On the role of nucleotides and lipids in the polymerization of the actin homolog MreB from a Gram-positive bacterium
- Université Paris-Saclay, INRAE, AgroParisTech, Micalis Institute, France
- Leibniz Institute of Polymer Research, and the Max-Bergmann-Center of Biomaterials, Germany
- Institute for Integrative Biology of the Cell (I2BC), Université Paris-Saclay, CEA, CNRS, France
- Laboratoire Physico Chimie Curie, Institut Curie, PSL Research University, CNRS UMR168, Sorbonne Université, 75005, France
Abstract
In vivo, bacterial actin MreB assembles into dynamic membrane-associated filamentous structures that exhibit circumferential motion around the cell. Current knowledge of MreB biochemical and polymerization properties in vitro remains limited and is mostly based on MreB proteins from Gram-negative species. In this study, we report the first observation of organized protofilaments by electron microscopy and the first 3D-structure of MreB from a Gram-positive bacterium. We show that Geobacillus stearothermophilus MreB forms straight pairs of protofilaments on lipid surfaces in the presence of ATP or GTP, but not in the presence of ADP, GDP or non-hydrolysable ATP analogs. We demonstrate that membrane anchoring is mediated by two spatially close short hydrophobic sequences while electrostatic interactions also contribute to lipid binding, and show that the population of membrane-bound protofilament doublets is in steady-state. In solution, protofilament doublets were not detected in any condition tested. Instead, MreB formed large sheets regardless of the bound nucleotide, albeit at a higher critical concentration. Altogether, our results indicate that both lipids and ATP are facilitators of MreB polymerization, and are consistent with a dual effect of ATP hydrolysis, in promoting both membrane binding and filaments assembly/disassembly.
Editor's evaluation
This important study makes the case that the assembly of MreB from Geobacillus, a Gram-positive organism differs substantially from MreB from the Gram-negative model organism, Escherichia coli. They make the compelling case that Geobacillus MreB assembly requires both interactions with membrane lipids and nucleotide binding: nucleotide hydrolysis is required for interaction with the membrane and interaction with lipids triggers polymerization. Altogether, these data make the strong case that MreB assembly dynamics can vary in significant, and organism-specific ways.
https://doi.org/10.7554/eLife.84505.sa0Introduction
Cytoskeletal proteins are known to polymerize into filaments that play critical roles in various aspects of cell physiology, including cell shape, mechanical strength and motion, cytokinesis, chromosome partitioning and intracellular transport. Prokaryotic cells contain homologs of the main eukaryotic cytoskeletal proteins, namely actin, tubulin and intermediate filaments (Cabeen and Jacobs-Wagner, 2010; Lin and Thanbichler, 2013; Shaevitz and Gitai, 2010), which were identified decades after their eukaryotic counterparts. In 2001, MreB proteins of the Gram-positive model bacterium Bacillus subtilis were found to form filamentous structures underneath the cytoplasmic membrane and to play a key role in the determination and maintenance of rod-shape (Carballido-Lopez, 2017; Jones et al., 2001). Soon after, the three-dimensional structure of one of the two MreB isoforms from the Gram-negative thermophilic bacterium Thermotoga maritima (MreBTm) was solved (van den Ent et al., 2001), confirming its structural homology with actin (Bork et al., 1992). Besides, MreBTm in solution was shown to assemble into filaments similar to filamentous actin (F-actin; van den Ent et al., 2001).
Research in the field of eukaryotic actin historically focused on elucidating structure-function relationships from in vitro studies. The availability of large amounts of soluble actin purified from several cell types since the 1940s enabled decades of mechanistic studies on actin polymerization (Pollard, 2016). In contrast, MreB from mesophilic bacteria proved particularly difficult to purify in a soluble form, thwarting efforts to perform in vitro assays. Instead, research on MreB primarily focused on cellular studies, driven by the advent of fluorescent microscopy in bacterial cell biology. Over the past two decades, the subcellular localization and dynamics of MreB have been described in several Gram-negative and Gram-positive species (Billaudeau et al., 2017; Billaudeau et al., 2019; Dion et al., 2019; Harris et al., 2014; Hussain et al., 2018; Olshausen et al., 2013; Oswald et al., 2016; Ouzounov et al., 2016; Renner et al., 2013; Schirner et al., 2015). In vivo, MreB proteins form discrete, nanometer-long, membrane-associated polymeric assemblies along the cell cylinder that move processively around the rod circumference together with proteins of the cell wall (CW) elongation machinery (Domínguez-Escobar et al., 2011; Garner et al., 2011; van Teeffelen et al., 2011), forming the so-called Rod complex. The motility of the Rod complex is driven by CW synthesis (Domínguez-Escobar et al., 2011; Garner et al., 2011; van Teeffelen et al., 2011) and MreB assemblies self-align circumferentially, along their direction of motion (Billaudeau et al., 2019; Hussain et al., 2018). Recently, it was proposed that the specific intrinsic curvature of MreB polymers increases their affinity for the greatest concave (negative) membrane curvature within the cell, that is the inner surface of the rod circumference, accounting for their orientation (Hussain et al., 2018). The current model is that self-aligned MreB filaments restrict the diffusion of CW biosynthetic proteins in the membrane and orient their motion to insert new peptidoglycan strands in radial hoops perpendicular to the long axis of the cell, promoting the cylindrical expansion of rod-shaped cells (Domínguez-Escobar et al., 2011; Garner et al., 2011; Hussain et al., 2018). However, many questions remain to be answered. What prompts the assembly of MreB on the inner leaflet of the cytoplasmic membrane? What is the architecture of the membrane-associated MreB polymeric assemblies and how is it controlled? How is their distribution along the cell cylinder regulated? What is the length of individual MreB filaments within these assemblies and how is it controlled? Are the filaments stable? Do they exhibit turnover like actin filaments? In vivo, the length of MreB filamentous assemblies can be affected by the intracellular concentration of the protein (Billaudeau et al., 2019; Salje et al., 2011), but seems to have little impact on MreB function (Billaudeau et al., 2019). No turnover of MreB assemblies was detected in vivo, at least relative to their motion around the cell circumference (Domínguez-Escobar et al., 2011; van Teeffelen et al., 2011). Therefore, MreB polymers are believed to be quite stable despite their motion in the cell. To elucidate in detail the molecular mechanisms underlying the functions of MreB, it remains necessary to understand their biochemical and polymerization properties. The majority of biochemical and structural studies on MreB proteins originally focused on the highly soluble Gram-negative MreBTm (Bean and Amann, 2008; Esue et al., 2005; Esue et al., 2006; Popp et al., 2010b; van den Ent et al., 2001; van den Ent et al., 2010). The tendency to aggregation upon purification hampered most in vitro studies of MreBs from other species. Over the last decade, purification and polymerization assays were nevertheless reported for MreBs from several Gram-negative bacteria, from the Gram-positive B. subtilis (MreBBs) and from wall-less Chlamydophila and Spiroplasma species (Dersch et al., 2020; Gaballah et al., 2011; Harne et al., 2020; Maeda et al., 2012; Mayer and Amann, 2009; Nurse and Marians, 2013; Pande et al., 2022; Salje et al., 2011; Takahashi et al., 2022; van den Ent et al., 2014).
Direct binding to the cell membrane was shown for MreB from the Gram-negative Escherichia coli (MreBEc) and T. maritima (Salje et al., 2011) and, more recently, for MreB from Spiroplasma citri (MreB5Sc; Harne et al., 2020). The N-terminal amphipathic helix of MreBEc was found to be necessary for membrane binding and also to cause the full-length purified protein to aggregate (Salje et al., 2011). Although this N-terminal amphipathic helix is dispensable for polymerization, it is required for proper function of MreBEc in vivo (Salje et al., 2011). MreBTm and MreB5Sc are devoid of such an N-terminal amphipathic helix, but instead possess a small hydrophobic loop that protrudes from the monomeric globular structure and mediates membrane binding (Pande et al., 2022; Salje et al., 2011). Additionally, an acidic C-terminal tail was shown to mediate a charge-based interaction of MreB5Sc with the membrane (Pande et al., 2022).
Altogether, in vitro work on MreBs from Gram-negative bacteria has shown that MreB polymerizes into straight double filaments in the presence of nucleotides, both in solution and on lipid membrane surfaces (Harne et al., 2020; Salje et al., 2011; Takahashi et al., 2022; van den Ent et al., 2014; van den Ent et al., 2010), and that filaments can assemble into larger sheets by lateral interactions (Esue et al., 2005; Esue et al., 2006; Harne et al., 2020; Nurse and Marians, 2013; Popp et al., 2010b; van den Ent et al., 2001; van den Ent et al., 2014). Furthermore, work on Caulobacter crescentus MreB (MreBCc) and MreBEc indicated an antiparallel arrangement of the straight pairs of protofilaments (van den Ent et al., 2014), in sharp contrast to the helical parallel pairs of protofilaments (double helix) characteristic of F-actin (Pollard, 1990). While the parallel arrangement of a protofilament doublet generates polarity and allows for the characteristic treadmilling of F-actin (Stoddard et al., 2017), the antiparallel arrangement in MreB protofilaments suggests a bidirectional polymerization/depolymerization mechanism (van den Ent et al., 2014). The directionality and the kinetics of MreB polymerization, as well as the role of nucleotides in this process remain to be shown. ATPase activity has been reported in solution for MreBTm, MreBEc, MreBBs and Spiroplasma MreBs (Esue et al., 2005; Esue et al., 2006; Mayer and Amann, 2009; Nurse and Marians, 2013; Pande et al., 2022; Popp et al., 2010b; Takahashi et al., 2022). However, the need for nucleotide binding and hydrolysis in polymerization remains unclear. Early reports suggested a strict dependency on hydrolysable nucleotides (ATP or GTP) for polymerization of MreBTm (Esue et al., 2006; van den Ent et al., 2001), and later for MreBEc (Nurse and Marians, 2013), while others claimed that polymerization occurred similarly in the presence of ADP (Bean and Amann, 2008; Gaballah et al., 2011; Mayer and Amann, 2009; Pande et al., 2022; Popp et al., 2010b; Takahashi et al., 2022), of the non-hydrolysable ATP analogue AMP-PNP (adenylyl-imidodiphosphate) (Bean and Amann, 2008; Mayer and Amann, 2009; Pande et al., 2022; Salje et al., 2011; Takahashi et al., 2022), or even in the absence of nucleotide (Mayer and Amann, 2009).
No electron microscopy (EM) images of protofilaments or atomic views of MreB from a Gram-positive bacterium have been reported to date. The two in vitro studies so far reported on B. subtilis MreB investigated polymerization by light scattering and sedimentation assays (Mayer and Amann, 2009) and by fluorescence microscopy (Dersch et al., 2020), respectively, but provided no evidence that MreBBs polymerized into protofilaments in the conditions tested. In Gram-positive bacteria, MreB proteins presumably have no N-terminal amphipathic helix (Salje et al., 2011), and the genome usually encodes several MreB isoforms (in contrast to Gram-negative that usually get by with a single mreB paralog), that may be related to their thicker and more complex CW structure (Chastanet and Carballido-Lopez, 2012). Inter- and intra-species differences in MreBs may exist at the structural or biochemical level, leading to differences in molecular interactions or biological functions.
In this study, we aimed to decipher fundamental structural and biochemical properties of MreB from a Gram-positive bacterium. We successfully purified a soluble form of MreB from the Gram-positive thermophilic Geobacillus stearothermophilus (MreBGs) and elucidated its crystal structure, confirming the classical actin/MreB fold and the presence of the small hydrophobic loop shown to mediate membrane binding in MreBTm and MreB5Sc (Pande et al., 2022; Salje et al., 2011). Polymerization assays showed that MreBGs forms straight pairs of protofilaments in the presence of lipids and nucleotide triphosphate (either ATP or GTP), and that these are dynamic. We also show that the interaction with lipids is mediated by electrostatic interactions (Pande et al., 2022) and by two spatially close hydrophobic motifs in the MreBGs monomers that comprise the small hydrophobic loop and the N-terminal end. Free in solution, MreBGs assembled into large sheets regardless of the bound nucleotide, albeit at a higher MreBGs concentration than the one required for polymerization into pairs of protofilaments on a lipid surface. Taken together, our results show a key role for ATP as facilitator of MreB polymerization on the membrane, and suggest that ATP hydrolysis promotes both MreB membrane binding and filament assembly/disassembly.
Results
Crystal structure of G. stearothermophilus MreB
To solve the structure of a MreB protein from a Gram-positive bacterium but overcome the notorious aggregation issues of MreB from mesophilic bacteria, we cloned and purified MreB from the thermophilic G. stearothermophilus (MreBGs). We chose G. stearothermophilus because of its proximity to the Bacillus genus and because of the highly conserved sequence of MreBGs compared to MreB from the model Gram-positive bacterium B. subtilis. MreBBs is more closely related to MreBGs (85.6% identity and 92.6% similarity) than to MreB of Gram-negative for which biochemical or structural data are available (either the thermophilic T. maritima with 55.8% identity, or the mesophilic C. crescentus, 56.9% identity and E. coli, 55.2% identity) (Figure 1—figure supplement 1).
MreBGs was purified to homogeneity following a two-step procedure (see Materials and methods). The protein could be purified in a soluble form (Figure 1—figure supplement 2) that remained functional for polymerization at concentrations below 13.4 µM (0.5 mg/mL). When stored frozen at higher concentrations or when conserved overnight at 4 °C, MreBGs rapidly aggregated (Figure 1—figure supplement 2A) and could not be recovered in a monomeric state, consistent with the known tendency of MreB proteins to aggregate.
