Role of oxidation of excitation-contraction coupling machinery in age-dependent loss of muscle function in Caenorhabditis elegans
- Department of Physiology and Cellular Biophysics, The Clyde and Helen Wu Center for Molecular Cardiology, United States
- PhyMedExp, Montpellier University, INSERM, CNRS, CHRU Montpellier, France
- Medical Intensive Care Unit, Montpellier University and Montpellier University Health Care Center, France
Abstract
Age-dependent loss of body wall muscle function and impaired locomotion occur within 2 weeks in Caenorhabditis elegans (C. elegans); however, the underlying mechanism has not been fully elucidated. In humans, age-dependent loss of muscle function occurs at about 80 years of age and has been linked to dysfunction of ryanodine receptor (RyR)/intracellular calcium (Ca2+) release channels on the sarcoplasmic reticulum (SR). Mammalian skeletal muscle RyR1 channels undergo age-related remodeling due to oxidative overload, leading to loss of the stabilizing subunit calstabin1 (FKBP12) from the channel macromolecular complex. This destabilizes the closed state of the channel resulting in intracellular Ca2+ leak, reduced muscle function, and impaired exercise capacity. We now show that the C. elegans RyR homolog, UNC-68, exhibits a remarkable degree of evolutionary conservation with mammalian RyR channels and similar age-dependent dysfunction. Like RyR1 in mammals, UNC-68 encodes a protein that comprises a macromolecular complex which includes the calstabin1 homolog FKB-2 and is immunoreactive with antibodies raised against the RyR1 complex. Furthermore, as in aged mammals, UNC-68 is oxidized and depleted of FKB-2 in an age-dependent manner, resulting in ‘leaky’ channels, depleted SR Ca2+ stores, reduced body wall muscle Ca2+ transients, and age-dependent muscle weakness. FKB-2 (ok3007)-deficient worms exhibit reduced exercise capacity. Pharmacologically induced oxidization of UNC-68 and depletion of FKB-2 from the channel independently caused reduced body wall muscle Ca2+ transients. Preventing FKB-2 depletion from the UNC-68 macromolecular complex using the Rycal drug S107 improved muscle Ca2+ transients and function. Taken together, these data suggest that UNC-68 oxidation plays a role in age-dependent loss of muscle function. Remarkably, this age-dependent loss of muscle function induced by oxidative overload, which takes ~2 years in mice and ~80 years in humans, occurs in less than 2–3 weeks in C. elegans, suggesting that reduced antioxidant capacity may contribute to the differences in lifespan among species.
Editor's evaluation
This manuscript will appeal to all with an interest in comparative physiology and the molecular biology of age-associated changes in muscle function. The authors draw parallels between aging skeletal muscle in humans and C. elegans, with evidence in support of age-dependent oxidation of the C. elegans ryanodine receptor ortholog, UNC-68, causing loss of the calstabin ortholog, FKB-2. This in turn results in calcium leak, reduced body wall calcium transients and muscle weakness, changes that are similar to those that occur in aging human skeletal muscle despite the dramatic differences in the lifespan of the two organisms.
https://doi.org/10.7554/eLife.75529.sa0Introduction
Approximately 50% of humans over the age of 80 develop muscle weakness, which contributes to falls and hip fractures, a leading cause of mortality in the elderly (Marzetti and Leeuwenburgh, 2006; Ganz et al., 2007; Santulli et al., 2013). Strikingly, despite an approximately 2000-fold difference in the lifespans of humans and Caenorhabditis elegans (Herndon et al., 2002; Ljubuncic and Reznick, 2009), both exhibit oxidative overload induced age-dependent reductions in muscle function and motor activity that ultimately contribute to senescence and death. Due to its short lifespan and well-characterized genome, C. elegans has been used as a model to study the genetics of aging and lifespan determination (Guarente and Kenyon, 2000; Kenyon, 2010), including the age-dependent decline in locomotion (Herndon et al., 2002; Hsu et al., 2009). Age-dependent reduction in locomotion in C. elegans has been attributed to degeneration of the nervous system (Liu et al., 2013) and the body wall musculature (Kirkwood, 2013). Here, we investigated the role of the ryanodine receptor (RyR)/intracellular calcium (Ca2+) release channel homolog, UNC-68, in age-dependent loss of muscle function in C. elegans.
Mammalian RyR1 is the major intracellular Ca2+ release channel in skeletal muscle required for excitation-contraction (E-C) coupling (Zalk et al., 2015). In mammals, peak intracellular Ca2+ transients evoked by sarcolemmal depolarization decrease with age (Gonzalez et al., 2003), and this decrease is associated with a reduced SR Ca2+ release (Jiménez-Moreno et al., 2008) that directly determines the force production of skeletal muscle. Our group has shown that a mechanism underlying age-dependent loss of muscle function is RyR1 channel oxidation which depletes the channel complex of the stabilizing subunit calstabin1 (calcium channel stabilizing binding protein type 1, or FKBP12), resulting in intracellular Ca2+ leak and muscle weakness (Andersson et al., 2011; Umanskaya et al., 2014). RyR1 is a macromolecular complex comprised of homotetramers of four ~565 kDa RyR monomers (; Zalk et al., 2007). Cyclic AMP (cAMP)-dependent protein kinase A (PKA) (Marx et al., 2000), protein phosphatase 1 (Kass et al., 2003), phosphodiesterase PDE4D3 (Lehnart et al., 2005), Ca2+-dependent calmodulin kinase II (CaMKII) (Currie et al., 2004; Kushnir et al., 2010), and calstabin1 (Bellinger et al., 2008) are components of the RyR1 macromolecular complex (Santulli and Marks, 2015). Calstabin1 is part of the RyR1 complex in skeletal muscle, and calstabin2 (FKBP12.6) is part of the RyR2 complex in cardiac muscle (Santulli et al., 2017). Calstabins are immunophilins (Marks, 1996) with peptidyl-prolyl isomerase; however, this enzymatic activity does not play a role in regulating RyR channels and rather they stabilize the closed state of RyRs and prevent a Ca2+ leak via the channel (Marx et al., 2000; Brillantes et al., 1994).
RyR belongs to a small family of large intracellular Ca2+ release channels, the only other member being the inositol 1,4,5-triphosphate receptor (IP3R) (Harnick et al., 1995; Jayaraman et al., 1995; Jayaraman and Marks, 2000). RyR may have evolved from IP3R-B, which encoded an IP3R-like channel that could not bind IP3 and was replaced by RyR at the Holozoa clade (Alzayady et al., 2015). Invertebrates have one gene for each of RyR and IP3R, while vertebrates have three (RyR1-3 and IP3R1-3). RyRs and IP3Rs are intracellular Ca2+ release channels on the SR/ER and are tetramers that along with associated proteins comprise the largest known ion channel macromolecular complexes (Marx et al., 2000; DeSouza et al., 2002). Defects in Ca2+ signaling linked to stress-induced remodeling that results in leaky RyR channels have been implicated in heart failure (Dridi et al., 2020c; Marks, 2003), cardiac arrhythmias (Dridi et al., 2020c; Lehnart et al., 2006; Lehnart et al., 2004; Vest et al., 2005; Wehrens et al., 2003), diabetes (Santulli et al., 2015), muscle weakness (Kushnir et al., 2020; Dridi et al., 2020b; Matecki et al., 2016; Dridi et al., 2020d), and neurodegenerative disorders (Dridi et al., 2020b; Lacampagne et al., 2017; Liu et al., 2012).
RyR has evolved unique SPRY domains (des Georges et al., 2016) that are absent in IP3R, one of which (SPRY2) allows RyR1 to directly interact with the L-type calcium channel (Cav1.1) in mammalian skeletal muscle (Cui et al., 2009). This interaction couples excitation of the sarcolemma to muscle contraction to overcome the dependence on extracellular Ca2+. RyR1 is remarkably well conserved, suggesting that independence from extracellular Ca2+ evolved to support locomotion in higher organisms.
UNC-68 is the RyR gene homolog in the C. elegans genome (Maryon et al., 1996). Worms lacking both exon 1.1 and promoter1 (Marques et al., 2020), and UNC-68 (e540) null mutants exhibit severely defective swimming behavior and locomotive characterized by the ‘unc’, or ‘unco-ordinated’ phenotype (Brenner, 1974). Treatment with ryanodine, a chemical ligand of RyR, induces contractile paralysis in wild-type (WT) C. elegans, whereas UNC-68 (e540) null mutants are unaffected by ryanodine (Maryon et al., 1996; Brenner, 1974; Sakube et al., 1997). Ca2+ transients triggered by action potentials in C. elegans body wall muscles require UNC-68.
We previously reported that in aged mice (2 years old equivalent to ~80-year old humans) RyR1 oxidation depletes calstabin1 from the channels and renders them leaky to Ca2+, which contributes to the loss of muscle function and impaired muscle strength (Umanskaya et al., 2014). In the present study, we show that UNC-68 is comprised of a macromolecular complex that is remarkably conserved compared to RyR1 and includes the channel-stabilizing subunit, FKB-2. Like calstabin, FKB-2 regulates UNC-68 by directly associating with the channel. Similar to what we previously observed in mice (Andersson et al., 2011), we found age-dependent oxidation of UNC-68 which causes depletion of FKB-2 from the UNC-68 channel complex, and reduces Ca2+ transients in aged nematodes. This aging phenotype was accelerated in FKB-2 (ok3007) worms, an FKB-2 deletion mutant that results in leaky UNC-68. Competing FKB-2 from UNC-68 with rapamycin or FK506 (Timerman et al., 1993) resulted in reduced body wall muscle Ca2+ transients and defective locomotion. Conversely, pharmacological and genetic oxidation of UNC-68 with the reactive oxygen species (ROS)-generating drug paraquat (Lee et al., 2003) caused FKB-2 dissociation from the channel and reduced contraction-associated Ca2+ transients. Reassociating FKB-2 with UNC-68 using the RyR-stabilizing drug S107 improved Ca2+ transients and locomotion in aged nematodes. We have recently reported the binding site for S107 and its second generation Rycal, ARM210, using cryogenic electron microscopy (Melville et al., 2022). The compound binds in a cleft in the cytosolic shell and prevents a remodeled RyR channel from sitting in a ‘primed state’ sensitive to activation (Melville et al., 2022; Miotto et al., in-revision Science Advances 2022). A clinical trial using ARM210 to fix the leak in RyR1 channels is currently underway at the NIH (NCT04141670).
