al., 2019; USF MCB4404L et al., 2017; Scott et al., 2019), three rubisco chaperones (Aigner et al., 2017; Feiz et al., 2014; Mueller-Cajar, 2017; Wheatley et al., 2014), and four genes of uncertain function (Figure 1C). We aimed to test whether these genes are sufficient to establish a functioning CCM in E. coli.
Results
As E. coli is a heterotroph, consuming organic carbon molecules to produce energy and biomass, it does not natively rely on rubisco. Therefore, in order to evaluate the effect of heterologous CCM expression, we first designed an E. coli strain that depends on rubisco carboxylation for growth. To grow on glycerol as the sole carbon source, E. coli must synthesize ribose 5-phosphate (Ri5P) for nucleic acids. Synthesis of Ri5P via the pentose phosphate pathway forces co-production of ribulose 5-phosphate (Ru5P). Deletion of ribose 5-phosphate isomerase (rpiAB genes, denoted Δrpi), however, makes Ru5P a metabolic ‘dead-end’ (Figure 2A). Expression of phosphoribulokinase (prk) and rubisco enables a ‘detour’ pathway converting Ru5P and CO2 into two units of the central metabolite 3-phosphoglycerate (3PG), enabling Ru5P metabolism and growth (Figure 2A). Additionally, cytosolic carbonic anhydrase activity is incompatible with the bacterial CCM (Price and Badger, 1989b). We therefore constructed a strain, named CCMB1 for ‘CCM Background 1’, lacking rpiAB and all endogenous carbonic anhydrases (Materials and methods, Appendix 1).
Figure 2 with 5 supplements see all
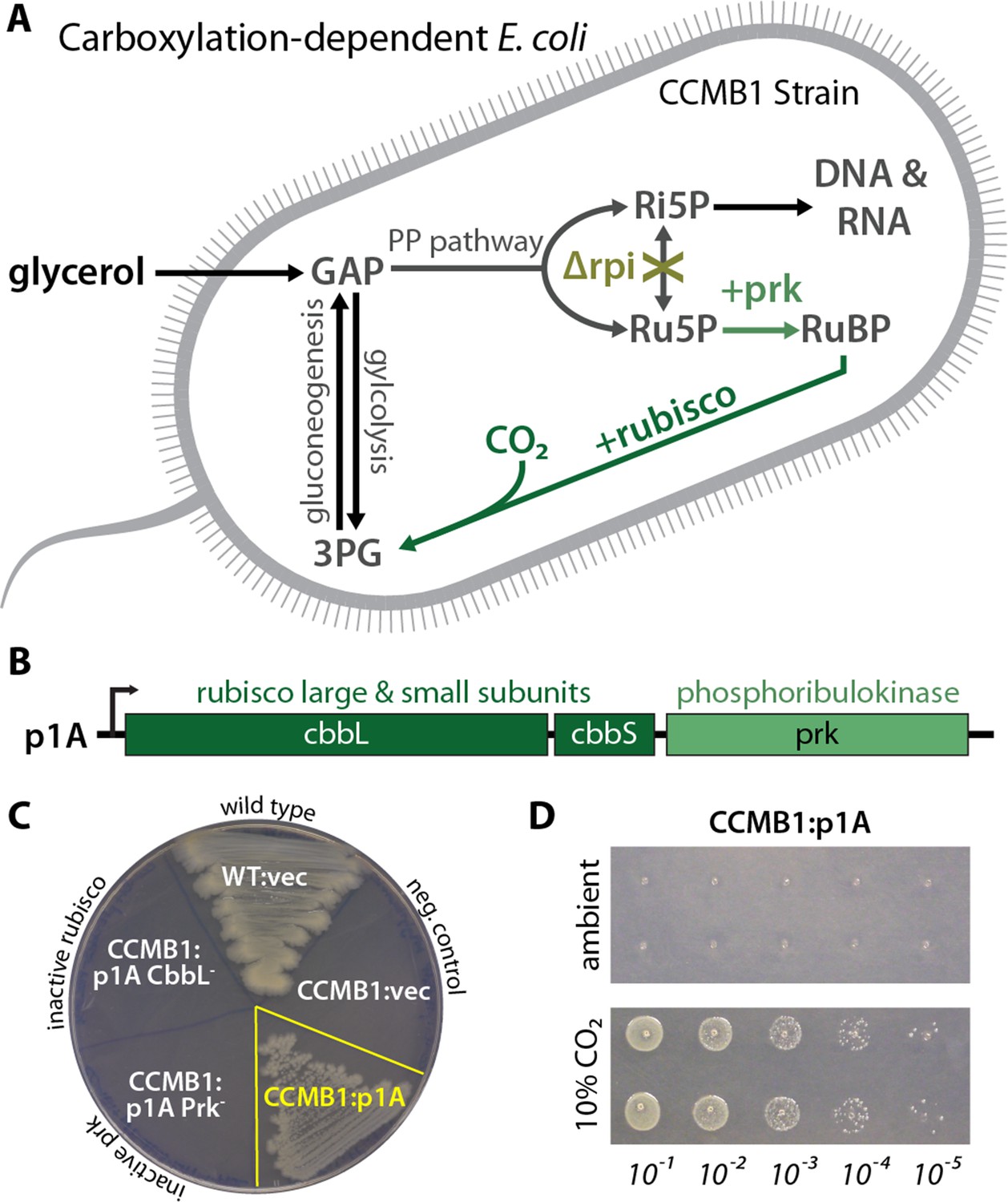
CCMB1 depends on rubisco carboxylation for growth on glycerol.
(A) Ribose-5-phosphate (Ri5P) is required for nucleotide biosynthesis. Deletion of ribose-phosphate isomerase (Δrpi) in CCMB1 blocks ribulose-5-phosphate (Ru5P) metabolism in the pentose phosphate (PP) pathway. Expression of rubisco (H. neapolitanus CbbLS) and phosphoribulokinase (S. elongatus PCC7942 prk) on the p1A plasmid (B) permits Ru5P metabolism, thus enabling growth on M9 glycerol media in 10% CO2 (C). Mutating the rubisco active site (p1A CbbL-) abrogates growth, as does mutating ATP-binding residues of Prk (p1A Prk-). (D) CCMB1:p1A grows well under 10% CO2, but fails to grow in ambient air. Cells were grown on M9 glycerol media throughout. The algorithmic design of CCMB1 is described in Figure 2—figure supplement 4 and Appendix 1. The mechanism of rubisco-dependence is diagrammed in Figure 2—figure supplement 3. Figure supplement 2 demonstrates growth of CCMB1:p1A on various media, Figure 2—figure supplement 5 demonstrates complementation by a variety of bacterial rubiscos and Figure 2—figure supplement 1 demonstrates anaerobic growth of CCMB1:p1A, establishing that oxygenation is not required for growth. Acronyms: ribulose 1, 5-bisphosphate (RuBP), 3-phosphoglycerate (3PG).
As predicted, CCMB1 required rubisco and prk for growth on glycerol minimal media in 10% CO2 (Figure 2B–C). CCMB1:p1A failed to grow on glycerol media in ambient air, however, presumably due to insufficient carboxylation at low CO2 (Figure 2D). As such, CCMB1:p1A displays the ‘high-CO2 requiring’ phenotype that is the hallmark of CCM mutants (Baker et al., 1998; Marcus et al., 1986; Price and Badger, 1989a). Four additional bacterial rubiscos were tested and displayed the same pattern, enabling CCMB1 to grow reproducibly in high CO2 but not in ambient air (Figure 2—figure supplement 1). When expressing rubisco and prk from the p1A plasmid, CCMB1 also grew reproducibly in an anoxic mix of 10:90 CO2:N2 (Figure 2—figure supplement 2) implying that carboxylation is sufficient for growth on glycerol media and rubisco-catalyzed oxygenation of RuBP is not required.
We expected that expressing a functional CO2-concentrating mechanism would cure CCMB1 of its high-CO2 requirement and permit growth in ambient air. We therefore generated two plasmids, pCB and pCCM, that together express all 20 genes from the H. neapolitanus CCM cluster (Figure 1C). pCB encodes 10 carboxysome genes (Bonacci et al., 2012; Cai et al., 2008), including rubisco large and small subunits, along with prk. The remaining H. neapolitanus genes, including putative rubisco chaperones (Aigner et al., 2017; Mueller-Cajar, 2017; Wheatley et al., 2014) and an inorganic carbon transporter (Desmarais et al., 2019; Scott et al., 2019), were cloned into the second plasmid, pCCM.
CCMB1 co-transformed with pCB and pCCM initially failed to grow on glycerol media. We therefore conducted selection experiments, described fully in Appendix 2, that ultimately resulted in the isolation of mutant plasmids conferring growth in ambient air. Briefly, CCMB1:pCB + pCCM cultures were grown to saturation in 10% CO2. These cultures were washed and plated on glycerol minimal media (Materials and methods). Colonies became visible after 20 days of incubation in ambient air, but only when induction and both plasmids were provided (Figure 3—figure supplement 1). Deep-sequencing of plasmid DNA revealed mutations in regulatory sequences (e.g. a promoter and transcriptional repressor) but none in sequences coding for CCM components (Supplementary file 1). Individual post-selection plasmids pCB’ and pCCM’ were reconstructed by PCR, resequenced, and transformed into naive CCMB1 (Materials and methods). As shown in Figure 3, pCB’ and pCCM’ together enabled reproducible growth of CCMB1 in ambient air, suggesting that the 20 genes expressed are sufficient to produce a heterologous CCM without any genomic mutations.
Figure 3 with 2 supplements see all
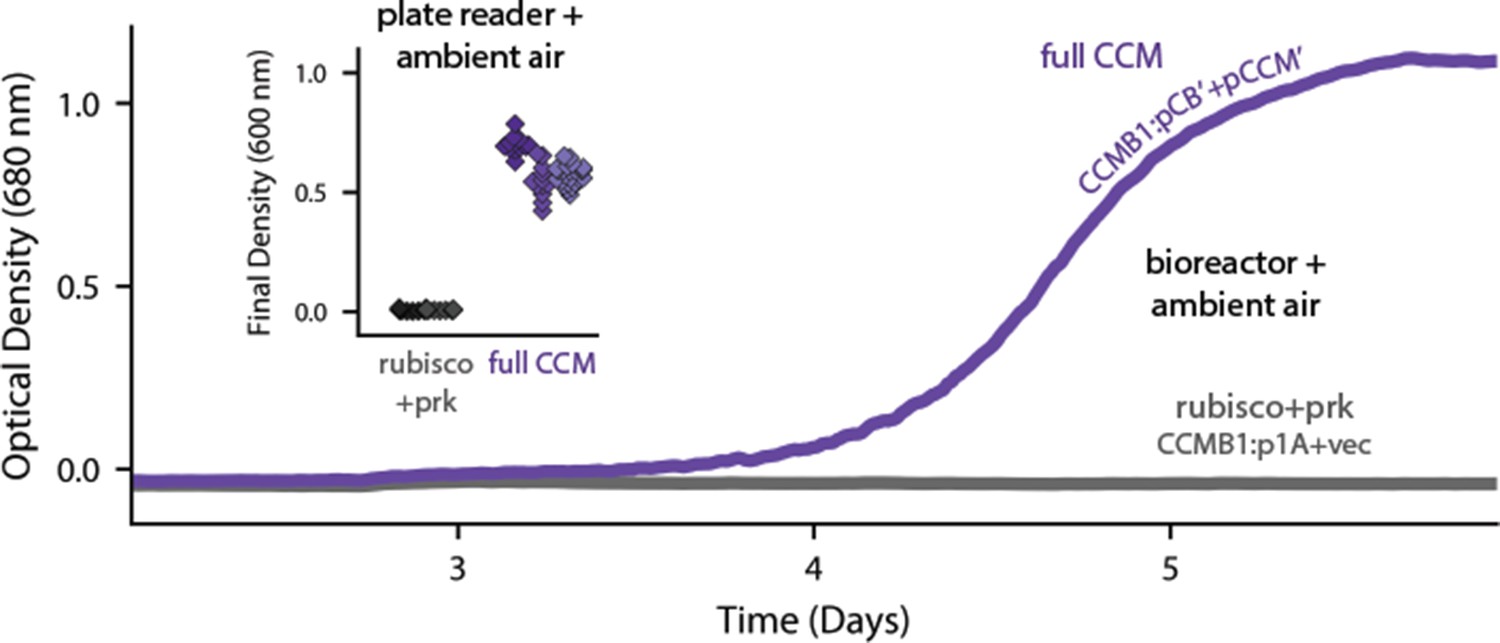
Expression of 20 CCM genes permits growth of CCMB1 in ambient air.