The purified MreBGs protein was crystallized and the structure of its apo form was solved at 1.8 Å resolution (Protein Data Bank Identifier (PDB ID) 7ZPT). The crystals belong to the monoclinic P21 space group and contain one molecule of MreBGs per asymmetric unit (Supplementary file 1). Monomers of apo MreBGs display the canonical fold of actin-like proteins, characterized by four subdomains IA, IIA, IB and IIB (Figure 1A). One of the most similar structures to apo MreBGs is the apo form of MreBTm (PDB ID 1JCF; van den Ent et al., 2001), with a root mean square deviation (rmsd) of 1.92 Å over 305 superimposed Cα atoms and a Z-score of 16.0. Superimposition of the two proteins (Figure 1A) revealed that MreBGs is in a slightly more open conformation than MreBTm, mainly due to a movement of domain IB, which is the less conserved within the actin superfamily of proteins. Loop β6-α2, which connects subdomains IA and IB and closes the nucleotide-binding pocket, is partially disordered in apo MreBGs. In domain IA, the hydrophobic loop α2-β7, which has been shown to be involved in MreBTm membrane binding (Salje et al., 2011) and is 2 residues longer in MreBGs (Figure 1—figure supplement 1), displays a distinct conformation, packed on the N-terminal extremity of the polypeptide chain.
Figure 1 with 2 supplements see all
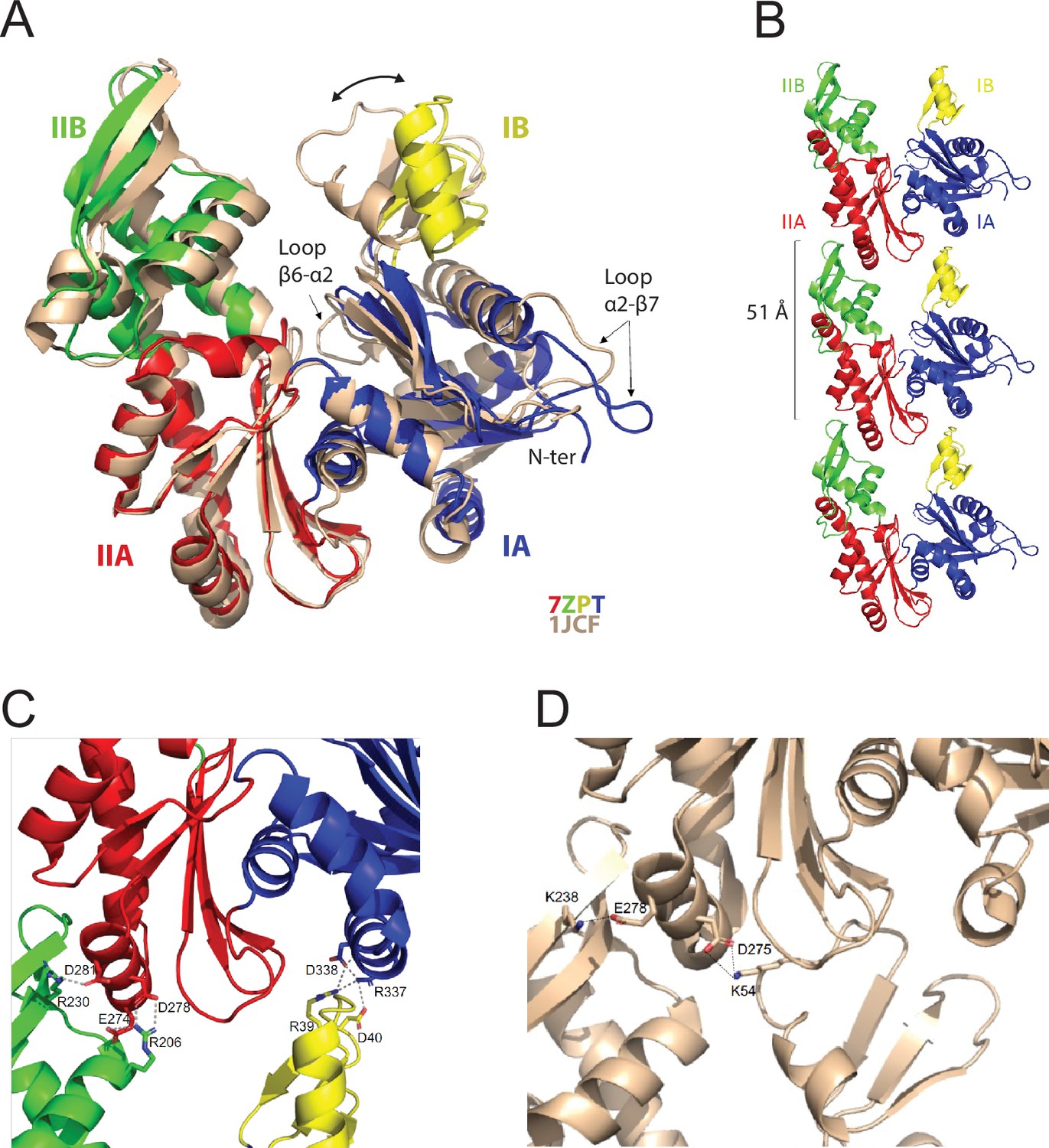
Crystal structure of the apo protofilament of MreB from G. stearothermophilus.
(A) Crystal structure of apo MreBGs (PDB ID 7ZPT), colored by subdomains, superimposed on the crystal structure of apo MreBTm (PDB ID 1JCF), in beige. The sequence similarity between the two proteins is 55.8%. Subdomain IA (blue) of MreBGs is formed by residues 1–32, 66–145 and 315–347; subdomain IB (yellow) by residues 33–65; IIA (red) by residues 146–181 and 246–314 and IIB (green) by residues 182–245. Superimposition of the two forms highlights the distinct positions of loops β6-α2 and α2-β7 as well as the movement of domain IB (two-headed arrow) resulting in slightly distinct subunit interaction modes as shown in panel C. (B) Protofilament structure of apo MreBGs. Three subunits of the protofilament formed upon crystal packing are displayed as cartoon and colored by subdomains. The subunit repeat distance is indicated. (C) Close view of the MreBGs intra-protofilament interface. The two subunits are colored by subdomains as in panel A, and shown as cartoons. Residues involved in putative salt bridges (gray dashed lines) are displayed as sticks colored by atom type (N in blue and O in red) and labeled. (D) Close view of the MreBTm intra-protofilament interface (PDB ID 1JCF). The two subunits are colored in beige as in panel A, and shown as cartoons. Residues involved in putative salt bridges (gray dashed lines) are displayed as sticks colored by atom type (N in blue and O in red) and labeled.
Crystal packing analysis revealed straight protofilaments characterized by a subunit repeat distance of 51 Å (Figure 1B), similar to that observed in crystal structures of other actin homologs (Harne et al., 2020; Pande et al., 2022; Roeben et al., 2006; van den Ent et al., 2014). However, because of the open conformation of MreBGs (Figure 1A), domain IB interacts with domain IA (Figure 1C) and not with domain IIA as observed for example for MreBTm (Figure 1D; van den Ent et al., 2001).
MreBGs polymerizes into straight pairs of protofilaments in the presence of lipids
We next investigated the polymerization of MreBGs by EM of negatively stained samples. In vivo, MreBBs forms membrane-associated nanofilaments (Billaudeau et al., 2019; Hussain et al., 2018; Jones et al., 2001), and MreB filaments from Gram-negative bacteria have been shown to have an intrinsic affinity for membranes (Garenne et al., 2020; Maeda et al., 2012; Salje et al., 2011; van den Ent et al., 2014). We hypothesized that the presence of lipids might be important for the assembly of MreBGs polymers, and thus performed polymerization reactions in the presence and in the absence of lipids. In the presence of ATP, MreBGs readily formed pairs of protofilaments on a monolayer of total E. coli lipid extract, while these were virtually not observed in the absence of lipids (Figure 2A). Using a semi-quantitative workflow analysis of TEM grids (Figure 2—figure supplement 1; Materials and methods), we found that in the presence of both ATP and lipids, MreBGs formed a lawn of double protofilaments in 100% of fields, while only 4% and 8% of the fields contained polymers (often at very low density) in the absence of either ATP or lipids, respectively (Figure 2A). To test if polymers had formed in solution but failed to bind to the hydrophobic EM grids, we instead used glow-discharged hydrophilic grids, which are commonly used to adsorb soluble proteins. Again, MreBGs filaments were not significantly detected in solution (Figure 2—figure supplement 2A). In the presence of lipids, the frequency of polymers was drastically reduced on the glow-discharged grids, consistent with impaired adhesion of a lipid monolayer to a hydrophilic surface (Figure 2—figure supplement 2A). We next hypothesized that the critical concentration for polymerization might be higher in the absence of lipids and thus raised the concentration of MreBGs in the reaction (Figure 2—figure supplement 2B). Again, virtually no pairs of protofilaments were detected in solution in these conditions. The only polymeric structures observed in the bulk solution, albeit very infrequently and only at high MreB concentration, were some large multilayered sheets forming ribbon-like structures among aggregates (Figure 2—figure supplement 2B and C). Taken together, these observations indicated that MreBGs polymerization is strongly enhanced by both ATP and lipids.
Figure 2 with 5 supplements see all
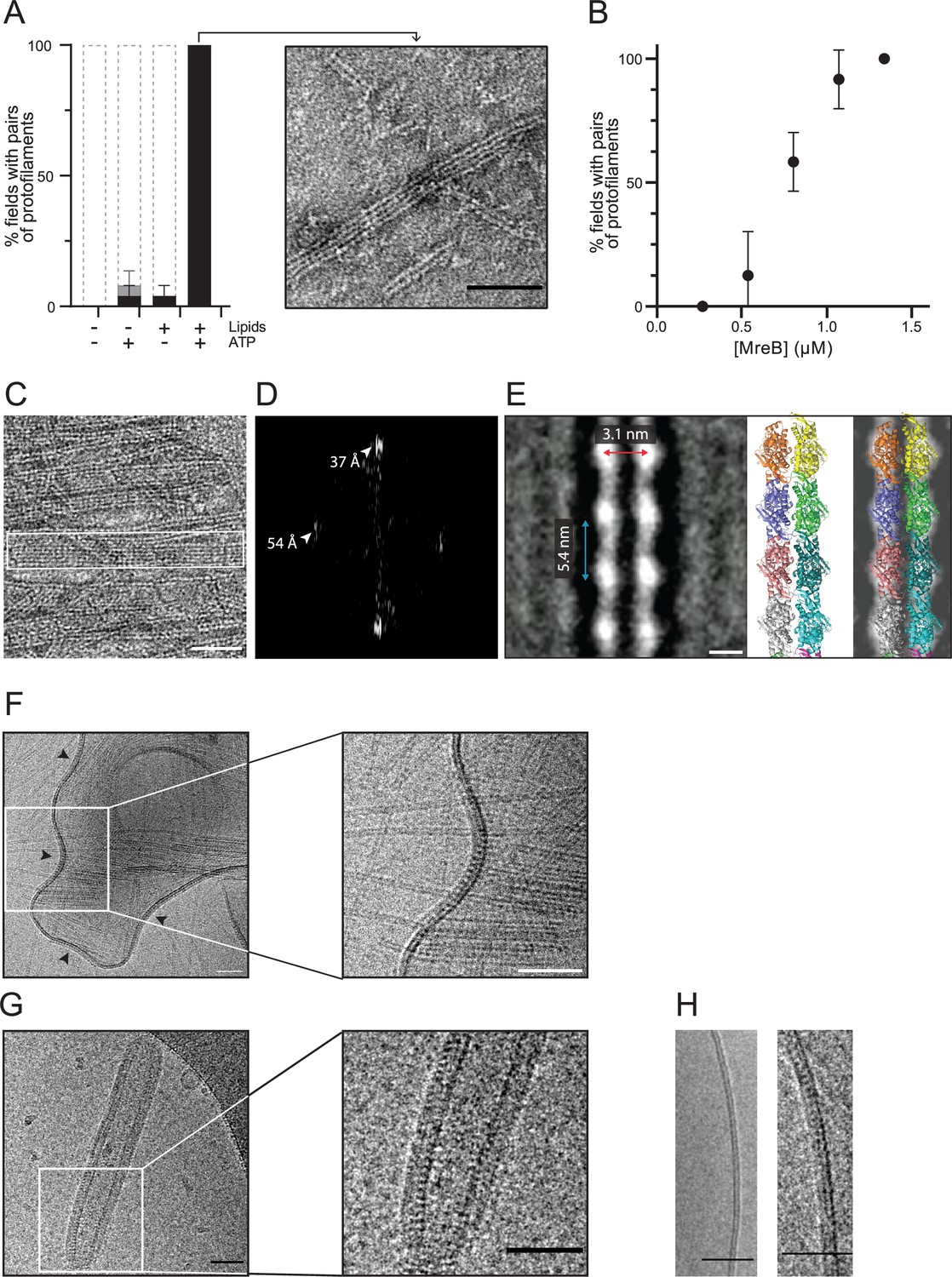
MreBGs forms double protofilaments in the presence of ATP and lipids.