Our study provides an underlying mechanism for age-dependent loss of muscle function in C. elegans including progressive oxidation of UNC-68, which depletes the stabilizing binding protein, FKB-2 and, renders the channel leaky within 2 weeks compared to 2 years in mice and 80 years in humans and a potential therapy.
Results
Conserved evolution and architecture of UNC-68
Phylogenic analysis of RyR and FKBP among species reveals remarkable evolutionary conservation (Figure 1A–B). UNC-68, the C. elegans intracellular calcium release channel, shares ~40% homology with the human RyR1 (Figure 1C). C. elegans FKB-2 has ~60% sequence identity with the skeletal muscle isoform calstabin1 (FKBP12) (Figure 1D). Based on these observations, we hypothesized that in C. elegans, UNC-68 comprises a macromolecular complex, similar to that of mammalian RyRs. To test this hypothesis, lysates were prepared from populations of freeze-cracked WT C. elegans, and UNC-68 was immunoprecipitated using mammalian anti-RyR antibody (5029) as previously described (Kushnir et al., 2018). The immunoprecipitates were immunoblotted to detect UNC-68, as well as other components of the RyR macromolecular complex including the catalytic subunit of protein kinase A (PKAcat), protein phosphatase 1 (PP1), FKB-2, and phosphodiesterase 4 (PDE-4) using mammalian anti-RyR, anti-PKA, anti-PP1, anti-calstabin, and anti-PDE-4 antibodies, respectively (Figure 1E). The previously published C. elegans anti-PDE-4 (Charlie et al., 2006) was used to detect PDE-4 on the channel. Our data show that UNC-68 comprises a macromolecular complex, similar to that found in the mammalian muscle, that includes PKAcat, PP1, PDE-4, and FKB-2. UNC-68 was depleted of FKB-2 in the FKB-2 (ok3007) null mutant (Figure 1E and G). In the FKB-2 null C. elegans, UNC-68 and the rest of the macromolecular complex could not be immunoprecipitated using an anti-FKBP antibody (Figure 1F and H). Taken together these data indicate remarkable evolutionary conservation of the RyR macromolecular complex.
Figure 1
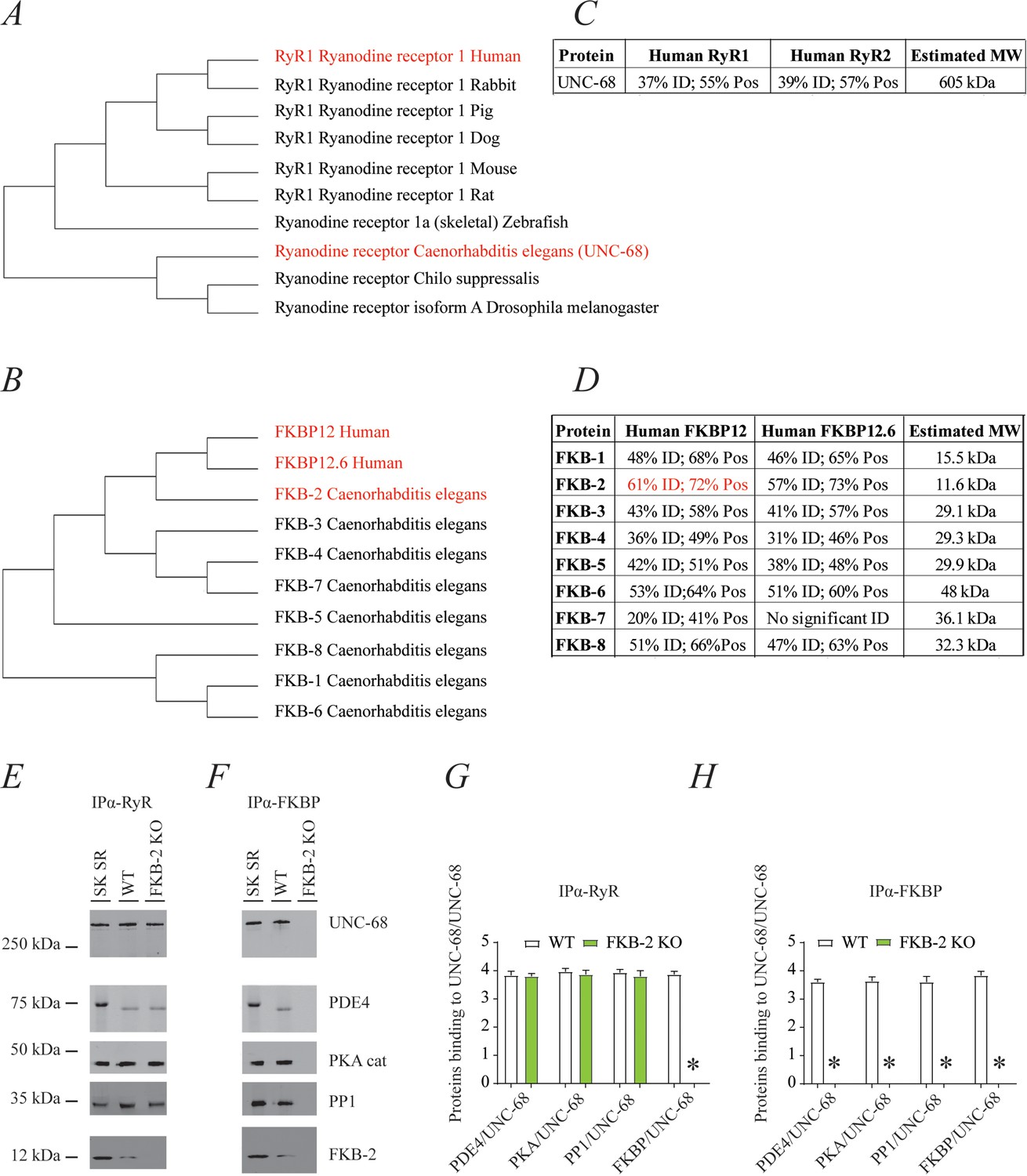
UNC-68 comprises a macromolecular complex comparable to its mammalian homolog ryanodine receptor (RyR); RyR (A) and FKBP (B) evolution among species was inferred by the maximum likelihood method based on the JTT matrix-based model.
(C) Homology comparison between UNC-68 and the two human RyR isoforms (RyR1 and RyR2). (D) Homology comparison between the different FKB isoforms (1–8) and the human FKBP isoforms (FKBP12 and FKB12.6). UNC-68 (E) and FKB-2 (F), respectively, were immunoprecipitated and immunoblotted using anti-RyR, anti-phosphodiesterase 4 (PDE4), anti-protein kinase A (catalytic subunit; PKAcat), anti-protein phosphatase 1 (PP1), and anti-calstabin (FKBP) antibodies in murine skeletal sarcoplasmic reticulum preparations (SK SR), wild-type (WT) populations of Caenorhabditis elegans, and populations of FKB-2 (ok3007). Images show representative immunoblots from triplicate experiments. (G and H) Quantification of bands intensity shown in E and F. Data are means ± SEM. One-way ANOVA shows * p<0.05 WT vs. FKB-2 KO. SK SR, sarcoplasmic reticulum fraction from mouse skeletal muscle. Figure 1—source data 1.
-
Figure 1—source data 1
Full incut gels of Figure 1.
- https://cdn.elifesciences.org/articles/75529/elife-75529-fig1-data1-v2.pdf
Age-dependent biochemical and functional remodeling of UNC-68
RyR1 channels are oxidized, leaky, and Ca2+ transients are reduced in aged mammalian skeletal muscle (Andersson et al., 2011). These changes occur by 2 years of age in mice (Andersson et al., 2011) and by 80 years of age in humans. Similarly, FKB-2 deficient worms exhibited an age-dependent decline in body wall muscle peak Ca2+ transients starting at day 7 post-hatching (Figure 2A–B).
Figure 2
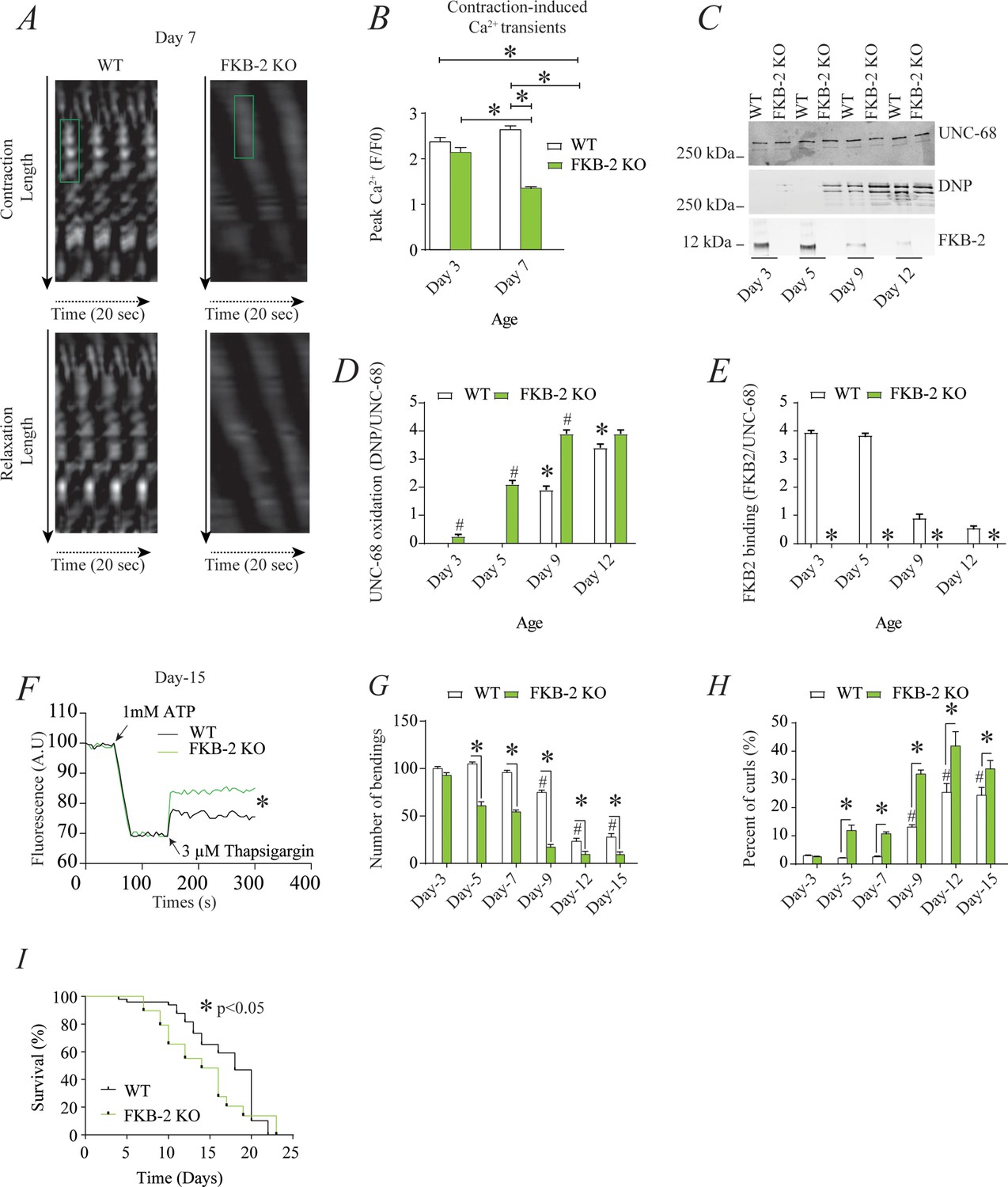
Remodeling of UNC-68 and age-dependent reduction in intracellular calcium (Ca2+) transients is accelerated in FKB-2 (ok3007) (A) Representative trace of Ca2+ transients from GCaMP2 wild type (WT) and FKB-2 KO (at day 7).