Time course data give representative growth curves from a bioreactor bubbling ambient air. CCMB1:pCB’ + pCCM’ grows well (purple, ‘full CCM’), while rubisco and prk alone are insufficient for growth in ambient air (grey, CCMB1:p1A+vec). Inset: a plate reader experiment in biological triplicate (different shades) gave the same result. Expressing the full complement of CCM genes led to an increase in culture density (optical density at 600 nm) of ≈0.6 units after 80 hr of cultivation. Bootstrapping was used to calculate a 99.9% confidence interval of 0.56–0.64 OD units for the effect of expressing the full CCM during growth in ambient air. Figure 3—figure supplement 1 and Appendix 2 describe the selection procedures in detail while Figure 3—figure supplement 2 shows triplicate growth curves and evaluates statistical significance.
To verify that growth in ambient air depends on the CCM, we generated plasmids carrying targeted mutations to known CCM components (Figure 4). An inactivating mutation to the carboxysomal rubisco (CbbL K194M) prohibited growth entirely. Mutations targeting the CCM, rather than rubisco itself, should ablate growth in ambient air while permitting growth in high CO2(Desmarais et al., 2019; Mangan et al., 2016; Marcus et al., 1986; Price and Badger, 1989a; Rae et al., 2013). Consistent with this understanding, an inactive mutant of the carboxysomal carbonic anhydrase (CsoSCA C173S) required high-CO2 for growth. Similarly, disruption of carboxysome formation by removal of the pentameric shell proteins or the N-terminal domain of CsoS2 also eliminated growth in ambient air. Removing the pentameric proteins CsoS4AB disrupts the permeability barrier at the carboxysome shell (Cai et al., 2009), while truncating CsoS2 prohibits carboxysome formation entirely (Oltrogge et al., 2020). Finally, an inactivating mutation to the inorganic carbon transporter also eliminated growth in ambient air (Desmarais et al., 2019).
Figure 4 with 2 supplements see all
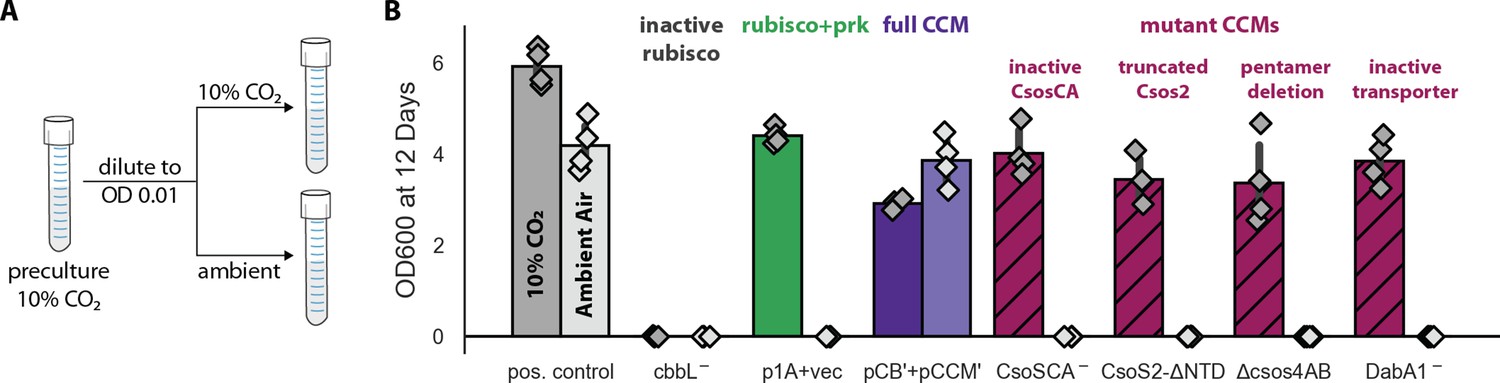
Growth in ambient air depends on the known components of the bacterial CCM.
We generated plasmid variants carrying inactivating mutations to known components of the CCM. (A) Pre-cultures were grown in 10% CO2 and diluted into pairs of tubes, one of which was cultured in 10% CO2 and the other in ambient air (Materials and methods). Strains were tested in biological quadruplicate and culture density was measured after 12 days to ensure an endpoint measurement of capacity to grow. (B) Targeted mutations to CCM components ablated growth in ambient air while permitting growth in 10% CO2, as expected. The left bar (darker color) gives the mean endpoint density in 10% CO2 for each strain. The right bar (lighter color) gives the mean in ambient air. Error bars give a 95% confidence interval for the mean. From left to right, in pairs: a positive control for growth (a complemented carbonic anhydrase knockout in grey, see Materials and methods) grew in 10% CO2 and ambient air, while a negative control CCMB1 strain carrying catalytically inactive rubisco (CCMB1:pCB’ CbbL-+pCCM’) failed to grow in either condition; CCMB1 expressing rubisco and prk but no CCM genes (green, CCMB1:p1A+vec) grew only in 10% CO2; CCMB1:pCB’+pCCM’ grew in 10% CO2 and ambient air, recapitulating results presented in Figure 3. The following four pairs of maroon bars give growth data for strains carrying targeted mutations to CCM genes: an inactivating mutation to carboxysomal carbonic anhydrase (CCMB1:pCB’ CsoSCA-+pCCM’), deletion of the CsoS2 N-terminus responsible for recruiting rubisco to the carboxysome (CCMB1:pCB’ CsoS2 ΔNTD+pCCM’), deletion of pentameric vertex proteins (CCMB1:pCB’ ΔcsoS4AB + pCCM’), and inactivating mutations to the DAB carbon uptake system (CCMB1:pCB’ DabA1- + pCCM’). All four CCM mutations abrogated growth in air while permitting growth in 10% CO2. The positive control is the CAfree strain expressing human carbonic anhydrase II (Materials and methods). Figure 4—figure supplement 1 describes statistical analyses, a 4-day replicate experiment, and additional mutants testing the contribution of rubisco chaperones to the CCM. Figure 4—figure supplement 2 gives measurements of media pH after growth in 10% CO2 and ambient air. Detailed description of all plasmid and mutation abbreviations is given in Supplementary file 1.
These experiments demonstrate that pCB’ and pCCM’ enable CCMB1 to grow in ambient air in a manner that depends on the known components of the bacterial CCM. To confirm that these cells produce carboxysome structures, we performed thin section electron microscopy. Regular polyhedral inclusions of ≈100 nm diameter were visible in micrographs (Figure 5A), implying production of morphologically normal carboxysomes. Furthermore, we were able to purify carboxysome structures from CCMB1:pCB’+pCCM’ using established methods. Carboxysomes from CCMB1:pCB’+pCCM’ were similar in appearance to those from the native host, although more heterogeneous in size and shape (Figure 5B). The rubisco complex was visible inside isolated carboxysomes and confirmed to co-migrate with the structure via SDS-PAGE analysis (Figure 5—figure supplement 1).
Figure 5 with 2 supplements see all

CCMB1:pCB’+pCCM’ produces carboxysomes when grown in air.
(A) Polyhedral bodies resembling carboxysomes are evident in electron micrographs of CCMB1:pCB’+pCCM’ cells grown in air (full CCM, both images on the right) but were not observed in a negative control lacking pCB and pCCM plasmids (left, Methods). All panels have equal scale. (B) Carboxysome structures purified from CCMB1:pCB’+pCCM’ grown in ambient air (Materials and methods, right) resemble structures isolated from the native host (left) in size and morphology. Figure 5—figure supplement 2 gives full size and additional images clearly showing rubisco inside isolated carboxysomes. SDS-PAGE gels in Figure 5—figure supplement 1 demonstrate co-migration of rubisco large and small subunits with carboxysomes structures through the purification procedure.
We next conducted isotopic labeling experiments to determine whether CCMB1:pCB’ + pCCM’ fixes CO2 from ambient air into biomass. Cells were grown in minimal media with 13C-labeled glycerol as the sole organic carbon source, such that CO2 from ambient air was the dominant source of 12C. The isotopic composition of amino acids in total biomass hydrolysate was analyzed via mass spectrometry (Materials and methods). Serine is a useful sentinel of rubisco activity because E. coli produces it from the rubisco product 3PG (Stauffer, 2004; Szyperski, 1995). 3PG is also an intermediate of lower glycolysis (Bar-Even et al., 2012), and so the degree of 12C labeling on serine reports on the balance of fluxes through rubisco and lower glycolysis (Figure 6A). We therefore expected excess 12C labeling of serine when rubisco is active in CCMB1. Consistent with this expectation, serine from CCMB1:pCB’+pCCM’ cells contained roughly threefold more 12C than the rubisco-independent control (Figure 6B). We estimated the contribution of rubisco to 3PG synthesis in vivo by comparing labeling patterns between the rubisco-dependent experimental cultures and controls (Appendix 2). Based on these estimates, rubisco carboxylation was responsible for at least 10% of 3PG synthesis in all four biological replicates (Figure 6C, Materials and methods), confirming fixation of CO2 from ambient air. As such, this work represents the first functional reconstitution of any CCM.
Figure 6 with 3 supplements see all

CCMB1:pCB’+pCCM’ fixes CO2 from ambient air into biomass.
Biological replicate cultures were grown in ambient air in M9 media containing 99% 13C labeled glycerol such that 12CO2 from air is the dominant source of 12C. In (A) 13C is depicted as open circles and partial 12C incorporation is indicated in green. As serine is a direct metabolic product of 3PG, we expect 12C enrichment on serine when rubisco is active in CCMB1 cells. 3PG also derives from glycolytic metabolism of glycerol, so complete 12C labeling of serine was not expected. (B) The 12C composition of serine from CCMB1:pCB’ + pCCM’ (‘Experiment’) is roughly threefold above the control strain (CAfree:vec+pFA-HCAII), which grows in a rubisco-independent manner (Materials and methods). Figure 6—figure supplement 1 gives 12C composition of all measured amino acids. (C) The fraction of 3PG production due to rubisco was predicted via Flux Balance Analysis and estimated from isotopic labeling data (Materials and methods, Appendix 3). Estimates of the rubisco flux fraction exceeded 10% for all four biological replicates and the mean estimate of ≈14% accords reasonably with predictions ranging from 16 to 24%. Appendix 3 and Figure 6—figure supplements 2–3 detail the flux inference procedure and give additional evidence for in vivo carboxylation from the fragmentation of serine.
Reconstitution in E. coli enabled us to investigate which H. neapolitanus genes are necessary for CCM function in the absence of any regulation or genetic redundancy (i.e. genes with overlapping function) present in the native host. We focused on genes involved in rubisco proteostasis and generated plasmids lacking acRAF, a putative rubisco chaperone, or carrying targeted mutations to CbbQ, an ATPase involved in activating rubisco catalysis (Aigner et al., 2017; Mueller-Cajar, 2017; Sutter et al., 2015; Wheatley et al., 2014). Although acRAF deletion had a large negative effect in H. neapolitanus (Desmarais et al., 2019), neither acRAF nor CbbQ were strictly required for CCMB1 to grow in ambient air. Consistent with our screen in the native host (Desmarais et al., 2019); however, acRAF deletion produced a substantial growth defect (Figure 4—figure supplement 1, panel C), suggesting that the rate of rubisco complex assembly is an important determinant of carboxysome biogenesis.