(A) Polymerization of MreBGs into pairs of protofilaments depends on the presence of lipids and ATP. MreBGs was set to polymerize in standard conditions in the presence or absence of ATP and lipid total extract from E. coli. Polymer formation is expressed as percent of fields containing high (black) or low (grey) density of polymers (see Figure 2—figure supplement 1 for details of the quantification method of MreB polymers on TEM grids). Values are average of two independent experiments. Error bars are standard deviations. Inset shows an example of a field of dual protofilaments on a negative stained TEM image. Scale bar, 50 nm. (B) Polymer formation as a function of MreBGs concentration. MreBGs was set to polymerize in standard conditions at a concentration ranging from 0.27 to 1.34 µM (0.01–0.05 mg/mL). Values are the average of two independent experiments. Error bars are standard deviations. (C, D) MreBGs polymers assemble into sheets. (C) EM image of MreBGs set to polymerize in standard conditions. Scale bar, 50 nm. Fourier transform (D) was obtained from the area indicated by a white box in (C) and revealed a longitudinal subunit repeat of the filaments of 54 Å and a lateral spacing of ~37 Å (arrowheads). (E) (Left) 2D averaging of images of negatively stained dual protofilaments of MreBGs from 1 554 individual particles. Scale bar, 3 nm. Two copies of the atomic structure of the protofilaments found in the MreBGs crystals shown to scale (Middle, for illustration the two protofilaments are displayed arbitrarily in an antiparallel conformation but could fit in a parallel conformation as well) and docked into the 2D averaged EM image (Right). (F, G) MreBGs polymers assemble on lipid bilayers and distort liposomes as shown by cryo-electron microscopy (cryo-EM). Cryo-EM micrographs of liposomes (0.37 mg/mL) made from E. coli lipid total extracts incubated with purified MreBGs (1.34 µM; 0.05 mg/mL) in the presence of ATP (2 mM), and low (F, 100 mM) or high (G, 500 mM) concentration of KCl. Arrowheads point to MreB accumulations. Scale bars, 50 nm. (H). Cryo-EM micrographs showing the cross-section of the membrane of liposomes in the absence (Left) and in the presence (Right) of ATP-bound MreBGs at 500 mM KCl. Scale bars, 50 nm.
On a lipid monolayer, polymers were observed at a concentration of MreB above 0.55 µM (0.02 mg/mL), for a theoretical critical concentration of ~0.45 µM (Figure 2B), which is very similar to the critical concentration reported for ATP-MreBTm (Bean and Amann, 2008). The simplest and most abundant assemblies are paired protofilaments (Figure 2A, Figure 2—figure supplements 1 and 3), as previously observed for MreBTm and MreBCc assembled on lipid monolayers (Salje et al., 2011; van den Ent et al., 2014), and for Spiroplasma MreBs in solution (Pande et al., 2022; Takahashi et al., 2022). Pairs of MreBGs protofilaments are generally straight, and single protofilaments were never observed. Paired protofilaments of different lengths, ranging from below 50 nm up to several micrometers, as well as partial lateral association into two-dimensional sheets of dual protofilaments are often observed on the same EM grid (Figure 2A and C, and Figure 2—figure supplement 3). Importantly, pairs of filaments and sheets always lay flat, indicating that they are oriented relative to the membrane surface. The diffraction patterns of the sheets showed a longitudinal repeat of 54 Å and a lateral spacing of ~37 Å (Figure 2C and D). 2D averaging of negatively stained EM images of 1 554 individual pairs of filaments (Figure 2E and Figure 2—figure supplement 4) confirmed a longitudinal subunit repeat of 54 Å and refined the lateral subunit repeat to 31 Å, and could accommodate well two scaled protofilaments found in the MreBGs crystals (Figure 2E). However, it is not possible to derive the orientation of the two protofilaments (i.e. parallel or antiparallel) from the EM density obtained by 2D averaging.
Cations modulate distortion of liposomes by MreBGs filaments
MreBGs filaments also formed on lipid bilayers as observed by cryo-electron microscopy (cryo-EM). To this end, we prepared large unilamellar vesicles (LUVs) from E. coli lipid total extract, and incubated them with MreBGs and ATP. LUVs alone were spherical (Figure 2—figure supplement 5A), but vesicles decorated with MreBGs filaments appeared strongly deformed, confirming that MreBGs was bound to the membrane. At 100 mM KCl (our standard polymerization condition), LUVs displayed inward bending (negatively curved areas) where MreBGs filaments accumulated (arrows in Figure 2F and Figure 2—figure supplement 5B), as previously reported for MreBTm and MreBCc (Salje et al., 2011; van den Ent et al., 2014). 100 mM KCl is a salt concentration commonly used in polymerization studies of actin and actin-like proteins, including MreB (Deng et al., 2016; Garner et al., 2004; Polka et al., 2009; Rivera et al., 2011). Yet, while cytoplasmic K+ concentrations are around 50–250 mM in multicellular eukaryotic cells (Rodríguez-Navarro, 2000; Schmidt-Nielsen, 1975), they reach 200–300 mM in yeast (Ariño et al., 2010) and vary greatly depending on the osmolality of the medium in bacteria (Cayley et al., 1991; Epstein and Schultz, 1965; Rhoads et al., 1976). In B. subtilis, the basal intracellular concentration of KCl fluctuates between 100 mM and 800 mM (Eisenstadt, 1972; Whatmore et al., 1990). We therefore tested how higher salt concentrations affect the properties of MreB polymers. At 500 mM KCl, MreBGs readily polymerized into straight pairs of filaments as well, which also distorted liposomes but did not induce negative curvature. Instead, they faceted and tubulated the liposomes (Figure 2G and Figure 2—figure supplement 5C), suggesting that high salt concentration increases the stiffness of MreB filaments and/or of the membrane. Specific binding of cations at discrete sites along the filament has been shown to stiffen actin filaments, determining their bending rigidity (Kang et al., 2012). Our result suggests that physiological salt concentrations may also play a fundamental role in the mechanical properties of MreB filaments. MreBGs largely coated the liposomes and displayed a regular pattern along the cross-section of tubulated vesicles (Figure 2G–H). This view is compatible with longitudinal sections of 2D-sheets of straight filaments aligned in parallel to the longitudinal axis of the cylinder, as previously suggested for the arrangement of MreBTm in rigid lipid tubes (van den Ent et al., 2014).
ATP or GTP drive efficient formation of double filaments on a membrane surface
The role of nucleotide binding and hydrolysis in MreB polymerization remains unclear. In actin, ATP binding or hydrolysis are not required for polymerization (De La Cruz et al., 2000; Kasai et al., 1965). ATP hydrolysis only occurs subsequent to the polymerization reaction, destabilizing the filaments upon release of the γ-phosphate (Korn, 1982; Korn et al., 1987). In contrast, MreBTm was reported to require either ATP or GTP to polymerize (Esue et al., 2006; Nurse and Marians, 2013; van den Ent et al., 2001). MreB from E. coli, C. crescentus, S. citri, and Leptospira interrogans also formed polymers in the presence of ATP, but the requirement of ATP for polymerization was not established (Barkó et al., 2016; Harne et al., 2020; Maeda et al., 2012; Nurse and Marians, 2013; Salje et al., 2011; van den Ent et al., 2014). However, MreB filaments or sheets of filaments were also observed in the presence of ADP (Gaballah et al., 2011; Pande et al., 2022; Popp et al., 2010b; Takahashi et al., 2022) or AMP-PNP (Pande et al., 2022; Salje et al., 2011; Takahashi et al., 2022). These observations indicated that ATP binding and hydrolysis is not strictly required for filament formation in vitro. An analysis of nucleotide-bound crystal structures of MreBCc also suggested that ATP binding may trigger the transition to the double-protofilament conformation (Pande et al., 2022). Furthermore, liposome binding studies of MreB5Sc pointed to an allosteric effect of ATP binding and hydrolysis for effective polymerization and membrane binding (Pande et al., 2022).
We then wondered about the specificity of MreBGs toward nucleotides and their role in polymerization on a lipid membrane. MreBGs formed straight pairs of protofilaments and sheets in the presence of either ATP or GTP, as shown by negative stain EM (Figure 3A). Noteworthy, the average length of double filaments displayed an approximately twofold increase in the presence of GTP compared to ATP (Figure 3—figure supplement 1A). The significance of this observation is unclear at present but it may reflect differential affinity, dissociation rate or hydrolytic activity of the two nucleotide triphosphates (NTPs). Next, we asked whether formation of pairs of filaments required nucleotide hydrolysis and tested if nucleotides diphosphate or non-hydrolysable ATP analogues would also support polymer assembly. Virtually no double filaments were observed when ATP/GTP was replaced by ADP, GDP, AMP-PNP, or ApCpp (5′‐adenylyl methylenediphosphate), either in the presence or in the absence of lipids, in our standard polymerization conditions (Figure 3A and Figure 3—figure supplement 1B). However, differential affinity of MreBGs for these nucleotides, or a higher critical concentration for MreBGs to polymerize in their presence could also explain these results. Both actin (Cooke and Murdoch, 1973; Iyengar and Weber, 1964; Kinosian et al., 1993) and MreBCc (van den Ent et al., 2014) have the highest affinity for ATP, followed by ADP and then by AMP-PNP. Similarly, the critical concentration of actin polymerized with ADP is about 18-fold higher than with ATP (Fujiwara et al., 2007; Pollard, 1986), while the critical concentration of Thermotoga and Spiroplasma ADP-MreBs was reported to be approximately threefold and twofold higher, respectively, than that of ATP-MreBs (Bean and Amann, 2008; Takahashi et al., 2022). In contrast, the critical concentration of B. subtilis MreB deduced from light scattering experiments was 0.9 µM regardless of the nucleotide species bound, and even in the absence of nucleotide (Mayer and Amann, 2009). To exclude that the absence of polymerization was due to reduced nucleotide binding, we first increased the concentration of ADP and AMP-PNP from 2 mM to 50 mM. Again, no polymers were detected in the negatively stained samples (Figure 3—figure supplement 1C). Next, we performed a competition experiment by mixing ATP (1 mM) with increasing amounts of AMP-PNP (1, 10 and 25 mM) in the polymerization reaction. Increasing amounts of AMP-PNP efficiently decreased the presence of MreBGs filaments on the EM grids (Figure 3B). We concluded that AMP-PNP binds to MreBGs but does not support efficient polymerization on a lipid surface. Next, we tested if the critical concentration of MreBGs could be higher in the presence of ADP or AMP-PNP than in the presence of ATP. At the highest MreBGs concentration that we could test in our experimental conditions (6.98 µM), about 15-fold the estimated ATP-MreBGs critical concentration (0.45 µM, Figure 2B), efficient formation of double filaments on the lipid monolayer was still observed only in the presence of ATP (Figure 3—figure supplement 1D). Although still scarce, ADP-MreBGs double filaments were nevertheless more frequent at higher MreB concentration (Figure 3—figure supplement 1D), indicating a higher critical concentration required for filament formation.
Figure 3 with 3 supplements see all
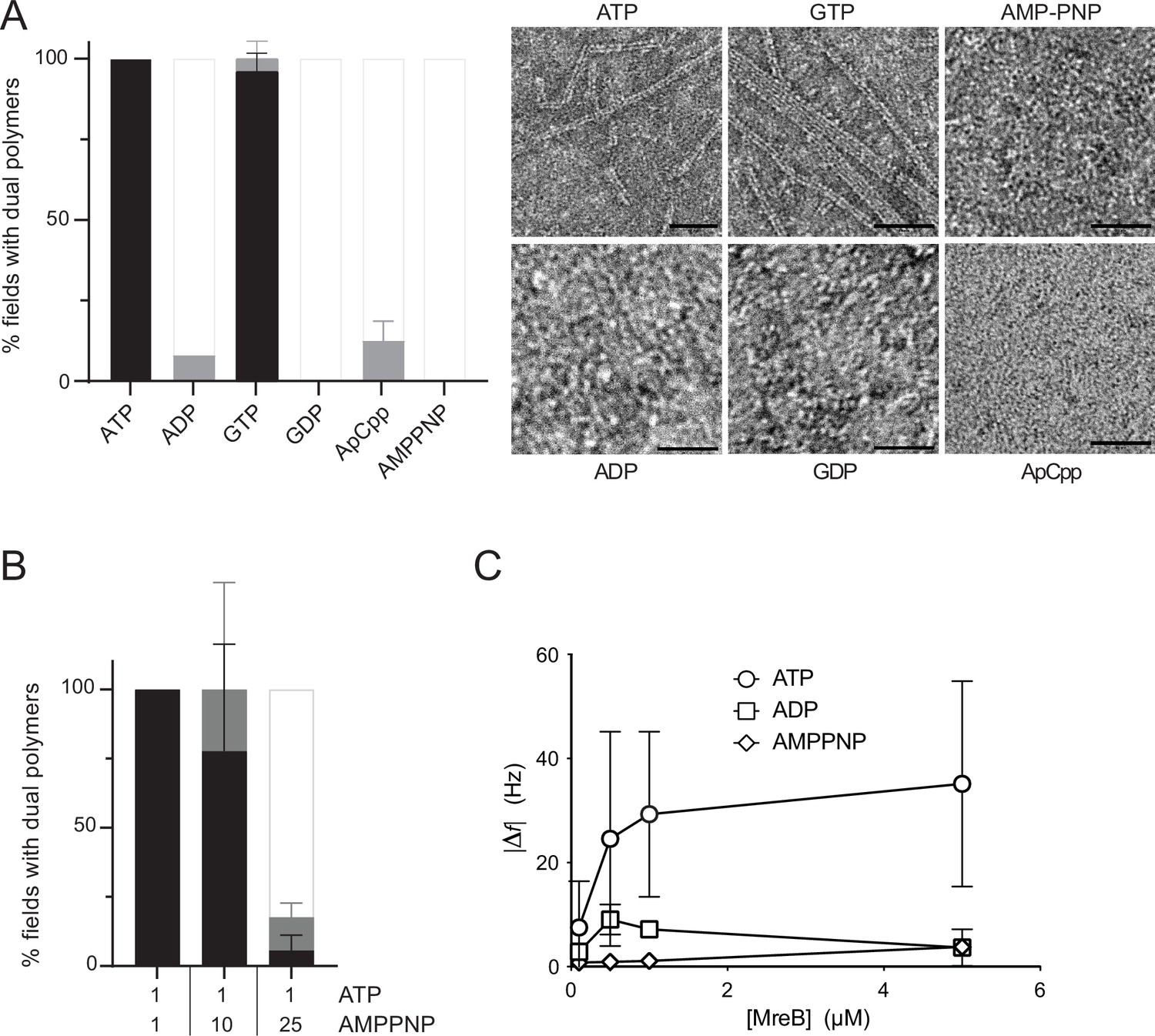
Double protofilaments of Geobacillus MreB efficiently form in the presence of hydrolysable nucleotides.