Green box denotes peak fluorescence from worm’s muscle during contraction. (B) Ca2+ transients in age-synchronized populations of WT and FKB-2 (ok3007) nematodes (at day 3 and 7); (C) UNC-68 was immunoprecipitated from age-synchronized populations of mutant (FKB-2 KO) and WT nematodes (at day 3, 5, 9, and 12) and immunoblotted using anti-RyR, anti-calstabin, and dinitrophenyl (DNP; marker of oxidation) antibodies. (D and E) Quantification of the average band intensity from triplicate experiments: band intensity was defined as the ratio of each complex member’s expression over its corresponding /UNC-68’s expression. Data are means ± SEM. * p<0.05 WT vs. FKB-2 KO in panel D, # p<0.05 WT vs. FKB-2 KO in panel E, * p<0.05 WT at day 3 vs. WT at day 5 and day 9. (F) Ca2+ leak assay performed with microsomes from WT and FKB-2 KO worms (day 5). Ca2+ uptake into the microsomes was initiated by adding 1 mM of ATP. Then, 3 µM of thapsigargin was added to block the sarco/endoplasmic calcium ATPase activity. Increased fluorescence is proportional to the spontaneous Ca2+ leakage throughout UNC-68. (G) Graph showing number of bends recorded for WT vs. FKB-2 KO worms at six distinct ages (day 3, 5, 7, 9, 12, and 15). (H) The number of curling events was calculated as a percentage of the overall motility (curls/bends). N = ~60 worms per group, except for day 15 (as fewer worms were alive at this timepoint). Day 15 = ~40 worms. (I) Percentage of survival of WT (average survival; 18 days) and FKB-2 KO worms (average survival; 14 days); Gehan-Breslow-Wilcoxon test for survival comparison was performed for statistical significance. Data are means ± SEM from triplicate experiments. One-way ANOVA shows * p<0.05 WT vs. FKB-2 KO, # p<0.05 WT at day 3 vs. WT at day 5, 7, 9, 12, and 15. Figure 2—source data 1.
-
Figure 2—source data 1
Full incut gels of Figure 2.
- https://cdn.elifesciences.org/articles/75529/elife-75529-fig2-data1-v2.pdf
RyR1 oxidation has been linked to SR Ca2+ leak and impaired muscle function during extreme exercise and in heart failure and muscular dystrophies (Bellinger et al., 2008; Bellinger et al., 2009; Allen et al., 2008). Furthermore, we have previously reported that oxidation of RyR1 and the subsequent intracellular Ca2+ leak are underlying mechanisms of age-related loss of skeletal muscle specific force (force normalized to the cross-sectional area of muscle) (Andersson et al., 2011). WT UNC-68 was oxidized (Figure 2C–D) and depleted of FKB-2 (Figure 2C–E) and in an age-dependent manner. These changes mirror those occurring with extreme exercise in mice and humans (Bellinger et al., 2008) and in a murine model of Duchenne muscular dystrophy (mdx mice) characterized by impaired muscle function (Bellinger et al., 2009). Importantly, by 80 years of age, ~50% of humans develop severe muscle weakness that is a strong predictor of mortality due to falls, gait imbalance, and related factors (Degens, 2007). Similarly, UNC-68 was significantly more oxidized (day 3–9) in FKB-2 (ok3007) worms compared to WT (Figure 2C–D).
To further demonstrate that UNC-68 channels lacking FKB-2 are inherently ‘leaky’, we used an assay that can monitor the rate of Ca2+ released from the SR. Age synchronized worms' microsomes (day 5) were mixed with the Ca2+ dye Fluo-4 and baseline fluorescence measurements were taken before adding 1 mM of ATP. By activating the sarco/endoplasmic calcium ATPase (SERCA) with ATP, cytosolic Ca2+ is pumped into the microsomes, resulting in a decrease in Fluo-4 fluorescence. Once the fluorescence level plateaus, thapsigargin (SERCA antagonist) is added to block Ca2+ reuptake into the SR. The rate at which the fluorescence increases directly correlates with the amount of Ca2+ passively leaking into the cytoplasm: a higher increase of fluorescence compared to WT control indicates leaky UNC-68 channels. Our data show that UNC-68 from FKB-2 KO worms had a higher rate of SR Ca2+ leak following thapsigargin administration compared to the WT channels (Figure 2F). This is corroborated by our previous findings, where disruption of RyR-calstabin binding increases the SR Ca2+ leak in mammalian tissues (Umanskaya et al., 2014).
In mammals, calstabin regulation of RyR is tightly coupled to beta-adrenergic signaling (Andersson et al., 2012), and it is known that calstabin KO mice must undergo exercise stress before demonstrating a distinct muscle phenotype (Bellinger et al., 2008). Our method of inducing exercise stress in the worm was to place it in M9 buffer and observe it swimming, a well-described behavioral assay (Lüersen et al., 2014). By using an extended time trial of 2 hr, the worms fatigue and exhibit exercise-induced stress similar to that observed in mammals. Our data show a defect in FKB-2 KO swimming behavior over the course of its lifespan when compared to the WT. FKB-2 KO worms had decreased bending activity earlier in life, beginning at day 5, and an increased proportion of curling, a sign of fatigue (Figure 2G–H). Throughout midlife, the FKB-2 KO worms lag significantly behind their age-matched WT counterparts, suggestive of decreased muscle function. Furthermore, FKB-2 KO worms exhibit reduced lifespan compared to WT (Figure 2I).
Pharmacologically mimicking aging phenotype affects Ca2+ transient and impairs exercise capacity
FKB-2 was competed off from the UNC-68 macromolecular complex using rapamycin or FK506 (Figure 3). Both rapamycin and FK-506 bind to calstabin and compete it off from RyR channels, resulting in leaky channels and release of SR Ca2+ in the resting state (Kaftan et al., 1996; Tang et al., 2002).
Figure 3
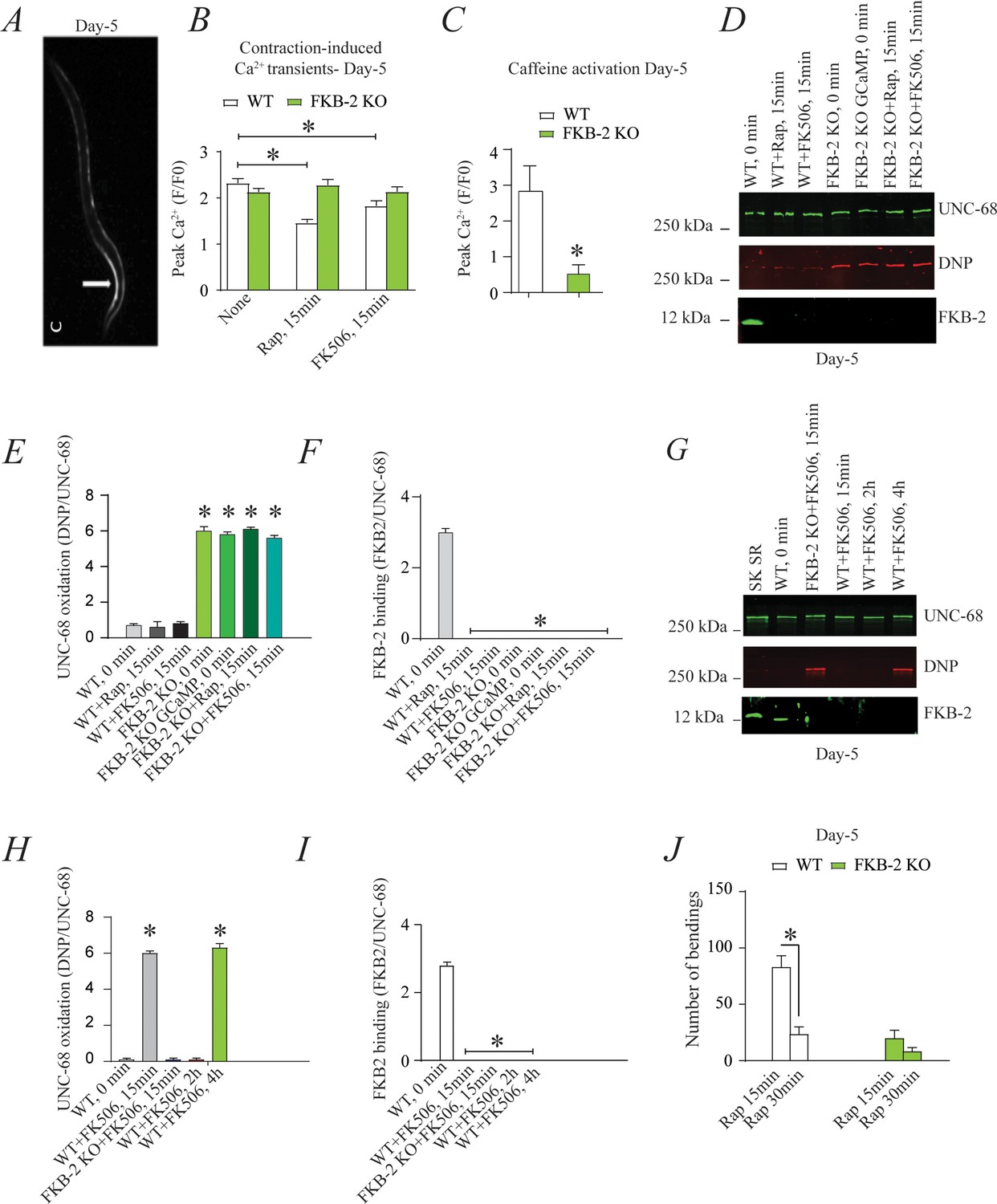
Depleting FKB-2 from UNC-68 causes UNC-68 oxidation (A) Representative image of caffeine activated calcium transient in GCaMP2 wild type (WT) at day 5; arrow denotes peak fluorescence in body wall muscle.