Discussion
Today, CCMs catalyze about half of global photosynthesis (Raven et al., 2017), but this was not always so. Land plant CCMs, for example, arose only in the last 100 million years (Flamholz and Shih, 2020; Raven et al., 2017; Sage et al., 2012). Although all contemporary Cyanobacteria have CCM genes, these CCMs are found in two convergently evolved varieties (Flamholz and Shih, 2020; Kerfeld and Melnicki, 2016; Rae et al., 2013), suggesting that the ancestor of present-day Cyanobacteria and chloroplasts did not have a CCM (Rae et al., 2013). So how did carboxysome CCMs come to dominate the cyanobacterial phylum?
Here, we demonstrated that the ɑ-carboxysome CCM from H. neapolitanus can be readily transferred between species and confers a large growth benefit, suggesting that these CCMs became so widespread by horizontal transfer between bacteria (Kerfeld and Melnicki, 2016; Rae et al., 2013). We constructed a functional bacterial CCM by expressing 20 genes in an E. coli strain, CCMB1, engineered to depend on rubisco carboxylation. In accordance with its role in native autotrophic hosts (Desmarais et al., 2019; Long et al., 2018; Marcus et al., 1986; Price and Badger, 1989a), the transplanted CCM required (i) ɑ-carboxysome structures containing both rubisco and carbonic anhydrase and (ii) inorganic carbon uptake at the cell membrane in order to enable CCMB1 to grow by fixing CO2 from ambient air (Figures 3–6). These results conclusively demonstrate that at most 20 gene products are required to produce a bacterial CCM. The ɑ-carboxysome CCM is apparently genetically compact and ‘portable’ between organisms. It is possible, therefore, that expressing bacterial CCMs in non-native autotrophic hosts will improve CO2 assimilation and growth. This is a promising approach to improving plant growth characteristics (Ermakova et al., 2020; Long et al., 2016; Wu et al., 2019) and also engineering enhanced microbial production of fuel, food products, and commodity chemicals from CO2 (Claassens et al., 2016; Gleizer et al., 2019).
Reconstitution also enabled us to test, via simple genetic experiments, whether particular genes play a role in the CCM (Figure 4—figure supplement 1). These experiments demonstrated that the rubisco chaperones are strictly dispensable for producing a functional bacterial CCM, although removing acRAF produced a substantial growth defect that warrants further investigation. Further such experiments can use our reconstituted CCM to delineate a minimal reconstitution of the bacterial CCM suitable for plant expression (Du et al., 2014; Long et al., 2018, Long et al., 2016; Occhialini et al., 2016; Orr et al., 2020), test hypotheses about carboxysome biogenesis (Bonacci et al., 2012; Oltrogge et al., 2020), and probe the relationship between CCMs and host physiology (Mangan et al., 2016; McGrath and Long, 2014; Price and Badger, 1989b). This last point deserves special emphasis as the growth physiologies of plants and bacteria are exceedingly different and it remains unclear whether microbial CCMs can function efficiently when expressed in macroscopic land plants (Flamholz and Shih, 2020).
Our approach to studying CCMs by reconstitution in tractable non-native hosts can be also applied to other CCMs, including β-carboxysome CCMs, the algal pyrenoid, and plausible evolutionary ancestors thereof. Historical trends in atmospheric CO2 likely promoted the evolution of CCMs (Fischer et al., 2016; Flamholz and Shih, 2020), so testing the growth of plausible ancestors of bacterial CCMs (e.g. carboxysomes lacking carbonic anhydrase activity) may provide insight into paths of CCM evolution and the composition of the ancient atmosphere at the time bacterial CCMs arose. In response to these same pressures, diverse eukaryotic algae evolved CCMs relying on micron-sized rubisco aggregates called the pyrenoids (Flamholz and Shih, 2020; Wang and Jonikas, 2020). Pyrenoid CCMs are collectively responsible for perhaps 70–80% of oceanic photosynthesis (Mackinder et al., 2016; Raven et al., 2017), yet many fundamental questions remain regarding the composition and operation of algal CCMs (Wang and Jonikas, 2020). Functional reconstitution of a pyrenoid CCM is a worthy goal which, once achieved, will indicate enormous progress in our collective understanding of the genetics, cell biology, biochemistry, and physical processes supporting the eukaryotic complement of oceanic photosynthesis. We hope such studies will further our principled understanding of, and capacity to engineer, the cell biology supporting CO2 fixation in diverse organisms.
Materials and methods
Growth conditions
Request a detailed protocolUnless otherwise noted, cells were grown on M9 minimal media supplemented with 0.4% v/v glycerol, 0.5 ppm thiamin (104 fold dilution of 0.5% w/v stock) and a trace element mix. The trace element mix components and their final concentrations in M9 media are: 50 mg/L EDTA, 31 mM FeCl3, 6.2 mM ZnCl2, 0.76 mM CuSO4·5H2O, 0.42 mM CoCl2·6H2O, 1.62 mM H3BO3, 81 nM MnCl2·4H2O. 100 nM anhydrotetracycline (aTc) was used in induced cultures. For routine cloning, 25 mg/L chloramphenicol and 60 mg/L kanamycin selection were used as appropriate. Antibiotics were reduced to half concentration (12.5 and 30 mg/L, respectively) for CCMB1 growth experiments and kanamycin was omitted when evaluating rubisco-dependence of growth as pF-derived plasmids carrying kanamycin resistance also express rubisco. Culture densities were measured at 600 nm in a table top spectrophotometer (Genesys 20, Thermo Scientific) and turbid cultures were measured in five- or tenfold dilution as appropriate in order to reach the linear regime of the spectrophotometer.
Agar plates were incubated at 37°C in defined CO2 pressures in a CO2 controlled incubator (S41i, New Brunswick). For experiments in which a frozen bacterial stock was used to inoculate the culture, cells were first streaked on agar plates and incubated at 10% CO2 to facilitate fast growth. Pre-cultures derived from colonies were grown in 2–5 mL liquid M9 glycerol media under 10% CO2 with a matching 1 mL control in ambient air. Negative control strains unable to grow in minimal media (i.e. active site mutants of rubisco) were streaked on and pre-cultured in LB media under 10% CO2.
Growth curves were obtained using two complementary methods: an eight-chamber bioreactor for large-volume cultivation (MC1000, PSI), and 96-well plates in a gas controlled plate reader plate (Spark, Tecan). For the 96-well format, cells were pre-cultured in the appropriate permissive media, M9 glycerol under 10% CO2 where possible. If rich media was used, for example for negative controls, stationary phase cells were washed in 2x the culture volume and resuspended in 1x culture volume of M9 media with no carbon source. Cultures were diluted to an OD of 1.0 (600 nm) and 250 μl cultures were inoculated by adding 5 μl of cells to 245 μl media. A humidity cassette (Tecan) was refilled daily with distilled water to mitigate evaporation during multi-day cultivation at 37 °C. Evaporation nonetheless produced irregular growth curves (e.g. Figure 3—figure supplement 2), which motivated larger volume cultivation in the bioreactor, which mixes by bubbling ambient air into each growth vessel. 80 mL bioreactor cultures were inoculated to a starting OD of 0.005 (600 nm) and grown at 37°C to saturation. Optical density was monitored continuously at 680 nm.
Anaerobic cultivation of agar plates was accomplished using a BBL GasPak 150 jar (BD) flushed six times with an anoxic mix of 10% CO2 and 90% N2. Tenfold titers of biological duplicate cultures were plated on M9 glycerol media with and without 20 mM NaNO3 supplementation. Because E. coli cannot ferment glycerol, NO3- was supplied as an alternative electron acceptor. Plates without NO3- showed no growth (Figure 2—figure supplement 1), confirming the presence of an anaerobic atmosphere in the GasPak.
Computational design of rubisco-dependent strains
Request a detailed protocolTo computationally design mutant strains in which growth is coupled to rubisco carboxylation flux, we used a variant of Flux Balance Analysis (Lewis et al., 2012) called ‘OptSlope’ (Antonovsky et al., 2016). Starting from a published model of E. coli central metabolism, the Core Escherichia coli Metabolic Model (Orth et al., 2010), we considered all pairs of central metabolic knockouts and ignored those that permit growth in silico in the absence of rubisco and phosphoribulokinase (Prk) activities. For the remaining knockouts, we evaluated the degree of coupling between rubisco flux and biomass production during growth in nine carbon sources: glucose, fructose, gluconate, ribose, succinate, xylose, glycerate, acetate, and glycerol. This approach highlighted several candidate rubisco-dependent knockout strains, including ΔrpiAB Δedd, which is the basis of the CCMB1 strain. Full discussion of our algorithmic approach to strain design is given in Appendix 1 along with detailed description of the proposed mechanisms of rubisco coupling in CCMB1 and a comparison to other rubisc-dependent E. coli strains. OptSlope source code is available at https://gitlab.com/elad.noor/optslope (Noor, 2019) and calculations specific to CCMB1 can be found at https://github.com/flamholz/carboxecoli (Flamholz and Noor, 2020; copy archived at swh:1:rev:76596e1e8614173d8ef64aa13e93674307cfa3de).
Genomic modifications producing the CCMB1 strain
Request a detailed protocolStrains used in this study are documented in Supplementary file 1. To produce CCMB1, we first constructed a strain termed ‘Δrpi’. This strain has the genotype ΔrpiAB Δedd and was constructed in the E. coli BW25113 background by repeated rounds of P1 transduction from the KEIO collection followed by pCP20 curing of the kanamaycin selection marker (Baba et al., 2006; Datsenko and Wanner, 2000). Deletion of edd removes the Entner-Doudoroff pathway (Peekhaus and Conway, 1998), forcing rubisco-dependent metabolism of gluconate via the pentose phosphate pathway (Figure 2—figure supplement 3). CCMB1 has the genotype BW25113 ΔrpiAB Δedd ΔcynT Δcan and was constructed from ΔrpiAB by deleting both native carbonic anhydrases using the same methods, first transducing the KEIO ΔcynT and then Δcan from EDCM636 (Merlin et al., 2003), which was obtained from the Yale Coli Genetic Stock Center. Transformation was performed by electroporation (ECM 630, Harvard Biosciences) and electrocompetent stocks were prepared using standard protocols. Strain genotypes were routinely verified by PCR, as described below.
Recombinant expression of rubisco, prk, and CCM components
Request a detailed protocolpFE21 and pFA31 are compatible vectors derived from pZE21 and pZA31 (Lutz and Bujard, 1997). These vectors use an anhydrotetracycline (aTc) inducible PLtetO-1 promoter to regulate gene expression. pF plasmids were modified from parent vectors to constitutively express the tet repressor (TetR) under the Pbla promoter so that expression is repressed by default (Liang et al., 1999). We found that an inducible system aids in cloning problematic genes like prk (Wilson et al., 2018). We refer to these vectors as pFE and pFA, respectively. The p1A plasmid (Figure 2A) derives from pFE and expresses two additional genes: the Form IA rubisco from H. neapolitanus and a prk gene from Synechococcus elongatus PCC 7942. The pCB plasmid is properly called pFE-CB, while pCCM is pFA-CCM. The two CCM plasmids are diagrammed in Figure 3—figure supplement 1. Cloning was performed by Gibson and Golden-Gate approaches as appropriate. Large plasmids (e.g. pCB, pCCM) were verified by Illumina resequencing (Harvard MGH DNA Core plasmid sequencing service) and maps were updated manually after reviewing results compiled by breseq resequencing software (Deatherage and Barrick, 2014). Plasmids used in this study are described in Supplementary file 1 and available on Addgene at https://www.addgene.org/David_Savage/.
Strain verification by PCR and phenotypic testing
Request a detailed protocolAs CCMB1 is a relatively slow-growing knockout strain, we occasionally observed contaminants in growth experiments. We used two strategies to detect contamination by faster-growing organisms (e.g. wild-type E. coli). As most strains grew poorly or not at all in ambient air, pre-cultures grown in 10% CO2 were accompanied by a matching 1 mL negative control in ambient air. Pre-cultures showing growth in the negative control were discarded or verified by PCR genotyping in cases where air-growth was plausible.