(A) ATP and GTP promote efficient assembly of MreBGs polymers on a lipid surface. MreB (1.34 µM; 0,05 mg/mL) was incubated in the presence of either ATP, ADP, GTP, GDP, or the non-hydrolysable AMP-PNP or ApCpp (2 mM), on a lipid monolayer. (Left) Quantification of MreBGs pairs of filaments. Polymer formation is expressed as percent of fields containing high (black), low (grey) density of polymers, or no polymers. Values are averages of at least two independent experiments. Error bars are standard deviations. (Right) representative TEM images. Scale bars, 50 nm. (B) AMP-PNP-MreBGs does not form double filaments on a lipid monolayer. AMP-PNP competes with ATP for binding to MreBGs, preventing polymerization. MreBGs was set to polymerize in standard conditions except that 2 mM ATP was replaced by a mix of ATP and AMP-PNP at the indicated concentrations (in mM). Polymer formation was quantified as in (A). Values are average of three independent experiments. Error bars are standard deviations. (C) Adsorption of MreBGs to a supported lipid bilayer (SLB) requires ATP. Frequency changes (|Δf|) in QCM-D experiments measured with varying amount (0.1–5 µM) of MreBGs on SLBs made of DOPC:DOPG 80:20 and in the presence of 2 mM of either ATP, ADP, or AMP-PNP. Values are an average of four independent experiments.
Taken together, these results show a noticeable dependence of MreBGs filaments on nucleotides and suggest that ATP/GTP binding and/or hydrolysis is required for efficient assembly of MreBGs into pairs of filaments on a membrane surface.
Nucleotide hydrolysis mediates binding of MreBGs to the membrane
Surprisingly, at high MreB concentrations, the large sheet- and ribbon-like structures observed to form in the bulk solution at very low frequency in the presence of ATP (Figure 2—figure supplement 2B and C) became frequent in the presence of ADP and very frequent in the presence of AMP-PNP (Figure 3—figure supplement 2A, B). The Fourier transforms of the sheet-like structures formed in these conditions showed sets of discrete reflections, with diffraction equatorial spots of about 6.5 nm (Figure 3—figure supplement 2C), larger than the repeats found on membrane-bound sheets, suggesting that membrane binding induces conformational changes in MreB filaments, as previously suggested (Shi et al., 2020). The presence of ADP- and AMP-PNP-MreBGs sheets in solution indicated that nucleotide hydrolysis is not strictly required for filament formation. However, the formation of extended sheets in solution concomitant with the absence of double protofilaments on a lipid surface in the presence of ADP or AMP-PNP, suggests that ATP hydrolysis promotes efficient filament formation at the membrane.
We then wondered whether ATP hydrolysis mediates MreBGs membrane binding or whether it triggers polymerization of membrane-bound monomers. To address this question, we turned to quartz crystal microbalance with dissipation monitoring (QCM-D) to measure the binding affinity of MreBGs to supported lipid bilayers (SLBs) of various lipid mixtures. QCM-D is a surface-sensitive technique that can be used to measure biomolecular interactions at aqueous interfaces in real time (Reviakine et al., 2011). Changes in frequency (Δf) and dissipation (ΔD) are recorded. The frequency is directly proportional to any mass added or removed (Sauerbrey, 1959), while dissipation changes are indicative of the viscoelastic properties of the attached layer. QCM-D was previously applied to study, for example, the binding affinity of the division proteins MinD and MinE of E. coli to SLBs (Renner and Weibel, 2012). E. coli and B. subtilis cytoplasmic membranes are mainly composed of phospholipids, with the negatively charged phosphatidylglycerol (PG) and the zwitterionic phosphatidylethanolamine (PE) being the dominant species (Bernat et al., 2016; Bishop et al., 1967; den Kamp et al., 1969; Laydevant et al., 2022; Nickels et al., 2017; Seydlová and Svobodová, 2008; Sohlenkamp and Geiger, 2016). Although lipid proportions vary widely depending on the strains and growth conditions, PE is largely dominant in E. coli while PG is more dominant in B. subtilis, indicating that phospholipids are more negatively charged in Gram-positive membranes. To mimic Bacillus membranes in our QCM-D assay, we used mixtures of the zwitterionic dioleoylphosphatidylcholine (DOPC) doped with the anionic dioleoylphosphatidylglycerol (DOPG) in various ratios (DOPC:DOPG 100:0, 90:10 or 80:20) to generate SLBs. DOPC was selected to replace PE because of its widespread role as a scaffold lipid in SLBs formation. We had to adopt a mixture that enabled us to form SLBs on planar substrates, as the inverted conical shape of PE makes the formation of planar SLBs difficult (PE tends to form non-bilayer structures because of its small headgroup). A typical SLBs signature experiment is shown in Figure 3—figure supplement 3A–B. Briefly, SLBs are formed after the adsorption of liposomes (Δf decrease, ΔD increase) onto activated silica surfaces. Once a critical surface concentration of liposomes is reached and the interactions between liposomes and the surface are suitable, the liposomes spontaneously rupture and coalesce into flat SLBs (Keller et al., 2000). After the formation of stable and flat SLBs (i.e. a stable baseline for frequency and dissipation; Figure 3—figure supplement 3A), we started to add MreBGs to the SLBs (Figure 3—figure supplement 3B, closed arrows). We recorded frequency and dissipation changes for the added MreBGs protein in varying concentrations on all SLBs. Binding was strongly dependent on ATP and was substantially affected by the lipid composition of SLBs (Figure 3C and Figure 3—figure supplement 3C and D). Increasing the levels of DOPG led to a higher amount of MreBGs binding, with DOPC:PG 80:20 giving the highest observed adsorption, suggesting that the presence of negatively charged lipids favors MreBGs binding to the membrane. In the presence of ATP, binding was detected almost instantaneously after adding MreBGs (Figure 3—figure supplement 3B, closed arrows) for all concentrations of MreB tested, either above or below the concentration in which polymers were observed by EM on a lipid monolayer (0.55 µM) (Figure 2B and Supplementary file 2). Interestingly, a shift in frequency was detected even at 0.1 µM MreB, well below the critical ATP-MreB concentration, suggesting that MreB monomers might bind too (Figure 3C and Figure 3—figure supplement 3B and E, F). The protein binding kinetics reached an equilibrium after approximately 5–10 min with a somewhat slower continued binding of additional MreBGs molecules (Figure 3—figure supplement 3B). Upon rinsing with the same buffer (Figure 3—figure supplement 3B, open arrows), MreBGs at low concentration (0.1 µM) was almost completely removed from the membrane while at higher concentration (1 µM) it remained stably absorbed. When replacing ATP with ADP or AMP-PNP, we were not able to detect any significant binding at either low or high concentrations of MreB, indicating a virtually complete loss of interaction (Figure 3C and Figure 3—figure supplement 3D and F). We further increased the concentration of ADP or AMP-PNP to exclude the possibility that the binding was simply affected by a decreased affinity of MreBGs for these nucleotides. Higher concentrations of ADP and AMP-PNP did not restore the binding of MreBGs to the SLBs (Figure 3—figure supplement 3G). Taken together, these results suggest that nucleotide hydrolysis drives MreBGs membrane binding. Because some binding was detected at 0.1 µM MreB in the presence of ATP but not of ADP and AMP-PNP, it is tempting to speculate that lipid binding might occur prior to polymerization into pairs of filaments on the membrane surface.
Finally, we used the Sauerbrey model (Sauerbrey, 1959) to calculate the average coverage and thickness of the layer of MreBGs attached to the SLB. The thickness of the MreB films ranged from 0.1 nm to approximately 4 nm on the SLBs with an 80:20 ratio of DOPC:DOPG, which corresponds to ~2.5% to 100% coverage assuming a monolayer filament thickness (Figure 3—figure supplement 3H and Materials and methods). These data suggest that MreBGs mainly form monolayers on the SLBs, with limited out-of-plane interactions (i.e. limited tendency to stack into multilayers), consistent with our EM observations of pairs of filaments and sheets lying flat on the lipid monolayer (Figure 2, Figure 3 and Figure 2—figure supplements 1 and 4) and with the pattern displayed by the filaments on cross-sections of vesicles (Figure 2G), and thus with the interaction of the membrane with a specific surface of the MreBGs filaments. Taken together, these observations suggest an oriented arrangement of MreBGs filaments on the membrane, with lateral interactions between filaments in the plane perpendicular to their membrane-binding surface.
The hydrophobic amino-terminus and α2-β7 loop of MreBGs are required for membrane anchoring alongside with electrostatic interactions
Membrane binding of MreBTm is mediated by a small loop containing two hydrophobic residues (L93 and F94), whereas binding of MreBEc and MreBCc is mediated by an amino-terminal extension (9 residues) predicted as an amphipathic helix, which is disordered in all crystal structures of MreBCc (Salje et al., 2011; van den Ent et al., 2014; Figure 1—figure supplement 1, green highlights). Albeit essential to MreB function in E. coli (Salje et al., 2011), this N-terminal extension is not required for polymerization in vitro (Salje et al., 2011; van den Ent et al., 2014). MreBBs was not predicted to carry an N-terminal amphipathic helix (Salje et al., 2011). A systematic search in a large panel of MreB proteins spanning over the entire bacterial kingdom revealed that N-terminal amphipathic helices are a conserved feature of the Proteobacteria phylum and most Gram-negative bacteria, but are absent in Firmicutes and Bacteroidetes species (Figure 4—figure supplement 1). Most Firmicutes, including Bacilli (MreBGs and MreBBs) and Clostridia, but with the notable exception of the wall-less Mollicutes, possess a shorter N-terminal sequence containing 4–7 hydrophobic amino-acids (Figure 1—figure supplement 1 and Figure 4—figure supplement 1). We noticed that in the crystal structure of the apo form of MreBGs this short hydrophobic N-terminal sequence is in close proximity to loop α2-β7 (Figure 1A), which in MreBTm carries the hydrophobic residues L93 and F94 involved in membrane binding (Salje et al., 2011). The α2-β7 loops of MreBBs and MreBGs contain additional hydrophobic residues (Figure 1—figure supplement 1), suggesting that they may also play a role in membrane interaction. Interestingly, analysis of the crystal structure of MreBGs also showed that a cluster of positively charged residues surrounds the protruding hydrophobic subdomain formed by the α2-β7 loop and the N-terminal sequence (Figure 4—figure supplement 2). Since our QCM-D data suggested that anionic lipids favor MreBGs binding to a lipid bilayer, we hypothesized that membrane binding may result from both ionic and hydrophobic interactions, with the positively charged residues that cluster around the membrane insertion hydrophobic region interacting with negatively charged lipids.
We then constructed and purified mutants deleted for either four hydrophobic residues of the α2-β7 loop (aa 95–98, GLFA), the N-terminal sequence (aa 2–7, FGIGTK), or both (Supplementary file 3). Folding of the protein was not affected by the deletions as shown by circular dichroism (CD) (Figure 4—figure supplement 3). To comparatively analyze membrane binding of these mutants, we first examined their polymerization at high-salt concentration (500 mM KCl). We reasoned that high salt would reduce the contribution of electrostatic interactions to membrane binding by screening the charges of the anionic lipids in the membrane, allowing to better assess the contribution of the short hydrophobic sequences to membrane anchoring. The three mutants and the wild-type MreBGs protein were set to polymerize in the presence of lipids and the formation of filaments was assessed by negative stain EM. The three mutants displayed a dramatic reduction in the formation of double filaments in the EM fields relative to the wild-type, with a gradation of defects, with the deletion of the GLFA sequence having the least effect and the double deletion having the greatest (Figure 4A). A similar gradient of phenotypes was observed at lower salt concentration (100 mM KCl) but the overall number of filaments detected on the membrane surface was increased as expected, consistent with electrostatic interactions also contributing to lipid binding (Figure 4—figure supplement 4A). Under the same polymerization conditions, no filaments were detected in solution for any of the mutants (Figure 4—figure supplement 4A), arguing against the possibility that the mutants could form filaments in solution but that they are impaired for membrane binding.