(B) Intracellular calcium (Ca2+) transients in day 5 age-synchronized populations of WT and FKB-2 (ok3007) nematodes treated with 15 μM and 50 μM rapamycin and FK506, respectively (treatment was applied for 15 min). (C) Fluorescence intensity following caffeine activation in age-matched GCaMP2: WT vs. GCaMP2: FKB-2 KO worms at day 5. (D) UNC-68 was immunoprecipitated and immunoblotted using anti-ryanodine receptor, anti-calstabin, and dinitrophenyl (DNP; marker of oxidation) antibodies in nematodes (at day 5) acutely treated with 15 μM and 50 μM rapamycin and FK506, respectively (treatment was applied for 15 min). (E–F) Quantification of the band intensity shown in (D): band intensity was defined as the ratio of either DNP (marker of UNC-68 oxidation) or FKB-2 binding over its corresponding /UNC-68’s expression. (G) UNC-68 was immunoprecipitated after 0, 15 min, 2 hr, and 4 hr FK506 exposure of the nematodes (at day 5). Representative immunoblots from triplicate experiments. (H–I) Quantification of the band intensity shown in (G): band intensity was defined as the ratio of either DNP (marker of UNC-68 oxidation) or FKB-2 binding over its corresponding /UNC-68’s expression. (J) Graph showing number of bends recorded for WT vs. FKB-2 KO worms (Day 5) treated for 20 and 30 min with 15 μM and 50 μM rapamycin and FK506, respectively. N ≥ 15 per group. Data are means ± SEM from triplicate experiments. One-way ANOVA shows * p<0.05 vs. WT for results shown in panel E, F, H, and I. Two-way ANOVA was used for results comparison in panel B, and t-test was used for results shown in C and J. SK SR; sarcoplasmic reticulum fraction from mouse skeletal muscle used as external control reference and was not quantified in the bar graphs. The time 0 min refers to untreated worms. Figure 3—source data 1.
-
Figure 3—source data 1
Full incut gels of Figure 3.
- https://cdn.elifesciences.org/articles/75529/elife-75529-fig3-data1-v2.pdf
Age-synchronized young C. elegans (5 days) were treated with rapamycin or FK506. Ca2+ transients were measured in partially immobilized transgenic nematodes expressing the genetically encoded Ca2+ indicator, Pmyo-3::GCaMP2, in the body wall muscle cells (Tallini et al., 2006; Liu et al., 2011; Figure 3A). Pharmacologic depletion of FKB-2 from UNC-68 by rapamycin or FK506 treatment (15 min exposure to each drug) caused reduced body wall muscle Ca2+ transients in WT C. elegans (Figure 3B). When FKB-2 was genetically depleted from the UNC-68 complex, as in the FKB-2 (ok3007) nematodes, treatment with rapamycin or FK506 had no effect on the Ca2+ transients (Figure 3B).
Continuous Ca2+ leak via UNC-68 would be expected to result in depleted SR Ca2+ stores; therefore, we utilized a common technique from the mammalian RyR literature to evaluate the SR Ca2+ stores. In brief, an activating concentration of caffeine is used to fully open the RyR channel, leading to a rapid release of Ca2+ from the SR into the cytoplasm. This increase can be approximated using a previously targeted, fluorescent Ca2+ sensitive dye or indicator. Caffeine was applied to day 5 cut worms (Figure 3C), and the amount of fluorescence given off by GCaMP2 was measured. The GCaMP2-WT worms demonstrated a strong Ca2+ transient within 10 s after caffeine administration, while GCaMP2-FKB-2 KO worms failed to produce a response, suggesting that their SR Ca2+ stores were too low to elicit one. Interestingly, GCaMP2-KO worms were observed as having very high background fluorescence, which may indicate an increase in cytosolic Ca2+ from passive UNC-68 leak.
Acute treatment with FK506 or rapamycin, for 15 min, each independently caused depletion of FKB-2 from the channel (Figure 3D, E and F) with no effect on the oxidation of UNC-68. Furthermore, longer treatment (2 and 4 hr) of WT worms with FK506 caused oxidation of UNC-68, demonstrating a relationship between depletion of FKB-2 and oxidation of UNC-68 (Figure 3G,H,I).
Indeed, rapamycin altered swimming behavior of WT but not FKB-2 KO worms in a time-dependent manner (Figure 3J). Taken together with our Ca2+ transient data, the observed muscle phenotype appears to be the result of UNC-68 channel leak. These data suggest that rendering UNC-68 channels leaky by removing FKB-2 depletes SR Ca2+, resulting in reduced Ca2+ transients and weakened muscle contraction.
Oxidation of UNC-68 causes reduced body wall muscle Ca2+ transients
To investigate the individual effect of age-dependent UNC-68 oxidation independent of the other confounding variables involved in aging (Herndon et al., 2002), we introduced a pharmacological intervention mimicking the aged state in young adult nematodes. Treating young adult nematodes (at 5 days of age) with the superoxide-generating agent paraquat (Lee et al., 2003) increased oxidation of UNC-68 and depletion of FKB-2 from the channel in a concentration-dependent manner (Figure 4A, B and C). Furthermore, contraction-associated Ca2+ transients decreased with paraquat treatment in a concentration-dependent manner (Figure 4D). Indeed, treatment with antioxidant N-Acetyl-L-cysteine improved Ca2+ transient in FKB-2 KO worms (Figure 4E). These data indicate that both UNC-68 oxidation and FKB-2 depletion independently contribute to the observed aging body wall muscle deterioration.
Figure 4
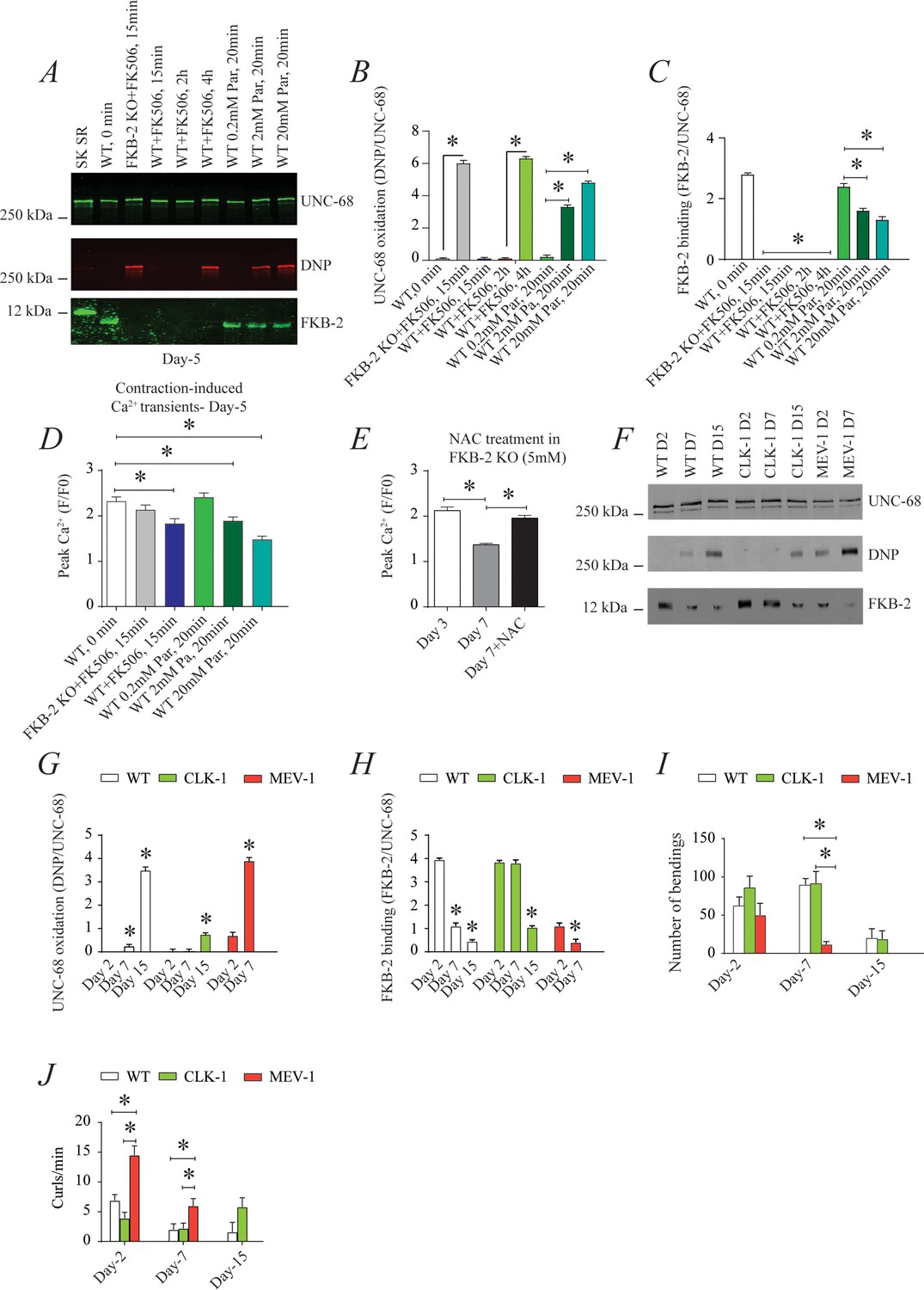
UNC-68 oxidation causes defective intracellular calcium (Ca2+) handling; (A) UNC-68 was immunoprecipitated and immunoblotted using anti-ryanodine receptor (RyR), anti-calstabin, and dinitrophenyl (DNP; marker of oxidation) antibodies in nematodes acutely treated for 0, 15 min, 2 hr, or 4 hr with FK506 or paraquat (treatment was applied for 20 min) at increasing concentration (day 5).