PCR genotyping was performed using primer sets documented in Supplementary file 1. Three primer pairs were used to probe a control locus (zwf) and two target loci (cynT and rpiA). The zwf locus is intact in all strains. cynT and rpiA probes test for the presence of the CCMB1 strain (genotype BW25113 ΔrpiAB Δedd ΔcynT Δcan). Notably, the CAfree strain (BW25113 ΔcynT Δcan) that we previously used to test the activity of DAB-type transporters (Desmarais et al., 2019) is a cynT knockout but has a wild-type rpiA locus, so this primer set can distinguish between wild-type, CAfree, and CCMB1. This was useful for some experiments where CAfree was used as a control (e.g. Figures S7-8). Pooled colony PCRs were performed using Q5 polymerase (NEB), annealing at 65°C and with a 50 s extension time.
Selection for growth in novel conditions
Request a detailed protocolCCMB1:pCB did not initially grow in M9 media supplemented with glycerol, which was unexpected because pCB carries rubisco and prk genes. We therefore performed a series of selection experiments to isolate plasmids conferring growth at elevated CO2 and then in ambient air. Here we describe the methodology; the full series of experiments is described in Appendix 2 and illustrated in Figure 3—figure supplement 1. CCMB1 cultures carrying appropriate plasmids were first grown to saturation in rich LB media in 10% CO2. Stationary phase cultures were pelleted by centrifugation for 10 min at 4000 x g, washed in 2x the culture volume, and resuspended in 1x culture volume of M9 media with no carbon source. After resuspension, multiple dilutions were plated on selective media (e.g. M9 glycerol media) and incubated in the desired conditions (e.g. in ambient air) with a positive control in 10% CO2 on appropriate media. When colonies formed in restrictive conditions, they were picked into permissive media, grown to saturation, washed and tested for re-growth in restrictive conditions by titer plating or streaking. Plasmid DNA was isolated from verified colonies and transformed into naive CCMB1 cells to test whether plasmid mutations confer improved growth (i.e. in the absence of genomic mutations).
We first selected for CCMB1:pCB growth on M9 glycerol media in 10% CO2 and then in M9 gluconate media under 10% CO2. The resulting plasmid, pCB-gg for ‘gluconate grower,’ was isolated and deep sequenced (Harvard MGH DNA Core plasmid sequencing service). Plasmid maps were manyally updated based on results from the breseq software (Deatherage and Barrick, 2014). Following this first round of selection, CCMB1 was co-transformed with pCB-gg and pCCM and selected for growth in ambient air. Washed stationary phase cultures of CCMB1:pCB-gg+pCCM were plated on M9 glycerol media in ambient CO2. Parallel negative control selections were plated on uninduced plates (no aTc) and using CCMB1:p1A+pCCM, which lacks carboxysome genes. Plates were incubated in a humidified incubator for 20 days until colonies became visible.
Forty colonies were picked and tested for re-growth in ambient air by titer plating. Pooled plasmid DNA was extracted from verified colonies and electroporated into naive CCMB1 to test plasmid-linkage of growth. Colony #4 re-transformant #13 grew robustly was chosen due to replicable growth. Pooled plasmid DNA extracted from this strain was resequenced by a combination deep sequencing and targeted Sanger sequencing of the TetR locus and origins of replication, as these regions share sequence between pCB and pCCM. The individual post-selection plasmids, termed pCB’ and pCCM’, were reconstructed from pooled plasmid extract by PCR and Gibson cloning. These plasmids, termed pCB’ and pCCM’, were again verified by resequencing. Naive CCMB1 was transformed with the reconstructed post-selection plasmids pCB’ and pCCM’ and tested for growth in ambient air in plate reader (Spark, Tecan) and bioreactor (MC1000, PSI) assay formats.
Design of mutant CCM plasmids
Request a detailed protocolTo verify that growth in ambient air depends on CCM components, we generated variants of pCB’ and pCCM’ carrying targeted null mutations. CCMB1 was then co-transformed with two plasmids: a mutant plasmid (of either pCB’ or pCCM’) and its cognate, unmodified plasmid. Mutant plasmids are listed here along with expected growth phenotypes, with full detail in Supplementary file 1. pCB’ CbbL K194M, or pCB’ cbbL-, contains an inactivating mutation to the large subunit of the carboxysomal Form 1A rubisco (Andersson et al., 1989; Cleland et al., 1998). This mutation was expected to abrogate rubisco-dependent growth entirely.
Mutations targeting the CCM, rather than rubisco itself, are expected to ablate growth in ambient air but permit growth in high CO2. The following plasmid mutations were designed to specifically target essential components of the CCM. pCB’ CsoSCA C173S, or pCB’ CsoSCA-, carries a mutation to an active site cysteine residue responsible for coordinating the catalytic Zn2+ ion in β-carbonic anhydrases (Sawaya et al., 2006). pCB’ CsoS2 ΔNTD lacks the N-terminal domain of CsoS2, which is responsible for recruiting rubisco to the carboxysome during the biogenesis of the organelle (Oltrogge et al., 2020). Similarly, pCB’ CbbL Y72R carries an arginine residue instead of the tyrosine responsible for mediating cation-π interactions between the rubisco large subunit and the N-termus of CsoS2. This mutation eliminates binding between the rubisco complex and the N-termus of CsoS2 (Oltrogge et al., 2020). pCB’ ΔcsoS4AB lacks both pentameric shell proteins, CsoS4AB, which was shown to disrupt the permeability barrier at the carboxysome shell (Cai et al., 2009). pCCM’ DabA1 C462A, D464A, or pCCM’ DabA1-, carries inactivating mutations to the putative active site of the inorganic carbon transporter component DabA1 (Desmarais et al., 2019).
Two more mutant plasmids were designed to test the roles of rubisco chaperones in producing a functional CCM. pCCM’ CbbQ K46A, E107Q, denoted pCCM’ CbbQ-, carries mutations that inactivate the ATPase activity of the CbbQ subunit of the CbbOQ rubisco activase complex (Tsai et al., 2015). pCCM’ ΔacRAF lacks the putative rubisco chaperone acRAF. acRAF is homologous to a plant rubisco folding chaperone (Aigner et al., 2017) and likely involved in the folding of the H. neapolitanus Form IA rubisco (Wheatley et al., 2014). Experimental evaluation of growth phenotypes for the above-described mutants is detailed below and results are given in Figure 4—figure supplement 1.
Phenotyping of matched cultures in 10% CO2 and ambient air
Request a detailed protocolTo interrogate the phenotypic effects of mutations to the CCM, we tested the growth of matched biological replicate cultures of CCM mutants (e.g. disruption of carboxysome components or transporter function) in M9 glycerol medium in 10% CO2 and ambient air (Figure 4A). Individual colonies were picked into a round-bottom tube with 4 mL of M9 glycerol media with full strength antibiotic and 100 nM aTc. 1 mL of culture was then transferred to a second tube. The 3 mL pre-culture was incubated in 10% CO2, while the 1 mL culture was incubated in ambient air as a negative control. Control strains unable to grow in minimal media (e.g. those expressing inactive rubisco mutants) were pre-cultured in LB media. High-CO2 pre-cultures were grown to saturation, after which optical density (OD600) was measured in five-fold dilution. Experimental cultures were inoculated to a starting OD600 of 0.01 in 3 mL of M9 glycerol media with 12.5 mg/L chloramphenicol and 100 nM aTc. Each pre-culture was used to inoculate a matched pair of experimental cultures, one incubated in 10% CO2 and another in ambient air (Figure 4A). The endpoint culture density was measured at 600 nm. All experiments were performed in biological quadruplicate. As a positive control we used a complemented double carbonic anhydrase knockout (CAfree:pFE-sfGFP+pFA-HCAII) as its growth in air depends on expression of the human carbonic anhydrase II (Desmarais et al., 2019).
Carboxysome purification and imaging
Request a detailed protocolRoughly 1.2 L of CCMB1:pCB’+pCCM’ was grown in M9 glycerol media in ambient air in two identical bioreactors (MC1000, PSI) as described above. Sixteen distinct 80 mL cultures were grown, comprising eight technical replicates of two biological replicates deriving from distinct colonies. Cells were harvested before the onset of stationary phase, with optical densities ranging from ≈0.5 to ≈2.0 (600 nm, Genesys 20, Thermo Scientific) and pooled before subsequent purification steps. Wild type H. neapolitanus cells were grown in a 10 L bioreactor (Eppendorf BioFlo 115) modified to function as a chemostat. A continuous culture was grown in DSMZ-68 medium at a dilution rate of 0.03–0.05/hour. The culture was grown at 30°C, sparged with ambient air and the pH was held constant at 6.4 by addition of KOH. Chemostat effluent was collected in a 20 L glass flask and cells harvested every 2–3 day by centrifugation at 6000 x g for 15 min. A cell pellet of approximately 10 L of culture was used for subsequent purification.
Cells were chemically lysed in B-PER II (Thermo Fischer) diluted to 1x with TEMB buffer (10 mM Tris pH 8.0, 10 mM MgCl2, 20 mM NaHCO3 and 1 mM EDTA) supplemented with 0.1 mg / mL lysozyme, 1 mM phenylmethylsulfonyl fluoride (PMSF) and 0.1 ul of benzonase/mL (Sigma-Aldrich). E. coli cells (CCMB1:pCB’+pCCM’) were lysed for 30 min under mild shaking while H. neapolitanus cells were stirred vigorously with a magnetic stirrer for 1 hr. Lysed cells were centrifuged 12,000 x g for 15 min to remove cell debris. The clarified lysate (supernatant) was centrifuged 40,000 x g for 30 min to pellet carboxysomes and obtained pellets were gently resuspended in 1.5 mL TEMB buffer. Resuspended pellets were loaded on top of a 25 mL 10–50% sucrose step gradient (10, 20, 30, 40% and 50% w/v sucrose, made in TEMB buffer) and ultracentrifuged at 105,000 x g for 35 min (SW 32 Ti Swinging-bucket, Beckman Coulter). Gradients were fractionated, analysed by SDS-PAGE and carboxysome containing fractions pooled. Due to the low concentration of carboxysomes in the CCMB1:pCB’+pCCM’ sample, fraction numbers corresponding to H. neapolitanus gradient were pooled. Pooled fractions were ultracentrifuged 100,000 x g for 90 min and pellets were gently resuspended in TEMB to obtain the final purified carboxysome sample. The co-migration of carboxysomes with rubisco confirmed by coomassie and silver stained SDS-page gels of the final sample. Purified carboxysomes were visualized by negative stain TEM. Sample was applied to glow discharged formvar/carbon coated copper grids. Grids were then washed with deionized water and stained with 2% aqueous uranyl acetate. Imaging was performed on a JEOL 1200 EX TEM (H. neapolitanus) or a Tecnai 12 TEM at 120 KV (FEI) (CCMB1:pCB’+pCCM’). Images were collected using UltraScan 1000 digital micrograph software (Gatan Inc).
Thin sectioning and electron microscopy of whole cells
Request a detailed protocolCCMB1:pCB’+pCCM’ was grown in ambient air in 3 mL of M9 glycerol medium and induced with 100 nM aTc. A carboxysome-negative control, CAfree:pFE-sfGFP+pFA-HCAII, was grown in the same conditions. Sample preparation and sectioning were performed by the University of California Berkeley Electron Microscope Laboratory. Cell pellets were fixed for 30 min at room temperature in 2.5% glutaraldehyde in 0.1 M cacodylate buffer pH 7.4. Fixed cells were stabilized in 1% very low melting-point agarose and cut into small cubes. Cubed sample was then rinsed three times at room temperature for 10 min in 0.1 M sodium cacodylate buffer, pH 7.4 and then immersed in 1% osmium tetroxide with 1.6% potassium ferricyanide in 0.1 M cacodylate buffer for an hour in the dark on a rocker. Samples were later rinsed three times with a cacodylate buffer and then subjected to an ascending series of acetone for 10 min each (35%, 50%, 75%, 80%, 90%, 100%, 100%). Samples were progressively infiltrated with Epon resin (EMS, Hatfield, PA, USA) while rocking and later polymerized at 60°C for 24 hr. 70 nm thin sections were cut using an Ultracut E (Leica) and collected on 100 mesh formvar coated copper grids. The grids were further stained for 5 min with 2% aqueous uranyl acetate and 4 min with Reynold's lead citrate. The sections were imaged using a Tecnai 12 TEM at 120 KV (FEI) and images were collected using UltraScan 1000 digital micrograph software (Gatan Inc).