Figure 4 with 5 supplements see all
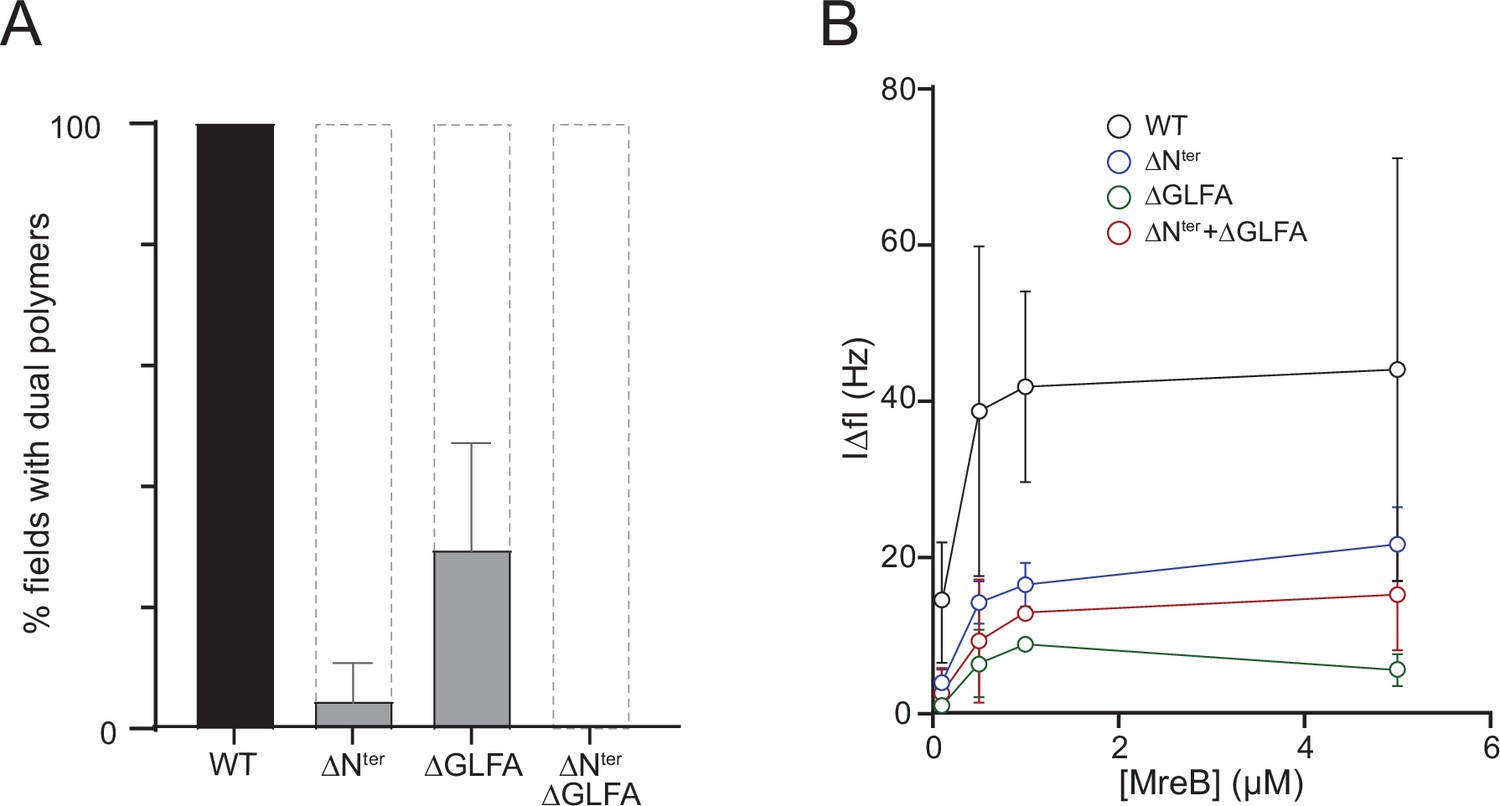
in theThe N-terminus and the α2β7 hydrophobic loop of MreBGs promote membrane binding and polymerization on a lipid surface.
(A) Both the hydrophobic α2-β7 loop and the N-terminus sequence of MreBGs are required for efficient polymerization on a lipid monolayer. Frequency and density of polymer formation in high salt (500 mM KCl) polymerization conditions, observed on negatively stained TEM images for the wild type (WT) and the mutants of the α2-β7 loop (ΔGLFA), the N-terminus (ΔNter) or both domains (ΔNter+ ΔGLFA) of MreBGs. Images were categorized based on the absence or the presence of low or high density of polymers. Values are the average of two independent experiments. Error bars are standard deviations. (B) The α2-β7 loop and the N-terminus domains of MreBGs enhance its adsorption to supported lipid bilayers. Frequency change (IΔfI) measured for the binding of various concentrations (0.1–5 µM) of purified wild-type (WT) and mutant forms of MreBGs to SLBs. Incubations were performed in polymerization buffer containing 500 mM KCl and 2 mM ATP. SLBs contained an 80:20 molecular ratio of DOPC:DOPG. Values are an average of at least two independent experiments.
In QCM-D experiments, membrane adsorption in the presence of ATP was strongly reduced in the three mutants relative to the wild-type protein, mirroring the polymerization assays at either high- or low-salt concentration (Figure 4B and Figure 4—figure supplement 4B). High salt reduced membrane binding relative to low salt in all conditions (Figure 4—figure supplement 4C). In the presence of ADP, binding was not observed for any of the mutants, as previously observed with the wild-type protein (Figure 4—figure supplement 4D). Taken together, these results show that the spatially close hydrophobic N-terminus and α2-β7 loop are membrane anchors of MreBGs filaments and further confirm that electrostatic interactions are also involved in lipid binding.
γ-phosphate dissociation after ATP/GTP hydrolysis by MreBGs is related to filament turnover
Our results suggest that MreBGs has an intrinsic affinity for lipids, with nucleotide hydrolysis involved in the switch of the protein from a soluble to a lipid-affine form, potentially through structural modifications. In order to test the impact of nucleotide binding, we co-crystallized MreBGs with ATP and solved the crystal structure of the complex at 2.3 Å resolution (PDB ID 8AZG). The crystals diffracted in space group P21212 (Supplementary file 1) with one molecule per asymmetric unit. The electron density map clearly revealed the presence of three phosphates, demonstrating that the ATP had not been hydrolyzed into ADP (Figure 4—figure supplement 5A). This is probably due to the absence of a Mg2+ ion in the catalytic site. No residual electron density peak could be interpreted as a Mg2+ ion with appropriate coordination. As a consequence, the binding mode of the nucleotide is distinct from other MreB structures, with the γ-phosphate located in the Mg2+ binding site (Figure 4—figure supplement 5B). The structure of the ATP-bound form of MreBGs is highly similar to the apo form, with a rmsd of 1.41 Å over 313 aligned Cα atoms (Figure 4—figure supplement 5C). However, ATP binding induced a small closure of the nucleotide-binding pocket, and loop β6-α2, which was disordered in the apo structure, is fully visible in the electron density map. The hydrophobic loop α2-β7 and the N-terminus also display an alternative conformation. Despite multiple co-crystallization trials, in the ATP-bound state the crystal packing only revealed monomers and never straight protofilaments as in the apo structure.
MreBs of several Gram-negative bacteria have been shown to slowly hydrolyze ATP in solution (Bean and Amann, 2008; Esue et al., 2005; Esue et al., 2006; Gaballah et al., 2011; Mayer and Amann, 2009; Nurse and Marians, 2013; Pande et al., 2022; Popp et al., 2010b). Our EM and QCM-D results suggested that ATP hydrolysis by MreBGs may be required for efficient membrane binding and polymerization, and thus that hydrolysis may occur in solution also. We monitored ATPase activity by measuring the release of inorganic phosphate (Pi) in the presence of ATP for a wide range of MreB concentrations, in the presence and in the absence of lipids. In our standard polymerization conditions, in the absence of lipids and at 53 °C (a temperature close to the optimal growth temperature of G. stearothermophilus), we observed a constant release of Pi over time, with an equilibrium rate of Pi dissociation of 0.198±0.008 Pi/min/MreB molecule (Figure 5A, Figure 5—figure supplement 1D). The rate was reduced approximately threefold, to 0.068±0.02 Pi/min/MreB molecule in the absence of lipids (Figure 5A, Figure 5—figure supplement 1D). Similar rates were observed when the incubations were performed at a higher KCl concentration (500 mM), with 0.158±0.003 and 0.081±0.004 Pi/min/MreB with and without lipids, respectively (Figure 5—figure supplement 1A-E). As expected, Pi release rates were decreased when incubations were performed at 37 °C instead of 53 °C, but the rate of Pi release remained comparatively higher in the presence of lipids (Figure 5—figure supplement 1A). These rates of Pi release in the presence of ATP (~1 Pi/MreB in 5 min at 53 °C) are comparable to those observed for MreBTm and MreBEc in vitro (Esue et al., 2005; Esue et al., 2006; Nurse and Marians, 2013), and also remarkably similar to the rate of the (very slow) dissociation of γ-phosphate after ATP hydrolysis within actin filaments, which has a half-time of ∼6 min (Carlier and Pantaloni, 1986). Interestingly, the release of Pi was constant over the length of our ATPase experiments (Figure 5—figure supplement 1B). However, similar density and lengths of negatively stained MreBGs polymers were observed over the EM grids for all incubation (polymerization) times tested, ranging from a few minutes to several hours. This suggests that after a fast polymerization step, either (i) filaments remain stable and release Pi constantly but very slowly, or (ii) Pi release reflects the turnover of the filaments. To test these hypotheses, we performed a series of semi-quantitative TEM experiments in the presence of a limiting concentration of ATP. We reasoned that if the population of filaments is in steady-state, polymerization should be unaffected at early time points but polymer density would decrease as ATP gets depleted and, conversely, if the polymers are static, they should not be affected over time. As shown in Figure 5B a similar density of double protofilaments was observed on the lipid monolayer at early time points at both saturating and limiting concentrations of ATP. However, polymers progressively disappeared over time in the presence of limiting concentrations of ATP, but not when ATP was in excess. These observations suggest that MreB polymerization on a lipid surface is a dynamic process, with steady-state polymerization/depolymerization rates.
Figure 5 with 1 supplement see all
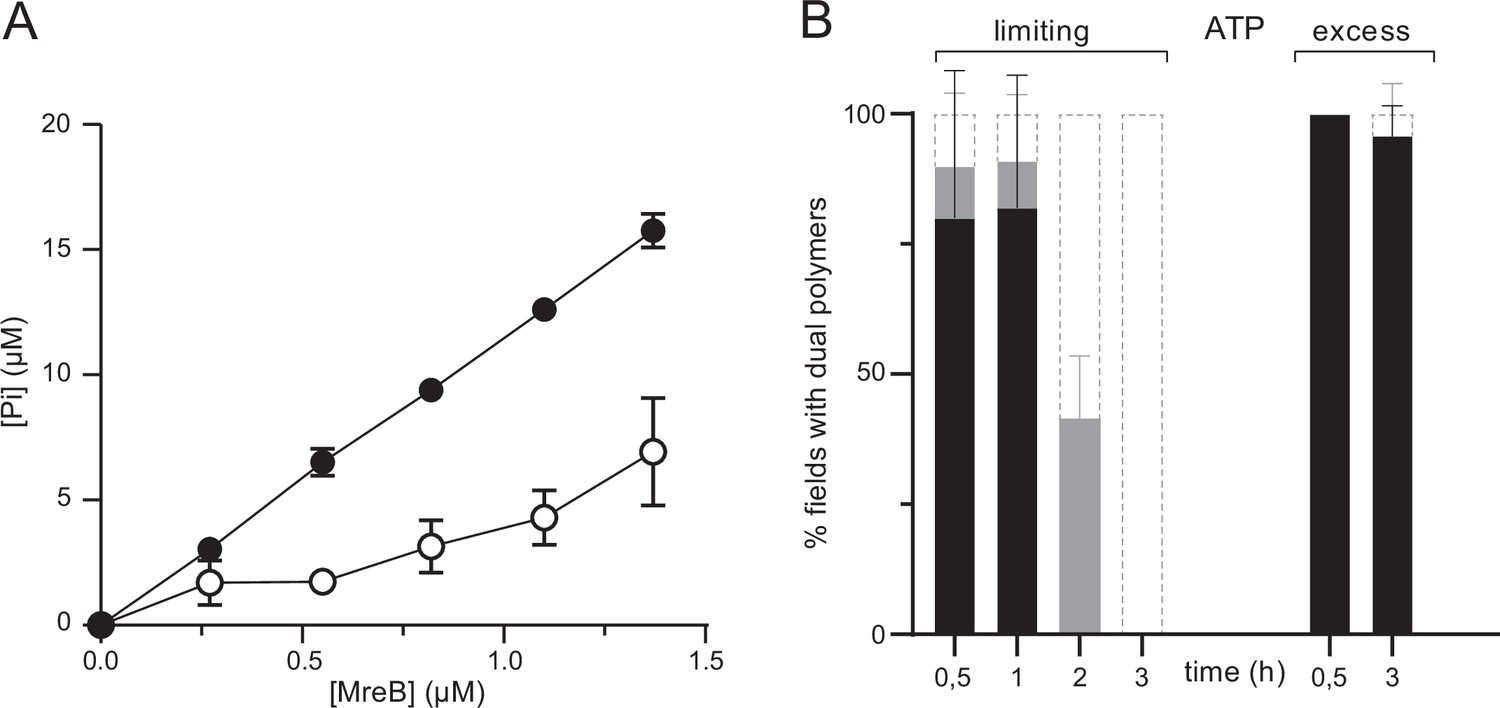
ATPase activity of MreBGs.
(A) The ATPase activity of MreBGs is stimulated in the presence of lipids. ATPase activity, measured by monitoring inorganic phosphate (Pi) release, of MreBGs at different concentrations (0.26–1.34 µM) in the presence of 0.5 mM ATP, 100 mM KCl, and in the presence (filled circles) or absence (empty circles) of 0.05 mg/mL liposomes, after 1 hr incubation at 53 °C. Values are averages of at least two independent experiments. Error bars are standard deviations. (B) Kinetics of MreBGs polymer formation in the presence of limiting ATP. MreBGs was set to polymerize in the presence of standard (2 mM; ‘excess’) or low (13 µM; ‘limiting’) concentration of ATP. Samples were prepared for TEM observation at different incubation times, up to 3 hr. Polymer formation is expressed as percent of fields containing high (black) or low (grey) density of polymers, or no polymers. Values are averages of two independent experiments. Error bars are standard deviations.