(B–C) Quantification of the band intensity shown in (A): band intensity was defined as the ratio of either DNP (marker of UNC-68 oxidation) or FKB-2 binding over its corresponding /UNC-68’s expression. (D) Contraction-associated Ca2+ transients measured in young age-synchronized WT nematodes treated for 15 with FK506 or for 20 min with increasing concentrations of paraquat (day 5). (E) Contraction-associated Ca2+ transients measured in FKB-2 KO nematodes treated with the antioxidant N-acetylcysteine (NAC) at 5 mM (day 7). (F) UNC-68 was immunoprecipitated and immunoblotted using anti-RyR, anti-calstabin, and DNP (marker of oxidation) antibodies in WT, the long lived (CLK-1) and the short lived (MEV-1) nematodes at day 2, 7, and 15. (G–H) Quantification of the average band intensity from triplicate experiments: band intensity was defined as the ratio of each complex member’s expression over its corresponding /UNC-68’s expression. (I) Graph showing number of bends recorded for WT vs. CLK-1 and MEV-1 worms at three distinct ages (day 2, 7, and 15). (J) The number of curling events was calculated as a percentage of the overall motility (curls/bends). N ≥ 20 per group. Data are means ± SEM from triplicate experiments. One-way ANOVA shows * p<0.05. Two-way ANOVA was used in panel I and J. SK SR; sarcoplasmic reticulum fraction from mouse skeletal muscle used as external control reference and was not quantified in the bar graphs. The time 0 min refers to untreated worms. Figure 4—source data 1.
-
Figure 4—source data 1
Full incut gels of Figure 4.
- https://cdn.elifesciences.org/articles/75529/elife-75529-fig4-data1-v2.pdf
To better clarify the role of oxidative stress in age-dependent UNC-68 remodeling and Ca2+ leak, we used two mutant mitochondrial electron transport chain (ETC) worms: the complex I mutant, CLK-1, and the complex II mutant, MEV-1. CLK-1 worms contain a Complex I-associated mutation such that they cannot synthesize their own ubiquinone (UQ), a redox active lipid that accepts and transfers electrons from Complex I or II to Complex III in the ETC. The reduction in Complex I activity of CLK-1 is associated with long-lived worms (Yang et al., 2011; Labuschagne et al., 2013; Kayser et al., 2004). In contrast, MEV-1 worms contain a Complex II (succinate dehydrogenase) cytochrome B560 mutation (Ishii et al., 1998; Senoo-Matsuda et al., 2001; Senoo-Matsuda et al., 2003), preventing electron transfer from succinate to fumarate and causing mitochondrial ROS production, which is associated with decreased lifespan, averaging only 9 days (Senoo-Matsuda et al., 2001). Interestingly, we have seen increased UNC-68 oxidation and FKB-2 depletion in the short-lived mutant (MEV-1) compared to WT and long-lived mutant (CLK-1) worms (Figure 4F, G and H). Indeed, MEV-1 worms exhibited reduced exercise capacity compared to WT and CLK-1 worms (Figure 4I–J).
UNC-68 Ca2+ channel is a potential therapeutic target in aging
The small molecule Rycal S107 inhibits SR Ca2+ leak by reducing the stress-induced depletion of calstabin from the RyR channel complex (Bellinger et al., 2009; Lehnart et al., 2008). Here, we show that treatment with S107 (10 μM) for 5 hr reassociated FKB-2 with UNC-68 without significant effect on the channel oxidation (Figure 5A, B and C). Furthermore, treatment with S107 improved peak Ca2+ in an FKB-2-dependent manner, as demonstrated by the fact that treating the FKB-2 KO worms did not change peak Ca2+ (Figure 5D–E). Interestingly, S107 treatment reduced age-dependent impairment of exercise capacity in WT worms at day 15 (Figure 5F). Of note, S107 has no effect on the WT worms' lifespan (Figure 5G). Furthermore, the treatment of the short-lived worms, MEV1, with S107 restored the FKB-2 association with UNC-68, despite the persistence of the channel oxidation (Figure 5H,I,J).
Figure 5
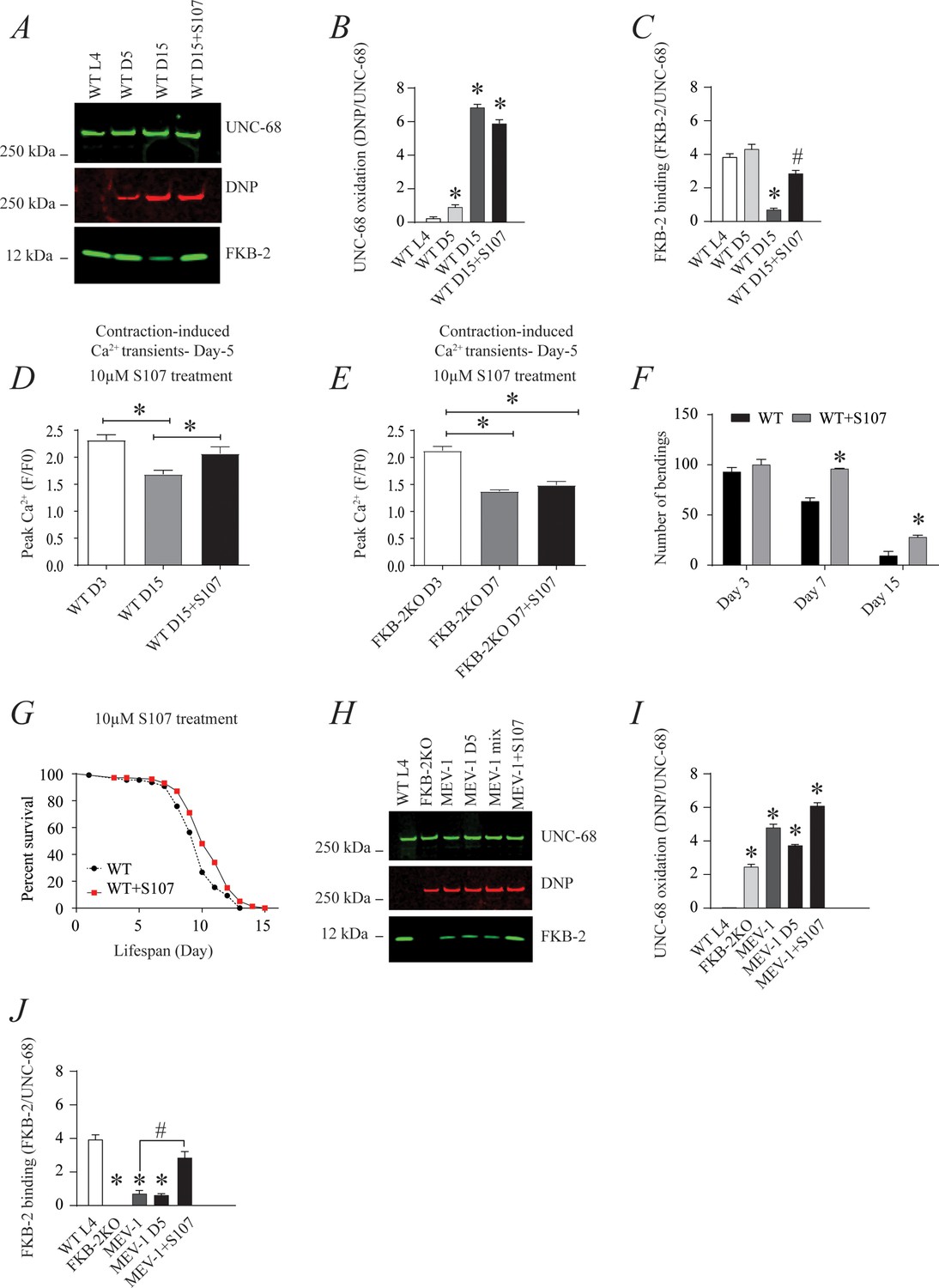
The ryanodine receptor (RyR)-stabilizing drug S107 increases body wall muscle calcium (Ca2+) transients in aged Caenorhabditis elegans; (A) UNC-68 was immunoprecipitated and immunoblotted with anti-RyR, anti-calstabin, and dinitrophenyl (DNP; marker of oxidation) in aged nematodes (Day L4, 5, and 15) with 10 µM of S107 (5 hr).
(B–C) Quantification of the band intensity shown in (A): band intensity was defined as the ratio of either DNP (marker of UNC-68 oxidation) or FKB-2 binding over its corresponding /UNC-68 expression. Data are mean ± SEM. * p<0.05 vs. wild-typd L4 (WTL4), # p<0.05 WT D15 vs. WT D15 + S107. (D–E) Contraction-associated Ca2+ transients were measured in age-synchronized WT (day 3 and 15) (D) and (E) FKB-2 KO worms (day 3 and 7). Contraction-associated Ca2+ transients in S107-treated worms were performed at day 15 for WT and day 7 for FKB-2 worms. (F) Graph showing number of bends recorded for WT vs. WT treated with S107 worms at different ages (day 3, 7, and 15). (G) Percent of survival of WT vs. WT treated with S107 nematodes; Gehan-Breslow-Wilcoxon test for survival comparison was performed for statistical significance. (H) UNC-68 was immunoprecipitated and immunoblotted with anti-RyR, anti-calstabin, and DNP (marker of oxidation) in short-lived nematodes (MEV-1) with S107 treatment (5 hr). (I–J) Quantification of the band intensity shown in (H): band intensity was defined as the ratio of either DNP (marker of UNC-68 oxidation) or FKB-2 binding over its corresponding /UNC-68 expression. N ≥ 20 per group. Data are mean ± SEM from triplicate experiments. One-way ANOVA shows * p < 0.05 vs WT L4 unless otherwise indicated. In panel F, a t-test was used to compare WT and WT + S107 for each day. #p<0.05 MEV-1, vs. MEV-1 +S107 in panel J. Figure 5—source data 1.
-
Figure 5—source data 1
Full incut gels of Figure 5.