Sample preparation and LC-MS analysis
Request a detailed protocolProtein-bound amino acids were analyzed in total biomass hydrolysate of cultures grown in minimal media with 99% 13C glycerol (Cambridge Isotopes) as the sole organic carbon source. Biological quadruplicate cultures of the experimental strain, CCMB1:pCB’ + pCCM’, and the rubisco-independent control strain, CAfree:pFE-sfGFP + pFA-HCAII, were grown in 80 mL volumes in a bioreactor bubbling ambient air into each growth vessel (MC1000, PSI). After harvesting biomass, samples were prepared and analyzed as described in Antonovsky et al., 2016. Briefly, the OD600 was recorded and 2 OD x mL of sample were pelleted by centrifugation for 15 min at 4000 x g. The pellet was resuspended in 1 mL of 6 N HCl and incubated for 24 hr at 110°C. The acid was subsequently evaporated under a nitrogen stream using a custom gas manifold (Nevins et al., 2005), resulting in a dry hydrolysate. Dry hydrolysates were resuspended in 0.6 mL of MilliQ water, centrifuged for 5 min at 14,000 x g, and supernatant was analyzed by liquid chromatography-mass spectrometry (LC-MS).
Hydrolyzed amino acids were separated using ultra performance liquid chromatography (UPLC, Acquity, Waters) on a C-8 column (Zorbax Eclipse XBD, Agilent) at a flow rate of 0.6 mL/min, and eluted off the column using a hydrophobicity gradient. Buffers used were: (A) H2O + 0.1% formic acid and (B) acetonitrile + 0.1% formic acid with the following gradient: 100% of A (0–3 min), 100% A to 100% B (3–9 min), 100% B (9–13 min), 100% B to 100% A (13–14 min), 100% A (14–20 min). The UPLC was coupled online to a triple quadrupole mass spectrometer (TQS, Waters). Data were acquired using MassLynx v4.1 (Waters). Amino acids and metabolites used for analysis were selected according to the following criteria: amino acids that had peaks at a distinct retention time and m/z values for all isotopologues and also showed correct 13C labeling fractions in control samples that contained protein hydrolyzates of WT cells grown with known ratios of uniformly 13C-labeled (U-13C) glucose to 12C-glucose. We further analyzed the serine M+2 isotopologue (parent ion in positive ionization mode with 108.1 m/z) using multiple reaction monitoring (MRM). This approach by selecting the channels of a daughter ion (fragment) with the formula [C2H6NO]+: (A) 61.1 m/z, where the undetected fragment contains a carboxylic acid carbon which is a 13C isotope and (B) 62.1 m/z, where the undetected fragment contains a carboxylic acid carbon which is 12C (Piraud et al., 2003). We looked at the ratio of the peak integrals of A/B to infer the distribution of 13C/12C for that particular carboxyl carbon. Since the carboxylic acid on L-serine derives from the rubisco carboxylation product 3-phosphoglycerate, measuring the 13C/12C distribution at this position reports directly on carboxylation by rubisco in vivo (Figure 6 and supplements) and with lower background than the total mass measurement described above.
Isotopic analysis of the composition of biomolecules
Request a detailed protocolThe total 13C fraction of each metabolite was determined as the weighted average of the fractions of all the isotopologues for that metabolite:
Here, N is the number of carbons in the compound (e.g. N = 3 for serine) and fi is the relative fraction of the i-th isotopologue, that is containing i 13C carbon atoms. Each metabolite’s total 12C fraction was calculated as . Our quantitative approach to inferring the rubisco carboxylation flux from these data is described fully in Appendix 3; source code and data are available at https://github.com/flamholz/carboxecoli.
Appendix 1
Strain design and testing
Strain design via the OptSlope algorithm
To computationally design mutant strains in which growth is coupled to rubisco carboxylation flux, we used a variant of Flux Balance Analysis (Lewis et al., 2012) called ‘OptSlope’ (Antonovsky et al., 2016). Optslope searches for metabolic knockout mutants in which biomass production is coupled to flux through a reaction of choice (e.g. rubisco) at all growth rates. This coupling is evident in plots of the feasible biomass production rate against feasible rubisco fluxes. In ‘rubisco-coupled’ designs, maximal biomass production requires non-zero rubisco carboxylation flux and increasing biomass production demands increased carboxylation (diagrammed in Figure 2—figure supplement 4). The slope of this relationship is the ‘coupling slope’ and quantifies the degree of coupling.
Starting from a published model of E. coli central metabolism, the Core Escherichia coli Metabolic Model (Orth et al., 2010), we considered all pairs of central metabolic knockouts and ignored those that permit growth in silico in the absence of rubisco carboxylation (EC 4.1.1.39) and phosphoribulokinase (EC 2.7.1.19) activities. For the remaining knockouts, we evaluated the degree of coupling between rubisco flux and biomass production during growth in nine carbon sources: glucose, fructose, gluconate, ribose, succinate, xylose, glycerate, acetate, and glycerol. This approach highlighted several candidate rubisco-dependent knockout strains, including ΔrpiAB Δedd. Consistent with the coupling mechanisms described below, OptSlope predicted rubisco-dependent growth of ΔrpiAB Δedd strains on all carbon sources except ribose. The OptSlope algorithm is available and documented at https://gitlab.com/elad.noor/optslope, outlined in Figure 2—figure supplement 4, and described fully in Antonovsky et al., 2016. Calculations specific to CCMB1 can be found online at https://github.com/flamholz/carboxecoli.
Proposed mechanisms of growth coupling in CCMB1
The proposed mechanism of rubisco-coupling depends on the carbon source, but in all cases coupling is explained by an inability to metabolize ribulose-5-phosphate (Ru5P) due to the removal of ribose-phosphate isomerase activity (ΔrpiAB). When gluconate or xylose is the growth substrate, Ru5P is produced directly from the carbon source. Although wild-type E. coli can metabolize gluconate via the ED pathway, the ED dehydratase knockout (Δedd) in CCMB1 blocks this route and forces 1:1 production of Ru5P from gluconate. Expression of prk and rubisco opens a new route of Ru5P metabolism, thus enabling CCMB1 to grow in gluconate or xylose media (Figure 2—figure supplement 3, panel A).
When glycerol is the growth substrate, it is taken into the cell and converted to glyceraldehyde 3-phosphate, which is metabolized through lower glycolysis or gluconeogenesis. The gluconeogenesis route produces hexoses that enter the pentose phosphate pathway, which is required to synthesize ribose 5-phosphate (Ri5P) for nucleotide and histidine biosynthesis. Depending on the growth rate, products of Ri5P make up 5–25% of E. coli biomass (Bremer and Dennis, 2008; Taymaz-Nikerel et al., 2010). However, the pentose phosphate pathway forces co-production of Ri5P, Ru5P and xylulose 5-phosphate (Xu5P). In the absence of ribose phosphate isomerase activity, there is no pathway for metabolism of Xu5P or Ru5P. This defect is complemented by the expression of rubisco and prk, which together form a ‘detour pathway’ converting Ru5P into the lower-glycolytic metabolite 3-phosphoglycerate (Figure 2—figure supplement 3, panel A).
Potential for phosphoglycolate salvage in E. coli
Plants, cyanobacteria and other autotrophs uniformly express ‘photorespiratory’ pathways to process the rubisco oxygenation product 2-phosphoglycolate, or 2PG (Eisenhut et al., 2008). Although these are typically called photorespiratory pathways after their discovery in plants, where they were named based on the reproducible observation of light-induced CO2 production in many C3 plant species (Zelitch, 1979), we refer to them as ‘phosphoglycolate salvage pathways’ following Claassens et al., 2020 because they are also found in chemolithoautotrophic organisms lacking photosynthesis. 2PG salvage appears to be essential in plants, cyanobacteria, and chemolithoautotrophic proteobacteria (Claassens et al., 2020; Eisenhut et al., 2008; Somerville and Ogren, 1980, Somerville and Ogren, 1979).
The E. coli genome encodes enzymes of a ‘glycerate pathway’ that could serve as a means of phosphoglycolate salvage. Indeed, this pathway was recently shown to be the primary route of phosphoglycolate salvage in C. necator, a proteobacterial chemolithoautotroph that notably lacks carboxysome genes (Claassens et al., 2020). The glycerate pathway proceeds by dephosphorylating 2PG to glycolate, oxidizing glycolate to glyoxylate, and converting two units of glyoxylate into tartronate semialdehyde via a decarboxylating lyase reaction. Tartronate semialdehyde can then be reduced to glycerate, which could enter into lower glycolysis and the TCA cycle (Figure 2—figure supplement 3). We attempted to delete the gph gene in CCMB1 as it encodes the 2PG phosphatase that catalyzes the first step of this putative pathway. However, the Δgph knockout was challenging to transform by electroporation, consistent with a proposed role in DNA repair (Teresa Pellicer et al., 2003). We reasoned that 2PG salvage might be required in CCMB1, as photorespiratory genes are essential in cyanobacteria (Eisenhut et al., 2008) and chemolithoautotrophic bacteria (Claassens et al., 2020; Desmarais et al., 2019) even though both groups often express carboxysome CCMs. We therefore proceeded leaving the genes of the putative glycolate pathway intact.
Verification of dependence on rubisco carboxylation for growth
To verify the dependence of CCMB1 on rubisco and phosphoribulokinase activities in minimal media, we constructed the p1A plasmid, expressing the large and small subunits of the H. neapolitanus rubisco (cbbLS) along with a phosphoribulokinase gene, prk, from the cyanobacterium S. elongatus PCC 7942 (Figure 2B). We further constructed mutant variants of p1A carrying inactive rubisco or prk genes. Rubisco was inactivated by mutating the large subunit active site lysine to methionine, producing p1A CbbL K194M, or p1A CbbL- for short (Andersson et al., 1989; Cleland et al., 1998). Prk was inactivated by mutating ATP-binding residues in the Walker A motif, producing p1A Prk K20M S21A, termed p1A Prk- for short (Cai et al., 2014; Higgins et al., 1986). CCMB1:p1A grew on glycerol and gluconate minimal media when provided 10% CO2 (Figure 2—figure supplement 5). CCMB1:p1A CbbL- and CCMB1:p1A Prk- both failed to grow on minimal media supplemented with glycerol or gluconate, demonstrating a dependence on both enzymes. So long as high CO2 was provided, neither activity was required for growth in rich LB media, which contains abundant nucleic acids precursors (Sezonov et al., 2007). We further tested five distinct bacterial rubiscos and found that they all permit robust growth in M9 glycerol media with elevated CO2 (5%, Figure 2—figure supplement 1). Although we included the Form IC rubisco from C. necator (also known as R. eutropha), the most CO2-specific bacterial rubisco known to date (Lee et al., 1991), none of the rubiscos tested permitted CCMB1 to grow in ambient air.
The observed high-CO2 requirement of CCMB1:p1A growth was expected for two independent reasons: (i) all known rubiscos display low net carboxylation rates in ambient air due to relatively low CO2 (≈0.04%) and relatively high O2 (≈21%), as shown in Figure 1B and discussed in Flamholz et al., 2019; Iñiguez et al., 2020, and (ii) CCMB1 entirely lacks carbonic anhydrase activity as the open reading frames of both cynT and can genes were purposely disrupted (Materials and methods). Carbonic anhydrase knockouts of many microbes, including E. coli and S. cerevisiae, are high-CO2 requiring, likely due to cellular demand for HCO3-(Aguilera et al., 2005; Desmarais et al., 2019; Du et al., 2014; Merlin et al., 2003). As the CCM cluster includes a carbonic anhydrase gene (the carboxysomal carbonic anhydrase, csoSCA) and the CCM is expected to qualitatively improve the carboxylation rate and CO2-specificity of the encapsulated rubisco, we hypothesized that a functional CCM would enable CCMB1 to grow in ambient air.