We have shown that MreBGs polymerizes into pairs of protofilaments in the presence of lipids and either ATP or GTP (Figure 3A and Figure 3—figure supplement 1A). MreBTm was also reported to polymerize in solution in the presence of ATP or GTP (Bean and Amann, 2008; Esue et al., 2006; Nurse and Marians, 2013; Popp et al., 2010a; van den Ent et al., 2001), and to release Pi at similar rates upon GTP and ATP hydrolysis (Esue et al., 2006). We found that MreBGs also releases Pi after hydrolysis of GTP as efficiently as after hydrolysis of ATP, both in the presence and in the absence of lipids (Figure 5—figure supplement 1C). Finally, the specific ATPase activity of the three mutants impaired for membrane binding and of the wild-type MreBGs reflected their polymerization and QCM-D assays at either high- or low-salt concentration (Figure 5—figure supplement 1D, E). In the absence of lipids, all proteins released Pi at comparative rates, suggesting that a basal level of ATP degradation occurs in solution. In the presence of lipids, the rate of Pi release of all mutants was reduced relative to the wild-type MreBGs, and overall Pi release rates were significantly reduced at high salt (500 mM KCl) relative to low salt (100 mM KCl) (Figure 5—figure supplement 1D–E). At 100 mM KCl, the mutant deleted for both the GLFA and the N-terminus hydrophobic sequences was the only one severely impacted in Pi release rate (Figure 5—figure supplement 1D), as it was in the formation of filaments on a lipid surface (Figure 4—figure supplement 4A). We concluded that the release of Pi by the wild-type and membrane-binding mutants in the presence of lipids was consistent with the density of double filaments observed in the EM fields.
Taken together, these results suggest that the presence of lipids is not required for the ATPase/GTPase activity of MreBGs. However, Pi release is enhanced in the presence of lipids, advocating for some conformational changes upon binding of MreBGs to the membrane and/or upon polymerization on the lipid surface. Furthermore, we show that pairs of filaments formed on a lipid surface are at steady-state, undergoing a dynamic, ATP-driven assembly/disassembly process.
Discussion
Here, we show that bacterial actin MreB from the Gram-positive bacterium G. stearothermophilus polymerizes into pairs of protofilaments on lipid surfaces and that this process is dynamic, with steady-state polymerization/depolymerization of the population of filaments. If individual double filaments are antiparallel (van den Ent et al., 2014), they are structurally nonpolar and thus their disassembly could result either from dynamic instability or from steady-state fluctuations (growing and shrinking) of the filaments ends. The requirement for a membrane is consistent with the observation that in vivo MreB polymeric assemblies are membrane-associated (i.e. localize at the cell periphery), in line with their role as scaffold of the CW elongation machinery. Membrane binding of MreBGs is direct and mediated by the hydrophobic α2-β7 loop protruding from the protein in domain IA, consistent with the prediction by Salje and colleagues that binding to membranes via such a hydrophobic loop and/or an amphipathic helix may be conserved for all MreBs (Salje et al., 2011). However, we found that MreBGs membrane binding is also mediated by the hydrophobic N-terminus, which together with the spatially closed α2-β7 loop would constitute a membrane anchor, as well as by electrostatic interactions, possibly through the positively charged residues around the membrane insertion hydrophobic region. The absence of an amphipathic helix and the presence instead of a hydrophobic N-terminus in many MreB sequences (Figure 4—figure supplement 1) suggest that most MreB use one or the other amino-terminal structure to bind to membranes.
Another important finding concerns the role of NTP in MreBGs membrane binding and formation of double protofilaments on a lipid surface. In the absence of lipids, MreBs from Gram-negative bacteria have been reported to assemble into sheets and bundles in the presence of either ATP or non-hydrolysable nucleotides (Barkó et al., 2016; Esue et al., 2005; Esue et al., 2006; Harne et al., 2020; Maeda et al., 2012; Nurse and Marians, 2013; Popp et al., 2010b; Salje et al., 2011; van den Ent et al., 2001), as we found here for MreBGs, indicating that membrane binding and nucleotide hydrolysis are not a prerequisite for polymerization. However, our data indicate that ATP binding and hydrolysis promote effective membrane binding and transition to the double-protofilament conformation, as recently suggested for Spiroplasma MreB5 (Pande et al., 2022). Efficient formation of double filaments on the lipid monolayer was not observed in the presence of ADP, even at the highest MreBGs concentration that we could test, about 15-fold the estimated ATP-MreBGs critical concentration. The critical concentration of actin polymerized with ADP is 18-fold higher than that of actin polymerized with ATP (Fujiwara et al., 2007; Pollard, 1986). This suggests that depolymerisation of the membrane-bound double filaments is stimulated by Pi release, as F-actin depolymerisation.
Taken together, our data suggest a model for ATP-driven polymerization of MreBGs on a lipid surface (Figure 6). Comparison of the crystal structures of apo and ATP-bound MreBGs shows that only minor conformational changes occur upon nucleotide-binding, in agreement with what was observed when comparing crystal structures of MreBCc and MreB5Sc in different nucleotide-bound states (Harne et al., 2020; Pande et al., 2022; van den Ent et al., 2014). This invariability of folding regardless of the bound ligands has also been observed in crystal structures of actin and other members of the actin superfamily (Schüler, 2001). ATP hydrolysis might promote small but dynamic structural changes that cannot be observed in crystal structures, which are locked in a conformation imposed by the packing. Different modes of membrane binding depending on the nucleotide state have indeed been suggested for MreB based on molecular dynamics simulations (Shi et al., 2020). It is tempting to speculate that MreB has limited affinity for lipids, and nucleotide hydrolysis may induce a conformational change that allows or stabilizes its interaction with the membrane. This is consistent with our findings that ATP is required for membrane interaction in QCM-D experiments and for detection of pairs of filaments on a lipid surface, and that at high concentration of MreB, ADP- and AMP-PNP-MreB sheets are frequent in solution while pairs of filaments found on the membrane are, respectively, scarce and virtually absent. Membrane interaction could occur by either a monomeric or an oligomeric form of MreB (Figure 6). It was previously speculated that the weak binding energy of individual membrane binding domains could be overcome by multimerization (Salje et al., 2011), which would favor the latter. Such oligomerization would nonetheless remain limited to very short filaments, as a nucleation step, since no filaments were detected in solution in these conditions. Binding to lipids would induce a second conformational change that triggers polymerization into double protofilaments (Figure 6). The double filament conformation might in turn favor Pi release, which would ultimately promote filament disassembly. In this scenario, the ADP-Pi-MreB intermediate would be the long-lived intermediate state within MreB filaments. The rate of Pi release from MreBGs is consistent with previous reports on MreBTm, MreBEc and MreB5Sc (Bean and Amann, 2008; Esue et al., 2005; Esue et al., 2006; Nurse and Marians, 2013) and is strikingly similar to that from F-actin. It will be interesting to measure the half-time of ATP hydrolysis by MreBGs and confirm if it is uncoupled and much faster than the half-time of Pi release, like in F-actin (~2 s and ~6 min, respectively; Pollard, 2016). In the absence of lipids, the decreased Pi release rate observed may reflect either a reduced Pi dissociation from soluble MreB-ADP-Pi or a reduced ATPase activity of the protein.
Figure 6
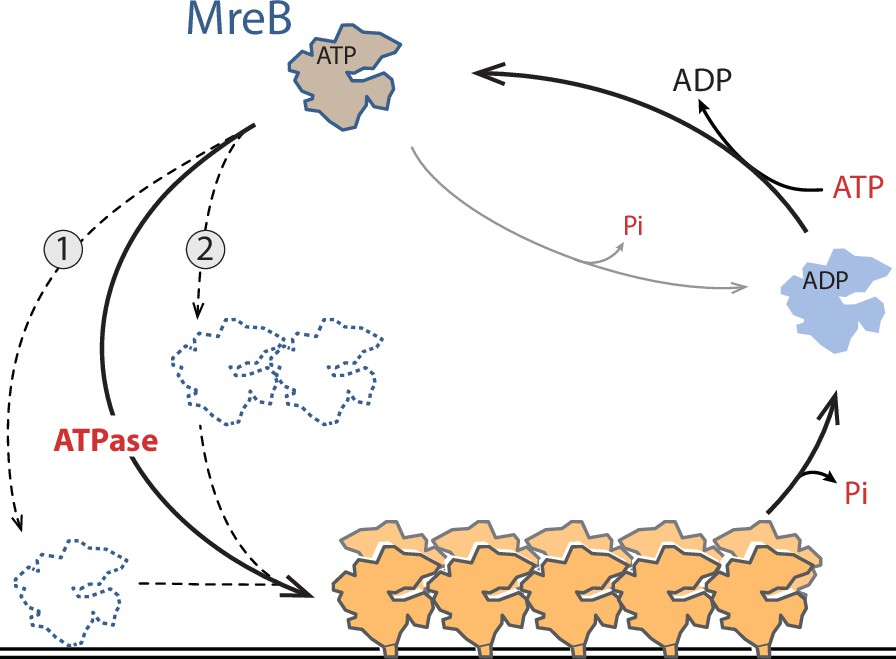
Model for ATP-driven MreBGs membrane binding and polymerization into pairs of filaments.
ATP hydrolysis stimulates MreBGs adsorption to lipids, possibly by promoting a conformational change that renders the hydrophobic α2-β7 loop and N-terminal protruding region prone for insertion into the membrane, either as a monomeric (1) or a nucleated (2) MreBGs form (dotted lines). Lipid binding would trigger formation of double-protofilaments on the lipid surface, which in turn would promote Pi release. Membrane-associated pairs of filaments may be mostly in the ADP-Pi form, and Pi release may destabilize them and ultimately promote disassembly.
Our cryo-EM data show that MreBGs filaments are straight and rigid at high KCl concentration, strongly deforming and faceting lipid vesicles, whereas at lower salt concentration, they bend the liposomes into negatively curved vesicles as previously reported for MreBTm and MreBCc (Salje et al., 2011; van den Ent et al., 2014; Figure 2F–G and Figure 2—figure supplement 5). A recent model postulates that MreB polymers are intrinsically curved and have a strong affinity for negatively curved membranes while avoiding to be positively bent (Hussain et al., 2018; Wong et al., 2019). The presence of a high concentration of KCl could stiffen MreB filaments, as shown to occur for actin filaments (Kang et al., 2012), and/or the lipid membrane, leading to a virtual disappearance of curved liposomes in vitro in these conditions. Because in bacterial cells the intracellular KCl concentration varies, ranging from 100 mM to 800 mM in B. subtilis (Eisenstadt, 1972; Whatmore et al., 1990), it questions the ability of MreB to induce or sense membrane curvature in vivo. In any case, the intrinsic affinity of MreB protofilaments for negative concave membrane curvature remains to be demonstrated. The identity of the bound nucleotide may also regulate the bending properties of MreB filaments, as it has been shown for actin filaments, with ADP-bound actin filaments being less stiff than ATP-bound filaments (Isambert et al., 1995). In agreement with this, molecular simulations suggest that MreB filaments exhibit nucleotide-dependent intersubunit bending, yet with straight, ADP-bound and curved, ATP-bound filaments (Colavin et al., 2014), opposite to actin. The kinetics of polymerization of MreB filaments, the apolarity of growth of presumably antiparallel doublets, and whether their disassembly results from dynamic instability or from growing and shrinking of the filaments at steady-state as well as the characterization of the polyelectrolyte properties of MreB filaments and their interactions with lipids are also important questions for future studies.
Materials and methods
General procedures and growth conditions
Request a detailed protocolDNA manipulations were carried out by standard methods. G. stearothermophilus was grown at 55 °C and E. coli at 37 °C in rich lysogeny broth (LB). Kanamycin was used at 25 µg/mL. All strains used in this study are listed in Supplementary file 4. All lipids, E. coli lipid Total Extract (TE), 1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC), and 1,2-dioleoyl-sn-glycero-3-phospho-(1´-rac-glycerol) (DOPG), were purchased from Avanti Polar Lipids Inc (Alabaster, AL, USA).
Cloning, expression and purification of MreB variants from G. stearothermophilus
Request a detailed protocolTwo mreB paralogs were identified in the genome of G. stearothermophilus ATCC 7953, corresponding to mreB and mbl of B. subtilis based on their synteny. The mreB ortholog displays a strong 92.6% similarity (85.6% overall identity) with mreB of B. subtilis (Figure 1—figure supplement 1). mreB from G. stearothermophilus ATCC 7953 was amplified by PCR using primers cc430 and cc431 (Supplementary file 5) and G. stearothermophilus growing cells as template. A second DNA fragment was generated by PCR on a derivative of plasmid pET28a (devoid of the first three codons following the NcoI restriction site), using primers cc433/cc432 (Supplementary file 5). The two resulting fragments were assembled by isothermal assembly and transformed into E. coli (Gibson et al., 2009). The resulting plasmid, pCC110, which carries a wild-type version of mreBGs in translational fusion with a 5’ extension encoding a 6-histidine tag, was used as a template to generate pCC116, pCC117 and pCC115, carrying the mreBGs gene deleted for codons 2–7 (FGIGTK), 102–105 (GLFA), or both, respectively. For this, pCC110 was PCR-amplified using primers cc582/cc583 (to generate pCC116) or cc584/cc585 (to generate pCC117) (Supplementary file 5) and the PCR products were treated with DpnI prior to transformation into E. coli. To generate pCC115, isothermal assembly was performed with two PCR products generated using primers cc582/cc585 and cc583/cc584 and pCC110 as template, and the product was transformed into E. coli. Following extraction and sequencing, the four resulting pCC plasmids were transformed into the T7 express E. coli expression host (Table. S4).
The his-tagged proteins were produced in T7 express E. coli cells grown in LB broth supplemented with kanamycin. Expression of recombinant MreB was induced by the addition of IPTG at the final concentration of 1 mM, when cultures reached an optical density at 600 nm of 0.6. Expression was performed over-night at 15 °C, with maximum aeration. Bacteria were harvested by centrifugation (5000 g for 7 min at 4 °C).