- https://cdn.elifesciences.org/articles/75529/elife-75529-fig5-data1-v2.pdf
Discussion
Taken together, our data show that the C. elegans intracellular Ca2+ release channel UNC-68 comprises a macromolecular complex which is highly conserved throughout evolution from nematodes to humans. In nematodes, the UNC-68 macromolecular complex is comprised of a similar array of regulatory subunits as the mammalian RyR1 channels: a phosphodiesterase PDE-4, a protein kinase PKA, a protein phosphatase PP1, and the immunophilin, FKB-2. Binding of FKB-2 (the C. elegans homolog of the mammalian RyR stabilizing protein calstabin) to the UNC-68 channel is required to prevent a pathological leak of intracellular Ca2+, similar to the manner observed in mammalian muscle (Andersson et al., 2011). C. elegans exhibit reduced Ca2+ transients, as well as oxidized UNC-68 channels and depleted FKB-2 by ~2 weeks of age. Genetic FKB-2 deficiency causes an accelerated aging phenotype; Ca2+ transients are reduced in younger populations of FKB-2 (ok3007) nematodes and UNC-68 is oxidized at an earlier time point in these mutants relative to WT. Treating aged WT nematodes with the RyR-stabilizing drug, S107, reassociates FKB-2 with UNC-68 and increases the Ca2+ transients, indicating that UNC-68 dysfunction is likely an underlying mechanism of age-dependent decrease in Ca2+ transients in C. elegans body wall muscle. The mechanism causing age-dependent UNC-68 dysfunction involves the loss of UNC-68/FKB-2 from the UNC-68 channel complex due to oxidation of the channel. Of note, this may create a vicious cycle of intracellular Ca2+ leak and oxidative overload in which leaky channels cause mitochondrial Ca2+ accumulation and high levels of ROS production which further oxidize UNC-68 and further exacerbate the Ca2+ leak over the course of the lifespan ( Dridi et al., 2020a) as has been demonstrated in mice (Andersson et al., 2011).
C. elegans exhibit an aging muscle phenotype similar to age-dependent loss of muscle function in humans (Andersson et al., 2011). This is characterized by impaired locomotion, reduction in muscle cell size associated with loss of cytoplasm and myofibrils, and progressive myofibril disorganization (Herndon et al., 2002). However, specific body wall muscle proteins involved in the C. elegans aging phenotype have not been determined. Here, we show that UNC-68 is oxidized in aged nematodes and depleted of the channel-stabilizing protein, FKB-2. Our group has reported similar remodeling of RyR1 in skeletal muscle from aged mice (Andersson et al., 2011) and in murine models of muscular dystrophies (Bellinger et al., 2009), all of which exhibit intracellular Ca2+ leak and reduced muscle specific force production.
Though the oxidative stress theory of aging was first proposed in 1956 (Hagen, 2003; Harman, 1956), there is still substantial controversy surrounding the role of ROS in aging. For example, deletion or overexpression of the ROS detoxification enzyme superoxide dismutase has little effect on lifespan in C. elegans (Gems and Doonan, 2009; Van Raamsdonk and Hekimi, 2012). However, loss of sesn-1, the gene encoding sestrin, an evolutionarily conserved protein required for regenerating hyperoxidized forms of peroxiredoxins and for ROS clearance, causes reduced lifespan (Yang et al., 2013). Furthermore, ROS levels measured in vivo in C. elegans increase with age (Back et al., 2012). Other oxidative/antioxidative genes are involved in ROS production and may play a crucial role in the UNC-68 oxidation (Supplementary file 1).
While the free radical theory of aging has taken a hit due to multiple observations that contradict the notion of a link between reduced oxidative load and longevity, the preponderance of data shows a correlation between oxidative damage and reduced lifespan (Shields et al., 2021). Moreover, there is no doubt that reduced muscle function is detrimental to survival (Wilkinson et al., 2018). The present study shows that a key effector of age-dependent oxidative overload, RyR1 channel leak and the resulting muscle dysfunction, occur approximately 2000 times faster in C. elegans compared to Homo sapiens and 50 times faster than in Mus musculus. Since the target system, RyR1/UNC68, is remarkably conserved and underlies dramatically similar physiological functions (namely SR Ca2+ release required for muscle contraction) the cause for the accelerated kinetics of aging must be determined elsewhere and in an unrelentingly constant manner as exemplified by the rigid control of species lifespan. There is however, only one known case of a significant prolongation of average lifespan in a species: Homo sapiens. Indeed, the average lifespan in the U.S. has doubled in the past century (Schanzenbach et al., 2016) largely due to improved sanitation and related public health measures that protect against communicable diseases, the present pandemic notwithstanding. This suggests that both environmental and intrinsic biological constraints can determine average lifespan. Since we are a species that can remodel our environment to a greater extent than others, we have been able to double our average lifespan by improving the environment, although now global warming threatens to reverse this achievement. The unanswered question remains what are the intrinsic biological constraints on a given species' longevity? Although, oxidative stress has been thought to be a major contributor to the skeletal muscle aging phenotype (Andersson et al., 2011), other biological factors, including changes to the nervous, hormonal, circulatory, and respiratory systems likely also play important roles.
It would be interesting to know if the increased UNC-68 oxidation-induced FKB-2 depletion and subsequent reduction in body wall muscle Ca2+ transients are a result of globally increased ROS levels or increased ROS levels in UNC-68-surrounding microdomains. For example, we have previously shown that inducing RyR leak in enzymatically dissociated skeletal muscle cells causes increased mitochondrial membrane potential and mitochondrial ROS production (Andersson et al., 2011). Based on these data, we have proposed a model in which RyR1 leak (due to age-dependent oxidation of the channel and subsequent dissociation of calstabin) causes mitochondrial Ca2+ overload, resulting in ROS production, thus leading to further oxidation of RyR1 and exacerbation of the SR Ca2+ leak. This creates a vicious cycle between RyR1 and mitochondria that contributes to age-dependent loss of muscle function.
We also demonstrate that the putative null mutant, FKB-2 (ok3007), prevents FKB-2 from co-immunoprecipitating with UNC-68. The aging phenotype that we characterize in WT nematodes (biochemically modified UNC-68 and reduced Ca2+ transients) is accelerated in FKB-2 (ok3007). There are eight FKBs that are homologous to mammalian calstabin in the C. elegans genome; FKB-1 and FKB-8 both have ~50% sequence identity to calstabin. Further studies could elucidate the possibility that in the absence of FKB-2, another FKB may stabilize UNC-68, in particular the aforementioned FKB-8 (its gene is in close proximity to that of FKB-2 on chromosome 2) and FKB-1 (most similar to FKB-2 in terms of molecular weight). Such a mechanism could provide transitory compensation for the lack of FKB-2, in which other FKB isoform(s) bind to UNC-68 with lower affinity. Because this binding is weak, and the channel is unstable, this compensation ends up failing at day 7 of age and the Ca2+ leak is exacerbated. This hypothesis is partially supported by the unaltered Ca2+ peaks in FKB2-KO worms at day 5 of age despite a complete depletion of FKB-2 binding protein. Such a compensatory mechanism was not observed with acute rapamycin and FK506 treatment potentially because, first, the Ca2+ leak was acute and there was no time for a compensatory response, and, second, these drugs could act on all FKB isoforms.
Another key question is why UNC-68 becomes oxidized within 2 weeks, whereas the same post-translational modification requires 2 years in mice and 80 years in humans (Ljubuncic and Reznick, 2009)? Given the high degree of conservation of RyR and other members of the complex (Figure 1), it is feasible that genetic screens in organisms such as C. elegans and Drosophila will yield additional crucial mediators that are common among species and explain disparities in age-dependent loss of muscle function such as genes-genes interactions, epigenetics or architecture, and gating of key proteins involved in aging such as RyR. Indeed, despite the conserved evolution of UNC-68, the channel contains higher numbers of methionine (3.5%) and serine (7.2%) (Supplementary file 2) compared to the human RyR1 (2.9 and 5.9%, respectively). Methionines are a primary target of oxidative stress that might cause defects in the channel gating and alter Ca2+ release. Disparities in RyR1 serine residues among species, which are phosphorylated by protein kinases in response to stress, can cause conformational changes to the channel, exposing more residues to oxidation and could be a potential mechanism contributing to the accelerated UNC-68 oxidation in C. elegans.
Regarding the conservation of EC coupling machinery, the UNC-68 is localized to a specific portion of a vesicular network surrounding the myofilament lattice which suggests that the general architecture of the SR is conserved in metazoans. RyRs in vertebrate striated muscle cluster at internal couplings with Ttubules and, peripheral couplings adjacent to the surface membrane, visible as a ‘feet’ in electron micrographs (Maryon et al., 1998). The 12–14 nm gap between the surface and SR vesicle membranes in C. elegans is identical to analogous gaps in vertebrate triad junctions suggesting that UNC-68 bridges these gaps as seen in vertebrates. These similarities in muscle architecture further support our findings regarding the similar muscle aging phenotype between mammals and nematodes and the validity of C. elegans as a useful model to study age-dependent loss of muscle function.
Finally, UNC-68 null mutants are defective in locomotion, but still propagate coordinated contraction waves by an unknown mechanism (Maryon et al., 1996). The only intracellular Ca2+ release channels known in the SR vesicles, other than RyRs, are IP3Rs. The C. elegans genome contains a single IP3R gene, the lfe-1 (or itr-1) (Clandinin et al., 1998). However, it has been reported that antibodies to lfe-1 specifically stain the nerve ring but do not stain the myofilament lattice. Furthermore, lfe-1 mutants exhibit normal motility suggesting the IP3Rs channels are not involved in the regulation of the body wall muscle contraction. Moreover, UNC-68 has been reported to be expressed in neurons (Sakube et al., 1997) which may complicate the interpretation of its function in skeletal muscle. However, it seems that the neuronal expression is minor and does not modulate skeletal muscle function. Indeed, transformation of UNC-68 null mutant animals with the WT UNC-68 gene or the WT UNC-68 coding sequence fused to the myo-3 promoter rescued motility defects and sensitivity to ryanodine-induced paralysis (Maryon et al., 1998). myo-3 is expressed in body wall muscles, as well as in enteric muscles (the enteric muscles do not affect motility). Furthermore, no staining of neurons has been observed with an anti-UNC-68 antibodies, which suggests that the major role of UNC-68 is supporting skeletal muscle contraction (Maryon et al., 1998).