To verify that CCMB1 depends specifically on rubisco carboxylation and not oxygenation for growth, we grew CCMB1:p1A on glycerol minimal medium in anoxic high-CO2 conditions (10:90 CO2:N2, Figure 2—figure supplement 2, Materials and methods). E. coli predominantly respires glycerol and, therefore, grows extremely slowly on glycerol in anaerobic and low O2 conditions (Stolper et al., 2010). We therefore supplied 20 mM NO3- as an alternate terminal electron acceptor (Unden and Dünnwald, 2008) in anaerobic growth conditions (see ‘Growth conditions’ in Materials and methods). CCMB1:p1A grew on glycerol media in anaerobic conditions when NO3- was provided. Growth is qualitatively weaker than a wild-type control, but this is consistent with the growth differences observed in aerobic conditions supplemented with 10% CO2 (Figure 2—figure supplement 2). To make this point clear, we note that E. coli generally grows much more robustly in the presence of O2 than in its absence, as O2 is the preferred terminal electron acceptor (Unden and Dünnwald, 2008). This can be seen by comparing the growth of the wildtype control between aerobic (10% CO2, balance air) and anoxic conditions in Figure 2—figure supplement 2. We therefore suggest that the weak growth of CCMB1:p1A in anoxic conditions is best explained as a combination of two effects: (i) impaired growth due to removal of rpiAB genes, and (ii) impaired growth due to the absence of O2. Alternatively, it is possible that oxygenation by rubisco plays some positive role in supporting growth, although we find this unlikely as CCMB1:p1A fails to grow in ambient air which contains abundant O2, as documented in Figure 2 and supplements. Irrespective of this nuanced issue, anaerobic growth of CCMB1:p1A on glycerol minimal media implies that growth can be supported by rubisco carboxylation alone and does not require the rubisco-catalyzed oxygenation of RuBP.
Comparison to other rubisco-dependent E. coli strains
One straightforward approach to generating a rubisco selection system is to knock out rubisco in a facultative autotroph, that is a strain that can be grown in a rubisco-independent fashion for the purposes of performing genetic manipulations. This approach has been applied in the facultative chemolithoautotrophs R. capsulatus and R. eutropha and is reviewed in Mueller-Cajar and Whitney, 2008; Wilson and Whitney, 2017. Here, we focus on approaches using genetically modified E. coli strains to select for rubisco activity.
In addition to the CCMB1 several other strategies for coupling the growth of E. coli to rubisco activity have been tested. One such approach involves the deletion of glyceraldehyde 3-phosphate dehydrogenase (gapA gene), a core component of lower glycolysis (Morell et al., 1992; Mueller-Cajar et al., 2007). This lesion is proposed to block the production of lower glycolytic metabolites, a defect that is rescued by the expression of prk and rubisco which together convert pentose-phosphate pathway intermediates (ribulose 5-phosphate) into the lower glycolytic intermediate, 3-phosphoglycerate. This strain has been used to select mutant variants of the model Form II rubisco from R. rubrum (Mueller-Cajar et al., 2007). Analysis via the OptSlope algorithm predicts that the growth rate of the ΔgapA strain does not depend on the rate of carboxylation by rubisco (Figure 1—figure supplement 1, panel C), which might explain why all rubisco variants isolated had lower maximum carboxylation rates (kcat,C) or lower CO2 specificities (SC/O) than the wild-type sequence.
Another approach involves co-expression of rubisco and prk in wildtype E. coli. Expression of prk alone entails non-productive consumption of pentose-phosphate intermediates (converting ribulose 5-phosphate into ribulose 1, 5-bisphosphate) and greatly restricts growth (Parikh et al., 2006; Wilson and Whitney, 2017). Co-expression of rubisco alleviates the negative effects of prk by converting the ‘useless’ ribulose 1, 5-bisphosphate, which is not natively found in E. coli, into the lower glycolytic intermediate, 3-phosphoglycerate. This approach to constructing a rubisco dependent E. coli strain suffers from a crucial drawback - disruption of prk produces a strain that grows in a rubisco-independent manner. Indeed, transposon insertions in prk were commonly observed and do not require rubisco for growth (Parikh et al., 2006; Wilson et al., 2018). This problem is alleviated in an improved strain, RDE2, which makes use of a prk-neoR fusion gene designed such that most mutations disrupting prk also disrupt the downstream antibiotic resistance marker. This approach greatly reduced the incidence of prk silencing, but did not remove it entirely (Wilson et al., 2018), suggesting that this approach is impractical for long-term selection experiments. Furthermore, many of the rubisco mutants produced in these strains did not display improved kinetics, but rather higher expression in E. coli (Zhou and Whitney, 2019). Expressing prk under a strong promoter was subsequently shown to be more deleterious to growth, which enabled selection for modest improvements in the kinetics of a bacterial rubisco (Zhou and Whitney, 2019). The resulting strain, termed RDE3, nonetheless produced a non-negligible number of false positives.
Considering the weaknesses of the above approaches illustrates that it is desirable (i) for the growth rate to be coupled to the rate of carboxylation by rubisco, and (ii) for this coupling to be based on E. coli’s native metabolism such that escape is implausible. This realization motivated the OptSlope algorithm, which we have successfully applied to design rubisco-dependent E. coli strains for different purposes. We previously used this approach to design strains appropriate for long-term selection experiments that successfully isolated partially and fully autotrophic E. coli strains, that is strains deriving most or all of biomass carbon from an inorganic source (Antonovsky et al., 2016; Gleizer et al., 2019). The strain reported here, CCMB1, is not autotrophic. Rather, as described above and in Figure 2—figure supplement 3, CCMB1 relies on rubisco and Prk activities to provide a ‘detour’ pathway around a lesion we introduced into its metabolism by removal of ribose-phosphate isomerase activity. Since CCMB1 carries a lesion in the pentose phosphate pathway, rather than glycolysis as in our previous studies (Antonovsky et al., 2016; Gleizer et al., 2019), it is more convenient for routine work as it produces overnight colonies when grown on rich media in high CO2. Since our purpose was to select for a functional CCM, and not for full autotrophy, we chose to work with CCMB1 for simplicity.
One deficiency of CCMB1, however, is that it relies on rubisco to consume ribulose 1, 5-bisphosphate and ‘reconnect’ its metabolism (Figure 2—figure supplement 3). It is therefore not required that ribulose 1, 5-bisphophate be consumed by carboxylation. Rather, rubisco-catalyzed oxygenation of ribulose 1, 5-bisphophate could, in principle, complement the ΔrpiAB lesion as the E. coli genome encodes enzymes that might together form a pathway metabolizing the oxygenation product, 2-phosphoglycolate (Figure 2—figure supplement 3, panel C). It is unlikely that this pathway is a significant contributor to the growth of CCMB1 in minimal media for several reasons that we discuss below. We also note that recently reported ‘glycerate biosensor’ strains might be used in future work to select specifically for rubisco carboxylation without concern for latent 2-phosphoglycolate metabolism (Aslan et al., 2020).
On the topic of the potential for an ersatz phosphoglycolate salvage pathway in E. coli, previous research suggests that the first gene of this putative pathway, 2-phosphoglycolate phosphatase, is not constitutively expressed (Teresa Pellicer et al., 2003). Moreover, if rubisco oxygenation was a significant contributor to the growth of complemented CCMB1 strains, we would expect growth to be linked to the presence and abundance of O2. Rather, we find that these strains uniformly fail to grow in ambient air, which contains 21% O2 (Figure 2 and supplements) and grow reproducibly in anoxic media with elevated CO2 (Figure 2—figure supplement 2) as discussed above. Finally, as shown in Figure 2—figure supplement 1, panel B, increasing CO2 levels also increased growth rate and yield for CCMB1:p1A, implying that CO2 is growth-limiting in M9 glycerol media. Altogether we conclude that CCMB1 depends on rubisco predominantly via the carboxylation of RuBP and not by its oxygenation. Nonetheless, we were careful throughout to re-transform plasmids isolated via selection experiments into naive CCMB1 in order to verify that growth phenotypes are linked to plasmid DNA. Retransformation provides some assurance that the CCM functions in the absence of any sizable genomic mutations or rearrangements that might, for example, induce expression of 2-phosphoglycolate salvage enzymes.
Appendix 2
Selection for growth in ambient air
Design of plasmids for CCM expression
As described in the Methods section, expression vectors used throughout this study were derived from pZE21 and pZA31 vectors of Lutz and Bujard, 1997. These two vectors have compatible origins of replication, measured copy numbers in E. coli, and use an anhydrotetracycline (aTc) inducible promoter to regulate gene expression. pFE and pFA plasmid backbones used herein were modified from parent vectors to constitutively express the tet repressor so that expression of heterologous gene products is repressed by default (Liang et al., 1999). The carboxysome expression plasmid, pCB, has a pFE backbone, while the second CCM expression plasmid, pCCM, has a pFA backbone. These genes expressed from these plasmids are diagrammed and discussed in Figure 3—figure supplement 1.
The expression unit of pCB derives from pHnCB10, a plasmid we previously showed enables production of carboxysome structures in E. coli (Bonacci et al., 2012). The operon expressing the carboxysome was cloned from pHnCB10 into pFE to generate pCB. Notably, this operon includes a carboxysome shell protein, csos1D, that is not natively found in the primary carboxysome operon in H. neapolitanus (Bonacci et al., 2012; Cai et al., 2008; Klein et al., 2009; Roberts et al., 2012). We chose to include csos1D with the carboxysome because its inclusion was previously observed to result in production of carboxysomes with more regular icosahedral morphology (Bonacci et al., 2012), which we hypothesized to be a correlate of proper assembly. Moreover, when we began this work, the transporters associated with proteobacterial CCMs had not yet been identified (Desmarais et al., 2019; USF MCB4404L et al., 2017; Scott et al., 2019) and we planned to express cyanobacterial transporters as in Du et al., 2014. As such, it seemed sensible to include all known carboxysome components on a single plasmid, a design we retained for convenience even after the inorganic carbon transporters were identified.
The expression unit of pCCM was cloned directly from the H. neapolitanus genome and encodes an operon adjacent to the carboxysome operon that expresses several CCM-related genes including a second copy of csos1D, which was left undisturbed to avoid any effects on gene expression. As diagrammed in Figure 3—figure supplement 1 and detailed in Supplementary file 1, this operon encodes 10 genes including at least six with plausible roles in the CCM: a DAB type inorganic carbon transporter (dabAB1), three genes known or hypothesized to interact with rubisco (acRAF, cbbOQ) and a parA family gene that is likely involved in partitioning H. neapolitanus carboxysomes daughter cells (MacCready et al., 2018; Savage et al., 2010). For this reason, we chose to express this operon instead of the smaller DAB2 transport operon that we characterized in previous work (Desmarais et al., 2019).
Phenotypic verification of pCB and pCCM plasmids
We used reporter strains to verify the primary activities of pCB and pCCM. Previous work has shown that a carbonic anhydrase knockout strain we call CAfree is complemented by heterologous expression of carbonic anhydrases or bicarbonate transporters (Desmarais et al., 2019; Du et al., 2014; Merlin et al., 2003). To characterize pCCM and the DAB1 transporter it encodes, we utilized an additional plasmid, pFA-DAB1, which expresses dabAB1 and one unnamed interstitial gene (mrpA family, PFAM 00361) on their own, that is in the absence of the seven other genes natively found in the same operon. We found that both pCCM and pFA-DAB1 complement CAfree for growth in ambient air, implying that the DAB1 transport complex is functional when heterologously expressed in E. coli on its own or in the context of the full operon (Figure 1—figure supplement 2).