Pellets were resuspended in buffer A (20 mM Tris pH 7, 500 mM KCl) supplemented with EDTA-free complete protease inhibitor (Roche) and 250 µg/mL of lysozyme. Cells were disrupted by sonication on a Vibra-Cell VC505 processor (Sonics & Materials, Inc, Newton, CT, USA) for 10 min with 10 seconds on/off cycles at 50% power, and the supernatant was collected after clarification by centrifugation (40 000 g for 20 min at 4 °C). The 6-histidine-tagged MreB variants followed a two-step purification procedure. The proteins were first purified by affinity chromatography on a Ni-nitrilotriacetic acid (Ni-NTA) agarose resin (Thermo Fisher Scientific). The column was washed with buffer A supplemented with 20 mM imidazole, and proteins were eluted with a step gradient of imidazole (100 mM to 400 mM) in buffer A. The collected fractions were analyzed by electrophoresis, using a 12% polyacrylamide precast gel (Mini-PROTEAN TGX stain free, Bio-Rad). Fractions containing the purest form of the proteins were loaded on a size exclusion chromatography HiLoad 16/60 Superdex 200 pg column (GE Healthcare Life Sciences / Cytiva), pre-equilibrated with buffer B (buffer A supplemented with 1 mM DTT and 2 mM EDTA) connected to an AKTA FPLC system (GE Healthcare Life Sciences). Fractions corresponding to the elution peaks were analyzed by electrophoresis to assess the presence of MreB, pooled and concentrated with an ultrafiltration spin column (Vivaspin, 10 000 MWCO; Sartorius), up to a maximum of 0.5 mg/mL (13.4 µM), as determined from the absorption at 280 nm measured using a Nanodrop spectrophotometer (Thermo Fisher Scientific). The recombinant proteins were aliquoted and immediately frozen and stored at –20 °C.
Preparation of lipid monolayers and negative stain electron microscopy
Request a detailed protocolUnless stated otherwise, the standard reaction buffer supporting polymerization contained 20 mM Tris pH7, 100 mM KCl, 2 mM ATP, and 5 mM Mg2, in which MreB was set to polymerize for 1–2 h at 25–37°C (Supplementary file 2). Polymerization of MreB on lipids was induced by creating a lipid monolayer on droplets containing MreB (typically 1.34 µM (0.05 mg/mL)) in the reaction buffer. Lipids from E. coli TE were dissolved to 2 mg/mL in chloroform in a glass vial and stored at –20 °C. Lipid preparations were diluted in chloroform to a final concentration of 0.5 mg/mL on the day of the experiment. Approximately 200 nL of lipid preparation were dropped on top of the droplets containing MreB in the reaction buffer, and incubated for polymerization.
For TEM observations, carbon-coated electron microscopy grids (CF300-Cu from Electron Microscopy Sciences or 1753-F from Ted Pella, Inc) were used without treatment to preserve their hydrophobic properties and allow an optimum adsorption of the lipid monolayer. To observe hydrophilic particles, grids were glow-discharged by a 30 s plasma treatment (PDC001, Harrick Plasma). Following polymerization, grids are placed, carbon side down, on top of lipid-coated reaction droplets and gently lifted after 2 min incubation. Grids were stained with either a solution of 2% uranyl formate or 1% uranyl acetate and air-dried prior to TEM observation. TEM images were acquired on a charge-coupled device camera (AMT) on a Hitachi HT 7700 electron microscope operated at 80kV (Milexia – France) or a Tecnai G2 LaB6 (Thermo Fisher FEI) microscope operated at 200 kV or a Tecnai Spirit (Thermo Fisher FEI) microscope operated at 80 kV.
Fourier Transformation of MreB sheets was done using ImageJ to obtain diffraction patterns. For 2D processing, a set of images was collected at a magnification of 50000 with a pixel size of 2.13 Å per pixel and a defocus varying from –2 to –1 µm, using a Tecnai G2 LaB6 (Thermo Fisher FEI) microscope operated at 200 kV and a F-416 TVIPS 4K4K camera. To obtain 2D class averages, particles were classified and aligned, using SPIDER (Frank et al., 1996). A total of 1554 particles were windowed out into 99x99 pixels images by using the Boxer interface of EMAN (Ludtke et al., 1999) and appended into a single SPIDER file, then normalized against the background. One round of reference-free alignment and classification was performed before references were selected from the first-class averages. Several rounds of multireference alignment and classification were then performed, and new references were selected from the class averages until no further improvement was obtained.
Quantification of MreB filaments on EM images
Request a detailed protocolWe set up a workflow to quantitatively compare the ability of MreB to form polymers in different conditions from EM images. To circumvent the issue of the highly heterogeneous distribution of polymers on the EM grids, which could bias an analysis based on visual inspection, we acquired, for each experimental replica, images on 12 random locations covering the entire grid (Figure 2—figure supplement 1). For each field, the density of pairs of protofilaments observed was scored (none, ‘-’; low density, ‘+’ or high density, ‘++’).
Preparation of liposomes and cryo-electron microscopy
Request a detailed protocolE. coli TE was dissolved in chloroform, aliquoted, dried under a stream of argon, and desiccated for 1 h under vacuum. The liposome solution was made by resuspending desiccated TE in polymerization buffer (20 mM Tris-HCl pH 7, 100 mM or 500 mM KCl, 2 mM ATP, 5 mM MgCl2) on the day of the experiment, to a final lipid concentration of 0.1 mg/mL. 1.34 µM (0.05 mg/mL) of purified MreB was mixed with the liposome solution and incubated 2 hr at room temperature. Four μL of sample were applied to a glow-discharged holey lacey carbon-coated cryo-electron microscopy grids (Ted Pella, USA). Most of the solution was blotted away from the grid to leave a thin (<100 nm) film of aqueous solution. The blotting was carried out on the opposite side from the liquid drop and plunge-frozen in liquid ethane at −181 °C using an automated freeze plunging apparatus (EMGP, Leica, Germany). The samples were kept in liquid nitrogen and imaged using a Tecnai G2 (FEI, Eindhoven, Netherlands) microscope operated at 200 kV and equipped with a 4k4k CMOS camera (F416, TVIPS), or a Glacios 200kV (Thermo Fisher) microscope equipped with a Falcon IV direct detector. The imaging was performed at a magnification of 50 000 with a pixel size of 2.13 Å using a total dose of 10 electrons per Å2.
Preparation of liposomes and QCM-D measurements
Request a detailed protocolDOPC and DOPG lipid mixtures were prepared in chloroform as described above except that desiccation was performed under vacuum overnight. The lipids were rehydrated in 10 mM Tris pH 7.0, 100 mM NaCl, 5 mM MgCl2 buffer at a final concentration of 5 mg/mL using three consecutive cycles of freezing in liquid nitrogen and thawing in an ultrasonic bath (Merck). The rehydrated lipid solutions were extruded 21 times through a 100 nm diameter pore size polycarbonate membrane (Avanti Polar Lipids Inc). The extruded solutions were stored at 4 °C and consumed within a week after preparation.
A QCM-D E4 (QSense AB, Biolin Scientific AB, Gothenburg, Sweden) was used to measure MreB binding to planar supported lipid bilayers (SLBs) as previously reported for MinD and MinE (Renner and Weibel, 2012). Briefly, during QCM-D measurements, frequency and dissipation changes are recorded based on the piezoelectric properties of the crystal probe (Rodahl et al., 1995). The quartz crystals (QSense AB, Biolin Scientific AB, Gothenburg, Sweden) were coated with a custom 50 nm-thick layer of silicon dioxide by chemical vapor deposition (GeSiM GmbH, Dresden, Germany). Prior to each measurement, quartz crystals were thoroughly cleaned in a 1:1:5 volumetric ratio of concentrated ammonium hydroxide (Sigma-Aldrich), 30% hydrogen peroxide (Sigma Aldrich), and ultrapure water (Merck) at 70 °C for 3 min. Prior to liposome deposition, the quartz crystals were then placed and oxidized in a plasma cleaner (Harrick Plasma, Ithaca, NY) for 2 min at high radio frequency. The oxidized (activated) crystals were placed into the QCM-D measurement chambers and immediately covered with 10 mM Tris buffer, 100 mM NaCl and 5 mM MgCl2. Subsequently, after a stable baseline was established, a liposome working solution (0.2 mg/mL) was pumped into the measurement chambers at 200 µL/min. After 2–20 min of incubation, a characteristic profile of supported planar lipid bilayer formation was observed (Figure 3—figure supplement 3A; Keller et al., 2000). After 5 min, the SLBs were rinsed with 10 mM Tris buffer containing 100 mM NaCl and 5 mM MgCl2 to remove unbound vesicles at 100 µL/min. The buffer was next exchanged to the reaction buffer (20 mM Tris-HCl pH 7, 100 mM or 500 mM KCl, 1 mM DTT and 2 mM EDTA). After a stable baseline was observed, MreB (ATP, ADP, AMP-PNP) in reaction buffer (20 mM Tris-HCl pH 7, 100 mM or 500 mM KCl, 1 mM DTT, 5 mM MgCl2) was added at 0.1, 0.5, 1 and 5 µM (low to high concentration) to the SLB at a pump speed of 100 µL/min for 5 min. The adsorption of MreB wild-type and mutants was measured for at least 20 min, until a plateau was approximately reached, before exchanging and rinsing with reaction buffer for 5 min at 100 µL/min. In a series of experiments (from low to high MreB concentration), MreB was almost completely displaced by the rinsing step, allowing multiple adsorption steps on a single SLB. However, at higher MreB concentrations the rinsing was only partially effective (Figure 3—figure supplement 3B). To avoid history effects on a SLB, we also reversed the MreB concentration steps (from high to low concentration). We calculated the thickness from frequency shifts using the Sauerbrey model included in the commercial analysis software tool QTools (QSense AB, Biolin Scientific AB, Gothenburg, Sweden). Each measurement was repeated at least twice with 2–3 repeats.
Circular dichroism
Request a detailed protocolThe secondary structure of recombinant WT and mutant forms of MreB were analyzed by circular dichroism (CD). Far-UV spectra were recorded on a J-810 spectropolarimeter (Jasco). Spectra were recorded from 260 to 200 nm at 20 °C in 1 mm path-length quartz cuvette at a protein concentration of 10 μM in 50 mM NaPO4 buffer at pH 7. Each CD spectrum was obtained by averaging 4 scans collected at a scan rate of 200 nm/min. Baseline spectra obtained with buffer were subtracted for all spectra.
NTPase activity assay
Request a detailed protocolATPase and GTPase activity of MreB were assayed by measuring the release of free inorganic phosphate (Pi) in a colorimetric assay using malachite green (Kodama et al., 1986; Mao et al., 2017). Pi produced was measured after a fix (end-point) or various (kinetic) incubation times in the reaction buffer (20 mM Tris, 100 mM or 500 mM KCl, 5 mM MgCl2) with appropriate supplements (e.g. 0.5 mM ATP or GTP, 0.05 mg/mL liposomes). The liposome solution was made on the day of the experiment by resuspending desiccated TE in water to 1 mg/mL. The reaction was initiated by the addition of MreB to the reaction mixture and ended by addition of 1 reaction volume of malachite revelation buffer (0.2% (w/v) ammonium molybdate, 0.7 M HCl, 0.03% (w/v) malachite green, 0.05% (v/v) Triton X-100). Incubations were performed at 53°C and 37°C for 1 h (end point) or less (kinetics). The quantity of Pi produced was determined by measuring the absorbance at 650 nm on a 96-well plate spectrophotometer (Synergy 2, Biotek). A mock reaction devoid of protein constituted the blank. A standard curve was made with a range of KH2PO4 diluted in the reaction buffer.
Crystallization, structure determination and refinement
Request a detailed protocolFreshly purified MreBGs containing an N-terminal 6xHis-tag (stored in 20 mM Tris pH7, 500 mM KCl, 2 mM EDTA, 1 mM DTT) was concentrated by centrifugation using a Vivaspin 5000 MWCO membrane tube (Sartorius). All crystallization assays were performed at 293 K by sitting-drop vapor diffusion using facilities from the crystallization platform of I2BC. Crystals of apo MreBGs were obtained from a 100:100 nL mixture of protein at 3 mg/mL (80 µM) with a crystallization solution composed of 33% polyethylene glycol (PEG) 300 in 0.1 M MES pH 6.7. For co-crystallization assays, 10 mM ATP-Mg was added to 6 mg/mL (160 µM) of protein. Crystals of the complex were obtained with a crystallization solution containing 16% PEG 8000, 20% Glycerol and 0.04 M potassium phosphate. All crystals were flash-frozen in liquid nitrogen before data collection. Diffraction-quality crystals attained their full sizes in roughly 10–14 days.
Diffraction data were recorded on beam line PROXIMA 1 (synchrotron SOLEIL, France) at a wavelength of 0.9786 Å. Data were processed with the XDS package (Kabsch, 2010). All structures were solved by molecular replacement using the MOLREP program (Vagin and Teplyakov, 1997) using the crystal structure of MreBCc (PDB ID 4CZJ; van den Ent et al., 2014), and the models were refined using PHENIX (Liebschner et al., 2019). The models were further improved by iterative cycles of manual rebuilding using COOT (Emsley et al., 2010). Final structural models were deposited in the Protein Data Bank (PDB; Berman et al., 2000). Statistics for all the data collections, refinement of the different structures and the PDB codes are summarized in Supplementary file 1. All structural figures were generated with PyMOL (The PyMOL Molecular Graphics System, version 1.2r3pre, Schrödinger, LLC n.d.). Protein structure comparison was performed using the PDBeFold service at European Bioinformatics Institute (http://www.ebi.ac.uk/msd-srv/ssm) (Krissinel and Henrick, 2004). Protein interfaces, surfaces and assemblies were analyzed using the PDBePISA service at European Bioinformatics Institute (http://www.ebi.ac.uk/pdbe/prot_int/pistart.html) (Krissinel and Henrick, 2007).