Taken together, our data indicate that the C. elegans homolog of RyR, UNC-68, is comprised of a macromolecular complex and regulated by the immunophilin, FKB-2. We have identified age-dependent reduction in body wall muscle Ca2+ transients in nematodes that is coupled to oxidation and remodeling of UNC-68. SR Ca2+ stores are depleted in FKB2-KO worms, suggesting passive UNC-68 leak. This observation is supported by the Ca2+ leak assay results, which show that FKB-2 regulation is critical in preventing UNC-68 channels from aberrantly ‘leaking’ Ca2+ into the cytoplasm. With reduced SR Ca2+, UNC-68 fails to release the burst of Ca2+ required for normal E-C coupling, leading to impaired muscle function. Loss of muscle function is evident in the FKB-2 KO worms during swimming trials, as middle-aged worms performed worse than their age-matched WT controls. Furthermore, our data strongly suggest a role for FKB-2 and UNC-68 in the age-dependent changes in Ca2+ signaling, as treatment with the pharmacological RyR stabilizer S107 increases body wall muscle Ca2+ transients. The advantage of targeting leaky RyR channels rather than using antioxidants would be the avoidance of the adverse effects of blocking beneficial oxidative signals.
Materials and methods
Key resources table
| Reagent type (species) or resource | Designation | Source or reference | Identifiers | Additional information |
|---|---|---|---|---|
| Strain, strain background (worms) | ok3007 | Caenorhabditis Genetics Center (University of Minnesota) | WormBase ID: WBVar00094093 | Genomic position: I: 2918075.12918967 |
| Strain, strain background (worms) | Pmyo-3:GCaMP2 worms | Kindly provided by Zhao-Wen Wang, University of Connecticut Health Center | ||
| Strain, strain background (worms) | mev-1 | Caenorhabditis Genetics Center (University of Minnesota) | WormBase ID: WBGene00003225 | Genomic position III: 10334277.10335168 |
| Strain, strain background (worms) | clk-1 | Caenorhabditis Genetics Center (University of Minnesota) | WormBase ID: WBGene00000536 | Genomic position III: 5277894.5279344 |
| Antibody | anti-RyR1 (Rabbit polyclonal) | Marks’ lab, Columbia University, NY, USA | Cat. #: 5,029 Aa 1327–1339 | WB (1:1000), (10 μl) |
| Antibody | anti-PDE4 (Rabbit monoclonal) | Kindly provided by Kenneth Miller, Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma | WB (1:1000), (10 μl) | |
| Antibody | anti-PP1 (Rabbit polyclonal) | Santa Cruz | Cat. #: sc6104 | WB (1:1000), (10 μl) |
| Antibody | anti-FKBP12 (Mouse monoclonal) | Santa Cruz | Cat. #: sc6104 | WB (1:2500), (10 μl) |
| Antibody | anti-FKBP12 (Rabbit polyclonal) | Abcam | Cat. #: ab2918 | WB (1:2000), (10 μl) |
| Commercial assay or kit | Oxyblot protein oxydation detection kit | Millipore | Cat. #: S7150 | WB (1:1000), (10 μl) |
| Chemical compound, drug | Rapamacin | Sigma Aldrich | Cat. #: 37,094 | |
| Chemical compound, drug | FK506 | Sigma Aldrich | Cat. #: Y0001926 | |
| Chemical compound, drug | Paraquat | Sigma Aldrich | Cat. #: 36,541 | |
| Chemical compound, drug | S107rycal drug | Marks’ lab, Columbia University, NY, USA | ||
| Software, algorithm | GraphPad | GraphPad | V8.0 |
C. elegans strains and culture conditions
Request a detailed protocolWorms were grown and maintained on standard nematode growth medium (NGM) plates on a layer of OP50 Escherichia coli at 20°C, as described (Brenner, 1974). N2 (Bristol) and fkb-2 (ok3007) were provided by the Caenorhabditis Genetics Center (University of Minnesota). fkb-2 (ok3007) was backcrossed six times. The transgenic strain expressing Pmyo-3: GCaMP2 was kindly provided by Zhao-Wen Wang, University of Connecticut Health Center (Liu et al., 2011). Pmyo-3: GCaMP2 was subsequently crossed into fkb-2 (ok3007) for measurement of contraction-associated Ca2+ transients.
Age synchronization
Request a detailed protocolAdult worms at the egg-laying stage were treated with alkaline hypochlorite solution to obtain age-synchronized populations, and eggs were plated on NGM plates, as described (Porta-de-la-Riva et al., 2012). For experiments requiring aged worms, age-synchronized animals at the L4 stage were collected in M9 buffer and plated on NGM plates containing 5-fluoro-2’-deoxyuridine (FUDR, Sigma, 50 μM) to prevent egg-laying (Mitchell et al., 1979).
Immunoprecipitation and immunoblotting
Request a detailed protocolNematodes were grown under standard conditions. For protein biochemistry experiments, a procedure to crack nematodes in a solubilizing and denaturing buffer was adapted (Francis and Waterston, 1985). Briefly, worms were washed and collected with M9 buffer, centrifuged for 2 min at 1000 rpm three times to wash. Worms were allowed to settle to the bottom of the collection tube by sitting on ice for ~5 min. Fluid was removed and the worm pellet was snap frozen in liquid nitrogen. Frozen pellets containing whole nematodes were rapidly thawed under warm running water. A volume of nematode solubilization buffer equal to the volume of the worm pellet was added (nematode solubilization buffer: 0.3% ethanolamine, 2 mM EDTA, 1 mM PMSF in DMSO, 5 mM DTT, 1× protease inhibitor), and tubes were microwaved (25 s for 100 μl pellet; time was increased for greater volumes). Lysates were then quickly drawn into a syringe through a 26-gauge needle and forced back through the needle into a new collection tube on ice. Samples were centrifuged at 1000 rpm for 2 min to remove insoluble material, and the supernatant was transferred to a new tube on ice. Lysates were snap frozen and stored in at –80°C.
A anti-mammalian RyR antibody (4 μg 5029 Ab [Jayaraman et al., 1992]) was used to immunoprecipitate UNC-68 from 100 μg of nematode homogenate. Samples were incubated with the antibody in 0.5 ml of a modified RIPA buffer (50 mM Tris-HCl pH 7.4, 0.9% NaCl, 5.0 mM NaF, 1.0 mM Na3VO4, 1% Triton- X100, and protease inhibitors) for 1 hr at 4°C. The immune complexes were incubated with protein A Sepharose beads (Sigma, St. Louis, MS) at 4°C for 1 hr, after which time the beads were washed three times with buffer. Proteins were size-fractionated by SDS-PAGE (6% for UNC-68, 15% for FKB-2) and transferred onto nitrocellulose membranes for 1 hr at 200 mA (SemiDry transfer blot, Bio-Rad). After incubation with blocking solution (LICOR Biosciences, Lincoln NE) to prevent non-specific antibody binding, immunoblots were developed using antibodies against RyR (5029, 1:5000), PKAcat (Santa Cruz Biotechnology, sc-903, 1:1000), PDE4 (kindly provided to us by Kenneth Miller, Oklahoma Medical Research Foundation, Oklahoma City, Oklahoma), PP1 (sc6104, 1:1000), or an anti-calstabin antibody (Santa Cruz 1: 2500). To determine channel oxidation, the carbonyl groups on the protein side chains were derivatized to 2,4-dinitrophenylhydrazone (DNP-hydrazone) by reaction with 2,4-dinitrophenylhydrazine (DNPH) according to manufacturers (Millipore) instructions. The DNP signal on immunoprecipitated UNC-68 was determined by immunoblotting with an anti-DNP antibody (Millipore, 1:1000). All immunoblots were developed and quantified using the Odyssey Infrared Imaging System (LICOR Biosystems, Lincoln, NE) and infrared-labeled secondary antibodies. In addition, immunoblotting and immunoprecipitation of the UNC-68 macromolecular complex were conducted using another anti-calstabin antibody (1:2000, Abcam) and the same methods as described.
Imaging contraction-associated body wall muscle Ca2+ transients
Request a detailed protocolSpontaneous changes in body wall muscle Ca2+ were measured in nematodes expressing GCaMP2 by fluorescence imaging using a Zeiss Axio Observer inverted microscope with an electron-multiplying CCD camera (Photometrics Evolve 512) and an LED light source (Colibri). Nematodes were partially immobilized by placing them individually into a 5–10 μl drop of M9 buffer, suspended between a glass slide and coverslip. 20-s videos of individual nematodes were recorded.
Analyzing contraction-associated body wall muscle Ca2+ transients
Request a detailed protocolContraction-associated body wall muscle Ca2+ transients were analyzed using an interactive data language-based image quantification software that was developed for this purpose in our laboratory. For each 20-s video, signals from the body wall muscles in nematodes expressing GCaMP2 fluorescence were analyzed using an edge-detection algorithm from each frame as ‘line-scan’ images, with the nematode perimeter on the y-axis and time (s) on the x-axis (Xie et al., 2013; Yuan et al., 2014). These images were then quantified based on the average of the peak Ca2+ fluorescence signal on the worm muscle wall.
Drug treatment
Request a detailed protocolTo pharmacologically deplete FKB-2 from UNC-68, nematodes were treated for 15 min with 15 μM rapamycin or imaging 50 μM FK506, respectively. To re-associate FKB-2 and UNC-68, aged nematodes were treated with 10 μM S107 for 3–5 hr. Oxidative stress was induced in the worms using 20 mM paraquat, a known generator of superoxide (Wu et al., 2017). Nematodes were grown in standard conditions, age-synchronized as described, washed and collected with M9 buffer, then centrifuged for 2 min at 1000 rpm three times. Worms were allowed to settle to the bottom of the collection tube by sitting on ice for ~5 min. Fluid was removed, the worm pellet was gently resuspended in M9 containing the appropriate drug concentration and gently rocked on a shaker at RT for the indicated time periods. Collection tubes were centrifuged for 2 min at 1000 rpm and M9 containing drug was removed and replaced with M9. Biochemistry or Ca2+ measurements were then conducted as previously described (Umanskaya et al., 2014).
Measuring SR Ca2+ stores using caffeine activation
Request a detailed protocolAge-synchronized GCaMP2: WT and GCaMP2: FKB-2 KO were grown on NGM plates at 20°C they were separated from their progeny and left undisturbed until day 5. Individual worms were placed in a drop of M9 on a coverslip. The liquid was carefully wicked away using KIMTECH wipes until only a sliver of moisture surrounded the worm. The worm was quickly glued down to the coverslip using a tiny drop of DermaWorm applied to the head and tail of the worm before the worm desiccated. 80 μl of M9 buffer was added immediately afterward to polymerize the glue. Once the worm was secure, a clean lateral cut to the immediate tail region was made using a 20 G 1½ needle (adapted from Wang ZW et al., Neuron 201148). An additional 170 μl of M9 buffer was applied for a total of 250 μl. The completed preparation was placed on the platform of a Zeiss confocal microscope; after 1 min at baseline, 25 mM of caffeine was added to an equal volume of M9 solution. The resulting body wall transients were recorded for 1 min.