Given that pCB encodes a prk and the same rubisco as the p1A plasmid (the carboxysomal rubisco from H. neapolitanus, cbbLS genes), we expected that it would complement CCMB1 for growth on M9 glycerol media in 10% CO2 as shown for CCMB1:p1A (Figure 2). CCMB1:pCB did not initially grow in glycerol minimal media in high CO2 or ambient air, however. Since CCMB1 requires rubisco and Prk activities for growth in glycerol media (Figure 2 and supplements) we performed a series of selection experiments to isolate plasmids conferring growth at elevated CO2 and, subsequently, in ambient air. In the first round of experiments, we selected for growth of CCMB1:pCB in minimal media in 10% CO2. This produced a plasmid, termed pCB-gg, that encodes carboxysome genes and permits CCMB1 to grow in 10% CO2 on glycerol and gluconate media. We subsequently co-transformed CCMB1 with pCB-gg and pCCM to select for growth in ambient air. Plasmids isolated and reconstructed from this second round of selection experiments were termed pCB’ and pCCM’, which are those described in the main text and figures. Experimental protocols are described in the Materials and methods and the full series of selection experiments is diagrammed in Figure 3—figure supplement 1. Here, we describe selection experiments in fuller detail.
Selection for growth of CCMB1:pCB in elevated CO2
We first selected for CCMB1:pCB growth on M9 glycerol media in 10% CO2 and then in M9 gluconate media under 10% CO2. This was achieved by plating washed stationary phase cultures on M9 media, incubating in a humidified CO2-controlled incubator, and waiting for colonies to appear. The resulting plasmid, pCB-gg.9 for ‘gluconate grower #9,’ was isolated and deep sequenced. pCB-gg.9 was found to carry two regulatory mutations: an amino acid substitution to the tet repressor (TetR E37A) and a nucleotide substitution in the Tet operator regulating the carboxysome operon (tetO2 +8T, Supplementary file 1).
Selection for growth of CCMB1:pCB gg.9+pCCM in ambient air
Following the first round of selection, CCMB1 was co-transformed with pCB-gg.9 and pCCM. Transformants grew in M9 glycerol media in 10% CO2 but failed to grow on in ambient air. We therefore performed another selection experiment, plating CCMB1:pCB-gg.9+pCCM on M9 glycerol media in ambient CO2. Parallel negative control selections were conducted on uninduced plates (no aTc) and using CCMB1:p1A+pCCM, which lacks carboxysome genes. Colonies formed on induced CCMB1:pCB-gg.9+pCCM plates after 20 days, but not on control plates lacking induction or carboxysome genes, respectively (Figure 3—figure supplement 1 panel F).
Forty colonies were picked and tested for re-growth in ambient CO2 by tenfold titer plating. 10 of 40 regrew. Six examples are given in Figure 3—figure supplement 1 panel G. Pooled plasmid DNA was extracted from verified colonies and electroporated into naive CCMB1 to test plasmid-linkage of growth. Plasmid DNA from colony #4 produced the most robust growth in ambient air (Figure 3—figure supplement 1 panel H). The growth of re-transformants was further evaluated by picking 16 biological replicate colonies and evaluating their growth in ambient air in liquid M9 glycerol media. Re-transformant #13 of pooled plasmid DNA from colony #4 was regrew robustly in all six technical replicates.
Isolation and reconstruction of pCB’ and pCCM’
Pooled plasmid DNA from colony #4 re-transformant #13 was resequenced by a combination deep sequencing and targeted Sanger sequencing of the TetR locus and origins of replication, as these regions share sequence between both parent plasmids (pCB and pCCM). The pCB sequence isolated from this retransformant was found to carry the same mutations as pCB-gg and pCCM had acquired the high-copy ColE1 origin of replication from pCB (Supplementary file 1). The individual mutant plasmids were reconstructed from pooled plasmid extract by PCR and Gibson cloning. These reconstructed post-selection plasmids, termed pCB’ and pCCM’, were verified once again by Illumina sequencing. Naive CCMB1 was then transformed with the reconstructed post-selection plasmids and tested for growth in ambient air. Post-selection plasmids conferred reproducible growth in ambient air in multiple growth conditions (Figure 3), implying that genomic mutations that formed during selections were not required to produce growth in ambient air.
Appendix 3
Inference of rubisco flux in vivo
Here, we describe our approach to estimating the rubisco flux in vivo in CCMB1:pCB’+pCCM’ cells. As a reminder, in the experiment we performed, the organic carbon source (glycerol) is 99% 13C labeled such that inorganic carbon (e.g. 12CO2, H12CO3-) from the media is the dominant source of 12C. Our approach to estimating rubisco flux takes advantage of three observations about the central metabolism of E. coli.
First, we note that serine is a direct metabolic product of the rubisco carboxylation product, 3-phosphoglycerate (3PG). As such, we assume that the isotopic composition of serine, which we measure (Materials and methods), is equal to that of 3PG. Second, based on the known pathways of E. coli central metabolism, we presume that there are only two routes to producing 3PG in CCMB1 cells - production through lower glycolytic metabolism of glycerol via phosphoglycerate kinase (pgk gene) and production by rubisco. Since 12C dominantly derives from an inorganic source in our experiment (Figure 6) we can estimate the rubisco flux by considering the 12C labeling of serine. However, though nearly all the inorganic carbon outside the cell is 12C (natural abundance is ≈99%), the isotopic composition of intracellular inorganic carbon will reflect a balance of import and intracellular decarboxylation of compounds deriving from the carbon source, which is 99% 13C glycerol. The final observation is that arginine is synthesized from glutamate via a carboxylation reaction (i.e. addition of a carbamoyl phosphate deriving from HCO3-). As such, we can infer the isotopic composition of intracellular inorganic carbon by examining the difference in labeling between arginine and glutamate (Gleizer et al., 2019). Altogether, these observations give us a framework, described in the forgoing sections, for deriving all the information necessary to estimate the rubisco carboxylation flux in vivo.
Estimating the effective intracellular 12CO2 fraction
E. coli cells grown in 13C glycerol will simultaneously respire glycerol, producing intracellular 13CO2, and take up extracellular 12C in the form of 12CO2 and H12CO3-. The isotopic composition of the intracellular inorganic carbon (Ci) pool will therefore reflect the balance of uptake and respiration. As rubisco carboxylation draws from the intracellular CO2 pool, we must estimate the isotopic composition of the Ci pool to evaluate the contribution of rubisco to 3PG and serine production. We used the carbamoyl phosphate moiety as a marker for the isotopic distribution of the intracellular Ci pool, as described in Gleizer et al., 2019. Briefly, carbamoyl phosphate synthesis is initiated by phosphorylation of bicarbonate, and the molecule is ultimately condensed with ornithine in the biosynthesis of L-arginine. A comparison of the mass isotopologue distribution of L-arginine, which contains one carbon deriving from carbamoyl phosphate, with the mass isotopologue distribution of L-glutamate, an ornithine precursor, can thus be used to estimate the fraction of 13CO2 in the cytosol.
We estimated the effective 13C labeling of intracellular inorganic carbon () as follows:
Here, is the relative fraction of 13CO2 out of the total CO2 pool (or, more formally, the Ci pool), and farg,i and fglu,i are the fraction of the i-th isotopologue of arginine and glutamate respectively. We assumed fast equilibration of the intracellular Ci pool because all strains used in labeling experiments express a carbonic anhydrase (either carboxysomal or cytosolic). An equivalent equation can be defined for the arginine-proline comparison (Gleizer et al., 2019), and we took the mean of inferences from arg-glu and arg-pro comparisons as an estimate of . The intracellular fraction of 12CO2 was then calculated from mass balance as . For brevity, we refer to these fractions as and , respectively.
Calculation of the intracellular rubisco carboxylation flux
When CCMB1 cells are grown on 99% 13C glycerol, 3-phosphoglycerate (3PG) can be produced via two routes: (i) rubisco-catalyzed carboxylation of RuBP and (ii) glycolytic metabolism of glycerol via dihydroxyacetone phosphate, or DHAP (Booth, 2005). We denote these two fluxes as Jrubisco and Jpgk, where pgk (phosphoglycerate kinase) is the glycolytic enzyme producing 3PG (Bar-Even et al., 2012). Serine is a direct metabolic product of 3PG (Szyperski, 1995; Stauffer, 2004) and was therefore assumed to have the same 12C composition as 3PG. Rubisco-catalyzed carboxylation of RuBP adds one CO2 to the 5-carbon substrate, producing two 3PG molecules containing a total of six carbon atoms. Therefore, 1/6 of carbon atoms on 3PG produced via rubisco carboxylation must derive from an inorganic source (Figure 6—figure supplement 2). Carboxylation draws CO2 from the intracellular inorganic carbon pool, whose 12C composition was inferred as described above.
Based on these assumptions, the 12C composition of 3PG, and therefore serine, equals a flux-weighted sum of contributions from rubisco and pgk. As such, the relative 3PG production flux that is due to rubisco, Jrubisco/(Jrubisco+Jpgk), can be inferred via the following calculation:
where the first equation is written for the control and the second for experimental cultures where rubisco is active (CCMB1:pCB’+pCCM’). and denote the 12C composition of serine in the control and experiment, respectively. Identical notation is used for RuBP and DHAP. As there are only two routes of 3PG production, the above equations can be simplified to solve for the relative flux through rubisco:
To calculate the rubisco flux in vivo we must attach values to several parameters in the above equation. was inferred on a per-sample basis, with the mean values being and for the control and experiment respectively (Figure 6—figure supplement 3, panel C). Because glycerol is converted into 3PG and serine via DHAP in wild-type E. coli (Booth, 2005), we expect that , as derived above. LC-MS measurements give and (Figure 6C). Valine is also a metabolic product of DHAP (Szyperski, 1995) and was found to have a similar 12C fraction in control cells (Figure 6—figure supplement 1). Since glycerol is immediately converted to DHAP in E. coli, we further assumed that .
RuBP is produced in CCMB1 when rubisco and prk are expressed. Since glycerol is the sole carbon source and there are no carboxylation reactions between DHAP and RuBP in CCMB1, we assumed . This assumption is supported by LC-MS measurements of histidine in control cells (Figure 6—figure supplement 1). Like RuBP, histidine is synthesized from a pentose-phosphate pathway intermediates (Szyperski, 1995; Winkler and Ramos-Montañez, 2009), and measured , which is very similar to . Using mean values to illustrate the calculation gives , implying that 15% of 3PG production is due to rubisco.
105 random samples were drawn from the experimentally determined parameter ranges to estimate a 99% confidence interval on the rubisco flux fraction. As the 12C composition of inorganic carbon () and serine are mechanistically linked via rubisco, these values were assumed to co-vary. Distributions were estimated on a per-sample basis by assuming 0.1% error in direct measurement of serine and 1% error in the inference of . These calculations gave a median flux estimate of 15.2% with 99% of values falling between 5.0% and 23.3%. The sample with the lowest inferred rubisco flux had a median estimate of 12.3% with 99% of values falling between 3.5% and 23.9%, implying that rubisco is responsible for a nonzero fraction of 3PG production in all samples. This and above calculations can be found on our GitHub repository in the linked Jupyter notebook.
Predicting rubisco carboxylation flux via Flux Balance Analysis
A stoichiometric model of complemented CCMB1 was generated from the Core Escherichia coli Metabolic Model (Orth et al., 2010) by adding the prk and rubisco carboxylation reactions and then deleting rpi and edd reactions. Parsimonious Flux Balance Analysis (pFBA) was applied to the resulting model to calculate intracellular metabolic fluxes that maximize the rate of biomass production. As many distinct flux distributions can yield the same (maximal) rate of biomass production, pFBA uses the minimum sum of fluxes objective to define a unique flux solution (Holzhütter, 2004). The COBRApy implementation of pFBA introduces an additional free parameter, the permissible fraction of the maximal biomass production rate fopt (Ebrahim et al., 2013). When fopt < 1.0, the biomass production can be less-than-optimal if this would further decrease the sum of fluxes. Additionally, we varied lower bound on the ATP maintenance cost, as this value affects the intracellular flux distribution and is not well-constrained in modified strains.
pFBA was run with fopt ranging from 0.8 to 1.0 and the lower bound on ATP maintenance ranging between 0% and 25% of ATP production to account for the fact that CCMB1 has not undergone selection to maximize biomass production. For each resulting flux distribution, the fraction of 3PG production flux due to rubisco was calculated as the fraction of 3PG molecules produced via rubisco carboxylation divided by the total flux to 3PG. These calculations predict that 16–22% of 3PG production is due to rubisco. The model was rerun after removing all possibility for product secretion by deleting all carbon-containing exchange reactions other than exchange of the carbon sources, CO2. This modification should give an upper bound on the fraction of 3PG production due to rubisco, as carbon cannot be shunted away from biomass production to overflow products. The ‘no overflow’ model predicted that 24% of 3PG production is due to rubisco independent of fopt. The overall range of predictions from 16–24% is plotted in Figure 6 and supplements. All calculations were performed using Python and COBRApy (Ebrahim et al., 2013), and source code can be found at https://github.com/flamholz/carboxecoli.