Data availability
Protein structures data have been deposited in PDB under the accession codes 7ZPT and 8AZG.
-
RCSB Protein Data BankID 7ZPT. Crystal structure of MreB from Geobacillus stearothermophilus ATCC7953.
-
RCSB Protein Data BankID 8AZG. Crystal structure of MreB from Geobacillus stearothermophilus ATCC7953 in complex with ATP.
References
-
Alkali metal cation transport and homeostasis in yeastsMicrobiology and Molecular Biology Reviews 74:95–120.https://doi.org/10.1128/MMBR.00042-09
-
Large-scale purification and in vitro characterization of the assembly of MreB from Leptospira interrogansBiochimica et Biophysica Acta 1860:1942–1952.https://doi.org/10.1016/j.bbagen.2016.06.007
-
Lipid composition in A strain of Bacillus subtilis, A producer of iturin A lipopeptides that are active against uropathogenic bacteriaWorld Journal of Microbiology & Biotechnology 32:157.https://doi.org/10.1007/s11274-016-2126-0
-
Contrasting mechanisms of growth in two model rod-shaped bacteriaNature Communications 8:15370.https://doi.org/10.1038/ncomms15370
-
The chemical composition of the cytoplasmic membrane of Bacillus subtilisEuropean Journal of Biochemistry 2:448–453.https://doi.org/10.1111/j.1432-1033.1967.tb00158.x
-
The bacterial cytoskeletonAnnual Review of Genetics 44:365–392.https://doi.org/10.1146/annurev-genet-102108-134845
-
BookThe actin-like Mreb 'cytoskeletonIn: Graumann PL, editors. In: Bacillus: Cellular and Molecular Biology. Caister Academic Press. pp. 223–262.https://doi.org/10.21775/9781910190579
-
The actin-like MreB proteins in Bacillus subtilis: a new turnFrontiers in Bioscience 4:1582–1606.https://doi.org/10.2741/s354
-
Interaction of actin with analogs of adenosine triphosphateBiochemistry 12:3927–3932.https://doi.org/10.1021/bi00744a022
-
Polymerization and structure of nucleotide-free actin filamentsJournal of Molecular Biology 295:517–526.https://doi.org/10.1006/jmbi.1999.3390
-
Phospholipid composition of Bacillus subtilisJournal of Bacteriology 99:298–303.https://doi.org/10.1128/jb.99.1.298-303.1969
-
Potassium content during growth and sporulation in Bacillus subtilisJournal of Bacteriology 112:264–267.https://doi.org/10.1128/jb.112.1.264-267.1972
-
Features and development of CootActa Crystallographica. Section D, Biological Crystallography 66:486–501.https://doi.org/10.1107/S0907444910007493
-
Cation Transport in Escherichia coli: V. Regulation of cation contentThe Journal of General Physiology 49:221–234.https://doi.org/10.1085/jgp.49.2.221
-
The assembly of MreB, a prokaryotic homolog of actinThe Journal of Biological Chemistry 280:2628–2635.https://doi.org/10.1074/jbc.M410298200
-
SPIDER and WEB: processing and visualization of images in 3D electron microscopy and related fieldsJournal of Structural Biology 116:190–199.https://doi.org/10.1006/jsbi.1996.0030
-
Flexibility of actin filaments derived from thermal fluctuations: effect of bound nucleotide, phalloidin, and muscle regulatory proteinsThe Journal of Biological Chemistry 270:11437–11444.https://doi.org/10.1074/jbc.270.19.11437
-
The relative affinities of nucleotides to g-actin and their effectsBiochimica et Biophysica Acta 86:543–553.https://doi.org/10.1016/0304-4165(64)90094-7
-
XDSActa Crystallographica. Section D, Biological Crystallography 66:125–132.https://doi.org/10.1107/S0907444909047337
-
Polymerization of actin free from nucleotides and divalent cationsBiochimica et Biophysica Acta 94:494–503.https://doi.org/10.1016/0926-6585(65)90058-0
-
Formation of supported membranes from vesiclesPhysical Review Letters 84:5443–5446.https://doi.org/10.1103/PhysRevLett.84.5443
-
Nucleotide binding to actin: cation dependence of nucleotide dissociation and exchange ratesThe Journal of Biological Chemistry 268:8683–8691.
-
Actin polymerization and its regulation by proteins from nonmuscle cellsPhysiological Reviews 62:672–737.https://doi.org/10.1152/physrev.1982.62.2.672
-
Secondary-structure matching (SSM), a new tool for fast protein structure alignment in three dimensionsActa Crystallographica. Section D, Biological Crystallography 60:2256–2268.https://doi.org/10.1107/S0907444904026460
-
Inference of macromolecular assemblies from crystalline stateJournal of Molecular Biology 372:774–797.https://doi.org/10.1016/j.jmb.2007.05.022
-
Growth-phase dependence of bacterial membrane lipid profile and labeling for in-cell solid-state NMR applicationsBiochimica et Biophysica Acta. Biomembranes 1864:183819.https://doi.org/10.1016/j.bbamem.2021.183819
-
Macromolecular structure determination using X-rays, neutrons and electrons: recent developments in PhenixActa Crystallographica. Section D, Structural Biology 75:861–877.https://doi.org/10.1107/S2059798319011471
-
Nucleotide-independent cytoskeletal scaffolds in bacteriaCytoskeleton 70:409–423.https://doi.org/10.1002/cm.21126
-
EMAN: semiautomated software for high-resolution single-particle reconstructionsJournal of Structural Biology 128:82–97.https://doi.org/10.1006/jsbi.1999.4174
-
Assembly of MreB filaments on liposome membranes: a synthetic biology approachACS Synthetic Biology 1:53–59.https://doi.org/10.1021/sb200003v
-
Assembly properties of the Bacillus subtilis actin, MreBCell Motility and the Cytoskeleton 66:109–118.https://doi.org/10.1002/cm.20332
-
Bacillus subtilis lipid extract, a branched-chain fatty acid model membraneThe Journal of Physical Chemistry Letters 8:4214–4217.https://doi.org/10.1021/acs.jpclett.7b01877
-
Purification and characterization of Escherichia coli MreB proteinThe Journal of Biological Chemistry 288:3469–3475.https://doi.org/10.1074/jbc.M112.413708
-
MreB Orientation Correlates with Cell Diameter in Escherichia coliBiophysical Journal 111:1035–1043.https://doi.org/10.1016/j.bpj.2016.07.017
-
Filament organization of the bacterial actin MreB is dependent on the nucleotide stateJournal of Cell Biology 221:221.https://doi.org/10.1083/jcb.202106092
-
The structure and assembly dynamics of plasmid actin AlfA imply a novel mechanism of DNA segregationJournal of Bacteriology 191:6219–6230.https://doi.org/10.1128/JB.00676-09
-
Rate constants for the reactions of ATP- and ADP-actin with the ends of actin filamentsThe Journal of Cell Biology 103:2747–2754.https://doi.org/10.1083/jcb.103.6.2747
-
Actin and Actin-Binding ProteinsCold Spring Harbor Perspectives in Biology 8:a018226.https://doi.org/10.1101/cshperspect.a018226
-
Polymeric structures and dynamic properties of the bacterial actin AlfAJournal of Molecular Biology 397:1031–1041.https://doi.org/10.1016/j.jmb.2010.02.010
-
Filament structure, organization, and dynamics in MreB sheetsThe Journal of Biological Chemistry 285:15858–15865.https://doi.org/10.1074/jbc.M109.095901
-
MinD and MinE interact with anionic phospholipids and regulate division plane formation in Escherichia coliThe Journal of Biological Chemistry 287:38835–38844.https://doi.org/10.1074/jbc.M112.407817
-
Cation transport in Escherichia coli. VIII. Potassium transport mutantsThe Journal of General Physiology 67:325–341.https://doi.org/10.1085/jgp.67.3.325
-
Architecture and assembly of a divergent member of the ParM family of bacterial actin-like proteinsThe Journal of Biological Chemistry 286:14282–14290.https://doi.org/10.1074/jbc.M110.203828
-
Deciphering key features in protein structures with the new ENDscript serverNucleic Acids Research 42:W320–W324.https://doi.org/10.1093/nar/gku316
-
Quartz crystal microbalance setup for frequency and Q -factor measurements in gaseous and liquid environmentsReview of Scientific Instruments 66:3924–3930.https://doi.org/10.1063/1.1145396
-
Potassium transport in fungi and plantsBiochimica et Biophysica Acta 1469:1–30.https://doi.org/10.1016/s0304-4157(99)00013-1
-
Crystal structure of an archaeal actin homologJournal of Molecular Biology 358:145–156.https://doi.org/10.1016/j.jmb.2006.01.096
-
Direct membrane binding by bacterial actin MreBMolecular Cell 43:478–487.https://doi.org/10.1016/j.molcel.2011.07.008
-
Verwendung von schwingquarzen zur wagung dunner schichten und zur mikrowagungZeitschrift for physik 155:206–222.https://doi.org/10.1007/BF01337937
-
Lipid-linked cell wall precursors regulate membrane association of bacterial actin MreBNature Chemical Biology 11:38–45.https://doi.org/10.1038/nchembio.1689
-
Comparative physiology of cellular ion and volume regulationThe Journal of Experimental Zoology 194:207–219.https://doi.org/10.1002/jez.1401940114
-
ATPase activity and conformational changes in the regulation of actinBiochimica et Biophysica Acta 1549:137–147.https://doi.org/10.1016/s0167-4838(01)00255-2
-
Development of membrane lipids in the surfactin producer Bacillus subtilisFolia Microbiologica 53:303–307.https://doi.org/10.1007/s12223-008-0047-5
-
The structure and function of bacterial actin homologsCold Spring Harbor Perspectives in Biology 2:a000364.https://doi.org/10.1101/cshperspect.a000364
-
Bacterial membrane lipids: diversity in structures and pathwaysFEMS Microbiology Reviews 40:133–159.https://doi.org/10.1093/femsre/fuv008
-
Evolution of polymer formation within the actin superfamilyMolecular Biology of the Cell 28:2461–2469.https://doi.org/10.1091/mbc.E15-11-0778
-
MOLREP: an automated program for molecular replacementJournal of Applied Crystallography 30:1022–1025.https://doi.org/10.1107/S0021889897006766
-
Bacterial actin MreB assembles in complex with cell shape protein RodZThe EMBO Journal 29:1081–1090.https://doi.org/10.1038/emboj.2010.9
-
The effects of osmotic upshock on the intracellular solute pools of Bacillus subtilisJournal of General Microbiology 136:2527–2535.https://doi.org/10.1099/00221287-136-12-2527
Article and author information
Author details
Ines Li de la Sierra-Gallay

Rut Carballido-Lopez
Funding
European Research Council (ERC-SG)
- Rut Carballido-Lopez
European Research Council (ERC-CG)
- Rut Carballido-Lopez
Agence Nationale de la Recherche (ANR-11-LABX0038)
- Aurélie Bertin
Agence Nationale de la Recherche (ANR-10-IDEX-0001-02)
- Aurélie Bertin
VolkswagenStiftung
- Lars D Renner
Agence Nationale de la Recherche (ANR-10-INSB-05-05)
- Sylvie Nessler
European Research Council (311231)
- Rut Carballido-Lopez
European Research Council (772178)
- Rut Carballido-Lopez
The funders had no role in study design, data collection and interpretation, or the decision to submit the work for publication.
Acknowledgements
We thank Davy Martin for CD acquisitions, Human Rezai and Juan Hermoso for useful discussions, and Xavier Henry for useful contributions upstream this work. This project has received funding from the European Research Council (ERC) under the European Union’s Seventh Framework Program (FP7) and under the Horizon 2020 research and innovation program (grant agreement No 311231 and grant agreement No 772178, respectively, to RC-L). We also thank the Labex Cell(n)Scale (ANR-11-LABX0038), Paris Sciences et Lettres (ANR-10-IDEX-0001–02), and the Cell and Tissue Imaging (PICT-IBiSA), Institut Curie, member of the French National Research Infrastructure France-BioImaging (ANR-10-INBS-04). LDR acknowledges funding by the VolkswagenStiftung. This work benefited from the expertise of Christine Péchoux at the MIMA2 facility (Université Paris-Saclay, INRAE, AgroParisTech, GABI, 78350, Jouy-en-Josas, France) for TEM observations https://doi.org/10.15454/1.5572348210007727E12, and of the crystallization platform of I2BC, supported by the French Infrastructure for Integrated Structural Biology (FRISBI, ANR-10-INSB-05–05). We acknowledge the synchrotrons ESRF (Grenoble, France) and SOLEIL (Saint-Aubin, France) for provision of synchrotron radiation facilities and we would like to thank the staffs of beamlines PROXIMA-2A and PROXIMA-1 at SOLEIL, and 1D23-2 at ESRF for assistance and advices during data collection.
Copyright
© 2023, Mao et al.
This article is distributed under the terms of the Creative Commons Attribution License, which permits unrestricted use and redistribution provided that the original author and source are credited.
Metrics
-
- 1,950
- views
-
- 262
- downloads
-
- 12
- citations
Views, downloads and citations are aggregated across all versions of this paper published by eLife.
Citations by DOI
-
- 12
- citations for umbrella DOI https://doi.org/10.7554/eLife.84505
Download links
A two-part list of links to download the article, or parts of the article, in various formats.
Downloads (link to download the article as PDF)
Open citations (links to open the citations from this article in various online reference manager services)
Cite this article (links to download the citations from this article in formats compatible with various reference manager tools)
On the role of nucleotides and lipids in the polymerization of the actin homolog MreB from a Gram-positive bacterium
eLife 12:e84505.
https://doi.org/10.7554/eLife.84505




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}