Calcium leak assay
Request a detailed protocolMicrosomes were prepared by centrifuging the C. elegans lysates (5 days synchronized populations) at 45,000× g for 30 min. Pellets were resuspended in lysis buffer containing 300 mM sucrose. Microsomes (5 µg/ml) were diluted into a 20 mM HEPES buffer (pH 7.2) containing 7 mM NaCl, 1.5 mM MgCl2, 120 mM K-gluconate, 5 mM K-phosphate, 8 mM K-phosphocreatine, 1 µM EGTA, and 2 µM CaCl2 mixed with 3 µM Fluo-4 and added to multiple wells of a 96-well plate. Calcium (Ca2+) loading of the microsomes was initiated by adding 1 mM ATP. After Ca2+ uptake and a new Fluo-4 signal baseline was observed, 3 μM Thapsigargin was added to inhibit the calcium uptake by the calcium pump (SERCA). The ‘leak’ of Ca2+ out of the SR is measured by the increase in intensity of the Fluo-4 signal (measured in a Tecan infinite F500 fluorescence plate reader).
Swimming behavior
Request a detailed protocolStandard M9 buffer was mixed with 2% agar and poured into 96-well plates to create a planar surface for analyzing worm swimming behavior. Once the mixture had polymerized, approximately 180 μl of M9 was pipetted on top of the agar bed and age-synchronized worms from one of two groups (WT or FKB-2 KO) were placed individually into each well. To assess differences in exercise fatigue, worms were allowed to swim freely in M9 buffer for 2 hr; swimming bends and curls (Lüersen et al., 2014) were recorded by eye for 1 min. Representative videos were taken of each group, and investigators were blinded over the course of each experiment. All recordings were made in duplicate.
Statistical analysis
Request a detailed protocolAll results are presented as mean ± SEM. Statistical analyses were performed using an unpaired two-tailed Student’s t test (for two groups) and one-way ANOVA with Tukey-Kramer test (for three or more groups), unless otherwise indicated. For survival statistical comparison, we used Gehan-Breslow-Wilcoxon test. p-values <0.05 were considered significant. All statistical analyses were performed with Prism 8.0.
Data availability
All data are described/available in the manuscript.
References
-
Skeletal muscle fatigue: cellular mechanismsPhysiological Reviews 88:287–332.https://doi.org/10.1152/physrev.00015.2007
-
Exploring real-time in vivo redox biology of developing and aging Caenorhabditis elegansFree Radical Biology & Medicine 52:850–859.https://doi.org/10.1016/j.freeradbiomed.2011.11.037
-
A dihydropyridine receptor alpha1s loop region critical for skeletal muscle contraction is intrinsically unstructured and binds to A SPRY domain of the type 1 ryanodine receptorThe International Journal of Biochemistry & Cell Biology 41:677–686.https://doi.org/10.1016/j.biocel.2008.08.004
-
Age-related skeletal muscle dysfunction: causes and mechanismsJournal of Musculoskeletal & Neuronal Interactions 7:246–252.
-
Protein kinase A and two phosphatases are components of the inositol 1,4,5-trisphosphate receptor macromolecular signaling complexThe Journal of Biological Chemistry 277:39397–39400.https://doi.org/10.1074/jbc.M207059200
-
Late Ventilator-Induced Diaphragmatic Dysfunction After ExtubationCritical Care Medicine 48:e1300–e1305.https://doi.org/10.1097/CCM.0000000000004569
-
Mitochondrial oxidative stress induces leaky ryanodine receptor during mechanical ventilationFree Radical Biology & Medicine 146:383–391.https://doi.org/10.1016/j.freeradbiomed.2019.11.019
-
Antioxidant defense and aging in C. elegans: is the oxidative damage theory of aging wrong?Cell Cycle (Georgetown, Tex.) 8:1681–1687.https://doi.org/10.4161/cc.8.11.8595
-
Oxidative stress, redox imbalance, and the aging processAntioxidants & Redox Signaling 5:503–506.https://doi.org/10.1089/152308603770310149
-
Aging: a theory based on free radical and radiation chemistryJournal of Gerontology 11:298–300.https://doi.org/10.1093/geronj/11.3.298
-
The human type 1 inositol 1,4,5-trisphosphate receptor from T lymphocytes. Structure, localization, and tyrosine phosphorylationThe Journal of Biological Chemistry 270:2833–2840.https://doi.org/10.1074/jbc.270.6.2833
-
FK506 binding protein associated with the calcium release channel (ryanodine receptorThe Journal of Biological Chemistry 267:9474–9477.
-
Calcineurin is downstream of the inositol 1,4,5-trisphosphate receptor in the apoptotic and cell growth pathwaysThe Journal of Biological Chemistry 275:6417–6420.https://doi.org/10.1074/jbc.275.9.6417
-
Sarcoplasmic reticulum Ca2+ release declines in muscle fibers from aging miceBiophysical Journal 94:3178–3188.https://doi.org/10.1529/biophysj.107.118786
-
The effects of complex I function and oxidative damage on lifespan and anesthetic sensitivity in Caenorhabditis elegansMechanisms of Ageing and Development 125:455–464.https://doi.org/10.1016/j.mad.2004.04.002
-
Intracellular calcium leak as a therapeutic target for RYR1-related myopathiesActa Neuropathologica 139:1089–1104.https://doi.org/10.1007/s00401-020-02150-w
-
Leaky Ca2+ release channel/ryanodine receptor 2 causes seizures and sudden cardiac death in miceThe Journal of Clinical Investigation 118:2230–2245.https://doi.org/10.1172/JCI35346
-
Gait-specific adaptation of locomotor activity in response to dietary restriction in Caenorhabditis elegansThe Journal of Experimental Biology 217:2480–2488.https://doi.org/10.1242/jeb.099382
-
Cellular functions of immunophilinsPhysiological Reviews 76:631–649.https://doi.org/10.1152/physrev.1996.76.3.631
-
unc-68 encodes a ryanodine receptor involved in regulating C. elegans body-wall muscle contractionThe Journal of Cell Biology 134:885–893.https://doi.org/10.1083/jcb.134.4.885
-
Muscle-specific functions of ryanodine receptor channels in Caenorhabditis elegansJournal of Cell Science 111 (Pt 19):2885–2895.https://doi.org/10.1242/jcs.111.19.2885
-
Skeletal muscle apoptosis, sarcopenia and frailty at old ageExperimental Gerontology 41:1234–1238.https://doi.org/10.1016/j.exger.2006.08.011
-
Basic Caenorhabditis elegans methods: synchronization and observationJournal of Visualized Experiments 10:e4019.https://doi.org/10.3791/4019
-
An abnormal ketamine response in mutants defective in the ryanodine receptor gene ryr-1 (unc-68) of Caenorhabditis elegansJournal of Molecular Biology 267:849–864.https://doi.org/10.1006/jmbi.1997.0910
-
Essential Roles of Intracellular Calcium Release Channels in Muscle, Brain, Metabolism, and AgingCurrent Molecular Pharmacology 8:206–222.https://doi.org/10.2174/1874467208666150507105105
-
Calcium release channel RyR2 regulates insulin release and glucose homeostasisThe Journal of Clinical Investigation 125:4316.https://doi.org/10.1172/JCI84937
-
Intracellular calcium release channels: an updateThe Journal of Physiology 595:3041–3051.https://doi.org/10.1113/JP272781
-
BookThe Changing Landscape of American Life ExpectancyWashington, DC: The Hamilton Project.
-
A complex II defect affects mitochondrial structure, leading to ced-3- and ced-4-dependent apoptosis and agingThe Journal of Biological Chemistry 278:22031–22036.https://doi.org/10.1074/jbc.M211377200
-
Beneficial and Detrimental Effects of Reactive Oxygen Species on Lifespan: A Comprehensive Review of Comparative and Experimental StudiesFrontiers in Cell and Developmental Biology 9:628157.https://doi.org/10.3389/fcell.2021.628157
-
Role of FKBP12.6 in cADPR-induced activation of reconstituted ryanodine receptors from arterial smooth muscleAmerican Journal of Physiology. Heart and Circulatory Physiology 282:H1304–H1310.https://doi.org/10.1152/ajpheart.00843.2001
-
The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulumThe Journal of Biological Chemistry 268:22992–22999.
-
Imaging atrial arrhythmic intracellular calcium in intact heartJournal of Molecular and Cellular Cardiology 64:120–123.https://doi.org/10.1016/j.yjmcc.2013.09.003
-
The role of DMQ9 in the long-lived mutant clk-1Mechanisms of Ageing and Development 132:331–339.https://doi.org/10.1016/j.mad.2011.06.009
-
SESN-1 is a positive regulator of lifespan in Caenorhabditis elegansExperimental Gerontology 48:371–379.https://doi.org/10.1016/j.exger.2012.12.011
-
Functional role of Calstabin2 in age-related cardiac alterationsScientific Reports 4:7425.https://doi.org/10.1038/srep07425
-
Modulation of the ryanodine receptor and intracellular calciumAnnual Review of Biochemistry 76:367–385.https://doi.org/10.1146/annurev.biochem.76.053105.094237
Article and author information
Author details
Funding
National Heart, Lung, and Blood Institute (R01HL145473)
- Andrew Marks
National Institute of Diabetes and Digestive and Kidney Diseases (R01DK118240)
- Andrew Marks
National Heart, Lung, and Blood Institute (R01HL142903)
- Andrew Marks
National Heart, Lung, and Blood Institute (R01HL061503)
- Andrew Marks
National Heart, Lung, and Blood Institute (R01HL140934)
- Andrew Marks
National Institute of Arthritis and Musculoskeletal and Skin Diseases (R01AR070194)
- Andrew Marks
National Heart, Lung, and Blood Institute (T32HL120826)
- Andrew Marks
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
This work was supported by grants from the NIH to ARM (T32HL120826, R01HL145473, R01DK118240, R01HL142903, R01HL061503, R01HL140934, R01AR070194).
Copyright
© 2022, Dridi et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,096
- views
-
- 205
- downloads
-
- 11
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 11
- citations for umbrella DOI https://doi.org/10.7554/eLife.75529
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
Role of oxidation of excitation-contraction coupling machinery in age-dependent loss of muscle function in Caenorhabditis elegans
eLife 11:e75529.
https://doi.org/10.7554/eLife.75529




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}