Data availability
All source data for all figures is available in the linked github repository along with accompanying Jupyter notebooks generating the data-driven portions of all figures.
References
-
Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: the keio collectionMolecular Systems Biology 2:2006.0008.https://doi.org/10.1038/msb4100050
-
The diversity and coevolution of Rubisco, plastids, pyrenoids, and chloroplast-based CO 2 -concentrating mechanisms in algaeCanadian Journal of Botany 76:1052–1071.https://doi.org/10.1139/b98-074
-
Insertion mutation of the form I cbbL gene encoding ribulose bisphosphate carboxylase/oxygenase (RuBisCO) in Thiobacillus neapolitanus results in expression of form II RuBisCO, loss of Carboxysomes, and an increased CO2 requirement for growthJournal of Bacteriology 180:4133–4139.https://doi.org/10.1128/JB.180.16.4133-4139.1998
-
Rethinking glycolysis: on the biochemical logic of metabolic pathwaysNature Chemical Biology 8:509–517.https://doi.org/10.1038/nchembio.971
-
The Path of Carbon in Photosynthesis XXI The Cyclic Regeneration of Carbon Dioxide AcceptorJournal of the American Chemical Society 76:1760–1770.https://doi.org/10.1021/ja01636a012
-
Mapping the carbon reduction cycle: a personal retrospectivePhotosynthesis Research 76:35–52.https://doi.org/10.1023/A:1024929725022
-
Rubisco is not really so badPlant, Cell & Environment 41:705–716.https://doi.org/10.1111/pce.13149
-
Following the path of carbon in photosynthesis: a personal storyPhotosynthesis Research 73:29–49.https://doi.org/10.1023/A:1020427619771
-
Oxygen inhibition and other properties of soybean ribulose 1,5-diphosphate carboxylaseThe Journal of Biological Chemistry 247:2171–2176.
-
Temperature response of rubisco kinetics in Arabidopsis thaliana: thermal breakpoints and implications for reaction mechanismsJournal of Experimental Botany 70:231–242.https://doi.org/10.1093/jxb/ery355
-
Determination of the average partial pressure of CO2 in chloroplasts from leaves of several C3 plantsFunctional Plant Biology 18:287–305.https://doi.org/10.1071/PP9910287
-
Transcript analysis of the Halothiobacillus neapolitanus cso operonArchives of Microbiology 189:141–150.https://doi.org/10.1007/s00203-007-0305-y
-
Microcompartments in prokaryotes: carboxysomes and related polyhedraApplied and Environmental Microbiology 67:5351–5361.https://doi.org/10.1128/AEM.67.12.5351-5361.2001
-
Harnessing the power of microbial autotrophyNature Reviews Microbiology 14:692–706.https://doi.org/10.1038/nrmicro.2016.130
-
Mechanism of rubisco: the carbamate as general baseChemical Reviews 98:549–562.https://doi.org/10.1021/cr970010r
-
Identification of mutations in laboratory-evolved microbes from next-generation sequencing data using breseqMethods in Molecular Biology 1151:165–188.https://doi.org/10.1007/978-1-4939-0554-6_12
-
DABs are inorganic carbon pumps found throughout prokaryotic phylaNature Microbiology 4:2204–2215.https://doi.org/10.1038/s41564-019-0520-8
-
On the road to C4 rice: advances and perspectivesThe Plant Journal : For Cell and Molecular Biology 101:940–950.https://doi.org/10.1111/tpj.14562
-
Evolution of oxygenic photosynthesisAnnual Review of Earth and Planetary Sciences 44:647–683.https://doi.org/10.1146/annurev-earth-060313-054810
-
Revisiting Trade-offs between rubisco kinetic parametersBiochemistry 58:3365–3376.https://doi.org/10.1021/acs.biochem.9b00237
-
Cell biology of photosynthesis over geologic timeCurrent Biology 30:R490–R494.https://doi.org/10.1016/j.cub.2020.01.076
-
The principle of flux minimization and its application to estimate stationary fluxes in metabolic networksEuropean Journal of Biochemistry 271:2905–2922.https://doi.org/10.1111/j.1432-1033.2004.04213.x
-
Species variation in kinetic properties of ribulose 1,5-bisphosphate carboxylase/oxygenaseArchives of Biochemistry and Biophysics 227:425–433.https://doi.org/10.1016/0003-9861(83)90472-1
-
Assembly, function and evolution of cyanobacterial carboxysomesCurrent Opinion in Plant Biology 31:66–75.https://doi.org/10.1016/j.pbi.2016.03.009
-
Identification and structural analysis of a novel carboxysome shell protein with implications for metabolite transportJournal of Molecular Biology 392:319–333.https://doi.org/10.1016/j.jmb.2009.03.056
-
Catalytic properties of recombinant octameric, Hexadecameric, and heterologous cyanobacterial/bacterial ribulose- 1,5-bisphosphate carboxylase/oxygenaseArchives of Biochemistry and Biophysics 291:263–269.https://doi.org/10.1016/0003-9861(91)90133-4
-
Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methodsNature Reviews Microbiology 10:291–305.https://doi.org/10.1038/nrmicro2737
-
Activities of constitutive promoters in Escherichia coliJournal of Molecular Biology 292:19–37.https://doi.org/10.1006/jmbi.1999.3056
-
Cyanobacterial CO2-concentrating mechanism components: function and prospects for plant metabolic engineeringCurrent Opinion in Plant Biology 31:1–8.https://doi.org/10.1016/j.pbi.2016.03.002
-
High CO(2) Requiring mutant of anacystis nidulans R(2)Plant Physiology 82:610–612.https://doi.org/10.1104/pp.82.2.610
-
Why is carbonic anhydrase essential to Escherichia coli?Journal of Bacteriology 185:6415–6424.https://doi.org/10.1128/JB.185.21.6415-6424.2003
-
Rubisco: maladapted or misunderstoodAustralian Journal of Botany 40:431.https://doi.org/10.1071/BT9920431
-
The diverse AAA+ machines that repair inhibited rubisco active sitesFrontiers in Molecular Biosciences 4:31.https://doi.org/10.3389/fmolb.2017.00031
-
Multivalent interactions between CsoS2 and rubisco mediate α-carboxysome formationNature Structural & Molecular Biology 27:281–287.https://doi.org/10.1038/s41594-020-0387-7
-
Directed evolution of RuBisCO hypermorphs through genetic selection in engineered E. coliProtein Engineering, Design and Selection 19:113–119.https://doi.org/10.1093/protein/gzj010
-
What's for dinner?: entner-doudoroff metabolism in Escherichia coliJournal of Bacteriology 180:3495–3502.https://doi.org/10.1128/JB.180.14.3495-3502.1998
-
ESI-MS/MS analysis of underivatised amino acids: a new tool for the diagnosis of inherited disorders of amino acid metabolism fragmentation study of 79 molecules of biological interest in positive and negative ionisation modeRapid Communications in Mass Spectrometry 17:1297–1311.https://doi.org/10.1002/rcm.1054
-
Functions, compositions, and evolution of the two types of carboxysomes: polyhedral microcompartments that facilitate CO2 fixation in cyanobacteria and some proteobacteriaMicrobiology and Molecular Biology Reviews 77:357–379.https://doi.org/10.1128/MMBR.00061-12
-
The possible evolution and future of CO2-concentrating mechanismsJournal of Experimental Botany 68:3701–3716.https://doi.org/10.1093/jxb/erx110
-
Photorespiration and the evolution of C4 photosynthesisAnnual Review of Plant Biology 63:19–47.https://doi.org/10.1146/annurev-arplant-042811-105511
-
Compilation of Henry's law constants (version 4.0) for water as solventAtmospheric Chemistry and Physics 15:4399–4981.https://doi.org/10.5194/acp-15-4399-2015
-
The structure of beta-carbonic anhydrase from the carboxysomal shell reveals a distinct subclass with one active site for the price of twoJournal of Biological Chemistry 281:7546–7555.https://doi.org/10.1074/jbc.M510464200
-
Escherichia coli physiology in Luria-Bertani brothJournal of Bacteriology 189:8746–8749.https://doi.org/10.1128/JB.01368-07
-
Biochemical characterization of predicted precambrian RuBisCONature Communications 7:10382.https://doi.org/10.1038/ncomms10382
-
Genome-derived minimal metabolic models for Escherichia coli MG1655 with estimated in vivo respiratory ATP stoichiometryBiotechnology and Bioengineering 107:369–381.https://doi.org/10.1002/bit.22802
-
The enzymatic formation of phosphoglyceric acid from ribulose diphosphate and carbon dioxideThe Journal of Biological Chemistry 218:795–810.
-
Along the trail from fraction I protein to rubisco (ribulose bisphosphate carboxylase-oxygenase)Photosynthesis Research 73:243–250.https://doi.org/10.1023/A:1020467601966
-
An improved Escherichia coli screen for rubisco identifies a protein-protein interface that can enhance CO2-fixation kineticsJournal of Biological Chemistry 293:18–27.https://doi.org/10.1074/jbc.M117.810861
-
BookImproving CO2 Fixation by Enhancing Rubisco PerformanceIn: Alcalde M, editors. Directed Enzyme Evolution: Advances and Applications. Springer International Publishing. pp. 101–126.https://doi.org/10.1007/978-3-319-50413-1
-
BookPhotorespiration: Studies with Whole TissuesIn: Gibbs M, Latzko E, editors. Photosynthesis II: Photosynthetic Carbon Metabolism and Related Processes. Springer Berlin Heidelberg. pp. 353–367.https://doi.org/10.1007/978-3-642-67242-2
-
Directed evolution of an improved rubisco; In vitro analyses to decipher fact from fictionInternational Journal of Molecular Sciences 20:5019.https://doi.org/10.3390/ijms20205019
Article and author information
Author details
Sumedha Ravishankar

Funding
U.S. Department of Energy (DE-SC00016240)
- David F Savage
European Research Council (NOVCARBFIX 646827)
- Ron Milo
National Science Foundation (MCB-1818377)
- David F Savage
Shell (EBI CW163755)
- David F Savage
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We dedicate this paper to the memory of Arren Bar-Even, who was a great friend and teacher and whose wit and intellect inspired us throughout this work. Thanks to Matt Davis for P1 transduction materials and advice, Hernan Garcia and Han Lim for pZ plasmids, Maggie Stoeva, Anna Engelbrektson, Anchal Mehra, Sophia Ewens and Tyler Barnum for help with anaerobic cultivation, Reena Zalpuri and Danielle Jorgens at the University of California Berkeley Electron Microscope Laboratory for advice and assistance with electron microscopy, and Rob Egbert and Adam Arkin for KEIO strains. We are grateful to Griffin Chure, Eric Estrin, Woody Fischer, Evan Groover, Darcy McRose, Sabeeha Merchant, Dipti Nayak, Luke Oltrogge, Naiya Phillips, and Ari Satanowski for detailed comments on the manuscript, and to Dan Arlow, Yinon Bar-On, Dan Davidi, Jack Desmarais, Hernan Garcia, Oliver Mueller-Cajar, Rob Nichols, Kris Niyogi, Dan Portnoy, Morgan Price, Noam Prywes, Jeremy Roop, Rachel Shipps, Patrick Shih, and Dan Tawfik, for support, advice and helpful discussions throughout.
Copyright
© 2020, Flamholz et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 12,793
- views
-
- 1,759
- downloads
-
- 130
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 130
- citations for umbrella DOI https://doi.org/10.7554/eLife.59882
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Functional reconstitution of a bacterial CO2 concentrating mechanism in Escherichia coli
eLife 9:e59882.
https://doi.org/10.7554/eLife.59882




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}